

Abstract
Humans have lived in tenuous battle with malaria over millennia. Today, while much of the world is free of the disease, areas of South America, Asia, and Africa still wage this war with substantial impacts on their social and economic development. The threat of widespread resistance to all currently available antimalarial therapies continues to raise concern. Therefore, it is imperative that novel antimalarial chemotypes be developed to populate the pipeline going forward. Phenotypic screening has been responsible for the majority of the new chemotypes emerging in the past few decades. However, this can result in limited information on the molecular target of these compounds which may serve as an unknown variable complicating their progression into clinical development. Target identification and validation is a process that incorporates techniques from a range of different disciplines. Chemical biology and more specifically chemo‐proteomics have been heavily utilized for this purpose. This review provides an in‐depth summary of the application of chemo‐proteomics in antimalarial development. Here we focus particularly on the methodology, practicalities, merits, and limitations of designing these experiments. Together this provides learnings on the future use of chemo‐proteomics in antimalarial development.
Keywords: antimalarial, chemical probe, malaria, target engagement, target identification
Abbreviations
- ABPP
activity‐based protein profiling
- ACT
artemisinin combination therapy
- AfBPP
affinity‐based protein profiling
- ALDH1
aldehyde dehydrogenase family 1
- ALLN
N‐acetyl‐Leu‐Leu‐Norleu‐al
- ART
artemisinin
- ATC
aspartate transcarbamoylase
- AzT
TAMRA azide
- AzTB
TAMRA biotin azide
- CDK2
cyclin dependent kinase
- CEPT
choline/ethanolamine phosphotransferase
- CETSA
cellular thermal shift assay
- CK1
casein kinase
- CnBr
cyanate ester
- CQ
chloroquine
- crapOME
contaminant repository for affinity purification
- CSP
circumsporozoite protein
- CuAAC
copper‐catalyzed azide‐alkyne cycloaddition
- DARTS
drug‐affinity responsive target stability
- DFO
desferrioxamine
- DFP
deferiprone
- DHODH
dihydroorotate dehydrogenase
- DV
digestive vacuole
- emPAI
exponentially modified protein abundance index
- HA
hemagglutinin A
- HDP
hemoglobin derived products
- HEA
hydroxyethyl amine
- HKMT
histone lysine methyltransferases
- HQ
hydroxychloroquine
- ICAT
isotope‐coded affinity tagging
- IEDDA
inverse‐electron demand Diels–Alder
- ITDR
isothermal drug response
- Itraq
isobaric tagging for relative and absolute quantification
- IVE‐GWAS
in vitro evolution—genome wide association studies
- LC/MS/MS
liquid chromatography‐tandem mass spectrometry
- MFQ
mefloquine
- MS
mass spectrometry
- Myr‐CoA
Myristoyl‐Coenzyme A
- NHS
N‐hydroxysuccinamide
- NMT
N‐myristoyltransferase
- PAL
photoaffinity labeling
- PfATC
Plasmodium falciparum aspartate transcarbamoylase
- PfCDPK1
Plasmodium falciparum calcium‐dependent protein kinase
- PfDHFR‐TS
Plasmodium falciparum dihydrofolate reductase‐thymidylate synthase
- PfENT4
Plasmodium falciparum Equilibrative Nucleoside Transporter
- PfMDR1
Plasmodium falciparum multidrug resistance protein
- PfOAT
Plasmodium falciparum ornithine aminotransferase
- PfPNP
Plasmodium falciparum purine nucleoside phosphorylase
- PfPyKII
Plasmodium falciparum pyruvate kinase II
- PfSPP
Plasmodium falciparum Signal Peptide Peptidase
- PI4K
phosphatidylinositol 4‐kinase
- PKG
cGMP‐dependent kinase
- PM
plasmepsin
- PMIX
plasmepsin IX
- PMX
plasmepsin X
- PQ
primaquine
- PROTACs
proteolysis‐targeting chimeras
- QR2
quinine oxidoreductase 2
- RBC
red blood cell
- RING
really interesting new gene
- Sal A
Salinipostin A
- SAR
structure–activity relationship
- SILAC
stable isotope labeling with amino acids in culture
- SPAAC
strain‐promoted azide‐alkyne cycloaddition
- SPR
surface plasmon resonance
- SPROX
stability of proteins from rates of oxidation
- TER
tetraethylrhodamine
- TMT
tandem mass tagging
1. INTRODUCTION
Malaria is a parasitic protozoan disease that causes a huge burden on human well‐being worldwide. 1 Over 200 million people are infected with the disease annually, resulting in approximately 619,000 deaths in 2021. 2 Restricted primarily to tropical regions, malaria is caused by the transmission of Plasmodium parasites by the bite of the female Anopheles mosquito. While a number of Plasmodium species have the potential to cause human disease, P. falciparum and P. vivax have the most significant impact on mortality and morbidity. 2 The management of malaria consists of mosquito intervention methods and antimalarial chemotherapy which collectively, have resulted in a significant reduction in morbidity and mortality over the past 20 years. 3 Unfortunately, resistance to most currently available antimalarials has been observed in P. falciparum, including the front‐line Artemisinin Combination Therapies (ACTs). 4 As such, some drug classes used to treat the disease are no longer recommended for clinical use. 5 , 6 To curb the onset of resistance, world health authorities have prescribed that new antimalarials have a novel chemotype and target a mechanism of action not previously reported. 7 More recently, a pioneering RTS,S/AS01 vaccine based on the circumsporozoite protein (CSP) has been approved for use in children in areas of high transmission. 2 However, the efficacy of the vaccine is just 36% over 4 years of monitoring. 8 Therefore, antimalarial chemotherapies will remain at the forefront of disease treatment and control.
To discover new antimalarial chemotypes, there has been an explosion in mass phenotypic high throughput screening of large compound libraries in the past 20 years. 9 , 10 , 11 These screens have been primarily performed on the asexual erythrocytic stage of P. falciparum as this form of the parasite is the most tractable in the laboratory, 12 although more recently, assays and platforms become available to screen both the sexual (both gametocyte and gamete) 13 and liver sporozoite and schizont stages of the P. falciparum lifecycle. 14 , 15 Additionally, methods have been established to screen against the latent P. vivax hypnozoite. 16 , 17 The mass phenotypic screening effort has resulted in the identification of starting points that have led to the development of several clinical candidates, such as cipargamin (KAE609), ganaplacide (KAF156), and MMV048, undergoing Phase II trials. 18 , 19 , 20 , 21 While phenotypic‐based screening has become the mainstay for the identification of new antimalarial chemotypes, target‐based screening has also uncovered starting points against genetically validated targets, for example, the dihydroorotate dehydrogenase (DHODH) inhibitor and Phase II clinical candidate DSM265. 22 , 23
Both phenotypic and target‐based drug discovery methods present their own unique challenges. For phenotypic drug discovery, once a hit molecule is identified, a key development task is to deconvolute the mechanism of action. 24 While antimalarials can be developed without a fully described mechanism of action, 25 establishing the target is highly desirable for the following reasons. 26 First, the target can help define the target product profile by understanding the target pharmacology, and secondly, visualizing the compound in complex with a protein target can expedite the development via structural‐based design. For target‐based drug discovery, target engagement within the parasite is important to demonstrate the compound is indeed killing the parasite via the target. Target engagement is also crucial in validating the target of the phenotypic hit once it has been uncovered by target identification methods. Nevertheless, for either target or phenotypic approaches, the process of target identification and engagement is a vital aspect of antimalarial research and development.
The most extensively used approaches toward antimalarial target identification involve omic methods, which have been comprehensively reviewed elsewhere. 27 , 28 , 29 , 30 , 31 , 32 Briefly, these include genomic, metabolomic, and proteomic methods. Genomic methods in target deconvolution involve in vitro evolution of resistance to the compound of interest followed by genome‐wide association studies (IVE‐GWAS) or nucleotide expression profiling with microarray followed by compound treatment. 33 IVE‐GWAS relies on culturing resistance to the compound of interest, which may not be possible if the compound elicits its pharmacological response via inhibition of multiple protein targets, pathways that are not genome‐encoded or host‐derived proteins. Metabolomics is another popular target deconvolution method whereby alterations to the parasite metabolome are detected following drug treatment, identifying pathways that are indirectly inhibited. 34 , 35 Generally, this method provides a top‐down analysis and requires further studies to elucidate the protein target(s). Global transcriptomics and proteomic methods follow a similar rationale with protein and mRNA levels monitored following drug treatment. 36 , 37
Chemo‐proteomic methods have recently emerged as a useful alternative to directly and unbiasedly detect the protein target(s) of antimalarial compounds discovered from phenotypic screening. Chemo‐proteomic techniques are an example of direct target identification methods in which the effects of compound–target interactions are measured directly, not through downstream events. Examples of such techniques include affinity binding techniques using pulldown probes and thermal stability profiling. Additionally, many of the unbiased chemo‐proteomic methods are adapted to biased methods to demonstrate compound engagement with a parasite protein target to assist with on‐target validation of the antimalarial under development.
This perspective will focus on the application of chemical biology methods in antimalarial target identification and target engagement. This appraisal of the field distinguishes itself from recent overviews 38 , 39 , 40 by providing a detailed description of key examples using chemical biology techniques in antimalarial target identification and target engagement while discussing the advantages and limitations. This review aims to act as a guide for the development and application of chemical biology techniques in antimalarial target deconvolution and more broadly in antimalarial drug development.
2. PARASITE AND HOST‐SPECIFIC CONSIDERATIONS IN CHEMO‐PROTEOMICS
2.1. A complex lifecycle
A unique aspect of Plasmodium biology is its complex, multi‐host lifecycle. Human infection begins with the injection of infective sporozoites from Anopheles mosquitoes and these parasites make their way to the liver and invade hepatocytes, where they undergo schizogony or form dormant hypnozoites in the case of P. vivax and P. ovale. 41 Liver schizonts release large numbers of merozoites into the bloodstream which invade red blood cells and begin the asexual blood cycle. A portion of these erythrocytic forms diverges into the sexual blood stage, forming gametocytes that can be consumed by the mosquito in a blood meal and develop in this definitive host which completes the cycle. Dramatic changes in parasite morphology and size occur across the different stages, and indeed within these stages. Consequently, the parasite proteome differs widely, as do potential drug targets. 42 The importance of developing drugs that target all of these stages has been clearly underlined therefore efficient phenotypic screening methods and henceforth target identification is essential for drug development. 43 Comparatively, proteomic sample preparation of Plasmodium is arduous and expensive in large quantities. 44 Therefore, a formidable challenge for Plasmodium proteomic research has been the development of robust culturing conditions at significant scale for quality data. 44 In the following Sections 2.1.1, 2.1.2, 2.1.3, 2.1.4, 2.1.5, parasite stage and host‐specific considerations are outlined for application in chemo‐proteomic methods.
2.1.1. Liver stage
One of the most difficult malaria lifecycle stages to study is the liver stage as the cells are not easily maintained for long periods and at scales sufficient for chemoproteomic analysis. To begin, infective sporozoites must be isolated and purified from the salivary glands of female Anopheles' mosquitoes, requiring specialized insectary facilities. 45 Additionally, the numbers of liver cells invaded by sporozoites is small and some species exhibit cell‐specific invasion. The rodent species P. berghei has been widely used to study the liver stages due to its ability to be cultured in human lung, 46 human hepatoma, 47 HeLa, 48 and mouse hepatocyte cell lines. 49 Culture of human infective species such as P. falciparum, P. vivax, and P. ovale has been achieved in primary liver hepatocytes, however, these host cells cannot be kept in continuous culture. 50 , 51 , 52 The human hepatoma cancer cell HepG2‐A16 was subsequently used as a method to culture liver stage P. vivax, but cannot support the development of P. falciparum. 53 , 54 More recently, the HC‐04 hepatocyte line has been developed to culture both P. falciparum and P. vivax liver stages. 55 , 56 To study the dormant liver stages produced by P. vivax and P. ovale, the specialized hepatocyte line imHC is used as HC‐04 hepatocytes proliferate unrestrictedly and detach from the culture dish, limiting their use for long‐term hypnozoites. 57 The lack of chemo‐proteomic studies on liver stage parasites reflects experimental challenges, for example, difficult culturing conditions and target deconvolution in the presence of abundant host cell proteins. More sensitive methods, therefore, need to be developed to study target identification/engagement in this stage. However, chemo‐proteomic experiments looking at parasite effector proteins in the host hepatocyte may be possible.
2.1.2. Asexual blood stage
The P. falciparum and P. knowlesi erythrocytic stages are the most easily maintained stage in vitro with the development of robust culturing conditions that enable continuous culture. 58 , 59 However, standard static cultures cannot be routinely kept above 10% parasitemia, therefore, considerable scale is required for large proteomic experiments. 60 In contrast, no continuous culturing conditions exist for P. vivax parasites and samples must be derived directly from human infections, further complicating the species’ chemo‐proteomic study. 61 Consequently, the majority of the chemo‐proteomic research has been performed on the P. falciparum asexual blood stages.
