

Abstract
Efficient removal of per‐ and polyfluoroalkyl substances (PFAS) from contaminated waters is urgently needed to safeguard public and environmental health. In this work, novel magnetic fluorinated polymer sorbents were designed to allow efficient capture of PFAS and fast magnetic recovery of the sorbed material. The new sorbent has superior PFAS removal efficiency compared with the commercially available activated carbon and ion‐exchange resins. The removal of the ammonium salt of hexafluoropropylene oxide dimer acid (GenX) reaches >99 % within 30 s, and the estimated sorption capacity was 219 mg g−1 based on the Langmuir model. Robust and efficient regeneration of the magnetic polymer sorbent was confirmed by the repeated sorption and desorption of GenX over four cycles. The sorption of multiple PFAS in two real contaminated water matrices at an environmentally relevant concentration (1 ppb) shows >95 % removal for the majority of PFAS tested in this study.
Keywords: Contaminated Water Remediation, Fluorinated Polymer Sorbent, Magnetic Separation, PFAS Removal
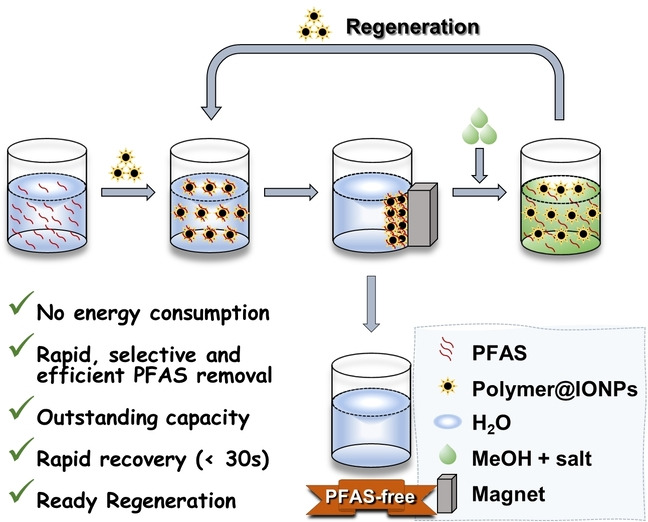
Magnetic polymer sorbents (polymer@IONPs) were prepared for rapid, selective and efficient removal of multiple per‐ and polyfluoroalkyl substances (PFAS) from real contaminated waters at environmentally relevant concentrations. Fast magnetic recovery of the sorbent within 30 s was achieved. Regeneration of the sorbent for repeated use was demonstrated over four cycles. The technology is promising for large‐scale implementation of PFAS capture.
Introduction
Per‐ and polyfluoroalkyl substances (PFAS) are a class of synthetic compounds that have been used in industry and consumer products worldwide since the 1950s. [1] The unique chemical structure of PFAS, containing a chain of carbon atoms bonded to fluorine atoms, makes them highly stable, persistent in the environment and liable to bioaccumulate in organisms. [2] Recent research suggests that exposure to high levels of certain, specific PFAS compositions and chain lengths may lead to adverse health outcomes, including disruption of immune and thyroid function, [3] liver and kidney diseases, [4] and harm to reproductive cells. [5] Hence, efficient removal of PFAS from the environment is urgently needed, and a number of new approaches have been proposed by the scientific community.
Granular activated carbon (GAC) is the most commonly used sorbent for the removal of long‐chain PFAS, [6] with relatively high removal efficiency (>90 %) achieved due to its highly porous structure and strong hydrophobic interactions with PFAS.[ 6a , 7 ] However, slow sorption of PFAS (often taking over 30 h), difficulty in regeneration (e.g. requiring thermal treatment at high temperatures),[ 6b , 7a , 8 ] and competitive sorption from other co‐contaminants significantly reduce its effectiveness for PFAS sorption.[ 6 , 9 ] Powdered activated carbon (PAC) with smaller particle size utilizes the same mechanism of PFAS sorption as GAC, but with a faster sorption rate (taking ≈5–12 h) and higher capacity making it a more reasonable choice for practical removal of PFAS compared with GAC.[ 6b , 7a , 8a ] However, as with GAC, its limitations including reduced removal efficiency of PFAS due to the presence of other contaminants and poor capacity for regeneration of the sorbent at room temperature should not be neglected.[ 6 , 10 ] The commercially available ion‐exchange resin (IEX) sorbs PFAS through electrostatic interactions, and can either be used singly or regenerated for repeated use, [6] with high PFAS removal efficiency (>90 %). [11] However, the presence of common ions, organic molecules and dissolved solids may interfere with PFAS sorption by IEX.[ 6 , 12 ] The limitations of these established technologies has promoted a number of research groups to propose new more effective sorbents.
The inclusion of fluorinated moieties within polymer sorbents has appeared as a promising approach for selective removal of PFAS via fluorous interactions. [13] In early work, Koda et al. prepared “fluorous‐core” star polymers for encapsulation of perfluorooctanoic acid (PFOA) from mixtures of ethanol/deuterium oxide (1 : 1, v/v). [13a] The authors characterized the binding of PFOA with the star polymers using 19F NMR spectroscopy and reported selective sorption in less than ten mins, driven by fluorous interactions between the perfluoroalkyl segments of PFOA and the polymer. With the addition of chloroform, the bound PFOA was released enabling the sorbent to be regenerated. [13a] By further crosslinking the polymers to produce star polymer gels, sorption of PFOA with 74 % efficiency was achieved by simple mixing and filtration. [13b] More recently, by applying similar design principles, fluorinated hydrogels proposed by Quan et al. and β‐cyclodextrin‐based fluorinated polymers reported by Xiao et al. showed >99 % removal efficiency for PFOA at environmentally relevant concentrations.[ 13c , 13d ] Both fluorinated sorbents have capacity for regeneration demonstrated by sorption/desorption experiments over several cycles. Previously our group prepared a novel amphiphilic poly(ethylene glycol)‐perfluoropolyether (PFPE) block polymer for removal of PFOA from aqueous solutions. [13e] The removal efficiency of PFOA was 97.8 % within five mins determined by 19F NMR diffusion‐ordered spectroscopy (DOSY).
Fluorous interactions have been shown to be important in the design of PFAS sorbents, however, electrostatic interactions additionally play a role in further improving sorption performance. In 2020, Kumarasamy et al. reported the design and preparation of ionic fluorogels containing cationic quaternized ammonium groups. [13f] The fluorogels not only show >99 % removal for PFOA but also had >80 % removal efficiency for 17 other legacy and emerging PFAS. More recently, we quantitatively investigated the contributions of fluorous and electrostatic interactions to the sorption of PFAS. [14] The sorption of PFOA by cationic PFPE‐containing polymer was significantly higher (≈100 %) compared with the non‐ionic PFPE‐containing polymer (≈15.2 %). Such enhanced sorption is due to the presence of electrostatic attraction between PFOA and the cationic polymer that precludes fast chemical exchange. [14] To be more specific, the presence of cationic groups can efficiently capture anionic PFAS molecules through tight electrostatic binding, while the fluorinated segments contribute selective recognition of PFAS via fluorous interactions. The above studies provide key design rules and highlight that the incorporation of both electrostatic and fluorous interactions is important to achieve optimum sorption performance.
