

Abstract
Boryltin compounds featuring the metal in the+1 or 0 oxidation states can be synthesized from the carbene‐stabilized tin(II) bromide (boryl)Sn(NHC)Br (boryl={B(NDippCH)2}; NHC=C{(N i PrCMe)2}) by the use of strong reducing agents. The formation of the mono‐carbene stabilized distannyne and donor‐free distannide systems (boryl)SnSn(IPrMe)(boryl) (2) and K2[Sn2(boryl)2] (3), using Mg(I) and K reducing agents mirrors related germanium chemistry. In contrast to their lighter congeners, however, systems of the type [Sn(boryl)] n are unstable with respect to disproportionation. Carbene abstraction from 2 using BPh3, and two‐electron oxidation of 3 both result in the formation of a 2 : 1 mixture of the Sn(II) compound Sn(boryl)2, and the hexatin cluster, Sn6(boryl)4 (4). A viable mechanism for this rearrangement is shown by quantum chemical studies to involve a vinylidene intermediate (analogous to the isolable germanium compound, (boryl)2Ge=Ge), which undergoes facile atom transfer to generate Sn(boryl)2 and trinuclear [Sn3(boryl)2]. The latter then dimerizes to give the observed hexametallic product 4, with independent studies showing that similar trigermanium species aggregate in analogous fashion.
Keywords: boryl ligand, cluster, germanium, sub-valent compounds, tin
The carbene‐stabilized distannyne and donor‐free distannide systems (boryl)SnSn(IPrMe)(boryl) and K2[Sn2(boryl)2], can be synthesized from (boryl)Sn(NHC)Br using Mg(I) or K reducing agents. In contrast to their germanium congeners, systems of the type [Sn(boryl)] n are unstable with respect to disproportionation: NHC abstraction from (boryl)SnSn(IPrMe)(boryl) and 2‐electron oxidation of K2[Sn2(boryl)2] both yield a 2 : 1 mixture of Sn(boryl)2 and the hexatin cluster, Sn6(boryl)4. A viable mechanism for this rearrangement is shown by quantum chemical studies to involve the vinylidene‐type intermediate (boryl)2Sn=Sn.

Introduction
The organometallic chemistry of the group 14 elements in their lower oxidation states (≤+2) has been revolutionized by the development of a range of sterically encumbered ancillary ligands, which allows access to discrete molecular species that retain coordinative and electronic unsaturation. Among these, the heavier group 14 analogues of alkynes (dimetallynes, REER, E = Si − Pb) have become a topical area within the field of molecular main group chemistry.[ 1 , 2 , 3 ] In part, this is because these compounds (and related systems) posed a challenge to long‐held ideas about the feasibility of multiple bonding between heavier main group elements. [4] In addition, due to their non‐linear (trans‐bent) structures and the resulting morphologies/energies of their frontier orbitals, dimetallynes can interact with a number of small molecule substrates in a manner comparable to transition metal complexes.[ 3 , 5 ] This chemistry includes seminal examples of the activation of dihydrogen, [6] and of organic substrates such as alkynes [7] and alkenes[ 6g , 8 ] (in some cases reversibly). [9]
Among the families of bulky monodentate ligands that have been employed to kinetically stabilize dimetallynes with respect to aggregation are strong σ‐donors such as aryl, silyl or amido substituents (e. g., I and II; Figure 1).[ 1 , 2 , 6c , 6d , 6e , 6g , 7b , 10 , 11 ] As an alternative, we have recently been interested in the use of boryl ligands, −{B(NDippCH)2} to access main group systems featuring unusual electronic or geometric structure, and unprecedented modes of reactivity. [12] With respect to compounds of the stoichiometry (RE) n (E=group 14 element), it is noteworthy that the use of boryl substituents leads to the isolation of a compound featuring the alternative vinylidene structure, (boryl)2GeGe, in contrast to the digermyne isomers XGeGeX observed for X=aryl, amido etc. (III; Figure 1).[ 6 , 12h ] With this in mind, and given the varied structural and reaction chemistry reported for distannynes, we were interested in examining the consequences of the use of boryl ancillary ligands in the related chemistry of tin.
Figure 1.

Systems of the stoichiometry (RE) n (E=group 14 element) relevant to the current study.
Results and Discussion
The N‐heterocyclic carbene stabilized (boryl)tin(II) bromide, {(HCDippN)2B}Sn(IPrMe)Br, 1 (Dipp=2,6‐ i Pr2C6H3; IPrMe = C{(N i PrCMe)2}), is readily synthesized from (IPrMe)SnBr2 and {(HCDippN)2B}Li(thf)2 [13] in diethyl ether solution and can be isolated in ca. 60 % yield after recrystallization from toluene. 1 has been characterized by standard spectroscopic and analytical methods and its structure in the solid state determined by X‐ray crystallography. 1 is thus confirmed to be monomeric, featuring a three‐coordinate tin centre; the angles defined at the metal centre are close to 90°, as expected for a heavier p‐block element, with the slightly wider B−Sn‐Br and B−Sn−C angles (98.1(1) and 97.4(2)° vs. 94.6(1) for ∠C−Sn‐Br) presumably reflecting the high steric demands of the boryl ligand (Figure 2).
Figure 2.

