

Abstract
Campylobacter jejuni and Campylobacter coli infections are the leading cause of foodborne gastroenteritis in high‐income countries. Campylobacter colonizes a variety of warm‐blooded hosts that are reservoirs for human campylobacteriosis. The proportions of Australian cases attributable to different animal reservoirs are unknown but can be estimated by comparing the frequency of different sequence types in cases and reservoirs. Campylobacter isolates were obtained from notified human cases and raw meat and offal from the major livestock in Australia between 2017 and 2019. Isolates were typed using multi‐locus sequence genotyping. We used Bayesian source attribution models including the asymmetric island model, the modified Hald model, and their generalizations. Some models included an “unsampled” source to estimate the proportion of cases attributable to wild, feral, or domestic animal reservoirs not sampled in our study. Model fits were compared using the Watanabe–Akaike information criterion. We included 612 food and 710 human case isolates. The best fitting models attributed >80% of Campylobacter cases to chickens, with a greater proportion of C. coli (>84%) than C. jejuni (>77%). The best fitting model that included an unsampled source attributed 14% (95% credible interval [CrI]: 0.3%–32%) to the unsampled source and only 2% to ruminants (95% CrI: 0.3%–12%) and 2% to pigs (95% CrI: 0.2%–11%) The best fitting model that did not include an unsampled source attributed 12% to ruminants (95% CrI: 1.3%–33%) and 6% to pigs (95% CrI: 1.1%–19%). Chickens were the leading source of human Campylobacter infections in Australia in 2017–2019 and should remain the focus of interventions to reduce burden.
Keywords: Bayesian analysis, Campylobacter, source attribution
1. BACKGROUND
Campylobacter infections are a major cause of zoonotic foodborne gastroenteritis worldwide and the leading cause in high‐income countries (Li et al., 2019), with a global burden of over 166 million cases and 3.7 million disability adjusted life years circa 2010 (Kirk et al., 2015). In Australia, foodborne Campylobacter causes an estimated 179,000 cases and 3200 hospitalizations each year (Kirk et al., 2014). Although the genus Campylobacter comprises 41 species (as of June 2022) (Euzéby, 2021), two species, Campylobacter coli and Campylobacter jejuni, cause >90% of human campylobacteriosis cases (Kaakoush et al., 2015). The most widely used sub‐species classification for Campylobacter research is seven‐gene multi‐locus sequence typing (MLST) (Dingle et al., 2001). Campylobacter coli and C. jejuni are typed in the same scheme, and this “Campylobacter” scheme contains more than 11,800 sequence types (STs) (Jolley et al., 2018). A range of warm‐blooded animals—including wild bird species and food production animals such as chickens, pigs, sheep, and cattle—can act as Campylobacter reservoirs. Human cases are often foodborne (Vally et al., 2014) and occasionally waterborne (Kaakoush et al., 2015), with some direct zoonotic transmission (Varrone et al., 2020) and rare person‐to‐person transmission (Kuhn et al., 2021). In Australia, most Campylobacter infections are sporadic, that is, not associated with recognized outbreaks (Kaakoush et al., 2015; Moffatt et al., 2020), making it difficult to identify the source of infection for most individual cases.
Bayesian source attribution models (e.g., Hald et al., 2004; Liao et al., 2019; Miller et al., 2017; Mullner, Jones, et al., 2009; Wilson et al., 2008) have been used to estimate the proportion of human campylobacteriosis attributable to sources by comparing the relative abundance of Campylobacter STs observed in sources and cases. Some methods attempt to account for the relative transmissibility and virulence of different strains (e.g., Hald et al., 2004; Miller et al., 2017; Mullner, Jones, et al., 2009), while others attempt to model organism genetic recombination and mutation to improve estimates of relative abundance of rare types in sources (e.g., Liao et al., 2019; Wilson et al., 2008). In many source attribution studies, including the present study, the sources of interest are animal reservoirs (e.g., chickens and pigs) rather than individual classes of food products, risk factors for infection, transmission routes, or systemic food safety failures. Nevertheless, source attribution estimates have been used to identify the primary animal reservoirs, inform effective food safety policy and interventions (Mullner, Spencer, et al., 2009; Sears et al., 2011), and identify human subpopulations with differing patterns of attribution (Lake et al., 2021; Liao et al., 2019).
The aim of this study was to estimate the proportion of Campylobacter infections attributable to chickens, pigs, and ruminants (cattle and sheep) in Australia during 2017–2019. We compared attribution estimates across a range of modeling frameworks, including models that considered a fourth “unsampled” source to reflect potential reservoirs for which no data were available.
2. METHODS
2.1. Data collection
Study data were collected as part of the broader CampySource project, a collaboration between Australian academic institutions, government agencies, and industries. A detailed description of study methods and data collection can be found elsewhere (Cribb et al., 2022; Varrone et al., 2018; Walker et al., 2019; Wallace et al., 2021, 2020). The sequence readset for each food isolate (Bioproject Accession: PRJNA591966), case isolates sampled as part of the case–control study (Bioproject Accession: PRJNA592186), and the case isolates included in the national snapshot (Bioproject Accession: PRJNA560409) are available through GenBank (Clark et al., 2016).
Cases were enrolled as part of a case–control study and a national snapshot of campylobacteriosis for which detailed sampling methods have been described previously (Cribb et al., 2022; Varrone et al., 2018; Wallace et al., 2021). We defined a case as a person with acute diarrhea where Campylobacter spp. was cultured from stool. Campylobacteriosis is a nationally notifiable disease in Australia, with a legislative requirement for all confirmed cases to be notified to the relevant health department. Cases were identified through a combination of state or regional notifiable disease surveillance systems and pathology service databases. Cases were excluded from the case–control study if they had traveled overseas at all in the previous two weeks; if they had traveled to another jurisdiction within Australia for the entirety of the previous two weeks; if they or their household members had diarrhea in the four weeks previous to their Campylobacter infection; if they were unable to answer the questionnaire in English; if they could not be contacted over telephone; or if enteric pathogens other than Campylobacter spp., Blastocystis hominis, or Dientamoeba fragiles were detected in their stool.
We collected and sequenced 531 Campylobacter isolates from human cases between February 2018 and October 2019 in three Australian jurisdictions: The Hunter New England (HNE) public health district of New South Wales (NSW), Queensland (Qld), and the Australian Capital Territory (ACT). We collected and sequenced an additional 184 human isolates from Victoria (Vic), Western Australia (WA), Tasmania (Tas), South Australia (SA), and the Northern Territory (NT) over the period of 2017–2019. Of the 715 case isolates described above, 164 were collected over a short time (October 2018—February 2019) across all sampled jurisdictions and have been published previously as part of national snapshot of campylobacteriosis (Wallace et al., 2021). After removing five isolates with indeterminate MLST assignments (two from Vic and three from Qld), 710 human case isolates were included in the analysis. Cases from three jurisdictions (HNE, Qld, and ACT) were interviewed regarding potential exposures for a case–control study (Varrone et al., 2018). We considered responses on age, gender, rurality of residence, notification date, and jurisdiction as potential model covariates.
Detailed sampling methods for food products have been described previously (Walker et al., 2019; Wallace et al., 2020). Jurisdictional health departments sampled retail meat products for Campylobacter testing between October 2016 and March 2019 in the ACT (Canberra), NSW (Hunter Valley and Greater Sydney regions), Qld (Brisbane, Toowoomba, Rockhampton, Townsville, and Cairns hospital and health districts), and Vic (Bendigo and Melbourne; chicken only), with isolates collected after March 2017 considered for sequencing. Samples included pre‐packaged (fresh or frozen) meats and delicatessen products. The prevalence of Campylobacter spp. contamination was expected to be low for muscle meat from pigs and ruminants (New South Wales Food Authority, 2018). Therefore, we sampled offal (kidney and liver) to maximize the total number of isolates from these sources. For chicken, a combination of muscle meat (with and without skin or bones) and offal (giblet and liver) products were sampled. We attempted to sequence all isolates from pig and ruminant samples, but due to budgetary constraints we only sequenced a subsample of isolates from chicken, aiming for a total of 500 isolates from this source. In Qld, Campylobacter spp. isolates from chicken were subsampled for genotyping to optimize coverage using a judgmental‐stratified approach across the following strata in order of isolate selection priority: time (calendar year and quarter), region (hospital and health district), Campylobacter species (C. jejuni, C. coli), abattoir/processor, temporal occurrence across calendar quarter, and meat type. Subsampling selected at least one isolate within each stratum defined by combinations of Campylobacter species, calendar quarter, and region, but otherwise proportional to abundance. Where there was a choice of isolates in a stratum, they were selected (in order of priority) to maximize the total number of abattoirs/processors sampled, spread multiple isolates from a single abattoir/processor across the calendar quarter, and maximize the diversity of chicken meat types sampled.
2.2. Campylobacter Isolation, sequencing, and genotyping
We isolated and confirmed C. coli and C. jejuni from the meat and offal samples according to ISO 10272−1:2017 (International Organization for Standardization, 2017) and AS 5013.06.2015 (Standards Australia, 2015) with minor modifications (Walker et al., 2019). Isolation and genomic analysis of isolates from food (Wallace et al., 2020) and clinical specimens (Wallace et al., 2021) has been described in detail elsewhere. Briefly, C. coli and C. jejuni isolates were grown from patient fecal samples, with samples stored at 2−8°C and processed for Campylobacter culture within 48 h of collection. We extracted Campylobacter DNA from food and patient isolates using the QiaSymphony DSP DNA Mini kit (Qiagen) according to manufacturer's instructions. We used the Nextera XT DNA Library Prep kit (Illumina, San Diego, CA, USA) to prepare DNA for whole genome sequencing which was performed on the Illumina Next‐Seq500 with 150 base‐pair paired‐end reads using the NextSeq 500 Mid Output kit (300 cycles) (Illumina). MLST was performed on de novo assembled contigs, searching with a BLAST‐based tool (Seemann, n.d.) against the PubMLST allele database (Jolley et al., 2018).
