

Abstract
Background
Sickle cell disease is caused by a defect in the β-globin subunit of adult hemoglobin (HbA). Sickle hemoglobin (HbS) polymerizes under hypoxic conditions producing deformed red blood cells (RBCs) that hemolyze and cause vaso-occlusion, resulting in progressive organ damage and early death. Elevated fetal hemoglobin (HbF) in RBCs protects against sickle cell disease complications. OTQ923 is a CRISPR-Cas9–edited CD34+ hematopoietic stem and progenitor cell (HSPC) product harboring a targeted disruption of the HBG1 and HBG2 (γ-globin) gene promoters that increases HbF expression in RBC progeny.
Methods
We performed a tiling CRISPR-Cas9 screen of the HBG1/HBG2 promoters by electroporating healthy-donor CD34+ cells with Cas9 complexed with one of 72 gRNAs and assessed the fraction of HbF-immunostaining erythroblasts (F-cells) in erythroid differentiated progeny. The gRNA resulting in the highest levels of F-cells was selected for clinical development. We enrolled participants with severe sickle cell disease in a multi-center, Phase I/II clinical study and assessed for safety and tolerability of OTQ923.
Results
In preclinical experiments, CD34+ HSPCs from healthy donors and individuals with sickle cell disease, edited with CRISPR-Cas9 using gRNA-68, had sustained on-target editing with no off-target mutations and produced high levels of HbF after in vitro differentiation or xenotransplantation into immunodeficient mice. Three participants received autologous OTQ923 after myeloablative conditioning and have been followed for 6–18 months. All participants engrafted and achieved stable HbF induction (21.8 to 25%), with HbF broadly distributed in RBCs (F-cells 71 to 84.5%). Sickle cell disease manifestations were reduced during the follow-up period.
Conclusions
CRISPR-Cas9 disruption of the HBG1 and HBG2 gene promoters is an effective strategy for HbF induction. Infusion of autologous OTQ923 into three participants with severe sickle cell disease resulted in sustained induction of RBC HbF and clinical improvement in disease severity. (ClinicalTrials.gov number, NCT04443907.)
Introduction
Sickle cell disease is an autosomal recessive disorder caused by mutations in the HBB gene, which encodes the β-globin subunit of adult hemoglobin (HbA, α2β2).1 The most common sickle cell disease mutation causes a homozygous p.Glu6Val substitution, resulting in the production of sickle hemoglobin (HbS, α2βS2).2 HbS polymerizes at low oxygen concentrations, causing red blood cells (RBCs) to become sickle-shaped, rigid, and fragile, resulting in microvascular occlusion, hemolysis, and inflammation. Individuals with sickle cell disease experience chronic anemia, recurrent pain, progressive multi-organ damage, and early mortality.2,3 Sickle cell disease symptoms appear in infancy as γ-globin gene (HBG1 and HBG2) transcription switches to β-globin (HBB), causing a shift from fetal hemoglobin (HbF; α2γ2) to HbA in RBCs.
Medical therapies for sickle cell disease, including hydroxyurea, blood transfusions, and recently approved drugs (L-glutamine, crizanlizumab, and voxelotor) are only partially effective.4–8 The only potentially curative option is allogeneic hematopoietic stem cell transplantation (HSCT), ideally from a human leukocyte antigen (HLA)-matched donor, which is available for fewer than 20% patients.9 Allogeneic HSCT is associated with immunologic complications, including graft rejection and graft-versus-host disease, which tend to be worse with HLA-mismatched donors.9
HbF induction is a proven strategy for treating sickle cell disease.2,10–12 A naturally occurring benign genetic condition termed hereditary persistence of fetal hemoglobin, which results in pancellular elevation of HbF in postnatal RBCs, ameliorates symptoms of co-inherited sickle cell disease.2,3,13 Five paralogous β-like globin genes are located in the β-globin gene cluster on human chromosome 11p. HBE encoding ε-globin, HBG2 (Gγ) and HBG1 (Aγ) encoding γ-globin, HBD encoding δ-globin and HBB encoding β-globin are sequentially located on chromosome 11p in that order and get activated successively during human ontogeny from embryonic to adult life. Around the time of birth, the site of red blood cell production shifts from the fetal liver to the bone marrow, and this transition is associated with a switch from γ-globin (HBG1/HBG2) to β-globin (HBB) production.2 Consequently, HbF declines and HbA (or HbS in case of individuals with sickle cell disease) increases. The γ-to-β-globin switch is an intriguing paradigm of the developmental regulation of gene expression and is clinically important because β-hemoglobinopathies can be treated by inhibiting this switch. 2 This perinatal γ-globin–to–β-globin switch is mediated by transcriptional repressor proteins, BCL11A and ZBTB7A/LRF, that bind cognate cis-regulatory elements in the HBG1/HBG2 promoters.14 Inhibiting the binding of these repressors to their targets in adult RBC precursors can reactivate expression of γ-globin and HbF.14 Transduction of sickle cell disease patient hematopoietic stem cells (HSCs) with a lentiviral vector encoding an erythroid-expressed short hairpin RNA against BCL11A,10 or targeted disruption of a BCL11A erythroid-specific enhancer by the clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 nuclease system followed by autologous HSCT,11 resulted in elevated HbF and reduced sickle cell disease symptoms. Additional strategies to increase HbF through genetic manipulation of HSCs also exist.2,13 It is not yet known which strategies are the safest and most effective for clinical application.
OTQ923 is an autologous, ex vivo CRISPR-Cas9–edited, CD34+ HSC product with a targeted disruption in the HBG1 and/or HBG2 promoters by a ribonucleic acid–protein (RNP) complex consisting of Streptococcus pyogenes Cas9 protein complexed to a single guide RNA (gRNA-68) discovered in an unbiased screen for cis-regulatory elements that repress HBG1/HBG2 transcription. gRNA-68 targets a site 246 bp upstream of the transcriptional start in each of the nearly identical tandem HBG1 and HBG2 genes (Fig. 1A), resulting in multiple editing outcomes, including a ~5-kb intergenic deletion that produces a single hybrid gene with the HBG2 promoter sequence fused to the downstream HBG1 (Fig. 1B). In preclinical studies, transfecting CD34+ HSCs with gRNA-68–Cas9 RNP produced near-pancellular HbF induction in erythroid progeny, like that observed in hereditary persistence of fetal hemoglobin. Here we describe the outcomes of the first three participants with sickle cell disease who were infused with OTQ923 in a Phase I/II clinical study (NCT04443907).