Molecular structures of {(HCDippN)2B}Sn(IPrMe)Br (1, upper), {(HCDippN)2B}SnSn(IPrMe){B(NDippCH)2} (2, centre) and K2[Sn2{B(NDipp CH)2}2] (3, lower) in the solid state as determined by X‐ray crystallography. Hydrogen atoms and solvate molecules omitted, and selected groups shown in wireframe format for clarity; thermal ellipsoids set at the 40 % probability level. Key bond lengths (Å) and angles (°): (for 1) Sn−B 2.295(5), Sn−C 2.307(5), 2.6325(8), B−Sn‐Br 98.1(1), B−Sn‐C 97.4(2), C−Sn‐Br 94.6(1); (for 2) Sn−Sn 2.581(1); Sn−B 2.243(7), 2.189(5); Sn−C 2.183(5); Sn‐Sn−B 109.1(2), 120.4(1); (for 3) Sn−Sn 2.749(1); Sn−B 2.288(3); Sn K 3.599(1), 3.575(1); Sn‐Sn−B 96.5(1).
1 serves as a convenient precursor for tin‐centred reduction chemistry. As such, reaction with Jones’ Mg(I) reagent, [{HC(MeCMesN)2}Mg]2, [14] yields the unsymmetrical mono NHC‐stabilized distannyne {(HCDippN)2B}SnSn(IPrMe){B(NDippCH)2} 2, while more forcing conditions (excess KC8 or potassium naphthalenide) yield the more reduced (formally Sn(0)) compound K2[Sn2{B(NDippCH)2}2], 3 (Scheme 1). This reactivity finds precedent in the chemistry of the related borylgermanium compounds {(HCDippN)2B}GeGe(IPrMe){B(NDippCH)2} and K2[Ge2{B(NDippCH)2}2], which can be generated from {(HCDippN)2B}Ge(IPrMe)Cl under similar reduction conditions. [12h] Solution‐phase multinuclear NMR data for 2 and 3 are consistent with a higher degree of molecular symmetry for the latter, viz one Dipp CH signal in the 1H NMR spectrum, and a single 11B NMR resonance at δB=55.2 ppm (see two CH signals and two 11B resonances at δB=43.5, 54.2 ppm for 2). Integration of the 1H NMR signals associated with the IPrMe ligand in 2 is consistent with the presence of one carbene fragment per dimeric Sn2(boryl)2 unit. Studies of both compounds by X‐ray crystallography confirm the structures implied spectroscopically, and yield Sn−Sn distances of 2.581(1) and 2.749(1) Å, for 2 and 3, respectively (Figure 2). The former distance is somewhat shorter that reported for the formal Sn=Sn double bond in ArDippSnSnArDipp (2.6675(4) Å), while the latter is in line with the slightly longer distances reported for doubly reduced terphenyl‐substituted distannynes (e. g., K2[Sn2ArDipp 2]: 2.7754(3) Å). [10a] In common with these systems, 3 also adopts a trans configuration about the Sn2 unit (∠Sn‐Sn−B=96.5(1)°), with the potassium counter‐ions being π‐bound between the flanking Dipp groups.
Scheme 1.

Reduction of {(HCDippN)2B}Sn(IPrMe)Br (1) by potassium or magnesium reducing agents to give {(HCDippN)2B}SnSn(IPrMe){B(NDippCH)2} (2) or K2[Sn2{B(NDippCH)2}2] (3); oxidation of 3 or carbene abstraction from 2 to yield a mixture of the tin cluster Sn6{B(NDippCH)2}4 (4) and diborylstannylene Sn{B(NDippCH)2}2 via a formal disproportionation process. [B]={B(NDippCH)2}.
In contrast to the corresponding germanium system, which is inert to carbene abstraction under mild conditions, [12h] removal of the remaining NHC ligand from 2 by the use of BPh3 proves to be synthetically viable (presumably due to the weaker nature of the Sn−C bond). Moreover, the products of this reaction are found to be identical to those obtained from 3 by the action of two equivalents of a trityl oxidant (Scheme 1). In each case, the two species formed (in a ratio of 2 : 1) are the known diborylstannylene Sn{B(NDippCH)2}2 [12f] and a new compound Sn6{B(NDippCH)2}4 (4), the structure of which was determined unambiguously by X‐ray crystallography (Figure 3). The molecular structure of 4 features four tin atoms arranged in distorted tetrahedral fashion, with opposite edges of the tetrahedron being elongated and bridged symmetrically by a [Sn{B(NDippCH)2}2] unit.[ 15 , 16 ] The molecule sits on a C 2 axis, with the Sn−Sn bonded distances within the tetrahedron (2.848(1), 2.831(1) Å) and the corresponding contacts involving the [Sn{B(NDippCH)2}2] units (2.841(1), 2.834(1) Å) all falling in the range expected for Sn−Sn single bonds. The Sn−B distances (2.259(6), 2.269(6) Å) are similar to those measured for Sn{B(NDippCH)2}2 itself. [12f]
Figure 3.