2.3. Source attribution modeling approach
We employed a generalization of existing Bayesian source attribution methods (Liao et al., 2019; Mullner, Jones, et al., 2009) to estimate attribution proportions including covariates for the cases and adjusting for differences between types, as applied previously (McLure et al., 2022). We modeled the proportion of human cases in subpopulation s attributable to transmission from each source j () given the number of cases in each subpopulation s due to each pathogen type i (), the number of isolates of each type observed in each putative source (), and weights for the relative exposure of humans to these putative sources . The proportion () of cases in subpopulation s that were due to pathogen type i from source j was modeled as:
with constraints and , where was the ability of source j to act as a source of infection for group s, was the relative abundance of type i in source j, and was the relative ability of subtype i to transmit from a source and lead to a reported case (which we call type transmission potential). The proportion of cases in subpopulation s attributed to source j was modeled as :
while the proportion of cases due to each type, , was modeled as :
We considered two models for the relative abundance of sequence types in sources (). The first was the Dirichlet model of Liao et al. (2019), which adopts independent symmetric Dirichlet priors for the relative abundance of sequence types in sources and models the numbers of isolates of each type observed in each source () as independent multinomial distributions:
The second approach was the asymmetric island model, first proposed for source attribution by Wilson et al. (2008) and developed further and implemented in an R package, islandR by Liao et al. (2019). The asymmetric island model uses the observed number of MLST types and frequency of alleles at each locus to estimate mutation rate (new allele generation), recombination (new sequence generation types from novel allele combinations extant in the sources), and transmission between sources, to estimate the relative abundance of all types in each source ().
Parameters were estimated either by joint inference of all parameters (joint Dirichlet model) or in two steps (two‐step Dirichlet and asymmetric island model). In two‐step approaches, the relative abundance of sequence types in each source () was estimated first, with all other parameters then estimated repeatedly using draws from the posterior distribution of In estimating the remaining parameters, the transmission potential of each type () and the exposure weights () were assumed to be the same for each subpopulation s, but the ability of each source to transmit to humans () was allowed to vary and modeled as:
where F was a model matrix defining a linear predictor based on binary, categorical, or ordinal covariates for each subgroup s of the cases, and β was a matrix of parameters for each source j. A reference source was assigned, and the associated parameters in the matrix β were fixed to 0, while the remaining parameters were given unit normal priors.
The number of human cases in subpopulation s due to pathogen type i was modeled as independent multinomial variables, that is, . The type transmission potential terms, , were constrained with a log‐normal prior:
Since Campylobacter is primarily foodborne (Vally et al., 2014), the exposure weights were approximated by the relative exposure to contaminated food products derived from each source, modeled as , where was the apparent consumption (per capita, per year) of food derived from source j (Australian Bureau of Agricultural and Resource Economics and Sciences, 2020), and was the prevalence of the pathogen in muscle meat derived from source j. The prevalence of Campylobacter in the muscle meat of each source j was modeled with the binomial model and a flat prior: where was the number of total tests and the number of positive tests. Data for prevalence in chicken muscle meat were derived from the CampySource study (Walker et al., 2019). CampySource collected offal rather than muscle meat samples for pigs and ruminants. Since Campylobacter prevalence was likely to be higher on offal than muscle meat, and Australians consume relatively little offal, we estimated prevalence in these meats from a separate survey (New South Wales Food Authority, 2018).
When employing the Dirichlet model for the relative abundance of sequence types in sources, an “unsampled source” was modeled by including an additional source without any observed samples: that is, . Consumption statistics could not be used to determine the exposure weight for the unsampled source. We therefore assumed the “consumption” for the unsampled source was equal to the least consumed product (pork).
The Modified Hald model proposed by Müllner, Jones, et al. (2009) can be seen as a special case of the modeling framework presented here, using the Dirichlet model of relative abundance of sequence types in sources and no covariates for cases. The attribution model proposed by Liao et al. (2019) can also be seen as a special case of our framework, using the asymmetric island model for the prevalence of types in sources and assuming all type transmission potential terms, , were equal to 1.
2.4. Source attribution models
We considered ten base models (Table 1), varying the assumptions for the type transmission potential terms and the model for the relative abundance of sequence types in sources (Dirichlet vs. asymmetric island). With the asymmetric island models (M7 and M10), we assumed the mutation and recombination rates of Campylobacter were the same across sources. With the Dirichlet models, we compared models with and without an “unsampled source” and primarily used a flat Dirichlet (1,1,…,1) prior for the relative abundance of sequence types in sources. However, we used an even less informative Dirichlet (0.1, 0.1,…,0.1) prior as a sensitivity analysis.
TABLE 1.
Source attribution models considered, with model numbers for those that converged
| Model | Type transmission potential terms (q) a | Model of relative abundance of STs in each source | prior | Joint vs. two‐step inference? b | With unsampled source? | |
|---|---|---|---|---|---|
| M1 | Varied | Dirichlet | Dirichlet (1,…,1) | Joint | Yes |
| M2 | No | ||||
| M3 | Dirichlet (0.1,…,0.1) | Yes | |||
| M4 | No | ||||
| M5 | Dirichlet (1,…,1) | Two‐step | Yes | ||
| M6 | No | ||||
| M7 | Asymmetric island | ||||
| * | Equal | Dirichlet | Dirichlet (1,…,1) | Joint | Yes |
| * | No | ||||
| * | Dirichlet (0.1,…,0.1) | Yes | |||
| * | No | ||||
| M8 | Dirichlet (1,…,1) | Two‐step | Yes | ||
| M9 | No | ||||
| M10 | Asymmetric island | ||||
Abbreviation STs, sequence types.
The fitting procedure for these models failed to converge, so results are omitted.
Type transmission potential terms were either all set to 1 or estimated using a log‐normal hyperprior with unknown variance.
In joint models, all parameters were estimated simultaneously in fully joint Bayesian inference; otherwise, the posterior distribution of the relative abundance of STs in each source was estimated separately, and 100 draws from this posterior were used to estimate the remaining parameters in a second inference step.
We then included covariates (age, rurality, gender, jurisdiction and season) into each of the base models. We used four age categories (0‐4, 5–18, 19–64, and 65 and over). Rurality was categorized as urban or rural, with those reporting residence in an inner city, urban, suburban, or town area categorized as urban, and those reporting residence in rural or remote areas categorized as rural. Season was classified by the date the case was reported as: Summer (December to February), Autumn (March to May), Winter (June to August) and Spring (September to November). Cases with a missing value for gender, age, rurality, jurisdiction, or season were excluded from only analyses involving the missing covariates.
2.5. Relative attributable proportion
To compare the risk associated with different animal sources, we calculated a quantity we call the relative attributable proportion (RAP). For each source, j, the RAP was estimated by the proportion of cases attributed to that source divided by the domestic annual consumption of meat products from that source (Australian Bureau of Agricultural and Resource Economics and Sciences, 2020), normalized against one of the sources as a reference:
We chose the most commonly consumed meat product in Australia—chicken—as the reference source, which therefore had an RAP of one.
2.6. Implementation
All analyses were conducted in the R software environment (R Core Team, 2021), with data cleaning and visualizations using tidyverse packages (Wickham et al., 2019) and ggVennDiagram (Gao, 2022). In the two‐step Dirichlet and asymmetric island models, the relative abundance of sequence types in sources was estimated using the R package, islandR, created by Liao et al. (2019). Inference for the asymmetric island model was done with Markov Chain Monte Carlo (MCMC) using the Metropolis–Hasting Algorithm, with 1000 iterations of warmup and thinning post‐warmup draws to one in every 100 iterations. Using 100 draws from the posterior distribution of the relative abundance of sequence types in sources, inference for remaining parameters for all models was performed using Hamiltonian MCMC using the No U‐Turn Algorithm implemented with the Stan language (Stan Development Team, 2020b) via the R package, Rstan (Stan Development Team, 2020a). The Hamiltonian MCMC step used four independent chains, 2000 warmup iterations, and 100 post‐warmup draws, for a total of 40,000 post‐warmup draws per model. In fully joint models, all inference was conducted using Hamiltonian MCMC with four chains, 2000 warmup iterations, and 2000 post‐warmup draws, for a total of 8000 post‐warmup draws per model. Convergence checking for Hamiltonian MCMC was done using the “R hat” statistic (Vehtari et al., 2021) across the four chains.
Model posterior predictive fits were compared using the Watanabe–Akaike information criterion (WAIC) (Watanabe & Opper, 2010) using the R package, loo (Vehtari et al., 2017), with a difference greater than five standard errors considered to be substantive evidence of superior model predictions. The WAIC uses log‐likelihood of each datapoint averaged across the posterior to assess how well models match points in the training data. For comparing joint models to one another (including models with and without covariates for cases), each isolate from either a case or source was considered a datapoint for calculating WAIC. However, for models where inference was conducted in two steps, only the data from the second step (i.e., case isolates) could be considered, and the WAIC on these data was the primary measure of model fit used to compare across all models.
3. RESULTS
3.1. Isolates and sequence types in cases and sources
After removing nine isolates with indeterminate ST assignments (five case isolates and four chicken isolates), the final dataset comprised genotyped isolates from 710 human cases, 480 chicken meat and offal samples, 88 ruminant (sheep and cattle) offal samples, and 44 pig offal samples. Campylobacter coli was more common in chickens (59%, 283/480) and pigs (64%, 28/44), and C. jejuni was more common in human cases (82%, 585/710) and ruminants (82%, 72/88). The 1322 isolates represented 175 different STs, 74 C. coli and 101 C. jejuni. STs shared across multiple sources, or sources and cases, were usually more common than those found only in cases or a single source. While 66 of 118 human case STs were not found in any sampled food source (18/31 C. coli STs and 48/87 C. jejuni STs), these types only accounted for 19% of all isolates from cases. Conversely the eight STs found in cases and every source (STs 21, 42, 50, 538, 827, 1181, 2083, and 7323) together accounted for 31% (221/710) of isolates from cases, 47% (41/88) of isolates from ruminants, 30% (146/480) of isolates from chickens, and 34% (15/44) of isolates from pigs. Fifteen C. coli STs (18 isolates) and two C. jejuni STs (two isolates) were found in pigs but no other sources. Three C. jejuni STs (three isolates) were only found in ruminants but no other sources. Similarly, 29 C. coli STs (101 isolates) and 31 C. jejuni STs (91 isolates) were only found in chicken (Figure A1, Table 2, Tables A1 and A2).
TABLE 2.
Number of distinct sequence types (STs) and isolates from humans and food sources. Percentages denote of the fraction of all STs/isolates from that origin. See Table A1 for C. coli numbers and Table A2 for C. jejuni numbers
| Origin | STs | Isolates | Unique* STs (%) | Isolates from Unique* STs (%) | STs without cases (%) | Isolates from STs without cases (%) |
|---|---|---|---|---|---|---|
| Human | 118 | 710 | 66 (55.9) | 134 (18.9) | N/A | N/A |
| Chicken | 88 | 480 | 60 (68.2) | 192 (40) | 38 (43.2) | 90 (18.8) |
| Pig | 33 | 44 | 17 (51.5) | 20 (45.5) | 21 (63.6) | 25 (56.8) |
| Ruminant | 25 | 88 | 3 (12) | 3 (3.4) | 2 (8) | 2 (2.3) |
Unique types for sources are types that were found in that source and no other source (except potentially in humans), while unique types for humans are those found in cases but not in any of the three sources.