Figure 1. OTQ923 molecular approach and preclinical characterization.
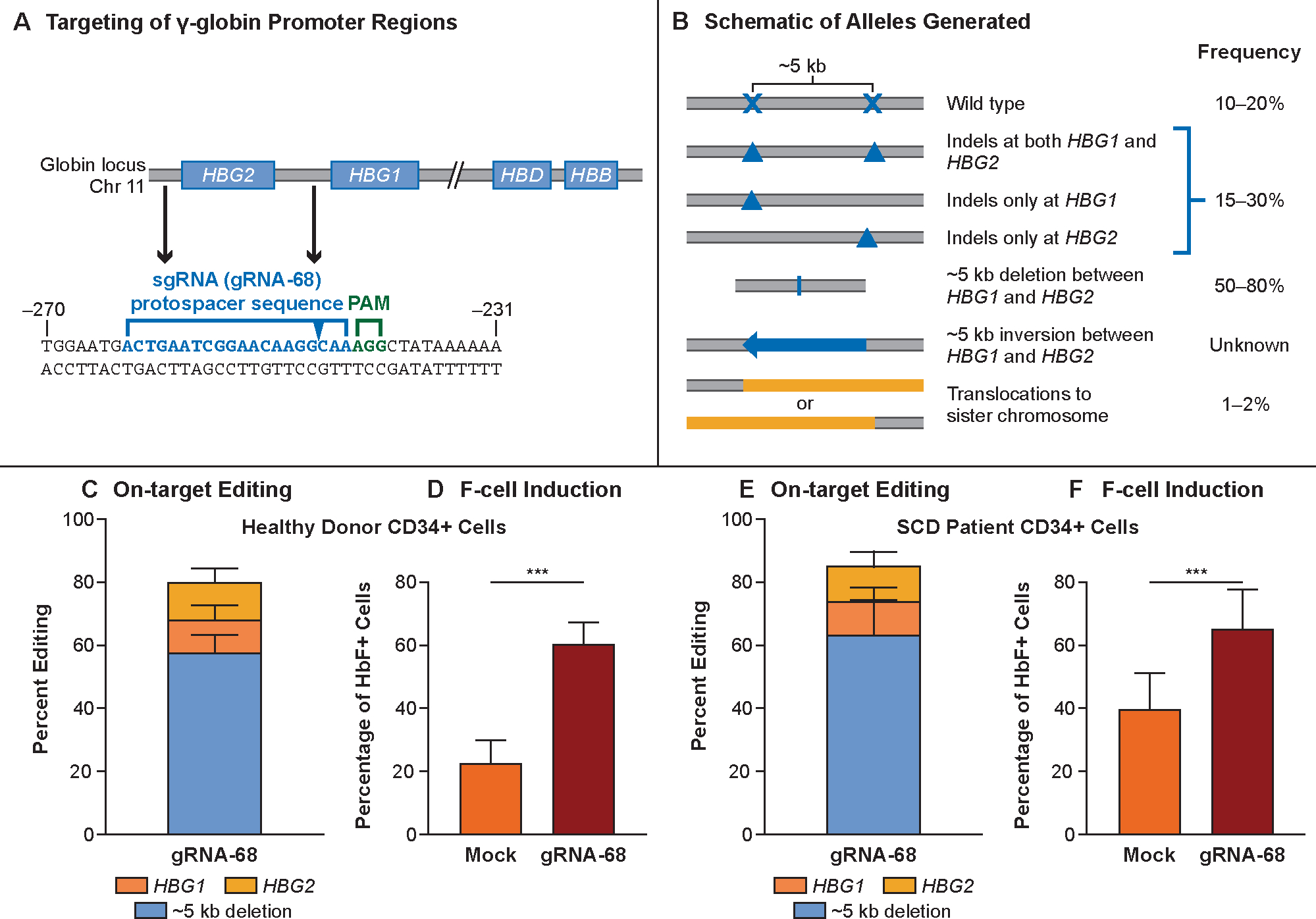
Panel A shows the targeting site of the ribonucleic acid–protein complex (RNP) consisting of Cas9 complexed with guide RNA-68 (gRNA-68). Genes in the β-like globin cluster on chromosome 11 are shown as boxes, with RNP targeting sites in HBG1 and HBG2 promoters indicated. The protospacer sequence of gRNA-68 is shown below in blue, and the protospacer adjacent motif (PAM) sequence is shown in green. The sequence is numbered in accordance with the start of transcription being at +1. The predicted cutting site is indicated by the blue arrow in the protospacer sequence. Panel B shows the editing outcomes as determined by qPCR,30 targeted NGS, and UnIT (Supplemental Methods). Most edits generated a deletion of ~5 kb caused by simultaneous double-stranded DNA breaks at the two target sites, thereby generating a single functional HBG2–HBG1 fusion gene with the HBG2 promoter sequence fused to the downstream HBG1 (48.8–87.4% alleles). Editing also resulted in small indels at either targeting site that accounted for 7.1–36.9% of alleles. No inversion of the ~5-kb fragment was detected. Panel C shows the distribution of editing outcomes in OTQ923 or control cells generated from healthy-donor CD34+ cells (n=4). Panel D shows the percentages of F-cells in erythroblasts generated by in vitro differentiation of OTQ293 or control cells described in Panel C (n=4 healthy donors). Panel E shows the amount of HbF protein estimated by HPLC in erythroblasts generated by in vitro differentiation of OTQ293 or control cells derived from healthy donors (n=2 healthy donors). Panel F shows the distribution of editing outcomes in OTQ923 cells or control cells generated from individuals with sickle cell disease (n=4). Panel G shows the percentages of F-cells in erythroblasts generated by in vitro differentiation of OTQ293 or control cells described in Panel E (n=3 sickle cell disease donors). Panel H shows the amount of HbF protein estimated by HPLC in erythroblasts generated by in vitro differentiation of OTQ293 or control cells derived from sickle cell disease patients (n=3). All graphs show data as the mean ± SD.
Methods
A tiled gRNA CRISPR-Cas9 screen to identify potential regulatory regions within the HBG1/HBG2 promoters
To identify new DNA targets for genetic induction of HbF, we performed a CRISPR-Cas9 screen by electroporating healthy-donor CD34+ cells with Cas9 complexed with one of 72 gRNAs tiled across the HBG1/HBG2 promoters (Table S1). The on-target editing efficiencies and the fraction of HbF-immunostaining erythroblasts (F-cells) were determined after in vitro erythroid differentiation, and the gRNAs resulting in the highest levels of F-cells were selected for further development (Supplemental Methods). Edited human HSCs were xenotransplanted into sublethally irradiated mice to assess the durability of editing and the extent of multi-lineage reconstitution.
