

Abstract
In the ligand prediction category of CASP15 the challenge was to predict the positions and conformations of small molecules binding to proteins that were provided as amino acid sequences or as models generated by the AlphaFold2 program. For most targets we used our template based ligand docking program ClusPro ligTBM, also implemented as a public server available at https://ligtbm.cluspro.org/. Since many targets had multiple chains and a number of ligands, several templates and some manual interventions were required. In a few cases no templates were found, and we had to use direct docking using the Glide program. Nevertheless, ligTBM was shown to be a very useful tool, and by any ranking criteria our group was ranked among the top five best performing teams. In fact, all the best groups used template based docking methods. Thus, it appears that the AlphaFold2 generated models, in spite of the high accuracy of the predicted backbone, have local differences from the X-ray structure that make the use of direct docking methods more challenging. The results of CASP15 confirms that this limitation can be frequently overcome by homology based docking.
Introduction
It is now generally recognized that the addition of machine learning methods, particularly the development and public release of the neural net program AlphaFold2 (AF2),1 opened a new chapter in protein structure prediction by generating models with near-experimental accuracy for a substantial fraction of the proteome. An important, possibly even the most important, related question is whether the AF2 generated models are accurate enough for docking small ligands, an essential step in the computational approach to drug discovery.2–6 Ligand docking methods have been previously tested in public experiments. A well-known example is D3R (Drug Design Data Resource) Grand Challenge (https://drugdesigndata.org/), a blinded prediction contest for the computational chemistry community.7–10 D3R included predicting the poses of ligands binding to proteins provided as X-ray structures. In contrast, in the ligand prediction category of CASP15 the target proteins were provided as amino acid sequences. Participants could also use AF2 models provided by the CASP organizers for some of the targets. Thus, the results of the CASP15 experiment provide important contributions toward answering the question whether models generated by AF2 or other tools are good enough for ligand docking and drug discovery.
Our ClusPro team (group number 350) participating in CASP15 submitted 77 models for 17 ligand docking targets. As detailed in Methods, our protocol involved three basic steps. Step 1, the prediction of the target protein structure, was required if an AF2 model was not provided by the CASP organizers. For protein structure prediction in most cases we used the standard AF211 or AlphaFold Multimer (AFM)12 protocol, the latter for generating structures for multi-chain targets. In cases with limited MSA we have also applied a combination of AF2, AFM, and our ClusPro protein-protein docking server to better explore the conformational space.13 In Step 2 we generated large conformational ensembles of the ligands to be docked if they had multiple feasible conformations. For most ligands we used the standard tool ETKDG from RDKIT14 to generate 1000 models. For macrocyclic ligands we used BRIKARD, an analytical approach inspired from robotics to sample the conformational space of constrained molecular systems.15,16 Step 3 used ClusPro LigTBM, a template-based ligand docking program developed in our lab.17 The method searches for known complexes with ligands that have partial coverage of the target ligand, performs conformational sampling and template-guided energy refinement to produce a variety of possible poses, and then scores the refined poses. LigTBM performed extremely well in the last round of the D3R (Drug Design Data Resource) Grand Challenge.10,18 Grand Challenge 4 (GC4) included predicting the poses of 20 ligands binding to beta secretase 1 (BACE 1). We also implemented the algorithm as the automated server, available to the research community as part of the ClusPro docking tools. LigTBM did not provide results if no appropriate template was found. In such cases we identified the ligand binding site using our FTMap protein mapping algorithm,19 also implemented as a server, and then used the well know Glide program for docking.20
On the CASP15 website ClusPro is shown as the 5th best performing group based on the prediction accuracy of the 1st pose. However, differences among the top five groups were fairly small. In fact, the evaluators also used some other success criteria. Considering all predictions within 10% of the best prediction ClusPro was ranked the third, and in terms of the Z-score we were placed as the best performer. We note that the top groups generally provided good predictions for the same targets, and there were a few targets that were not well predicted by any of the groups. Here we will focus on the targets on which we did well, and on some targets that turned out to be very difficult for us. As a general comment we emphasize that all successful methods used template-based docking methods rather than traditional direct docking tools. This issue will be further discussed after presenting our methods and results.
