

SUMMARY
Tumor necrosis factor α (TNF-α) is a major pro-inflammatory cytokine, important in many diseases, that sensitizes nociceptors through its action on a variety of ion channels, including voltage-gated sodium (NaV) channels. We show here that TNF-α acutely upregulates sensory neuron excitability and current density of threshold channel NaV1.7. Using electrophysiological recordings and live imaging, we demonstrate that this effect on NaV1.7 is mediated by p38 MAPK and identify serine 110 in the channel’s N terminus as the phospho-acceptor site, which triggers NaV1.7 channel insertion into the somatic membrane. We also show that the N terminus of NaV1.7 is sufficient to mediate this effect. Although acute TNF-α treatment increases NaV1.7-carrying vesicle accumulation at axonal endings, we did not observe increased channel insertion into the axonal membrane. These results identify molecular determinants of TNF-α-mediated regulation of NaV1.7 in sensory neurons and demonstrate compartment-specific effects of TNF-α on channel insertion in the neuronal plasma membrane.
Graphical Abstract
In brief
TNF-α is a major cytokine involved in inflammatory pain. Tyagi et al. show that TNF-α causes increased membrane insertion of NaV1.7, a sodium channel linked to pain disorders, in a p38 MAPK-dependent manner; however, this process is differentially regulated in DRG axons vs. somas.
INTRODUCTION
Inflammatory pain results from the sensitization of nociceptors in response to tissue injury and is mediated by a variety of cytokines, kinins, growth factors, and eicosanoids acting on multiple targets including voltage-gated sodium (NaV) channels.1–3 NaV1.7 is an obligate mediator of pain because it sets the gain on peripheral nociceptor excitability by amplifying subthreshold depolarizations.4–6 NaV1.7 has been implicated in the development of inflammatory pain—nocifensive responses to injection of various inflammatory mediators were significantly dulled or abolished in mice after knockout of NaV1.7 in small dorsal root ganglion (DRG) neurons.7 Inflammatory mediators are known to increase transport of NaV1.7 channels to neuronal membranes, resulting in increased surface levels and an increase in current density.8–10 This leads to nociceptor activation by decreasing the threshold for action potential firing,11 which sensitizes DRG neurons in the setting of inflammation. The molecular and cellular mechanisms that underlie temporal and spatial regulation of NaV1.7 channels in sensory neurons under inflammatory conditions, however, are not well understood.
One of the most prominent pro-inflammatory cytokines is tumor necrosis factor α (TNF-α).12,13 The role of TNF-α in producing and maintaining hyperalgesia is well established; intraplantar injection of TNF-α produced hypersensitivity to mechanical and thermal stimuli in mice and rats,14–17 and TNF-α neutralizing agents attenuated mechanical allodynia and thermal hyperalgesia in animal models of pain.18,19 Additionally, the role of TNF-α in driving the development and sequelae of long COVID is beginning to be elucidated20,21—these patients commonly suffer from chronic pain as part of their post-infection syndrome.22,23 A better understanding of the underlying pathophysiological mechanisms behind these pain conditions is necessary to develop more effective treatments.
There is evidence that TNF-α mediates nociceptor sensitization and leads to painful states by (1) triggering further pro-inflammatory cytokine release and (2) direct binding to the TNF receptor (TNFR), followed by intracellular signal transduction.12,24 Additionally, the interplay between TNF-α and several NaV channels in inflammatory pain is well substantiated. Both TNF-α and NaV channel expressions are upregulated in multiple models of inflammatory pain.25–28 Further, TNF-α has been directly implicated in modifying NaV expression and activity in DRG neurons—increasing both tetrodotoxin-sensitive (TTX-S) and TTX-resistant (TTX-R) sodium currents in DRG neurons.29–33 Multiple studies have provided evidence that chronic exposure of DRG neurons to TNF-α over the timescale of days can drive increased transcription and translation of NaV1.7 channels.34 Together with data showing attenuated inflammatory pain in NaV1.7-knockout mice,7 this further supports a role of the TNF-α/NaV1.7 interplay in inflammatory pain.
However, neuronal sensitization and pain can develop acutely following inflammatory cytokine release (within minutes of an insult such as tissue injury). These responses happen too quickly to be explained by transcription and translation of new NaV channels. In line with these observations, acute administration of TNF-α mimics the acute release of this cytokine by resident macrophages following tissue injury and results in immediate pain.12,15,35 This acute response suggests that rapid sensitization of nociceptors is mediated by a signaling cascade, for example phosphorylation, that can act on existing effectors without relying on de novo channel biogenesis. In support of this hypothesis, a previous study showed that acute treatment of DRG neurons with TNF-α increased the current density of NaV1.8 channels in a p38 MAPK-mediated manner—albeit without determining whether this was a direct effect of phosphorylating the channel or as an indirect outcome of phosphorylating an intermediary factor.31 Recently, acute treatment of DRG neurons with TNF-α was reported to increase NaV1.7 activity by means of a protein-protein interaction with p65, a subunit of the nuclear factor κB (NF-κB) complex.36 However, the mechanism of this interaction was not investigated, and TNF-α ‘s effect on the distribution of NaV1.7 channels in peripheral neurons remains unexplored.
An important gap in our knowledge is how effectors that impact cellular responses under inflammatory conditions, for example NaV1.7 channels, are dynamically regulated in time and space. In mammalian physiology, this is particularly relevant at axonal ends, where action potential firing is initiated in response to stimuli. Recently, we developed OPAL (optical pulse-chase axonal long distance), a live-imaging approach that enables interrogation of the dynamics of membrane protein delivery and distribution in specific neuronal compartments. Using tagged full-length NaV channel constructs coupled with spinning-disk confocal microscopy, it is now possible to evaluate the effects of inflammatory mediators on these channels in both somas and distal axons of sensory neurons.8,9,37 Together with electrophysiological recordings, these techniques allow us to study the effects of TNF-α on the biophysical properties of NaV1.7, as well as the temporal and spatial regulation of channel insertion at the plasma membrane of somas and axons of sensory neurons.
In this study, we investigate the mechanisms of the response of DRG neurons to the acute application of TNF-α, which mimics the effects of cytokine release immediately after tissue injury. We demonstrate that acute (minutes) application of TNF-α on DRG neurons induces an increase in NaV1.7 current density and neuronal excitability and show that the phosphorylation of a specific serine residue in the N terminus of the channel by p38 MAPK is critically important for this effect on the channel. Using our live-cell imaging assay, we reveal a surprising finding—that the acute action of TNF-α is differentially regulated in somas and axons. Phosphorylation of the serine 110 residue in the N terminus of the channel causes a rapid insertion of the channel in the somatic membrane. Although NaV1.7 channels accumulate within axons close to their endings, we did not detect insertion of the channels in the axonal membrane even after 1 h of incubation with TNF-α—revealing a compartment-specific effect of the cytokine. Our findings delineate multiple points in the inflammatory pain process that could be targets for therapeutic intervention and offer evidence that the regulation of voltage-gated ion channels in DRG neurons is compartment specific.
RESULTS
TTX-S and TTX-R sodium current density increases after acute TNF-α exposure
DRG neurons express TTX-S (NaV1.1, NaV1.6, and NaV1.7) and TTX-R (NaV1.8 and NaV1.9) sodium channels.4,38 TTX-S and TTX-R sodium currents were recorded in DRG neurons with and without preincubation with 20 ng/mL TNF-α for 20 min using the whole-cell patch-clamp technique (Figure 1A). Current density was calculated as peak sodium current (nA) normalized to the whole-cell capacitance (pF). TTX-S current density was significantly larger (145%) in the presence of TNF-α ( , 0.61 ± 0.06 nA/pF, n = 21) than in the absence of TNF-α (DMSO control; □, 0.42 ± 0.05 nA/pF, n = 22) (Figures 1B and 1C; p < 0.05). Similarly, TTX-R current density was significantly larger (147%) in the presence of TNF-α (
, 0.61 ± 0.06 nA/pF, n = 21) than in the absence of TNF-α (DMSO control; □, 0.42 ± 0.05 nA/pF, n = 22) (Figures 1B and 1C; p < 0.05). Similarly, TTX-R current density was significantly larger (147%) in the presence of TNF-α ( , 0.36 ± 0.02 nA/pF, n = 21) than in the absence of TNF-α (DMSO control;
, 0.36 ± 0.02 nA/pF, n = 21) than in the absence of TNF-α (DMSO control;  , 0.24 ± 0.02 nA/pF, n = 22) (Figures 1B and 1C; p < 0.05). TNF-α exposure also produced a small hyperpolarizing shift in the voltage dependence of activation of both TTX-S (~5 mV) and TTX-R (~4 mV) sodium currents (Figure 1D). These results indicate that an acute, 20 min incubation of TNF-α is enough to increase both TTX-S and TTX-R sodium currents in DRG neurons.
, 0.24 ± 0.02 nA/pF, n = 22) (Figures 1B and 1C; p < 0.05). TNF-α exposure also produced a small hyperpolarizing shift in the voltage dependence of activation of both TTX-S (~5 mV) and TTX-R (~4 mV) sodium currents (Figure 1D). These results indicate that an acute, 20 min incubation of TNF-α is enough to increase both TTX-S and TTX-R sodium currents in DRG neurons.
Figure 1. Native TTX-S and TTX-R neuronal sodium currents, as well as human NaV1.7 currents, are increased by a short application of TNF-α.

(A) Representative TTX-S and TTX-R current traces recorded from DRG neurons in the absence (left) and presence (right) of 20 ng/mL TNF-α for 20 min
(B) Current density-voltage relationships for TTX-S and TTX-R currents in the absence or presence of TNF-α.
(C) Inward current density through TTX-S and TTX-R sodium channels both increased in the presence of TNF-α (n = 21) vs. vehicle (n = 22). *p < 0.05 by unpaired Student’s t test.
(D) Conductance-voltage relationships of TTX-S and TTX-R sodium currents recorded from DRG neurons. Application of TNF-α hyperpolarizes the voltage dependence of activation of both TTX-S and TTX-R sodium channels. Conductance-voltage (G-V) relationships were fit by Boltzmann equations. The midpoint (V1/2) and slope (k) values for the voltage-dependent activation curves of TTX-S channels were −28.27 ± 1.13 and 5.02 ± 0.21 mV in the absence of TNF-α and −33.50 ± 1.37 and 4.21 ± 0.29 mV in the presence of TNF-α, respectively. The V1/2 and k values for the voltage-dependent activation curves of TTX-R channels were −19.86 ± 0.90 and 5.02 ± 0.14 mV in the absence of TNF-α and −23.50 ± 1.19 and 5.02 ± 0.17 mV in the presence of TNF-α, respectively.
(E) Representative hNaV1.7 current traces recorded from DRG neurons in the absence (left) and presence (right) of 20 ng/mL TNF-α for 20 min. Endogenous TTX-R currents were minimal.
(F) Current density-voltage relationships for hNaV1.7 in the absence and presence of TNF-α.
(G) Inward current density through hNaV1.7 increased in the presence of TNF-α (n = 13 untreated, 11 treated) vs. vehicle (n = 6 untreated, 6 treated). *p < 0.05 by unpaired Student’s t test.
(H) Conductance-voltage relationships of hNaV1.7 sodium currents recorded from DRG neurons. G-V relationships were fit by Boltzmann equations. Application of TNF-α hyperpolarizes the voltage-dependence of activation of hNaV1.7. The V1/2 and k values for the voltage-dependent activation curves of neurons treated with DMSO control were −26.4 ± 1.1 and 6.2 ± 0.3 mV. The V1/2 and k values for the voltage-dependent activation curves of neurons treated with TNF-α were −32.5 ± 1.3 and 4.6 ± 0.5 mV.
Data are presented as mean ± SEM.
TNF-α regulates human NaV1.7 channels
The similar pharmacological susceptibility to TTX of NaV1.7 and other TTX-S currents in DRG neurons makes isolating NaV1.7 currents from total sodium currents difficult. To overcome this hurdle, we used biolistic transfection to transfect plasmids encoding human Nav1.7 (hNaV1.7) into rat DRG neurons that had been cultured for 7 days in vitro (7 DIV). After 7 DIV, the expression of endogenous TTX-R NaV channels in DRG neurons is significantly reduced.39–41 In this state, current through hNaV1.7 (which has been modified to be TTX-R42) constitutes the overwhelming majority of current when TTX is present in the bath (Figure 1E). DRG neurons were co-transfected with plasmids encoding the channel and EGFP to identify positively transfected cells. We acquired whole-cell patch-clamp recordings from transfected cells that were identified by green fluorescence. hNaV1.7 current was recorded from DRG neurons preincubated with 20 ng/mL TNF-α or DMSO control for 20 min (Figures 1E and 1F). Peak current density of hNaV1.7 was significantly larger (293%) in DRG neurons in the presence of TNF-α (, 0.41 ± 0.07 nA/pF, n = 11) than in the absence of TNF-α (□, 0.14 ± 0.02 nA/pF, n = 13) (Figures 1F and 1G, p < 0.05). Endogenous TTX-R current density was too small to be measured.39 Thus, the effect of TNF-α on TTX-R currents could not be identified in these neurons (Figures 1E–1G). TNF-α exposure also produced a small (~6 mV) hyperpolarizing shift in the voltage dependence of activation of hNaV1.7 (V1/2: 26.40 ± 1.09 vs. 32.46 ± 1.27 mV for DMSO control vs. TNF-α, respectively; p < 0.05, Figure 1H). Acute application of TNF-α did not affect the inactivation properties of hNaV1.7 or of endogenous TTX-S and TTX-R currents (Figure S1).