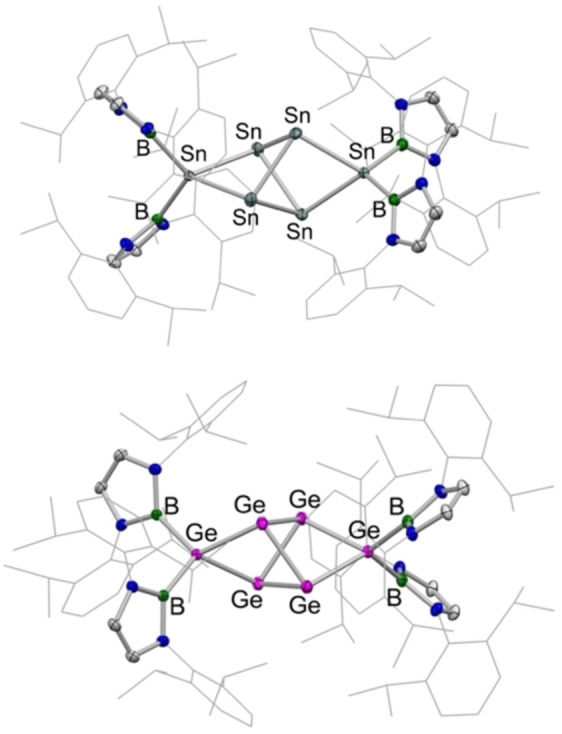
Molecular structures of Sn6{B(NDippCH)2}4 (4, upper) and Ge6{B(NDippCH)2}4 (6, lower) in the solid state as determined by X‐ray crystallography. Hydrogen atoms and solvate molecules omitted and selected groups shown in wireframe format for clarity; thermal ellipsoids set at the 40 % probability level. Key bond lengths (Å) and angles (°): (for 4) Sn−B 2.259(6), 2.269(6); Sn(B)‐Sn 2.841(1), 2.834(1); Sn−Sn 2.848(1), 2.831(1); (for 6) Ge−B 2.095(3), 2.105(3); Ge(B)‐Ge 2.500(1), 2.506(1); Ge−Ge 2.490(1), 2.468(1).
The formation of 4 in this fashion by the two‐electron oxidation of K2[Sn2{B(NDippCH)2}2], 3, contrasts with the analogous reaction of K2[Ge2{B(NDippCH)2}2], which yields the digermavinylidene {(HCDippN)2B}2GeGe, via transient formation of the isomeric diboryldigermyne {(HCDippN)2B}GeGe{B(NDippCH)2} (which is calculated to be ca. 2–3 kcal mol−1 higher in energy). [12h] With this in mind, the conversion of 3 to 4 was investigated by quantum chemical methods (Figure 4; see Supporting Information for computational details). A mechanism is proposed involving initial formation of the corresponding diboryldistannyne by oxidation of 3 (or removal of the carbene donor from 2) followed by 1,2‐migration of a boryl group to form a distannavinylidene (Int 1). As with its germanium counterpart, this system lies lower in energy than the distannyne isomer (ΔG=−5.4 kcal mol−1) and is accessed via a low activation barrier (TS1, ΔG≠=6.7 kcal mol−1 compared to 9.5 kcal mol−1 in the germanium case). [12h] Unlike its germanium counterpart, however, Int1 cannot be isolated – an observation we ultimately attribute to the weaker nature of the Sn=Sn interaction. Tin atom transfer between two distannylvinylidene units can then occur to produce one of the experimentally observed products, Sn{B(NDippCH)2}2, and a second intermediate, Int 2, which features a triangular Sn3 unit. The conversion of Int1 to Int2 is endergonic with a moderate barrier (ΔG=7.5 kcal mol−1, ΔG≠=18.3 kcal mol−1) and is likely rate limiting. However, subsequent dimerization of Int 2 to yield the other experimentally observed product (4) is a massively exergonic process and constitutes the thermodynamic driving force of the overall process (ΔG=−75.5 kcal mol−1).
Figure 4.

DFT calculated mechanism for the disproportionation of diboryldistannyne (formed by oxidation of 3 or NHC abstraction from 2) into diborylstannylene Sn{B(NDippCH)2}2 and tin cluster Sn6{B(NDippCH)2}4. [B]={B(NDippCH)2}.
The hypothesis that Sn6{B(NDippCH)2}4 (4) is formed by the dimerization of a tri‐tin system also gains some experimental credence from aspects of the corresponding germanium chemistry. While digermvinylidene {(HCDippN)2B}2GeGe does not undergo spontaneous disproportionation in the manner of its putative tin counterpart (presumably due to the stronger Ge=Ge bond, compared to Sn=Sn), the reaction of K2[Ge2{B(NDippCH)2}2] with (IPrMe)GeCl2 yields an oily green compound, which is proposed to be the trigermanium system Ge3{B(NDippCH)2}2(IprMe) (5; Scheme 2) primarily on the basis of multinuclear NMR measurements and subsequent reactivity (see below). A combination of 1H and 11B NMR measurements suggests that the structure of 5 possesses two (equivalent) boryl groups and a single IPrMe carbene ligand. While 5 proved too labile to be isolated as a pure bulk substance for definitive structural characterization, it is susceptible to carbene abstraction using BPh3 to give a deep green product, which can be obtained as single crystals. Most revealingly (in the context of the tin chemistry outlined above) a combination of multinuclear NMR, micro‐analytical and crystallographic studies shows that this compound is Ge6{B(NDippCH)2}4 (6), i. e. the germanium analogue of Sn6{B(NDippCH)2}4 (4) (Figure 3). [17] Structurally, 4 and 6 are very similar, differing primarily in the E−E and E−B bond lengths, at a level expected based on the differing covalent radii of germanium and tin (Δr=1.39–1.20=0.19 Å). [18] The formation of 6 in this way, suggests more broadly that systems of the stoichiometry E3{B(NDippCH)2}2 (E = Ge, Sn) are indeed prone to dimerization with accompanying boryl ligand migration.
Scheme 2.

Synthesis of Ge6{B(NDippCH)2}4 (6), the germanium analogue of Sn6{B(NDippCH)2}4 (4). [B]={B(NDippCH)2}.