Clinical study oversight
This study was sponsored by Novartis Pharmaceuticals. The trial was designed by Novartis Pharmaceuticals in collaboration with the authors. The research protocol was approved by the institutional review boards or ethics committees at the participating sites, and the study conduct was monitored by an independent data safety and monitoring committee. Each participant provided informed consent. Data collection and analysis were performed by Novartis Pharmaceuticals in collaboration with the authors. The first author wrote the first draft of the manuscript and subsequently revised it in consultation with the last and the penultimate authors. All authors had full access to the trial data and critically reviewed the manuscript. No one who is not an author contributed to writing the manuscript. The investigators vouch for the accuracy and completeness of data generated at their respective sites, and the investigators and Novartis Pharmaceuticals vouch for the fidelity of the trial to the protocol. Confidentiality agreements were in place between the sponsor and each investigative site during the trial.
Trial design and eligibility
This ongoing study enrolls adults between 18 and 40 years of age with a confirmed diagnosis of sickle cell disease and at least one of the following indicators of disease severity: three episodes of vaso-occlusive crisis or two episodes of acute chest syndrome within the previous 2 years, recurrent priapism, a history of stroke, a chronic transfusion requirement, or RBC alloimmunization. Detailed inclusion/exclusion criteria are included in the clinical trial protocol available at NEJM.org.
OTQ923 production and infusion
Trial participants received monthly RBC exchange transfusions for at least 2 months before CD34+ cell collection. CD34+ HSCs were mobilized with plerixafor,15 collected via apheresis,16 cryopreserved, and shipped to a central manufacturing facility (Fig. S1). After thawing, the apheresis product was enriched for CD34+ cells by immunomagnetic selection and electroporated with the CRISPR-Cas9–gRNA-68 RNP complex to produce OTQ923. Before OTQ923 infusion, participants underwent conditioning with pharmacokinetically targeted myeloablative busulfan (cumulative exposure goal: 80–100 mg*h/L) (Table S2). Additional details of the preclinical characterization, manufacture, and infusion of OTQ923 are provided in the Supplementary Appendix.
Assessments of clinical outcomes
Adverse events and blood product transfusions were monitored to assess the safety and tolerability of OTQ923. The time to neutrophil engraftment (absolute neutrophil count of ≥500 cells/μL for 3 consecutive days), time to platelet engraftment (platelet count of ≥50,000 cells/μL without platelet transfusion in the preceding 7 days), and HbF expression, as measured by immuno-flow cytometry (for F-cells) and capillary electrophoresis (for %HbF), were determined after OTQ923 infusion. On-target editing was assessed longitudinally in peripheral blood (PB) and bone marrow (BM) cells by next-generation sequencing (NGS) to detect small indels and by droplet digital polymerase chain reaction (ddPCR) to quantify the ~5-kb deletion.
Results
Identification of a previously unknown transcriptional repressor element in the HBG1/HBG2 promoters
The CRISPR-Cas9 screen revealed several gRNAs that increased F-cells in erythroid progeny of edited healthy-donor CD34+ cells (Fig. S2). Editing with gRNA-68 increased the percentage of HbF immunostaining cells (F-cells) to levels equal or superior to those achieved by other gRNAs, including a positive control targeting the BCL11A +58 erythroid-specific enhancer. The gRNA-68–targeted region includes no known HBG1/HBG2 regulatory elements, suggesting that our screen has uncovered a previously unknown regulatory region.
Preclinical validation of OTQ923
The preclinical cell product was manufactured by editing CD34+ HSCs from healthy donors and from individuals with sickle cell disease by using gRNA-68. In healthy-donor HSCs, on-target HBG1/HBG2 editing was 80.5% ± 9.8% (mean ± SD, n=4) resulting in 60.5% ± 6.8% F-cells (n=4) after in vitro erythroid differentiation, compared to 22.9% ± 3.5% F-cells in mock-edited controls (CD34+ cells electroporated with Cas9 protein only). In HSCs from individuals with sickle cell disease, on-target HBG1/HBG2 editing was 85.8% ± 14.7% (n=4), resulting in 65.4% ± 12.1% F-cells (n=3), as compared to 39.6% ± 12.4% F-cells in mock-edited controls (Figs. 1C, D, F and G). HbF protein levels assessed by ion-exchange HPLC in the hemolysates after in vitro erythroid differentiation were 20.9% ± 0.2% and 26.2% ± 2.9% in gRNA-68 HSCs compared to 12.0% ± 0.3% and 16.1% ± 2.3% in mock-edited controls from healthy donor (n=2) and individuals with sickle cell disease (n=3), respectively (Figs. 1E and H). The most common editing outcome was the ~5-kb deletion, as noted in previous reports.17,18 After transplantation into immunodeficient NSG mice, healthy donor–derived OTQ923 showed durable long-term engraftment and multi-lineage reconstitution similar to that of mock-edited control cells (Fig. S3). These findings indicated that genome editing with gRNA-68 did not impair the developmental potential of long-term repopulating HSCs. The indel patterns were consistent and reproducible across donors (Fig. S4), suggesting that the DNA repair events were not random.
Potential off-target editing sites were nominated using computational methods and SITE-Seq,19 which measures off-target indels caused by incubating purified DNA with RNP. Of the 279 unique potential off-target sites identified in these screening assays, none were detected in OTQ923 by PCR and NGS analysis with a sensitivity of 1% at a median coverage of 34840× (Table S3 and S4). No abnormalities were identified in OTQ923 with a targeted NGS cancer gene panel (Pan-Heme v1.0) (Tables S5 and S6).
Participant outcomes
As of March 27, 2023 (data cut-off), a total of 16 patients have consented to the study. Of these, 3 have been treated with OTQ923, 4 have had a partial dose of OTQ923 manufactured, and the remaining 9 participants were either found to be ineligible after signing consents, withdrew participation from the study, or are yet to start apheresis. Here we report the first three participants who have received OTQ923.