TNF-α application increases the excitability of DRG neurons expressing hNaV1.7
Because the brief incubation (20 min) of TNF-α significantly increased the current density of NaV1.7, we hypothesized that acute treatment with TNF-α may increase the excitability of DRG neurons. To test this hypothesis, we examined the excitability of DRG neurons expressing hNaV1.7 acutely exposed to 20 ng/mL TNF-α by current-clamp electrophysiology.
As described earlier, DRG neurons had to be cultured for 7 DIV to become firmly attached to the substrate to endure the pressure of the gold projectiles during biolistic transfection. DRG neurons that have been cultured for 7 DIV undergo significant time-dependent cellular changes compared to acutely isolated neurons or neurons in culture for a short period of time.39 By contrast, transfection of neurons by electroporation permits the delivery of NaV1.7 to acutely isolated DRG neurons and thus enables studies of neurons that are closer to their native state. Therefore, this method makes it possible to investigate neuronal firing after the expression of hNaV1.7 because of less time-dependent change in channel expression after 1 DIV. Additionally, we confirmed that electroporation does not increase the amount of TNF-α present in DRG neuron supernatant by ELISA (Figure S2).
Representative current-clamp traces show action potentials (APs) elicited by 200 (Figure 2A) and 500 pA (Figure 2B) stimuli by step current injection from the resting membrane potential of these neurons. Compared to DMSO control (■), acute treatment with TNF-α () did not change resting membrane potential (−53.9 ± 1.3 mV), cell capacitance (72.6 ± 6.5 pF), or input resistance (346.3 ± 62.0 MU). However, the current threshold (; 170.0 ± 19.7 pA, n = 16) and voltage threshold (−32.9 ± 0.9 mV, n = 16) in the presence of TNF-α were significantly lower than those in the absence of TNF-α (■; 475.0 ± 77.1 pA and −29.0 ± 1.1 mV; n = 14, p < 0.05). Interestingly, spontaneous subthreshold oscillations were nearly absent in control-treated neurons but were pronounced in the TNF-α-treated neurons.
Figure 2. Acute exposure to TNF-α causes hyperexcitability of DRG neurons.

Current-clamp recordings of DRG neurons expressing hNaV1.7 treated with TNF-α (red) or vehicle (DMSO; black).
(A) Action potentials elicited by injection of 200 pA current in DRG neurons expressing hNaV1.7 treated with TNF-α (right) vs. vehicle (left).
(B) Action potentials elicited by injection of 500 pA current in DRG neurons expressing hNaV1.7 treated with TNF-α (right) vs. vehicle (left).
(C) Quantification of repetitive firing of action potentials provoked by current injections of increasing amplitude.
(D) Recordings from neurons subjected to 500 ms ramp current injection from Resting Membrane Potential (RMP) to 1 nA (2 pA/ms).
(E) Recordings from neurons subjected to 500 ms ramp current injection from RMP to 2 nA (4 pA/ms).
(F) Quantification of action potentials (APs) evoked by ramp current injection from 0.5 to 2 nA by 500 ms ramp current injections of four different speeds.
(G) Latency of AP generation was quantified as the time required from stimulus onset to the peak of the first AP. AP latency of neurons treated with TNF-α was lower than those treated with vehicle. Data are presented as mean ± SEM. *p < 0.01 by unpaired Student’s t tests with Bonferroni correction for multiple comparisons.
APs were elicited from 100 to 500 pA with a 500 ms step current injection from the resting membrane potential. The number of APs (crossing 0 mV) in the presence of TNF-α () was also significantly increased compared to that in the absence of TNF-α (■) (Figure 2C). As expected, these results indicate that TNF-α acutely increased the excitability of DRG neurons by lowering the voltage and current thresholds of hNaV1.7.
To examine the effect of TNF-α on the properties of APs fired by DRG neurons expressing hNaV1.7, a 500 ms ramp current (0–1 and 0–2 nA) was injected from the resting membrane potential, and only overshooting APs were counted (i.e., AP peaks that cross 0 mV) (Figures 2D–2G). Representative traces show the APs of the DRG neurons elicited by the 2 (ramp to 1 nA; Figure 2D) and 4 pA/ms (ramp to 2 nA) ramp stimuli (Figure 2E). Following ramp injections, only a few neurons in the control condition displayed subthreshold oscillations before the onset of the first AP. In contrast, after TNF-α treatment, subthreshold oscillations were evident in the majority of cells. TNF-α significantly increased the number of APs of the DRG neurons for all stimuli (1–4 pA/ms) (Figure 2F; n = 14 for DMSO control, n = 16 for TNF-α, p < 0.05). Time-to-first AP peak was significantly faster in the presence of TNF-α for all stimuli (Figure 2G; p < 0.05). These results indicate that acute exposure to TNF-α increases the excitability of DRG neurons.
Figure 4. The phosphorylation site S110 in the NaV1.7 N terminus is critical for acute upregulation of human NaV1.7 current by TNF-α.
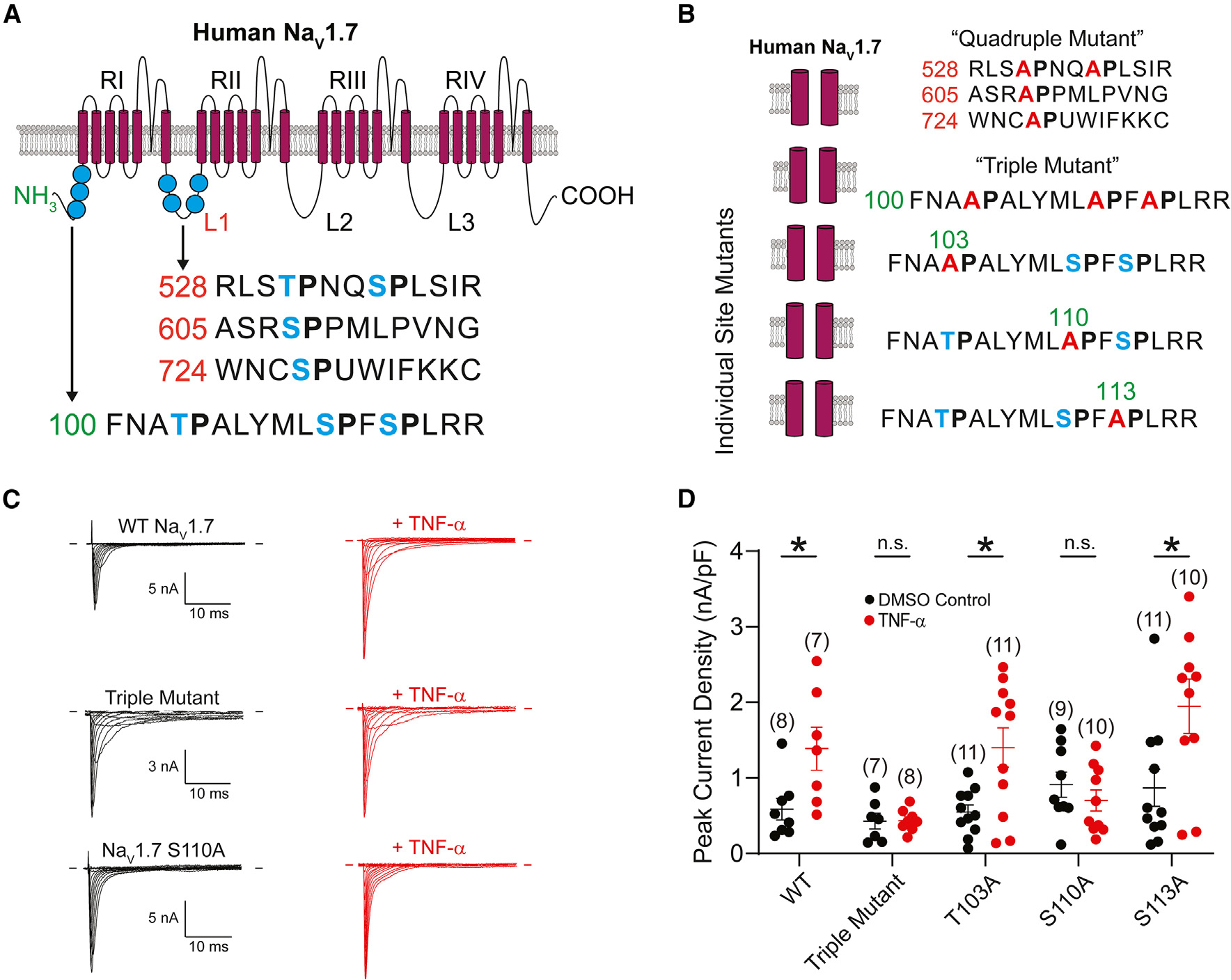
(A) Schematic depicting the membrane topology of human NaV1.7. Putative phosphorylation sites in the L1 (red numbers) and N terminus (green numbers) marked in blue. The schematic was adapted from Tyagi et al.48 with the permission of the authors.
(B) Phosphorylation site mutants generated in this study. Putative phosphorylation sites are marked in blue, and alanine substitutions are depicted in red.
(C) Representative NaV1.7 current traces in the absence (left) and presence (right) of TNF-α from NaV1.8-null mouse neurons expressing (from top to bottom) WT, triple mutant, and S110A NaV1.7 channels.
(D) Peak current densities from NaV1.8-null neurons expressing NaV1.7 phosphorylation site mutants. Data are presented as mean ± SEM. * p < 0.05 by unpaired t test.
TNF-α-mediated upregulation of NaV1.7 current is dependent on p38 MAPK and not NF-κB
Previous studies have reported TNFR1 activation mediates the effects of TNF-α on several cell processes by activating NF-κB43 or p38 MAPK.44 A recent study reported a role for NF-κB in the acute modulation of NaV1.7 by TNF-α.36 We investigated whether the activation of NF-κB or p38 MAPK contributes to the acute TNF-α-induced enhancement of hNaV1.7 current. DRG neurons were pretreated with 20 μM SC-514, a selective NF-κB inhibitor, for 1 h or for 30 min with 20 μM SB203580, a selective p38 MAPK inhibitor, before and during incubation with 20 ng/mL TNF-α (Figure 3).
Figure 3. Acute enhancement of hNaV1.7 current by TNF-α is dependent on the p38 MAPK.
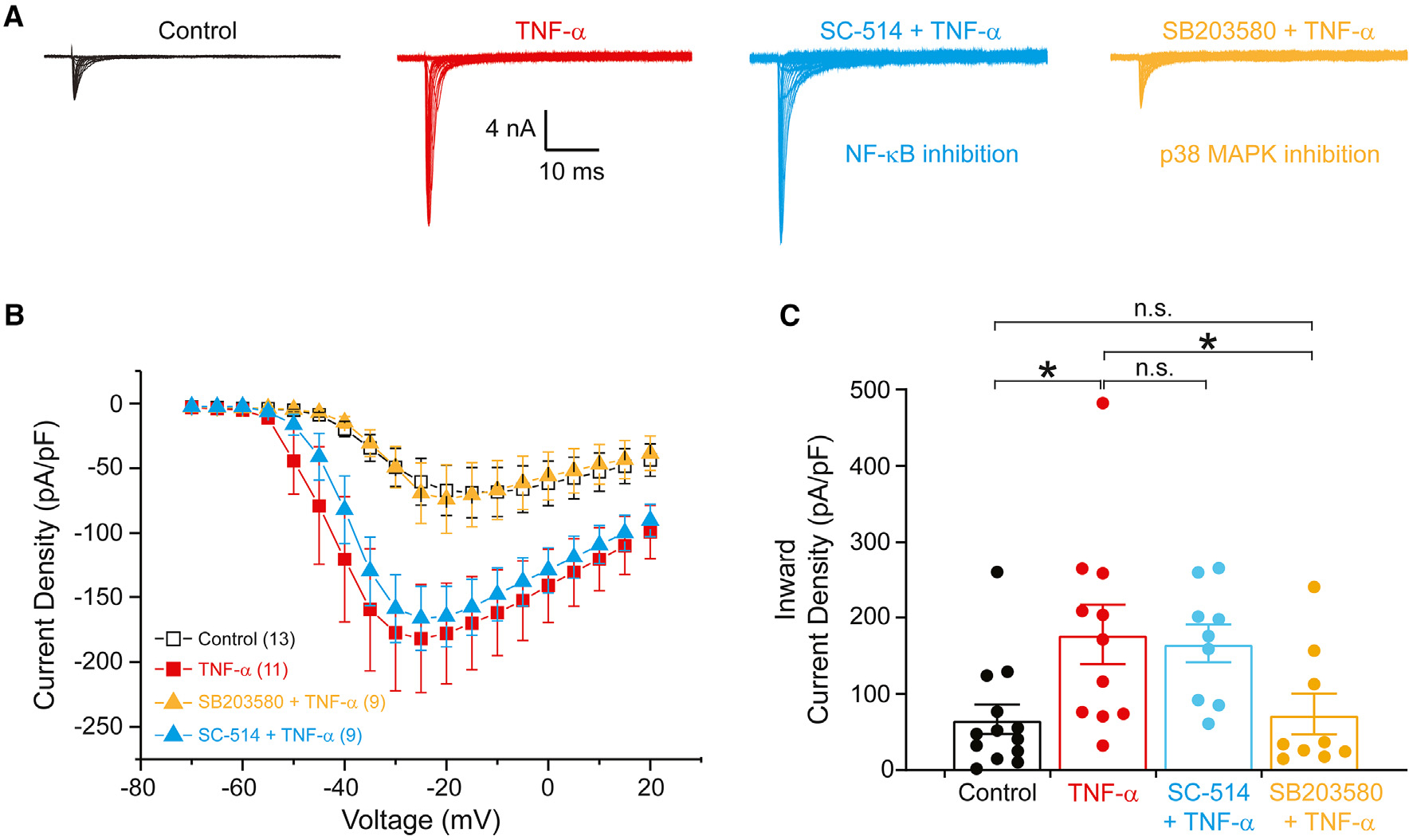
(A) Representative hNaV1.7 currents recorded from DRG neurons in the absence (black) and presence (red) of 20 ng/mL TNF-α for 20 min. Blue traces are from TNF-α-exposed neurons preincubated with 20 μM SC-514 (an inhibitor of NF-κB) for 1 h. Gold traces are from TNF-α-exposed neurons preincubated with 20 μM SB203580 (an inhibitor of the p38 MAPK) for 30 min
(B) Current density-voltage relationships for hNaV1.7 sodium currents in the absence and presence of TNF-α and with preincubation with inhibitors of NF-κB or p38 MAPK.
