

Abstract
The Bis‐T series of compounds comprise some of the most potent inhibitors of dynamin GTPase activity yet reported, e. g., (2E,2′E)‐N,N′‐(propane‐1,3‐diyl)bis(2‐cyano‐3‐(3,4‐dihydroxyphenyl)acrylamide) (2), Bis‐T‐22. The catechol moieties are believed to limit cell permeability, rendering these compounds largely inactive in cells. To solve this problem, a prodrug strategy was envisaged and eight ester analogues were synthesised. The shortest and bulkiest esters (acetate and butyl/tert‐butyl) were found to be insoluble under physiological conditions, whilst the remaining five were soluble and stable under these conditions. These five were analysed for plasma stability and half‐lives ranged from ∼2.3 min (propionic ester 4), increasing with size and bulk, to greater than 24 hr (dimethyl carbamate 10). Similar profiles where observed with the rate of formation of Bis‐T‐22 with half‐lives ranging from ∼25 mins (propionic ester 4). Propionic ester 4 was chosen to undergo further testing and was found to inhibit endocytosis in a dose‐dependent manner with IC50 ∼8 μM, suggesting this compound is able to effectively cross the cell membrane where it is rapidly hydrolysed to the desired Bis‐T‐22 parent compound.
Keywords: dynamin, endocytosis, prodrug, Bis-T-22
The Bis‐T series of compounds comprise some of the most potent inhibitors of dynamin GTPase activity yet reported, such as Bis‐T‐22 (2). The catechol moieties are believed to limit cell permeability, rendering these compounds largely inactive in cells. To solve this problem, a prodrug strategy was envisaged, and eight ester analogues were synthesised. Prodrugs, e. g., 4, of 2 effect a rapid and full block of endocytosis, whereas the parent 2 is cell impermeable.

Introduction
Dynamin belongs to the superfamily of large (∼100 kDa) GTP‐binding proteins that are intricately involved in various membrane trafficking and membrane fission or fusion processes.[ 1 , 2 , 3 ] They are act as mechanochemical enzymes that hydrolyse GTP to constrict and deform biological membranes.[ 4 , 5 , 6 ]
In mammals classical dynamin exist in three isoforms: dynamin I (dynI) is primarily neuron‐specific; dynamin II (dynII) is ubiquitous; and dynamin III (dynIII) exists in various tissues including the brain, testes, lung, heart and potentially cells of hematopoietic origin.[ 1 , 2 , 3 , 4 ] The classical dynamins mediate membrane fission during clathrin‐mediated endocytosis (CME) and play additional cellular roles in the exocytic fusion pore, actin dynamics and cytokinesis.[ 7 , 8 , 9 , 10 ] Dynamin mutants and/or overexpression has been linked to various cancers, often with poor prognosis.[ 11 , 12 ] Endocytic pathway defects have been associated with a multitude of human neurological conditions.[ 13 , 14 ] Dynamin's association with microtubules has been linked with endocytosis defects in Alzheimer's Disease. Inhibition of dynamin has also been linked to a possible treatment for epilepsy. [15]
The dynamins are typically characterized as possessing five domains: a GTPase domain,[ 16 , 17 ] a pleckstrin homology (PH) domain,[ 18 , 19 ] a middle domain, a GTPase effector (GED) domain[ 16 , 20 , 21 ] and a proline rich domain (PRD).[ 17 , 18 , 22 , 23 ] Each domain is a potential drug target.
Dynamin inhibitors have been shown as valuable tools for investigating endocytic pathways.[ 24 , 25 ] Inhibitors from our laboratories span the MiTMAB,[ 26 , 27 , 28 ] RTIL, [29] Iminodyn, [30] Pthaladyn, [31] dyngo®, [32] Rhodadyn, [33] DynolesTM,[34–36] Quinodyn, [37] Naphthaladyn, [38] phenothiazine drugs [39] and Bis‐T‐22,[ 40 , 41 ] compound families. Other groups have reported dynasore analogues, [42] some selective serotonin reuptake inhibitors, [43] and high throughput screening hits as dynamin inhibitors.[ 42 , 44 , 45 ] Of these, the MiTMAB,[ 27 , 46 ] Bis‐T [47] and dynole [48] classes have anti‐tumour activity that show selectivity for cancer cells with minimal to no effect on non‐tumourigenic fibroblasts. These dynamin inhibitors induce apoptosis via the intrinsic pathway, exclusively following cytokinesis failure and polyploidisation. [27] In addition, dynamin inhibitors alter cell signalling pathways and have been shown to enhance the effects of existing chemotherapeutic approaches.[ 49 , 50 ] This suggests that targeting dynamin may be a novel anti‐cancer strategy. In support of this, dynole‐34‐2 and a phenothiazine drug were recently shown to be effective as combination therapies in mouse models of leukemia and head and neck cancer respectively,[ 49 , 51 ] or as a mono‐therapy in glioblastoma. [52]
The dimeric tyrphostins (the Bis‐T series) were among the first reported dynamin inhibitors and are the focus of the current study. Their binding site on dynamin is unknown, and they are not GTP competitive. [41] Members of this compound series have shown potential therapeutic utility in animal models of kidney disease.[ 53 , 54 , 55 ] Bis‐T‐22 (2) is equally the most active in the dynamin inhibitor in the series, with an IC50 for dynamin I inhibition of 1.7±2 μM) (compound 22 in Table 1 of [41] ). It contains two highly polar catechol groups which have been shown to be essential for dynamin inhibition.[ 40 , 41 ] Bis‐T‐22 (2) displayed varying degrees of cell permeability, limiting its potential as a future therapeutic. Herein we describe a simple prodrug strategy enhancing the cell permeability of the Bis‐T series and explore Bis‐T‐22’s mechanism of action.
Table 1.
Physico‐chemical properties of Bis‐T‐22 (2) and prodrugs 3–10.
|
Compound |
Log P[a] |
Aqueous Stability[b] |
|---|---|---|
|
2 (Bis‐T‐22) |
2.61 |
ND[c] |
|
3 |
2.77 |
ND |
|
4 |
4.30 |
Y |
|
5 |
7.63 |
Y |
|
6 |
8.00 |
Y |
|
7 |
6.93 |
Y |
|
8 |
11.19 |
ND |
|
9 |
8.08 |
ND |
|
10 |
3.11 |
Y |
[a] Calculated using the Drug Likeness tool in ICM, Molsoft Ca; [b] Stock solutions of the compound were incubated in tris buffer (pH 7.4) at 37 °C for 24 h, stability judged by HPLC analysis, with compound identified as stable if <10 % Bis‐T‐22 was observed. [c] ND=not determined
Results and Discussion
In our early investigations with Bis‐T‐22 (2), we were unable to convincingly demonstrate that this compound inhibits dynamin and clathrin‐mediated endocytosis (CME) in all cultured cell lines (Figure 1). Endocytosis block was essentially complete with 100 μM 2 in Swiss 3T3 (Figure 1), HER14 and B104 cells (not shown), but 2 was largely ineffective in COS7, A431, B35 and HeLa cells at up to 300 μM (Figure 1, and SI Figure S1). This obviously limits the use of this compound, despite its excellent dynamin IC50 value (dyn I IC50=1.7 μM; dyn II IC50=0.5 μM), [41] for the study of endocytosis in a wide range of cell types. We rationalised that this variability in activity was possibly due to the inability of 2 to permeate the cell membrane, and thus block endocytosis, although other factors may explain the variability, such as cell line differences in active efflux transporter mechanisms or xenobiotic metabolism via cytochrome P450 oxidases and transferase enzymes like glutathione S‐transferases. To address the hypothesis that the deficit with 2 was in cell permeability, we sought to explore a prodrug approach and enhance compound 2’s cell permeability.[ 56 , 57 , 58 ]
Figure 1.