Conclusions
In conclusion we have shown that boryltin compounds featuring the metal in the formal oxidation states +1 and 0 can be accessed from a carbene‐stabilized tin(II) bromide precursor by the use of strong reducing agents. The formation of the mono‐carbene stabilized distannyne and donor‐free distannide systems (boryl)SnSn(IPrMe)(boryl) (2) and K2[Sn2(boryl)2], (3) mirrors related germanium chemistry occurring under similar conditions. In contrast to their lighter congeners, however, systems of the type [Sn(boryl)] n are unstable with respect to disproportionation. Carbene abstraction from 2 using triphenylborane and two‐electron oxidation of 3 both result in the formation of a 2 : 1 mixture of the diborylstannylene, Sn(boryl)2, and the hexatin cluster, Sn6(boryl)4 (4). A viable mechanism for this rearrangement is shown by quantum chemical studies to proceed via a vinylidene intermediate (analogous to the isolable germanium compound, (boryl)2Ge=Ge), which undergoes facile atom transfer in the case of tin to generate Sn(boryl)2 and trinuclear [Sn3(boryl)2]. The latter then dimerizes to give the observed hexametallic product 4. Independent studies also show that similar trigermanium species aggregate in analogous fashion.
Experimental Section
Syntheses of new compounds [19]
{(HCDippN)2B}Sn(IPrMe)Br, 1: To a suspension of (IPrMe)SnBr2 (0.70 g, 1.5 mmol) in Et2O (5 mL) at −35 °C was slowly added a pre‐cooled (−35 °C) solution of {(HCDippN)2B}Li ⋅ 2THF (0.82 g, 1.5 mmol) also in Et2O (5 mL). After stirring at this temperature for 30 min, the reaction mixture was slowly warmed to room temperature and stirred for another 1 h. Volatiles were removed under vacuum and the residue was extracted with toluene (5 mL). The filtrate was concentrated and triturated with pentane. After storing at −30 °C overnight, the pale yellow crystalline solid was isolated and dried under vacuum. Single crystals suitable for X‐ray crystallography were obtained from benzene. Yield: 0.72 g, 61.6 %. Anal. Calc. for C37H56BBrN4Sn: C 57.99 %, H 7.37 %, N 7.31 %; Meas.: C 57.68 %, H 7.48 %, N 7.00 %. 1H NMR (400 MHz, C6D6, 298 K): δH 1.00 (d, J HH=7.0 Hz, 6H, CH(CH3 ) 2 of carbene), 1.07 (d, J HH=7.0 Hz, 6H, CH(CH3 ) 2 of carbene), 1.10 (d, J HH=6.9 Hz, 6H, CH(CH3 ) 2 of Dipp), 1.21 (d, J HH=6.9 Hz, 6H, CH(CH3 ) 2 of Dipp), 1.30 (d, J HH=6.9 Hz, 6H, CH(CH3 ) 2 of Dipp), 1.56 (s, 6H, CCH3 of carbene), 1.59 (d, J HH=6.9 Hz, 6H, CH(CH3 ) 2 of Dipp), 3.42 (sept, J HH=6.9 Hz, 2H, CH(CH3)2 of Dipp), 3.59 (sept, J HH=6.9 Hz, 2H, CH(CH3)2 of Dipp), 5.40 (sept, J HH=7.0 Hz, 2H, CH(CH3)2 of carbene), 6.37 (s, 2H, CH of boryl), 7.12 (t, J HH=4.6 Hz, 2H, p‐ArH of Dipp), 7.24 (d, J HH=4.6 Hz, 4H, m‐ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 298 K): δB 38.8. 13C{1H} (151 MHz, C6D6, 298 K): δC–10.0 (C C H3 of carbene), 20.9 and 22.3, (CH( C H3)2 of carbene), 23.5, 24.3, 26.2 and 26.6 (CH( C H3)2 of Dipp), 28.7 and 28.9 ( C H(CH3)2 of Dipp), 54.4 ( C H(CH3)2 of carbene), 122.7 ( C H of boryl), 123.6 (p‐Ar of Dipp), 123.7 (m‐Ar of Dipp), 126.4 ( C CH3 of carbene), 127.5 (m‐Ar of Dipp), 141.4 (o‐Ar of Dipp), 146.3 and 146.6 (i‐Ar of Dipp), 171.0 (R2 C of carbene). 119Sn{1H} NMR (112 MHz, C6D6, 298 K): δB 78.
{(HCDippN)2B}SnSn(IPrMe){B(NDippCH)2}, 2: A mixture of 1 (0.20 g, 0.26 mmol), IPrMe (0.047 g, 0.26 mmol) and [{HC(MeCMesN)2}Mg]2 (0.093 g, 0.13 mmol) was dissolved in toluene (5 mL) at room temperature. The colour of the reaction mixture immediately started to change to dark brown. After stirring for 30 min, volatiles were removed in vacuo, and the residue and was extracted with pre‐cooled pentane (−30 °C). After concentration, the filtrate was stored at −30 °C for ca. 2 d. The resulting black crystalline compound was isolated, washed with small amount of cold pentane and dried in vacuo. Yield: 0.120 g, 77.1 %. Crystals for X‐ray crystal structure determination were obtained from a saturated solution in hexane at 4 °C. Anal. Calc. for C63H94B2N6Sn2+0.5 C5H12: C 63.93 %, H 8.19 %, N 6.83 %; Meas.: C 64.50 %, H 7.76 %, N 6.48 %. 1H NMR (400 MHz, C6D6, 298 K): δH 0.83 (d, J HH=7.0 Hz, 12H, CH(CH3 ) 2 of carbene), 1.17 (d, J HH=6.8 Hz, 24H, CH(CH3 ) 2 of Dipp), 1.20 (br, overlapping, 24H, CH(CH3 ) 2 of Dipp), 1.59 (s, 6H, CCH3 of carbene), 3.32 (br, 4H, CH(CH3)2 of Dipp), 3.44 (br, 4H, CH(CH3)2 of Dipp), 5.45 (sept, J HH=7.3 Hz, 2H, CH(CH3)2 of carbene), 6.30 (br, 2H, CH of boryl), 6.44 (br, 2H, CH of boryl), 7.10 (m, 8H, m‐ArH of Dipp), 7.19 (m, 4H, p‐ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δB 43.5, 54.2. 13C{1H} (151 MHz, C6D6, 298 K): δC 10.1 (C C H3 of carbene), 20.4 and 21.3 (CH( C H3)2 of carbene), 24.3, 25.7 and 26.1 (CH( C H3)2 of Dipp), 28.5 and 28.6 ( C H(CH3)2 of Dipp), 54.7 ( C H(CH3)2 of carbene), 122.3 ( C H of boryl), 123.5 and 123.8 (m‐Ar of Dipp), 126.8 ( C CH3 of carbene), 126.7 and 127.4 (p‐Ar of Dipp), 141.6 and 142.5 (o‐Ar of Dipp), 146.2 and 146.4 (i‐Ar of Dipp), 182.1 (R2 C : of carbene).