(C) Comparison of maximal hNaV1.7 current density between neurons treated with DMSO control (n = 13), TNF-α (n = 11), or TNF-α and inhibitors of NF-κB (SC-514, n = 9) and p38 MAPK (SB203580, n = 9). Data are presented as mean ± SEM. *p < 0.05 vs. control and SB203580 + TNF-α by one-way ANOVA followed by the Bonferroni post-hoc test.
Data are presented as mean ± SEM.
Representative hNaV1.7 current traces were recorded from DRG neurons in the absence and presence of 20 ng/mL TNF-α for 20 min with preincubation of 20 μM SB203580 or 20 μM SC-514 (Figure 3A). The current density of hNaV1.7 was significantly larger (257%) in DRG neurons in the presence of TNF-α (, 0.18 ± 0.04 nA/pF, n = 11) compared to in the absence of TNF-α (□, 0.07 ± 0.02 nA/pF, n = 13, p < 0.05) (Figures 3B and 3C). Preincubation of DRG neurons with SC-514 did not inhibit the effect of treatment with TNF-α on hNaV1.7 current density ( , 0.17 ± 0.02 nA/pF, n = 9), whereas preincubation with SB203580 completely inhibited the effect of TNF-α on the hNaV1.7 current density (
, 0.17 ± 0.02 nA/pF, n = 9), whereas preincubation with SB203580 completely inhibited the effect of TNF-α on the hNaV1.7 current density ( , 0.07 ± 0.02, n = 9, p < 0.05). Because of the discrepancy in our results compared to those previously published, which used a different NF-κB inhibitor, pyrrolidine dithiocarbonate (PDTC), we performed head-to-head comparisons of the NF-κB and p38 MAPK inhibitors on hNaV1.7 currents in the DRG-derived ND7/23 cell line using a high-throughput automated patch-clamp electrophysiology assay (Figure S3). We found that preincubation with either NF-κB inhibitor, SC-514 or PDTC, did not affect the TNF-α-mediated increase in NaV1.7 current density, while the p38 MAPK inhibitor SB203580 did. These results indicate that acute application of TNF-α significantly increases the current density of hNaV1.7 through the p38 MAPK signaling pathway but not through the NF-κB signaling pathway.
, 0.07 ± 0.02, n = 9, p < 0.05). Because of the discrepancy in our results compared to those previously published, which used a different NF-κB inhibitor, pyrrolidine dithiocarbonate (PDTC), we performed head-to-head comparisons of the NF-κB and p38 MAPK inhibitors on hNaV1.7 currents in the DRG-derived ND7/23 cell line using a high-throughput automated patch-clamp electrophysiology assay (Figure S3). We found that preincubation with either NF-κB inhibitor, SC-514 or PDTC, did not affect the TNF-α-mediated increase in NaV1.7 current density, while the p38 MAPK inhibitor SB203580 did. These results indicate that acute application of TNF-α significantly increases the current density of hNaV1.7 through the p38 MAPK signaling pathway but not through the NF-κB signaling pathway.
The S110A residue is necessary for the TNF-α-mediated upregulation of NaV1.7
We have previously shown that NaV1.6 and NaV1.8 channels are phosphorylated by p38 MAPK at specific serine-proline (SP) sites in the intracellular loop (L1) of the channel that links domains I and II.45–47 We searched the sequence of NaV1.7 to identify putative p38 phospho-acceptor sites and found 7 such motifs, 3 in the N terminus of the channel and 4 in the first cytoplasmic loop (L1; Figure 4A). To determine if any of these residues were critical for acute TNF-α-mediated upregulation of NaV1.7, we mutated each of the serine residues to an alanine (rendering them resistant to phosphorylation; Figure 4B). This string of mutations significantly reduced the effect of TNF-α on NaV1.7 (Figure S4). To narrow down the location of the critical residue(s), we generated a channel construct with just the L1 phosphorylation sites mutated (“quadruple mutant,” T531A, S535A, S608A, S727A) as well as a separate construct with just the N-terminal sites mutated (“triple mutant,” T103A, S110A, S113A). While the quadruple mutant demonstrated a normal response to treatment with TNF-α, the triple mutant significantly reduced the effect of treatment (Figure S4).
To confirm that the triple mutant channel did not respond to modulation by TNF-α, we transfected DRG neurons harvested from NaV1.8-null mice with plasmids encoding the triple mutant and mCherry. In the whole-cell voltage-clamp configuration, we recorded NaV1.7 currents in the absence or presence of 20 ng/mL TNF-α for 20 min (Figure 4C). The peak current density in DRG neurons expressing the triple mutant was not significantly different in the population of neurons incubated with TNF-α compared to those treated with DMSO control (0.44 ± 0.05 vs. 0.43 ± 0.10 nA/pF; n = 8 and 7, respectively; p > 0.05; Figures 4C and 4D). Since NaV1.8-null DRG neurons expressing wild-type (WT) hNaV1.7 were responsive to incubation with TNF-α (mean peak current density 1.39 ± 0.29 vs. 0.59 ± 0.14 nA/pF in control; n = 7 and 8, respectively; p < 0.05; Figures 4C and 4D), we hypothesized that at least one (or perhaps a combination) of these three phosphorylation sites is involved in the TNF-α-mediated increase in NaV1.7 current density.
To identify the specific phosphorylation site(s) responsible for TNF-α-mediated acute upregulation of NaV1.7, we generated individual mutations—T103A, S110A, or S113A—and assessed their current density in transfected DRG neurons from NaV1.8-null mice. NaV1.7 currents were recorded from red fluorescent cells incubated for 20 min with either 20 ng/mL TNF-α or DMSO. Neurons expressing NaV1.7 T103A had a higher peak current density when exposed to TNF-α than to control (1.40 ± 0.26 vs. 0.55 ± 0.09 nA/pF; n = 11 and 11, respectively; p < 0.05; Figure 4D). Similarly, neurons expressing NaV1.7 S113A exhibited a marked increase in current density in neurons after TNF-α incubation compared to control (1.95 ± 0.36 vs. 0.87 ± 0.25 nA/pF; n = 10 and 11, respectively; p < 0.05; Figure 4D). DRG neurons expressing NaV1.7 S110A, however, recapitulated the results seen in neurons expressing the triple mutant. Neurons incubated with TNF-α did not have a significant increase in peak current density compared to those incubated with DMSO control (0.70 ± 0.14 vs. 0.91 ± 0.17 nA/pF; n = 10 and 9, respectively, p > 0.05; Figure 4D), demonstrating that the S110 residue is necessary for acute upregulation of NaV1.7 current by TNF-α.
TNF-α drives vesicles carrying NaV1.7 to the distal axon
Though our electrophysiological investigations indict a p38 MAPK-dependent increase in NaV1.7 activity in response to acute TNF-α exposure, these studies reflect changes in the electrogenisome in neuronal cell bodies specifically. In vivo, DRG neurons have long axons that extend to peripheral axonal terminals, which are the actual site of AP electrogenesis.49
Recently, we developed high-fidelity molecular tools and live-imaging methodology that enable the study of ion channel regulation at these distal sites.8,9,50,51 We used this approach to further elucidate the spatiotemporal mechanisms underlying the regulation of NaV1.7 by TNF-α.
These studies required a full-length hNaV1.7 construct linked to a HaloTag enzyme (Halo-NaV1.7), a self-labeling enzymatic tag that reacts covalently with a specific cognate ligand, which can be conjugated to a variety of synthetic fluorophores. We first verified that acute TNF-α application still increased Halo-NaV1.7 current when expressed in DRG neurons (Figure S5) and that mutant Halo-NaV1.7 (Halo-NaV1.7 S110A) retained WT gating properties (Figure S6).
To determine if TNF-α affected the trafficking of vesicles carrying NaV1.7 to the distal axon, we assessed channels using OPAL imaging8,9 (Figure 5A). The OPAL method utilizes microfluidic chambers to establish cultures in which neuronal cell bodies are confined to one chamber and their axons grow through microgrooves into an axon-only chamber. This enables fluorescent labeling of HaloTag in either the soma or axonal chamber. We labeled Halo-NaV1.7 channels present in the soma of DRG neurons and then visualized anterogradely trafficking fluorescent vesicles in axons before and after application of TNF-α or DMSO vehicle (Figures 5B and 5C). We found that application of TNF-α for 20 min significantly increased vesicular flux to the distal axon (8.44 ± 1.09 before application vs. 11.21 ± 1.21 after application; n = 29, 3 independent cultures; p < 0.01 by paired t test; Figures 5B–5D). The increase in flux generated by TNF-α addition was significantly greater compared to DMSO control (n = 26, 3 independent cultures; p < 0.05 by unpaired t test; Figures 5D and 5E).
Figure 5. Acute exposure to TNF-α drives packaging of NaV1.7 and vesicular flux to the axons of DRG neurons.
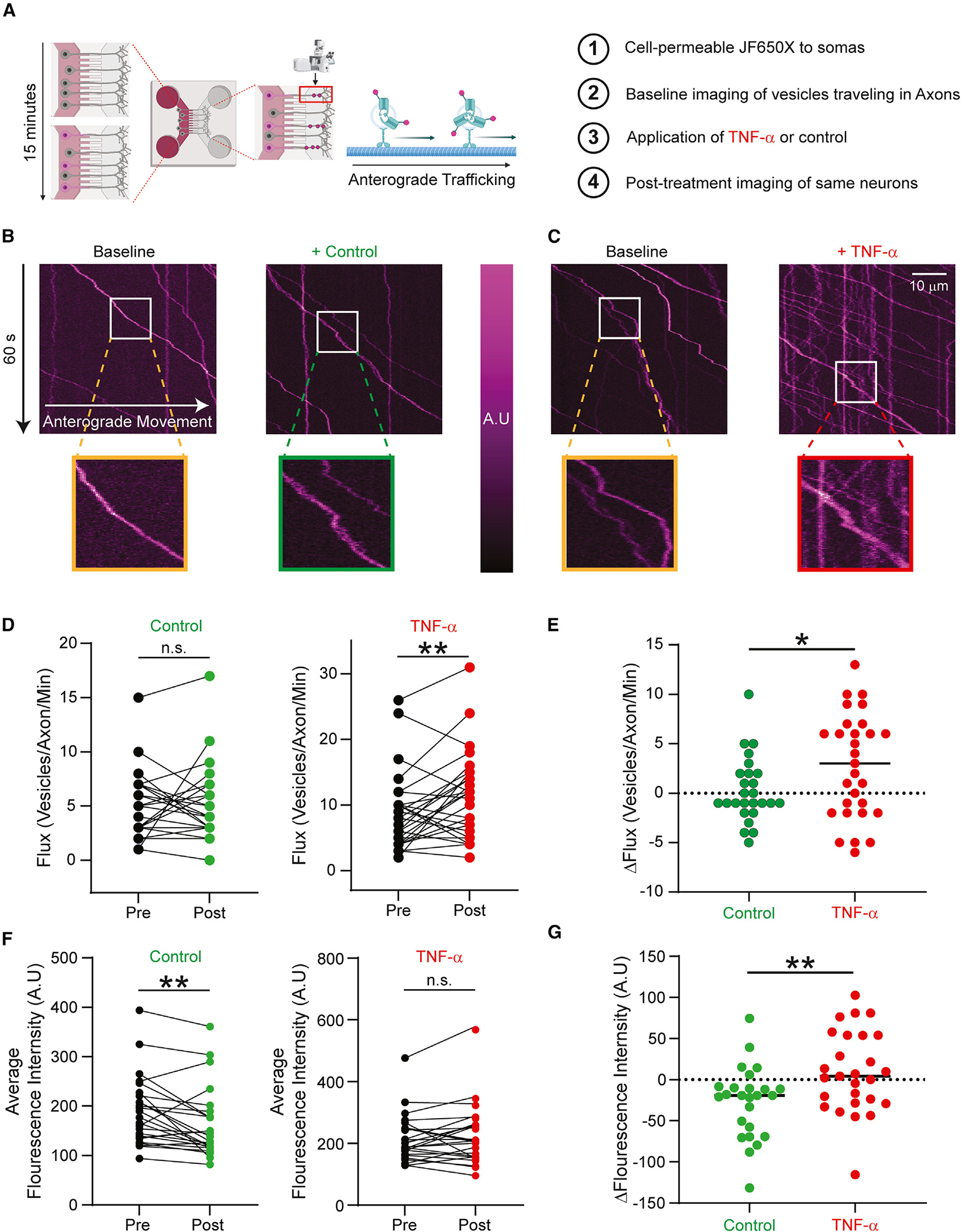
(A) Schematic of OPAL imaging. Cell-permeable JF650X-HaloTag is added to the somatic chamber of MFCs containing DRG neurons expressing Halo-NaV1.7. Time lapses of vesicular trafficking in axons were taken before and after perfusion of 20 ng/mL TNF-α or vehicle (imaging saline).