Methods
Using templates to generate “refined” protein models using AlphaFold2 and AlphaFold Multimer
An ensemble of protein models were generated using AlphaFold Multimer,12 template-based modeling,21,22 and a combination of Alphafold2 and the protein docking web server Cluspro.13 For AlphaFold Multimer, 25 models were generated using an unmodified version of the algorithm, with the maximum template release date set to May 14th, 2022. Additionally, 5 models of the target were generated using AlphaFold Multimer with template searching disabled, and MSAs generated from the MMSeqs2 API.23 Template-based protein models were constructed using the ClusPro-TBM protocol.21 Finally, docked structures were generated by running Alphafold2 with the pTM parameter set to obtain a model of each chain in a protein–protein complex. Then the single chain predictions were docked using ClusPro.24 For homomeric structures the multimer docking mode of ClusPro was used to generate additional docked complexes.24 All structures were collected in an ensemble, and each structure of the ensemble was provided as a template to AlphaFold Multimer to generate final “refined” complexes. AlphaFold2 and AlphaFold Multimer produce a predicted local distance difference test (pLDDT) score for each residue. Values greater than 90 indicate high confidence, whereas values below 50 indicate low confidence in the accuracy of the residue coordinates.
Generating macrocycle conformations using BRIKARD
The structure of the ligand (2:3 Fe-bisucaberin) involved in complex with target Q9I116 (CASP ID T1118) was sampled using the program BRIKARD.16 BRIKARD uses an exact analytical approach inspired from robotics to exhaustively sample the conformational space of constrained molecular systems such as complex fused ring macrocycles and protein loops. BRIKARD accepts as input a MOL2 model of the molecule to be sampled, either in open form with specified closure geometry or as produced by some modeling program such as RDKIT,14 using canonical values for bond lengths and bond angles. The full flexibility of the torsional degrees of freedom including peptide bonds, is explored while bond lengths and angles are maintained at their canonical values. The resulting diverse ensemble is energy minimized to relieve strain, and the lowest energy structures are clustered and ranked by energy.
Template based ligand docking using ligTBM
ClusPro ligTBM is a template-based docking program introduced at D3R (Drug Design Data Resource) Grand Challenge.17 The input data are the receptor structure as a PDB file and the ligand chemical structure provided as a SMILES string. A similarity search for the complex is performed in the Protein Data Bank (PDB) database to find templates of highly-homologous protein chains with similar ligands. We first use BLAST to search for the sequence-similar (e-value = 10−20, sequence identity ≥ 30%) proteins. For each ligand in the found structures we calculate the Maximum Common Substructure (MCS) and the Tanimoto score based on Daylight molecular fingerprint as implemented in RDKit.14 The ligands with Tanimoto score ≥ 0.4 and MCS coverage ≥ 50% located within 8 Å of the selected chain are retained, thus forming protein-ligand template structures. An ensemble of initial conformations is generated for the ligand. For each of the templates found, the next steps are carried out independently. In the general case we used the ETKDG method from RDKit to generate 1,000 conformers for the target ligand.14 For macrocyclic ligands we used the program BRIKARD as described. For each template, we aligned all conformers to the template’s MCS and retained only one conformer with the lowest Root Mean Square Deviation (RMSD) of the MCS. The resulting protein-ligand structures were subjected to restrained all-atom energy minimization to remove possible clashes and “relax” the ligand. The poses were ranked based on their coverage and similarity to the template.17
Protein preparation and direct docking using Glide
The structures of the receptors were prepared using the Schrödinger protein preparation wizard,25 which provides minor structure optimization, ensuring the best performance of the following Glide runs.20 We used the standard setting of the wizard, added and optimized the positions of the hydrogen atoms and altered residue ionization/tautomer states using PROPKA26 at pH 7, removed the water molecules further than 3 Å away from the HET atoms, and performed restrained structure minimization with the OPLS4 force field27 converging the heavy atoms to RMSD of 0.3 Å. The prepared structures were used to generate the Glide receptor grids. The center of each grid box was placed in the location identified by FTMap19 clusters in the complex with the inner and outer box sizes equal to 20 Å and 40 Å, respectively. We used an extra-precision XP Glide protocol for the docking and scoring. The best Glide models were selected based on the Glide XP and Emodel Glide scores.20 The models were energy minimized using backbone-constrained minimization in OpenMM with Amber parametrization (ff19SB, GAFF2, AM1-BCC, Open Babel protonation at pH = 7.4, and OBC2+ACE GBSA).28 Ligand heavy atoms were fixed while hydrogen atoms were free to move.