3.2. Overall attribution proportions
In almost all models, chickens were estimated to be the most common source for both C. jejuni (9/10 models) and C. coli (all models). The proportion of cases attributed to chickens was lower for C. jejuni than for C. coli (Figure 1). Dirichlet models with an unsampled source—in which we consider attribution to an unknown fourth source for which the relative abundance of sequence types in unknown due to an absence of samples (see Section 2 for details)—had reduced attribution to ruminants and pigs but similar attribution to chickens in most models. Performing joint inference compared to two‐step inference resulted in only slightly higher attribution to chickens and unsampled sources and reduced attribution to ruminants and pigs.
FIGURE 1.
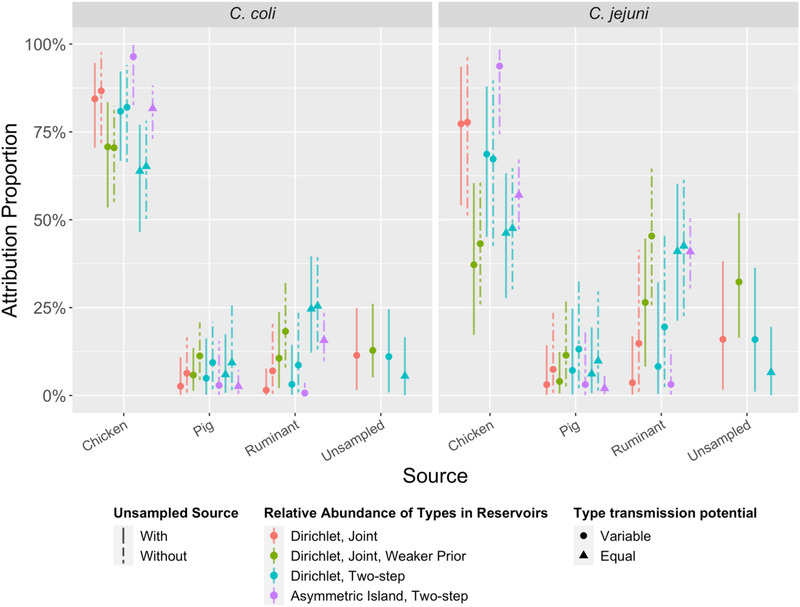
Source attribution proportions of C. jejuni and C. coli cases to three sampled sources (chicken, pig, and ruminant) in 10 models (M1–M10, left to right). Four models (M1, M3, M5, M8) also include a fourth, “unsampled source.” The asymmetric island model is intrinsically unable to accommodate an unsampled source. See Table 1 for the list of assumptions for each model.
Including covariates for cases in the joint attribution models (M1‐M4) did not lead to substantive improvements in model posterior predictions as measured with WAIC. Although the point estimates for the proportions of C. coli and C. jejuni cases attributed to chickens were lower in rural than urban areas for all models except M10, the 95% credible interval for the differences included zero (no difference; Figure A3). This absence of substantive improvement in model fit was observed across all covariates (gender, age group, jurisdiction, and season) and models. As models with covariates could only be fit to the subset of the cases enrolled via the case–control study (n = 531), models without covariates were preferred.
3.3. Variability of transmission potential by sequence type
Our data indicate that transmission potential (ability to transmit and cause disease) varied between sequence types in our study. For instance, ST48 was more common among cases (4.2%) than in any of the sources (ruminants: 1.1%, chickens: 0.8%, and pigs: 0.0%), indicating high transmission potential (Table A3). Conversely, ST827 was more common in every source (ruminants: 9.1%, chickens: 9.6%, and pigs: 4.5%) than in cases (1.8%) (Table A4), and ST832 accounted for 3.3% and 2.3% of chicken and pig isolates but none of the cases, indicating low transmission potential. Models allowing transmission potential to vary between STs (M1–M7) had a much better fit to the relative abundance of STs in cases (Figure A4) and substantially better WAIC values than models that assumed that all STs had equal transmission potential (M8–M10) (Table A5). These models with variable transmission potential (M1–M7) attributed more cases to chicken meat and the unsampled source and fewer cases to ruminants compared to those that assumed all STs had equal transmission potential (M8–M10) (Figure 1). Campylobacter coli STs were generally estimated to have lower transmission potential than C. jejuni STs, although there was substantial variation within each species (Figure 2). STs isolated from cases but none of the animal meat sources (e.g., ST2398) were estimated to occur rarely in the sampled sources but have a relatively high transmission potential. When sequence type transmission potential and relative abundance of STs in sources were estimated in two steps (M5–M7), transmission potential was more variable between STs than in models where all parameters were jointly estimated (M1–M4). When comparing models, estimates of transmission potential and abundance varied most for those types that were found in cases but not in sources (e.g., ST2398). In joint Dirichlet models, using a weaker prior or including an unsampled source increased estimates of the relative abundance of these types in sources (Table A4) and correspondingly reduced the estimates of their transmission potential (Table A6). For the Dirichlet models, transmission potential varied at most by a factor of 220 across sequence types and varied by less than a factor of 45 across the most common sequence types in cases (Figure 2 and Table A6). However, under the asymmetric island model, types such as ST2398 (observed in cases but not sources) were estimated to have a trillion times greater transmission potential than other types (Figure 2 and Table A6), which is implausible
FIGURE 2.
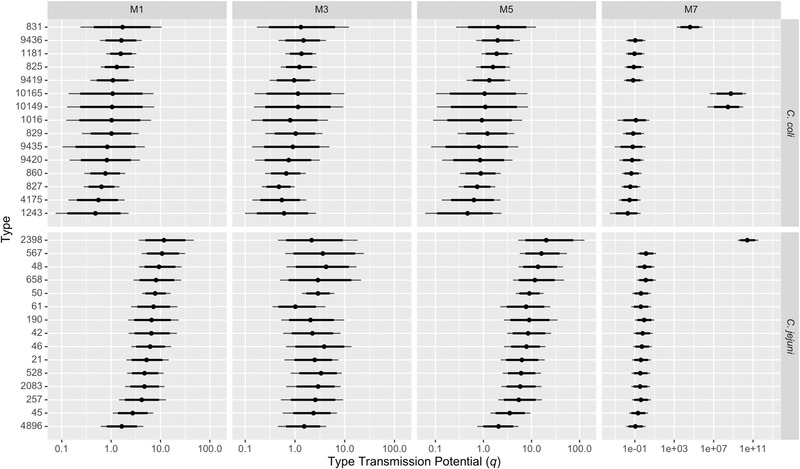
Posterior median and 95% credible intervals for type transmission potential (relative ability of a sequence type to transmit from a source and lead to a reported campylobacteriosis case) of the 15 most common C. coli and C. jejuni multi‐locus sequence types (STs) in human cases for four models. STs have been ordered by posterior median type transmission potential in model M1. Note the x‐axis is on a log scale, with wider limits for M7 (asymmetric island model). See Table 1 for details of the four models.
3.4. Model diagnostics and comparisons
The joint Dirichlet model with variable type transmission potential had very good convergence metrics (Rhat < 1.008). When type transmission potential terms were set to be equal, the joint Dirichlet model failed to converge (Rhat > 1), further highlighting the importance of including type transmission potential terms. Two‐step inference with variable or equal type transmission potential converged adequately (Rhat < 1.04).
Comparison of observed and estimated relative abundance of sequence types in sources (Tables A1 and A2) suggested the default priors for these quantities in the Dirichlet model may be too strong, with better concordance under a weaker prior. Use of a weaker prior for the Dirichlet model leads to substantively poorer model fit to case data as measured with WAIC (Table A5). However, the weaker prior improved fit to source data such that the WAIC calculated over cases and sources was lower (though not substantively) for models with a weaker prior (Table A5). Using the weaker prior had little effect on the attribution proportions for C. coli, but substantially increased the proportion of C. jejuni cases attributed to ruminants and unsampled sources while reducing attribution to chickens.
The asymmetric island models produced source attribution estimates with narrower credible intervals than Dirichlet models, both at the species (Figure 1) and ST level (Figure A2). This was particularly notable for sequence types such as ST2398 (which was found in cases but not in sources) for which models M7 and M10 attributed 97% (95% credible interval [CrI]: 83%−100%) and 65% (95% CrI: 55%−74%) of cases to chicken (Figure A2). The asymmetric island model also attributed a larger proportion of cases to chicken meat and a smaller proportion to ruminants and pigs.
Model M1 had the best predictions for the relative abundance of STs in cases as measured with WAIC (Table A5). M1 had substantively better model predictions than all other models except M2 (same as M1 but without an unsampled source) and M7 (asymmetric Island model with variable type transmission potential) (Table A5). When WAIC was calculated for predictions of sequence type abundance in cases and sources, M3 had the best predictions, although the difference between M3 and M1 was not substantive (less than five standard errors) (Table A5). As the only difference between M1 and M3 is that the latter has a weaker prior on relative abundance of sequence types in sources, this indicates that the strength of this prior involves a trade‐off between prediction in cases compared to sources. As we were primarily interested in cases, M1 was our preferred model. With model M1, the proportions of cases attributed to each source were 80% (95% CrI: 61%–92%) to chickens, 2% (CrI: 0.3%–12%) to ruminants, 2% (CrI: 0.2%–11%) to pigs, and 14% (CrI: 0.3%–32%) to the “unsampled source” (Figure 1). With model M2, the proportions of cases attributed to each source were similar to M1 for chicken (81% [CrI: 59%–96%]) but with greater attribution to ruminants (12% [CrI: 1.3%–33%]) and pigs (6% [CrI: 1.1%–19%]) (Figure 1). Attribution proportions with M7 were similar to M1 for ruminants (2% [CrI: 0.3%–9.6%]) and pigs (2% [CrI: 0.1%–14%]), but with more attribution to chicken (96% [CrI: 80%–99%]) (Figure 1).
3.5. Relative attributable proportion by source
When comparing the relative attributable proportion (RAP) for each source—calculated by dividing proportion of cases attributed to a source by per capita domestic consumption of meat from that source—all models estimated chickens had a higher RAP than pigs (Figure A5). Half of the models estimated that RAP of chicken was higher than ruminants, and the remaining models (which included the three worst fitting models, M8–M10) were inconclusive (i.e., 95% credible intervals for RAP include 1, i.e., equal RAP). Our best fitting model (M1) estimated that chickens had an RAP 22 times that of pigs (95% CrI: 3.6–240) and 27 times that of ruminants (95% CrI: 4.0–190). However, models without an “unsampled source” attributed a greater percentage of cases to ruminants and pigs when compared to similar models with an “unsampled source,” increasing the estimates of RAP for these sources. For instance, model M2 (which had similar fit statistics to M1) estimated that chickens had an RAP 7.3 times that of pigs (95% CrI: 2.1–47) and 4.8 times that of ruminants (95% CrI: 1.3–51) (Figure A5).