Continuous in vitro cultures of Plasmodium are characteristically asynchronous in their lifecycle. 58 Protein expression can be highly stage‐specific and is fundamentally linked with stage‐specific activity observed in most antimalarial compounds. Once the stage of arrest is established, the specificity of proteomic data is enhanced with samples generated from synchronized parasites obtained through a range of methods. Sorbitol ring synchronization leads to purified ring stage cultures via stage‐specific permeability pathways. 62 Mid‐trophozoite, schizonts, and gametocytes, on the other hand, can be purified using a magnetic resin that attracts the iron‐containing hemozoin complexes resulting from hemoglobin digestion in the parasite. 63 Finally, centrifugation‐based purifications such as Percoll gradients can also be used to separate these stages according to their relative density. 64
2.1.3. Transmission stages
Gametocyte culturing conditions are analogous to the asexual erythrocytic stage and therefore can also be suitable for proteomic research. 13 , 65 , 66 , 67 , 68 , 69 Until recently, several issues have made their production at scale more difficult. The small numbers of asexual parasites that commit to this pathway (~5%) and the progressive loss of a culture's ability to produce gametocytes have been a bottleneck to production. 70 , 71 However, a recently reported CRISPR/Cas9‐engineered gametocyte producer line enables high sexual commitment rates (75%) for larger scale production. 72 The inducible overexpression of the sexual commitment factor GDV1 greatly improves the control and yield of sexual forms for use in transmission research. 72 Methods to separate early (I–III) and late (IV and V) gametocytes have been established. 13 , 69 Therefore, as with the other stages of the lifecycle, phenotyping, and establishment of early versus late‐stage gametocyte activity should be aligned with an effective proteomic study.
In vitro methods to culture and purify the remainder of the transmission stages that occur in the mosquito have been developed but at small scales. Exflagellation, or the formation of gametes from gametocytes, can be achieved through parasite resuspension in fetal bovine serum at pH 8. 73 In vitro and ex vivo culturing of ookinetes is most common in P. berghei. 74 , 75 This is because the efficiency of conversion of P. falciparum parasites to mature ookinetes in vitro is very low compared to in vivo. 76 , 77 Maturation from ookinete to oocyst requires a complex coculture system with Drosophila cells and Matrigel substrate. 78 Overall, the chemo‐proteomic study of the mosquito stages, particularly in P. falciparum, suffers from challenges in obtaining sufficient material and at the correct stage for a sensitive quantitative study. Improvements in culturing conditions, workup, and instrument sensitivity will aid in future proteomic work.
2.1.4. Sub‐proteomics
In some cases, phenotypic indications such as timing and stage of antimalarial activity can provide clues as to the mechanism of action. For example, antimalarials with a delayed death phenotype (activity >48 h) are known to target the development of apicoplast organelles in daughter parasites. 79 , 80 , 81 If such a hypothesis is known, the resolution and specificity of chemoproteomic results can be improved by obtaining sub‐proteomic extracts from isolated organelles. This has been achieved for the analysis of the food vacuole, 82 micronemes, 83 , 84 and nucleus 85 with differential centrifugation followed by density gradient separation. There are also analogous methods to isolate the mitochondria and apicoplast through nitrogen cavitation followed by density gradient separation, but these have not yet been applied to proteomic research. 86
2.1.5. Human host erythrocytes
As an obligate parasite, a major challenge in Plasmodial proteomic research is the contamination of host proteins. The blood and liver stages are encased in their host cell as well as a parasitophorous vacuole membrane. High abundance erythrocyte proteins, in particular hemoglobin, can mask the low abundance of parasite proteins. 44 To avoid this, erythrocytic parasites can be purified with saponin lysis which selectively disrupts the erythrocyte membrane while leaving both the parasite and parasitophorous membrane. 87 However, this does not fully resolve these issues as the parasites themselves break down hemoglobin and store by‐products such as hemozoin and hemoglobin‐derived products (HDPs) which can also complicate the sensitivity of proteomic studies. 88 Therefore, for the proteomic study of Plasmodium careful consideration of protein extraction conditions should be taken. For example, traditional lysis buffers containing urea, thiourea, and DTT are thought to disrupt the food vacuole and thus release HDP, while freeze‐thaw lysis does not. 88 However, the removal of erythrocyte proteins may not always be desirable and proteomics can be performed on parasitized red blood cells to identify potential human target proteins. For example, it is predicted that around 280 proteins of parasite origin are exported to the host erythrocyte with roles in immune avoidance and host cell remodeling. 89 , 90 , 91 13%–23% of these exported proteins are known to be essential, although no drugs are known to target these proteins as of yet, these could potentially be targets of antimalarials and require target deconvolution studies. 91
3. CHEMICAL PROBES
One of the most widely applied chemical biology reagents in antimalarial target identification is the chemical probe. For the purposes of target identification, a chemical probe is a reagent used to purify or pulldown target proteins from complex mixtures by means of affinity or activity‐based protein profiling (AfBPP or ABPP). AfBPP leverages the intrinsic affinity of a compound of interest, acting like a bait. ABPP uses a slightly revised principle, relying on a reactive warhead that targets specific residues in the target active site. Often this is used to assess enzymatic families that have conserved catalytic residues, such as serine hydrolases, cysteine proteases, aspartyl, and glutamyl glycosidases. 92 Unlike AfBPP, the target protein becomes covalently linked to the reactive warhead of the ABPP chemical probe, and therefore is irreversibly modified.
The construction of a chemical probe is achieved by conjugating a target‐interacting warhead via a linker to a solid support or functional tag (Figure 1). The sophistication of the compound conjugation or labeling method has rapidly expanded from simple resin immobilization to employing click chemistry, photo‐crosslinking, and even bioorthogonal chemistry. These methods utilize a specialized set of specific chemical reactions there are highly efficient for the conjugation of small molecules. The positioning of the linker or functional label on the compound structure is key to maintaining the target protein binding affinity or activity. 93 Typically, the correct positioning of the label requires prior knowledge of the structure–activity relationship (SAR) performed by rational design, or if the protein target is known, visualization of the compound in complex with the protein target may guide the appropriate location to append the label. Confirmation of the probe's activity is often desired; however, the addition of large linkers may preclude cellular permeability and therefore this measurement may not be useful. Instead, a lower molecular weight handle can be utilized as a surrogate to ascertain the activity of the probe.
Figure 1.
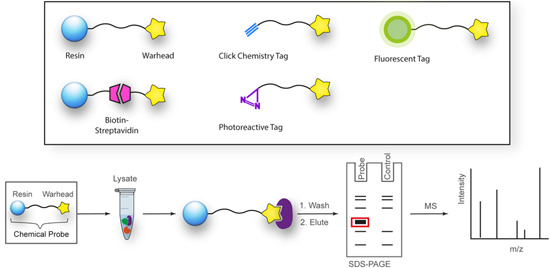
Structures and workflow of chemical probes used for target deconvolution. A range of chemical probe types can be employed for target elucidation, including resin immobilized probes, biotin‐streptavidin‐linked probes, fluorescent tag‐linked probes, and finally, probes with click chemistry and photoreactive tags. Chemical probes are constructed by linking the drug moiety to a solid support resin. The cellular lysate is applied to the resin to identify binding proteins. Rigorous washing steps reduce the levels of nonspecific, leaving only high‐affinity binders attached to the resin. The proteins are separated by SDS‐PAGE and are characterized either by western blot analysis or mass spectrometry. [Color figure can be viewed at wileyonlinelibrary.com]
Once the pulldown is complete, protein characterization methods differ depending on the level of knowledge of the target. Target validation in parasites typically utilizes a biased approach whereby an antibody to the target or an engineered parasite line expressing the labeled target is used for detection by Western blot. For unbiased approaches, these methods rely extensively on mass spectrometry and quantitative proteomics to identify targets (described in Section 5). 94 Poly‐pharmacology is a common feature of phenotypically discovered drugs, and indeed the most effective antimalarials for combatting drug resistance. 95 An unbiased approach can be used to identify such features, therefore, it is well suited for the identification of antimalarial targets. 95 Similarly, chemical probes have the ability to identify off‐target binding which can have important implications for understanding human toxicity that may be observed. 96 For example, an antimalarial chemical probe has led to a proposal for the toxicity observed with chloroquine (discussed later). 97 Chemical probes are pharmacologically relevant, concentration‐dependent, and can be used with almost any cell type.
A limitation of AfBPPs and ABPPs is the propensity toward detecting false positives. For AfBPP, since many antimalarials (and indeed most other drug‐like compounds) have some degree of hydrophobicity, they are therefore predisposed to nonspecific protein interactions. 98 Furthermore, AfBPPs and ABPPs are typically used at high concentrations that are not physiologically relevant increasing the likelihood of detecting false‐positive proteins. Distinguishing between nonspecific binding and true low‐affinity binders can be difficult, underpinning the importance of high‐affinity probes in addition to careful probe design and inclusion of appropriate vehicle and negative controls. 99 , 100 On the other hand, the reactive warhead on ABPP probes may lead to the modification of nontarget proteins. Due to these challenges, poorly characterized and nonselective probes have marred the reliability of research in this field. 101 A need for emphasis on high‐quality chemical probes prompted the release of minimum standards for chemical probes by the Chemical Probes Portal. 102 Here, it is recommended that probes should have well‐characterized in vitro activity, with a suitably structurally analogous inactive control, profiling of potential off‐target activity, and finally, evidence of cell permeability. 102 Finally, while this method is widely applicable to cell types, it is largely limited to soluble proteins. 103 While membrane proteins on rare occasions are suitable for both AfBPP and ABPP, they first require treatment of cells with an ionic nondenaturing detergent to release them from the surrounding membrane. Optimal solubilization conditions are difficult to predict without prior knowledge of the target or the parasite phenotype upon antimalarial treatment, as discussed in Section 2.
3.1. Affinity and activity based protein profiling
3.1.1. Chemical probe immobilization techniques
Resin immobilization
Resin immobilized chemical probes are the simplest and most classical design. Resins are typically polymeric solid supports such as Sepharose (agarose) functionalized with a suitable reactive group, such as N‐hydroxysuccinamide (NHS) or cyanate ester (CNBr). The warhead is covalently attached via a linker to resin beads in an orientation that allows it access to the active site of target proteins. 104 The workflow (Figure 1) generally involves the incubation of these chemical probes with cellular or tissue extracts, followed by extensive washing to remove nonspecific interactions. For AfBPP, remaining strong binders are eluted from the resin and separated by SDS‐PAGE at which point enriched protein bands can be identified compared to an inactive control probe. Elution conditions can include excess unlabeled drug to further assure the specificity of the binding proteins. ABPP results in irreversible protein binding therefore elution is not possible. Proteins are prepared for proteomics with on‐bead trypsin digestion, or alternatively, chemically, enzymatically, and photolytically cleavable linkers can be used to release the protein from its solid support.
Streptavidin immobilization
The activity of the probe may be impeded by the process of immobilization. To accommodate indirect affinity purification with such molecules, functional tags such as biotin can be used. 105 A high‐affinity interaction between biotin and streptavidin (K d ≈ 10−14 M) enables enrichment and immobilization when the latter is immobilized to an agarose resin. 106 , 107 In some cases, 108 these biotin‐labeled probes are cell permeable and can be developed for use in live cells where the probe is captured following the cellular lysis. 98
Bioorthogonal immobilization
A major advancement in the field of chemical probes is the development of robust bioorthogonal reactions; those that proceed within a cellular context without altering normal biochemistry. 109 These reactions require complete chemoselectivity against a horde of cellular functional groups and must proceed rapidly at low temperatures in aqueous media. 110 Coined by Sharpless et al., 111 “click chemistry” reactions have dominated this space. These reactions “follow nature's lead” and join modular units through highly specific and biocompatible chemical reactions. 111 Common click chemistry reactions include the copper‐catalyzed azide‐alkyne cycloaddition (CuAAC), strain‐promoted azide‐alkyne cycloaddition (SPAAC), and the inverse‐electron demand Diels–Alder (IEDDA) using a strained alkene and tetrazine. 112
The copper‐mediated CuAAC reaction uses terminal azide and alkyne functionalities to form a 1,2,3‐triazole linkage based on the Huisgen Cycloaddition (Figure 2A). However, the use of cytotoxic copper reagents can be undesirable and are therefore not applicable for use in live Plasmodium. 113 Therefore, copper‐free methods have come into prominence for this purpose. The first of these is SPAAC, which uses a strained cyclooctene ring to promote the formation of the triazole linkage (Figure 2B). 114 Other common copper‐free methods also employ facile cycloaddition chemistry, such as IEDDA reactions. An example of this type of reaction employs activated or strained alkenes such as norbornene with a tetrazines functionality (Figure 2C). 115 Bioorthogonal probes are particularly useful where steric restrictions of target binding preclude conjugation to a larger group in situ and lead to a significant reduction in probe activity. 116 A functionalized biotin molecule can be conjugated to the click chemistry partner to enable streptavidin affinity capture. 117 Fluorescent tags can also be conjugated to enable in‐gel fluorescence and live‐cell imaging. 118 Importantly, the same bioorthogonal probe can be used for both experiments, expanding its utility.
Figure 2.