Results
In our predictions, we utilized a combination of template-based modeling and free docking, with template based-approach playing the dominant role due to the nature of target ligands. Here, we talk some of the successes of TBM (H1114, T1124, T1146, T1152, T1170, T1186, T1158v4) and direct docking (T1158v1-v2), and discuss some of the more challenging targets (T1127, T1158v3). Table 1 is a summary of the results for all targets discussed in the paper.
Table 1.
Summary of ClusPro results in CASP15.
| Target ID | Target name | Uniprot ID | Ligand ID | Ligand code | RMSD, Å |
|---|---|---|---|---|---|
|
| |||||
| T1124 | MfnG | A0A0D4WTP2 | 1 | SAH | 0.764 |
| 2 | SAH | 0.92 | |||
| 3 | TYR | 4.781 | |||
| 4 | TYR | 5.505 | |||
|
| |||||
| T1146 | Putative ribosephosphate isomerase | Q6MQ80 | 1 | NAG | 0.56 |
|
| |||||
| T1152 | protein-carbohydrate complex | --- | 1 | NAG | 1.622 |
|
| |||||
| T1170 | RuvB | Q5M2B1 | 1 | ADP | 6.479 |
| 2 | ADP | 6.469 | |||
| 3 | ADP* | --- | |||
| 4 | AGS | 0.919 | |||
| 5 | AGS | 0.943 | |||
| 6 | AGS | 1.039 | |||
| 7 | Mg | 0.926 | |||
| 8 | Mg | 0.999 | |||
| 9 | Mg | 0.734 | |||
| 10 | AGS | 1.191 | |||
|
| |||||
| T1186 | beta lactamase | A0ESG7 | 1 | LIG | 1.121 |
|
| |||||
| T1158v1 | MRP4(E1202Q) | --- | 1 | XPG | 2.368 |
|
| |||||
| T1158v2 | MRP4(E1202Q) | --- | 1 | P2E | 2.981 |
|
| |||||
| T1158v3 | MRP4(E1202Q) | --- | 1 | XH0 | 4.226 |
|
| |||||
| T1158v4 | MRP4(E1202Q) | --- | 1 | ATP | 0.918 |
| 2 | ATP | 1.561 | |||
| 3 | 2MG | 0.443 | |||
| 4 | 2MG | 0.374 | |||
|
| |||||
| T1127v2 | L-ornithine N5-acetyltransferase NATA1 | Q9ZV05 | 1 | COA | 3.47 |
| 2 | COA | 3.402 | |||
| 3 | EPE† | --- | |||
| 4 | EPE† | --- | |||
| 5 | MPD† | --- | |||
Experimental results have shown no presence of this ligand; ligand 10 was determined at the site instead.
Ligands were defined as not relevant.
T1124
Target T1124, was represented by a homodimeric structure of the MfnG protein from Streptomyces drozdowiczii. The goal for this target was to produce a model of the homodimeric receptor in complex with two ligands, S-adenosyl-L-homocysteine (SAH) and tyrosine (TYR). In this case, we relied on AlphaFold Multimer (v2) to generate the initial model of the receptor (the highest plDDT model was selected) and manually resolved the clash between residues TRP254 and GLN274 in the final model of the protein dimer.
Since we were required to predict the positions of 2 SAH ligands and 2 TYR ligands in a homodimeric structure, we assumed that one copy of each ligand is present in each subunit, and used LigTBM to identify template protein-ligand complexes for SAH (PDB ID: 6M83) and TYR (PDB ID: 4KIG). The template structure used for modeling SAH contained an actual SAH molecule, which made pose generation straightforward. For TYR, 3-(4-Hydroxy-phenyl)pyruvic acid (ENO) was identified as a template, and TYR was superimposed on ENO using the MCS match between the two ligands using the LigTBM protocol. The high similarity of the templates allowed for high accuracy of the SAH modeled poses, with our top-1 submissions featuring 0.764/0.920 Å RMSD for the two SAH molecules. Our selection of the template for TYR, however, proved to be suboptimal, and while the placement of the TYR ring was broadly correct, the overall pose of the ligand was inadequate, featuring 5.715/5.678 Å RMSD.