An important additional consideration when designing PFAS sorbents is how to effectively collect and recycle the PFAS‐containing materials. The most commonly reported methods for removal of PFAS, high‐speed centrifugation and cartridges filled with non‐magnetic sorbents, both involve high energy consumption (Scheme 1a). In this current work, a magnetic separation method was employed as an alternative strategy for recovery of the sorbent after PFAS sorption (Scheme 1b). Magnetic iron oxide nanoparticles (IONPs, Fe3O4) were synthesized and grafted with a series of non‐ionic or cationic PFPE‐containing polymers prepared using reversible addition‐fragmentation chain‐transfer (RAFT) polymerization. These PFPE‐containing polymers were prepared with similar content of fluorine (≈20 wt %), but different contents of cationic quaternized ammonium groups (0–39 wt %) by controlling the degree of polymerization (DP) of the two monomers, 2‐dimethylaminoethyl acrylate (DMAEA) and oligo(ethylene glycol)methyl ether acrylate (OEGA). Our results reveal that P2‐9+@IONPs, the grafted polymer sorbent with DP of OEGA and the quaternized ammonium monomer of 2 and 9, respectively, containing the highest amount of cationic groups (39 wt %) showed the best performance in the removal of multiple PFAS. The results demonstrate that the binding between P2‐9+@IONPs and one major emerging PFAS pollutant, ammonium salt of hexafluoropropylene oxide dimer acid (GenX), is both rapid and highly efficient. Regeneration of the sorbent was confirmed by conducting both sorption and desorption experiments over multiple cycles. The sorption of multiple PFAS and magnetic regeneration of our new sorbents works efficiently in ground water matrices at environmentally relevant concentration (1 ppb), highlighting the potential of using fluorinated magnetic sorbents for the treatment of environmental PFAS‐contaminated solutions.
Scheme 1.
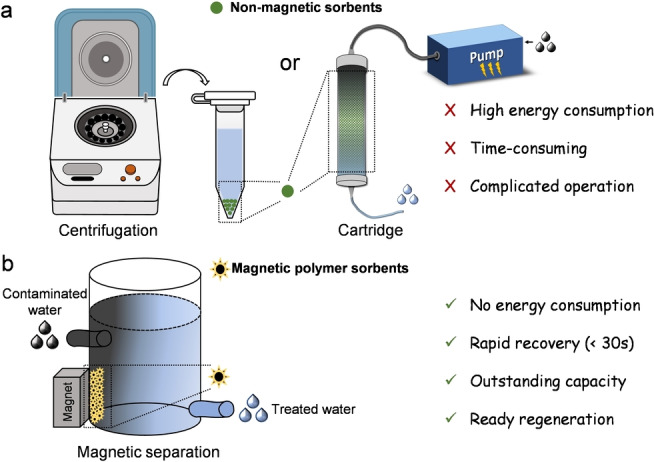
Schematic illustration of different methods for PFAS removal and sorbent recovery. a) Conventional centrifugation for recovery of non‐mangetic material and cartridge filled with non‐magnetic sorbents for PFAS removal. b) Recovery of magnetic polymer grafted IONPs via magnetic separation.
Results and Discussion
Design and Synthesis of Magnetic Polymer Sorbents
Before synthesizing magnetic polymer sorbents, four fluorinated cationic polymers having similar fluorine content (≈20 wt %) but different amounts of cationic segments were prepared. The incorporation of perfluoropolyether (PFPE) provides important fluorine‐fluorine hydrophobic interactions between the sorbent and the perfluoroalkyl segment of PFAS. [13e] The presence of cationic groups in the polymer sorbents further increases their capture efficiency of PFAS through electrostatic interactions, especially when PFAS are at low concentrations. [14] Magnetic iron oxide nanoparticles (IONPs, Fe3O4) were produced using the thermal decomposition method and grafted with the fluorinated polymers, for simple, rapid recycling of the sorbents after sorption of PFAS. The synthetic scheme for the preparation of magnetic fluorinated polymer sorbents is shown in Scheme 2.
Scheme 2.
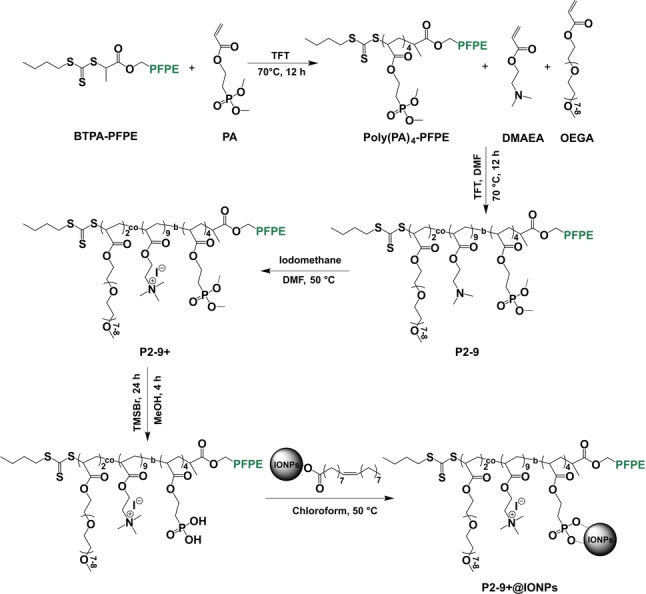
Typical synthetic scheme for magnetic fluorinated polymer sorbents.
The PFPE‐containing macro‐chain‐transfer agent was synthesized by the N‐(3‐(dimethylamino)propyl)‐N′‐ethylcarbodiimide hydrochloride/4‐(dimethylamino)pyridine (EDCl/DMAP) coupling esterification reaction between 2‐(butylthiocarbonothioylthio)propionic acid (BTPA) and hydroxy‐terminated PFPE. Both 1H and 19F NMR spectra shown in Figure S1 and Figure S2 indicate the successful preparation of PFPE macro‐chain‐transfer agent (BTPA‐PFPE).[ 13e , 14 ] Successful synthesis of 2‐(dimethoxyphosphoryl)ethyl acrylate (PA) using dimethyl (2‐hydroxyethyl)phosphonate and acryloyl chloride as the reactants was also confirmed by 1H and 31P NMR spectra shown in Figure S3 and Figure S4. [15]
Four candidate PFPE‐containing polymers were prepared by reversible addition‐fragmentation chain‐transfer (RAFT) polymerization (Scheme 2). [16] Two steps were conducted to obtain the targeted PFPE polymers. Firstly, the polymer precursor, poly(PA)4‐PFPE was synthesized using PA as the monomer and BTPA‐PFPE as the macro‐chain‐transfer agent. After purification, the 1H, 19F and 31P NMR spectra in Figure S5, Figure S6 and Figure S7 support the successful synthesis of the polymer precursor. Secondly, poly(PA)4‐PFPE was used for further chain extension with oligo(ethylene glycol)methyl ether acrylate (OEGA) and/or 2‐(dimethylamino)ethyl acrylate (DMAEA) as the monomers. Four polymers, poly(PA4‐b‐OEGA8)‐PFPE, poly(PA4‐b‐OEGA6‐DMAEA3)‐PFPE, poly(PA4‐b‐OEGA4‐DMAEA6)‐PFPE and poly(PA4‐b‐OEGA2‐DMAEA9)‐PFPE (denoted as P8‐0, P6‐3, P4‐6 and P2‐9, respectively) were successfully synthesized as confirmed by the 1H and 19F NMR spectra in Figure 1a, Figure S8, Figure S9 and Figure S10. Taking P2‐9 as an example (Figure 1a), the peaks due to methylene protons (2H, −CH2O−) adjacent to the ester groups of OEGA, DMAEA and PA (peaks i, l and n) contribute to a broad peak at 3.90–4.30 ppm in the 1H spectrum. The molar mass dispersity (Đ) for each polymer was measured by size exclusion chromatography, demonstrating narrow molecular weight distributions of the four polymers (Đ=1.07 for P8‐0, P6‐3 and P4‐6, and Đ=1.06 for P2‐9).