The effect of 100 μM Bis‐T‐22 (2) on the uptake of a transferrin red dye (TxR‐Tfn) by endocytosis. A/B Control using Swiss 3T3 cells (no compound), A. TxR‐Tfn stain evident; B. TxR‐Tfn colocalised with DAPI (nuclear staining); C/D with 100 μM 2 added; E/F Control using COS7 cells (no compound), E. TxR‐Tfn stain evident; F. TxR‐Tfn colocalised with DAPI (nuclear staining); G/H with 100 μM 2 added.
Our initial investigations provided no detail on the mechanism by which 2 inhibited dynamin. In principle there are multiple points in the dynamin endocytosis cycle that a small molecule could block dynamin action. In the formative stages, dynamin associates with the plasma membrane via its PH (pleckstrin homology, lipid binding) domain; at the later power stroke phase, dynamin hydrolyses GTP to GDP (providing the energy required to drive vesicle scission). We investigated the effect of 2 in both of these scenarios (Figure 2).
Figure 2.
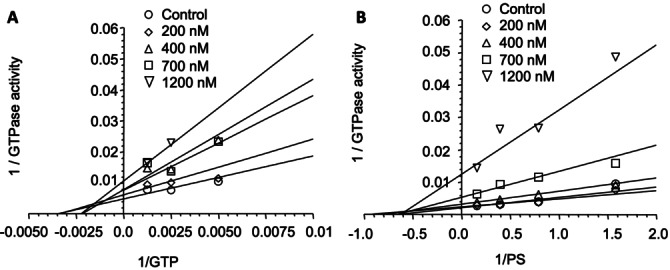
The effect of 2 on dynamin activity with increasing GTP concentration; B. Effect of 2 on dynamin activity with increasing phospholipid (PS) concentration.
Our analysis reveals that Bis‐T‐22 (and by association, this class of compounds) effects dynamin inhibition in a non‐GTP and non‐lipid dependent manner. It is possible that Bis‐T‐22 affects dynamin assembly, regardless of the exact mode of action, this inhibition offers a different point of intervention in dynamin‐dependent endocytosis than other reported compounds from our laboratories. The naphthaladyns, [38] quinodyns [37] and pthaladyns [31] are GTP competitive, the pyrimidyns are mixed competitive with GTP and lipids, [59] the MiTMABs are PH domain (lipid) competitive, [26] while the dynoles are presumed to be allosteric inhibitors and the dyngos also block dynamin assembly. [32]
Thus, developing a cell permeable Bis‐T‐22 analogue would afford the biology community an additional orthogonal chemical scaffold and mode of action inhibitor to explore the effect of dynamin inhibition on biological systems. To this end, we developed a number of Bis‐T‐22 (2) analogues incorporating commonly utilised prodrug moieties, which comprised alkyl esters (acetic acid 3, propionic acid 4, butyric acid 5, isobutyric acid 6, hexanoic acid 8 and trimethyl acetic acid 9), an isobutyl carbonate (7) and a dimethylcarbamate (10). The synthesis of these derivatives from the parent compound Bis‐T‐22 (2) is described in Scheme 1.
Scheme 1.

Reagents and conditions: i) neat, 25 °C, 2 h ii) piperidine (cat), EtOH, reflux, 2 h iii) acid anhydride (for 3, 4, 5, 6 and 8) or isobutyl chloroformate (7), DMAP, pyridine, 0 to 25 °C, 2 h iv) trimethylacetic anhydride (9) or dimethylcarbamyl chloride (10), K2CO3, DMF, 25 °C, 18 h.
In a typical synthesis, treatment of propane‐1,3‐diamine with methyl cyanoacetate gave the required 1,3‐dicyanoamide (1), which under Knoevenagel condensation conditions, with excess 3,4‐dihydroxybenzaldehyde gave 2. [41] Treatment of 2 with the appropriate acid anhydride or isobutyric chloroformate and DMAP generated esters 3, 4, 5, 6, 8 and carbonate 7, respectively. Bis‐T‐22 (2) was also reacted with trimethylacetic anhydride and dimethyl carbomylchloride in the presence of K2CO3 to afford ester 9 and carbamate 10, respectively. Compounds 3–10 were then submitted for biological evaluation and, as expected, displayed no in vitro dynamin inhibitory activity at 300 or 500 μM. This is consistent with the previously described pharmacophore requirements for this compound class.[ 40 , 41 ]
To evaluate their suitability as potential endocytosis inhibitors, a number of physicochemical properties of prodrugs 3–10 were determined (Table 1). It can be seen that all the prodrugs display greater lipophilicity (Log P) than Bis‐T‐22 (2). Compounds 3, 8 and 9 were insoluble in the buffer solution, and were not investigated further. Given our desire to develop cell permeable prodrugs, we examined the aqueous stability of the remaining analogues (4–7 and 10) by HPLC analysis (24 h, pH 7.4 phosphate buffer). No detectable decomposition was noted (Table 1).
To be useful as prodrugs of 2, analogues must be readily converted to the active parent compound in vivo. Thus, stock solutions (100 μM) of 4–7 and 10 in 5 % rat blood plasma at 37 °C for 24 h. Aliquots were taken at regular time intervals and the half‐lives determined by HPLC peak area analysis of the prodrug, Bis‐T‐22 (2), and an internal standard (benzophenone). Under these conditions, prodrugs 4–7 were quantitatively hydrolysed to 2 and, as expected, the stability of the prodrug increased with the steric bulk of the ester group (Table 2). Dimethylcarbamate 10 exhibited significant enzymatic stability with no evidence of hydrolysis even on increasing the rat plasma concentration to 80 % and 24 h incubation. This is consistent with the prolonged lifetime exhibited by prodrugs which contain this moiety.[ 60 , 61 ]
The hydrolysis of propionic acid ester 4 was then followed by HPLC analysis (Figure 3). Upon the addition of plasma, compound 4 was rapidly converted into a number of well resolved intermediate adducts, presumably containing one or more prodrug moieties (Figure 3C). As the concentration of the intermediates increased, hydrolysis of 4 slows (Figure 3E >10 min) due to preferential hydrolysis of the more sterically accessible intermediates. This corresponds to a steady increase in the formation of 2 until hydrolysis of 4 was complete (Figure 3E ≈60 min).
Figure 3.

HPLC traces illustrating the cleavage of prodrug 4 to generate Bis‐T‐22 in 5 % rat blood plasma. Panels (A) and (B) show 100 μM solutions of prodrug 4 (Rt = 14.8 min) and Bis‐T‐22 (2) (Rt=5 min) respectively. Panels (C) and (D) show compound 4 after a 10 min and 60 min incubation in 5 % rat plasma respectively. All HPLC traces contain benzophenone as an internal standard (Rt=10.5 min). Panel (E) shows the relationship between the hydrolysis of 4 and the formation of Bis‐T‐22 (2) (data calculated based on the HPLC determined peak area relative to internal standard).
Encouraged by the calculated increase in membrane permeability of 4 and its rapid in vivo conversion to 2, we sought to ascertain the efficacy of prodrug 4 as an in‐cell endocytosis inhibitor. We examined the effect of 30 μM of 4 and 100 μM Bis‐T‐22 (2) on EGF‐TxR uptake in HeLa cells (Figure 4). EGF is internalised into cells by clathrin‐mediated endocytosis (CME), which is a dynamin‐dependent process. Both the control (DMSO vehicle only) and the Bis‐T‐22 (2) treated cells showed significant EGF‐TxR internalisation, indicating no effect on CME. In contrast, the cells treated with prodrug 4 displayed a marked reduction in CME and a significant increase in green fluorescence.
Figure 4.