4. DISCUSSION
We estimate that approximately 80% of campylobacteriosis in Australia during 2017–2019 was attributable to transmission from chickens, with greater attribution to chickens for C. coli than C. jejuni. Our models attributed to meat sources (e.g., chicken vs. ruminant), not transmission routes (e.g., consumption of contaminated chicken meat compared to contact with chicken feces). However, as most campylobacteriosis in Australia is believed to be foodborne (Vally et al., 2014), we can approximate the risk posed by the consumption of meat from a source with the relative attributable proportion, that is, by dividing the proportion of cases attributed to that source by its domestic consumption. Applying this method, we estimate consuming chicken meat posed a 22–27 times greater risk of campylobacteriosis than consuming meat from pigs and ruminants, in accord with the findings of the case–control study (Cribb et al., 2022).
Our modeling of sporadic campylobacteriosis cases aligns with outbreak investigation findings in Australia, the majority of which have linked cases to chicken or dishes containing chicken (Moffatt et al., 2020). Source attribution studies in other high income countries have also identified chicken as the leading source of Campylobacter infections, for example, Switzerland (Kittl et al., 2013), New Zealand (Liao et al., 2019; Mullner, Spencer, et al., 2009), Germany (Rosner et al., 2017), Denmark (Boysen et al., 2014), the Netherlands (Mughini‐Gras et al., 2018), and the United Kingdom (Thépault et al., 2017). In our study, only a small number of C. coli and C. jejuni campylobacteriosis cases were attributed to pigs, similar to studies conducted in Denmark (Boysen et al., 2014), Switzerland (Kittl et al., 2013), and the Netherlands (Mughini‐Gras et al., 2018) and findings for C. jejuni in Germany (Rosner et al., 2017). However, our attributions contrast with findings in Germany where pigs have been identified as a major source of C. coli campylobacteriosis. Our attribution proportion estimates for ruminants were sensitive to model assumptions (e.g., inclusion/exclusion of an “unsampled source”), with up to one quarter of C. coli and one‐half of C. jejuni infections attributable to ruminants in some models, but less than 5% of infections in other models. However, the model with best fit to data (M1) and the two models with similar fit (M2 and M7) all attributed <12% to ruminants. We did not find inclusion of case covariates such as rurality, age, sex, or jurisdiction substantially improved model predictions. However, point estimates of attribution to chicken meat were higher in urban than rural populations, in agreement with studies in New Zealand (Liao et al., 2019).
The number of isolates from ruminants and pigs was low despite extensive sampling, as the prevalence of Campylobacter was lower than anticipated. Observed ST diversity in each source indicated sampling was far from reaching saturation for pigs and ruminants. Up to one third of cases were attributed to an “unsampled source” when this was included in models. However, in the model with the best fit, only 14% of cases were attributed to the unsampled source. Models without an unsampled source attributed more cases to all three sampled sources, but primarily ruminants and pigs. Attribution to the unsampled source may genuinely indicate one or more unidentified sources (e.g., companion animals, environmental sources including from wildlife, water, overseas acquisition), but may also be driven by uncertainty introduced by low isolate numbers from ruminants and pigs and the high ST diversity.
Although all models identified chickens as the primary source of campylobacteriosis, there were substantial differences in attribution dependent on model assumptions. The most influential assumption concerned the relative transmission potential of sequence types. In our model, sequence type transmission potential captured any ST organism‐intrinsic differences affecting the risk of developing a (notified) infection following exposure to a contaminated food source, for example, ST‐specific differences in abundance on contaminated food, survival, or virulence. Our findings indicated transmission potential varied between STs. Models that made the strong assumption that all types had equal transmission potential had attribution estimates with narrower credible intervals, but with substantially poorer fit to the data. Unlike the Dirichlet model, the asymmetric island model incorporates locus‐level information about STs relatedness by considering shared alleles. However, including differences in type transmission potential in the asymmetric island model resulted in biologically implausible estimates of transmission potential for STs that were not detected in sources. In our models, ST48 was estimated to have high transmission potential, in agreement with a study in New Zealand (Miller et al., 2017). Given emerging Campylobacter virulence factor research (e.g., Bolton, 2015; Zhang et al., 2016), it may prove valuable to investigate whether these factors are associated with ST transmission potential in source attribution studies. If estimates of type transmission potential are consistent over time and across studies, transmission potential could be used to differentiate STs of substantial concern (e.g., ST48) or limited concern (e.g., ST827 and ST832).
Models that allowed for differences in transmission potential between STs suggested C. coli STs generally had lower transmission potentials than C. jejuni STs, possibly explaining why C. coli predominated in chickens and yet C. jejuni predominated in human cases. Salmonellosis source attribution models have often accounted for variable serotype transmission potential (e.g., Hald et al., 2004; Miller et al., 2017; Mughini‐Gras et al., 2014; Mullner, Jones, et al., 2009). Our findings are consistent with our previous study that found that assuming Salmonella serotypes had equal transmission potential distorted attribution estimates, particularly underestimating attribution to sources harboring a mix of serotypes with low and high transmission potential, namely, broiler chickens (McLure et al., 2022).
Many source attribution studies rely on secondary analysis of multiple datasets, often adopting different sampling approaches for different food animals, including targeted sampling (e.g., outbreak investigations) and sampling at different stages of the food production process (e.g., farm, processor, or retail). In contrast, our study applied a common sampling approach, sampling specific animal sources, all from retail meats over the course of two years. This included samples from the four major food animals consumed in Australia (chickens, pigs, cattle, and sheep). We included cases from every Australian state and two most populous territories (the ACT and NT). We collected food isolates from four jurisdictions including Australia's three most populous states (NSW, Qld, and Vic). We did not find any differences in the human cases between jurisdiction in this or previous studies (Wallace et al., 2021) and therefore believe our estimates of source attribution are generalizable to the general Australian population. By adopting a model comparison approach, we were able to identify findings consistent across models and assumptions, including high attribution to chicken.
Our study has some limitations. Given the generally low prevalence of Campylobacter on muscle meat from pigs and ruminants, we chose to sample offal meats, which have previously been found to have much higher prevalence of Campylobacter contamination (New South Wales Food Authority, 2018). If the STs present on muscle meat and offal are substantially different, our samples may not have been representative of the STs encountered when consuming meat from ruminants and pigs. However, the STs in muscle and offal meats from chicken were similar (Wallace et al., 2020) supporting the validity of our sampling approach. Despite extensive sampling efforts, the number of isolates from pigs and ruminants limited the statistical power of our study (Walker et al., 2019; Wallace et al., 2020), particularly with respect to detection sensitivity of differences in attribution by state, rurality, or other covariates. There is a general lack of theoretical work to inform questions of power and minimum sample sizes at the design phase of source attribution studies (Smid et al., 2013). While Campylobacter has been isolated from a wide range of wild and domestic animal species, this study only included samples from the primary retail meat production animals in Australia. However, 62%–89% of cases in Australia are believed to be foodborne (Vally et al., 2014) primarily from meat, in concordance with our best fitting models that attributed 86% to chickens, pigs, and ruminants, and 14% to an unsampled source. Attribution to unsampled source must be interpreted with caution, as it could represent genuine unsampled reservoir(s) (e.g., wildlife and/or companion animals, water) but might also be an artifact of limited sample sizes. In particular, the large attribution proportions for the unsampled sources predicted by the model with weak priors on the relative abundance of sequence types in sources (M3) should be interpreted as an uncertain attribution rather than strong evidence of one or more major unsampled sources. Some of the attribution to an unsampled source may be due to travel associated cases, though this was unlikely to be a major factor in our study, as about 97% of Australian campylobacteriosis cases are believed to be domestically acquired (Kirk et al., 2014) and 68% of the cases in our study were from a case–control study that specifically excluded travel‐associated cases. As cases were recruited from notifiable disease databases, our study only included cases that sought medical care and had a stool culture, presumably biasing our sample toward moderate‐to‐severe cases. Though not representative, these cases are the priority for disease prevention. The sampling periods cases and food were only partially overlapping (February 2018 to October 2019 vs. March 2017 to March 2019). However, this was unlikely to have biased our analyses as the relative frequency of STs in sources and cases were unlikely to have changed substantially over the sampling period and the multi‐year sampling periods allowed us to average over any seasonal differences. Application of the asymmetric island model to attribute relative abundance of STs in sources performed worse than anticipated, leading to a poor model fit when all STs were assumed to have equal transmission potential (the standard assumption when using the asymmetric island model), but resulting in biologically implausible estimates of transmission potential when this assumption was relaxed. While the asymmetric island model in theory could have incorporated more of the genomic data available (e.g., core genome MLST or single nucleotide polymorphisms), we anticipate that increased granularity in typing would only exacerbate the problems with estimating transmission potential. Similarly, the Dirichlet model could not be used on the full genomic data as it performs poorly when most isolates are assigned a unique type. Further work is required to better account for differences in type transmission potential when using the asymmetric island model, for example, improving capabilities for fully joint inference and modeling correlations in transmission potential between related types.
Our best fitting model estimated that chickens account for about 80% of campylobacteriosis and that even after adjusting for the relatively high rates of chicken meat consumption chicken poses a risk of campylobacteriosis approximately 5–30 times higher than ruminants and 7–72 times higher than pigs. Chickens should therefore remain the priority target for reducing burden of Campylobacteriosis in Australia, including increased promotion of safe food handling practices for all raw meats and reduction of meat contamination during production and processing.
AUTHOR CONTRIBUTIONS
Conceptualization: Martyn D. Kirk, Kathryn Glass, and Nigel French. Methodology: Kathryn Glass and Angus McLure. Software: Angus McLure. Formal analysis: Angus McLure and Dieter Bulach. Investigation: Cameron R.M. Moffatt, Mary Valcanis, Amy Jennison, and James J. Smith. Resources: Amy Jennison, Mary Valcanis, and Dieter Bulach. Data curation: Rhiannon Wallace, Dieter Bulach, and Danielle M. Cribb. Visualization: Amy Jennison. Supervision: Kathryn Glass. Project administration: Danielle M. Cribb. Funding acquisition: Kathryn Glass, Martyn D. Kirk, Nigel French, Mary Valcanis, Dieter Bulach, and Emily Fearnley. Amy Jennison prepared the original draft and all authors reviewed and edited the manuscript. The final version of the manuscript was approved by all authors.