Figure 1.
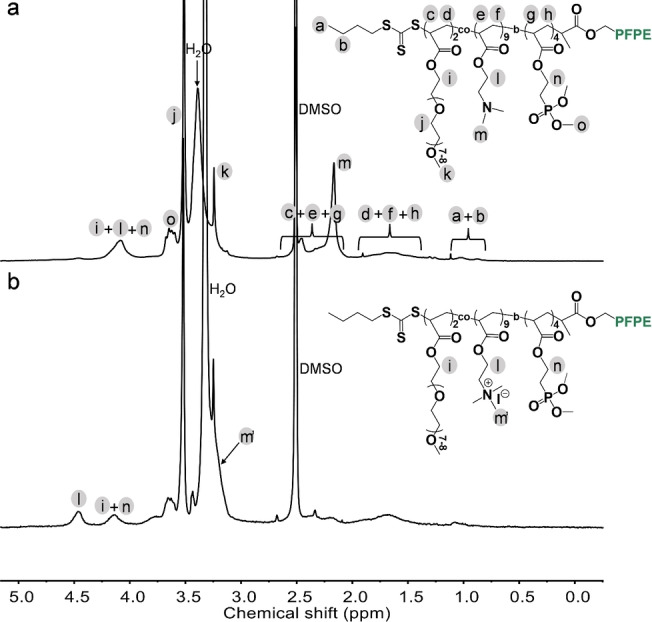
1H NMR spectra of P2‐9 (a) and P2‐9+(b) in DMSO‐d6.
Quaternized polymers P6‐3+, P4‐6+ and P2‐9+ were prepared by adding excess iodomethane to the corresponding polymers P6‐3, P4‐6 and P2‐9, respectively. The degree of quaternization of the three polymers was determined to be ≈100 % using 1H NMR.[ 14 , 17 ] Typically, the peak at 2.16 ppm (peak m) in Figure 1a and Figure S9 due to the two methyl groups (6H, 2×−CH3) of DMAEA fully disappeared and shifted to ≈3.20 ppm (peak m’) in Figure 1b and Figure S11. Meanwhile, after quaternization, the peak due to methylene protons adjacent to the ester groups of DMAEA shifted to 4.46 ppm (peak l), while the other peaks (peaks i and n) due to the methylene protons (2H, −CH2O−) adjacent to the ester groups of OEGA and PA remain unchanged. The fluorine contents of the four polymers after quaternization were calculated from the NMR spectra to be approximately 20 wt %. The content of quaternized DMAEA (cationic monomer) was also calculated as 0 % for P8‐0, 12.6 % for P6‐3+, 25.6 % for P4‐6+ and 39.0 % for P2‐9+ based on weight. Deprotection of the phosphonate was then conducted by silylation and methanolysis reactions. Peak o in the 1H NMR spectrum due to the methyl groups (6H, 2×−CH3, Figure 1b and Figure S11) of the phosphonate group disappears after deprotection, confirming the successful conversion from phosphonate to phosphonic acid (Figure S12). The hydrodynamic diameters of the four polymers after phosphonate deprotection were <30 nm (number‐based) by dynamic light scattering (DLS) in Milli‐Q water (Table S1), indicating that the PFPE polymers were well dispersed in solution.
Oleic acid coated IONPs (OA@IONPs) were synthesized by the thermal decomposition method. [18] After purification, IONPs with an average diameter of 21.4 nm and a narrow size distribution were obtained (transmission electron microscopy, TEM, Figure 2a and 2b). The presence of oleic acid on the surface of the nanoparticles was confirmed by Fourier‐transform infrared spectroscopy (FT‐IR), with typical stretching vibrations of the carbonyl group (C=O, 1731 cm−1) and alkyl carbon‐hydrogen bond (C−H, 2914 and 2851 cm−1) being observed (Figure 2c). [19] The content of oleic acid on the surface of IONPs was determined to be ≈30 wt % using thermogravimetric analysis (TGA, blue curve, Figure 2d).
Figure 2.
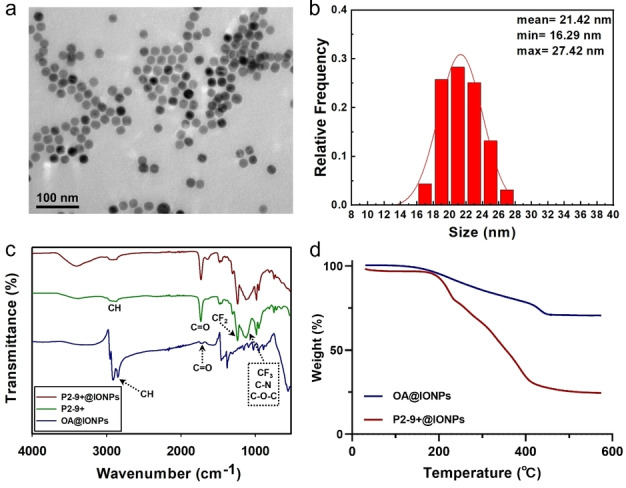
Characterizations of IONPs before and after grafting P2‐9+. a) TEM image; b) Size histogram; c) FTIR spectra of OA@IONPs, P2‐9+ polymer and P2‐9+ polymer grafted IONPs (P2‐9+@IONPs); d) TGA of OA@IONPs (in blue) and P2‐9+@IONPs (in red).
Successful grafting of the PFPE‐containing polymers on the IONPs through ligand exchange was confirmed by FTIR, TGA, TEM and zeta potential (ζ) measurements. The FTIR spectrum of P2‐9+@IONPs indicates the presence of characteristic peaks belonging to the PFPE polymer (CF2 at 1241 cm−1 and CF3 at 1129 cm−1, Figure 2c and Figure S13) and confirms the successful grafting of polymer on the surface of the IONPs. [20] The weight percent of polymer grafted on IONPs was measured by TGA (Figure 2d, Figure S14 and Table 1), and was 86 %, 73 %, 70 % and 74 % for P8‐0, P6‐3+, P4‐6+ and P2‐9+, respectively. Neither obvious change in morphology nor size of the IONPs after grafting of the polymer was observed (Figure S15), indicating the IONPs are stable after grafting with the polymer chains. The zeta potential (ζ) of the magnetic polymer sorbents increased with increasing degree of polymerization of the cationic groups (Table 1). The ζ were negative for the two polymer sorbents with no to low amounts of quaternized cationic groups (i.e. P8‐0@IONPs and P6‐3+@IONPs). P4‐6+@IONPs and P2‐9+@IONPs with higher degrees of polymerization of the cationic groups were positively charged at ≈30 mV. The negative ζ of P8‐0 and P6‐3+ grafted IONPs is most probably due to the ionization of the excess and exposed phosphonic acid groups of the polymers not bound on the surface of the IONPs, [21] and is confirmed by measuring ζ of the polymers before grafting IONPs (Table 1).