Panel A&D, control DMSO vehicle‐treated HeLa cells, no compound with images recorded in A the red channel, and D in the green channel. B&E The effects of 100 μM Bis‐T‐22 2 (B, red channel; E, green channel) on the uptake of EGF‐TxR dye in HeLa cells. C&F the effects of 30 μM prodrug 4 (C, red channel; F, green channel) on internalisation of EGF‐Tx in HeLa cells. The control cells show level of autofluorescence in the green channel and the product Bis‐T‐22 is highly fluorescent in the green channel.
To unequivocally demonstrate that the green fluorescence was due to the presence of Bis‐T‐22 (2) within the cells, we recorded the fluorescence emission spectra of Bis‐T‐22 (2) and prodrug 4 in the region where the green signal is measured (515–555 nm). We chose a 30 min preincubation time plus an additional 10 min ligand stimulation to potentially produce up to 80 % conversion from prodrug to active drug, based on the data in Figure 4F. Bis‐T‐22 (2) displayed significant green fluorescence whereas 4 exhibited minimal fluorescence in the region of interest (Figure 5). The results are consistent with the efficient diffusion of prodrug 4 across the cell membrane and is effectively cleaved, in vivo, to liberate the active compound, Bis‐T‐22 (2).
Figure 5.
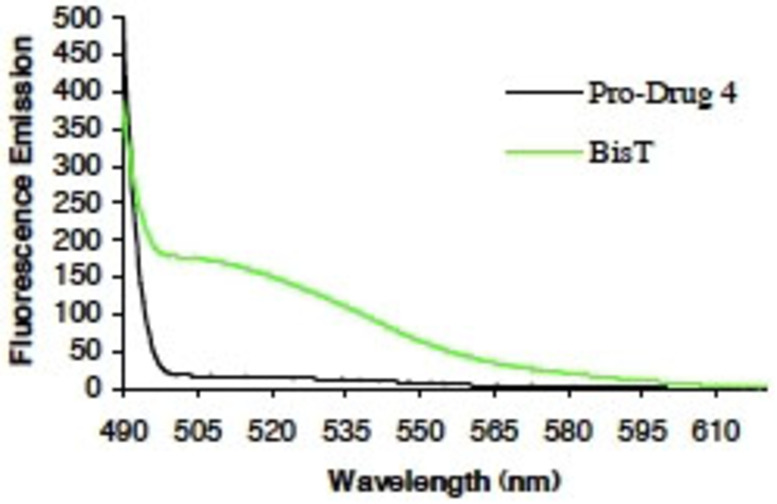
Fluorescence emission spectra of Bis‐T‐22 (2) and prodrug 4. Bis‐T‐22 (2), but not 4, exhibits significant and broad fluorescence in the green region (515–555 nm) of the spectra.
To further examine the in‐cell impact of prodrug 4 on CME, a quantitative study was carried out using a second ligand, transferrin, and a second cell line (COS7 cells) (Figure 6). Transferrin uptake (i. e. CME) was determined by measuring fluorescence intensity in cells treated with 4 relative to control cells which were treated with DMSO vehicle only. Data was collected from 100 randomly selected cells. Prodrug 4 inhibited transferrin CME in a concentration‐dependent manner, with an in‐cell IC50 (8.0±0.5 μM), similar to the in vitro IC50 of Bis‐T‐22 (2) (1.7±0.2 μM) (Figure 4). As the actual concentration of dynamin within the cell is unknown, a direct comparison of IC50 in vitro and in‐cell is not appropriate. This combined with our observation of significant in cell fluorescence, which we have demonstrated arises from prodrug 4 hydrolysis to 2 (cf Figures 4 and 5) suggests that diffusion of 4 across the cell membrane is efficient, with little extracellular hydrolysis taking place. Furthermore, the level of endocytosis is closely correlated (r2=−0.95) to the intensity of green fluorescence exhibited by the affected cell. This is consistent with the efficient cellular internalisation and rapid hydrolysis of 4 to yield the active parent compound, Bis‐T‐22 (2).
Figure 6.
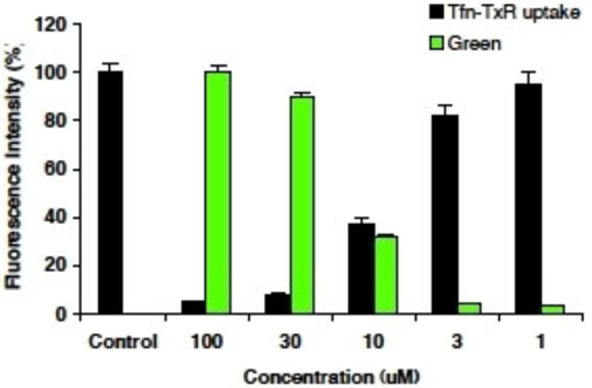
The effect of prodrug 4 on internalisation of Texas‐red EGF (Tfn) in COS7 cells. The values represent the mean data for 100 randomly selected drug treated cells relative to the control cells. Prodrug 4 pre‐incubated with cells for 30 min prior to addition of Tfn, 10 min incubation with Tfn present prior to HPLC analysis. Note a red‐tagged ligand is required since Bis‐T‐22 interferes in the green channel, preventing CME analysis.
In light of its successful CME inhibition in COS7 and HeLa cells, we sought to examine the effectiveness of prodrug 4 in other cell lines. At 30 μM prodrug 4, but not the parental Bis‐T‐22, inhibited transferrin and EGF uptake (where applicable) in HER14, B104, B35, Swiss 3T3 and A431 cells (data not shown).
Conclusion
We have shown that small molecule inhibitors of dynamin GTPase activity can be used to block CME in cells if rendered cell‐permeable. Bis‐T‐22 (2) is an in vitro dynamin inhibitor with an IC50 of 1.7 μM, however it has low or no effect on CME in multiple cell lines tested (not shown). Consequently, a prodrug strategy was employed in an attempt to improve the in‐cell availability of Bis‐T‐22 (2). A number of prodrug candidates were prepared and their physical properties determined (lipophilicity (log P), enzymatic and chemical stability).
The propionic acid ester 4 displayed a calculated increase in membrane permeability and was rapidly hydrolysed by non‐specific esterases in plasma or in cells to liberate the active parent, Bis‐T‐22 (2). This was also indicated by a large increase in fluorescence in the green channel as the active Bis‐T‐22 (2) was formed. Thus, it was prioritised for further evolution and was found to inhibit CME of two different ligands in two different cell lines. The observed potency in COS‐7 cells was 8.0±0.5 μM. In conclusion, Bis‐T‐22 (2) and its propionic acid ester prodrug 4 represent novel small molecule inhibitors of endocytosis.
Experimental Section
Chemistry
General methods
THF was freshly distilled from sodium‐benzophenone. Flash chromatography was carried out using silica gel 200–400 mesh (60 Å). 1H and 13C NMR were recorded at 300 MHz and 75 MHz respectively using a Bruker Avance 300 MHz spectrometer in CDCl3 and DMSO‐d6 . GCMS was performed using a Shimadzu GCMS‐QP2100. The instrument uses a quadrupole mass spectrometer and detects samples via electron impact ionization (EI). The University of Wollongong, Australia, Biomolecular Mass Spectrometry Laboratory analyzed samples for HRMS. The spectra were run on the VG Autospec‐oa‐tof tandem high resolution mass spectrometer using CI (chemical ionization), with methane as the carrier gas and PFK (perfluorokerosene) as the reference. Microanalysis was conducted at the Microanalyis Unit at the Australian National University, Canberra Australia. All samples returned satisfactory analyses. Compound purity was confirmed by a combination of LC–MS (HPLC), micro and/or high resolution mass spectrometry and NMR analysis. All analogues are≥95 % purity.