(B) Representative kymographs from an axon containing multiple anterogradely trafficking vesicles carrying Halo-NaV1.7 before and 20 min after perfusion of vehicle. Insets show the signal intensity of the magnified tracks.
(C) Representative kymographs from an axon containing multiple anterogradely trafficking vesicles carrying Halo-NaV1.7 before and 20 min after perfusion of TNF-α. Insets show the signal intensity of the magnified tracks.
(D) Flux of vesicles carrying Halo-NaV1.7 significantly increased following acute application of TNF-α. Vehicle perfusion did not affect the flux of anterogradely moving Halo-NaV1.7-positive vesicles. **p < 0.01, paired Student’s t test.
(E) Neurons exposed to TNF-α had significantly increased flux of vesicles carrying Halo-NaV1.7 compared to vehicle. *p < 0.05 by unpaired Student’s t test.
(F) Quantitation of the average fluorescence intensity of anterogradely moving vesicles carrying Halo-NaV1.7. Fluorescence intensity of Halo-NaV1.7 vesicles was significantly lower following vehicle perfusion; intensity was not different following TNF-α application. **p < 0.01, paired Student’s t test. Scale bars: 10 μm
(G) The change in fluorescence intensity of vesicles carrying Halo-NaV1.7 following TNF-α application was significantly higher than the one produced by vehicle. *p < 0.05, unpaired Student’s t test.
Data are presented as mean ± SEM.
Because each Halo-NaV1.7 channel is capable of binding only one HaloTag ligand, quantifying the fluorescence intensity of a vesicle is a surrogate measure for the number of labeled channels carried in that vesicle. Though exposure to TNF-α did not increase the signal intensity of vesicles carrying NaV1.7, vehicle perfusion resulted in a modest decrease in intensity. Comparing TNF-α administration to DMSO vehicle demonstrates that acute application of TNF-α resulted in a relative increase in fluorescence per vesicle (Figures 5F–5G; p < 0.01 by unpaired t test). This indicates that anterogradely moving vesicles carrying NaV1.7 channels to the distal axon contain more channels per vesicle after acute exposure to TNF-α.
Acute exposure to TNF-α results in increased channel insertion in neuronal somatic membranes but not axons
We hypothesized that the increase in NaV1.7 current density seen in response to acute TNF-α exposure was due to increased channel insertion in the neuronal membrane driven by the p38 MAPK cascade. To investigate this possibility, we extended our previously described insertion assay9 to both somas and axons of DRG neurons grown in microfluidic chambers (MFCs; Figure 6A). We used dual-color fluorescence microscopy to quantify the insertion rate of NaV1.7 channels in response to acute treatment with TNF-α or vehicle (Figures 6C and 6E).
Figure 6. Insertion of NaV1.7 in response to acute TNF-α exposure is differentially regulated in somas and axons.

(A) Schematic of insertion assay. DRG neurons plated in MFC chambers enabled the compartment-specific investigation of insertion dynamics. First, channels at the cell surface were labeled with cell-impermeable JF549i-HaloTag ligand. Excess ligand was thoroughly washed away, and the second cell-impermeable ligand (JF-635i) was added to the bath. Newly inserted channels would be labeled by fluorophore present in the bath and fluoresce in the far-red channel of the spinning-disk confocal microscope.
(B) Representative images of DRG neuronal somas before (left) and after (right) exposure to TNF-α for 20 min. Top: somas of DRG neurons expressing WT Halo-NaV1.7. Bottom: somas from DRG neurons expressing Halo-NaV1.7 S110A.
(C) Acute application of TNF-α drives insertion of WT Halo-NaV1.7 channels to the somatic membrane of DRG neurons above the baseline rate of insertion. Mutation of the S110 residue reduces this effect.
(D) Representative images captured from the distal axons of DRG neurons before (left) and after (right) exposure to TNF-α for 20 min. Top: axons of DRG neurons expressing WT Halo-NaV1.7. Bottom: axons of DRG neurons expressing Halo-NaV1.7 S110A. Because only newly inserted channels were labeled with Halo-JF635i ligand, the baseline fluorescence intensity of distal axons was very low in the far-red wavelength. Representative images have been background subtracted so that the distal axonal signal can be better visualized by readers. The image adjustment was identical for both vehicle- and TNF-α-treated conditions.
(E) TNF-α does not affect the insertion kinetics of WT Halo-NaV1.7 channels at the distal axons of DRG neurons. There were no statistically significant differences between the insertion of WT channels vs. those channels carrying the S110A mutation.
(F) Representative images captured from ND7/23 cells transfected with hNaV1.7-NTHalo before (left) and after (right) exposure to vehicle or 20 ng/mL TNF-α for 20 min.
(G) Acute application of TNF-α drives insertion of hNaV1.7-NTHalo to the somatic membrane relative to vehicle. Data from each cell are normalized to the fluorescence intensity at baseline (preperfusion). *p < 0.05 and **p < 0.01 by unpaired Student’s t test. n.s. (not significant), p > 0.05 by unpaired Student’s t test. Scale bars: 10 μm.
Data are presented as mean ± SEM.
In neuronal somata, our results recapitulated those obtained by electrophysiological recordings—TNF-α exposure increased NaV1.7 surface levels within 20 min (70% increase with TNF-α [n = 15, 3 independent cultures] vs. 32% increase with vehicle [n = 15, 3 independent cultures]; p < 0.05 by unpaired t test; Figures 6B and 6C). This effect was reduced in neurons expressing Halo-NaV1.7 S110A channels (70% increase for WT channels vs. 26% increase for mutant channels [n = 15, 3 independent cultures]; p > 0.05; Figures 6B and 6C). These results indicate that acute TNF-α exposure results in increased NaV1.7 insertion into the somatic membrane and that the mechanism of this process depends on S110 in the channel N terminus.
We then conducted the same assay in distal axons. Surprisingly, we found that in contrast to results in the soma, acute TNF-α application did not increase insertion of NaV1.7 channels in the distal axon membrane within 20 min (5.1% with TNF-α [n = 20, 3 independent cultures] vs. 4.9% with vehicle [n = 21, 3 independent cultures]; p > 0.05 by unpaired t test; Figures 6D and 6E) or even 1 h (68.6% with TNF-α vs. 51.1% with vehicle; p > 0.05; Figure 6E). These data reveal a neuronal-compartment-specific mechanism of TNF-α-mediated regulation of NaV1.7 channels.
To determine whether the N terminus of the channel is sufficient to mediate TNF-α-driven somatic membrane insertion, we generated a reporter construct that encodes the N terminus of NaV1.7 fused in frame with a transmembrane segment affixed with the HaloTag enzyme (hNaV1.7-NTHalo). We performed our insertion assay on DRG-neuron-derived ND7/23 cells transfected with hNaV1.7-NTHalo and found that acute treatment of ND7/23 cells with 20 ng/mL TNF-α significantly increases the membrane insertion of the N terminus by ~4.5-fold within 20 min relative to vehicle (normalized fluorescence increase: TNF-α [3,736.0 a.u., n = 24] vs. vehicle [813.3 a.u., n = 21]; p < 0.01; Figures 6F and 6G), consistent with the effect that was seen with the full-length channel. Together with our electrophysiological data, this indicates that the N terminus of hNaV1.7 is sufficient and that the S110 residue is necessary to mediate the acute effect of TNF-α on the level of the channel in a p38-dependent manner.
DISCUSSION
TNF-α is one of the most prominent cytokines involved in the development of inflammatory pain, and its effects on NaV1.7 channels are particularly relevant because of the essential role of NaV1.7 in peripheral neuronal excitability and pain sensation.52–54 Several groups have demonstrated that TNF-α increases sensory neuronal hyperexcitability and evokes pain symptoms in rodent models.19,29,30,33,55 These TNF-α-mediated changes in neuronal excitability have been shown to persist through a range of TNF-α concentrations, from 0.01 34,56 to 100 ng/mL.30,57 Some of this hyperexcitability has been linked to the ability of TNF-α to drive de novo biogenesis of pro-nociceptive voltage-gated ion channels, including NaV1.7.34,55 Importantly, these studies investigated neuronal excitability following chronic exposure (6 h to days) to TNF-α, though inflammatory pain can be felt within minutes of tissue injury. In this study, we provide data showing that acute treatment with TNF-α causes pro-excitatory changes in NaV1.7 channel post-translational regulation in a manner dependent on the phosphorylation of the channel at a specific serine residue in the N terminus by p38 MAPK. These data provide a mechanistic basis for acute inflammatory pain.
We show that acute (20 min) exposure of DRG neurons to TNF-α dramatically increases both endogenous TTX-S sodium current as well as human NaV1.7 current as recorded from the neuronal cell body (Figures 1 and 2). We also demonstrate that these changes in NaV1.7 activity manifest as DRG neuronal hyperexcitability (Figure 3), indicating that TNF-α-induced upregulation of NaV1.7 may result in a decreased nociceptive threshold during the acute phase of inflammatory pain. Additionally, TNF-α leads to the development of spontaneous subthreshold oscillations in membrane potential, which have been shown to contribute to pain pathogenesis58–60 and whose amplitude depends on NaV1.7.61 We demonstrate that the acute TNF-α-induced upregulation of NaV1.7 is mediated by p38 MAPK (Figure 3) and have identified serine 110 in the N terminus of the channel as the necessary phospho-acceptor site for this action (Figure 4). We show that TNF-α drives vesicles carrying NaV1.7 to the distal axon (Figure 5) but that there is no increase in actual channel insertion at the distal axon (Figure 6). By contrast, we reveal that TNF-α increases NaV1.7 channel insertion in the somatic membrane within 20 min. Finally, we show that the N terminus of the channel is sufficient to mediate this effect of TNF-α on the channel (Figure 6). Surprisingly, our observations suggest that the insertion of NaV1.7 is differentially regulated in DRG axons compared to cell bodies.
One of the classical signaling cascades that the TNF-α-TNFR1 interaction initiates is the activation of the p38 MAPK.31,62 Our results show that the selective and widely used inhibitor of the p38 MAPK, SB203580, results in a reduction of the acute TNF-α-mediated increase in NaV1.7 currents in DRG neurons (Figure 3). A previous study reported that TNF-α application for 5 min increases rat NaV1.7 current through protein-protein interaction with phosphorylated p-65 (p-p65), a subunit of the NF-κB complex.36 Based on experiments utilizing PDTC, an NF-κB inhibitor, they concluded that TNF-α’s acute effects on NaV1.7 channels are dependent on an NF-κB signaling cascade. We show, by using PDTC as well as another selective inhibitor of NF-κB, SC-514,63 that NF-κB blockade does not block the acute modulation of NaV1.7 by TNF-α. The reason for the difference between our study and that of Xie et al. 36 is not clear.
We have previously shown that rat NaV1.8 is a substrate for activated-p38 MAPK and that two serine residues in the L1 loop (S551 and S556) are the phosphate-accepting sites.46 Further, activation of p38 MAPK results in an increase in NaV1.8 current density, consistent with the TNF-α effect that has been reported.31 We screened the sequence of NaV1.7 for putative phosphorylation sites and hypothesized that cytoplasmic SP or TP motifs may control p38-mediated regulation of NaV1.7 channels as well. We systematically tested 7 putative phosphorylation sites (Figures 4A and S4) and identified S110 in the NaV1.7 N terminus as necessary for the acute action of TNF-α on the channel. Interestingly, the p38 phospho-acceptor sites in NaV1.7 (N terminus) and NaV1.8 (L1) are part of PXSP motifs, minimal linear motifs that have been implicated in protein-protein interactions, which suggests that potentially similar mechanisms are involved in the TNF-α-induced upregulation of these two channel isoforms at the somatic plasma membrane.
Resident macrophages in sensory ganglia have been shown to proliferate and acutely release TNF-α after peripheral nerve injury.64,65 TNF-α-mediated upregulation of NaV1.7 current may be a key step in the development of neuropathic pain following nerve injury. Our studies show that the mechanism of acute NaV1.7 modulation by TNF-α is increased channel insertion in the somatic membrane (Figure 6). When investigated by electrophysiology, these changes manifest as increased neuronal excitability. Our observations, however, suggest that the insertion of NaV1.7 is differentially regulated in DRG axons compared to cell bodies. Because TNF-α acutely increases the flux of NaV1.7-carrying vesicles and also the number of channels in each vesicle, the differential control of channel surface insertion at the distal axonal membrane appears to be due to decreased insertion kinetics rather than a lack of channels available to be inserted in the axonal compartment.