Table 1.
Molecular characterization of the four fluorinated magnetic polymer sorbents.
|
Polymer Sorbents |
DP OEGA/DMAEA [a] |
M n,NMR [a] [g mol−1] |
M n,SEC [b] [g mol−1] |
Đ [b] |
19F Content[c] [wt %] |
Quaternized DMAEA[d] [wt %] |
Polymer[e] [wt %] |
ζ [f] [mV] |
|---|---|---|---|---|---|---|---|---|
|
P8‐0 |
8/0 |
6900 |
8100 |
1.07 |
19.8 |
0 |
– |
−23.9 |
|
P8‐0@IONPs |
86 |
−33.3 |
||||||
|
P6‐3+ |
6/3 |
6400 |
7100 |
1.07 |
20.1 |
12.6 |
– |
−23.9 |
|
P6‐3+@IONPs |
73 |
−3.9 |
||||||
|
P4‐6+ |
4/6 |
5800 |
6900 |
1.07 |
20.4 |
25.6 |
– |
31.0 |
|
P4‐6+@IONPs |
70 |
28.8 |
||||||
|
P2‐9+ |
2/9 |
5300 |
6900 |
1.06 |
20.8 |
39.0 |
– |
33.2 |
|
P2‐9+@IONPs |
74 |
34.2 |
[a] DP and M n,NMR for P8‐0 was calculated by integrating the terminal methyl group belonging to BTPA (peak a) and the terminal methyl protons from OEGA (peak k) in Figure S8; DP and M n,NMR for P6‐3, P4‐6 and P2‐9 were calculated by integrating the two terminal methoxy protons belonging to PA (peak o), terminal methyl protons from OEGA (peak k) and methylene protons next to the ester group (peak i+l+n) in Figure S8. [b] M n,SEC and Đ were acquired by size exclusion chromatography in N,N‐dimethylacetamide using a RI detector before quaternization. [c] Weight percentage of fluorine of polymer after quaternization. [d] Weight percentage of quaternized DMAEA side chains of polymer, including the paired iodide ion. [e] Weight percentage of polymer from polymer grafted IONPs, determined by TGA. [f] Determined by DLS after quaternization and phosphonate deprotection.
Equilibrium Multiple PFAS Sorption Using Magnetic Polymer Sorbents
The four magnetic polymer sorbents, along with two commercially available materials, activated carbon (AC, 20–40 mesh particle size) and anion exchange resin (IEX, Amberlite IRA‐410, 20–25 mesh particle size), were used to treat solutions containing 11 different PFAS (Figure 3a and Figure S16), including long‐chain perfluoroalkyl carboxylic acids (PFCAs, C n F2n+1COOH, n≥7), long‐chain perfluoroalkyl sulfonic acids (PFSAs, C n F2n+1SO3H, n≥6,), short‐chain PFCAs (n<7), short‐chain PFSAs (n<6) and ammonium salt of hexafluoropropylene oxide dimer acid (GenX).[ 1c , 22 ] The initial concentration of each PFAS was 100 μg L−1 (ppb). 200 mg L−1 (ppm) sodium chloride (NaCl) and 20 ppm humic acid (HA) were added to simulate a natural PFAS contaminated environment. Upon completion of sorption, a neodymium magnet (N50) was used to fully separate the magnetic polymer sorbents from the treated solution by exposure for 10 min (Figure 3b), while centrifugation was performed for the two commercially available sorbents. The results in Figure 3c show that the IONPs grafted with the non‐ionic PFPE‐containing polymer, i.e. P8‐0@IONPs, showed low to moderate removal for short‐chain PFAS (i.e. perfluorobutanoic acid (PFBA), perfluoropentanoic acid (PFPeA), perfluorohexanoic acid (PFHxA), perfluoroheptanoic acid (PFHpA) and perfluorobutanesulfonic acid (PFBS)) and ammonium salt of hexafluoropropylene oxide dimer acid (GenX), but the removal reached >98 % for long‐chain PFAS (e.g. perfluorooctanoic acid (PFOA), perfluorononanoic acid (PFNA), perfluorodecanoic acid (PFDA), perfluorohexanesulfonic acid (PFHxS) and perfluorooctanesulfonic acid (PFOS)). This is mainly due to the stronger fluorous interactions between the PFPE segments of the sorbent and the fluorinated tail of long‐chain PFAS compared with that of short‐chain PFAS. [23] The effective removal of long‐chain PFAS using the neutral polymer is consistent with the work previously reported by our group. [13e] For PFCAs and PFSAs with the same number of carbons in the fluorinated tail, sorption of PFCAs is less efficient than that of PFSAs using neutral P8‐0@IONPs. For example, the removal of PFPeA (n=4) by P8‐0@IONPs was 4.7 %, much lower than that of PFBS (n=4) with a removal efficiency at 65.9 %, and can be attributed to the less hydrophobic nature of PFCA than PFSA that results in weaker hydrophobic interactions between PFAS and the PFPE segments of the neutral sorbent. [24]
Figure 3.
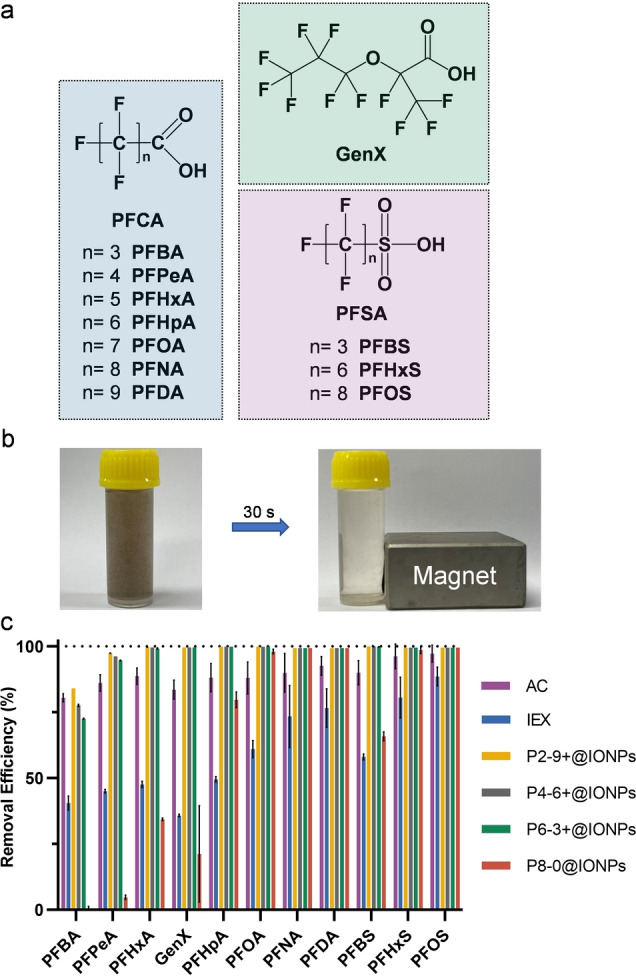
Equilibrium PFAS removal using magnetic polymer sorbents and commercially available AC and IEX. a) Chemical structures of 11 PFAS used in this work. b) Photograph of P2‐9+@IONPs before (left) and after (right) magnetic separation for 30 s. c) Removal efficiency of 11 PFAS treated by different sorbents for 24 h. Polymer concentration, 0.5 mg mL−1 (excluding IONPs); AC and IEX, 0.5 mg mL−1; PFAS initial concentration, 100 ppb each. Water constituents, milli‐Q water with the presence of NaCl (200 ppm) and humic acid (20 ppm), pH=5.0. The results are the average of three replicates, and one standard deviation is shown.