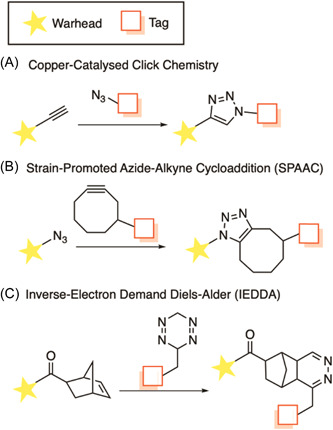
Common bioorthogonal reactions used in the construction of chemical probes. [Color figure can be viewed at wileyonlinelibrary.com]
3.2. Photo‐affinity based protein profiling
A major disadvantage of traditional AfBPPs is that their effectiveness is dependent on the activity or affinity of the probe as well as the abundance of the protein target. 100 UV‐mediated covalent photo‐crosslinking or photo‐affinity labeling (PAL) has been developed as a method to circumvent this problem. 119 PAL involves adding a photo‐reactive tag to the probe structure and upon UV irradiation, a reactive radical is generated that allows covalent linkage to proteins in close proximity to the chemical probe—ideally a protein for which the probe has the highest affinity. The photo‐reactive tag consists of groups that can generate reactive diradicals, carbenes, or nitrenes yielded from benzophenones, diazirine, and aryl azide, respectively (Figure 3). 120 To allow for in‐gel fluorescence or affinity capture, often PAL ligands also feature a second functional tag such as a click chemistry handle or a biotin/streptavidin binding partner.
Figure 3.
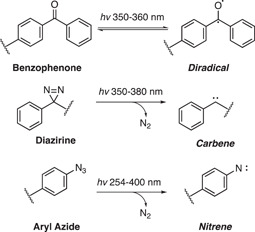
Common photoaffinity ligands. Benzophenones, diazirines, and aryl azides generate highly reactive species upon excitation with UV light which facilitate photocrosslinking to adjacent proteins when included in a probe structure.
The choice of PAL handle comes with several caveats. Benzophenones have some distinct biochemical advantages in that they are more chemically stable than the other groups and can be handled in ambient light. 121 Additionally, the excitation is reversible in the absence of a suitable C‐H bond to insert into, therefore a sample can be repeatedly excited to improve yields. 122 However, an increase in excitation time can have implications in increasing nonspecific labeling. 123 The size of the benzophenone group can also be difficult to incorporate into the structure without diminishing affinity. 124 Therefore, the comparatively small size of aryl diazirine and aryl azide groups has led to an uptick in their usage. 105 Aryl diazirines produce better photo‐crosslinking yields than aryl azides, perhaps due to the increased reactivity of the carbene over the nitrene. 125 Benzophenones and aryl diazirines are also maximally activated at relatively high wavelengths, causing minimal damage to proteins. 126 However, unlike benzophenones, both aryl diazirines and aryl azides can be susceptible to UV‐induced rearrangement and photolysis which reduces the efficiency of labeling. 127 Nonspecific labeling can be considered a broad problem for all PAL probes as pulldowns are generally performed in great excess. 128 This becomes particularly problematic where target abundance is low, and nonspecific binding obscures its detection. 129 Demonstrating a labeling profile that is specific versus a negative control probe and is disrupted by free compound competition is very important to ensure that specific binding. 129
3.3. Affinity based protein profiling examples
AfBPP has been commonly employed for both target identification and target engagement of antimalarials under development. Reliable pull‐down of the target from parasites is typically reliant on having a highly potent and target‐selective compound as a template for the design of the AfBPP in addition to the appropriate controls to exclude promiscuous and abundant proteins. To provide confidence in the pulled‐down protein are indeed genuine, a bioorthogonal technique should be used to provide supporting evidence. Several AfBPP examples are given below that successfully pulldown the target which is confirmed by a target validation method. These examples include AfBPPs based on MMV048, purvalanol B, purfalcamine, and WM382.
3.3.1. Quinoline antimalarials
The first published use of chemical probes in antimalarial target identification aimed to discover binding or reactive protein targets of quinoline antimalarials. Quinoline antimalarials include hydroxychloroquine (HQ), chloroquine (CQ), primaquine (PQ), and mefloquine (MFQ), 97 which have been in clinical use since the mid‐20th century without a well‐defined mechanism of action. 130 Structural similarity between the quinolines and purine nucleotides led to a hypothesis that they may target purine (ATP) interacting proteins. 97 Therefore, two types of probes were employed, a promiscuous ATP‐Sepharose probe for application in competition experiments as well as quinoline‐Sepharose conjugates. PQ was affixed with its primary amine functionality to NHS‐activated Sepharose (Figure 4A), and HQ with its free hydroxyl group to epoxy‐activated Sepharose (Figure 4B). The ATP‐Sepharose probes were incubated with infected RBC cellular extracts to pull down the RBC and P. falciparum purine proteome. Eluting with PQ, CQ, and MFQ did not result in the identification of any P. falciparum proteins. However, the drugs were highly selective for two human proteins from RBC extracts, aldehyde dehydrogenase (ALDH1) and quinine oxidoreductase 2 (QR2). The same experiments were performed with the PQ and HQ conjugated probes, again selectively eluting only human proteins ALDH1 and QR2. Subsequent in vitro target validation identified potent inhibition of QR2 by CQ and PQ, and only weak inhibition of ALDH1 by CQ. Together, this implicated human QR2 as a probable target of CQ and PQ, whose role is the detoxification of quinones which can cause oxidative damage. 131 The malaria parasite itself is sensitive to oxidative damage, 132 and inhibition of QR2 by quinolines may create an inhospitable environment for parasite growth. While the inhibition of ALDH1 likely does not represent the quinolines' antimalarial target, the authors believe that affinity to ALDH1 may explain chloroquine's reported retinopathy. ALDH1 may have a metabolic role in protecting the eye from UV damage, 133 and treatment with chloroquine indeed results in the hyperaccumulation of retinaldehyde in the retina. 134 , 135 , 136
Figure 4.
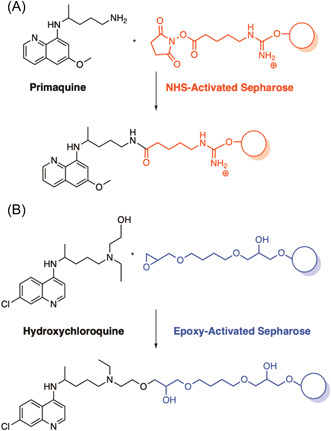
Resin immobilized probes of the known antimalarials primaquine and hydroxychloroquine for the identification of cellular targets. Pulldown of the resin immobilized probes in infected erythrocyte lysate resulted in the enrichment of two human proteins, ALDH1 and QR2. Biochemical validation confirmed QR2 as a probable target and indicated that inhibition of ALDH1 may be the result of an off‐target effect. [Color figure can be viewed at wileyonlinelibrary.com]
3.3.2. MMV048
The antimalarial candidate, MMV048 (Figure 5) 137 was developed from a 2‐aminopyridine class identified from a phenotypic high‐throughput screen of the commercial SoftFocus kinase library. 19 As such the exact molecular target of MMV048 was unknown, although presumed to be a kinase. 137 For target deconvolution, a related analog MMV666845 was chosen as it possesses a primary amine functionality (Figure 5). 137 This moiety was covalently immobilized to Sepharose beads by an undisclosed method and was subsequently treated with parasite‐infected RBC lysate. Eluting bound proteins with unlabeled MMV048 identified one high‐affinity binding protein, phosphatidylinositol 4‐kinase (PI4K). 137 Similar to the quinolones, competitive inhibition with unlabeled MMV048 with “kinobeads” derivatized with a broad set of ATP competitive kinase inhibitors that covered approximately 50% coverage of the Plasmodium proteome was also performed, resulting in a dose‐dependent competitive elution of PI4K in the presence of MMV048. 137 In vitro resistance evolution experiments also identified mutations in PI4K validating it as a target. 137 More recently, kinobeads and lipid‐kinobeads with coverage of 54 P. falciparum kinases were used to uncover that sapanisertib had the strongest competition for PKG (PF3D7_1436600), PI4Kβ (PF3D7_0509800), and PI3K (PF3D7_0515300) using P. falciparum lysate. 138 PfPKG and PfPI4Kβ were confirmed as targets of sapanisertib using an ATP competitive biochemical inhibition using recombinant protein, further demonstrating the utility of kinobeads in target identification of antimalarials with kinase‐like chemotypes.
Figure 5.
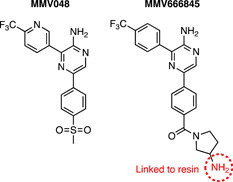
A resin immobilized chemical probe of MMV048 used in the identification of Plasmodium falciparum cellular targets. An active analog of MMV048 with an amine functionality was chosen to link to the Sepharose resin. Phosphatidylinositol 4‐kinase (PI4K) was identified as a probable target, confirmed with competition experiments with MMV048 and subsequent in vitro resistance evolution. [Color figure can be viewed at wileyonlinelibrary.com]
3.3.3. Torin 2
Torin 2 is a known competitive ATP inhibitor of regulatory the kinase mTOR with indications in the treatment of some cancers. 139 In a screen of known chemical entities, it was shown to have potent activity against both gametocytes and asexual P. falciparum. 140 Due to the absence of an mTOR homolog within the P. falciparum genome, target deconvolution was performed using resin immobilized AfBPP. 140 Torin 2 lacks a suitable functional group for attachment to the resin, therefore, the analog WWH030 with a piperazine carboxamide moiety which had minimal impact on gametocytocidal activity was used as the AfBPP (Figure 6A). WWH030 was conjugated to NHS Sepharose, along with a structural similar Torin 1 compound with weak parasite activity which was employed as control AfBPP (Figure 6B). Thirty‐one proteins were specifically pulled down with the Torin 2 chemical probe in gametocyte lysate which was complemented by DARTS target identification (discussed later in Section 3.2), identifying 3 common putative targets: phosphoribosylpyrophosphate synthetase (PF3D7_1325100, ribose‐phosphate diphosphokinase), aspartate transcarbamoylase (PF3D7_1344800, PfATC) and a putative transporter (PF3D7_0914700). 140 PfATC is an enzyme involved with pyrimidine biosynthesis, a pathway targeted directly and indirectly by a number of antimalarials. To validate its role in Torin 2 antimalarial activity, dose–response assays were performed against recombinant PfATC, reported at 68 µM. 141 Transgenic parasites overexpressing ATC were used to validate Torin 2, which revealed a more than 18‐fold reduction in activity compared to the control. 141 The other two putative targets have not been further validated to date. However, Torin 2 analogs have since been developed with greater selectivity for parasites over the human mTor enzyme, improved solubility, and metabolic profile. 142 These analogs have been shown to exert antiparasitic activity through inhibition of phosphatidylinositol 4‐kinase (Pf PI4KIIIβ). 142
Figure 6.
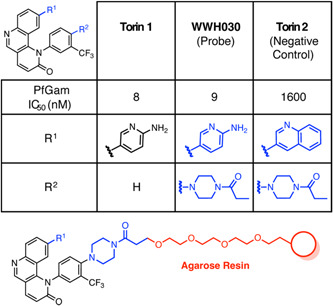
Summary of the human mTOR inhibitor Torin 2 P. falciparum activities and AfBPP design. An equipotent and structurally related compound WWH030 was used to construct a chemical probe for Torin 2 as it possessed a suitable handle. The negative control was constructed from the significantly less active Torin 2. Pulldown in P. falciparum gametocytes revealed putative targets, including phosphoribosyl pyrophosphate synthetase, aspartate carbamoyltransferase, ATCase, and (PF3D7_0914700). [Color figure can be viewed at wileyonlinelibrary.com]
3.3.4. Purvalanol B
Purvalanol B was identified from a screen of a known human drug library and subsequently investigated using an AfBPP approach. 143 The drug is known to target the human cyclin‐dependent kinase 2 (CDK2), 144 a member of an important family of cell cycle regulators implicated in cancers and neurodegenerative diseases. 145 However, this compound has also been found to have an antiproliferative effect on a range of human protozoan parasites, including P. falciparum. 143 After examination of the x‐ray structure of purvalanol B in complex with human CDK2, it was established that the carboxylic acid group would make an appropriate handle for functionalization in an AfBPP as it would have minimal effect on binding (Figure 7). 144 Previous SAR indicated that the addition of a methyl group at the N6 position on analog 95 M significantly diminished CDK2 inhibitory activity so was used as a control in the AfBPP study (Figure 7). 146 The authors also found that large functionalities at N9 reduced CDK2 inhibition, therefore, they placed the linker at this position for use as an additional control (95‐N9). 146 Pulldown in P. falciparum resulted in a singular protein, casein kinase 1 (CK1). The authors found that purvalanol B did not significantly inhibit mammalian CK1, but potently inhibited PfCK1 (IC50 0.30 μM) despite the high sequence conservation. Unfortunately, this discovery has not resulted in further exploration of a CK1‐targeted antimalarial, however, the study has prompted the investigation of inhibitors in other protozoan parasitic species examined such as Leishmania and Trypanosoma. 147 , 148 , 149
Figure 7.