Participant 1 was a 22-year-old male with the βS/βS genotype who had been receiving hydroxyurea therapy and chronic RBC exchange transfusions for 6 years at study enrollment because of recurrent vaso-occlusive crisis episodes (Table 1). He discontinued hydroxyurea 2 months before CD34+ cell mobilization while continuing to receive exchange transfusions. Before OTQ923 infusion, his total hemoglobin level was 10.0 g/dL, with 0.4% HbF, and he had 4% F-cells (Fig. 2A). His last RBC transfusion was 26 days after OTQ923 infusion (Fig. 2B). Between 12- and 18-months post infusion, his total hemoglobin was maintained at 10.3–11.9 g/dL, with 25–26.8% HbF and 78.1–87.8% F-cells (Fig. 2A).
Table 1.
Participant Demographics and Outcomes.
| Participant 1 | Participant 2 | Participant 3 | |
|---|---|---|---|
| Demographics and disease severity | |||
| Age (at screening)/sex | 22 years/male | 21 years/male | 24 years/female |
| Sickle cell disease genotype | βS/βS | βS/βS | βS/βS |
| Sickle cell disease - related symptoms before enrollment | Six episodes of acute chest syndrome over the past 10 years and a history of a silent cerebral infarct, retinopathy, and priapism. | Four vaso-occlusive episodes, 3 episodes of acute chest syndrome, and a silent cerebral infarct during the preceding 20 years. | Twenty five vaso-occlusive pain episodes in the 2 years prior to enrollment. |
| Sickle cell disease treatment ongoing at study enrollment | Chronic blood transfusions and hydroxyurea. | Hydroxyurea. | Chronic blood transfusions and hydroxyurea. |
| Apheresis collection and OTQ923 manufacture | |||
| Mobilization cycles (Lasting 2–3 d each) | 3 | 2 | 3 |
| Cell dose manufactured, ×106 cells/kg | 2.8, a combination of 2 manufacturing batches, each with 84% editing efficiency | 5.99, a combination of 3 batches with editing efficiencies of 78%, 75%, and 73%, respectively | 5.04, a combination of 2 batches with editing efficiencies of 87% and 82%, respectively |
| Follow-up and outcomes | |||
| Neutrophil engraftment | Day +26 | Day +20 | Day +18 |
| Platelet engraftment | Day +44 | Day +29 | Day +29 |
| AEs experienced since OTQ923 infusion | 36 | 16 | 45 |
| AEs related to OTQ923 | 0 | 0 | 0 |
| Follow-up since OTQ923 infusion | 18 months | 12 months | 6 months |
| Sickle cell disease - related complications since OTQ923 infusion† | One vaso-occlusive crisis episode with acute chest syndrome occurring at 17 months after infusion. Recurrent intermittent priapism. No new stroke, or silent cerebral infarct. Continued mild hemolysis. Worsening osteonecrosis of femur. | One vaso-occlusive crisis episode occurring at 12 months after infusion. No acute chest syndrome, stroke or priapism. Continued mild hemolysis. Persistent osteonecrosis of femoral head. | One vaso-occlusive crisis occurring at 9 months after infusion.* Continued mild hemolysis. Persistent osteonecrosis of femoral head. |
The observation period for post-treatment sickle cell–related events starts on the day of first OTQ923 infusion and ends on the day of last follow-up.
AEs, adverse events.
This event happened after the data cut off and hence the rest of the follow up is only up to 6 months.
Figure 2. Total hemoglobin and its fractions, F-cell percentage, and tracking of edited alleles in the first two participants.
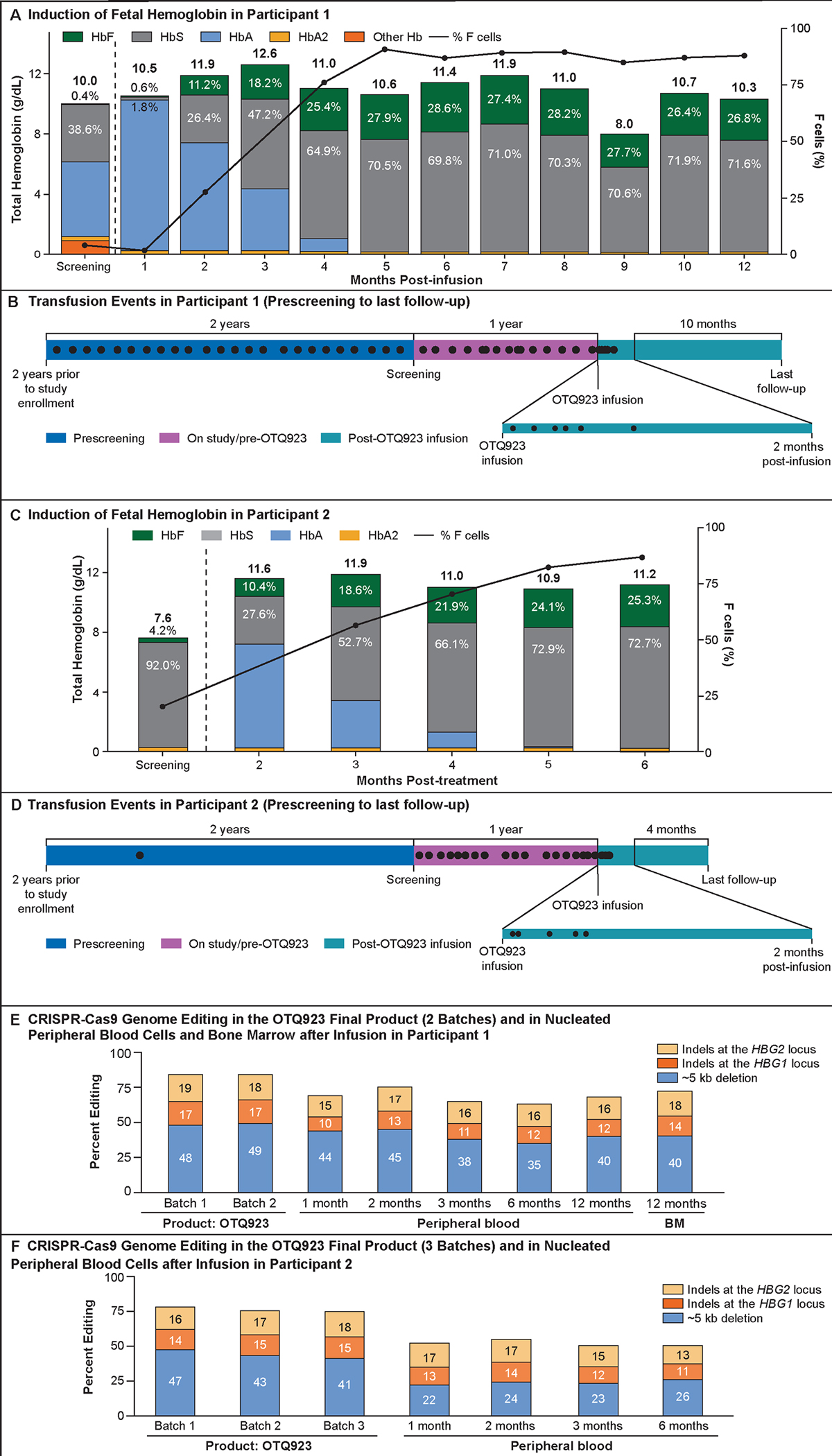
Panel A shows the total hemoglobin and hemoglobin fractionation over time for Participant 1. Participant 1 experienced a transient decline in hemoglobin from 11.0 to 8.0 g/dL between months 8 and 9, coincident with an intercurrent viral infection and associated bone marrow suppression, which recovered spontaneously at the subsequent timepoint. Panel B shows the number of blood transfusions received by Participant 1 over time. Panel C shows the percentage of allelic editing in OTQ923 product (2 batches), nucleated peripheral blood cells (up to 18 months after OTQ923 infusion), or bone marrow (at 12 months after OTQ923 infusion) from Participant 1. Panel D shows the total hemoglobin and hemoglobin fractionation over time for Participant 2. Panel E shows the number of transfusions received by Participant 2 over time. Panel F shows the percentage of allelic editing in OTQ923 product (3 batches), nucleated peripheral blood cells (up to 12 months after OTQ923 infusion), or bone marrow (at 12 months after OTQ923 infusion) from Participant 2. Panel G shows the total hemoglobin and hemoglobin fractionation over time for Participant 3. Panel H shows the number of transfusions received by Participant 3 over time. Panel I shows the percentage of allelic editing in OTQ923 product (2 batches), or nucleated peripheral blood cells (up to 6 months after OTQ923 infusion) from Participant 3. In panels A, D and G, green or gray labels indicate the proportions of fetal hemoglobin (HbF) or sickle hemoglobin (HbS), respectively, as percentages of the total hemoglobin at those respective time points. Total hemoglobin values are reported above each bar. The line graph (black line) represents the percentage of F-cells over time after OTQ923 infusion and indicates near-pancellular HbF expression. In panels B, E and H each black dot represents a transfusion. The time periods prior to enrollment, after enrollment on the study but prior to OTQ923 infusion, and after OTQ923 infusion are shown in different colors. In panels C, F and I orange bars represent the percentages of indels detected by next-generation sequencing (bright orange indicates indels at the HBG1 locus, pale orange indicates indels at the HBG2 locus). Blue bars represent the ~5-kb deletion that was detected in the infused product by quantitative PCR or by droplet digital PCR (ddPCR; also see Figure S5).