After incorporation of the quaternized ammonium groups, the removal of PFAS by the three quaternized polymer grafted IONPs, i.e. P2‐9+@IONPs, P4‐6+@IONPs and P6‐3+@IONPs, significantly increased (Figure 3c). This is especially true for the removal of short‐chain PFAS and GenX, and is presumably due to electrostatic attractions between the quaternized ammonium group of polymer and the charged head group of PFAS. The results agree well with our previous work, [14] demonstrating a more efficient and tighter binding of PFAS with cationic polymer sorbents compared with the neutral sorbents. Among the four PFPE‐containing polymer grafted IONPs sorbents, the P2‐9+@IONPs with the largest content of cationic groups (39 wt % of cationic monomer) show the highest removal for all 11 PFAS (84 % for PFBA, 97.3 % for PFPeA, and >99.5 % removal for the remaining nine PFAS, Figure 3c). The results are in line with the ζ values shown in Table 1, suggesting that for polymer sorbents with the same fluorine content, a larger content of cationic group leads to higher ζ and consequently higher levels of removal of PFAS.
The magnetic polymer sorbents with both quaternized ammonium and PFPE segments show higher PFAS removal compared with the two commercially available sorbents (Figure 3c). After treatment for 24 h, AC shows 80.5 % removal of PFBA, lower than that of P2‐9+@IONPs at 84.0 %, but higher than the other magnetic polymer sorbents and IEX (i.e. 77.6 % for P4‐6+@IONPs, 72.5 % for P6‐3+@IONPs, and 40.5 % for IEX). The removal efficiency of the other ten PFAS using our magnetic polymer sorbents was significantly higher than AC and IEX. In addition, the PFAS removal efficiency of our sorbents is superior to previously reported magnetic sorbents. [25] For example, Gong et al. prepared starch‐stabilized magnetic nanoparticles and demonstrated PFOA removal of ≈62 % in the absence of organic compounds, and a reduction of 95 % in removal efficiency was observed after addition of 4.8 mg L−1 HA to the solution. [25a] Our sorbents have a much higher level of PFOA removal at >99.8 % even in presence of 20 mg L−1 HA, demonstrating outstanding performance compared with previously reported work. The results highlight the importance of incorporation of both quaternized cationic and PFPE segments for more effective removal of both PFCAs and PFSAs with different chain lengths.
The stability of magnetic polymer sorbents was confirmed by collecting the 19F NMR spectroscopy of the residue solution after PFAS removal and magnetic cycling of sorbents. The 19F NMR spectrum is shown in Figure S17 and no detectable 19F signal from PFPE polymer can be observed, indicating that the polymer tightly binds on the surface of IONPs without leaching.
Sorption Kinetics of GenX Using P2‐9+@IONPs
The kinetics of sorption of GenX was investigated in this work as it is now used as an alternative to PFOA as a polymer processing aid. [26] P2‐9+@IONPs show the highest PFAS removal efficiency among all magnetic polymer sorbents and was chosen for the kinetics study. Sorption kinetics of GenX was tested for a solution with an initial concentration of 100 ppb using P2‐9+@IONPs as the sorbent in deionized water. The results in Figure 4 show that >99 % of GenX can be efficiently removed within 30 s with a further increase to ≈99.5 % in less than three mins, demonstrating the rapid capture of GenX with excellent removal efficiency. After three mins, no detectable signal from GenX was observed by liquid chromatography with tandem mass spectrometry (LC–MS/MS). Furthermore, desorption of GenX from P2‐9+@IONPs was not observed within the experimental time window, suggesting that the binding of GenX to P2‐9+@IONPs via electrostatic and fluorous interactions is strong and irreversible in aqueous solution. The findings are in line with the work published by Kumarasamy et al., [13f] where quantitative removal of GenX at an initial concentration of 200 ppb using ionic fluorogel was achieved within one minute.
Figure 4.
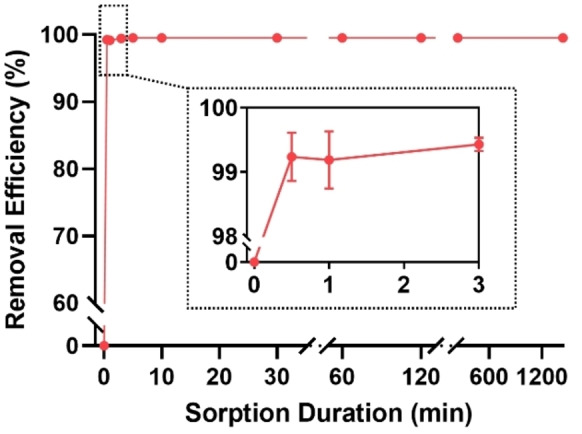
Sorption kinetics of GenX using P2‐9+@IONPs. Polymer concentration, 0.1 mg mL−1 (excluding IONPs). GenX initial concentration, 100 ppb. The results are the average of three replicates, and one standard deviation is shown.
Sorption Isotherms of GenX Using P2‐9+@IONPs
In order to understand the binding parameters between PFAS and the sorbent, a sorption isotherm study of GenX using P2‐9+@IONPs was conducted. After treating solutions with GenX concentrations from 0.1 to 50 ppm with a fixed sorbent concentration at 0.1 mg mL−1 in deionized water, the sorption data was fitted by two different sorption models, the Langmuir and Freundlich (Figure 5) using Equation (1) and Equation 2. [27]
| (1) |
| (2) |
Figure 5.
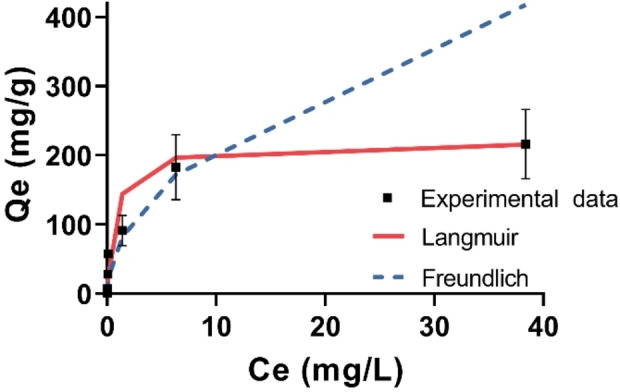
Binding isotherm study between GenX and P2‐9+@IONPs. Polymer concentration, 0.1 mg mL−1 (excluding IONPs). GenX concentration, 0.1–50 ppm. Sorption duration, 24 h. The results are the average of three replicates, and standard deviation is shown.