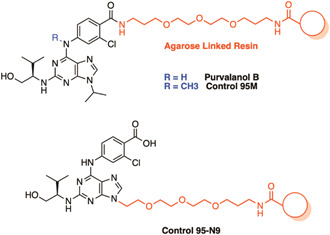
A resin immobilized chemical probe used in the identification of Plasmodium falciparum targets of the human cyclin‐dependent kinase 2 (CDK2) inhibitor purvalanol B. Purvalanol B and related inactive controls were immobilized via a PEG linker to an agarose resin for target identification in P. falciparum. Pulldown identified only one potential target, P. falciparum casein kinase 1 (PfCK1). [Color figure can be viewed at wileyonlinelibrary.com]
3.3.5. Purfalcamine
Purfalcamine was identified as a potent inhibitor of P. falciparum calcium‐dependent protein kinase 1 (PfCDPK1) from a target‐based screen of a kinase‐directed heterocyclic library. 150 To validate the parasite targets of purfalcamine, an agarose immobilized purfalcamine AfBPP was incubated with parasite lysate in the absence or presence of unlabeled purfalcamine (Figure 8). 150 The AfBPP pulled down a hypothetical protein (PF13_01116), a putative FAD‐dependent glycerol‐3‐phosphate dehydrogenase (PFC0275w), a conserved hypothetical protein (PFF0785w), and PfCDPK1. 150 The highly abundant pyruvate kinase was pulled down in both the competition and noncompetition conditions, therefore was not considered a specific target. Microscopic examination identified that purfalcamine caused cycle arrest at the schizont stage, 150 consistent with pfcdpk1 gene transcription supporting PfCDPK1 as the primary target. 151
Figure 8.
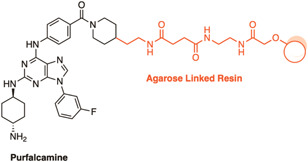
A resin immobilized chemical probe for validation of the cellular targets of purfalcamine. Pulldown with this probe identified several proteins with PfCDPK1 as the likely candidate. [Color figure can be viewed at wileyonlinelibrary.com]
3.3.6. Imidazopyridazine antimalarials
The imidazopyridazine antimalarial scaffold was discovered in another target‐based screen against PfCDPK1 employing two different compound libraries. 152 The first was a library containing a diverse set of 35,422 compounds, and the second was the BioFocus kinase library. This identified a number of scaffolds with sub‐nanomolar inhibitory activity against PfCDPK1, including the imidazopyridazine chemotype. While compounds of this class were indeed active against asexual P. falciparum parasites, the subsequent SAR studies indicated that the level of PfCDPK1 inhibition correlated poorly with the inhibition of parasite growth. 153 Subsequent chemical genetics altering the kinase sensitivity to inhibitors established that inhibition of PfCDPK1 did not alter parasite viability in asexual stages, ruling it out as a potential target. 154 This also called into question the validity of PfCDPK1 as a legitimate target for the previously mentioned 2,6,9‐purine purfalcamine. Phenotypic studies were therefore initiated on imidazopyridazine analogs, where it was discovered that two sub‐structural imidazopyridazine classes had distinct mechanisms of action depending on their aromatic linker. Compounds with a pyrimidine‐linker arrested parasites at late schizogony, whereas the non‐pyrimidine‐linker arrested parasites at trophozoite stage (Figure 9). 154
Figure 9.
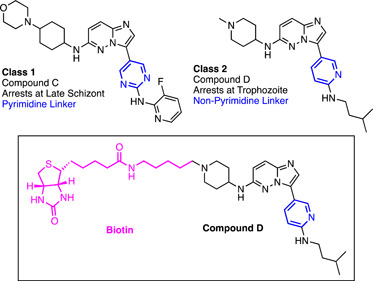
Imidazopyridazine compounds identified using a target‐based screen against PfCDPK1. Two classes of imidazopyridazine compounds were identified, differing in their aromatic linker. Class 1 imidazopyridazines possessed a pyrimidine linker and arrested parasites at the late schizont stage. Class 2 imidazopyridazines are linked via nonpyrimidine aromatic rings and arrest at the trophozoite stage. Biotinylation of compound D enabled streptavidin affinity pulldown for the identification of cellular targets. The probe identified the molecular chaperone PfHSP90 as a potential target for the compound. [Color figure can be viewed at wileyonlinelibrary.com]
The authors noted the schizontocidal activity of class 1 matched phenotype of a kinase closely related to PfCDPK1, cGMP‐dependent kinase (PKG). 154 Indeed, the antiparasitic SAR of class 1 compound closely correlated with PKG IC50. 154 Additionally, chemical genetics performed on PKG identified a link between the kinase's sensitivity to the inhibitor and parasite viability. 154 For target identification of class 2 nonpyrimidine targets, an affinity pulldown approach was taken. Compound D (Figure 9) was conjugated to biotin for affinity capture of targets with streptavidin‐agarose. This pulldown only identified one significant target—HSP90, a molecular chaperone containing an ATP binding site that is essential for mediating the transition from ring to trophozoite development. 155 Recombinant PfHSP90 binding assays were subsequently used to confirm compound interaction of 6.17 μM, similar to another HSP90 inhibitor 17‐AAG which also blocks parasite development at the trophozoite stage. 154 The authors considered this to be a promising target, although could not rule out other targets not able to be pulled down in this study. Indeed, a discrepancy between the potent 360 nM cellular activity and weak protein binding points to this possibility. Heat shock proteins are well‐known promiscuous binders as a function of their role in protein folding. 156 Accordingly, HSP90 is included in the CRAPome, a repository for common nonspecific binders in AfBPP for the human and yeast proteomes. 156 Without a negative control compound or competition experiment nonspecific interactions cannot be ruled out for this target.
3.3.7. Plasmepsin X
Plasmepsins are aspartic proteases, some of which are essential and are potential drug targets, including plasmepsin IX and X (PMIX and PMX) which are involved in the parasite invasion and egress pathway. 157 , 158 Following a high‐throughput screen of an aspartic protease inhibitor library, it was discovered that a novel scaffold inhibited P. falciparum growth with nanomolar potency. 159 By selecting for resistance, PMX was determined as a probable target of these compounds. 159 An AfBPP approach was implemented to validate PMX as the target of these compounds. First, a hemagglutinin A (HA) tagged PMX parasite line was developed that would be used to detect the target protein by western blot with anti‐HA antibodies. 159 A click chemistry AfBPP approach was then used to construct the chemical probes (Figure 10). The solid support was first synthesized, attaching the strained cyclooctyne amine‐BCN to NHS‐Sepharose via its terminal amine. Next, the lead active compound (WM382) was modified with an azide moiety by a PEG linker to give the probe called WM853. Copper‐free click chemistry was used to attach these two hemispheres together for the final pulldown. Due to the stage‐specific expression of PMX, the pulldowns were performed using the lysate of late schizont stage saponin‐treated parasites. Western blot identified efficient pulldown of PMX which was interrupted by the presence of free excess lead compound WM382. Interestingly, WM382 also inhibits PMIX at a lower affinity than PMX, 160 but was not pulled down in this study, highlighting the requirement for high‐affinity ligands for successful pulldown of genuine targets.
Figure 10.
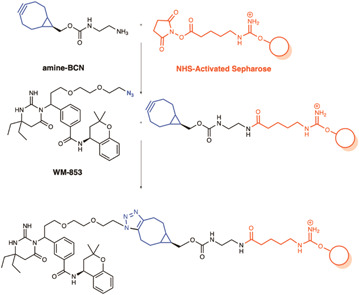
SPAAC probes used in the target validation of WM382 against plasmepsin X. An azide functionalized derivative of the lead compound (WM‐853) was used to attach the compound of interest to a Sepharose resin using SPAAC copper‐free conditions. These probes were incubated with lysate from an HA‐tagged PMX parasite line where PMX was identified as a binder by western blot with an anti‐HA antibody. Pulldown of PMX was competitively inhibited by the addition of the parent compound WM382. [Color figure can be viewed at wileyonlinelibrary.com]
3.4. Activity based protein profiling examples
ABPPs based on antimalarials that typically covalently engage their protein targets are by their very nature reactive and therefore potentially have several targets rather than one exclusive target. These ABPPs typically pulldown many protein targets, which can be difficult to deconvolute and determine whether each protein is a genuine binding protein. A key example in the following section is the endoperoxide antimalarials which are known to mechanistically cross‐link with many proteins, and therefore using the ABPP method it has been difficult to reliably detect target proteins.
3.4.1. N‐251 and N‐89
ABPPs were implemented to identify the targets of novel endoperoxide drugs N‐251 and N‐89. 161 In the ABPP design, a lysine linker was coupled to the hydroxyl group of N‐251, termed N‐346, for conjugation to the resin functionalized with an azlactone (Figure 11). 161 PfERC, Pf14‐3‐3, and PfHSP70 were the highest enriched proteins from the pulldown with the N‐346 ABPP. Subsequently, differential protein expression analysis confirmed that the expression of these proteins was altered by treatment with N‐251 and N‐89. 161 The latter two are unlikely targets as they are known to promiscuously bind compounds in their roles as kinase regulator and molecular chaperone, respectively. 162 PfERC is an essential ER‐resident protein, important for asexual parasite egress. 163 N‐251 and ‐89, but not the related endoperoxide, artemisinin, were subsequently confirmed to bind weakly to PfERC by surface plasmon resonance (K D 1.6 × 10–4 M and 3.8 × 10–3 M). 161 The binding of these compounds may represent a mechanism for these novel endoperoxides or may be the result of nonspecific binding.
Figure 11.
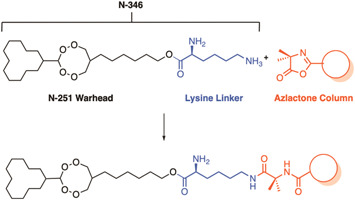
A resin immobilized chemical probe for the novel endoperoxide N‐251. The novel endoperoxide N‐251 was linked to an azlactone Sepharose resin via a lysine linker to create the probe N‐346. Treatment with cellular lysate resulted in the enrichment of PfERC, Pf14‐3‐3, and PfHSP70. Weak binding of N‐251 and N‐89 to PfERC was confirmed subsequently by surface plasmon resonance (SPR). [Color figure can be viewed at wileyonlinelibrary.com]
3.4.2. Artemisinin
Two concurrent seminal studies on the mechanism of Artemisinin by Wang et al. 164 and Ward and Hemingway et al. 165 utilized ABPPs with click chemistry handles. This method allowed for the in situ use of the probes which is important given the evidence of site‐specific activation of the endoperoxide moiety. 166 Both studies attached the click chemistry handles via a linker to the C10 position of the ART structure which proved not to significantly reduce antiparasitic activity. 164 , 165 Differences in linker structure and size separate the two studies (Figure 12), where Ward and Hemingway et al. denote their probes as P1 and P2 while Wang et al. denote their probe as AP1. AP1 features a much longer carbamate linker, terminating in an alkyne click chemistry handle (Figure 12A). 164 P1 and P2 feature a short amide linker, deliberately chosen by the authors to avoid issues with cytotoxicity that have been reported with longer amide linkers (Figure 12B). 167 Both alkyne and azide handles were conjugated for comparison of copper‐mediated and copper‐free click conditions due to previous studies proposing the potential for copper to activate artemisinin in a similar manner to iron in hemozoin. 168 A final difference between the two studies is the use of inactive control ABPPs by Ward and Hemingway et al. who constructed corresponding non‐peroxidic probes CP1 and CP2 which were inactive against parasites. Despite these differences, the workflow of the two studies was similar. Live parasites were exposed to the probes to allow for protein alkylation and proteins were subsequently extracted. The extracts were combined with a clickable functional group, either a fluorophore for in‐gel fluorescence analysis or biotin conjugate for streptavidin bead affinity capture. P1, P2, and AP1 all alkylated a large number of proteins by in‐gel fluorescence. 164 , 165 Control probes CP1 and CP2 did not show any alkylation by in‐gel fluorescence. 165 The authors found AP1 alkylation to be dose‐dependent, unobservable in uninfected erythrocytes, and antagonized by co‐incubation with free radical scavengers. 164 This supports the parasite‐specific activation of the scaffold and the formation of a free radical.
Figure 12.
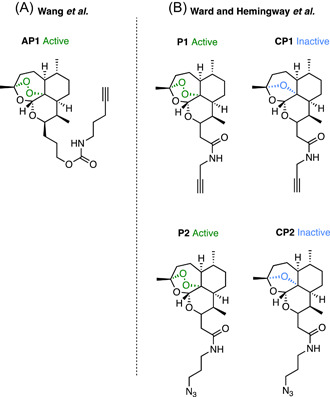
Artemisinin‐based click chemistry probes. (A) Alkyne and azide click chemistry probes by Hemingway and Ward et al. identified 42 common proteins in an affinity pulldown, with a majority containing a glutathione binding motif which may be particularly susceptible to radical alkylation. When P1 was retested by Maser et al. with additional controls far fewer proteins were pulled down, none of which were found in the original study. (B) Alkyne probe AP1 pulled down 125 high‐confidence proteins with similar pathway coverage to the P1 and P2 probes. [Color figure can be viewed at wileyonlinelibrary.com]
When coupled to the streptavidin beads, probes P1, P2, and AP1 similarly pulled down a wide range of targets involved in many essential pathways. 164 , 165 P1 identified 58 high‐confidence proteins, four of which were pulled down nonspecifically in low abundance by the inactive CP1 probe. 165 P2 pulled down 62 proteins, 42 of which were in common with P1, 165 while the control azide probe CP2 did not pull down any protein nonspecifically. 165 The copper‐free method with P2 appeared to detect these proteins with greater sensitivity due to the high efficiency of the strain‐promoted click reaction with the DIBO cyclooctyne. 165 P2 was also assessed with cell lysate and showed no significant difference in the labeling of proteins. 165 AP1, on the other hand, pulled down high confidence 124 protein targets, including a further 125 proteins pulled down in repeat experiments. 164 It has been suggested that the larger range of identified targets is due to the increase in linker size and thus lipophilicity. 165 This cannot be assessed without the comparison of an inactive control for AP1.