K2[Sn2{B(NDippCH)2}2], 3, and Li2[Sn2{B(NDippCH)2}2]: A mixture of 1 (0.20 g, 0.26 mmol) and KC8 (0.21 g, 1.6 mmol) was dissolved/suspended in toluene (5 mL) at room temperature. The reaction mixture was sonicated for 2 h with occasional manual stirring, resulting in a dark red solution. The reaction mixture can be filtered at this point and the filtrate concentrated and stored at −30 °C to give deep red crystals of 3 suitable for crystallography. 3 is not stable at room temperature and metathesis to give the more stable dilithium derivative allows for more convenient spectroscopic characterization: the dark red reaction mixture can alternatively be filtered into a Schlenk flask containing LiI (0.035 g, 0.29 mmol), and the resulting mixture stirred for 20 min. After filtration, the filtrate was concentrated (to ca. 1 mL) and layered with hexane (3 mL). Storing at 4 °C overnight gave dark purple crystals, which were isolated, washed with small amount of cold hexane and dried under vacuum. Yield of Li2[Sn2{B(NDippCH)2}2]: 0.035 g (0.05 mmol, 19.3 %). 1H NMR (400 MHz, C6D6, 298 K): δH 1.21 (m, 6H, CH(CH3 ) 2 of carbene overlapping with pentane), 1.28 (d, J HH=6.9 Hz, 12H, CH(CH3 ) 2 of Dipp), 1.35 (d, J HH=6.9 Hz, 12H, CH(CH3 ) 2 of Dipp), 1.69 (s, 6H, CCH3 of carbene), 3.94 (sept, J HH=6.9 Hz, 6H, CH(CH3)2 of Dipp overlapping with CH(CH3)2 of carbene), 6.58 (s, 2H, CH of boryl), 7.19 (m, 2H, p‐ArH of Dipp), 7.21 (m, 4H, m‐ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δB 55.2 (br). 13C{1H} (151 MHz, C6D6, 298 K): δC 9.0 (C C H3 of carbene), 24.6 (CH( C H3)2 of carbene), 24.8 (CH( C H3)2 of Dipp), 25.3 (CH( C H3)2 of Dipp), 28.6 ( C H(CH3)2 of Dipp), 50.0 ( C H(CH3)2 of carbene), 121.3 ( C H of boryl), 122.9 (m‐Ar of Dipp), 125.4 (p‐Ar of Dipp), 145.9 (o‐Ar of Dipp), 148.4 (i‐Ar of Dipp).
Sn6{B(NDippCH)2}4, 4: A mixture of 2 (0.10 g, 0.08 mmol) and BPh3 (20.3 mg, 0.08 mmol) was dissolved in toluene (5 mL) at room temperature. After stirring for 2 h, the reaction mixture was filtered into a layering Schlenk and concentrated (to ca. 1 mL). Pentane (15 mL) was then added on top of the filtrate and the mixture left for crystallization for several days. The supernatant was then decanted and the resulting deep red crystals washed with pentane and dried under vacuum. Yield: 20.0 mg, 21.1 %. Anal. Calc. for C52H72B2N4Sn3+0.4 C5H12: C 55.93 %, H 6.68 %, N 4.83 %; Meas.: C 56.56 %, H 6.31 %, N 4.78 %. 1H NMR (400 MHz, C6D6, 298 K): δH 0.77 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 0.98 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.16 (m, 12H, CH(CH3 )2 of Dipp overlapping), 1.23 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.26 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.49 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.57 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.61 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.57 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 2.48 (sept, J HH=6.8 Hz, 2H, CH(CH3)2 of Dipp), 2.58 and 2.63 (m, 4H, CH(CH3)2 of Dipp overlapping), 3.59 (sept, J HH=6.8 Hz, 2H, CH(CH3)2 of Dipp), 6.05 and 6.21 (d, J HH=2.2 Hz, 4H, CH of boryl), 7.19 (m, 4H, p‐ArH of Dipp), 7.30 (m, 8H, m‐ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δB 46.6. 13C{1H} (151 MHz, C6D6, 298 K): δC 21.5, 23.8, 27.0, 27.1, 27.2, 27.6 and 27.7 (CH( C H3)2 of Dipp), 27.9, 28.0, 28.2, 28.6 and 29.0 ( C H(CH3)2 of Dipp), 123.8 ( C H of boryl), 124.2 and 124.6 (p‐Ar of Dipp), 124.9 (m‐Ar of Dipp), 139.9, 141.3, 144.9 and 145.8 (o‐Ar of Dipp), 147.1 and 148.5 (i‐Ar of Dipp).