Where C e is the residual concentration of GenX at equilibrium (mg L−1), Q m (mg g−1) is the estimated maximum sorption capacity, Q e (mg g−1) is the amount of GenX bound on the sorbent at equilibrium, K L (L mg−1) is the Langmuir equilibrium constant representing binding affinity, [28] K F ((mg g−1)(L mg−1)1/n ) is the Freundlich constant, n is the intensity of sorption.
The parameters for the two models were calculated based on the linearized forms of equations 1 and 2 (Figure S18), and are listed in Table 2. The experimental data fits the Langmuir better than the Freundlich model, with a coefficient of determination (R2) of 0.9979 for the Langmuir model and R2 of 0.9029 for the Freundlich model. K L and Q m were calculated to be 4.8×105 M−1 and 219 mg g−1, respectively, suggesting a stronger binding affinity between GenX and our sorbent P2‐9+@IONPs with a superior or similar capacity compared with previously reported sorbents, e.g., ionic perfluoroalkane hydrogels (K L unreported, Q m=34 mg g−1), amine‐functionalized covalent organic framework (K L=6.3×104 M−1, Q m=200 mg g−1), and β‐cyclodextrin (β‐CD) polymer (K L=8.8×104 M−1, Q m=222 mg g−1) (Table S2). [29] The Q m value of P2‐9+@IONPs is also comparable with the ionic fluorogels prepared by Kumarasamy et al. (e.g. IF‐30+, Q m=217 mg g−1), [13f] but with a smaller K L value (4.8×105 M−1 vs. 1.5×107 M−1, Table S2). The stronger binding affinity of the ionic fluorogel could be due to its much higher fluorine content (≈50 vs. 20.8 wt %) that promotes the capture of GenX molecules. In summary, the magnetic polymer sorbent P2‐9+@IONPs shows high binding affinity and maximum sorption capacity that provides efficient removal of GenX. In addition, facile recovery of the sorbent P2‐9+@IONPs using a magnet makes this material suitable for application in commercial processes.
Table 2.
Langmuir and Freundlich constants for the sorption of GenX using P2‐9+@IONPs.
|
Langmuir |
Freundlich |
||||
|---|---|---|---|---|---|
|
K L [M−1] |
Q m [mg g−1] |
R2 |
K F [mg g−1] [L mg−1]1/n |
n |
R2 |
|
4.8×105 |
219 |
0.9979 |
69.9 |
2.0 |
0.9029 |
Regeneration of P2‐9+@IONPs
Regeneration and reusability are key factors in evaluating the practicality of PFAS sorbents. [30] In this work, the regeneration of P2‐9+@IONPs was examined after sorption of GenX, and was tested over four cycles. Both sorption and desorption of GenX were examined, with sorption/desorption times of ten mins for each cycle (Figure 6). Desorption was performed by replacing the aqueous solution with same volume of 400 mM of methanolic ammonium acetate solution. According to previous work, [31] organic solvents, e.g. methanol, play an important role in breaking fluorous interactions, while the addition of salt can efficiently extract the electrostatically‐bound PFAS. The results in Figure 6 demonstrate a high extent of removal of GenX (>99 %, >9.9 mg g−1) in the first sorption cycle, followed by full extraction of GenX in the methanolic salt solution. The sorption and desorption capacities were maintained over four cycles, suggesting that P2‐9+@IONPs can be regenerated and reused after multiple cycles, thus greatly improving the economic feasibility of the sorbent.
Figure 6.
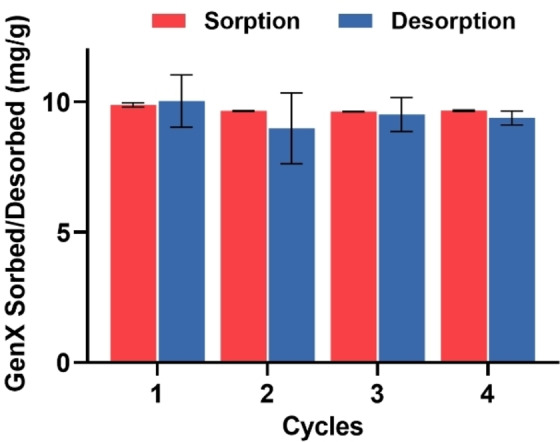
Regeneration ability of P2‐9+@IONPs. Polymer concentration, 1 mg mL−1; GenX concentration, 10 ppm. Sorption and desorption duration, 10 mins. The results are the average of triplicates, and standard deviation is shown.
Multiple PFAS Removal in Ground Wastewater Matrices
The composition of ground water, including dissolved organic and inorganic matter, is highly complex and may interfere with sorption of PFAS at low concentrations by sorbents, resulting in poor performance of removal of PFAS. In this work we employed two samples of different ground wastewater (GWW), namely GWW1 and GWW2, collected from a ground well and an effluent lagoon of two different activated sludge wastewater treatment plants, respectively. The total organic carbon (TOC) of the GWW samples was 5.6 mg L−1 and 8.1 mg L−1 for GWW1 and GWW2, respectively. The water samples were spiked with multiple PFAS targeting an environmentally relevant concentration of 1 ppb, and then treated with P2‐9+@IONPs. At predetermined times of 30 mins and 2 h, the water solutions were collected, followed by magnetic separation (10 mins), solid‐phase extraction (SPE) and LC‐MS/MS analysis for measurement of residue PFAS concentrations. The results in Figure 7 show that after treatment by the polymer sorbent for 30 mins, high removal efficiency was observed for the majority of the tested PFAS from the two GWWs, with >95 % removal of short‐chain PFAS including PFHxA, PFHpA and PFBS, >98 % removal of all long‐chain PFAS tested, and >96 % removal of GenX being quantified. After 2 h, no significant difference in removal efficiency of PFAS was observed (Figure S19), indicating rapid sorption of PFAS in the two GWWs. The results demonstrate that P2‐9+@IONPs can efficiently capture PFAS even with presence of complicated components in the water matrices, and can potentially be applied for water treatment of real contaminated water samples.
Figure 7.
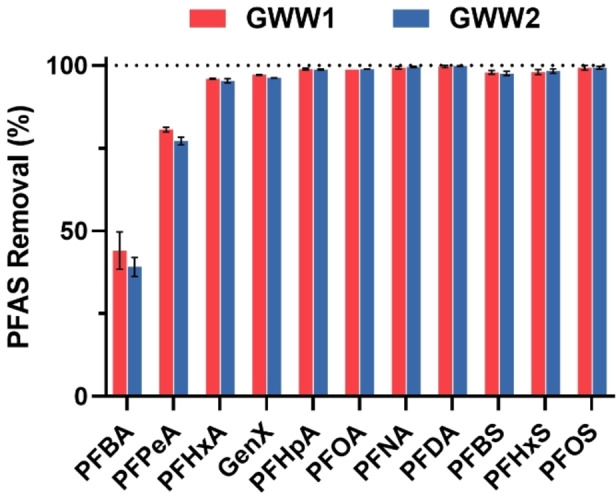
Multiple PFAS removal by P2‐9+@IONPs in two different ground wastewater matrices. TOC: GWW1, 5.6 mg L−1; GWW2, 8.1 mg L−1. pH: GWW1, 7.4; GWW2, 7.3. Polymer concentration, 0.5 mg mL−1 (excluding IONPs). PFAS initial concentration, 1 ppb each. Sorption duration, 30 mins. The results are the average of triplicates, and the standard deviation is shown.