T1146
Target T1146 required modeling an isomerase from Bdellovibrio bacteriovorus in complex with 2-acetamido-2-deoxy-beta-D-glucopyranose (NAG). While the original target description presumed 2 copies of NAG molecule interacting with the receptor, only a single NAG was observed in the final X-ray structure. Here we used the organizer-provided AlphaFold2 models as receptor structures and relied on the LigTBM modeling approach to identify the templates for ligand placement. For the submission we used the 4Q5K and 4Q68 templates for final pose prediction, however, the latter turned out to be irrelevant due to the updated experimental results. The remaining high-quality template contained an actual NAG molecule, which straightforwardly resulted into a high-quality final prediction with 0.560 Å RMSD.
T1152
Target T1152 represented another protein–carbohydrate complex and involved the interaction between the Clostridium thermocellum Spore coat assembly protein SafA with a beta-1,4-GlcNAc trisaccharide. The interesting aspect of this target was that a single trisaccharide molecule interacted with two protein receptors simultaneously. From the modeling perspective, this target represented another straightforward TBM-centric case. ClusPro LigTBM identified PDB ids 4B9H and 4B8V as suitable templates, the latter having higher resolution and containing a fusion protein containing 3 copies of the same domain interacting with quad-saccharide molecule. Using this structure as a template for ligand placement and relying on AF2 for a receptor model gave us an 1.622 Å RMSD prediction, while our best in the top-5 prediction featured an RMSD of 1.047 Å (Figure 1A).
Figure 1.
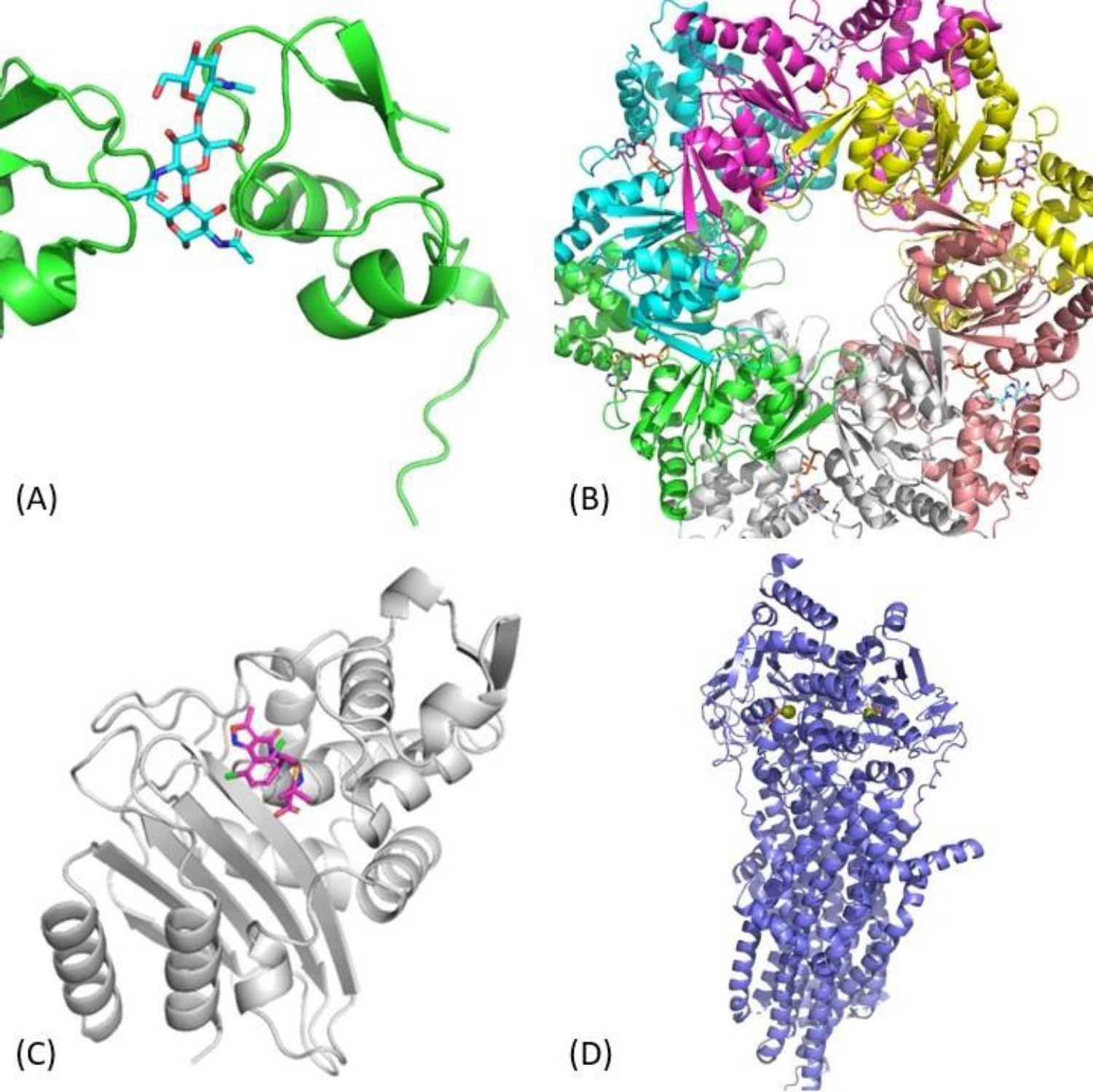
Results for LigTBM cases. (A) T1152: protein-carbohydrate complex (PDB ID 7R1L); (B) T1170: RuvB complex derived from Streptococcus thermophilus with bound ATP and AGS ligands and MG2+ ions (PDB ID 7PBR); (C) T1186: beta-lactamase with dicloxacillin; (D) T1158v4: Multidrug Resistance Protein 4 with ATP and MG2+ ions.