2‐Cyano‐N‐[3‐(2‐cyanoacetylamino)propyl]acetamide (1) [62]
Propane‐1,3‐diamine (1.50 g, 25 mmol) and methyl cyanoacetate (5.00 g, 50 mmol) were stirred at 25 °C for 2 hr. The resulting white solid was then mixed with 10 mL ethanol and collected by filtration. Recrystallisation from ethanol gave the title compound as a white solid. Yield 4.95 g (81 %) mp 146 °C (Lit [148] °C). [29] 1H NMR (DMSO‐d6 ): 1.55 (quin, J=6.9 Hz, 2H), 3.08 (q, J=6.9 Hz, 4H), 3.60 (s, 4H), 8.22 (br s, 2H); 13C NMR (DMSO‐d6 ): 25.3 (2C), 28.5, 36.9 (2C), 116.2 (2C), 162.1 (2C).
2‐Cyano‐N‐{3‐[2‐cyano‐3‐(3,4‐dihydroxyphenyl)acryloylamino]propyl}‐3‐(3,4‐dihydroxyphenyl)acrylamide (2)
To a solution of 1 (1.00 g, 4.8 mmol) and 3,4‐dihydroxybenzaldehyde (1.45 g, 10.5 mmol) in EtOH (25 mL) was added piperidine (2 drops). The resulting mixture was refluxed for 2 hr, cooled to room temperature, diluted with 1 M HCl (10 mL) and extracted with EtOAc (3×50 mL). The combined extracts were dried (MgSO4), evaporated and the residue recrystallised from MeOH to give the desired compound as a yellow solid. Yield 1.89 g (88 %) mp 274–275 °C (Lit [277] °C).10 1H NMR (DMSO‐d6 ): 1.68–1.77 (m, 2H), 3.24–3.28 (m, 4H), 6.87 (d, J=8.4 Hz, 2H), 7.28 (dd, J=2.1, 8.4 Hz, 2H), 7.53 (d, J=2.1 Hz, 2H), 7.93 (s, 2H), 8.19 (br s, 2H); 13C NMR (DMSO‐d 6, DEPTQ): 28.9, 37.4 (2C), 100.5 (2C), 115.9 (2C), 116.0 (2C), 117.3 (2C), 123.3 (2C), 125.3 (2C), 145.7 (2C), 150.7 (2C), 150.9 (2C), 161.7 (2C).
Acetic acid 2‐acetoxy‐5‐(2‐cyano‐2‐{3‐[2‐cyano‐3‐(3,4‐diacetoxyphenyl) acryloylamino]propylcarbamoyl}vinyl)phenyl ester (3)
To a cooled (0 °C) solution of 2 (Bis‐T‐22 (0.50 g, 1.1 mmol) and DMAP (0.014 g, 0.11 mmol) in pyridine (3 mL) was added acetic anhydride (0.88 g, 8.8 mmol). The mixture was stirred at 25 °C for 2 hrs and poured over crushed ice. The resulting precipitate was collected and washed with water. The crude product was recrystallised from acetone to yield to the title compound as an off‐white solid. Yield 0.430 g (63 %) mp 242–244 °C. 1H NMR (DMSO‐d6 ): 1.77 (m, 2H), 2.32 (s, 12H), 3.28 (m, 4H, obscured), 7.49 (d, J=8.5 Hz, 2H), 7.84 (d, J=1.9 Hz, 2H), 7.89 (dd, J=1.9, 8.5 Hz, 2H), 8.17 (s, 2H), 8.48 (br t, J=5.5 Hz, 2H).; 13C NMR (DMSO‐d6 ): 20.26 (2C), 20.35 (2C), 28.6, 37.6 (2C), 107.1 (2C), 116.0 (2C), 124.6 (2C), 125.0 (2C), 128.6 (2C), 130.5 (2C), 142.3 (2C), 144.7 (2C), 148.8 (2C), 160.7 (2C), 167.9 (2C), 168.1 (2C).
Propionic acid 5‐(2‐{3‐[3‐(3,4‐bis‐propionyloxyphenyl)‐2‐cyanoacryloylamino]propylcarbamoyl}‐2‐cyanovinyl)‐2‐propionyloxyphenyl ester (4)
Synthesised using the general procedure as for 3 and propionic anhydride. Afforded an off white solid. Yield 0.484 g (65 %) mp 146–148 °C. 1H NMR (DMSO‐d6 ): 1.14 (t, J=7.5 Hz, 12H), 1.77 (m, 2H), 2.63 (qd, J=2.6, 7.5 Hz, 8H), 3.28 (m, 4H, obscured), 7.50 (d, J=8.5 Hz, 2H), 7.84 (d, J=2.0 Hz, 2H), 7.90 (dd, J=2.0, 8.5 Hz, 2H), 8.17 (s, 2H), 8.47 (br t, J=5.6 Hz, 2H); 13C NMR (DMSO‐d6 ): 8.7 (2C), 8.8 (2C), 26.6 (2C), 26.7 (2C), 28.6, 37.5 (2C), 107.0 (2C), 116.0 (2C), 124.5 (2C), 125.0 (2C), 128.4 (2C), 130.4 (2C), 142.2 (2C), 144.7 (2C), 148.7 (2C), 160.6 (2C), 171.1 (2C), 171.3 (2C). C35H36N4O10: Calc C62.49, H5.39 N8.33 Anal C62.12, H5.49, N7.99.
Butyric acid 5‐(2‐{3‐[3‐(3,4‐bis‐butyryloxyphenyl)‐2‐cyanoacryloylamino]propylcarbamoyl}‐2‐cyanovinyl)‐2‐butyryloxyphenyl ester (5)
Synthesised using the general procedure as for 3 and butyric anhydride. Afforded an off‐white solid. Yield 0.440 g (55 %) mp 144–145 °C. 1H NMR (DMSO‐d6 ): 0.97 (t, J=7.3 Hz, 12H), 1.64 (sex, J=7.3 Hz, 8H), 1.78 (m, 2H), 2.59 (td, J=1.6, 7.3 Hz, 8H), 3.28 (m, 4H, obscured), 7.49 (d, J=8.5 Hz, 2H), 7.84 (d, J=1.6 Hz, 2H), 7.90 (dd, J=1.6, 8.5 Hz, 2H), 8.17 (s, 2H), 8.47 (br t, J=5.3 Hz, 2H); 13C NMR (DMSO‐d6 ): 13.21 (2C), 13.23 (2C), 17.7 (2C), 17.8 (2C), 28.6, 34.8 (2C), 34.9 (2C), 37.5 (2C), 107.1 (2C), 116.0 (2C), 124.6 (2C), 125.1 (2C), 128.4 (2C), 130.5 (2C), 142.2 (2C), 144.7 (2C), 148.7 (2C), 160.6 (2C), 170.3 (2C), 170.4 (2C).
Isobutyric acid 5‐(2‐{3‐[3‐(3,4‐bis‐isobutyryloxyphenyl)‐2‐cyanoacryloylamino]propylcarbam‐oyl}‐2‐cyanovinyl)‐2‐isobutyryloxyphenyl ester (6)
Synthesised using the general procedure as for 3 and isobutyric anhydride. Afforded an off white solid. Yield 0.435 g (54 %) mp 142–144 °C. 1H NMR (DMSO‐d6 ): 1.23 (d, J=6.8 Hz, 24H), 1.77 (m, 2H), 2.85 (m, 4H), 3.29 (m, 4H), 7.49 (d, J=8.4 Hz, 2H), 7.83 (br s, 2H), 7.91 (d, J=8.4 Hz, 2H), 8.17 (s, 2H), 8.47 (br t, J=4.9 Hz, 2H); 13C NMR (DMSO‐d6 ): 18.47 (4C), 18.50 (4C), 28.6, 33.1 (2C), 33.2 (2C), 37.5 (2C), 107.1 (2C), 116.0 (2C), 124.5 (2C), 125.2 (2C), 128.2 (2C), 130.5 (2C), 142.2 (2C), 144.8 (2C), 148.6 (2C), 160.6 (2C), 173.5 (2C), 173.7 (2C).