In addition, the full scale treatment of PFAS contaminated water using P2‐9+@IONPs is highly possible due to 1) low amount of sorbent required for efficient PFAS removal from real contaminated wastewater (i.e. 0.5 mg mL−1) and 2) the high feasibility of scale up synthesis of magnetic polymer sorbents. The extracted PFAS from the contaminated sources can subsequently be destroyed using destructive techniques, including advanced oxidative or reductive degradation. [32]
Conclusion
Magnetic fluorinated polymer sorbents are promising for the efficient removal of multiple PFAS from contaminated water sources. A series of fluorinated magnetic nanoparticles were prepared containing similar fluorine contents but different contents of cationic segments, namely P8‐0@IONPs, P6‐3+@IONPs, P4‐6+@IONPs and P2‐9+@IONPs. P2‐9+@IONPs, with the highest content of cationic segments, shows the most efficient removal of 11 PFAS compared with the other three polymeric IONPs, as well as two commercially available sorbents, i.e. activated carbon and anion exchange resin. A study of the kinetics of sorption of GenX by P2‐9+@IONPs demonstrates rapid and efficient removal of the PFAS (>99 %) within 30 s, with no desorption of GenX being observed after 24 h. The sorption isotherm studies show that the binding between P2‐9+@IONPs and GenX is described well by the Langmuir model. The ability to regenerate of P2‐9+@IONPs was confirmed by conducting both sorption and desorption of GenX for multiple cycles. Finally, the sorption of multiple PFAS in two different ground water matrices at an environmentally relevant concentration (1 ppb) using P2‐9+@IONPs demonstrates that the PFPE‐containing sorbent is a promising sorbent and can be practically used for PFAS remediation from real contaminated environments.
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors acknowledge the Australian Research Council (DP210101496) and the National Health and Medical Research Council (APP1157440 to C. Z.) for funding of this research. The Chemours Company is acknowledged for providing perfluoropolyether. Dr. Katie Macintosh from Council of the City of Gold Coast is also acknowledged for providing two ground wastewater matrices. The Australian National Fabrication Facility, Queensland Node is acknowledged for access to some items of equipment. Open Access publishing facilitated by The University of Queensland, as part of the Wiley ‐ The University of Queensland agreement via the Council of Australian University Librarians.
X. Tan, P. Dewapriya, P. Prasad, Y. Chang, X. Huang, Y. Wang, X. Gong, T. E. Hopkins, C. Fu, K. V. Thomas, H. Peng, A. K. Whittaker, C. Zhang, Angew. Chem. Int. Ed. 2022, 61, e202213071; Angew. Chem. 2022, 134, e202213071.
Contributor Information
Prof. Andrew K. Whittaker, Email: a.whittaker@uq.edu.au.
Dr. Cheng Zhang, Email: c.zhang3@uq.edu.au.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- 1.
- 1a. Key B. D., Howell R. D., Criddle C. S., Environ. Sci. Technol. 1997, 31, 2445–2454; [Google Scholar]
- 1b. Prevedouros K., Cousins I. T., Buck R. C., Korzeniowski S. H., Environ. Sci. Technol. 2006, 40, 32–44; [DOI] [PubMed] [Google Scholar]
- 1c. Buck R. C., Franklin J., Berger U., Conder J. M., Cousins I. T., de Voogt P., Jensen A. A., Kannan K., Mabury S. A., van Leeuwen S. P., Integr. Environ. Assess. Manage. 2011, 7, 513–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.
- 2a. Xiao F., Simcik M. F., Gulliver J. S., Water Res. 2012, 46, 6601–6608; [DOI] [PubMed] [Google Scholar]
- 2b. Brusseau M. L., Anderson R. H., Guo B., Sci. Total Environ. 2020, 740, 140017; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Sáez M., de Voogt P., Parsons J. R., Environ. Sci. Pollut. Res. Int. 2008, 15, 472–477; [DOI] [PubMed] [Google Scholar]
- 2d. Haukås M., Berger U., Hop H., Gulliksen B., Gabrielsen G. W., Environ. Pollut. 2007, 148, 360–371; [DOI] [PubMed] [Google Scholar]
- 2e. Ren J., Point A. D., Baygi S. F., Fernando S., Hopke P. K., Holsen T. M., Crimmins B. S., Sci. Total Environ. 2022, 819, 152974; [DOI] [PubMed] [Google Scholar]
- 2f. Death C., Bell C., Champness D., Milne C., Reichman S., Hagen T., Sci. Total Environ. 2021, 774, 144795. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Goudarzi H., Miyashita C., Okada E., Kashino I., Chen C.-J., Ito S., Araki A., Kobayashi S., Matsuura H., Kishi R., Environ. Int. 2017, 104, 132–138; [DOI] [PubMed] [Google Scholar]
- 3b. Lee J. E., Choi K., Ann. Pediatr. Endocrinol. Metab. 2017, 22, 6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Huang Q., Zhang J., Martin F. L., Peng S., Tian M., Mu X., Shen H., Toxicol. Lett. 2013, 223, 211–220; [DOI] [PubMed] [Google Scholar]
- 4b. Ferrari F., Orlando A., Ricci Z., Ronco C., Curr. Opin. Crit. Care 2019, 25, 539–549. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Yuan Y., Ding X., Cheng Y., Kang H., Luo T., Zhang X., Kuang H., Chen Y., Zeng X., Zhang D., Chemosphere 2020, 241, 125074; [DOI] [PubMed] [Google Scholar]
- 5b. Song X., Tang S., Zhu H., Chen Z., Zang Z., Zhang Y., Niu X., Wang X., Yin H., Zeng F., Environ. Int. 2018, 113, 50–54. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a.L. Cummings, A. Matarazzo, N. Nelson, F. Sickels, C. Storms, Recommendation on perfluorinated compound treatment options for drinking water, New Jersey Drinking Water Quality Institute Treatment Subcommittee Report, New Jersey, 2015;
- 6b. Zhang C., Yan K., Fu C., Peng H., Hawker C. J., Whittaker A. K., Chem. Rev. 2022, 122, 167–208. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Du Z., Deng S., Bei Y., Huang Q., Wang B., Huang J., Yu G., J. Hazard. Mater. 2014, 274, 443–454; [DOI] [PubMed] [Google Scholar]
- 7b. Eschauzier C., Beerendonk E., Scholte-Veenendaal P., De Voogt P., Environ. Sci. Technol. 2012, 46, 1708–1715; [DOI] [PubMed] [Google Scholar]