The overall coverage of parasite pathways between the ABPP types appears to be similar. Alkylated proteins converge on a subset of pathways, including glycolysis, nucleic acid biosynthesis, protein biosynthesis, invasion, protein transport, and redox antioxidant defense. 164 , 165 Notably, a substantial number of the proteases involved in hemoglobin digestive pathway in the DV were labeled, including plasmepsin I, plasmepsin II, and cathepsin D. 164 , 165 However, the incomplete labeling of proteases in this pathway (e.g., falcipain II and falcipain III) suggests a degree of selectivity to ART‐protein alkylation. 164 , 165 Analysis of targets pulled down by P1 and P2 indicated a correlation with proteins that had a glutathione binding motif. 164 The authors suggested this free thiol may be an easy target for the ART free radical which has previously been shown to form cysteine adducts. 169 The formation of these adducts may directly contribute to the specificity seen above to aspartic and cysteine proteases in the DV hemoglobin digestion pathway.
Interestingly, alkylated targets of P1 and P2 were shown to be differentially affected by the addition of the iron chelator, DFO. The plasmepsins and the majority of the glycolytic enzymes were not significantly affected by DFO treatment, whereas ornithine aminotransferase (PfOAT) was. 165 PfOAT was also identified as a target of AP1, and in vitro biochemical analysis showed that binding occurred only in the presence of added haemin. 164 This binding was further enhanced by the addition of reagents that reduced hemin to heme (Vitamin C, GSH, and Na2S2O4). 164 The addition of ferrous iron, on the other hand, had no impact on the binding of AP1 to PfOAT. 164 Additionally, the binding of AP1 to PfOAT appeared to be protein structure‐dependent as heat denaturation of PfOAT diminished binding. 164
Both studies also explored the mechanisms of ART activation. Ward and Hemingway et al. tested the effect of DFO pre‐treated cellular homogenates on P1 alkylation and found that it significantly reduced but not ablated pulled down proteins. 165 This data suggested that ART may be involved in a nonferrous iron‐mediated activation pathway, although this notion was questionable as the concentration of DFO used for chelation in live parasites and in free heme homogenates were significantly different. 170 Wang et al. then tested the effect of iron chelating agents DFO and DFP (deferiprone) in live parasites which did not result in a significant reduction in AP1 protein alkylation by in‐gel fluorescence. 164 However, a cysteine protease inhibitor N‐acetyl‐Leu‐Leu‐Norleu‐al (ALLN) that inhibits the production of heme via the parasite hemoglobin digestion pathway, 171 caused a significant decrease in the fluorescent labeling of proteins by AP1. Together, this points to heme as the predominant source of ART activation. 164 However, this fails to explain the activity of ART in the early ring stage, where hemoglobin digestion does not occur. 172 The authors surmised that hemoglobin biosynthesis that occurs at this stage could be a source of heme for ART activation. To test this, synchronized early ring parasites were pretreated with the hemoglobin synthesis inhibitor SA which proved to reduce the level of ART protein binding by AP1. 164 Hemoglobin digestion inhibitor ALLN also had no effect on AP1 alkylation in ring stages. 164
Maser et al. 173 later re‐evaluated the same probes from Ward and Hemingway et al. 165 (P1 and CP1, Figure 12B) with additional controls. The authors included a DMSO‐treated control, an ART‐treated control, the nonperoxidic control CP1 as well as multiple probe concentrations. 173 Remarkably, the proteins alkylated by this experiment had little in common with the targets identified by Ward and Hemmingway et al. 165 At a concentration of 100 ng/mL P1 alkylated 15 specific proteins, none of which were present in the original study. 173 1000 ng/mL P1 alkylated an additional eight unique proteins, only one of which was identified in the original study. 173 The targets identified by Maser et al. were more analogous to Wang et al. 164 with AP1 with 10 and 6 proteins in common for 100 and 1000 ng/mL concentrations of P1, respectively. 173 Some targets identified by the previous studies, such as DHFR, were identified in the negative controls of this study and therefore were removed from consideration. 173 The authors concluded that this variation in ART alkylation is the result of a stochastic binding pattern that may be more linked to radical proximity rather than any specificity. 173
What is clear from these studies is that the vast number of targets alkylated by ART contribute to its parasite lethality. Glutathionylated proteins appear to represent a large proportion of these targets, presumably due to their susceptibility to alkylation. The peroxide bond is responsible for its activity which is the site of free radical formation. ART also accumulates specifically in infected erythrocytes where it appears heme is responsible for the majority of its activation in later parasite stages. A limitation of these studies is that they do not explore potential noncovalent targets of ART, nor potential nonprotein targets such as heme. 173 It is also evident that the structure of the probes can vastly affect the results of the pulldowns. This highlights the importance of careful probe design and confirmation of potential targets with other means of target identification or biochemical analysis.
3.4.3. 1,2,4‐Trioxolanes
Based on the rational design of ART‐based probes, 165 synthetic endoperoxide 1,2,4‐trioxolane ABPPs were constructed. 174 The probes were designed with minimal linker size and thus lower lipophilicity on the basis of greater specificity and pharmacological relevance. 174 Both alkyne (TP1) and azide (TP2) functionalities were used to assess the utility of copper‐mediated and copper‐free methods (Figure 13). Finally, non‐peroxide probes were again synthesized as inactive controls (CPT1 and CPT2; Figure 13). To assess the specificity of the probes, in‐gel fluorescence was determined by clicking on an Alexa Fluor 488 tag. 174 As was previously observed with the ART probes, the azide probe TP2 had greater labeling intensity due to the efficiency of the copper‐free strain‐promoted cycloaddition reaction. 165 The protein alkylation profile of TP2 was then compared against the analogous ART probe P2 (Figure 12). 165 The results of the affinity purification were overwhelmingly similar between the two chemotypes. Of 62 total pulled down proteins, 53 of these were identical. 174 The roles of these proteins were again in heme digestion, energy supply, DNA synthesis, and antioxidant defense systems. 174 Interestingly, 70% of the proteins identified were glutathionylated, supporting the theory that the radical formed by heme activation reacts with the disulfide bond present at the site of this posttranslational modification. There were six proteins identified that appeared in one experimental replicate but not the other, 164 demonstrating the importance of experimental design in ABPP studies.
Figure 13.
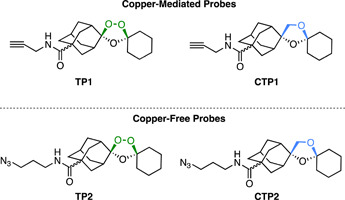
Structures of ozonide click‐chemistry probes TP1 and TP2 and their inactive nonperoxidic control compounds CTP1 and CPT2 synthesized by O'Neill et al. Probes based on an alkyne handle (above) were optimized for a copper‐mediated click reaction, whereas probes with an azide handle (below) use copper‐free methods. 53 common proteins were identified between the two probes with diverse roles, although the majority were glutathionylated. [Color figure can be viewed at wileyonlinelibrary.com]
Alongside the re‐evaluation of the ART probe P1 (Figure 12), Maser et al. constructed novel alkyne functionalized 1,2,4‐trioxolanes (OZ726 and OZ727, Figure 14). 173 Stringent controls were used including DMSO pretreatment, parent compound OZ03 pretreatment and the use of a non‐peroxidic control carbaOZ727. Interestingly, the degree of overlap between the targets of OZ726 and OZ727 was only 30% (6 of 20 proteins). 173 Indeed, there was no overlap between the proteins alkylated in this study to those of TP1 and TP2 (Figure 13). 174 This again illustrates how influential the structure of probes can be to the target profile. In direct comparison with the ART probe P1, the overlap in specificity was just 17% and 13% for OZ726 and OZ727, respectively. 173 When tested at a 10‐fold higher concentration, OZ726 alkylated 9 of the 11 proteins identified by P1 at the same concentration. 173 However, an additional 16 proteins were identified by OZ726 at this concentration that were not identified in any of the previous experiments. 173 As the authors concluded with P1, the alkylation of 1,2,4‐trioxolanes appears to be random, which is consistent with the irregular cellular localization of 1,2,4‐trioxolane fluorescent probes. 170 , 175 , 176
Figure 14.
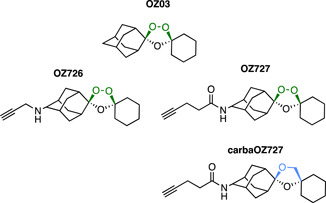
Structures of bioorthogonal ozonide probes by Maser et al. The alkyne‐based copper click chemistry probes identified stochastically alkylated targets with little overlap between similarly structured probes OZ726 and OZ727. [Color figure can be viewed at wileyonlinelibrary.com]
3.4.4. Salinipostin A
Salinipostin A (Sal A) is a marine natural product with low nanomolar activity against P. falciparum and has an unknown mechanism of action. Previous mechanistic studies had been unable to generate resistant parasites which suggests that the compound may act through multiple essential pathways. 177 An alkyne tag was added to a Salinipostin A analog, Sal alk, to enable a range of functionalization for ABPP and fluorescence co‐localization studies. (Figure 15). 178 First, a TAMRA fluorescent label, azide was conjugated to Sal alk via click chemistry and upon treatment with parasites confirmed that multiple targets bound to the structure by in‐gel fluorescence. 178 The labeling of many of these proteins was completed with the addition of unlabeled Sal A in a dose‐dependent manner. 178 A biotin azide was also conjugated to the alkyne handle for pulldown streptavidin‐resin using parasite lysate pre‐incubated with Sal A or vehicle control. The 10 proteins most highly enriched in these experiments, all possessed classical α/β serine hydrolase domains (Ser‐His‐Asp catalytic triad or a Ser‐Asp dyad). 178 piggyBac mutagenesis studies have determined that four of these are essential for parasite viability, 179 although have not yet been confirmed as genuine binders in subsequent studies.
Figure 15.
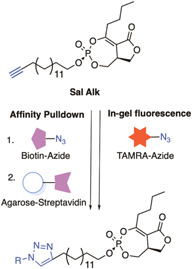
Multi‐functional click chemistry probes of Marine natural product Salinipostin A (Sal A). This multi‐functional probe helped to identify 10 enriched proteins with a common α/β serine hydrolase domain, 4 of which were found to be essential for parasite survival. [Color figure can be viewed at wileyonlinelibrary.com]
3.4.5. Myr‐CoA
The Plasmodium N‐Myristoyl Transferase (NMT) catalyzes the attachment of a myristate lipid tail from Myristoyl‐Coenzyme A (Myr‐CoA) to N‐terminal glycine on specific substrates in membrane trafficking (Figure 16A). 180 Despite its utility as a target in fungal and trypanosome infections, the genetic essentiality of NMT in P. falciparum had not yet been demonstrated. 181 , 182 Therefore, ABPPs were designed for use in an NMT substrate capture experiment. 183 The probe was constructed based on the structure of the enzyme substrate, Myr‐CoA, with an alkyne handle termed YnMyr‐CoA (Figure 16B). 183 A trifunctional capture reagent was also synthesized featuring a TAMRA fluorescent reporter, biotin affinity capture moiety, and a trypsin cleavable linker (Figure 16B). The cleavable linker allowed the specific identification of the site at which proteins were N‐myristoylated without external labeling, resulting in an unambiguous hydrophilic zwitterionic moiety that can be detected with tandem mass spectrometry (MS/MS). 183 In‐gel imaging was employed to demonstrate that peptide tagging was dose‐dependent which could be competitively inhibited by excess free myristate. The pulldown experiments with avidin purification identified over 30 NMT substrates that have diverse functions including motility, protein transport, parasite development, and phosphorylation pathways. These included N‐myristoylated proteins that had been genetically validated for essentiality in other eukaryotes but not in P. falciparum. The wide diversity of the pulled down proteins identifies NMT as a promising drug target in P. falciparum.
Figure 16.
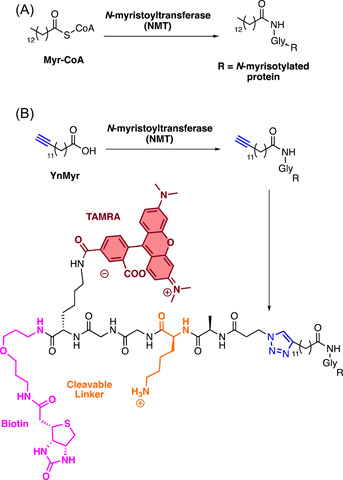
YnMyr probe developed for the recognition of P. falciparum N‐myristoylated protein targets. An analog of the MyrCoA with an alkyne handle (YnMyr) was constructed for capture with a trifunctional capture reagent. The terminal azide reagent contains a TAMRA fluorophore for in‐gel fluorescence, a biotin moiety for affinity capture, and a trypsin cleavable linker capable of acting as a tag for the identification of myristoylated proteins by tandem mass spectrometry (MS/MS). [Color figure can be viewed at wileyonlinelibrary.com]
3.5. Photo‐crosslinking probe examples
Photo‐crosslinking is generally introduced to an AfBPP or an ABPP to facilitate the covalent linkage of the probe with protein target(s) in parasites. This strategy, followed by a bioorthogonal method to validate the target, has been successfully used by several groups including examples based on the HEA class of protease inhibitors.