Isobutyloxylcarbonyl acid 5‐(2‐{3‐[3‐(3,4‐bis‐isobutyryloxycarbonylphenyl)‐2‐ cyanoacryloylamino] propylcarbamoyl}‐2‐cyanovinyl)‐2‐isobutyryloxycarbonylphenyl ester (7)
Synthesised using the general procedure as for 3 and isobutyl chloroformate to afford an off‐white solid. Yield 0.552 g (63 %) mp 137–139 °C. 1H NMR (DMSO‐d6 ): 0.93 (d, J=6.7 Hz, 24H), 1.76 (m, 2H), 1.98 (m, 4H), 3.29 (q, J=5.6 Hz, 4H), 4.05 (d, J=6.5 Hz, 8H), 7.65 (d, J=8.4 Hz, 2H), 7.95 (m, 4H), 8.21 (s, 2H), 8.54 (br t, J=5.2 Hz, 2H); 13C NMR (DMSO‐d6 ): 18.4 (8C), 27.22 (2C), 27.23 (2C), 28.5, 37.5 (2C), 74.8 (2C), 74.9 (2C). 107.7 (2C), 115.9 (2C), 124.2 (2C), 124.6 (2C), 128.8 (2C), 131.0 (2C), 142.2 (2C), 144.4 (2C), 148.4 (2C), 151.6 (2C), 151.8 (2C), 160.5 (2C).
Hexanoic acid 5‐(2‐{3‐[3‐(3,4‐bis‐hexanoyloxyphenyl)‐2‐cyanoacryloylamino]‐propylcarbamoyl}‐2‐cyanovinyl)‐2‐hexanoyloxyphenyl ester (8)
Synthesised using the general procedure as for 3 and hexanoic anhydride to afford an off‐white solid. Yield 0.621 g (67 %) mp 147–148 °C. 1H NMR (DMSO‐d6 ): 0.89 (m, 12H), 1.33 (m, 16H), 1.63 (m, 8H), 1.77 (m, 2H), 2.59 (t, J=6.6 Hz, 8H), 3.29 (m, 4H, obscured), 7.49 (d, J=8.4 Hz, 2H), 7.84 (d, J=1.9 Hz, 2H), 7.89 (dd, J=1.9, 8.4 Hz, 2H), 8.17 (s, 2H), 8.47 (br t, J=5.5 Hz, 2H); 13C NMR (DMSO‐d6 ): 13.7 (4C), 21.7 (4C), 23.87 (2C), 23.95 (2C), 28.6, 30.5 (4C), 33.0 (2C), 33.1 (2C), 37.5 (2C), 107.1 (2C), 116.0 (2C), 124.6 (2C), 125.0 (2C), 128.4 (2C), 130.4 (2C), 142.1 (2C), 144.7 (2C), 148.7 (2C), 160.6 (2C), 170.3 (2C), 170.5 (2C).
2,2‐Dimethylpropionic acid 5‐[2‐(3‐{3‐[3,4‐bis‐(2,2‐dimethylpropionyloxy)phenyl]‐2‐cyanoacryloyl amino}propylcarbamoyl)‐2‐cyanovinyl]‐2‐(2,2‐dimethylpropionyloxy)phenyl ester (9)
To a cooled (0 °C) solution of 2 (Bis‐T‐22) (0.50 g, 1.1 mmol) and potassium carbonate (0.22 g, 2.2 mmol) in DMF (3 mL) was added trimethylacetic anhydride (1.63 g, 8.8 mmol). The mixture was stirred at 25 °C for 18 hr and diluted with water (10 mL) and EtOAc (30 mL). The organic layer was washed with brine (2×20 mL), dried (MgSO4) and concentrated under reduced pressure. The crude product was purified by flash column chromatography eluting with petroleum spirit/EtOAc (1 : 1) to yield the title compound as a light orange oil, which solidified upon standing. Yield 0.543 g (63 %) mp 149–151 °C. 1H NMR (DMSO‐d 6): 1.30 (s, 36H), 1.77 (m, 2H), 3.28 (m, 4H, obscured), 7.47 (d, J=8.5 Hz, 2H), 7.81 (d, J=1.7 Hz, 2H), 7.91 (dd, J=1.7, 8.5 Hz, 2H), 8.17 (s, 2H), 8.47 (br t, J=5.5 Hz, 2H); 13C NMR (DMSO‐d6 ): 26.7 (12C), 28.5, 37.5 (2C), 38.7 (4C), 107.1 (2C), 116.0 (2C), 124.5 (2C), 125.4 (2C), 127.9 (2C), 130.5 (2C), 142.4 (2C), 145.0, 148.6 (2C), 160.7 (2C), 174.9 (2C), 175.1 (2C).
Dimethylcarbamic acid 5‐(2‐{3‐[3‐(3,4‐bis‐dimethylcarbamoyloxyphenyl)‐2‐cyanoacryloylamino] propylcarbamoyl}‐2‐cyanovinyl)‐2‐dimethylcarbamoyloxy phenyl ester (10)
Synthesised using the general procedure as for 9 and dimethylcarbomyl chloride. Recrystallisation from acetone afforded an off‐white solid. Yield 0.435 g (54 %) mp 141–142 °C; 1H NMR (DMSO‐d6 ): 1.77 (quin, J=6.5 Hz, 2H), 2.93 (s, 12H), 3.01 (d, J=3.2 Hz, 12H), 3.29 (m, 4H, obscured), 7.47 (d, J=8.2 Hz, 2H), 7.83 (m, 4H), 8.16 (s, 2H), 8.46 (br t, J=5.5 Hz, 2H); 13C NMR (DMSO‐d6 ): 28.6, 35.9 (4C), 36.3 (4C), 37.5 (2C), 106.4 (2C), 116.1 (2C), 124.1 (2C), 124.7 (2C), 128.1 (2C), 129.6 (2C), 143.3 (2C), 146.0 (2C), 149.0, 152.4 (2C), 152.6 (2C), 160.7 (2C).
Biology
Materials
Phosphatidylserine (PS), phenylmethylsulfonylfluoride (PMSF) and Tween 80 were from Sigma‐Aldrich (St Louis, CA). GTP was from Roche Applied Science (Germany), leupeptin was from Bachem (Bubendorf, Switzerland). Gel electrophoresis reagents, equipment and protein molecular weight markers were from Bio‐Rad (Hercules, CA). Collagenase was from Roche. Paraformaldehyde (PFA) was from Merck Pty Ltd (Kilsyth, Australia). Coverslips were from Lomb Scientific (Sydney, Australia). Penicillin/streptomycin, phosphate buffered salts, foetal calf serum (FCS) and Dulbecco's Minimal Essential Medium (DMEM) were from Invitrogen (Mount Waverley, Victoria, Australia). Alexa‐594 conjugated Tf (Tf‐A594), and DAPI were from Molecular Probes (Oregon, USA). All other reagents were of analytical reagent grade or better.
Drugs
Drugs were made in‐house or were purchased from Sigma‐Aldrich. The drugs were made up as stock solutions in 100 % DMSO and diluted in 50 % v/v DMSO/20 mM Tris/HCl pH 7.4 or cell media prior to further dilution in the assay. The final DMSO concentration in the GTPase or endocytosis assays was at most 3.3 % or 1 % respectively. The GTPase assay for dynamin I was unaffected by DMSO up to 3.3 %. Drugs were dissolved as 30 mM stocks in 100 % DMSO and were light yellow in colour. Stocks were stored at −20 °C for several months. They were diluted into solutions of 50 % DMSO made up in 20 mM Tris/HCl pH 7.4 and then diluted again into the final assay.