Ge3{B(NDippCH)2}2(IPrMe), 5: To a solution of K2[Ge2{B(NDippCH)2}2] (0.02 g, 0.02 mmol) in toluene at −35 °C was slowly added a solution of (IPrMe)GeCl2 (0.006 g, 0.02 mmol) also in toluene. After stirring for 10 min, the reaction mixture was slowly warmed to room temperature and stirred for another 20 min. Volatiles were removed under vacuum and the residue was extracted with pentane. The filtrate was then dried under vacuum and used for spectroscopic characterization. Yield: 0.015 g, 63.8 %. 1H NMR (400 MHz, C6D6, 298 K) δ 0.74 (s, 6H, CH(CH3 ) 2 of carbene), 1.16, 1.22 and 1.24 (overlapping, 24H, CH(CH3 )2 of Dipp), 1.44 (CCH3 of carbene), 3.58 (br, 4H, CH(CH3)2 of Dipp), 6.06 (br, 1H, CH(CH3)2 of carbene), 6.33 (s, 2H, CH of boryl), 7.01 (m, 4H, m‐ArH of Dipp), 7.11 (m, 2H, p‐ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 298 K): δB 36.5.
Ge6{B(NDippCH)2}4, 6: A mixture of 5 (15 mg, 0.013 mmol) and BPh3 (3.2 mg, 0.013 mmol) was dissolved in C6D6 (0.3 mL) at room temperature. After standing for 2 min, the reaction mixture was filtered into a NMR tube and concentrated (to ca. 0.1 mL). 0.5 mL of pentane was then added on top of the filtrate and the mixture left for crystallization overnight. The solution was then decanted and the resulting deep green crystals were washed with pentane and dried under vacuum. Yield: 7.1 mg, 55.0 %. Anal. Calc. for C52H72B2N4Ge3: C 62.92 %, H 7.31 %, N 5.64 %; Meas.: C 62.34 %, H 7.14 %, N 5.42 %. 1H NMR (400 MHz, C6D6, 298 K): δH 0.81 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 0.93 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.06 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.20 (m, 18H, CH(CH3 )2 of Dipp overlapping), 1.33 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 1.57 (d, J HH=6.8 Hz, 6H, CH(CH3 )2 of Dipp), 2.41 (sept, J HH=6.8 Hz, 2H, CH(CH3)2 of Dipp), 2.53, 2.57 (m, 4H, CH(CH3)2 of Dipp overlapping), 3.57 (sept, J HH=6.8 Hz, 2H, CH(CH3)2 of Dipp), 5.83, 5.91 (d, J HH=2.3 Hz, 4H, CH of boryl), 7.11 (m, 2H, ArH of Dipp), 7.17 (m, 4H, ArH of Dipp overlapping with C6D6), 7.30 (m, 6H, ArH of Dipp). 11B{1H} NMR (128 MHz, C6D6, 25 °C): δB 33.3. 13C{1H} (151 MHz, C6D6, 298 K): δC 22.6, 23.8, 27.1, 27.3 and 27.5 (CH( C H3)2 of Dipp), 27.9, 28.1, 28.8 and 29.2 ( C H(CH3)2 of Dipp), 123.5, 123.8 ( C H of boryl), 124.0, 124.9 (p‐Ar of Dipp), 125.0, 125.1 (m‐Ar of Dipp), 139.9, 141.7, 145.0, 145.5 (o‐Ar of Dipp), 147.2, 148.7 (i‐Ar of Dipp).
Quantum chemical calculations: All computational work reported here was carried out at the density functional theory (DFT) level, using ORCA (Revision 5.0.1).[ 20a , 20b , 20c ] The exchange correlation functional B3LYP[ 20d , 20e , 20f , 20g ] was employed in conjunction with the LanL2DZ basis set with the D4 dispersion correction.[ 20h , 20i , 20j , 20k ] All calculations were performed on model systems to reduce the computational cost, with iPr groups being replaced with Me groups. The nature of the stationary points, minima and transition states, was confirmed by full frequency calculations, and are characterized by zero or one imaginary frequency respectively along with full IRC (intrinsic reaction coordinate) calculations for transition states.
Supporting Information
Full synthetic and characterizing data, representative NMR spectra, details of quantum chemical calculations and xyz files for optimized structures.
Deposition Number(s) 2215109 (1), 2215110 (2), 2215111 (3), 2215112 (4), 2215113 (6) contain(s) the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service
Conflict of interest
The authors declare no conflict of interest.
1.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgments
We thank the EPSRC Centre for Doctoral Training in Inorganic Chemistry for Future Manufacturing (OxICFM CDT, EP/S023828/1, studentship to A.E.C.), the Leverhulme Trust (RP‐2018‐246, studentship to A.H.) and the Alexander von Humboldt Stiftung (Feodor Lynen Fellowship to M.A.E.).
Zheng X., Crumpton A. E., Protchenko A. V., Heilmann A., Ellwanger M. A., Aldridge S., Chem. Eur. J. 2023, 29, e202203395.
A previous version of this manuscript has been deposited on a preprint server (https://doi.org/10.26434/chemrxiv‐2022‐zk61r).
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.