3.5.1. ACT‐186128
Photo‐crosslinking probes were also used to identify the target of the novel antimalarial ACT‐186128 discovered in a phenotypic screen. 184 A photo‐AfBPP was developed for application in live cells employing a phenyl azide photo‐crosslinking moiety and a biotin tag for both fluorescent labeling and affinity purification (Figure 17). 185 Live‐cell imaging enabled by the association of the Alexa488‐streptavidin fluorescent reporter with the biotin moiety showed localization throughout the cytoplasm in all parasite life stages, consistent with its lack of stage specificity. 185 Pulldown with streptavidin beads was performed after treatment of both intact parasitized red blood cells and saponin‐liberated parasites with the photo‐AfBPP. The pulldown with intact infected RBCs identified one target, the Pf multidrug resistance protein 1 (PfMDR1). 185 The pulldown with saponin‐isolated parasites identified over 20 targets with the highest enriched candidates being PfMDR1, Equilibrative Nucleoside Transporter (PfENT4), hexose transporter, glideosome‐associated protein 50/secreted acidic phosphatase, and S‐adenosylmethionine synthetase. 185 The latter five were subsequently ruled out in biochemical validation studies, while PfMDR1 remained a viable target. 185
Figure 17.
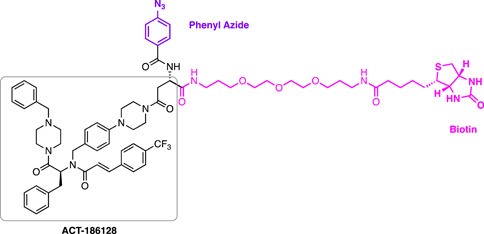
ACT‐186128 chemical probe. [Color figure can be viewed at wileyonlinelibrary.com]
3.5.2. Aspartyl protease inhibitors
Hydroxyethyl amine (HEA)‐based inhibitors have been used to target aspartyl proteases in the Plasmodium parasite 157 , 186 , 187 and were designed as a non‐cleavable transition state mimic for the functional profiling and identification of plasmepsins. 188 At the time, just 5 of a putative 10 plasmepsins (PMs) had been identified in P. falciparum, the digestive vacuole PMs I‐IV (which are known to be non‐essential and redundant in function) 189 and plasmepsin V (which is essential for protein export to the host erythrocyte). 190 , 191 To validate that these HEA inhibitors genuinely bind to PMs, photo‐AfBPPs were developed possessing an azide click chemistry handle, benzophenone photo‐crosslinking group, and a tetramethylrhodamine (TER) fluorescent reporter (Figure 18). 188 The TER reporter enabled in‐gel fluorescent quantification and target binding was demonstrated with recombinant protein. 188 Exposure of the probes to parasite homogenates followed by two‐dimensional (2D) gel electrophoresis and western blot analysis demonstrated that the probe bound to all four digestive PMs. 188
Figure 18.
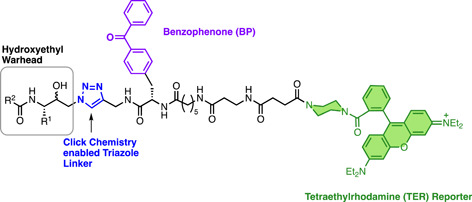
Structure of multifunctional hydroxyethyl chemical probes used for the target identification. Pulldown identified all four known plasmepsins (I–IV) as targets for the hydroxyethyl warhead. [Color figure can be viewed at wileyonlinelibrary.com]
3.5.3. Signal peptidase inhibitors
P. falciparum signal peptide peptidase (PfSPP) is an intra‐membrane aspartyl protease located within the parasite endoplasmic reticulum, responsible for the processing of membrane‐embedded signal peptides left behind by the secretory pathway. 192 PfSPP was hypothesized as a potential target for antimalarial therapy due to the sensitivity of P. falciparum to known human SPP and related aspartyl protease inhibitors such as (Z‐LL)2, LY‐411575, NITD679, and NITD731. 193 To validate SPP as the target protein of these inhibitors in P. falciparum, a multifunctional AfBPP was synthesized based on the peptidomimetic inhibitor (Z‐LL)2 (Figure 19). 193 The probe featured a biotin tag for affinity purification and a benzophenone moiety to facilitate covalent photo‐crosslinking. Photo‐labeling and affinity purification performed with parasite lysate successfully identified PfSPP binding via western blot analysis using anti‐PfSPP for detection. 193 Lysate pretreated with free (Z‐LL)2, LY‐411575, NITD679, and NITD731, and all demonstrated a competitive reduction in PfSPP pulldown by the probe. 193 Together, this validated that the known inhibitors of human SPP also targeted the Plasmodium homolog. 193
Figure 19.
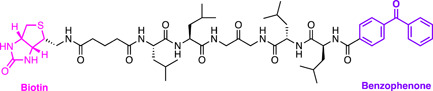
Structure of multifunctional (Z‐LL)2 probe used for the target validation study. A benzophenone moiety enabled photoaffinity labeling, while the biotin moiety enabled affinity pulldown which could be detected via western blot for PfSPP. [Color figure can be viewed at wileyonlinelibrary.com]
3.5.4. Albitiazolium
De novo phospholipid synthesis is an essential process for the growth and survival of Plasmodium parasites. 194 Therefore, the pathway has generated interest as a promising novel target for antimalarial chemotherapy. The primary phospholipid in P. falciparum membranes is phosphatidylcholine which consists of a choline phosphate head group that contains a quaternary ammonium moiety. 194 A series of highly potent antimalarial choline mimics, the bis‐thiazoliums, were developed based on the ability of quaternary ammonium salts to inhibit phospholipid metabolism. 195 Unfortunately, the lead compound stemming from this campaign, Albitiazolium, 196 has since been discontinued in Phase II pediatric trials due to a lack of efficacy. 197 Before this, its exact mechanism of action had been in question but was primarily considered to be impairing choline transport from the plasma. 198 Therefore, a bifunctional chemical probe UA1936 was developed featuring a phenyl azide moiety for covalent photo‐crosslinking as well as a benzyl azido which could be used as a clickable handle for affinity purification and fluorescent labeling (Figure 20). 199 An inactive AfBPP control, UA2050, was also included. 199 In live cells, fluorescent labeling using click chemistry with the benzyl azido group showed partial colocalization with ER and Golgi‐specific antibodies. 199 Pulldown was enabled with an alkyne agarose resin after incubation of the probe in whole saponin‐liberated parasites. These parasites were pretreated with either vehicle control or free Albitiazolium. 199 Two proteins were specifically enriched by the UA1936 AfBPP. One of these proteins is choline/ethanolamine phosphotransferase (CEPT) which performs the final step in phosphatidylcholine and phosphatidylethanolamine biosynthesis. This was unsurprising as Albitiazolium was previously found to inhibit CEPT activity. 198 The other is a protein (PFL1815c) with an uncharacterized function. Only CEPT was competitively displaced by treatment with Albitiazolium, confirming it as the target of this antimalarial compound class. 199
Figure 20.
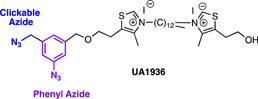
Structure of Albitiazolium bifunctional probe. Photo‐crosslinking and click chemistry affinity purification resulted in the identification of choline/ethanolamine phosphotransferase (CEPT) as a promising target. [Color figure can be viewed at wileyonlinelibrary.com]
3.5.5. Diaminoquinazoline
Diaminoquinazoline compounds are known inhibitors of human histone lysine methyltransferases (HKMT) and were pursued as potential epigenetic regulators of cancer. 200 , 201 , 202 , 203 , 204 , 205 The scaffold was found to have antimalarial activity in a large screen by GSK and subsequently included in the Tres Cantos Antimalarial Set (TCAMS). 9 Since then, the scaffold has been extensively optimized and found to have multistage activity. 206 , 207 , 208 , 209 Despite this, the precise targets of the scaffold remain unclear. There are 10 putative PHKMT enzymes but only one, PfSET7, has been successfully purified for biochemical analysis. 210 To unbiasedly detect targets of BIX‐01294, whose SAR had previously been outlined by the group (Figure 21), 208 , 209 a photo‐crosslinking chemical probe was constructed by Fuchter et al. 211 A diazirine photo‐crosslinking group was chosen due to its small size, as well as an alkyne click chemistry handle for functional analysis. A TAMRA azide (AzT) was ligated using click chemistry for in‐gel fluorescent characterization and demonstrated a competitive profile with the parent BIX‐01294 compound. A TAMRA biotin azide (AzTB) was also used for affinity purification which identified 104 significantly enriched proteins that were filtered through essentiality screening. Only three of these have been found to be essential in P. falciparum: PfnPrx which is involved in reversing DNA damage, 212 NAPL which is a nucleosome assembly protein, 213 and PfHSP110c which is a cytosolic heat shock protein that prevents the aggregation of asparagine‐rich proteins at febrile temperatures. 214 In P. berghei, which is significantly better characterized, 35 of the enriched proteins have been found to be essential. Of these, the most significantly enriched proteins had roles in translational and transcriptional regulation. Histone lysine methyltransferases were absent from the list, although this may reflect a bias for cytosolic proteins in these lysate‐based experiments, as well as inherent instability or a low abundance of these proteins.
Figure 21.
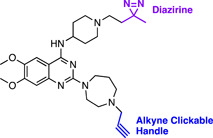
Structure of BIX‐01294 probe. 104 enriched protein targets were identified following photo‐crosslinking and affinity purification, only 35 of which were found to be essential. These targets included those with roles in translational and transcriptional regulation. Notably, histone lysine methyl transferases were absent from the list of targets, which are commonly inhibited by diaminoquinazoline compounds in humans.
4. STABILITY‐BASED METHODS
4.1. Cellular thermal shift assay
A recently adapted method of antimalarial target identification and validation is the cellular thermal shift assay (CETSA). 215 CETSA establishes the target engagement of small molecules in cells or tissues by leveraging an increase in protein thermal stability when bound to a ligand. 216 At elevated temperatures, proteins begin to unfold, exposing hydrophobic residues, and precipitate out of solution. However, ligand binding leads to an increase in protein stability and remain in solution at higher temperatures. 217 CETSA has traditionally been used as a target validation technique where stabilized proteins are detected via western blot (CETSA‐WB). 218 When CETSA is combined with mass spectrometry (CETSA‐MS), it becomes applicable for unbiased, proteome‐wide target identification. 219 A major advantage of CETSA over other techniques is the ability to verify and quantify the binding of high‐affinity targets in live or lysed cells.
The workflow for CETSA‐MS has several key steps (Figure 22). First, the melting behavior of the proteome must be characterized to identify a suitable temperature range for testing. Samples of whole cells, tissues, or lysate are exposed to a gradient of either temperature in the melt curve method or drug concentration in the isothermal drug response (ITDR) method. 220 Following the thermal challenge, whole cells and tissues are lysed and the soluble protein fraction is isolated. 220 Proteins from this fraction can be labeled for quantitative determination and then digested into peptide fragments for analysis by tandem mass spectrometry. 219
Figure 22.
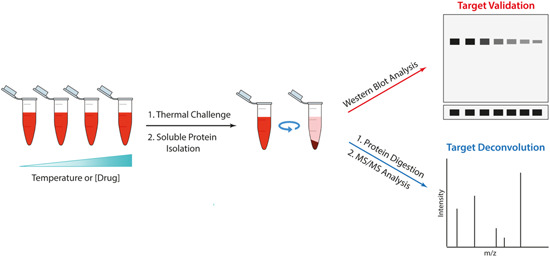
Experimental workflow for CETSA‐MS. Thermal challenge is applied to the samples of interest, modifying either temperature or drug concentration between samples. The soluble protein fraction is isolated, digested, and analyzed by MS/MS or western blot. [Color figure can be viewed at wileyonlinelibrary.com]
While CETSA is a robust method for a wide range of protein targets, not all proteins are amenable to the technique. In general soluble cellular proteins can be easily evaluated through CETSA, however, thermodynamic stabilization is less significant in transmembrane proteins. 220 Approximately 30% of the plasmodium genome is predicted to have at least one transmembrane domain (PlasmoDB). 221 Examples with membrane proteins have been reported but require treatment with detergent to first liberate the proteins. 217 , 222 Additionally, the nature of the protein‐ligand interaction can influence a lack of stabilization. Should a ligand bind to a domain that is not significantly affected by denaturation or exert its effects by modulating interaction with a secondary protein, a stabilization effect will not be seen. 220 Increases in thermal stability may not always be a result of direct binding. Proteins involved with complex metabolic pathways can be stabilized by increases in physiological ligands or proteins as a result of drug treatment. 216 Comparison of CETSA performed with whole‐cells and lysate can be used to control for this factor. 223 CETSA, such as AfBBPs and ABBPs, are prone to false positives. The use of high and nonphysiological concentrations of the compound can result in the detection of false‐positive binding proteins that may not be involved in the antimalarial mechanism of action. Careful selection of the compound concentration and the use of a structurally similar inactive control compound is helpful in decreasing the number of proteins detected and excluding false‐positive or nonphysiologically relevant proteins.