Participant 2 was a 21-year-old male with the βS/βS genotype who was experiencing multiple vaso-occlusive crisis episodes while on hydroxyurea (Table 1). Four months before CD34+ cell mobilization and collection, hydroxyurea was discontinued, and monthly exchange RBC transfusions were initiated. At screening evaluation, before the initiation of monthly RBC transfusions, the participant’s total hemoglobin level was 7.6 g/dL, with 4.2% HbF, and he had 20.4% F-cells (Fig. 2D). His last RBC transfusion was 17 days after OTQ923 infusion (Fig. 2E). Between 6- and 12-months post infusion, his total hemoglobin was maintained at 8.6–11.5 g/dL, with 23–25.3% HbF and 80.4–86.8% F-cells (Fig. 2D).
Participant 3 was a 24-year-old female with the βS/βS genotype who was experiencing frequent vaso-occlusive crises episodes necessitating hospital admissions, avascular necrosis of her hips, and iron overload (Table 1). She discontinued hydroxyurea 8 months before her first CD34+ cell mobilization while continuing to receive monthly exchange transfusions. Following initiation of exchange transfusions but before myeloablation, this participant’s total hemoglobin level was 8.3 g/dL, with 1.4% HbF, and she had 6.2% F cells. Her last RBC transfusion was 19 days after OTQ923 infusion (Fig. 1H). Between 4- and 6-months post infusion, her total hemoglobin was maintained around 10.5 g/dL, with 19–23.4 % HbF and 69.7–85.6% F-cells (Fig. 1G).
The indels in unsorted nucleated PB, sorted T cells, B cells and myeloid cells/neutrophils as well as BM cells obtained from the participants after OTQ923 infusion were diverse, with the most common one being the ~5-kb deletion (Figs. 2E, 2F and S5). The indel allele frequencies were consistent among the three patients and were comparable to those in preclinical samples (Figs. S4). Indel frequencies were also consistent between BM cells representative of the hematopoietic stem cells as well as sorted B cells and myeloid cells/neutrophils in each participant (Fig. S5). Indel frequencies fluctuated in the T cells during the first few months after infusion (Fig. S5).
Participants experienced several adverse events (Table S7) but all were considered to be related to either myeloablative busulfan conditioning or underlying sickle cell disease. None of the adverse events were considered to be related to OTQ923. Since undergoing OTQ923 infusion, all three participants have experienced at least one sickle cell disease-related event each. Participant 1 experienced a vaso-occlusive crisis associated with back and leg pain, which progressed to acute chest syndrome at 17 months after infusion. He subsequently started experiencing intermittent priapism associated with sexual activity, which is being treated with as needed pseudoephedrine. Participant 2 experienced a vaso-occlusive crisis associated leg pain at 12 months after infusion. Participants 1 and 2 required hospital admission for the management of their pain episodes. After the data cut, Participant 3 also experienced a vaso-occlusive crisis associated hip and lower back pain (9 months after infusion), which was managed conservatively in the outpatient setting. All three individuals also continue to experience focal intermittent episodic pain in their hips and lower extremities, which is similar in quality to the pain experienced previously from pre-existing femoral osteonecrosis and perhaps subjectively worse in Participant 1. Of note, Participant 1 was in a motor vehicle accident around 18 months after infusion and sustained major injuries including a liver laceration and a distal radial fracture, but recovered promptly with conservative management, specifically without blood transfusions or triggering a vaso-occlusive crisis. Likewise, Participant 2 underwent a planned total hip arthroplasty 6 months after infusion and recovered promptly without any complications or peri-procedural blood transfusions. Further, in the first two participants, at 1-year following infusion, cardiac chamber sizes and cardiac function on echocardiogram have either remained the same or slightly improved compared to their baseline before the infusion. Pulmonary function tests and renal function (estimated glomerular filtration rate) have remained stable as well.