Compartment-specific regulation of channel surface expression by TNF-α has implications for the physiology of acute neuroinflammation. In peripheral sensory neurons, AP electrogenesis and propagation is initiated in the distal axons.4,49,66 In spite of this, and in the face of an enormous metabolic cost, the DRG neuron soma is an excitable structure,67,68 and APs initiated in the periphery have been shown to invade the cell body.69 The role and reasons for this somatic excitability are unclear, though several pieces of evidence suggest that somatic excitability may be a critical component of nociception. For one, somatic hyperexcitability causes some DRG neurons to fire spontaneously.68,70,71 In small-diameter DRG neurons, increasing somatic Na+ conductance elevates the safety factor for spike propagation across the T-junction,72 and simulations suggest that increasing somatic NaV current density increases the safety factor for transmission of APs through the T-junction and into the proximal process.73 Additionally, cell bodies in the DRG have been implicated in the phenomena of cross-excitation and cross-sensitization, where soma-soma communication through gap junctions leads to the spread of pain sensation from the locus of injury to areas where nerves are apparently undamaged.74–76 Electrogenesis from within the DRG, driven by inflammatory mediators including TNF-α, might be an under-appreciated driver of peripheral pain syndromes including post-herpetic neuralgia, phantom limb pain, complex regional pain syndrome, and others.
Changes in the ion channel architecture of the neuronal cell body clearly potentiates pain signaling, and we propose that TNF-α-mediated insertion of NaV1.7 channels into the somatic membrane represents a potential mechanism for acute inflammatory pain. This hypothesis is supported by the observation that TNF-α-secreting resident macrophages in the DRGs themselves proliferate in a spared-nerve injury (SNI) model prior to development of allodynia.64
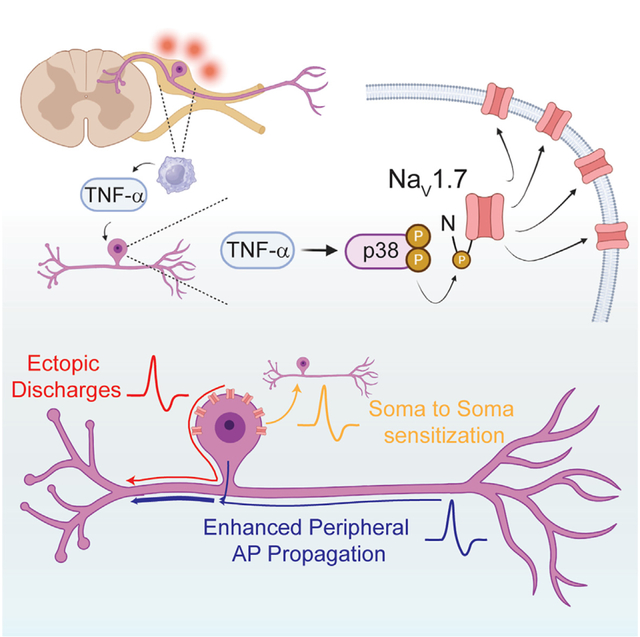
We propose that in the acute stages of inflammation, TNF-α released by resident macrophages acts locally on neuronal cell bodies and, through action of the p38 MAPK, phosphorylates a critically important S110 residue in the N terminus of the threshold channel NaV1.7 (Figure 7). This phosphorylation drives NaV1.7 channels to the somatic membrane surface, which manifests as somatic hyperexcitability. This hyperexcitability could leave DRG neurons more prone to ectopic discharge in the absence of a peripheral noxious stimulus and reduce overall nociceptive threshold by augmenting AP propagation from the periphery. Additionally, DRG ectopia in nociceptors may contribute to central spinal sensitization in dorsal horn neurons. Importantly, though these acute effects may explain how pain develops quickly in response to inflammation, clinically, neural exposure to TNF-α is much more prolonged, and transcriptional and translational regulation are enabled. However, the effects of TNF-α on NaV1.7 channels that we report here should persist even in the chronic stages of pain and may contribute to sensitization in long-term disease as well. More broadly, our work emphasizes the need for interrogation of both somatic and axonal ion channel expression when forming conclusions about the effect of compounds on channel function or of channel effect on neuronal physiology. Our studies suggest that differential control of trafficking and insertion may be a mechanism to alter electrical properties of neurons in response to physiologic changes or pathologic insults.
Figure 7. Summary of the acute effect of TNF-α on DRG neurons.

In response to tissue damage, macrophages release TNF-α in the DRG. Acutely, TNF-α binding to a TNFR initiates a phosphorylation cascade. The p38 MAPK is phosphorylated, which subsequently phosphorylates NaV1.7 at the N-terminal site S110. This leads to an increase in insertion kinetics of the channel at the somatic membrane. Increased somatic NaV1.7 could lead to increased ectopic discharge, enhanced peripheral AP propagation, and soma-to-soma sensitization.
Limitations of the study
TNF-α can trigger signaling cascades by binding to different TNF-α receptors—TNFR1 and TNFR2. Activation of these different receptor isoforms can trigger diverse effector functions.44 Though we did not indict a specific TNFR isoform in our study, we suspect that the acute effects we report here are mediated through TNFR1 because this isoform is expressed at a higher level than TNFR2 in rodent and human sensory neurons,77 the soluble form of TNF-α binds primarily to TNFR1,43 and TNFR1 has been shown to be required to modulate ion channels in both DRG and cortical neurons.29,56,78 Our imaging studies rely on overexpression of NaV1.7 channels, which could potentially be trafficked differently than native channels. However, these concerns are mitigated because imaging experiments in the soma recapitulate electrophysiologic findings in native neurons, and we have demonstrated that overexpression of targets does not cause proteins to be trafficked indiscriminately.8,79 While our studies suggest that a slow channel insertion step is responsible for the differential control of channel insertion in axons vs. somas, we do not delineate the underlying mechanism, which we will pursue in future studies. We also point out that our culture system does not recapitulate the pseudounipolar structure of sensory afferents and lacks innervated targets; therefore, we cannot delineate peripheral vs. central axonal ends. Our studies assay freely outgrowing distal axons rather than true axonal terminals within target tissues. Distal axons in our culture continue to grow during imaging, which may contribute to the slower insertion of the channels in this compartment. However, we have previously shown that treatment of similar DRG cultures with an inflammatory mediator cocktail increases the insertion of NaV1.7 channels in distal axons after 4 h of treatment.9 This supports the idea that delayed inflammatory-mediated increase in the insertion of the channels is not necessarily caused by the continued increase of neuronal membrane in the growth cone but is more likely caused by a slower insertion mechanism compared to that in the soma. Finally, though TNF-α is a critical cytokine involved in the development of inflammatory pain, it is one of many inflammatory mediators that act on DRG neurons to drive hyperexcitability. Some of these mediators may have synergistic or complementary effects, which were not explored in our experiments.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Sulayman Dib-Hajj (sulayman.dib-hajj@yale.edu).
Materials availability
Plasmids generated in this study are available from the lead contact upon request.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request
This paper does not report original code
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND STUDY PARTICIPANT DETAILS
Animals
Animals used in this this study were 2–4 d old Sprague-Dawley rat pups (Figures 1, 2, 3, 4, 6, and 7) or 4–6-week-old NaV1.8-null mice (Figure 5). Neonatal Animals were selected randomly and thus included males and females; adult mice of both sexes were used.
Maintenance, care, and experiments using animals were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the IACUC of the Veterans Administration Connecticut Healthcare System.
METHOD DETAILS
NaV1.7 constructs
All human NaV1.7 plasmids were rendered TTX-resistant (hNaV1.7-R) by substituting amino acid (a.a.) Tyr362 with serine (Y362S) using QuikChange Lightning site-directed mutagenesis.42
Halo constructs
We generated a codon-optimized Halo-NaV1.7 construct.9,79 The final construct topology is in order from the N terminus: 1–30 a.a. β4 signal peptide, 3× myc tag (EQKLISEEDL), Halo-tag enzyme (297 a.a.) (Promega), 3× HA tag (YPYDVPDYA), 21 a.a. transmembrane segment (β4 163–183), 7 a.a. linker (SGLRSAT), hNaV1.7-R. This channel retains WT gating properties and enables similar current density.79,80
Reagents
A 0.1 mg/mL stock solution of TNF-α was prepared in sterile water and diluted in DRG medium or external solution to the final concentration. DRG neurons were incubated with 20 ng/mL TNF-α for 20 min in a 37°C incubator to assess the acute effects of TNF-α on sodium currents and neuronal excitability. DRG neurons were pretreated with an IKK-β inhibitor (20 μM SC-514) for 1 h (IKK-β is an enzyme complex that forms part of NF-κB signaling pathway)81 or a p38 MAPK inhibitor (20 μM SB203580) for 30 min46 before and during the incubation of TNF-α (20 ng/mL) for 20 min.
Primary DRG culture
Animals used in this study were 2–4 d old Sprague-Dawley rat pups (Figures 1, 2, 3, 4, 6, and 7) or 4–6-week-old NaV1.8-null mice (Figure 5). DRGs from these animals were harvested and dissociated.82,83 Animals were selected randomly and thus included males and females. Dissected DRGs were incubated at 37°C for 20 min in complete saline solution (CSS) [in mM: 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, and 10 HEPES (pH 7.2), adjusted with NaOH], supplemented with 0.6 EDTA and collagenase A (1.5 mg/mL). DRG tissue was then incubated for 20 min at 37°C in CSS containing collagenase D (1.5 mg/mL), 0.6 EDTA, and papain (30 U/ml). DRG tissue was centrifuged and triturated in 1 mL of DRG culture medium DMEM/F12 (1:1) with penicillin (100 U/ml), streptomycin (0.1 mg/mL), 2 L-glutamine, and 10% FBS containing BSA (1.5 mg/mL) and trypsin inhibitor (1.5 mg/mL). The suspended neurons were then filtered through a 70 μm nylon mesh cell strainer, and then washed with DRG culture medium.
Biolistic transfection
For precipitation of the human-NaV1.7 plasmid onto gold particles,82 10 mg of human-NaV1.7 and 5 μg of EGFP DNA (NaV1.7: EGFP = 2 : 1) were added into 15 mg of 1 μm diameter gold particles. The gold coated tubing was stored in a desiccated box at −20°C. Helium pressure was used by the gun to shoot the gold particles into the cells. The cells were incubated at 37°C in 95% O2 and 5% CO2. Cell viability and recordings of hNaV1.7 current were assessed 24 h after the biolistic transfection. Transfected cells were identified by the green fluorescence from EGFP.
Electroporation of rat pup DRG neurons
After the preparation of DRG neuron suspension, the cells were centrifuged for 3 min (100×g) and the supernatant was removed. Cells were gently resuspended at room temperature in 100 μL of a mixed Nucleofector solution (82 μL NucleofectorTM solution +18 μL Supplement). Then, a 100 μL cell suspension with DNA (5 μg hNaV1.7 + 1 μg EGFP) was mixed by pipetting. After-ward, a 20 μL cell suspension was transferred into a strip provided in the Nucleofector kit. The sample was covered with the bottom of the strip, and air bubbles were avoided while pipetting. The strip was closed with the cap. The strip was inserted into its holder in the Nucleofector. Program DN-100 was used for rat DRG neurons. After transfection, 60 μL of pre-warmed BSA/TI solution was immediately added, and 80 μL were seeded per coverslip. DRG medium was added to a final volume of 1 mL per well 30 min after seeding, and the cells were incubated at 37°C in 95% O2 and 5% CO2. Cell viability and the recording of hNaV1.7 current were assessed 24 h after the transfection. Transfected cells were identified by the green fluorescence from EGFP.
Electroporation of NaV1.8-null mouse neurons
NaV1.8-null mouse neurons were transfected with a Nucleofector IIS and Amaxa Basic Neuron SCN Nucleofector Kit. Briefly, following harvest, the cell suspension was centrifuged (100 × g for 3 min), and the cell pellet was resuspended in 20 μL of Nucleofector solution, mixed with 2.5 mg of either WT human-NaV1.7 or mutant NaV1.7 constructs and 0.1 0.1 μg mCherry. After electroporation using Nucleofector IIS and protocol SCN-BNP 6, 100 μL of calcium-free DMEM (37°C) was added, and cells were incubated at 37°C for 5 min to recover. The cell mixture was then diluted with DRG media containing 1.5 mg/mL BSA (low endotoxin) and 1.5 mg/mL trypsin inhibitor, seeded 100 μL onto poly-D-lysine/laminin-coated coverslips (BD), and incubated at 37°C in a 95% air/5% CO2 (v/v) incubator for 45 min to allow neurons to attach to the coverslips. After 45 min, 0.9 mL of DRG media was added into each well, and the DRG neurons were maintained at 37°C in a 95% air/5% CO2 (v/v) incubator before voltage-clamp recordings.
Voltage-clamp electrophysiology
Voltage-clamp recordings of sodium currents in Figures 1, 2, 3, and 4 were obtained with an Axopatch 200B and a MultiClamp 700B amplifier, and the data were analyzed using the pClamp 10.7 and OriginPro 2015 software. Recordings in Figure 5 were obtained with a HEKA EPC-10 Amplifier.