4.1.1. CETSA examples
Quinine, mefloquine, and pyrimethamine
Unbiased CETSA‐MS has recently been adapted to the field of antimalarial target deconvolution. The first example was using quinine and its derivative mefloquine (Figure 23). 223 In this methodology, blood stage P. falciparum parasites and lysate samples were subjected to thermal melt or ITDR conditions; in all testing four separate experiment types. As CETSA had not been previously applied to P. falciparum, the melting properties of the proteome at trophozoite stage were characterized between 37°C and 73°C. T m values could then be calculated for 80% (1821 proteins) of the trophozoite proteome, representing 65% of the overall blood‐stage proteome. 223 Interestingly, proteins in infected RBCs had comparatively less thermal stability than their counterparts found in the lysate. 223 Only 362 human erythrocyte proteins were characterized by this process, due to the disproportionate presence of hemoglobin which complicates the detection of peptides by MS. 224 For the ITDR method, thermal challenge temperature was performed at 51°C to represent the average T m for the proteome and 57°C for the fraction of the proteome that had greater thermal stability. 223
Figure 23.
Chemical structures of antimalarials assessed by CETSA. Pyrimethamine, quinine, and mefloquine were used as examples to develop and validate CETSA‐MS as a target deconvolution method in P. falciparum. As expected, CETSA‐MS identified the target engagement of dihydrofolate reductase‐thymidylate synthase (PfDHFR‐TS) as the target for pyrimethamine whose target was known. CETSA‐MS identified purine nucleoside phosphorylase (PfPNP) as a probable target for quinine and a potential weak target for mefloquine.
To validate the method, ITDR and melt curve assays were performed in the presence of pyrimethamine (Figure 23), a known inhibitor of P. falciparum dihydrofolate reductase‐thymidylate synthase (PfDHFR‐TS). 225 As expected, samples treated with pyrimethamine exhibited a temperature and dose‐dependent stabilization of PfDHFR‐TS. 223 However, no such stabilization could be detected in treated infected RBCs. 223 It was postulated that this could be the result of decreased affinity in a cellular context or due to the presence of a competing ligand such as folate. Validation of the infected RBC method was performed with the broad‐spectrum cysteine protease inhibitor, E64d. 226 , 227 , 228 In this study, it was found that E64d stabilized four proteins, three of which were cysteine proteases (falcipain 2A, falcipain 3, and dipeptidyl aminopeptidase), while one was unexpectedly not a cysteine protease, the DSK2 protein homolog (PF3D7_1113400). 223 The lack of thermal stabilization in cell lysate might also represent the necessity of the cellular environment for target engagement. This can include cellular drug activation, the availability of important cofactors, or the accumulation of the drug in a specific cellular compartment. 223 Therefore, it is recommended to perform experiments with both lysate and whole cells to give greater confidence in the data.
ITDR was performed on cell lysate treated with quinine and MFQ (Figure 23) and purine nucleoside phosphorylase (PfPNP) was the only protein that showed a significant dose‐dependent stabilization. 223 Ribosomal subunits and translation initiation factor 2 were also detected on treatment with MFQ, which is consistent with previous reports of its interaction with the ribosomal complex. 229 In infected RBCs, PfPNP was similarly stabilized by quinine, but interestingly not MFQ. 223 Instead, whole‐cell ITDR experiments with MFQ identified pyruvate kinase II (PfPyKII), although this may represent an increase in its abundance when cells are treated above 37°C and represent a downstream effect of drug binding or a stress response. 223 Hsp70 and a GrpE protein homolog Mge1, two mitochondrial proteins, were also shown to be stabilized by MFQ but only at the highest dose. 223 It was postulated that this may again be a result of an indirect effect on the mitochondrial membrane via reactive oxygen species formed by MFQ. 223 Target engagement of quinine to PfPNP was confirmed by CETSA‐WB where dose‐dependent stabilization was again seen. 223 Indeed, in vitro binding experiments by surface plasmon resonance (SPR) confirmed a K d of 20 nM and 40 µM for quinine and MFQ, respectively. 223 The enzymatic conversion of inosine to hypoxanthine by PfPNP was also found to be inhibited by quinine (K i 138 nM) and mefloquine (K i 5.9 µM). 223 Overall, this data demonstrates PfPNP binds to quinine, but further investigation into the significance of PNP as the mechanism of action is required.
Plasmepsin IX and X inhibitors
Alongside the chemical probe described earlier, Favuzza et al. demonstrated target engagement of their plasmepsin protease targeting compounds using CETSA‐WB. 159 Resistance selection to the initial hit compound WM4 (Figure 24) indicated that PMX was the target. However, the potent tool compound WM382 appeared to have only a low level of cross‐resistance, indicating that it may have an additional target. 159 It was hypothesized this additional target could be the closely related aspartyl protease, PMIX. As no recombinant PMIX was available at the time, CETSA‐WB was implemented to biochemically validate compound binding with HA‐tagged PMIX and PMX parasites. CETSA‐WB performed schizont purified parasite lysate successfully demonstrated that WM382 indeed stabilized both PMIX and X, while the initial hit compound WM4 stabilizes only PMX (Figure 24). 159 The PMV inhibitor W601, used as a control did not induce the stabilization of either PMIX or PMX. 230
Figure 24.
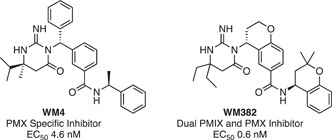
Structures of plasmepsin inhibitors and their specific targets. CETSA‐WB was used to confirm WM382 targets plasmepsin IX and X.
4.2. Drug‐affinity responsive target stability
Drug‐affinity responsive target stability (DARTS) is a relatively new chemo‐proteomic technique used in target identification and validation. Similar to CETSA, the advantage of this technique is that it does not require modification of the compound of interest. 231 DARTS is reliant on the decreased protease susceptibility generally observed upon binding of a ligand to its protein target. 232 Therefore, the addition of proteases to drug‐treated lysate enriches target proteins in the mixture. DARTS is applicable to any cell type and has a relatively simple workflow. 233 In DARTS, varying amounts of protease are used to determine a proteolysis curve, which is related to binding affinity. 234 Enriched proteins are then detected either by western blot or mass spectrometry. 231 Although DARTS is a robust method to demonstrate target engagement, the method has many of the same limitations as CETSA‐MS. One disadvantage is the binding affinity (K d) of the compound of interest may limit the effectiveness of the method, although DARTS has been successfully applied across a range of inhibitory concentrations. 234 Another, is that protein targets involved with complexes or metabolic processes may result in the stabilization of proteins not bound by the compound of interest. 234 A particular limitation of DARTS is that some proteins can be innately resistant to protease degradation. 235
4.2.1. DARTS examples
Torin 2
DARTS was used to complement the aforementioned AfBPP study to identify target proteins of the human mTOR inhibitor Torin 2 in gametocytes. 140 A western blot from the DARTS experiment identified several protein bands that were stabilized in the presence of Torin 2 but not by the inactive Torin 1 control. The western blot bands were subsequently analyzed by mass spectrometry and revealed three proteins also found in the AfBPP study, phosphoribosyl pyrophosphate synthetase, aspartate transcarbamoylase (ATC), and a putative transporter (PF3D7_0914700). 140 As mentioned, ATC was the only validated protein from these putative targets. 141
4.3. Stability of proteins from rates of oxidation
An additional stability‐based technique developed for the purposes of target identification and validation is stability of proteins from rates of oxidation (SPROX). SPROX leverages the fact that ligand‐protein complexes usually reduce the rate of methionine oxidation compared to nonligated proteins. 236 SPROX is considered more limited than CETSA and DARTS, as only proteins that contain multiple methionine residues may be targeted by this method, and the oxidation of methionines is not always mitigated by ligand binding. 237 Additionally, nontarget proteins are not selectively degraded during the method, just chemically altered. 237 Therefore, a negative enrichment of the target is not achieved and results may be more difficult to interpret. However, SPROX does possess a distinct advantage in proteins that act within multimeric complexes. Protein complexes are known to co‐aggregate in thermal profiling with similar melt curves, meaning that CETSA is unlikely to distinguish target engagement between these individual members. 238
4.3.1. SPROX examples
Clemastine
SPROX‐MS has been used alongside CETSA‐MS to determine the target of antihistamine clemastine. 239 Clemastine was discovered to have antimalarial activity against both liver stage P. berghei and erythrocytic stage P. falciparum. 240 However, Plasmodium does not encode any proteins with homology to the human target of clemastine, the histamine H1 receptor. Therefore, stability‐based techniques CETSA and SPROX were employed to determine the molecular target within the erythrocytic P. falciparum parasite. 239 CETSA was able to identify a destabilization in the PfTCP‐1 ring complex (TRiC) in the presence of clemastine. The TRiC is an eight membered heterologous chaperone complex required for de novo cytoskeletal protein folding of actin and tubulin. 241 , 242 However, CETSA was unable to distinguish between the complex members with all eight being destabilized, perhaps due to the aforementioned co‐aggregation effect. 239 With SPROX, compound‐dependent stabilization was observed in just one member of this complex, the delta subunit. 239 This was confirmed biochemically, with the K d to the delta subunit correlating well with the EC50 of the compounds in the parasites. 239 The effect of this inhibition was also confirmed phenotypically with tubulin misfolding observed, leading to the disorientation of mitotic spindles. 239
5. USE OF QUANTITATIVE PROTEOMICS
Proteomics‐based chemical biology techniques such as affinity pulldown and CETSA suffer from many practical issues with low‐affinity drugs and low‐abundance proteins. 98 When these techniques are coupled with quantitative proteomics, the identification of putative targets is significantly more robust. 94 Quantitative proteomics relies on differential heavy isotope labeling between peptide samples, which are then pooled and can be distinguished by mass spectrometry for relative quantification. Isotopically labeled peptide standards of known quantities may also be added to allow for absolute quantification. For example, heavy isotope labeling of either the control or active probes during affinity pulldown can help to establish specific protein interactions from nonspecific protein interactions the relative quantity of robust targets will be enriched only by the active probe. Enhanced detection of nonspecific binding can be important for low‐affinity binders as it reduces the reliance on excessively stringent washing techniques. 94 For CETSA, the ability to perform relative quantification on a number of samples along a temperature or concentration gradient is central to demonstrating target engagement. Together with the ability to quantify relative peptide abundance, pooling samples together in one run serves to reduce instrument running time and variation between sample runs. 243
5.1. Isotope‐coded affinity tagging
The first example of these methods is isotope‐coded affinity tagging (ICAT). Stable isotopes do not alter the steric or physicochemical properties of a protein, but differences in mass are readily distinguishable through mass spec. 244 Traditionally, ICAT uses isotopically labeled tags with a specific cysteine binding moiety and a biotin tag for purification (Figure 25). 245 Proteins are then labeled with an isotopically heavy or light tag, digested, mixed, and analyzed mass spectrally (Figure 25). 246 Proteins that are equally enriched in both heavy and light‐labeled samples are nonspecific binders, where specific enrichment in the active chemical probe sample is considered to correspond to the putative target. 245 This technique is heavily reliant on the proportion of cysteine‐containing proteins within the proteome of interest. Cysteine residues represent just 1.7 mol% of the P. falciparum proteome and are present in only 17.5% of predicted tryptic peptides. 247 , 248 Therefore, many proteins may not be effectively captured by this technique. Additionally, incomplete labeling may occur, further reducing this further. 249
Figure 25.