Protein production
Dynamin I was purified from sheep brain by extraction from the peripheral membrane fraction of whole brain [63] and affinity purification on GST‐Amph2‐SH3‐sepharose as described, [64] yielding 8–10 mg protein from 200 g sheep brain.
Receptor‐mediated endocytosis assay
Transferrin (Tfn‐TxR) and EGF (EGF‐TxR) uptake was analysed in Swiss 3T3, HER14, B104, A431, B35, HeLa and COS7 cells, where applicable. All cells were grown in Dulbecco's modified essential medium (DMEM) supplemented with 10 % foetal calf serum (FCS) and cell media was removed and replaced with DMEM for 16 hrs before the experiment.
The drugs were added for 30 mins before the addition of Tfn‐TxR (final concentration 5.0 μg/mL) or EGF‐TxR (final concentration 1.0 μg/mL) and then incubated at 37 °C for 10 mins. All media was removed and replaced with ice‐cold acid wash solution (0.2 M acetic acid, 0.5 M NaCl, pH 2.8) to remove surface bound ligand for 10 min. This was then replaced with 4 % paraformaldehyde (pH 7.5) and incubated for 10 min at 25 °C. This was replaced with phosphate buffered saline (PBS) containing DAPI to stain nuclei and incubated for 10 min. The cells were washed in PBS before the coverslips were mounted on slides in media containing DABCO to prevent fading and sealed with nail polish.
Fluorescence was monitored using a Leica DMLB bright field microscope and SPOT digital camera. For quantitative analysis the average integrated intensity of the EGF‐TxR signal per cell was calculated for each well using Metamorph (Molecular Devices), and the data expressed as a percentage of control cells (vehicle treated only). Data was then analysed in Excel.
Aqueous stability
To 9900 μL of phosphate buffer (made up from 0.05 M NaH2PO4 and 0.05 M Na2HPO4, pH 7.4) was added 100 μL of the appropriate drug (10 mM stock in 100 % DMSO). The resulting solution was mixed and incubated for 24 hrs at 37 °C. 248 μL aliquots were taken at 0, 60, 120, 240, 540, and 24 hr and frozen on dry ice until analysis. The aliquots were thawed in water bath and benzophenone added (2 μL of 10 mM stock in DMSO). The aliquots were mixed and analysed by HPLC as described below.
Rat blood plasma stability
To 9850 μL of phosphate buffer (made up from 0.05 M NaH2PO4 and 0.05 M Na2HPO4, pH 7.4) was added 100 μL of the appropriate drug (10 mM stock in 100 % DMSO) followed by 50 μL of rat blood plasma (100 μM final drug concentration, 5 % final rat blood plasma concentration). The resulting solution was mixed and incubated for 24 hr at 37 °C. 248 μL aliquots were taken at 0, 2.5, 5, 10, 15, 20, 30, 40, 50, 60, 90, 120, 180, 240, 300, 420, 540, 720, 1440 and 24 hrs and frozen on dry ice until analysis. The aliquots were thawed in water bath (c.a. 15–20 °C to prevent esterase activity) and benzophenone added (2 μL of 10 mM stock in DMSO). The aliquots were mixed and analysed by HPLC as described below.
Rat blood plasma was obtained by bleed from the lateral tail vein of adult (>150 g weight) male Wistar rats. Blood was allowed to completely clot at 37degC for 60 min followed by 2 hr at room temperature, then centrifuged (1000×g) for 10 min and the serum was transferred to a new tube where 0.1 % sodium azide was used as preservative before storage at −20 °C. These experimental procedures were approved by the Children's Medical Research Institute / Children's Hospital Westmead Animal Care and Ethics Committee (ACEC number C116/05).
Malachite Green GTPase assay
The Malachite Green method was used for the sensitive colorimetric detection of orthophosphate (Pi). It is based on the formation of a phosphomolybdate complex at low pH with basic dyes, causing a colour change. The assay procedure is based on stimulation of native sheep brain‐purified dynamin I by sonicated phosphatidylserine (PS) liposomes [65] However, in our earlier studies we used 200 nM dynamin I while in the present study we used 20 nM requiring a reformulation of assay volumes and the Malachite Green reagent for the present study.[ 28 , 41 ] Purified dynamin I (20 nM) (diluted in 6 mM Tris/HCl, 20 mM NaCl, 0.01 % Tween 80, pH 7.4) was incubated in GTPase assay buffer (5 mM Tris/HCl, 10 mM NaCl, 2 mM Mg2+, 0.05 % Tween 80, pH 7.4, 1 μg/mL leupeptin and 0.1 mM PMSF) and GTP 0.3 mM in the presence of test compound for 30 min at 37 °C. The final assay volume was 150 μL in round bottomed 96 well plates. Plate incubations were performed in a dry heating block with shaking at 300 rpm (Eppendorf Thermomixer). Dynamin I GTPase activity was maximally stimulated by addition of 4 μg/ml phosphatidylserine (PS) liposomes (although this concentration slightly varied from batch to batch of dynamin I). The reactions were all terminated with 10 μL of 0.5 M EDTA pH 8.0 and the samples were stable for several hours at room temperature. To each well was added 40 μL of Malachite Green solution (2 % w/v ammonium molybdate tetrahydrate, 0.15 % w/v malachite green and 4 M HCl: the solution was passed through 0.45 μm filters and stored in the dark for up to 2 months at room temperature) and colour was allowed to develop for 5 min (and was stable up to 2 hour). The absorbance of samples in each plate was determined on a microplate spectrophotometer at 650 nm. Phosphate release was quantified by comparison with a standard curve of sodium dihydrogen orthophosphate monohydrate (baked dry at 110 °C overnight) which was run in each experiment. GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA) was used for plotting data points and analysis of enzyme kinetics using non‐linear regression. The curves were generated using the Michaelis‐Menten equation v=Vmax[S]/(Km+[S]) where S=PS activator or GTP substrate. After the Vmax and Km values were determined, the data was transformed using the Lineweaver‐Burke equation 1/v=1/Vmax+(Km/V)×(1/[S]).Dynamin I GTPase data are reported as basal activity subtracted from PS‐stimulated activity.
Analytical methods
HPLC experimental details
Bis‐T‐22 and its prodrug derivatives were determined by gradient reverse‐phase HPLC procedure using a Pharmacia Biotech SMART system equipped with a variable wavelength UV detector operated at 254 nm, and a 50μL injection loop. The chromatographic separation was achieved on a GRACEVYDAC C18‐column (5 μm, 100 mm, 2.1 mm) using a H2O (+0.1 % TFA) and CH3CN gradient at a flow rate of 1.0 mL/min. Quantitation of the compounds was done by measuring the peak areas in relation to a benzophenone standard. Data was then analysed using Excel.
Conflict of interest
We have a commercial agreement to supply dynamin inhibitors via Abcam UK
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by grants from the National Health and Medical Research Council (Australia) and The Australia Research Council. Open Access publishing facilitated by The University of Newcastle, as part of the Wiley ‐ The University of Newcastle agreement via the Council of Australian University Librarians.
Odell L. R., Robertson M. J, Young K. A, McGeachie A. B., Quan A., Robinson P. J., McCluskey A., ChemMedChem 2022, 17, e202200400.