T1170
Target T1170 was based on an electron microscopy structure of a hexameric RuvB complex derived from Streptococcus thermophilus (Figure 1B). The complex was resolved with 3 adenosine diphosphate (ADP) molecules, 3 adenosine triphosphate-gamma-S (AGS) molecules, and 3 magnesium ions (MG). As such, the goal was to predict the structure of the multimeric complex, and then place all 9 ligands accurately within it. Our model of the receptor complex was based on AlphaFold2 prediction obtained with a manually-prepared template, which, in its turn, was based on the structure with PDB ID 3PFI, selected as the result of the template search performed using ClusProTBM. The ligand poses, were based on the templates identified using ClusPro LigTBM (PDB ID 6BLB for ADP and PDB ID 5DAC for AGS and MG). While the placement of ADP could be done straightforwardly using the 6BLB template, the pose of AGS/MG could not be carried over from 5DAC due to clashes of Adenosine moiety of AGS with our RuvB model. To resolve this issue, we therefore prepared a chimeric template for AGS using a combination of 5DAC and 6BLB by taking the thiophosphate moiety and MG coordinates from 5DAC and adenosine moiety from 6BLB. Finally EKTDG method was used to generate the conformation of a complete AGS molecule, which was placed using the chimeric template described above. The resulting top-1 model featured RMSDs in the range of 0.972–1.006 Å (all copies) for AGS, RMSDs in the range of 0.864–0.873 Å (all copies) for MG, and RMSD in the range of 6.469–6.479 for ADP.
T1186
Target T1186 was a beta-lactamase in complex with the covalent acyl-intermediate during cleavage of dicloxacillin, and represented another straightforward template-based modeling case. The top template from ligTBM search (PDB ID: 7K2Y) contained a ligand providing structural support for approximately a half of the target small molecule, and combining it with an AlphaFold2 model of the receptor resulted into a 1.121 Å RMSD top-1 model, and 0.814 Å best in top-5 model.