References
- 1. Phillips A. D., Wright R. J., Olmstead M. M., Power P. P., J. Am. Chem. Soc. 2002, 124, 5930–5931. [DOI] [PubMed] [Google Scholar]
- 2. Stender M., Phillips A. D., Wright R. J., Power P. P., Angew. Chem. Int. Ed. 2002, 41, 1785–1787. [DOI] [PubMed] [Google Scholar]
- 3. Power P. P., Nature 2010, 463, 171–177. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Lansing E., Kenneth Pitzer B. S., The Thermodynamic and Physical Properties of the Elements, Cornell University Press, 1947; [Google Scholar]
- 4b. Gusel'Nikov L. E., Nametkin N. S., Chem. Rev. 1979, 79, 529–577. [Google Scholar]
- 5.Selected reviews:
- 5a. Power P. P., Organometallics 2007, 26, 4362–4372; [Google Scholar]
- 5b. Rivard E., Power P. P., Inorg. Chem. 2007, 46, 10047–10064; [DOI] [PubMed] [Google Scholar]
- 5c. Power P. P., Acc. Chem. Res. 2011, 44, 627–637; [DOI] [PubMed] [Google Scholar]
- 5d. Fong H., Moret M.-E., Lee Y., Peters J. C., Organometallics 2013, 32, 3053–3062; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5e. Hanusch F., Groll L., Inoue S., Chem. Sci. 2021, 12, 2001–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Spikes G. H., Fettinger J. C., Power P. P., J. Am. Chem. Soc. 2005, 127, 12232–12233; [DOI] [PubMed] [Google Scholar]
- 6b. Peng Y., Brynda M., Ellis B. D., Fettinger J. C., Rivard E., Power P. P., Chem. Commun. 2008, 6042; [DOI] [PubMed] [Google Scholar]
- 6c. Li J., Schenk C., Goedecke C., Frenking G., Jones C., J. Am. Chem. Soc. 2011, 133, 18622–18625; [DOI] [PubMed] [Google Scholar]
- 6d. Hadlington T. J., Hermann M., Li J., Frenking G., Jones C., Angew. Chem. Int. Ed. 2013, 52, 10199–10203; [DOI] [PubMed] [Google Scholar]
- 6e. Hadlington T. J., Jones C., Chem. Commun. 2014, 50, 2321; [DOI] [PubMed] [Google Scholar]
- 6f. Wang S., Sherbow T. J., Berben L. A., Power P. P., J. Am. Chem. Soc. 2018, 140, 590–593; [DOI] [PubMed] [Google Scholar]
- 6g. Kelly J. A., Juckel M., Hadlington T. J., Fernández I., Frenking G., Jones C., Chem. Eur. J. 2019, 25, 2773–2785. [DOI] [PubMed] [Google Scholar]
- 7.See for example:
- 7a. Cui C., Olmstead M. M., Power P. P., J. Am. Chem. Soc. 2004, 126, 5062–5063; [DOI] [PubMed] [Google Scholar]
- 7b. Sasamori T., Sugahara T., Agou T., Guo J. D., Nagase S., Streubel R., Tokitoh N., Organometallics 2015, 34, 2106–2109; [Google Scholar]
- 7c. Sugahara T., Guo J. D., Sasamori T., Karatsu Y., Furukawa Y., Ferao A. E., Nagase S., Tokitoh N., Bull. Chem. Soc. Jpn. 2016, 89, 1375–1384. [Google Scholar]
- 8.See for example:
- 8a. Hadlington T. J., Li J., Hermann M., Davey A., Frenking G., Jones C., Organometallics 2015, 34, 3175–3185; [Google Scholar]
- 8b. Sasamori T., Sugahara T., Agou T., Sugamata K., Guo J.-D., Nagase S., Tokitoh N., Chem. Sci. 2015, 6, 5526–5530; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Bouška M., Tydlitát J., Jirásko R., Růžička A., Dostál L., Herres-Pawlis S., Hoffmann A., Jambor R., Eur. J. Inorg. Chem. 2018, 2018, 2038–2044. [Google Scholar]
- 9.
- 9a. Peng Y., Ellis B. D., Wang X., Fettinger J. C., Power P. P., Science. 2009, 325, 1668–1670; [DOI] [PubMed] [Google Scholar]
- 9b. Sugahara T., Guo J. D., Sasamori T., Nagase S., Tokitoh N., Chem. Commun. 2018, 54, 519–522. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Pu L., Phillips A. D., Richards A. F., Stender M., Simons R. S., Olmstead M. M., Power P. P., J. Am. Chem. Soc. 2003, 125, 11626–11636; [DOI] [PubMed] [Google Scholar]
- 10b. Sugiyama Y., Sasamori T., Hosoi Y., Furukawa Y., Takagi N., Nagase S., Tokitoh N., J. Am. Chem. Soc. 2006, 128, 1023–1031; [DOI] [PubMed] [Google Scholar]
- 10c. Perla L. G., Kulenkampff J. M., Fettinger J. C., Power P. P., Organometallics 2018, 37, 4048–4054. [Google Scholar]
- 11. Leung W. P., Chiu W. K., Mak T. C. W., Organometallics 2014, 33, 225–230. [Google Scholar]
- 12.