While their reticulocyte counts and serum bilirubin remain elevated, lactate dehydrogenase has trended down over time (Table 2). Around 8 months after cell infusion, Participant 1 experienced an upper respiratory tract viral infection, possibly COVID, which was associated with a drop in hemoglobin from 11 g/dL the month before to 8 g/dL (most likely due to viral bone marrow suppression). Hemoglobin subsequently recovered spontaneously to pre-infection levels in a few weeks after the patient had complete recovery of his infection symptoms. Participant 2 also experienced a drop in his hemoglobin level during month 7 evaluations related to surgical blood loss during total hip arthroplasty. His hemoglobin level too returned to pre-surgery level at the next follow up. Neither participant required a blood transfusion. In fact, Participant 2 has been undergoing monthly phlebotomy to reduce pre-existing iron overload. Morphologic assessment of the bone marrow was performed in the first two participants right before myeloablation and at one year after cell infusion. The bone marrow evaluations done before myeloablation showed cellular marrow with erythroid predominant trilineage hematopoiesis. At one year after infusion, the bone marrow in both participants showed cellular marrow with relatively balanced and orderly trilineage hematopoiesis, normal myeloid-erythroid progeny ratio, and no evidence of increased blasts or dysplasia.
Table 2.
Markers suggestive of ongoing hemolysis and hematopoietic recovery for the trial participants at various timepoints after OTQ923 infusion
| Participant 1 | Participant 2 | Participant 3 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Normal Range | Screening | Month 3 | Month 6 | Month 12 | Month 18 | Screening | Month 3 | Month 6 | Month 12 | Screening | Month 3 | Month 6 | |
| Absolute Reticulocytes (106/mm3) | 0.021–0.085 | 0.3364 | 0.4025 | 0.2419 | 0.2721 | 0.1943 | 0.3034 | 0.3478 | 0.3951 | 0.3802 | 0.3444 | 0.1841 | 0.2321 |
| Lactate dehydrogenase (LDH)(U/L) | 94–260 | 345 | 265 | 263 | 217 | 292 * | 397 | 387 | 284 | 243 | 359 | 341 | 325 |
| Total bilirubin (μmol/L) | 3–21 | 26 | 24 | 48 | 43 | 56 * | 60 | 32 | 65 | 72 | 67 | 31 | 31 |
Abnormal values are bolded.
These laboratory studies were collected a few weeks after the patient had a motor vehicle accident and sustained a liver laceration. Hence their significance needs to be interpreted in conjunction with the patient’s clinical status.
Discussion
Genetic modification of autologous HSCs shows great promise for treating sickle cell disease, although the best approaches are not yet established. Lentiviral vector–mediated addition of an anti-sickling β-like globin is effective,20 although this approach does not eliminate βS-globin expression and creates a potentially toxic excess of β-like globin chains which can lead to ineffective erythropoiesis and erythroid dysplasia.21 In contrast, induction of endogenous γ-globin transcription concomitantly reduces βS-globin, thereby maintaining the balance of α-globin and β-like globin chains.2 Disrupting the BCL11A erythroid-specific enhancer with CRISPR-Cas9 can increase HbF and alleviate sickle cell disease symptoms.11 However, disrupting a repressor element in the HBG1/HBG2 promoters represents a more targeted approach than eliminating erythroid BCL11A expression entirely, as the latter may impair erythropoiesis.22,23 Through a CRISPR-Cas9 screen, we identified a previously unknown repressor element in HBG1/HBG2 promoters that in our preclinical testing, upon disruption, caused F-cell induction with an efficiency similar to that of disrupting the BCL11A erythroid-specific enhancer.
A single infusion of OTQ923 into three individuals with severe sickle cell disease caused sustained increases in total hemoglobin, HbF, and F-cells, reducing the occurrence of vaso-occlusive crises, without any detected off-target effects. Engraftment of the genetically modified cells was observed in the time frame expected for genetically modified CD34+ selected grafts (18–26 days),10,11,20 with no skewing in the differentiation of edited HSCs to myeloid or B-cell lineages. T cells exhibited variable editing in the first few months, consistent with expansion of unedited T cells or those derived from the edited HSCs after infusion. While all three participants have experienced at least one vaso-occlusive crisis episode each since infusion, the frequency of such episodes is very limited. Similar incidence of vaso-occlusive crisis events has been noted in another ongoing trial with comparable HbF induction.10 More importantly, in the first two participants, cardiac, pulmonary and renal function appears to be stable or improved at annual evaluation after infusion. This preservation of organ function – at least in the short term – raises hope that sickle cell disease induced organ dysfunction may be prevented by this therapy. However, osteonecrosis persisted and perhaps even worsened after treatment. While exposure to busulfan could have contributed to worsening bone health,24,25 continued sickle cell disease-related damage cannot be ruled out. The short-term safety profile of OTQ923 therapy was as expected after myeloablative busulfan and autologous HSCT.
Despite improved hematological parameters and reduction in incidence of sickle cell disease symptoms, all participants exhibited ongoing mild hemolysis indicating that their RBC HbF levels were insufficient to inhibit HbS polymerization completely. We observed marked improvement in some hematological indices such as total hemoglobin and RBC count, but stress erythropoiesis ensued, although improved from before therapy, as indicated by a normalized myeloid-erythroid progeny ratio noted on bone marrow assessments. Hemolysis appears to be somewhat improved in all participants as suggested by improved biochemical markers of hemolysis such as LDH and serum bilirubin, but it is still persistent as evidenced by elevated reticulocyte count, comparable to other ongoing clinical trials.10,20 Transient viral bone marrow suppression in Participant 1 with resulting decline in hemoglobin level is further evidence of continued hemolysis. However, the bone marrow assessments showing balanced and orderly trilineage hematopoiesis without any evidence of dysplasia, along with a rise in total hemoglobin level from baseline in all three participants supports improved hematopoiesis after the infusion of genetically modified cells. This rise in total hemoglobin has led to clinical improvement in all three recipients. While the degree of HbF induction seen in these participants may be enough for sickle cell disease attenuation, it is not sufficient for complete disease amelioration.