The internal solution contained the following (in mM): 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES, at a final pH of 7.3 (310 mOsm/L with sucrose). The external solution for the sodium currents after electroporation contained the following (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 TEA-Cl, 0.1 CdCl2, 0.0003 TTX and 10 HEPES, pH 7.3 (320 mOsm/L with sucrose). The external solution for the sodium currents of native DRG neurons contained the following (in mM): 30 NaCl, 110 Choline-Cl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 TEA-Cl, 0.1 CdCl2 and 10 HEPES, pH 7.3 (320 mOsm/L with sucrose). The external solution for the sodium currents after biolistic transfection contained the following (in mM): 70 NaCl, 70 Choline-Cl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 TEA-Cl, 0.1 CdCl2, 0.0003 TTX and 10 HEPES, pH 7.3 (320 mOsm/L with sucrose). Current-clamp recordings were performed with MultiClamp 700B amplifiers using the following solutions: the internal solution contained (in mM): 140 KCl, 1 EGTA, 10 NaCl, 10 HEPES, 2 Mg-ATP, pH 7.3 (310 mOsm/L with sucrose), and the external solution contained (in mM): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 glucose, and 10 HEPES, pH 7.3 (320 mOsm/L with sucrose). For experiments with TNF-α incubation, TNF-α (20 ng/mL) was maintained in the extracellular solution. Cells in the vehicle control condition were incubated with an equal concentration of DMSO.
Small-diameter DRG neurons (≤30 μm in diameter) were selected because the majority of small-diameter DRG neurons are known to be nociceptive neurons (Hunt and Mantyh, 2001; Choi et al., 2007). Patch pipettes were pulled from soft glass capillaries using a programmable horizontal microelectrode puller. The pipette tips had a resistance of 0.9–1.2 and 1.8–2.1 MΩ when filled with the internal solution for the voltage-clamp and current-clamp, respectively. The coverslips containing adherent cells were mounted onto glass and maintained at 22°C using a temperature controller. The pClamp 10.7 software (Molecular Devices) was used for data acquisition and analysis. The sampling rate was 20–100 kHz and the sodium currents were filtered at 5 kHz (four pole Bessel filter). In the whole-cell configuration, if the series resistance was larger than 1.5 MΩ, the recording was discarded to reduce the voltage errors. The effective series resistance under 1.5 MΩ was compensated by 90% if the sodium current exceeded 2 nA. Recordings were started 10 min after establishing the whole-cell configuration to allow the currents to stabilize.
Action potentials (APs) were elicited by two types of current injections: long (500 ms) and ramp (0–0.5, 1, 1.5, or 2 nA in 500 ms). Long current injections were used to measure the current threshold and the number of APs evoked by injecting a 500 pA current. Data are presented as the means ± S.E., and the statistical significance was determined using the Student’s unpaired t-tests or one-way ANOVA followed by Bonferroni’s post-hoc test (p < 0.05).
Phosphorylation site Identification
Macroscopic currents were recorded in voltage-clamp mode using an EPC-10 amplifier and the PatchMaster Next program. Series resistance compensation of 80–90% was applied to reduce voltage error. Cells were excluded if the voltage error exceeded 5 mV. Recordings were sampled at 50 kHz through a low-pass Bessel filter of 2.9 kHz. After achieving the whole-cell configuration, a 5-min delay was applied to allow adequate time for the pipette solution and cytoplasmic milieu to equilibrate. Cells in the TNF-α treated condition were incubated with 20 ng/mL TNF-α for 20 min before recording. Those in the vehicle control condition were incubated with an equal concentration of DMSO.
DRGs from adult male (4–6 week old) NaV1.8-KO mice were harvested and dissociated.82,83 After trituration, neurons were co-transfected with 0.1 mg of mCherry and 2.5 mg of either WT hNaV1.7, hNaV1.7 T103A, hNaV1.7 S110A, hNaV1.7 S113A, or hNaV1.7 T103_S110A_S113A (all constructs with the Y362S substitution to render the channel TTX-resistant) using a Nucleofector IIS and Amaxa Basic Neuron SCN Nucleofector Kit. Cells were plated into 24-well plates containing cover slips coated with poly-L-lysine and laminin and maintained in DRG media at 37°C for 24 h after transfection before acquiring patch-clamp recordings.
To measure NaV1.7 currents, small diameter (<25 mM) DRG neurons with red fluorescence were selected for whole-cell voltage-clamp recording. Patch pipettes were fabricated from borosilicate glass (World Precision Instruments) using a P-97 puller (Sutter Instruments) and fire-polished for a resistance of 0.8–1.2 MΩ when filled with internal solution. The pipette internal solution was the same as above. For the WT NaV1.7 as well as the NaV1.7 Triple-Mutant construct, external bath solution contained (in mM): 30 NaCl, 90 Choline-Cl, 20 TEA-Cl, 3 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 5 Sucrose, 0.1 CdCl2, 0.001 TTX (pH 7.3 with NaOH, adjusted to 320 mOsm/L with sucrose). For the other NaV1.7 single-mutant constructs, external bath solution contained (in mM): 50 NaCl, 70 Choline-Cl, 20 TEA-Cl, 3 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 5 Sucrose, 0.1 CdCl2, 0.001 TTX (pH 7.3 with NaOH, adjusted to 320 mOsm/L with sucrose).
Cells were held at −80 mV. From this holding potential, cells were pre-pulsed to −100mV for 1 s to allow NaV1.7 channels to recover from inactivation, and then pulsed to potentials between −70 mV and +40 mV in 5 mV steps. Current density was measured by normalizing peak currents to cell capacitance.
Statistical significance was determined by unpaired Students t-test. False discovery rate (FDR) was quantified to control for multiple comparisons (a = 0.05).
High-throughput automated patch clamp electrophysiology
ND7/23 cells transfected with hNav1.7 channels were evaluated by automated voltage-clamp (Figure S3). Plasmids encoding hNav1.7 were introduced into ND7/23 cells via lipid-based transfection (Lipofectamine 3000). Cells were harvested into a cell suspension 48 h post-transfection for recordings by an automated electrophysiology robot (Qube 384). External bath solution was (in mM): 140 NaCl, 20 TEA-Cl, 3 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 5 Sucrose, 0.1 CdCl2, 0.001 TTX (pH 7.3 with NaOH, adjusted to 320 mOsm/L. TNF-α was maintained in the bath for conditions where it was present in the incubation step. Internal pipette solution was (in mM): 140 CsF, 1 EGTA, 10 HEPES, 10 NaCl (pH 7.3 with CsOH, adjusted to 310 mOsm/L with dextrose). ND7/23 cells were incubated with control, TNF-α, SC-514, and SB 203580 solutions as described above. PDTC-treated cells were preincubated with PDTC (10 nM) for 1 h before and during incubation with TNF-α for 20 min.
Halo-NaV1.7 channels were evaluated by automated voltage-clamp of transiently transfected HEK cells (Figure S6). Plasmids encoding Halo-NaV1.7 or Halo-NaV1.7 S110A (as control) were introduced into HEK 293 cells via lipid-based transfection (Lipofectamine 3000). Cells were harvested into a cell suspension 48 h post-transfection for recordings by an automated electrophysiology robot (Qube 384). Internal pipette solution was as above. External bath solution was (in mM): 145 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES (pH 7.4 with NaOH, adjusted to 305 mOsm/L with dextrose).
Step-by-step protocols for the operation of these high through-put machines is described elsewhere.84
Spinning disk microscopy
MFCs were bound to glass-bottomed dishes according to manufacturer’s instructions.8,9,79 Dissected DRGs were transfected as described above51,82 and plated into the somatic chamber of prepared MFCs containing DRG media with growth factors. Media in the axonal chambers was supplemented with 2x growth factors. After 24 h, medium was changed to serum free-medium in both chambers. After 5–7 days in vitro, DRG neurons were taken for imaging experiments.
Neurons were washed with DRG-Neuronal Imaging Saline (NIS) for these experiments. DRG-NIS contained (in mM): 136 NaCl, 3 KCl, 1 MgSO4, 2.5 CaCl2, 0.15 NaH2PO4, 0.1 Ascorbic Acid, 20 HEPES, 8 Dextrose (pH 7.4 with NaOH, adjusted to 320 mOsm/l).
Insertion assays
Insertion assays in these studies were adapted from a previously described method.9 Briefly, Halo-NaV1.7 channels at the surface of the neuron were labeled with cell-impermeable, red JF549i-HaloTag ligand (100 nM) for 15 min followed by a thorough wash step to remove excess label. Then, neurons were exposed to cell-impermeable, far-red JF635i-HaloTag ligand (10 nM) which was maintained throughout the experiment. With this schema, newly inserted channels would pick-up free HaloTag and fluoresce in the far-red wavelength. Both axons and somas were imaged at baseline, then both chambers were treated with TNF-α or vehicle for 20 min, and confocal z-stacks of far-red fluorescence were acquired every 20 min for 1 h while TNF-α remained in the bath solution. For analysis, z-stacks were processed by maximum-intensity projection in ImageJ software and the mean fluorescence intensity of each color was measured within the distal-most 20 mM of the axons or around the circumference of the soma. The background of the maximum intensity projection (modal gray value) was determined for each timepoint and field of view and was subtracted from each intensity measurement. Groups were compared by multiple unpaired t-tests. False discovery rate (FDR) was quantified to control for multiple comparisons (a = 0.05).
For insertion assays on ND7/23 cells, ND7/23 cells were plated on 35 mm glass bottom dishes and reverse transfected with plasmids encoding hNaV1.7-NTHalo using TransIT transfection reagent. The remainder of the insertion assay was conducted as described above.
Trafficking assays
Trafficking assays in these studies were variations on the Optical Pulse-chase Axonal Long-distance (OPAL) imaging method described previously.8,9,37,50,51,79 100 nm cell-permeable JFX650 ligand was applied to the somatic chamber of MFCs containing DRG neurons transfected with Halo-NaV1.7 constructs. After 15 min of incubation, the chambers were thoroughly washed with NIS, and MFCs were placed in a 37°C stage-top incubator for imaging. Multiple fields of view in the axonal chamber were selected, and anterogradely trafficking vesicles were imaged continuously for 1.5 min. After this initial image was acquired, solutions in both chambers were exchanged with solutions containing 20 ng/mL TNF-α or vehicle. After 20 min of incubation, the same fields were imaged enabling before-and-after comparison of anterograde trafficking in treated neurons.
Resulting movies were opened in ImageJ and the KymographClear toolset was used to create kymographs of the selected axons.85 Axons containing anterogradely-moving vesicles were traced manually using a 50 μm segmented line, and the signal under that line was converted into a two-dimensional image that could be further analyzed. KymoButler is a machine learning algorithm that traces vesicle tracks from kymographs.86 Vesicle flux was determined by counting the number of vesicles which crossed the midline of the kymograph. Vesicle intensity was determined as the average of fluorescence values of pixels along the vesicle track, minus the background signal of the kymograph (defined as the modal value for that kymograph). Vesicle velocity was defined as the average speed over the duration of the track, including pauses and stops. The fluorescence intensities of multiple vesicles within axons were averaged. Intra-group comparisons were made via paired Student’s t-tests. Between group comparisons were made via unpaired Student’s t-tests (a = 0.05).
ELISA
The concentrations of TNF-α in the medium of the cultured DRG neurons after electroporation were assayed with the Rat TNF-α ELISA MAXTM Deluxe Sets, according to the manufacturer’s protocols (Figure S2). Briefly, to assay the supernatant of the DRG neuron cultures, 50 μL of Assay diluent A were added to each well. Then, 50 μL of standards were added to each standard well. The plate was sealed and incubated at RT for 2 h with shaking. The plate was washed four times with Wash Buffer. Then, 100 μL of diluted Detection Antibody solution was added to each well, and the plate was sealed and incubated at room temperature for 1 h with shaking. After the plate was washed four times with Wash Buffer, 100 μL of diluted Avidin-HRP solution was added to each well. The plate was incubated at room temperature for 30 min and washed with Wash Buffer. Wells were soaked in Wash Buffer for 1 min to wash each well so to minimize background noise. Then, 100 μL of freshly mixed TMB Substrate Solution were added and incubated in the dark for 25 min. Positive wells turned blue and were read at 450 nm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were conducted in Python 3.9 equipped with the Pandas package. Graphs were generated with GraphPad Prism or OriginPro software. Statistical details of all experiments can be found in the results section and/or relevant figure legends. Statistical significance was set at α = 0.05. For all data, normality was evaluated by the Shapiro-Wilks test before undergoing parametric vs. non-parametric tests for group mean comparison. For multiple comparisons, p was corrected by the Bonferroni correction, or False discovery rate was quantified.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Tetrodotoxin | Calbiochem | 554412 |
| TNF-α | Invitrogen | RRTNFAI |
| SB203580 | Merck Millipore | 152121–47-6 |
| SC-514 | Tocris | 3318 |
| JF650X-HaloTag ligand | HHMI Janelia Research Campus | N/A |
| JF549i-HaloTag ligand | HHMI Janelia Research Campus | N/A |
| JF635i-HaloTag ligand | HHMI Janelia Research Campus | N/A |
| Lipofectamine 3000 | Invitrogen | L3000001 |
| TransIT-LT1 Transfection reagent | Mirus | MIR 2300 |
| Basic Nucleofector Kit for Primary Mammalian Neurons | Lonza | V4XP-3032 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Rat TNF-α ELISA MAXTM Deluxe Set | BioLegend | 438204 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| ND7/23 | Sigma | 92090903 |
| HEK293 | Sigma | 85120602 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Sprague-Dawley rat pups | Envigo | N/A |
| Nav1.8 KO mice | Dr. John Wood, UCL | N/A |
|
| ||
| Recombinant DNA | ||
|
| ||
| hNav1.7 | This Paper | N/A |
| hNav1.7 Septuple Mutant | This Paper | N/A |
| hNav1.7 Quadruple Mutant | This Paper | N/A |
| hNav1.7 Triple Mutant | This Paper | N/A |
| hNav1.7 T103A | This Paper | N/A |
| hNav1.7 S110A | This Paper | N/A |
| hNav1.7 S113A | This Paper | N/A |
| hNav1.7-Nterminus-Halo | This Paper | N/A |
| Halo-Nav1.7 | This Paper | N/A |
| Halo-Nav1.7 S110A | This Paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| PatchMaster Next | HEKA Elektronik | https://www.heka.com/downloads/downloads_main.html#down_patchmaster_next |
| Python | Python Software Foundation | https://www.python.org |
| Prism | GraphPad Software, Inc | https://www.graphpad.com/ |
| Andor Fusion | Oxford Instruments | N/A |
| Analyzer | Sophion | https://sophion.com/products/high-throughput-screening-on-qube/ |
| ViewPoint | Sophion | https://sophion.com/products/high-throughput-screening-on-qube/ |
| pClamp | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/acquisition-and-analysis-software/pclamp-software-suite |
| Adobe Illustrator | Adobe | N/A |
|
| ||
| Other | ||
|
| ||
| Helios Gene Gun | Bio-Rad | 1652431 |
| Nucleofector IIS | Lonza | N/A |
| Axopatch 200B | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/amplifiers/axon-instruments-patch-clamp-amplifiers |
| MultiClamp 700B | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system/amplifiers/axon-instruments-patch-clamp-amplifiers |
| HEKA EPC-10 | HEKA Elektronik | https://www.heka.com/ |
| P97 Pipette Puller | Sutter Instruments | N/A |
| Qube-384 | Sophion | https://sophion.com/products/high-throughput-screening-on-qube/ |
| Eclipse TE2000-U inverted microscope | Nikon | N/A |
| Dragonfly High Speed Confocal Microscope | Oxford Instruments | N/A |
Highlights.