T1158v1-v4
The T1158v1-v4 series of targets was based on co-crystals of Multidrug Resistance Protein 4 with a set of ligands (XPG, P2E, and MRP, respectively, for v1, v2, and v3, and 2xATP and 2xMG for v4). It is known that a homologous protein (Multidrug Resistance Protein 1) has two spatially separated binding sites, the substrate binding site and the ATP binding site, and undergoes conformational change from an open to a closed state upon ATP binding.29 With these considerations in mind, we assumed that non-ATP target ligands of T1158 v1, v2, and v3 bind to the substrate binding site of the open conformation, and the target ligands of T1158 v4, which include ATP, bind to the ATP binding site of the closed conformer. We, therefore, took two different approaches to modeling targets T1158v1-v3 and T1158v4. Since our template identification protocol provided no hits for T1158v1-v3, we turned to direct docking with Glide to predict the binding poses of the target ligands within the substrate binding site. We selected the first and fifth AF2-predicted structures provided by the organizers as receptor models, and used a bounding box limited to the extent of the substrate binding site for Glide docking runs. The final predictions were hand-picked from top Glide results and resulted into 0.368 Å, 2.981 Å, and 4.225 Å RMSDs for T1158v1, T1158v2 and T1158v3, respectively (Figure 2).
Figure 2.

Results for Glide docking of ligands to Multidrug Resistance Protein 4 (MRP4) (A) T1158v1: MRP4 with XPG; (B) T1158v2: MRP4 with P2E; (C) T1158v3: MRP4 with MRP
In modeling of ligand poses for T1158 v4 we followed a more conservative template-based approach as suitable templates were available. LigTBM identified a number of experimental structures containing exact target ligands and receptor templates with above 30% sequence identity. We retained the hits with resolutions higher than 3.5 Å, ending up with PDB IDs 6BHU, 7SVD, and 5W81 as potential templates for our ligand pose prediction. Additionally, since all of these templates take a closed conformation, we generated a closed-like conformation of the receptor by aligning the components of the receptor model involved in the hinge motion determining the open/closed conformation to the closed state templates. This, however, did not affect the ATP binding site. Our top-1 model featured 0.443/0.374 Å RMSDs for the two MG ions and 0.918/1.561 Å RMSDS for the ATP molecules.
T1127v2