Changing the manufacturing process may help enhance HbF expression by increasing the on-target editing frequency of repopulating HSCs. First, we observed a decline in the frequency of edited alleles in the reconstituted peripheral blood cells and bone marrow in the participants as compared to the infused product. Thus, an opportunity exists to improve the editing of the long-term repopulating HSCs, which would then lead to persistence of a higher degree of editing in the reconstituted blood cells resulting in a higher HbF. Second, it is also possible that the HBG1/HBG2 promoter site targeted by gRNA-68 is not a very potent repressor of HBG1/HBG2 transcription in vivo and hence disrupting this site only causes partial or incomplete de-repression of HbF production. At the same time, perhaps the indels produced by gRNA-68 may only be disrupting the regulatory element partially. Ongoing studies to better characterize the putative negative regulatory motif targeted by gRNA-68 may facilitate its more precise disruption to optimize HbF induction. Third, the use of an aryl hydrocarbon receptor antagonist could be further optimized to expand the HSCs. Lastly, combinatorial approaches targeting multiple regulatory elements in tandem could also improve HbF induction,26 though this approach is yet to be evaluated with Cas9-directed editing. A potential downside of combinatorial approaches is the risk of more potent DNA damage response by inducing multiple double-strand DNA breaks.
Previous studies of autologous HSCT for sickle cell disease used freshly collected cells for manufacturing the cellular drug product.10,11,20 In contrast, the current study used a cryopreserved apheresis product to facilitate future access to OTQ923 therapy for the majority of sickle cell disease patients who reside in low-resource settings.
In summary, we have shown that Cas9 disruption of a newly discovered negative regulatory region in the HBG1/HBG2 promoters of autologous HSCs from sickle cell disease patients results in RBC HbF induction and a partial correction of sickle cell disease. While busulfan was used as a myeloablative agent in this trial, as has been in the case for most other ongoing trials,10,11,20 given the associated toxicity profile of busulfan alternatives such as reduced intensity melphalan,27 alternative reduced toxicity alkylating agents such as treosulfan28 and non-genotoxic antibody drug conjugates29 should be evaluated in future clinical trials. Although the long-term durability of the response and the safety of this genetically modified product continue to be evaluated, our data suggest that this approach offers a safe and potentially disease attenuating option for patients with severe sickle cell disease.
Supplementary Material
Acknowledgements
The authors thank the patients who participated in the trial, their families, and the clinical associates and personnel at each site, especially Stacy High, Sanjay Verma, Pam Young and Christopher Omahen who assisted with the study.
We are grateful for the support from our Novartis colleagues, Craig Mickanin, Carsten Russ, Konrad Mueller, Karin Abitorabi, Sven Schuierer, Shuai Chen, Qi Zhang, Madhura Panditrao, Alberto Del Rio-Espinola, Daher Ibrahim Aibo, Azeddine Elhajouji, Andrea Knight, Jennifer Snead, Christian Schmedt, Michael Patel, Imelda Schulhmann, Thomas Peters, Jean Regard, Marjolaine Phan, Filippo Vegni, Clement Purcell, Lantz Mackey, Aidan Condon, Liu Yiang, Karolina Kulec, Victor Lin, Ayala Tovy, Roman Sloutsky, Sejong Chun, Cameron Trenor, Lloyd Klickstein Evan Beckman, Tewis Bouwmeester, Alice Shaw, David Steensma, Glenn Dranoff and James Bradner, in addition to our Intellia colleagues, Bo Han, Nishit Patel and Dan O’Connell.
Editorial assistance was provided by Manoshi Nath, MSc and Zoe Crossman, PhD and was funded by Novartis Pharmaceuticals Corporation.
The authors also thank Keith A. Laycock, PhD, ELS, who is an employee of St. Jude Children’s Research Hospital, for scientific editing of the manuscript. Authors sincerely acknowledge the contribution of Mitchell J. Weiss, MD, PhD who critically reviewed the manuscript and provided several constructive comments as well as Jonathan Yen, PhD, Parul Rai, MD, Gabriela Maron, MD and Amittha Wickrema, PhD for their inputs.
Funding Statement
The clinical trial was funded by Novartis Pharmaceuticals who was the sponsor of the study.
References
- 1.Piel FB, Steinberg MH, Rees DC. Sickle Cell Disease. N Engl J Med 2017;376(16):1561–1573. DOI: 10.1056/NEJMra1510865. [DOI] [PubMed] [Google Scholar]
- 2.Doerfler PA, Sharma A, Porter JS, Zheng Y, Tisdale JF, Weiss MJ. Genetic therapies for the first molecular disease. The Journal of clinical investigation 2021;131(8) (In eng). DOI: 10.1172/JCI146394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Paikari A, Sheehan VA. Fetal haemoglobin induction in sickle cell disease. Br J Haematol 2018;180(2):189–200. DOI: 10.1111/bjh.15021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rankine-Mullings AE, Nevitt SJ. Hydroxyurea (hydroxycarbamide) for sickle cell disease. The Cochrane database of systematic reviews 2022;9(9):CD002202. (In eng). DOI: 10.1002/14651858.CD002202.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller ST, Wright E, Abboud M, et al. Impact of chronic transfusion on incidence of pain and acute chest syndrome during the Stroke Prevention Trial (STOP) in sickle-cell anemia. J Pediatr 2001;139(6):785–9. DOI: 10.1067/mpd.2001.119593. [DOI] [PubMed] [Google Scholar]
- 6.Ataga KI, Kutlar A, Kanter J, et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. N Engl J Med 2017;376(5):429–439. DOI: 10.1056/NEJMoa1611770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vichinsky E, Hoppe CC, Ataga KI, et al. A Phase 3 Randomized Trial of Voxelotor in Sickle Cell Disease. N Engl J Med 2019;381(6):509–519. DOI: 10.1056/NEJMoa1903212. [DOI] [PubMed] [Google Scholar]
- 8.Niihara Y, Miller ST, Kanter J, et al. A Phase 3 Trial of l-Glutamine in Sickle Cell Disease. N Engl J Med 2018;379(3):226–235. DOI: 10.1056/NEJMoa1715971. [DOI] [PubMed] [Google Scholar]