Acute exposure to TNF-α increases NaV1.7 current density and neuronal excitability
Increased NaV1.7 current density occurs through p38 MAPK phosphorylation of residue Ser110
TNF-α exposure increases delivery of vesicles carrying NaV1.7 channels to distal axons
Insertion of NaV1.7 channels is differentially regulated in DRG axons compared to somas
ACKNOWLEDGMENTS
We thank Daniel Sosniak and Matthew Alsaloum for technical assistance. We thank Luke Lavis and Jonathan Grimm for the generous gifts of the JaneliaFluors. We thank Joshua Huttler for helpful discussions. This work was supported by Merit Review Awards B9253-C and BX004899 from the US Dept. of Veterans Affairs Rehabilitation Research and Development Service and Biomedical Laboratory Research and Development Service, respectively (S.G.W. and S.D.D.-H.). The Center for Neuroscience and Regeneration Research is a collaboration of the Paralyzed Veterans of America with Yale University. S.T. and G.P.H.-R. are supported by NIH/NIGMS Medical Scientist Training Program T32GM007205. S.T. is supported by NIH/NINDS T32NS041228. G.P.H.-R. is supported by NIH/NINDS 1F31NS122417–01. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. M.-R.G. is a Banting Fellow at Yale and is supported by the Canadian Institutes of Health Research (CIHR) grant number 471896. J.-S.C. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2016R1A2B4011333 and NRF-2018R1A6A1A03025108).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113685.
REFERENCES
- 1.Amir R, Argoff CE, Bennett GJ, Cummins TR, Durieux ME, Gerner P, Gold MS, Porreca F, and Strichartz GR (2006). The Role of Sodium Channels in Chronic Inflammatory and Neuropathic Pain. J. Pain 7. S1–29. [DOI] [PubMed] [Google Scholar]
- 2.Kidd BL, and Urban LA (2001). Mechanisms of inflammatory pain. Br. J. Anaesth. 87, 3–11. [DOI] [PubMed] [Google Scholar]
- 3.Talbot S, Foster SL, and Woolf CJ (2016). Neuroimmunity: Physiology and Pathology. Annu. Rev. Immunol. 34, 421–447. [DOI] [PubMed] [Google Scholar]
- 4.Bennett DL, Clark AJ, Huang J, Waxman SG, and Dib-Hajj SD (2019). The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 99, 1079–1151. [DOI] [PubMed] [Google Scholar]
- 5.Dib-Hajj SD, Yang Y, Black JA, and Waxman SG (2013). The NaV1.7 sodium channel: from molecule to man. Nat. Rev. Neurosci. 14, 49–62. [DOI] [PubMed] [Google Scholar]
- 6.Rush AM, Cummins TR, and Waxman SG (2007). Multiple sodium channels and their roles in electrogenesis within dorsal root ganglion neurons. J. Physiol. 579, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nassar MA, Stirling LC, Forlani G, Baker MD, Matthews EA, Dickenson AH, and Wood JN (2004). Nociceptor-specific gene deletion reveals a major role for Nav1.7 (PN1) in acute and inflammatory pain. Proc. Natl. Acad. Sci. USA 101, 12706–12711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Akin EJ, Higerd-Rusli GP, Mis MA, Tanaka BS, Adi T, Liu S, Dib-Hajj FB, Waxman SG, and Dib-Hajj SD (2019). Building sensory axons: Delivery and distribution of NaV1.7 channels and effects of inflammatory mediators. Sci. Adv. 5, eaax4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Higerd-Rusli GP, Tyagi S, Baker CA, Liu S, Dib-Hajj FB, Dib-Hajj SD, and Waxman SG (2023). Inflammation differentially controls transport of depolarizing Nav versus hyperpolarizing Kv channels to drive rat nociceptor activity. Proc. Natl. Acad. Sci. USA 120, e2215417120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toledo-Aral JJ, Moss BL, He ZJ, Koszowski AG, Whisenand T, Levinson SR, Wolf JJ, Silos-Santiago I, Halegoua S, and Mandel G (1997). Identification of PN1, a predominant voltage-dependent sodium channel expressed principally in peripheral neurons. Proc. Natl. Acad. Sci. USA 94, 1527–1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vasylyev DV, Han C, Zhao P, Dib-Hajj S, and Waxman SG (2014). Dynamic-clamp analysis of wild-type human Nav1.7 and erythromelalgia mutant channel L858H. J. Neurophysiol. 111, 1429–1443. [DOI] [PubMed] [Google Scholar]
- 12.Cook AD, Christensen AD, Tewari D, McMahon SB, and Hamilton JA (2018). Immune Cytokines and Their Receptors in Inflammatory Pain. Trends Immunol. 39, 240–255. [DOI] [PubMed] [Google Scholar]
- 13.Parameswaran N, and Patial S (2010). Tumor Necrosis Factor-α Signaling in Macrophages. Crit. Rev. Eukaryot. Gene Expr. 20, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cunha FQ, Poole S, Lorenzetti BB, and Ferreira SH (1992). The pivotal role of tumour necrosis factor alpha in the development of inflammatory hyperalgesia. Br. J. Pharmacol. 107, 660–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linley JE, Rose K, Ooi L, and Gamper N (2010). Understanding inflammatory pain: ion channels contributing to acute and chronic nociception. Pflu€gers Archiv 459, 657–669. [DOI] [PubMed] [Google Scholar]
- 16.Perkins MN, and Kelly D (1994). Interleukin-1 beta induced-desArg9-bradykinin-mediated thermal hyperalgesia in the rat. Neuropharmacology 33, 657–660. [DOI] [PubMed] [Google Scholar]
- 17.Woolf CJ, Allchorne A, Safieh-Garabedian B, and Poole S (1997). Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br. J. Pharmacol. 121, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindenlaub T, and Sommer C (2000). Partial sciatic nerve transection as a model of neuropathic pain: a qualitative and quantitative neuropathological study. Pain 89, 97–106. [DOI] [PubMed] [Google Scholar]
- 19.Sommer C, Lindenlaub T, Teuteberg P, Schä fers M., Hartung T., and Toyka KV. (2001). Anti-TNF-neutralizing antibodies reduce pain-related behavior in two different mouse models of painful mononeuropathy. Brain Res. 913, 86–89. [DOI] [PubMed] [Google Scholar]
- 20.Frere JJ, Serafini RA, Pryce KD, Zazhytska M, Oishi K, Golynker I, Panis M, Zimering J, Horiuchi S, Hoagland DA, et al. (2022). SARS-CoV-2 infection in hamsters and humans results in lasting and unique systemic perturbations after recovery. Sci. Transl. Med. 14, eabq3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Henkes S-S, Bosurgi L, Dutzmann J, Sedding D, Frese T, et al. (2022). The IL-1β, IL-6, and TNF cytokine triad is associated with post-acute sequelae of COVID-19. Cell Rep. Med. 3, 100663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kemp HI, Corner E, and Colvin LA (2020). Chronic pain after COVID-19: implications for rehabilitation. Br. J. Anaesth. 125, 436–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Global Burden of Disease Long COVID Collaborators; Wulf Hanson S., Abbafati C., Aerts JG., Al-Aly Z., Ashbaugh C., Ballouz T., Blyuss O., Bobkova P., Bonsel G., et al. (2022). Estimated Global Proportions of Individuals With Persistent Fatigue, Cognitive, and Respiratory Symptom Clusters Following Symptomatic COVID-19 in 2020 and 2021. JAMA 328, 1604–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wei S, Qiu C-Y, Jin Y, Liu T-T, and Hu W-P (2021). TNF-α acutely enhances acid-sensing ion channel currents in rat dorsal root ganglion neurons via a p38 MAPK pathway. J. Neuroinflammation 18, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Magalhães SF, Manzo LP, de Faria FM, de Oliveira-Fusaro MC, Nishijima CM, Vieira WF, Bonet IJM, dos Santos GG, Tambeli CH, and Parada CA (2021). Inflammatory pain in peripheral tissue depends on the activation of the TNF-α type 1 receptor in the primary afferent neuron. Eur. J. Neurosci. 53, 376–389. [DOI] [PubMed] [Google Scholar]
- 26.Leo M, Argalski S, Schä fers M., and Hagenacker T. (2015). Modulation of Voltage-Gated Sodium Channels by Activation of Tumor Necrosis Factor Receptor-1 and Receptor-2 in Small DRG Neurons of Rats. Mediat. Inflamm. 2015, 124942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu S-C, Chang Y-S, Kan H-W, and Hsieh Y-L (2018). Tumor necrosis Factor-α mediated pain hypersensitivity through Ret receptor in resiniferatoxin neuropathy. Kaohsiung J. Med. Sci. 34, 494–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Berta T, Xu Z-Z, Liu T, Park JY, and Ji R-R (2011). TNF-α contributes to spinal cord synaptic plasticity and inflammatory pain: Distinct role of TNF receptor subtype 1 and 2. Pain 152, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X, Pang R-P, Shen K-F, Zimmermann M, Xin W-J, Li Y-Y, and Liu X-G (2011). TNF-α enhances the currents of voltage gated sodium channels in uninjured dorsal root ganglion neurons following motor nerve injury. Exp. Neurol. 227, 279–286. [DOI] [PubMed] [Google Scholar]
- 30.Czeschik JC, Hagenacker T, Schäfers M, and Büsselberg D (2008). TNF-alpha differentially modulates ion channels of nociceptive neurons. Neurosci. Lett. 434, 293–298. [DOI] [PubMed] [Google Scholar]
- 31.Jin X, and Gereau RW (2006). Acute p38-mediated modulation of tetrodotoxin-resistant sodium channels in mouse sensory neurons by tumor necrosis factor-alpha. J. Neurosci. 26, 246–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozaktay AC, Kallakuri S, Takebayashi T, Cavanaugh JM, Asik I, DeLeo JA, and Weinstein JN (2006). Effects of interleukin-1 beta, interleukin-6, and tumor necrosis factor on sensitivity of dorsal root ganglion and peripheral receptive fields in rats. Eur. Spine J. 15, 1529–1537. [DOI] [PubMed] [Google Scholar]
- 33.Spicarova D, Nerandzic V, and Palecek J (2011). Modulation of spinal cord synaptic activity by tumor necrosis factor α in a model of peripheral neuropathy. J. Neuroinflammation 8, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tamura R, Nemoto T, Maruta T, Onizuka S, Yanagita T, Wada A, Murakami M, and Tsuneyoshi I (2014). Up-regulation of NaV1.7 sodium channels expression by tumor necrosis Factor-α in cultured bovine adrenal chromaffin cells and rat dorsal root ganglion neurons. Anesth. Analg. 118, 318–324. [DOI] [PubMed] [Google Scholar]
- 35.Richter F, Natura G, Löser S, Schmidt K, Viisanen H, and Schaible H-G (2010). Tumor necrosis factor causes persistent sensitization of joint nociceptors to mechanical stimuli in rats. Arthritis Rheum. 62, 3806–3814. [DOI] [PubMed] [Google Scholar]
- 36.Xie M-X, Zhang X-L, Xu J, Zeng W-A, Li D, Xu T, Pang R-P, Ma K, and Liu X-G (2019). Nuclear Factor-kappaB Gates Nav1.7 Channels in DRG Neurons via Protein-Protein Interaction. iScience 19, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker CA, Tyagi S, Higerd-Rusli GP, Liu S, Zhao P, Dib-Hajj FB, Waxman SG, and Dib-Hajj SD (2023). Paclitaxel effects on axonal localization and vesicular trafficking of NaV1.8. Front. Mol. Neurosci. 16, 1130123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dib-Hajj SD, Cummins TR, Black JA, and Waxman SG (2010). Sodium Channels in Normal and Pathological Pain. Annu. Rev. Neurosci. 33, 325–347. [DOI] [PubMed] [Google Scholar]
- 39.Black JA, Langworthy K, Hinson AW, Dib-Hajj SD, and Waxman SG (1997). NGF has opposing effects on Na+ channel III and SNS gene expression in spinal sensory neurons. Neuroreport 8, 2331–2335. [DOI] [PubMed] [Google Scholar]
- 40.Fjell J, Cummins TR, Dib-Hajj SD, Fried K, Black JA, and Waxman SG (1999). Differential role of GDNF and NGF in the maintenance of two TTX-resistant sodium channels in adult DRG neurons. Brain Res. Mol. Brain Res. 67, 267–282. [DOI] [PubMed] [Google Scholar]