An interesting example of the rather unexpected challenges for the template-based approach was Target T1127, a homodimeric structure for Arabidopsis thaliana L-ornithine N5-acetyltransferase (Uniprot AC/ID: Q9ZV05/NATA1_ARATH) in complex with two occurrences of Coenzyme A (COA). While LigTBM identified two promising templates for the protein–COA complex (PDB IDs 2B58 and 2B4B) involving an actual COA molecule and 28% receptor sequence identity (51% sequence similarity), our submissions only featured rather humble 3.4 Å RMSDs and differed significantly in the placement of the Sulfur-containing “tail” (Figure 3). Potential reasons for this underperformance could include subtle differences in the residue composition of the binding site, as well as the presence of different additional small molecules in the template target structures.
Figure 3.
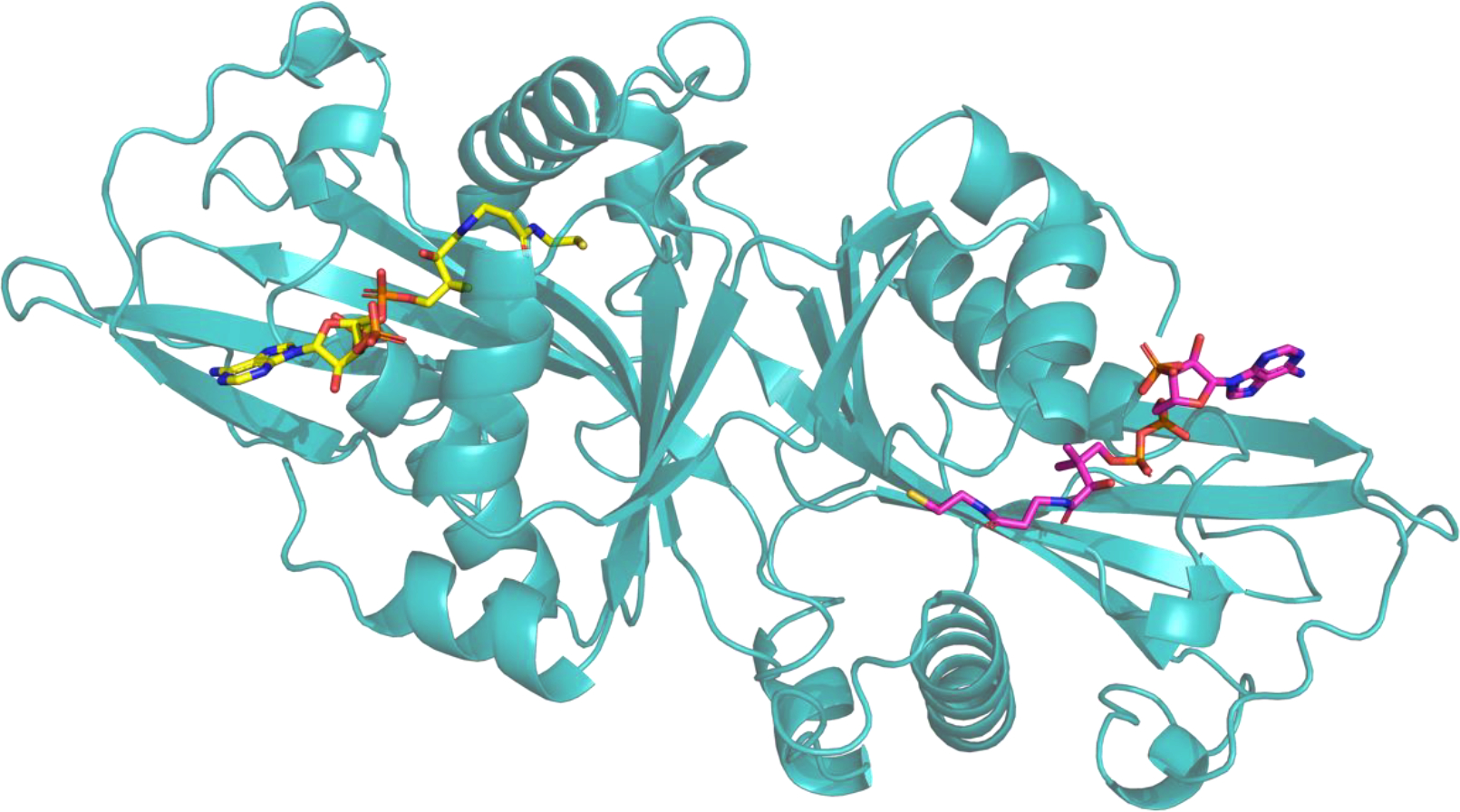
Result for target T1127v2: Homodimeric structure for Arabidopsis thaliana L-ornithine N5-acetyltransferase in complex with two Coenzyme A molecules.
Conclusions
In CASP15 we used the template based ligand docking program ClusPro ligTBM developed earlier in our lab.17 LigTBM was also implemented as a public server available at https://ligtbm.cluspro.org/. However, many of the targets had multiple chains and several ligands and other complexities such as bound ions that required manual interventions. Nevertheless, with some exceptions the essential protocol used by our group was the one described for the ligTBM program.17 Although a number of criteria were used for ranking the performance of the different groups, by any of these criteria our group was ranked among the top five participating teams. Thus, we conclude that using ligTBM was the right choice. In fact, all the best performing groups used template based docking method. Considering these results, it appears that the AlphaFold generated models, in spite of the high accuracy of the predicted backbone, have local differences from the X-ray structure that make the use of direct docking methods challenging. In agreement with this observation, some publications conclude that the results of direct docking to AF2 models are substantially poorer than docking to holo X-ray structures, but comparable or even slightly better than docking to apo structures.6 However, the results of CASP15 confirms that this limitation can be frequently overcome by homology based docking. Once a few ligands are placed using such methods, co-minimization of the protein and ligand can results in models more suitable for virtual screening with established docking tools.4
Acknowledgements
This investigation was supported by grants DMS 2054251 and AF 1645512 from the National Science Foundation, and R01GM140098, R35GM118078, RM1135136 and R01GM102864 from the National Institute of General Medical Sciences.