- 12a. Protchenko A. V., Birjkumar K. H., Dange D., Schwarz A. D., Vidovic D., Jones C., Kaltsoyannis N., Mountford P., Aldridge S., J. Am. Chem. Soc. 2012, 134, 6500–6503; [DOI] [PubMed] [Google Scholar]
- 12b. Protchenko A. V., Dange D., Harmer J., Tang C. Y., Schwarz A. D., Kelly M. J., Phillips N., Birjkumar K. H., Jones C., Kaltsoyannis N., Mountford P., Aldridge S., Nat. Chem. 2014, 6, 315–319; [DOI] [PubMed] [Google Scholar]
- 12c. Protchenko A. V., Dange D., Schwarz A. D., Blake M. P., Jones C., Mountford P., Aldridge S., J. Am. Chem. Soc. 2014, 136, 10902–10905; [DOI] [PubMed] [Google Scholar]
- 12d. Protchenko A. V., Blake M. P., Schwarz A. D., Jones C., Mountford P., Aldridge S., Organometallics 2015, 34, 2126–2129; [Google Scholar]
- 12e. Dange D., Davey A., Abdalla J. A. B., Aldridge S., Jones C., Chem. Commun. 2015, 51, 7128–7131; [DOI] [PubMed] [Google Scholar]
- 12f. Protchenko A. V., Bates J. I., Saleh L. M. A., Blake M. P., Schwarz A. D., Kolychev E. L., Thompson A. L., Jones C., Mountford P., Aldridge S., J. Am. Chem. Soc. 2016, 138, 4555–4564; [DOI] [PubMed] [Google Scholar]
- 12g. Usher M., Protchenko A. V., Rit A., Campos J., Kolychev E. L., Tirfoin R., Aldridge S., Chem. Eur. J. 2016, 22, 11685–11698; [DOI] [PubMed] [Google Scholar]
- 12h. Rit A., Campos J., Niu H., Aldridge S., Nat. Chem. 2016, 8, 1022–1026; [DOI] [PubMed] [Google Scholar]
- 12i. Dange D., Sindlinger C. P., Aldridge S., Jones C., Chem. Commun. 2017, 53, 149–152; [DOI] [PubMed] [Google Scholar]
- 12j. Protchenko A. V., Urbano J., Abdalla J. A. B., Campos J., Vidovic D., Schwarz A. D., Blake M. P., Mountford P., Jones C., Aldridge S., Angew. Chem. Int. Ed. 2017, 56, 15098–15102; [DOI] [PubMed] [Google Scholar]
- 12k. Protchenko A. V., Vasko P., Do D. C. H., Hicks J., Fuentes M. Á., Jones C., Aldridge S., Angew. Chem. Int. Ed. 2019, 58, 1808–1812; [DOI] [PubMed] [Google Scholar]
- 12l. Protchenko A. V., Vasko P., Fuentes M. Á., Hicks J., Vidovic D., Aldridge S., Angew. Chem. Int. Ed. 2021, 60, 2064–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Segawa Y., Yamashita M., Nozaki K., Science 2006, 314, 113–115. [DOI] [PubMed] [Google Scholar]
- 14. Green S. P., Jones C., Stasch A., Science 2007, 318, 1754–1758. [DOI] [PubMed] [Google Scholar]
- 15.For a (superficially) structurally related Sn6 cluster (of the type Sn6R6) see: Sindlinger C. P., Wesemann L., Chem. Sci. 2014, 5, 2739–2746. [Google Scholar]
- 16.For reviews of group 14 element cluster chemistry, see for example:
- 16a. Schnepf A., Chem. Soc. Rev. 2007, 36, 745–758; [DOI] [PubMed] [Google Scholar]
- 16b. Scharfe S., Kraus F., Stegmaier S., Schier A., Fässler T. F., Angew. Chem. Int. Ed. 2011, 50, 3630–3670. [DOI] [PubMed] [Google Scholar]
- 17.For a germanium cluster of similar Ge6X4 composition, see: Helmer J., Hepp A., Lips F., Dalton Trans. 2020, 49, 11843–11850. [DOI] [PubMed] [Google Scholar]
- 18. Cordero B., Gómez V., Platero-Prats A. E., Revés M., Echeverría J., Cremades E., Barragán F., Alvarez S., Dalton Trans. 2008, 2832–2838. [DOI] [PubMed] [Google Scholar]
- 19.Deposition NumberNummerierung?
- 19a. 2215109 (1), 2215110 (2), 2215111 (3), 2215112 (4), 2215113 (6) contain;
- 19b.the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
- 20.
- 20a. Neese F., Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73–78; [Google Scholar]
- 20b. Neese F., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2017, 8, e1327; [Google Scholar]
- 20c. Neese F., Wennmohs F., Becker U., Riplinger C., J. Chem. Phys. 2020, 152, 224108; [DOI] [PubMed] [Google Scholar]
- 20d. Becke A. D., J. Chem. Phys. 1993, 98, 5648–5652; [Google Scholar]
- 20e. Lee C., Yang W., Parr R. G., Phys. Rev. B 1988, 37, 785–789; [DOI] [PubMed] [Google Scholar]
- 20f. Vosko S. H., Wilk L., Nusair M., Can. J. Phys. 1980, 58, 1200–1211; [Google Scholar]
- 20g. Stephens P. J., Devlin F. J., Chabalowski C. F., Frisch M. J., J. Phys. Chem. 1994, 98, 11623–11627; [Google Scholar]
- 20h. Wadt W. R., Hay P. J., J. Chem. Phys. 1985, 82, 270; [Google Scholar]
- 20i. Wadt W. R., Hay P. J., J. Chem. Phys. 1985, 82, 284; [Google Scholar]
- 20j. Wadt W. R., Hay P. J., J. Chem. Phys. 1985, 82, 299; [Google Scholar]
- 20k. Caldeweyher E., Ehlert S., Hansen A., Neugebauer H., Spicher S., Bannwarth C., Grimme S., J. Chem. Phys. 2019, 150, 154122. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Data Availability Statement
The data that support the findings of this study are available in the supplementary material of this article.