ACKNOWLEDGMENTS
This research was part of the CampySource Project which was funded jointly by a National Health and Medical Research Council grant (NHMRC GNT1116294) and AgriFutures, Australian Government Department of Health, Food Standards Australia New Zealand, New South Wales Food Authority, Queensland Health, and ACT Health. Martyn D. Kirk was supported by a National Health and Medical Research Council fellowship (GNT1145997). Amy Jennison was supported through an Australian Research Council Discovery Project Grant (DP180100246). Danielle M. Cribb was supported by an Australian Government Research Training Program (AGRTP) scholarship.
The authors would like to thank Jonathan Marshall for valuable conversations about the asymmetric island model and for making changes to the islandR package to accommodate equal mutation and recombination rates across sources.
The authors would like to thank the extended CampySource Project team, reference panel, and additional contributors to the study. The CampySource Project Team comprised three working groups and a reference panel. The working groups focused on food and animal sampling, epidemiology and modeling, and genomics. The reference panel included expert representatives from government and industry.
The study includes the following partner organizations: the Australian National University, Massey University, University of Melbourne, Queensland Health, Queensland Health Forensic and Scientific Services, New South Wales Food Authority, New South Wales Health, Hunter New England Health, Victorian Department of Health and Human Services, Food Standards Australia New Zealand, Commonwealth Department of Health, and AgriFutures Australia–Chicken Meat Program. CampySource also collaborated with the following organizations: ACT Health, Sullivan Nicolaides Pathology, University of Queensland, Primary Industries and Regions South Australia, Department of Health and Human Services Tasmania, Meat and Livestock Australia, and New Zealand Ministry for Primary Industries. We would particularly like to acknowledge the public health and environmental health officers from our partner and collaborating organizations who carried out the project sampling.
The CampySource Project Team included: Nigel P French, Massey University, New Zealand; Mary Valcanis, The University of Melbourne; Dieter Bulach, The University of Melbourne; Emily Fearnley, South Australian Department for Health and Wellbeing; Russell Stafford, Queensland Health; Amy Jennison, Queensland Health; Trudy Graham, Queensland Health; Keira Glasgow, Health Protection NSW; Kirsty Hope, Health Protection NSW; Themy Saputra, NSW Food Authority; Craig Shadbolt, NSW Food Authority; Arie Havelaar, The University of Florida, USA; Joy Gregory, Department of Health and Human Services, Victoria; James Flint, Hunter New England Health; Simon Firestone, The University of Melbourne; James Conlan, Food Standards Australia New Zealand; Ben Daughtry, Food Standards Australia New Zealand; James J. Smith, Queensland Health; Heather Haines, Department of Health and Human Services, Victoria; Sally Symes, Department of Health and Human Services, Victoria; Barbara Butow, Food Standards Australia New Zealand; Liana Varrone, The University of Queensland; Linda Selvey, The University of Queensland; Tim Sloan‐Gardner, ACT Health; Deborah Denehy, ACT Health; Radomir Krsteski, ACT Health; Natasha Waters, ACT Health; Kim Lilly, Hunter New England Health; Julie Collins, Hunter New England Health; Tony Merritt, Hunter New England Health; Rod Givney, Hunter New England Health; Joanne Barfield, Hunter New England Health; Ben Howden, The University of Melbourne; Kylie Hewson, AgriFutures Australia–Chicken Meat Program; Dani Cribb, The Australian National University; Rhiannon Wallace, The Australian National University; Angus McLure, The Australian National University; Ben Polkinghorne, The Australian National University; Cameron Moffatt, The Australian National University and Queensland Health; Martyn Kirk, The Australian National University; and Kathryn Glass, The Australian National University.
Open access publishing facilitated by Australian National University, as part of the Wiley ‐ Australian National University agreement via the Council of Australian University Librarians.
1.
TABLE A1.
Number of distinct C. coli multi‐locus sequence types (STs) and isolates from humans and food sources
| Origin | STs | Isolates | Uniquea STs (%) | Isolates from Uniquea STs (%) | STs without cases (%) | Isolates from STs without cases (%) |
|---|---|---|---|---|---|---|
| Human | 31 | 125 | 18 (58) | 19 (15) | N/A | N/A |
| Chicken | 40 | 283 | 29 (73) | 101 (36) | 28 (70) | 73 (26) |
| Pig | 23 | 28 | 15 (65) | 18 (64) | 19 (83) | 23 (82) |
| Ruminant | 7 | 16 | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
Note: Percentages, where given, denote of the fraction of all C. coli STs/isolates with the same source (humans, chicken, pig, or ruminant).
Unique types for sources are types that were found in that source and no other source (but potentially in humans), while unique types for humans are those found in cases but not in any of the three sources.
TABLE A2.
Number of distinct C. jejuni multi‐locus sequence types (STs) and isolates from humans and food sources
| Origin | STs | Isolates | Unique* STs (%) | Isolates from Unique* STs (%) | STs without cases (%) | Isolates from STs without cases (%) |
|---|---|---|---|---|---|---|
| Human | 87 | 585 | 48 (55) | 115 (20) | N/A | N/A |
| Chicken | 48 | 197 | 31 (65) | 91 (46) | 10 (21) | 17 (8.6) |
| Pig | 10 | 16 | 2 (20) | 2 (13) | 2 (20) | 2 (13) |
| Ruminant | 18 | 72 | 3 (17) | 3 (4.2) | 2 (11) | 2 (2.8) |
Note: Percentages, where given, denote of the fraction of all C. jejuni STs/isolates with the same source (humans, chicken, pig, or ruminant).
Unique types for sources are types that were found in that source and no other source (but potentially in humans), while unique types for humans are those found in cases but not in any of the three sources.
TABLE A3.
Comparison of observed and estimated relative abundance (%) of two multi‐locus sequence types (STs) in the three sources and “unsampled source” (where relevant). ST50 and ST48 were the first and fourth most common STs in humans in our study. The relative abundance is the percentage of all Campylobacter isolates from a given source that belong to the indicated sequence type
| Model a | Sequence type 50 | Sequence type 48 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ruminant | Pig | Chicken | Unsampled | Ruminant | Pig | Chicken | Unsampled | |
| Observed | 24 | 11 | 5.2 | NA | 1.1 | 0.0 | 0.8 | NA |
| (21/88) | (5/44) | (25/480) | (0/0) | (1/88) | (0/44) | (4/480) | (0/0) | |
| M1 | 8.4 | 2.8 | 4.1 | 0.6 | 0.8 | 0.5 | 1.0 | 0.7 |
| (5.4–12.0) | (1.1–5.4) | (2.8–5.8) | (1.8 × 10−2 − 2.2) | (0.1–2.2) | (1.1 × 10−02 − 1.7) | (0.4–1.8) | (1.8 × 10−2 − 2.5) | |
| M2 | 8.4 | 2.8 | 4.1 | NA | 0.8 | 0.5 | 1.0 | NA |
| (5.3–12.2) | (1.0–5.1) | (2.8–5.8) | (0.1–2.2) | (1.2 × 10−2 − 1.8) | (0.4–1.8) | |||
| M3 | 20 | 8.6 | 5.2 | 0.8 | 1.4 | 0.2 | 1.0 | 2.8 |
| (13–28) | (2.9–17) | (3.3–7.3) | (2.3 × 10−16 − 8.3) | (0.1–4.5) | (6.9 × 10−17 − 1.8) | (0.3–2.0) | (7.1 × 10−14 − 13.9) | |
| M4 | 19.3 | 8.5 | 5.1 | NA | 1.5 | 0.2 | 0.9 | NA |
| (13–27) | (2.8–17) | (3.4–7.2) | (0.1–4.5) | (2.0 × 10−16 − 2.0) | (0.3–2.0) | |||
| M5 and M8 | 8.4 | 2.7 | 4.0 | 0.6 | 0.8 | 0.5 | 0.8 | 0.6 |
| (5.3–12) | (1.0–5.3) | (2.6–5.6) | (1.5 × 10−2 − 2.1) | (0.1–2.1) | (1.2 × 10−2 − 1.7) | (0.2–1.6) | (1.5 × 10−2 − 2.1) | |
| M6 and M9 | 8.4 | 2.7 | 4.0 | NA | 0.8 | 0.5 | 0.8 | NA |
| (5.3–12) | (1.0–5.3) | (2.6–5.6) | (0.1–2.1) | (1.2 × 10−2 − 1.7) | (0.2–1.6) | |||
| M7 and M10 | 16.6 | 8.8 | 4.7 | NA | 0.8 | 0.3 | 0.7 | NA |
| (15–18) | (7.3–11) | (4.4–5.1) | (0.8–0.9) | (0.2–0.4) | (0.6–0.7) | |||
Note: Observed values are the percentage of isolates of the given sequence type with numbers given in parentheses. The point estimates for the models are the posterior mean with 95% credible intervals.
The pairs of models M5 and M8, M6 and M9, and M7 and M10 use the same model for relative abundance of sequence types in sources. See Table 1 for a full description of the models.
TABLE A4.
Comparison of observed and estimated relative abundance (%) of multi‐locus sequence types (STs) in the three sources and “unsampled source” (where relevant). The relative abundance is the percentage of all Campylobacter isolates from a given source that belong to the indicated sequence type
| Model a | Sequence type 827 | Sequence type 2398 | ||||||
|---|---|---|---|---|---|---|---|---|
| Ruminant | Pig | Chicken | Unsampled | Ruminant | Pig | Chicken | Unsampled | |
| Observed | 9.1 | 4.5 | 9.6 | NA | 0.0 | 0.0 | 0.0 | NA |
| (8/88) | (2/44) | (46/480) | (0/0) | (0/88) | (0/44) | (0/480) | (0/0) | |
| M1 | 3.4 | 1.4 | 7.1 | 0.6 | 0.4 | 0.5 | 0.3 | 0.9 |
| (1.5–6.0) | (0.3–3.2) | (5.2–9.2) | (1.6 × 10−2 − 2.0) | (1.2 × 10−2 − 1.6) | (1.3 × 10−2 − 1.9) | (1.4 × 10−2 − 0.9) | (3.0 × 10−2 − 2.9) | |
| M2 | 3.4 | 1.4 | 7.1 | NA | 0.5 | 0.6 | 0.3 | NA |
| (1.6–5.8) | (0.3–3.3) | (5.2–9.3) | (1.5 × 10−2 − 1.7) | (1.7 × 10−2 − 2.0) | (2.7 × 10−2 − 0.9) | |||
| M3 | 7.1 | 3.5 | 9.2 | 0.5 | 0.2 | 0.2 | 2.9 × 10−2 | 5.0 |
| (3.2–12.3) | (0.5–9.4) | (6.8–12.0) | (1.0 × 10−16 − 4.3) | (1.3 × 10−16 − 1.9) | (1.4 × 10−16 − 2.4) | (1.3 × 10−16 − 0.3) | (3.9 × 10−6 − 15.3) | |
| M4 | 7.0 | 3.4 | 9.2 | NA | 0.9 | 0.7 | 0.1 | NA |
| (3.2–12.2) | (0.4–9.2) | (6.9–11.9) | (1.3 × 10−13 − 3.2) | (1.1 × 10−15 − 4.9) | (4.6 × 10−17 − 0.5) | |||
| M5 & M8 | 3.4 | 1.4 | 7.2 | 0.6 | 0.4 | 0.5 | 0.2 | 0.6 |
| (1.6–5.9) | (0.3–3.3) | (5.3–9.3) | (1.5 × 10−2 − 2.1) | (9.7 × 10−3 − 1.4) | (1.2 × 10−2 − 1.7) | (3.9 × 10−3 − 0.6) | (1.5 × 10−2 − 2.1) | |
| M6 and M9 | 3.4 | 1.4 | 7.2 | NA | 0.4 | 0.5 | 0.2 | NA |
| (1.6–5.9) | (0.3–3.3) | (5.3–9.3) | (9.7 × 10−3 − 1.4) | (1.2 × 10−2 − 1.7) | (3.9 × 10−3 − 0.6) | |||
| M7 and M10 | 7.3 | 5.7 | 7.8 | NA | 2.1 × 10−11 | 5.7 × 10−12 | 1.5 × 10−11 | NA |
| (7.0–7.6) | (5.0–6.3) | (7.5–8.0) | (8.9 × 10−12 − 4.4 × 10−11) | (1.4 × 10−12 − 1.3 × 10−11) | (6.8 × 10−12 − 3.1 × 10−11) | |||
Note: Observed values are the percentage of isolates of the given sequence type with numbers given in parentheses. The point estimates for the models are the posterior mean with 95% credible intervals.