References
- 1.Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wong F, Krishnan A, Zheng EJ, et al. Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery. Mol Syst Biol. 2022;18(9):e11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scardino V, Di Filippo JI, Cavasotto CN. How good are AlphaFold models for docking-based virtual screening? iScience. 2023;26(1):105920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Vass M, Shi D, et al. Benchmarking Refined and Unrefined AlphaFold2 Structures for Hit Discovery. J Chem Inf Model. 2023;63(6):1656–1667. [DOI] [PubMed] [Google Scholar]
- 5.Kersten C, Clower S, Barthels F. Hic sunt dracones: molecular docking in uncharted territories with structures from AlphaFold2 and RoseTTAfold. J Chem Inf Model. 2023;63(7):2218–2225. [DOI] [PubMed] [Google Scholar]
- 6.Holcomb M, Chang YT, Goodsell DS, Forli S. Evaluation of AlphaFold2 structures as docking targets. Protein Sci. 2023;32(1):e4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gathiaka S, Liu S, Chiu M, et al. D3R grand challenge 2015: Evaluation of protein-ligand pose and affinity predictions. J Comput Aided Mol Des. 2016;30(9):651–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gaieb Z, Liu S, Gathiaka S, et al. D3R Grand Challenge 2: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des. 2018;32(1):1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaieb Z, Parks CD, Chiu M, et al. D3R Grand Challenge 3: blind prediction of protein-ligand poses and affinity rankings. J Comput Aided Mol Des. 2019;33(1):1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parks CD, Gaieb Z, Chiu M, et al. D3R grand challenge 4: blind prediction of protein-ligand poses, affinity rankings, and relative binding free energies. J Comput Aided Mol Des. 2020;34(2):99–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jumper J, Hassabis D. Protein structure predictions to atomic accuracy with AlphaFold. Nat Methods. 2022;19(1):11–12. [DOI] [PubMed] [Google Scholar]
- 12.Evans R, O’Neill M, Pritzel A, et al. Protein complex prediction with AlphaFold-Multimer. BioRxiv. 2021:2021.2010. 2004.463034. [Google Scholar]
- 13.Ghani U, Desta I, Jindal A, et al. Improved docking of protein models by a combination of alphafold2 and cluspro. BioRxiv. 2021:2021.2009. 2007.459290. [Google Scholar]
- 14.Landrum G RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling. http://rdkitsourceforgenet. [Google Scholar]
- 15.Coutsias EA, Seok C, Jacobson MP, Dill KA. A kinematic view of loop closure. J Comput Chem. 2004;25(4):510–528. [DOI] [PubMed] [Google Scholar]
- 16.Coutsias EA, Lexa KW, Wester MJ, Pollock SN, Jacobson MP. Exhaustive conformational sampling of complex fused ring macrocycles using inverse kinematics. J Chem Theory Comput. 2016;12(9):4674–4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alekseenko A, Kotelnikov S, Ignatov M, et al. ClusPro LigTBM: Automated Template-based small molecule docking. J Mol Biol. 2020;432(11):3404–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kotelnikov S, Alekseenko A, Liu C, et al. Sampling and refinement protocols for template-based macrocycle docking: 2018 D3R Grand Challenge 4. J Comput Aided Mol Des. 2020;34(2):179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kozakov D, Grove LE, Hall DR, et al. The FTMap family of web servers for determining and characterizing ligand-binding hot spots of proteins. Nat Protoc. 2015;10(5):733–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friesner RA, Banks JL, Murphy RB, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. [DOI] [PubMed] [Google Scholar]
- 21.Porter KA, Padhorny D, Desta I, et al. Template-based modeling by ClusPro in CASP13 and the potential for using co-evolutionary information in docking. Proteins. 2019;87(12):1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Porter KA, Desta I, Kozakov D, Vajda S. What method to use for protein-protein docking? Curr Opin Struct Biol. 2019;55:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mirdita M, Steinegger M, Soding J. MMseqs2 desktop and local web server app for fast, interactive sequence searches. Bioinformatics. 2019;35(16):2856–2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kozakov D, Hall DR, Xia B, et al. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. [DOI] [PubMed] [Google Scholar]
- 26.Rostkowski M, Olsson MH, Søndergaard CR, Jensen JH. Graphical analysis of pH-dependent properties of proteins predicted using PROPKA. BMC Struct Biol. 2011;11:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lu C, Wu C, Ghoreishi D, et al. OPLS4: Improving force field accuracy on challenging regimes of chemical space. J Chem Theory Comput. 2021;17(7):4291–4300. [DOI] [PubMed] [Google Scholar]
- 28.Eastman P, Pande VS. OpenMM: A Hardware Independent Framework for Molecular Simulations. Comput Sci Eng. 2015;12(4):34–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Johnson ZL, Chen J. ATP binding enables substrate release from multidrug resistance protein 1. Cell. 2018;172(1–2):81–89 e10. [DOI] [PubMed] [Google Scholar]