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Ramachandran R., Schmid S. L., Curr. Biol. 2018, 28, R411–R416. [DOI] [PubMed] [Google Scholar]
- 2. Mettlen M., Chen P.-H., Srinivasan S., Danuser G., Schmid S. L., Annu. Rev. Biochem. 2018, 87, 871–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lee H., Yoon Y., Biochem. Soc. Trans. 2016, 44, 1725–1735. [DOI] [PubMed] [Google Scholar]
- 4. Khan I., Steeg P. S., Br. J. Cancer 2021, 124, 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sochacki K. A., Taraska J. W., Trends Cell Biol. 2018, 29, 241–256. [DOI] [PubMed] [Google Scholar]
- 6. Rosendale M., Van T. N. N., Grillo-Bosch D., Sposini S., Claverie L., Gauthereau I., Claverol S., Choquet D., Sainlos M., Perrais D., Nat. Commun. 2019, 10, 4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sundborger A. C., Fang S., Heymann J. A., Ray P., Chappie J. S., Hinshaw J. E., Cell Rep. 2014, 8, 734–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Z., Schmid S. L., J. Cell Biol. 2020, 219, e202005126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Conner S. D., Schmid S. L., Nature 2003, 422, 37–44. [DOI] [PubMed] [Google Scholar]
- 10. Reubold T. F., Faelber K., Plattner N., Posor Y., Ketel K., Curth U., Schlegel J., Anand R., Manstein D. J., Noé F., Haucke V., Daumke O., Eschenburg S., Nature 2015, 525, 404–408. [DOI] [PubMed] [Google Scholar]
- 11. Zhang J., Ding L., Holmfeldt L., Wu G., Heatley S. L., Payne-Turner D., Easton J., Chen X., Wang J., Rusch M., Lu C., Chen S.-C., Wei L., Collins-Underwood J. R., Ma J., Roberts K. G., Pounds S. B., Ulyanov A., Becksfort J., Gupta P., Huether R., Kriwacki R. W., Parker M., McGoldrick D. J., Zhao D., Alford D., Espy S., Bobba K. C., Song G., Pei D., Cheng C., Roberts S., Barbato M. I., Campana D., Coustan-Smith E., Shurtleff S. A., Raimondi S. C., Kleppe M., Cools J., Shimano K. A., Hermiston M. L., Doulatov S., Eppert K., Laurenti E., Notta F., Dick J. E., Basso G., Hunger S. P., Loh M. L., Devidas M., Wood B., Winter S., Dunsmore K. P., Fulton R. S., Fulton L. L., Hong X., Harris C. C., Dooling D. J., Ochoa K., Johnson K. J., Obenauer J. C., Evans W. E., Pui C.-H., Naeve C. W., Ley T. J., Mardis E. R., Wilson R. K., Downing J. R., Mullighan C. G., Nature 2012, 481, 157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Trochet D., Bitoun M., J. Exp. Clin. Cancer Res. 2021, 40, 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Prichard K., O'Brien N. S., Murcia S. R., Baker J., McCluskey A., Front. Cell. Neurosci. 2022, 15, 754110, doi: 10.3389/fncel.2021.754110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. González-Jamett A. M., Haro-Acuña V., Momboisse F., Caviedes P., Bevilacqua J. A., Cárdenas A. M., J. Neurochem. 2014, 128, 210–223. [DOI] [PubMed] [Google Scholar]
- 15. Li Y.-Y., Zhou J.-X., Fu X.-W., Bao Y., Xiao Z., Epilepsy Res. 2022, 182, 106915. [DOI] [PubMed] [Google Scholar]
- 16. Brodin L., Löw P., Shupliakov O., Curr. Opin. Neurobiol. 2000, 10, 312–320. [DOI] [PubMed] [Google Scholar]
- 17. Marks B., Stowell M. H. B., Vallis Y., Mills I. G., Gibson A., Hopkins C. R., McMahon H. T., Nature 2001, 410, 231–235. [DOI] [PubMed] [Google Scholar]
- 18. Hinshaw J. E., Annu. Rev. Cell Dev. Biol. 2000, 16, 483–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stowell M. H. B., Marks B., Wigge P., McMahon H. T., Nat. Cell Biol. 1999, 1, 27–32. [DOI] [PubMed] [Google Scholar]
- 20. Hinshaw J. E., Schmid S. L., Nature 1995, 374, 190–192. [DOI] [PubMed] [Google Scholar]
- 21. Sever S., Damke H., Schmid S. L., J. Cell Biol. 2000, 150, 1137–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lai M. M., Hong J. J., Ruggiero A. M., Burnett P. E., Slepnev V. I., Camilli P. D., Snyder S. H., J. Biol. Chem. 1999, 274, 25963–25966. [DOI] [PubMed] [Google Scholar]
- 23. Tan T. C., Valova V. A., Malladi C. S., Graham M. E., Berven L. A., Jupp O. J., Hansra G., McClure S. J., Sarcevic B., Boadle R. A., Larsen M. R., Cousin M. A., Robinson P. J., Nat. Cell Biol. 2003, 5, 701–710. [DOI] [PubMed] [Google Scholar]
- 24. Harper C. B., Martin S., Nguyen T. H., Daniels S. J., Lavidis N. A., Popoff M. R., Hadzic G., Mariana A., Chau N., McCluskey A., Robinson P. J., Meunier F. A., J. Biol. Chem. 2011, 286, 35966–35976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harper C. B., Popoff M. R., McCluskey A., Robinson P. J., Meunier F. A., Trends Cell Biol. 2013, 23, 90–101. [DOI] [PubMed] [Google Scholar]
- 26. Quan A., McGeachie A. B., Keating D. J., van Dam E. M., Rusak J., Chau N., Malladi C. S., Chen C., McCluskey A., Cousin M. A., Robinson P. J., Mol. Pharmacol. 2007, 72, 1425–1439. [DOI] [PubMed] [Google Scholar]
- 27. Joshi S., Perera S., Gilbert J., Smith C. M., Mariana A., Gordon C. P., Sakoff J. A., McCluskey A., Robinson P. J., Braithwaite A. W., Chircop M., Mol. Cancer Ther. 2010, 9, 1995–2006. [DOI] [PubMed] [Google Scholar]
- 28. Hill T. A., Odell L. R., Quan A., Abagyan R., Ferguson G., Robinson P. J., McCluskey A., Bioorg. Med. Chem. Lett. 2004, 14, 3275–3278. [DOI] [PubMed] [Google Scholar]
- 29. Zhang J., Lawrance G. A., Chau N., Robinson P. J., McCluskey A., New J. Chem. 2007, 32, 28–36. [Google Scholar]
- 30. Hill T. A., Mariana A., Gordon C. P., Odell L. R., Robertson M. J., McGeachie A. B., Chau N., Daniel J. A., Gorgani N. N., Robinson P. J., McCluskey A., J. Med. Chem. 2010, 53, 4094–4102. [DOI] [PubMed] [Google Scholar]
- 31. Odell L. R., Howan D., Gordon C. P., Robertson M. J., Chau N., Mariana A., Whiting A. E., Abagyan R., Daniel J. A., Gorgani N. N., Robinson P. J., McCluskey A., J. Med. Chem. 2010, 53, 5267–5280. [DOI] [PubMed] [Google Scholar]