The pairs of models M5 and M8, M6 and M9, and M7 and M10 use the same model for relative abundance of sequence types in sources. See Table 1 for a full description of the models.
TABLE A5.
Predictive performance of 10 source attribution models, measured with Watanabe–Akaike information criterion (WAIC) for the prediction of the relative abundance of sequence types in cases (all models: M1–M10) or cases and sources (jointly estimated models only: M1–M4)
| Cases | Cases and sources | |||||
|---|---|---|---|---|---|---|
| Model | WAIC | WAIC difference | Effective # parameters | WAIC | WAIC difference | Effective # parameters |
| M1 | 5819.2 (76.4) | 0.0 (0.0) | 102.8 (5.6) | 10,785.4 (95.8) | 112.7 (24.6) | 209.6 (7.5) |
| M2 | 5822.2 (76.8) | 3.0 (1.1) | 103.5 (5.7) | 10,792.0 (96.3) | 119.4 (24.1) | 210.1 (7.6) |
| M3 | 5870.4 (82.5) | 51.1 (9.6) | 131.2 (9.0) | 10,672.7 (112.1) | 0.0 (0.0) | 312.7 (14.5) |
| M4 | 5881.2 (84.6) | 62.0 (12.1) | 135.5 (9.4) | 10,708.8 (114.1) | 36.2 (5.1) | 318.7 (14.9) |
| M5 | 5828.4 (77.5) | 9.2 (1.6) | 109.8 (6.2) | — | — | — |
| M6 | 5831.1 (78.0) | 11.9 (2.2) | 110.9 (6.3) | — | — | — |
| M7 | 5896.1 (89.5) | 76.8 (92.4) | 172.8 (13.3) | — | — | — |
| M8 | 6690.1 (60.4) | 870.8 (51.5) | 108.4 (3.3) | — | — | — |
| M9 | 6721.2 (63.6) | 902.0 (52.6) | 116.3 (4.1) | — | — | — |
| M10 | 9950.8 (382.8) | 4131.5 (361.1) | 17.5 (2.0) | — | — | — |
Note: Standard errors are given in parentheses. WAIC is smaller in models with better fit. WAIC difference is calculated with respect to the best model (M1 for predictions in cases, M3 for predictions in cases and sources). However, we only consider differences substantive when greater than five times the standard error for the difference, for example, when considering predictions in cases, M1 is substantively better than all models except M2 and M7.
TABLE A6.
Estimates and 95% credible intervals for selected parameters related to type transmission potential in the seven models (M1–M7) that allowed for transmission potential variability between sequence types. See Table 1 for further details of models
| Model a | σ b | q 2398 | q 48 | q 50 | q 827 |
|---|---|---|---|---|---|
| M1 | 1.2 (1.0–1.5) | 18 (4.6–85) | 15 (4.7–48) | 12 (5.2–29) | 0.6 (0.3–1.5) |
| M2 | 1.2 (1.0–1.5) | 22 (5.6–110) | 15 (5.6–49) | 11 (4.7–25) | 0.6 (0.3–1.2) |
| M3 | 1.0 (0.6–1.3) | 4.8 (0.8–56) | 9.2 (1.2–43) | 6.0 (2.3–16) | 0.5 (0.2–1.0) |
| M4 | 1.2 (0.9–1.6) | 23 (4.1–210) | 16 (4.2–66) | 5.0 (2.1–13) | 0.3 (0.1–0.6) |
| M5 | 1.4 (1.2–1.7) | 40 (7.9–270) | 19 (6.2–70) | 10 (4.5–25) | 0.7 (0.3–1.8) |
| M6 | 1.4 (1.2–1.7) | 34 (6.6–170) | 17 (5.6–58) | 12 (5.4–29) | 0.7 (0.3–1.4) |
| M7 | 12 (10–13) | 7.6 × 1011 (2.2 × 1011 − 2.5 × 1012) | 28 (14–61) | 12 (6.4–25) | 3.4 × 10−2 (4.2 × 10−2 − 0.4) |
Models M8–M10 assumed all types had equal transmission potential. See Table 1 for a full description of models M1–M7.
The standard deviation (on the log scale) of the log‐normal distribution of type transmission potential parameters q.
FIGURE A1.
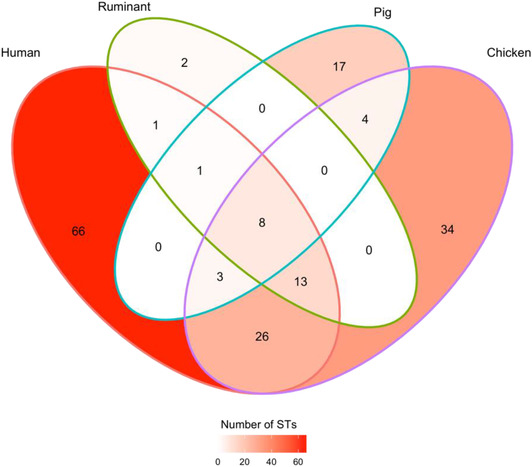
Venn diagram summarizing the number of multi‐locus sequence types (STs) found in cases and sources, or combinations of cases and sources. For instance, 66 STs were found only in cases, 26 STs were found in chicken and cases but not in pigs or ruminants, and 8 STs (representing 32% [423/1322] of study isolates) were found in cases and all sources.
FIGURE A2.
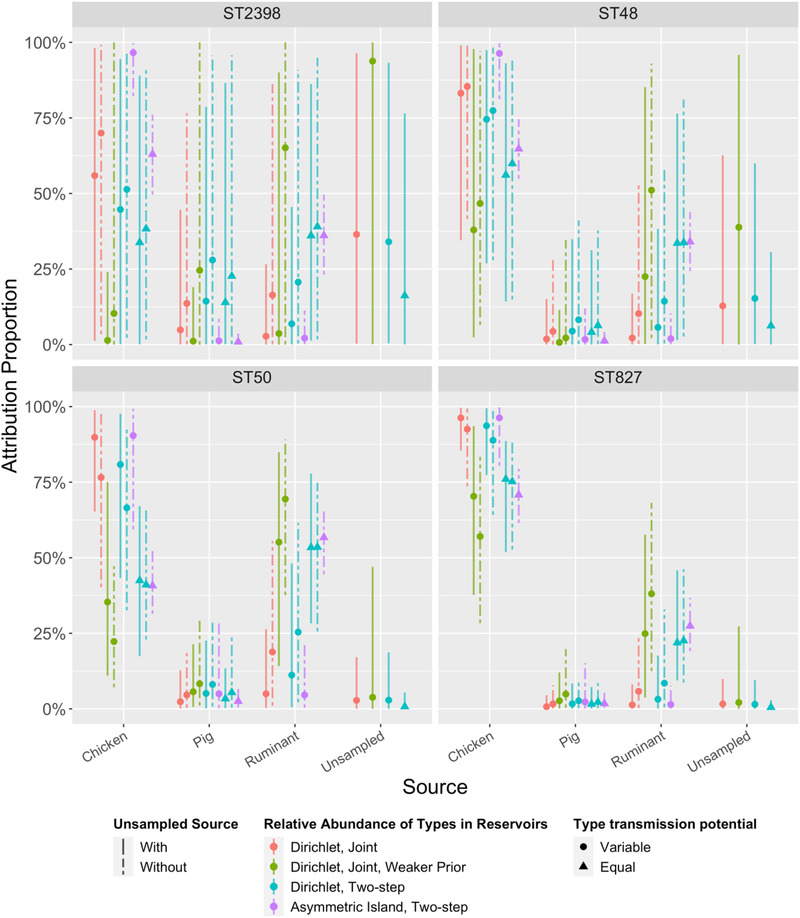
Source attribution proportions of four selected multi‐locus sequence types (STs) to three sampled sources in 10 models (M1–M10, left to right). Four models (M1, M3, M5, and M8) also include a fourth, “unsampled source.” See Table 1 for more details about the models. ST50 (C. jejuni) was the most observed type in cases, ruminants, and pigs and the fifth most common type in chickens. ST48 (C. jejuni) was fourth most common type in humans, but rare or absent in all sources. ST827 (C. coli) was the second most common type in chickens, and found in the other sources, but relatively uncommon in cases. ST2398 (C. jejuni) was more common in cases than ST827, but not detected in any sources. Estimates of relative abundance of these types in the respective sources can be found in Tables A3 and A4. Estimates of the transmission potential of the four types can be found in Table A6.
FIGURE A3.