- 9.de la Fuente J, Gluckman E, Makani J, et al. The role of haematopoietic stem cell transplantation for sickle cell disease in the era of targeted disease-modifying therapies and gene editing. Lancet Haematol 2020;7(12):e902–e911. DOI: 10.1016/S2352-3026(20)30283-0. [DOI] [PubMed] [Google Scholar]
- 10.Esrick EB, Lehmann LE, Biffi A, et al. Post-Transcriptional Genetic Silencing of BCL11A to Treat Sickle Cell Disease. N Engl J Med 2021;384(3):205–215. (In eng). DOI: 10.1056/NEJMoa2029392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Frangoul H, Altshuler D, Cappellini MD, et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and beta-Thalassemia. N Engl J Med 2021;384(3):252–260. DOI: 10.1056/NEJMoa2031054. [DOI] [PubMed] [Google Scholar]
- 12.Bauer DE, Orkin SH. Hemoglobin switching’s surprise: the versatile transcription factor BCL11A is a master repressor of fetal hemoglobin. Current opinion in genetics & development 2015;33:62–70. (In eng). DOI: 10.1016/j.gde.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bhoopalan SV, Yen JS, Levine RM, Sharma A. Editing human hematopoietic stem cells: advances and challenges. Cytotherapy 2022. DOI: 10.1016/j.jcyt.2022.08.003. [DOI] [PubMed] [Google Scholar]
- 14.Masuda T, Wang X, Maeda M, et al. Transcription factors LRF and BCL11A independently repress expression of fetal hemoglobin. Science (New York, NY) 2016;351(6270):285–9. DOI: 10.1126/science.aad3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uchida N, Leonard A, Stroncek D, et al. Safe and efficient peripheral blood stem cell collection in patients with sickle cell disease using plerixafor. Haematologica 2020;105(10):e497. (In eng). DOI: 10.3324/haematol.2019.236182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sharma A, Leonard A, West K, et al. Optimizing haematopoietic stem and progenitor cell apheresis collection from plerixafor-mobilized patients with sickle cell disease. Br J Haematol 2022;198(4):740–744. (In eng). DOI: 10.1111/bjh.18311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Metais JY, Doerfler PA, Mayuranathan T, et al. Genome editing of HBG1 and HBG2 to induce fetal hemoglobin. Blood advances 2019;3(21):3379–3392. (In eng). DOI: 10.1182/bloodadvances.2019000820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nature medicine 2016;22(9):987–90. (In eng). DOI: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cameron P, Fuller CK, Donohoue PD, et al. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat Methods 2017;14(6):600–606. (In eng). DOI: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 20.Kanter J, Walters MC, Krishnamurti L, et al. Biologic and Clinical Efficacy of LentiGlobin for Sickle Cell Disease. N Engl J Med 2022;386(7):617–628. DOI: 10.1056/NEJMoa2117175. [DOI] [PubMed] [Google Scholar]
- 21.Walters MC, Thompson AA, Kwiatkowski JL, et al. Lovo-cel (bb1111) Gene Therapy for Sickle Cell Disease: Updated Clinical Results and Investigations into Two Cases of Anemia from Group C of the Phase 1/2 HGB-206 Study. Blood 2022;140(Supplement 1):26–28. DOI: 10.1182/blood-2022-162288. [DOI] [Google Scholar]
- 22.Chang K-H, Sanchez M, Heath J, et al. Comparative Studies Reveal Robust HbF Induction By Editing of HBG1/2 Promoters or BCL11A Erythroid-Enhancer in Human CD34+ Cells but That BCL11A Erythroid-Enhancer Editing Is Associated with Selective Reduction in Erythroid Lineage Reconstitution in a Xenotransplantation Model. Blood 2018;132(Supplement 1):409–409. DOI: 10.1182/blood-2018-99-114780. [DOI] [Google Scholar]
- 23.Heath JdD E; Sanchez M; Haskett S; Wang T; Sousa P; Gotta G; Siwak J; Viswanathan R; Ta F; Brennan E; Tabbaa D; Marco E; Zuris J; Isik M; Ta T; Giannoukos G; Teixera S; Wilson C; Albright C; Chang K-H. Genome editing of HBG1/2 promoter leads to robust HbF induction in vivo, while editing of BCL11A erythroid enhancer results in erythroid defects. European Hematology Association. Amsterdam, The Netherlands 2019. [Google Scholar]
- 24.Bojko P, Hilger RA, Ruehm SG, Dirsch O, Seeber S, Scheulen ME. Femoral head necrosis in three patients with relapsed ovarian cancer receiving high-dose chemotherapy followed by autologous peripheral blood stem cell transplantation. Bone Marrow Transplant 2003;31(6):487–91. DOI: 10.1038/sj.bmt.1703886. [DOI] [PubMed] [Google Scholar]
- 25.Socie G, Cahn JY, Carmelo J, et al. Avascular necrosis of bone after allogeneic bone marrow transplantation: analysis of risk factors for 4388 patients by the Societe Francaise de Greffe de Moelle (SFGM). Br J Haematol 1997;97(4):865–70. DOI: 10.1046/j.1365-2141.1997.1262940.x. [DOI] [PubMed] [Google Scholar]
- 26.Liu B, Brendel C, Vinjamur DS, et al. Development of a double shmiR lentivirus effectively targeting both BCL11A and ZNF410 for enhanced induction of fetal hemoglobin to treat beta-hemoglobinopathies. Mol Ther 2022;30(8):2693–2708. DOI: 10.1016/j.ymthe.2022.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimley M, Asnani M, Shrestha A, et al. Safety and Efficacy of Aru-1801 in Patients with Sickle Cell Disease: Early Results from the Phase 1/2 Momentum Study of a Modified Gamma Globin Gene Therapy and Reduced Intensity Conditioning. Blood 2021;138(Supplement 1):3970–3970. DOI: 10.1182/blood-2021-147469. [DOI] [Google Scholar]
- 28.Strocchio L, Zecca M, Comoli P, et al. Treosulfan-based conditioning regimen for allogeneic haematopoietic stem cell transplantation in children with sickle cell disease. Br J Haematol 2015;169(5):726–36. DOI: 10.1111/bjh.13352. [DOI] [PubMed] [Google Scholar]
- 29.Czechowicz A, Palchaudhuri R, Scheck A, et al. Selective hematopoietic stem cell ablation using CD117-antibody-drug-conjugates enables safe and effective transplantation with immunity preservation. Nat Commun 2019;10(1):617. DOI: 10.1038/s41467-018-08201-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Traxler EA, Yao Y, Wang YD, et al. A genome-editing strategy to treat β-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nat Med 2016;22(9):987–90. (In eng). DOI: 10.1038/nm.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.