- 7c. Pauletto P. S., Bandosz T. J., J. Hazard. Mater. 2022, 425, 127810. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Yu Q., Zhang R., Deng S., Huang J., Yu G., Water Res. 2009, 43, 1150–1158; [DOI] [PubMed] [Google Scholar]
- 8b. Sonmez Baghirzade B., Zhang Y., Reuther J. F., Saleh N. B., Venkatesan A. K., Apul O. G., Environ. Sci. Technol. 2021, 55, 5608–5619; [DOI] [PubMed] [Google Scholar]
- 8c. Deng S., Nie Y., Du Z., Huang Q., Meng P., Wang B., Huang J., Yu G., J. Hazard. Mater. 2015, 282, 150–157. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Aggarwal D., Goyal M., Bansal R., Carbon 1999, 37, 1989–1997; [Google Scholar]
- 9b. Pastrana-Martínez L. M., Lopez-Ramon M. V., Fontecha-Camara M. A., Moreno-Castilla C., Water Res. 2010, 44, 879–885. [DOI] [PubMed] [Google Scholar]
- 10. Oyetade O. A., Varadwaj G., Nyamori V. O., Jonnalagadda S. B., Martincigh B. S., Rev. Environ. Sci. Bio/Technol. 2018, 17, 603–635. [Google Scholar]
- 11. Yang Y., Ding Q., Yang M., Wang Y., Liu N., Zhang X., Environ. Sci. Pollut. Res. Int. 2018, 25, 29267–29278. [DOI] [PubMed] [Google Scholar]
- 12. Deng S., Yu Q., Huang J., Yu G., Water Res. 2010, 44, 5188–5195. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Koda Y., Terashima T., Sawamoto M., J. Am. Chem. Soc. 2014, 136, 15742–15748; [DOI] [PubMed] [Google Scholar]
- 13b. Koda Y., Terashima T., Takenaka M., Sawamoto M., ACS Macro Lett. 2015, 4, 377–380; [DOI] [PubMed] [Google Scholar]
- 13c. Quan Q., Wen H., Han S., Wang Z., Shao Z., Chen M., ACS Appl. Mater. Interfaces 2020, 12, 24319–24327; [DOI] [PubMed] [Google Scholar]
- 13d. Xiao L., Ling Y., Alsbaiee A., Li C., Helbling D. E., Dichtel W. R., J. Am. Chem. Soc. 2017, 139, 7689–7692; [DOI] [PubMed] [Google Scholar]
- 13e. Tan X., Zhong J., Fu C., Dang H., Han Y., Král P., Guo J., Yuan Z., Peng H., Zhang C., Macromolecules 2021, 54, 3447–3457; [Google Scholar]
- 13f. Kumarasamy E., Manning I. M., Collins L. B., Coronell O., Leibfarth F. A., ACS Cent. Sci. 2020, 6, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tan X., Sawczyk M., Chang Y. X., Wang Y. Q., Usman A., Fu C. K., Kral P., Peng H., Zhang C., Whittaker A. K., Macromolecules 2022, 55, 1077–1087. [Google Scholar]
- 15. Qiao R., Esser L., Fu C., Zhang C., Hu J., Ramirez-Arcia P., Li Y., Quinn J. F., Whittaker M. R., Whittaker A. K., Davis T. P., Biomacromolecules 2018, 19, 4423–4429. [DOI] [PubMed] [Google Scholar]
- 16. Tian X., Ding J., Zhang B., Qiu F., Zhuang X., Chen Y., Polymer 2018, 10, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Truong N. P., Jia Z., Burges M., McMillan N. A., Monteiro M. J., Biomacromolecules 2011, 12, 1876–1882. [DOI] [PubMed] [Google Scholar]
- 18. Park J., An K., Hwang Y., Park J. G., Noh H. J., Kim J. Y., Park J. H., Hwang N. M., Hyeon T., Nat. Mater. 2004, 3, 891–895. [DOI] [PubMed] [Google Scholar]
- 19. Ibarra J., Melendres J., Almada M., Burboa M. G., Taboada P., Juarez J., Valdez M. A., Mater. Res. Express 2015, 2, 095010. [Google Scholar]
- 20. Shim J., Kim H. J., Kim B. G., Kim Y. S., Kim D. G., Lee J. C., Energy Environ. Sci. 2017, 10, 1911–1916. [Google Scholar]
- 21. Glowinska A., Trochimczuk A. W., Molecules 2020, 25, 4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brendel S., Fetter E., Staude C., Vierke L., Biegel-Engler A., Environ. Sci. Eur. 2018, 30, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ateia M., Maroli A., Tharayil N., Karanfil T., Chemosphere 2019, 220, 866–882. [DOI] [PubMed] [Google Scholar]
- 24. Li R., Alomari S., Islamoglu T., Farha O. K., Fernando S., Thagard S. M., Holsen T. M., Wriedt M., Environ. Sci. Technol. 2021, 55, 15162–15171. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Gong Y., Wang L., Liu J., Tang J., Zhao D., Sci. Total Environ. 2016, 562, 191–200; [DOI] [PubMed] [Google Scholar]
- 25b. Badruddoza A. Z. M., Bhattarai B., Suri R. P., ACS Sustainable Chem. Eng. 2017, 5, 9223–9232; [Google Scholar]
- 25c. Du Z., Deng S., Zhang S., Wang W., Wang B., Huang J., Wang Y., Yu G., Xing B., Environ. Sci. Technol. 2017, 51, 8027–8035; [DOI] [PubMed] [Google Scholar]
- 25d. Meng P., Fang X., Maimaiti A., Yu G., Deng S., Chemosphere 2019, 224, 187–194. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a. Li J., He J., Niu Z., Zhang Y., Environ. Int. 2020, 135, 105419; [DOI] [PubMed] [Google Scholar]
- 26b. Zhang S., Chen K., Li W., Chai Y., Zhu J., Chu B., Li N., Yan J., Zhang S., Yang Y., Environ. Int. 2021, 156, 106745. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Langmuir I., J. Am. Chem. Soc. 1918, 40, 1361–1403; [Google Scholar]
- 27b. Zhang C., Sui J. H., Li J., Tang Y. L., Cai W., Chem. Eng. J. 2012, 210, 45–52; [Google Scholar]
- 27c. Parida K. M., Sahu S., Reddy K. H., Sahoo P. C., Ind. Eng. Chem. Res. 2011, 50, 843–848. [Google Scholar]
- 28.
- 28a. Kamaruddin N. H., Bakar A. A. A., Mobarak N. N., Zan M. S. D., Arsad N., Sensors 2017, 17, 2277; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28b. Li M., Messele S. A., Boluk Y., Gamal El-Din M., Carbohydr. Polym. 2019, 221, 231–241. [DOI] [PubMed] [Google Scholar]
- 29.
- 29a. Huang P. J., Hwangbo M., Chen Z., Liu Y., Kameoka J., Chu K. H., ACS Omega 2018, 3, 17447–17455; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29b. Ji W., Xiao L., Ling Y., Ching C., Matsumoto M., Bisbey R. P., Helbling D. E., Dichtel W. R., J. Am. Chem. Soc. 2018, 140, 12677–12681; [DOI] [PubMed] [Google Scholar]
- 29c. Yang A., Ching C., Easler M., Helbling D. E., Dichtel W. R., ACS Mater. Lett. 2020, 2, 1240–1245. [Google Scholar]
- 30. Zhou G., Luo J., Liu C., Chu L., Ma J., Tang Y., Zeng Z., Luo S., Water Res. 2016, 89, 151–160. [DOI] [PubMed] [Google Scholar]
- 31. Ateia M., Alsbaiee A., Karanfil T., Dichtel W., Environ. Sci. Technol. Lett. 2019, 6, 688–695. [Google Scholar]
- 32. Olatunde O. C., Kuvarega A. T., Onwudiwe D. C., Heliyon 2020, 6, e05614. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.