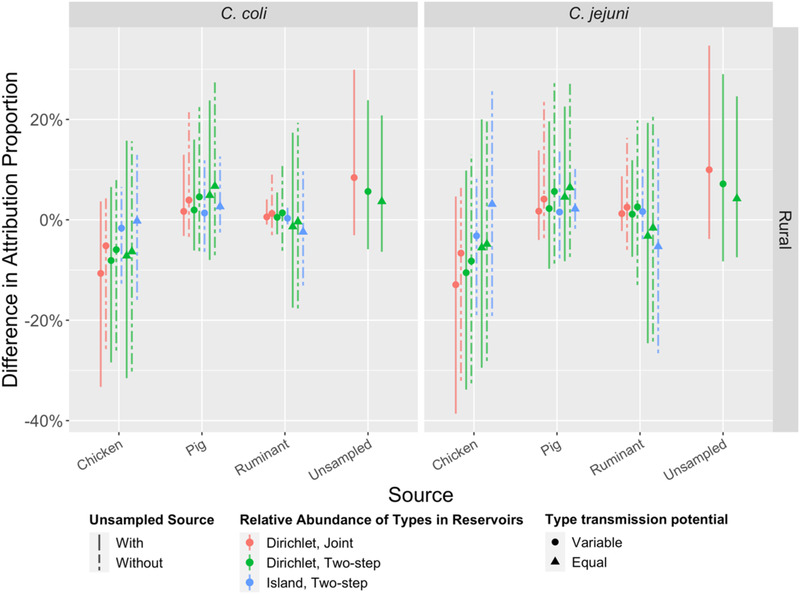
Percent difference in source attribution proportions between urban and rural populations (urban cases as references) for eight models (M1, M2, M5–M10). See Table 1 for model details. Vertical bars are 95% credible intervals (CrI). Note that all CrIs include 0% (no difference).
FIGURE A4.
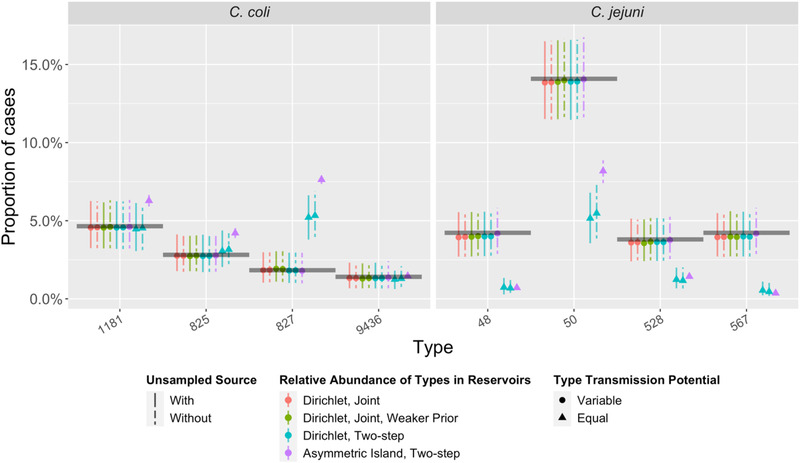
The observed proportion of cases due to the five most common multi‐locus sequence types (STs) from C. coli and C. jejuni (black horizontal lines) compared to predictions (colored points and vertical 95% credible intervals) under 10 source attribution models (M1–M10, left to right). Note that for some STs, the credible intervals for M10 (asymmetric island model) are so narrow that they are not visible.
FIGURE A5.
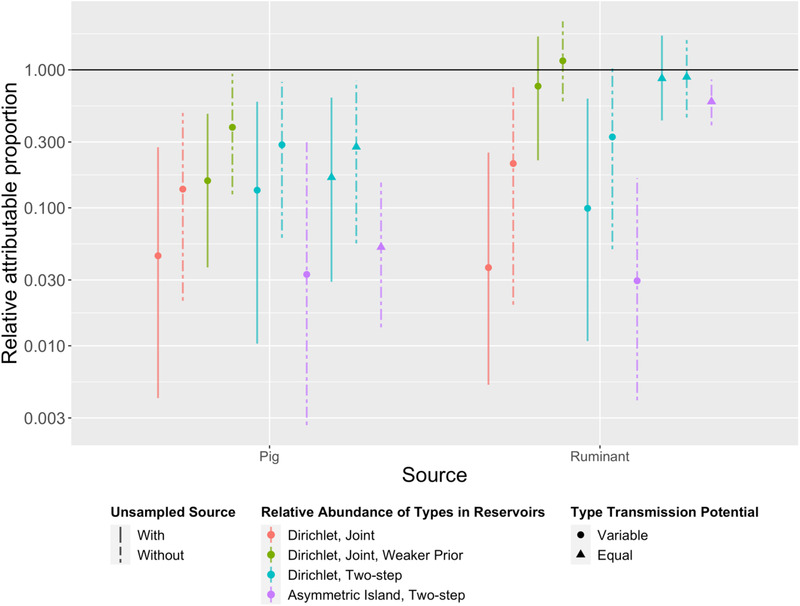
Estimates of relative attributable proportion (RAP) of campylobacteriosis under 10 source attribution models (M1–M10, left to right). RAP was calculated by dividing attribution proportion by the annual Australian consumption of meat products derived from that source and normalized against a reference source (chicken). All models indicated pig meat poses less risk (lower RAP) than chicken. Note the y‐axis is on a log scale, and that the “unsampled source” is omitted due to lack of respective consumption statistics or appropriate equivalent exposure measure.
McLure, A. , Smith, J. J. , Firestone, S. M. , Kirk, M. D. , French, N. , Fearnley, E. , Wallace, R. , Valcanis, M. , Bulach, D. , Moffatt, C. R. M. , Selvey, L. A. , Jennison, A. , Cribb, D. M. , & Glass, K. (2023). Source attribution of campylobacteriosis in Australia, 2017–2019. Risk Analysis, 43, 2527–2548. 10.1111/risa.14138
DATA AVAILABILITY STATEMENT
The sequence readset for each food isolate (Bioproject Accession: PRJNA591966), case isolates sampled as part of the case–control study (Bioproject Accession: PRJNA592186) and the case isolates included in the national snapshot (Bioproject Accession: PRJNA560409) are available through GenBank.
REFERENCES
- Australian Bureau of Agricultural and Resource Economics and Sciences . (2020). Agricultural commodity statistics. https://www.agriculture.gov.au/abares/research‐topics/agricultural‐outlook/data
- Bolton, D. J. (2015). Campylobacter virulence and survival factors. Food Microbiology, 48, 99–108. [DOI] [PubMed] [Google Scholar]
- Boysen, L. , Rosenquist, H. , Larsson, J. T. , Nielsen, E. M. , Sørensen, G. , Nordentoft, S. , & Hald, T. (2014). Source attribution of human campylobacteriosis in Denmark. Epidemiology and Infection, 142(8), 1599–1608. 10.1017/s0950268813002719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, K. , Karsch‐Mizrachi, I. , Lipman, D. J. , Ostell, J. , & Sayers, E. W. (2016). GenBank. Nucleic Acids Research, 44(D1), D67–D72. 10.1093/nar/gkv1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribb, D. M. , Varrone, L. , Wallace, R. L. , Mclure, A. T. , Smith, J. J. , Stafford, R. J. , Bulach, D. M. , Selvey, L. A. , Firestone, S. M. , French, N. P. , Valcanis, M. , Fearnley, E. J. , Sloan‐Gardner, T. , Graham, T. , Glass, K. , & Kirk, M. D. (2022). Risk factors for campylobacteriosis in Australia: Outcomes of a 2018–2019 case‐control study. 10.21203/rs.3.rs-1387051/v1 [DOI] [PMC free article] [PubMed]
- Dingle, K. E. , Colles, F. M. , Wareing, D. R. A. , Ure, R. , Fox, A. J. , Bolton, F. E. , Bootsma, H. J. , Willems, R. J. L. , Urwin, R. , & Maiden, M. C. J. (2001). Multilocus sequence typing system for Campylobacter jejuni . Journal of Clinical Microbiology, 39(1), 14–23. 10.1128/jcm.39.1.14-23.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Euzéby, J. P. (2021). List of prokaryotic names with standing in nomenclature: Genus Campylobacter list of prokaryotic names with standing in nomenclature. https://lpsn.dsmz.de/genus/campylobacter
- Gao, C.‐H. (2022). ggVennDiagram: A ‘ggplot2’ Implement of Venn Diagram. https://github.com/gaospecial/ggVennDiagram
- Hald, T. , Vose, D. , Wegener, H. C. , & Koupeev, T. (2004). A Bayesian approach to quantify the contribution of animal‐food sources to human salmonellosis. Risk Analysis, 24(1), 255–269. 10.1111/j.0272-4332.2004.00427.x [DOI] [PubMed] [Google Scholar]
- International Organization for Standardization . (2017). Microbiology of the food chain—Horizontal method for detection and enumeration of Campylobacter spp.—Part 1: Detection method. https://www.iso.org/standard/63225.html
- Jolley, K. A. , Bray, J. E. , & Maiden, M. C. J. (2018). Open‐access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Research, 3, 124. 10.12688/wellcomeopenres.14826.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaakoush, N. O. , Castaño‐Rodríguez, N. , Mitchell, H. M. , & Man, S. M. (2015). Global epidemiology of campylobacter infection. Clinical Microbiology Reviews, 28(3), 687–720. 10.1128/cmr.00006-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, M. D. , Ford, L. , Glass, K. , & Hall, G. (2014). Foodborne illness, Australia, circa 2000 and circa 2010. Emerging Infectious Diseases, 20(11), 1857–1864. 10.3201/eid2011.131315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk, M. D. , Pires, S. M. , Black, R. E. , Caipo, M. , Crump, J. A. , Devleesschauwer, B. , Döpfer, D. , Fazil, A. , Fischer‐Walker, C. L. , Hald, T. , Hall, A. J. , Keddy, K. H. , Lake, R. J. , Lanata, C. F. , Torgerson, P. R. , Havelaar, A. H. , & Angulo, F. J. (2015). World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: A data synthesis. PLoS Medicine, 12(12), e1001921. 10.1371/journal.pmed.1001921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kittl, S. , Heckel, G. , Korczak, B. M. , & Kuhnert, P. (2013). Source attribution of human Campylobacter isolates by MLST and Fla‐typing and association of genotypes with quinolone resistance. PLoS One, 8(11), e81796. 10.1371/journal.pone.0081796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, K. , Hvass, A. , Christiansen, A. , Ethelberg, S. , & Cowan, S. (2021). Sexual contact as risk factor for Campylobacter infection, Denmark. Emerging Infectious Diseases, 27(4), 1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake, R. J. , Campbell, D. M. , Hathaway, S. C. , Ashmore, E. , Cressey, P. J. , Horn, B. J. , Pirikahu, S. , Sherwood, J. M. , Baker, M. G. , Shoemack, P. , Benschop, J. , Marshall, J. C. , Midwinter, A. C. , Wilkinson, D. A. , & French, N. P. (2021). Source attributed case‐control study of campylobacteriosis in New Zealand. International Journal of Infectious Diseases, 103, 268–277. 10.1016/j.ijid.2020.11.167 [DOI] [PubMed] [Google Scholar]
- Li, M. , Havelaar, A. H. , Hoffmann, S. , Hald, T. , Kirk, M. D. , Torgerson, P. R. , & Devleesschauwer, B. (2019). Global disease burden of pathogens in animal source foods, 2010. PLoS One, 14(6), e0216545. 10.1371/journal.pone.0216545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, S.‐J. , Marshall, J. , Hazelton, M. L. , & French, N. P. (2019). Extending statistical models for source attribution of zoonotic diseases: A study of campylobacteriosis. Journal of The Royal Society Interface, 16(150), 20180534. 10.1098/rsif.2018.0534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLure, A. , Shadbolt, C. , Desmarchelier, P. M. , Kirk, M. D. , & Glass, K. (2022). Source attribution of salmonellosis by time and geography in New South Wales, Australia. BMC Infectious Diseases, 22(1), 4. 10.1186/s12879-021-06950-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller, P. , Marshall, J. , French, N. , & Jewell, C. (2017). sourceR: Classification and source attribution of infectious agents among heterogeneous populations. PLoS Computational Biology, 13(5), e1005564. 10.1371/journal.pcbi.1005564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffatt, C. R. M. , Fearnley, E. , Bell, R. , Wright, R. , Gregory, J. , Sloan‐Gardner, T. , Kirk, M. , & Stafford, R. (2020). Characteristics of Campylobacter gastroenteritis outbreaks in Australia, 2001 to 2016. Foodborne Pathogens and Disease, 17(5), 308–315. 10.1089/fpd.2019.2731 [DOI] [PubMed] [Google Scholar]