- 32. McCluskey A., Daniel J. A., Hadzic G., Chau N., Clayton E. L., Mariana A., Whiting A., Gorgani N. N., Lloyd J., Quan A., Moshkanbaryans L., Krishnan S., Perera S., Chircop M., von Kleist L., McGeachie A. B., Howes M. T., Parton R. G., Campbell M., Sakoff J. A., Wang X., Sun J., Robertson M. J., Deane F. M., Nguyen T. H., Meunier F. A., Cousin M. A., Robinson P. J., Traffic 2013, 14, 1272–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Robertson M. J., Hadzic G., Ambrus J., Pomè D. Y., Hyde E., Whiting A., Mariana A., von Kleist L., Chau N., Haucke V., Robinson P. J., McCluskey A., ACS Med. Chem. Lett. 2012, 3, 352–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hill T. A., Gordon C. P., McGeachie A. B., Venn-Brown B., Odell L. R., Chau N., Quan A., Mariana A., Sakoff J. A., (nee Fabbro) M. C., Robinson P. J., McCluskey A., J. Med. Chem. 2009, 52, 3762–3773. [DOI] [PubMed] [Google Scholar]
- 35. Robertson M. J., Deane F. M., Robinson P. J., McCluskey A., Nat. Protoc. 2014, 9, 851–870. [DOI] [PubMed] [Google Scholar]
- 36. Gordon C. P., Venn-Brown B., Robertson M. J., Young K. A., Chau N., Mariana A., Whiting A., Chircop M., Robinson P. J., McCluskey A., J. Med. Chem. 2012, 56, 46–59. [DOI] [PubMed] [Google Scholar]
- 37. MacGregor K. A., Abdel-Hamid M. K., Odell L. R., Chau N., Whiting A., Robinson P. J., McCluskey A., Eur. J. Med. Chem. 2014, 85, 191–206. [DOI] [PubMed] [Google Scholar]
- 38. Abdel-Hamid M. K., Macgregor K. A., Odell L. R., Chau N., Mariana A., Whiting A., Robinson P. J., McCluskey A., Org. Biomol. Chem. 2015, 13, 8016–8028. [DOI] [PubMed] [Google Scholar]
- 39. Daniel J. A., Chau N., Abdel-Hamid M. K., Hu L., von Kleist L., Whiting A., Krishnan S., Maamary P., Joseph S. R., Simpson F., Haucke V., McCluskey A., Robinson P. J., Traffic 2015, 16, 635–654. [DOI] [PubMed] [Google Scholar]
- 40. Odell L. R., Chau N., Mariana A., Graham M. E., Robinson P. J., McCluskey A., ChemMedChem 2009, 4, 1182–1188. [DOI] [PubMed] [Google Scholar]
- 41. Hill T., Odell L. R., Edwards J. K., Graham M. E., McGeachie A. B., Rusak J., Quan A., Abagyan R., Scott J. L., Robinson P. J., McCluskey A., J. Med. Chem. 2005, 48, 7781–7788. [DOI] [PubMed] [Google Scholar]
- 42. Macia E., Ehrlich M., Massol R., Boucrot E., Brunner C., Kirchhausen T., Dev. Cell 2006, 10, 839–850. [DOI] [PubMed] [Google Scholar]
- 43. Takahashi K., Miyoshi H., Otomo M., Osada K., Yamaguchi N., Nakashima H., Biochem. Biophys. Res. Commun. 2010, 391, 382–387. [DOI] [PubMed] [Google Scholar]
- 44. Mohanakrishnan A., Tran T. V. M., Kumar M., Chen H., Posner B. A., Schmid S. L., Biorxiv 2017, 153106. [Google Scholar]
- 45. Mohanakrishnan A., Tran T. V. M., Kumar M., Chen H., Posner B. A., Schmid S. L., PLoS One 2017, 12, e0185639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Joshi S., Braithwaite A. W., Robinson P. J., Chircop M., Mol. Cancer 2011, 10, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lees J. G., Gorgani N. N., Ammit A. J., McCluskey A., Robinson P. J., O'Neill G. M., Biochim. Biophys. Acta, Mol. Cell Res. 2015, 1853, 611–618. [DOI] [PubMed] [Google Scholar]
- 48. Chircop M., Perera S., Mariana A., Lau H., Ma M. P. C., Gilbert J., Jones N. C., Gordon C. P., Young K. A., Morokoff A., Sakoff J., O'Brien T. J., McCluskey A., Robinson P. J., Mol. Cancer Ther. 2011, 10, 1553–1562. [DOI] [PubMed] [Google Scholar]
- 49. Tremblay C. S., Chiu S. K., Saw J., McCalmont H., Litalien V., Boyle J., Sonderegger S. E., Chau N., Evans K., Cerruti L., Salmon J. M., McCluskey A., Lock R. B., Robinson P. J., Jane S. M., Curtis D. J., Nat. Commun. 2020, 11, 6211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. von Beek C., Alriksson L., Palle J., Gustafson A.-M., Grujic M., Melo F. R., Sellin M. E., Pejler G., PLoS One 2021, 16, e0256708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chew H. Y., Lima P. O. D., Cruz J. L. G., Banushi B., Echejoh G., Hu L., Joseph S. R., Lum B., Rae J., O'Donnell J. S., de Long L. M., Okano S., King B., Barry R., Moi D., Mazzieri R., Thomas R., Souza-Fonseca-Guimaraes F., Foote M., McCluskey A., Robinson P. J., Frazer I. H., Saunders N. A., Parton R. G., Dolcetti R., Cuff K., Martin J. H., Panizza B., Walpole E., Wells J. W., Simpson F., Cell 2020, 180, 895–914.e27. [DOI] [PubMed] [Google Scholar]
- 52. Luwor R., Morokoff A. P., Amiridis S., D'Abaco G., Paradiso L., Stylli S. S., Nguyen H. P. T., Tarleton M., Young K. A., O'Brien T. J., Robinson P. J., Chircop M., McCluskey A., Jones N. C., Cancer Invest. 2019, 37, 1–12. [DOI] [PubMed] [Google Scholar]
- 53. Sever S., Chang J., Gu C., Traffic 2013, 14, 1194–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Schiffer M., Teng B., Gu C., Shchedrina V. A., Kasaikina M., Pham V. A., Hanke N., Rong S., Gueler F., Schroder P., Tossidou I., Park J.-K., Staggs L., Haller H., Erschow S., Hilfiker-Kleiner D., Wei C., Chen C., Tardi N., Hakroush S., Selig M. K., Vasilyev A., Merscher S., Reiser J., Sever S., Nat. Med. 2015, 21, 601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gu C., Lee H. W., Garborcauskas G., Reiser J., Gupta V., Sever S., J. Am. Soc. Nephrol. 2017, 28, 446–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Albert A., Nature 1958, 182, 421–423. [DOI] [PubMed] [Google Scholar]
- 57. Ettmayer P., Amidon G. L., Clement B., Testa B., J. Med. Chem. 2004, 47, 2393–2404. [DOI] [PubMed] [Google Scholar]
- 58. Beaumont K., Webster R., Gardner I., Dack K., Curr. Drug Metab. 2003, 4, 461–485. [DOI] [PubMed] [Google Scholar]
- 59. Odell L. R., Abdel-Hamid M. K., Hill T. A., Chau N., Young K. A., Deane F. M., Sakoff J. A., Andersson S., Daniel J. A., Robinson P. J., McCluskey A., J. Med. Chem. 2016, 60, 349–361. [DOI] [PubMed] [Google Scholar]
- 60. Olsson O. A. T., Svensson L.-Å., Pharm. Res. 1984, 1, 19–23. [DOI] [PubMed] [Google Scholar]
- 61. Tunek A., Levin E., Svensson L.-Å., Biochem. Pharmacol. 1988, 37, 3867–3876. [DOI] [PubMed] [Google Scholar]
- 62. Gazit A., Osherov N., Gilon C., Levitzki A., J. Med. Chem. 1996, 39, 4905–4911. [DOI] [PubMed] [Google Scholar]
- 63. Robinson P. J., Sontag J.-M., Liu J.-P., Fykse E. M., Slaughter C., McMahontt H., Südhof T. C., Nature 1993, 365, 163–166. [DOI] [PubMed] [Google Scholar]
- 64. Quan A., Robinson P. J., Methods Enzymol. 2005, 404, 556–569. [DOI] [PubMed] [Google Scholar]
- 65. van der Bliek A., Redelmeier T., Damke H., Tisdale E., Meyerowitz E., Schmid S., J. Cell Biol. 1993, 122, 553–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.