- 41.Leffler A, Cummins TR, Dib-Hajj SD, Hormuzdiar WN, Black JA, and Waxman SG (2002). GDNF and NGF reverse changes in repriming of TTX-sensitive Na(+) currents following axotomy of dorsal root ganglion neurons. J. Neurophysiol. 88, 650–658. [DOI] [PubMed] [Google Scholar]
- 42.Yang Y, Huang J, Mis MA, Estacion M, Macala L, Shah P, Schul-man BR, Horton DB, Dib-Hajj SD, and Waxman SG (2016). Nav1.7-A1632G Mutation from a Family with Inherited Erythromelalgia: Enhanced Firing of Dorsal Root Ganglia Neurons Evoked by Thermal Stimuli. J. Neurosci. 36, 7511–7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grell M, Douni E, Wajant H, Löhden M, Clauss M, Maxeiner B, Georgopoulos S, Lesslauer W, Kollias G, Pfizenmaier K, and Scheurich P (1995). The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 83, 793–802. [DOI] [PubMed] [Google Scholar]
- 44.Baud V, and Karin M (2001). Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 11, 372–377. [DOI] [PubMed] [Google Scholar]
- 45.Gasser A, Cheng X, Gilmore ES, Tyrrell L, Waxman SG, and Dib-Hajj SD (2010). Two Nedd4-binding motifs underlie modulation of sodium channel Nav1.6 by p38 MAPK. J. Biol. Chem. 285, 26149–26161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hudmon A, Choi J-S, Tyrrell L, Black JA, Rush AM, Waxman SG, and Dib-Hajj SD (2008). Phosphorylation of Sodium Channel Nav1.8 by p38 Mitogen-Activated Protein Kinase Increases Current Density in Dorsal Root Ganglion Neurons. J. Neurosci. 28, 3190–3201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wittmack EK, Rush AM, Hudmon A, Waxman SG, and Dib-Hajj SD (2005). Voltage-Gated Sodium Channel Nav1.6 Is Modulated by p38 Mitogen-Activated Protein Kinase. J. Neurosci. 25, 6621–6630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tyagi S, Bendrick TR, Filipova D, Papadopoulos S, and Bannister RA (2019). A mutation in CaV2.1 linked to a severe neurodevelopmental disorder impairs channel gating. J. Gen. Physiol. 151, 850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dubin AE, and Patapoutian A (2010). Nociceptors: the sensors of the pain pathway. J. Clin. Invest. 120, 3760–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Akin EJ, Alsaloum M, Higerd GP, Liu S, Zhao P, Dib-Hajj FB, Waxman SG, and Dib-Hajj SD (2021). Paclitaxel increases axonal localization and vesicular trafficking of Nav1.7. Brain 144, 1727–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tyagi S, Sarveswaran N, Higerd-Rusli GP, Liu S, Dib-Hajj FB, Waxman SG, and Dib-Hajj SD (2023). Conserved but not critical: Trafficking and function of NaV1.7 are independent of highly conserved polybasic motifs. Front. Mol. Neurosci. 16, 1161028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McDermott LA, Weir GA, Themistocleous AC, Segerdahl AR, Blesneac I, Baskozos G, Clark AJ, Millar V, Peck LJ, Ebner D, et al. (2019). Defining the Functional Role of NaV1.7 in Human Nociception. Neuron 101, 905–919.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deng L, Dourado M, Reese RM, Huang K, Shields SD, Stark KL, Maksymetz J, Lin H, Kaminker JS, Jung M, et al. (2023). Nav1.7 is essential for nociceptor action potentials in the mouse in a manner independent of endogenous opioids. Neuron 111, 2642–2659.e13. [DOI] [PubMed] [Google Scholar]
- 55.Huang Y, Zang Y, Zhou L, Gui W, Liu X, and Zhong Y (2014). The role of TNF-alpha/NF-kappa B pathway on the up-regulation of voltage-gated sodium channel Nav1.7 in DRG neurons of rats with diabetic neuropathy. Neurochem. Int. 75, 112–119. [DOI] [PubMed] [Google Scholar]
- 56.Chen W, Sheng J, Guo J, Gao F, Zhao X, Dai J, Wang G, and Li K (2015). Tumor necrosis Factor-α enhances voltage-gated Na+ currents in primary culture of mouse cortical neurons. J. Neuroinflammation 12, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valdez-Morales EE, Sánchez-Navarro CA, Reyes-Pavón D, Barrios-Garcia T, Ochoa-Cortes F, Barajas-Espinosa A, Barragán-Iglesias P, and Guerrero-Alba R (2022). TNF-α enhances sensory DRG neuron excitability through modulation of P2X3 receptors in an acute colitis model. Front. Immunol. 13, 872760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu C-N, Devor M, Waxman SG, and Kocsis JD (2002). Subthreshold Oscillations Induced by Spinal Nerve Injury in Dissociated Muscle and Cutaneous Afferents of Mouse DRG. J. Neurophysiol. 87, 2009–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kovalsky Y, Amir R, and Devor M (2008). Subthreshold oscillations facilitate neuropathic spike discharge by overcoming membrane accommodation. Exp. Neurol. 210, 194–206. [DOI] [PubMed] [Google Scholar]
- 60.Kovalsky Y, Amir R, and Devor M (2009). Simulation in sensory neurons reveals a key role for delayed Na+ current in subthreshold oscillations and ectopic discharge: implications for neuropathic pain. J. Neurophysiol. 102, 1430–1442. [DOI] [PubMed] [Google Scholar]
- 61.Choi J-S, and Waxman SG (2011). Physiological interactions between Nav1.7 and Nav1.8 sodium channels: a computer simulation study. J. Neurophysiol. 106, 3173–3184. [DOI] [PubMed] [Google Scholar]
- 62.Li Y-P, Chen Y, John J, Moylan J, Jin B, Mann DL, and Reid MB (2005). TNF-α lpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. Faseb. J. 19, 362–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kumar S, McDonnell PC, Gum RJ, Hand AT, Lee JC, and Young PR (1997). Novel Homologues of CSBP/p38 MAP Kinase: Activation, Substrate Specificity and Sensitivity to Inhibition by Pyridinyl Imidazoles. Biochem. Biophys. Res. Commun. 235, 533–538. [DOI] [PubMed] [Google Scholar]
- 64.Guimarães RM, Aníbal-Silva CE, Davoli-Ferreira M, Gomes FIF, Mendes A, Cavallini MCM, Fonseca MM, Damasceno S, Andrade LP, Colonna M, et al. (2023). Neuron-associated macrophage proliferation in the sensory ganglia is associated with peripheral nerve injury-induced neuropathic pain involving CX3CR1 signaling. Elife 12, e78515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Konnova EA, Deftu A-F, Chung PCS, Kirschmann G, Decosterd I, and Suter MR (2023). Kir2.1 modulation in macrophages sensitises dorsal root ganglion neurons through TNF secretion after nerve injury. Preprint at bioRxiv. 10.1101/2023.06.21.545843. [DOI] [Google Scholar]
- 66.Esposito MF, Malayil R, Hanes M, and Deer T (2019). Unique Characteristics of the Dorsal Root Ganglion as a Target for Neuromodulation. Pain Med. 20, S23–S30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Amir R, and Devor M (2003). Electrical Excitability of the Soma of Sensory Neurons Is Required for Spike Invasion of the Soma, but Not for Through-Conduction. Biophys. J. 84, 2181–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wall PD, and Devor M (1983). Sensory afferent impulses originate from dorsal root ganglia as well as from the periphery in normal and nerve injured rats. Pain 17, 321–339. [DOI] [PubMed] [Google Scholar]
- 69.Chisholm KI, Khovanov N, Lopes DM, La Russa F, and McMahon SB (2018). Large Scale In Vivo Recording of Sensory Neuron Activity with GCaMP6. eNeuro 5, ENEURO.0417–17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Liu C-N, Wall PD, Ben-Dor E, Michaelis M, Amir R, and Devor M (2000). Tactile allodynia in the absence of C-fiber activation: altered firing properties of DRG neurons following spinal nerve injury. Pain 85, 503–521. [DOI] [PubMed] [Google Scholar]
- 71.Study RE, and Kral MG (1996). Spontaneous action potential activity in isolated dorsal root ganglion neurons from rats with a painful neuropathy. Pain 65, 235–242. [DOI] [PubMed] [Google Scholar]
- 72.Du X, Hao H, Gigout S, Huang D, Yang Y, Li L, Wang C, Sundt D, Jaffe DB, Zhang H, and Gamper N (2014). Control of somatic membrane potential in nociceptive neurons and its implications for peripheral nociceptive transmission. PAIN® 155, 2306–2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sundt D, Gamper N, and Jaffe DB (2015). Spike propagation through the dorsal root ganglia in an unmyelinated sensory neuron: a modeling study. J. Neurophysiol. 114, 3140–3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amir R, and Devor M (1996). Chemically Mediated Cross-Excitation in Rat Dorsal Root Ganglia. J. Neurosci. 16, 4733–4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim YS, Anderson M, Park K, Zheng Q, Agarwal A, Gong C, Saijilafu, Young L., He S., LaVinka PC., et al. (2016). Coupled Activation of Primary Sensory Neurons Contributes to Chronic Pain. Neuron 91, 1085–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Spray DC, and Hanani M (2019). Gap junctions, pannexins and pain. Neurosci. Lett. 695, 46–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jung M, Dourado M, Maksymetz J, Jacobson A, Laufer BI, Baca M, Foreman O, Hackos DH, Riol-Blanco L, and Kaminker JS (2023). Cross-species transcriptomic atlas of dorsal root ganglia reveals species-specific programs for sensory function. Nat. Commun. 14, 366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hensellek S, Brell P, Schaible H-G, Bräuer R, and Segond von Banchet G (2007). The cytokine TNFα increases the proportion of DRG neurones expressing the TRPV1 receptor via the TNFR1 receptor and ERK activation. Mol. Cell. Neurosci. 36, 381–391. [DOI] [PubMed] [Google Scholar]
- 79.Higerd-Rusli GP, Alsaloum M, Tyagi S, Sarveswaran N, Estacion M, Akin EJ, Dib-Hajj FB, Liu S, Sosniak D, Zhao P, et al. (2022). Depolarizing NaV and Hyperpolarizing KV Channels Are Co-Trafficked in Sensory Neurons. J. Neurosci. 42, 4794–4811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Higerd-Rusli GP, Tyagi S, Liu S, Dib-Hajj FB, Waxman SG, and Dib-Hajj SD (2023). The fates of internalized NaV1.7 channels in sensory neurons: Retrograde cotransport with other ion channels, axon-specific recycling, and degradation. J. Biol. Chem. 299, 102816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kishore R, Luedemann C, Bord E, Goukassian D, and Losordo DW (2003). Tumor Necrosis Factor–Mediated E2F1 Suppression in Endothelial Cells. Circ. Res. 93, 932–940. [DOI] [PubMed] [Google Scholar]
- 82.Dib-Hajj SD, Choi JS, Macala LJ, Tyrrell L, Black JA, Cummins TR, and Waxman SG (2009). Transfection of rat or mouse neurons by biolistics or electroporation. Nat. Protoc. 4, 1118–1126. [DOI] [PubMed] [Google Scholar]
- 83.Faber CG, Lauria G, Merkies ISJ, Cheng X, Han C, Ahn H-S, Persson A-K, Hoeijmakers JGJ, Gerrits MM, Pierro T, et al. (2012). Gain-of-function Nav1.8 mutations in painful neuropathy. Proc. Natl. Acad. Sci. USA 109, 19444–19449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ghovanloo M-R, Tyagi S, Zhao P, Kiziltug E, Estacion M, Dib-Hajj SD, and Waxman SG (2023). High-throughput combined voltage-clamp/current-clamp analysis of freshly isolated neurons. Cell Rep. Methods 3, 100385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mangeol P, Prevo B, and Peterman EJG (2016). KymographClear and KymographDirect: two tools for the automated quantitative analysis of molecular and cellular dynamics using kymographs. Mol. Biol. Cell 27, 1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jakobs MAH, Dimitracopoulos A, and Franze K (2019). KymoButler, a deep learning software for automated kymograph analysis. Elife 8, e42288. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request
This paper does not report original code
Any additional information required to reanalyze the data reported in this work paper is available from the lead contact upon request.