- Mughini‐Gras, L. , Barrucci, F. , Smid, J. H. , Graziani, C. , Luzzi, I. , Ricci, A. , Barco, L. , Rosmini, R. , Havelaar, A. H. , Van Pelt, W. , & Busani, L. (2014). Attribution of human Salmonella infections to animal and food sources in Italy (2002–2010): Adaptations of the Dutch and modified Hald source attribution models. Epidemiology and Infection, 142(5), 1070–1082. 10.1017/s0950268813001829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mughini‐Gras, L. , Kooh, P. , Augustin, J.‐C. , David, J. , Fravalo, P. , Guillier, L. , Jourdan‐Da‐Silva, N. , Thébault, A. , Sanaa, M. , & Watier, L. (2018). Source attribution of foodborne diseases: Potentialities, hurdles, and future expectations. Frontiers in Microbiology, 9, 1983. 10.3389/fmicb.2018.01983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullner, P. , Jones, G. , Noble, A. , Spencer, S. E. F. , Hathaway, S. , & French, N. P. (2009). Source attribution of food‐borne zoonoses in New Zealand: A modified Hald model. Risk Analysis, 29(7), 970–984. 10.1111/j.1539-6924.2009.01224.x [DOI] [PubMed] [Google Scholar]
- Mullner, P. , Spencer, S. E. F. , Wilson, D. J. , Jones, G. , Noble, A. D. , Midwinter, A. C. , Collins‐Emerson, J. M. , Carter, P. , Hathaway, S. , & French, N. P. (2009). Assigning the source of human campylobacteriosis in New Zealand: A comparative genetic and epidemiological approach. Infection, Genetics and Evolution, 9(6), 1311–1319. 10.1016/j.meegid.2009.09.003 [DOI] [PubMed] [Google Scholar]
- New South Wales Food Authority . (2018). Campylobacter in meat and offal: Microbiological quality of beef, lamb and pork meat cuts and offal. https://www.foodauthority.nsw.gov.au/sites/default/files/_Documents/scienceandtechnical/campylobacter_in_meat_and_offal.pdf
- R Core Team . (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing. https://www.R‐project.org/
- Rosner, B. M. , Schielke, A. , Didelot, X. , Kops, F. , Breidenbach, J. , Willrich, N. , Gölz, G. , Alter, T. , Stingl, K. , Josenhans, C. , Suerbaum, S. , & Stark, K. (2017). A combined case‐control and molecular source attribution study of human Campylobacter infections in Germany, 2011–2014. Scientific Reports, 7(1), 5139. 10.1038/s41598-017-05227-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears, A. , Baker, M. G. , Wilson, N. , Marshall, J. , Muellner, P. , Campbell, D. M. , Lake, R. J. , & French, N. P. (2011). Marked campylobacteriosis decline after interventions aimed at poultry, New Zealand. Emerging Infectious Diseases, 17(6), 1007–1015. 10.3201/eid1706.101272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann, T. (n.d.). mlst . GitHub. https://github.com/tseemann/mlst
- Smid, J. H. , Mughini Gras, L. , De Boer, A. G. , French, N. P. , Havelaar, A. H. , Wagenaar, J. A. , & Van Pelt, W. (2013). Practicalities of using non‐local or non‐recent multilocus sequence typing data for source attribution in space and time of human campylobacteriosis. PLoS One, 8(2), e55029. 10.1371/journal.pone.0055029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stan Development Team . (2020a). RStan: The R interface to Stan. http://mc‐stan.org/
- Stan Development Team . (2020b). Stan modeling language users guide and reference manual, 2.25. https://mc‐stan.org
- Standards Australia . (2015). Food microbiology, Method6: Examination for specific organisms—Campylobacter (AS 5013.6:2015). https://www.saiglobal.com/PDFTemp/Previews/OSH/as/as5000/5000/5013.6‐2004.pdf
- Thépault, A. , Méric, G. , Rivoal, K. , Pascoe, B. , Mageiros, L. , Touzain, F. , Rose, V. , Béven, V. , Chemaly, M. , & Sheppard, S. K. (2017). Genome‐wide identification of host‐segregating epidemiological markers for source attribution in Campylobacter jejuni . Applied and Environmental Microbiology, 83(7), 03085–03016. 10.1128/aem.03085-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vally, H. , Glass, K. , Ford, L. , Hall, G. , Kirk, M. D. , Shadbolt, C. , Veitch, M. , Fullerton, K. E. , Musto, J. , & Becker, N. (2014). Proportion of illness acquired by foodborne transmission for nine enteric pathogens in Australia: An expert elicitation. Foodborne Pathogens and Disease, 11(9), 727–733. [DOI] [PubMed] [Google Scholar]
- Varrone, L. , Glass, K. , Stafford, R. J. , Kirk, M. D. , & Selvey, L. (2020). A meta‐analysis of case‐control studies examining sporadic campylobacteriosis in Australia and New Zealand from 1990 to 2016. Australian and New Zealand Journal of Public Health, 44(4), 313–319. 10.1111/1753-6405.12998 [DOI] [PubMed] [Google Scholar]
- Varrone, L. , Stafford, R. J. , Lilly, K. , Selvey, L. , Glass, K. , Ford, L. , Bulach, D. , & Kirk, M. D. (2018). Investigating locally relevant risk factors for Campylobacter infection in Australia: Protocol for a case–control study and genomic analysis. BMJ Open, 8(12), e026630. 10.1136/bmjopen-2018-026630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehtari, A. , Gelman, A. , & Gabry, J. (2017). Practical Bayesian model evaluation using leave‐one‐out cross‐validation and WAIC. Statistics and Computing, 27, 1413–1432. [Google Scholar]
- Vehtari, A. , Gelman, A. , Simpson, D. , Carpenter, B. , & Bürkner, P.‐C. (2021). Rank‐normalization, folding, and localization: An improved ‘R hat’ for assessing convergence of MCMC. Bayesian Analysis, 16(2), 667–718. 10.1214/20-BA1221 [DOI] [Google Scholar]
- Walker, L. J. , Wallace, R. L. , Smith, J. J. , Graham, T. , Saputra, T. , Symes, S. , Stylianopoulos, A. , Polkinghorne, B. G. , Kirk, M. D. , & Glass, K. (2019). Prevalence of Campylobacter coli and Campylobacter jejuni in retail chicken, beef, lamb, and pork products in three Australian states. Journal of Food Protection, 82(12), 2126–2134. 10.4315/0362-028x.jfp-19-146 [DOI] [PubMed] [Google Scholar]
- Wallace, R. L. , Bulach, D. , McLure, A. , Varrone, L. , Jennison, A. V. , Valcanis, M. , Smith, J. J. , Polkinghorne, B. G. , Glass, K. , & Kirk, M. D. (2021). Antimicrobial resistance of Campylobacter spp. causing human infection in Australia: An international comparison. Microbial Drug Resistance, 27, 518–528. 10.1089/mdr.2020.0082 [DOI] [PubMed] [Google Scholar]
- Wallace, R. L. , Bulach, D. M. , Jennison, A. V. , Valcanis, M. , Mclure, A. , Smith, J. J. , Graham, T. , Saputra, T. , Firestone, S. , Symes, S. , Waters, N. , Stylianopoulos, A. , Kirk, M. D. , & Glass, K. (2020). Molecular characterization of Campylobacter spp. recovered from beef, chicken, lamb and pork products at retail in Australia. PLoS One, 15(7), e0236889. 10.1371/journal.pone.0236889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe, S. , & Opper, M. (2010). Asymptotic equivalence of Bayes cross validation and widely applicable information criterion in singular learning theory. Journal of Machine Learning Research, 11(12), 3571–3594. [Google Scholar]
- Wickham, H. , Averick, M. , Bryan, J. , Chang, W. , McGowan, L. D. A. , François, R. , Grolemund, G. , Hayes, A. , Henry, L. , & Hester, J. (2019). Welcome to the Tidyverse. Journal of Open Source Software, 4(43), 1686. [Google Scholar]
- Wilson, D. J. , Gabriel, E. , Leatherbarrow, A. J. H. , Cheesbrough, J. , Gee, S. , Bolton, E. , Fox, A. , Fearnhead, P. , Hart, C. A. , & Diggle, P. J. (2008). Tracing the source of campylobacteriosis. PLoS Genetics, 4(9), e1000203. 10.1371/journal.pgen.1000203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, T. , Luo, Q. , Chen, Y. , Li, T. , Wen, G. , Zhang, R. , Luo, L. , Lu, Q. , Ai, D. , Wang, H. , & Shao, H. (2016). Molecular epidemiology, virulence determinants and antimicrobial resistance of Campylobacter spreading in retail chicken meat in Central China. Gut Pathogens, 8(1), 48. 10.1186/s13099-016-0132-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sequence readset for each food isolate (Bioproject Accession: PRJNA591966), case isolates sampled as part of the case–control study (Bioproject Accession: PRJNA592186) and the case isolates included in the national snapshot (Bioproject Accession: PRJNA560409) are available through GenBank.