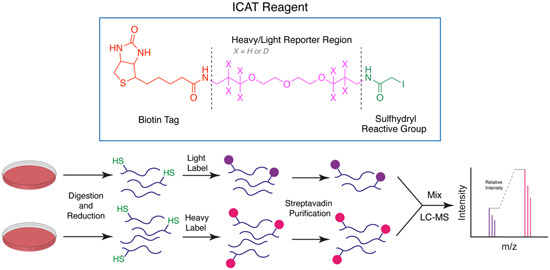
ICAT reagent and experiment workflow. The ICAT reagent contains a biotin tag for purification of labeled peptides, a heavy/light labeled PEG reporter region for mass spectral identification, as well as a sulfhydryl (cysteine) reactive group. ICAT is capable of differentially labeling two different samples which are first digested into tryptic peptides and reduced to expose sulfhydryl groups. The peptides are labeled with reagent and purified via streptavidin binding. The samples are then mixed and analyzed by mass spectrometry, where the relative intensity of the samples can be measured using differences in their mass. [Color figure can be viewed at wileyonlinelibrary.com]
5.2. Isobaric labeling methods
Isobaric labeling is another quantification method that uses chemical labels to modify amino acid side chains of peptide samples. The labels are identical in mass that, upon fragmentation, yield reporter tags with differential heavy 13C and 15N isotope labeling and therefore mass. The general structure of these labels is a reporter group, a mass balancing linker grouping and an amine reactive group, NHS (Figure 26). iTRAQ (isobaric tagging for relative and absolute quantification) and TMT (tandem mass tagging) are examples of isobaric labeling techniques and follow essentially the same principle with minor differences in their label structure. 250 , 251 , 252 iTRAQ is available to simultaneously quantify 4 or 8 samples concurrently, 253 whereas TMT can quantify a larger range of samples with 2, 6, 8, or 10 different labels. 254 Both of these methods can be adapted for absolute protein quantification by the addition of synthetic isobaric peptide standards. 251 , 255 Both the N‐terminus and lysine residues are labeled by isobaric labeling methods; therefore, they are widely applicable to a majority of peptides and proteins in all cell types. 243 Multiplexing other steps into the protocol, such as 2D liquid chromatography and TiO2‐mediated phosphopeptide enrichment, may be useful in quantifying post‐translationally modified proteins. 256
Figure 26.
iTRAQ and TMT are methods of isobaric labeling for quantitative proteomic measurements. [Color figure can be viewed at wileyonlinelibrary.com]
Isobaric labeling methods have been applied in combination with the aforementioned chemical biology techniques for the purposes of antimalarial target identification. TMT labeling has been used by Chibale et al. in the target identification of MMV048 following pulldown with a Sepharose‐linked probe and by Nordlund et al. 2019 for CETSA‐MS to identify targets of quinine, mefloquine, and pyrimethamine. 137 , 223 Additionally, 4‐plex iTRAQ has been used alone as a method of target identification by chemoproteomics to monitor the relative expression levels of P. falciparum proteins following treatment with Doxycycline. 257 Treatment resulted in the differential expression of 40 distinct proteins, many localized to the mitochondria and apicoplast organelles with functions of protein synthesis and processing.
5.3. Stable isotope labeling with amino acids in culture
The gold standard method of quantitative proteomics is called stable isotope labeling with amino acids in culture (SILAC). This method uses heavy isotope labeling of essential amino acids, supplied in culture to cells of interest to distinguish between conditions. 258 Relative quantification is achieved by supplementing one sample with natural amino acids, while the other receives amino acids with heavy 13C or 15N isotope labels. These amino acids are incorporated into newly synthesized proteins, therefore unlike other methods of quantitative proteomics, isotopic labeling is achieved before the experiment. SILAC provides gold standard relative quantification for several reasons. The workflow is compatible with the majority of cell types and beyond routine cell culture, requires no specialized treatment. 259 Labeling occurs in a complete manner and sample mixing occurs at the cellular stage, meaning that MS preparation steps such as purification and protein digestion are done on a uniform sample of pre‐labeled proteins. 260 Other labeling strategies involve the mixing of samples following the preparation of the peptides for MS, meaning that sample loss can occur in these handling steps, affecting the relative quantities of samples. 260
SILAC was developed for P. falciparum in 2004 by Nirmalan et al. to overcome challenges inherent to Plasmodium. 247 Amino acids are sourced via several avenues: digestion of erythrocyte hemoglobin, de novo synthesis as well as import from the host erythrocyte. Isoleucine is the only amino acid not present in human hemoglobin; therefore, it is obtained entirely from exogenous sources. 261 This makes it uniquely suited to quantitative heavy isotope labeling in Plasmodium. Unlike cysteine, isoleucine is also highly abundant, representing 9.2 mole% of the proteome and is present in 60% of tryptically digested peptides. 247 A difference of 7 Da is observed between 13C6–15N1 and 12C6–14N1 isotopes which provides excellent spectral separation. 262 The authors use this newly developed method to identify targets of pyrimethamine and tetracycline through differential proteome analysis of P. falciparum parasites. 247 The method has since been applied to the proteome analysis of chloroquine and artemisinin treatment, 262 but has not been used in combination with chemical biology‐based target identification. There are perhaps a few reasons for this. Although lysine and arginine isotopologues have been commercially developed for SILAC multiplexing of up to four samples, 263 no such technology is available with isoleucine for the above method in P. falciparum. This greatly limits experimental design and precludes use with CETSA altogether. The expense of SILAC's isotopic reagents also hinders its widespread use in the laboratory. 103
5.4. Label‐free methods
The most widely applied methods of quantitative proteomics in malaria drug discovery are label‐free methods. While label‐based methods are considered to be more accurate, expensive reagents, a limited number of samples, and limits in sample applicability make label‐free methods particularly attractive. 264 Label‐free methods rely on liquid chromatography‐tandem mass spectrometry (LC‐MS/MS) and are based on two methods of detecting peptide abundance: ion intensity and spectral number. 265
Spectral counting uses the number of MS/MS events corresponding to a single peptide to measure protein enrichment. 266 A higher abundance of protein will result in a greater number of tandem MS spectra generated. 267 More accurate quantification is enabled by correcting for overall protein length (NSAF, dNASF, and SIN). 268 , 269 , 270 or by the theoretical number of tryptic peptides (emPAI and APEX). 271 An advantage of spectral counting is its simplified workflow. Unlike in ion intensity measurement, computational pre‐processing of data is not required and therefore can be employed immediately. 272 However, the primary issue with spectral counting is the presence of nonunique peptides or those shared by multiple proteins. It has been estimated that around 50% of tryptic peptides identified in databases are nonunique and some proteins are entirely composed of nonunique tryptic peptides. 272 This has previously been accounted for by excluding them from the analysis altogether, distributing them to proteins based on another property such as the abundance of other corresponding unique peptides, or by ignoring that they are shared and counting them multiple times. 264 The latter method is now considered inappropriate as many proteins will not be accurately captured. 273 emPAI has been extensively used in quantitative proteomics for antimalarial research 154 , 164 , 165 , 174 , 183 , 199 as it is incorporated into the protein identification search engine Mascot. 274
The ion intensity method relies on measuring the area of an MS1 peak (AUC) at a given mass‐to‐charge ratio (m/z). A linear correlation exists between this area and peptide concentration (r 2 = 0.991–0.9978), allowing for direct comparison between identical peptides for accurate determination of their relative quantity. 275 , 276 Approximate quantification of peptides is enabled through several normalization methods. Like spectral counting methods, normalization can be achieved by accounting for the number of theoretical peptides (iBAQ and riBAQ), 277 , 278 the molecular mass of the protein (TPA), 279 or taking the intensity of the top three most intense peptides (TOP3). 280 MS/MS spectra must also be subsequently obtained to confirm the identity of each peptide. In complex peptide mixtures, it is necessary to carefully optimize the statistical and computational parameters of quantification. 281 In particular, technical variations in peptide retention time, co‐elution, and background noise are concerns of this method. 265 High‐resolution mass spectrometers and computational methods aid in aligning data between runs. 267 Programs MaxQuant and Progenesis QI have been used in antimalarial target ID to determine relative quantification through AUC. 173 , 185 , 211 This is considered a main disadvantage of AUC as it adds significant complexity to experimental optimization and quantification can be variable depending on which algorithm is used. 272 However, overall AUC is considered more accurate than spectral counting due to the higher stochasticity of [MS/MS] methods.
6. FUTURE DIRECTIONS
Chemoproteomic‐based target deconvolution is rapidly evolving with older technologies being incrementally improved and new technologies being developed. To optimize the application of chemical probes, it would be beneficial to develop a plasmodial database similar to the human CRAPome which acts as a repository for common contaminant proteins in affinity purification–mass spectrometry. 156 Meanwhile, other methods remain to be applied to full antimalarial target deconvolution, such as PROTACs.
6.1. Plasmodium CRAPome
As has been previously highlighted, nonspecific binding to the support resin is a prominent issue for affinity‐based protein purification techniques. While inactive control probes can effectively account for this issue, the availability of these controls can at times be limited. Negative controls taken at an individual level may also be sensitive to small variations in sample preparation and fail to give a complete picture of nonspecific binding. Fortunately, nonspecific binding is largely independent of the bait molecule and more likely due to the chosen resin. With this in mind, the CRAPome or the Contaminant Repository for Affinity Purification was created. This database collates and annotates published MS proteomic data derived from negative control probes which could be used to score query MS data. 156 This database currently exists only for the human, mouse, drosophila, yeast, and E. coli proteomes with a range of resin types. 282 Given that affinity‐based purification is extensively used as a target deconvolution method in Plasmodium, we feel that a similar database would be beneficial to the identification of high‐quality targets.
6.2. Proteolysis‐targeting chimeras
Proteolysis‐targeting chimeras (PROTACs) are bifunctional molecules that artificially enhance the clearance of a protein by recruiting cellular machinery that facilitates its degradation. 283 They can be considered a kind of modified chemical probe, containing a drug pharmacophore conjugated via a linker to an E3 ligase binding moiety (Figure 27). They bring into proximity the protein target of a compound with machinery that can ubiquitylate it and thereby destine it for proteasomal degradation. Combined with mass spectrometry PROTACs have great potential for the identification and validation of drug targets, acting as a form of chemical knockdown which is fast, dose‐dependent, and reversible. 284 They have been used previously to identify the targets of phenotypically discovered anticancer compounds in human cells, 285 , 286 but not yet in Plasmodium. Plasmodium possesses homologs of the eukaryotic and prokaryotic proteosomes as well as the cyanobacterial Clp protease. 287 There are several E3 ligase proteins present in the P. falciparum proteome, with RING (really interesting new gene) finger E3's being the most abundant. 288 It has been shown that Plasmodia rely heavily on protein degradation for development and stress response in all lifecycle stages. 289 While a functional ubiquitin proteasomal degradation system almost certainly exists in Plasmodium, future work in this space will require multi‐omic characterization of E3 ligases, the design of suitable E3 ligands, and by extension, the PROTAC molecules themselves. 290 The high molecular weight, lipophilicity, rotatable bonds, and polar surface area necessitated by heterodimeric bifunctional compounds can be a barrier to cell permeability and therefore their utility in cell‐based assays. 291 Therefore, there is some time before the widespread use of this technology in Plasmodium parasites.
Figure 27.
Mechanism of action of proteolysis‐targeting chimeras (PROTACs). PROTACs are heterodimeric bifunctional molecules that link an E3 ligand to a drug molecule. By doing this, they bring into proximity a target with complexes that polyubiquitylate it and target it for degradation by the proteasome. Coupled with mass spectrometry, these molecules can detect the targets of drug molecules identified by phenotypic screening. [Color figure can be viewed at wileyonlinelibrary.com]
7. CONCLUSIONS
With resistance arising to currently available antimalarials and many of those undergoing clinical development, the need for novel therapeutics continues. High‐throughput screening continues to be a prominent method by which these chemical entities are discovered. This necessitates robust and informative techniques for target identification to determine the drug mechanism of action. Today, target identification necessarily pulls techniques from many disciplines to deconvolute complex protein mixtures. Chemical biology represents an emerging field in target identification and engagement that is complementary to a range of others, such as drug resistance and metabolomics. It is an incredibly direct technique for target identification and engagement, providing an explicit link between the chemical entity and the biological effect. For these reasons, it has been extensively applied to the field of antimalarial drug discovery.
Chemical probes have proven to be diversly applicable across chemotypes and mechanisms of action. Therefore, it is unsurprising that antimalarial target identification has heavily relied on this technique. An evolution in chemical structure from the traditional Sepharose‐conjugated probes has enabled this expansion in functionality. Biocompatible functional tags such as biotin or alkyne/azide click chemistry handles have enabled their use in live cells, rather than using cell lysate with a resin‐tethered probe. Similarly, fluorescently tagged probes allow valuable information on compartmental localization in live cells to be gained. Photo‐crosslinking probes enhance the sensitivity of methods in Sections 3.2 and 3.4 and have been utilized in complex, multifunctional structures (Table 1) Overall, chemical probes are an information‐rich target identification method that must be employed with careful controls and verified through other distinct approaches.
Table 1.
Summary of chemo‐proteomic methods used in target identification and engagement.
| Method | Method strengths | Method weaknesses | Exemplar and target | Refs. | |
|---|---|---|---|---|---|
| AfBPP |
|
|
WM382/PfPMX | 159 | |
| ABPP |
|
|
Salinipostin A/ten Pf α/β serine hydrolases | 178 | |
| Photo‐affinity BPP |
|
|
(Z‐LL)2/PfSPP | 193 | |
| CETSA‐MS |
|
|
Quinine/PfPNP | 223 | |
| DARTS |
|
|
Torin 2/PfATC | 140 | |
| SPROX |
|
|
Clemastine/PfTRiC | 239 | |
Stability‐based techniques such as CETSA, DARTS, and SPROX have been used with success in antimalarial target identification. These are relatively new methods to measure target engagement that does not require the modification of the drug of interest. For CETSA, the ability to assess target engagement in the context of both cellular lysates and in live cells provides a biologically relevant result. However, their lack of applicability to all targets remains an issue. Membrane‐bound targets and those involved with complex multi‐protein oligomers or pathways can provide misleading results (Table 1).
The advent of chemical biology target deconvolution owes its roots to advancements in proteomics, in particular mass spectrometry modified to allow quantitative assessment. Isotopic labelings, including ICAT, iTRAQ, TMT, and SILAC as well as label‐free methods, allow for relative or absolute quantification of protein levels across different samples. As chemical biology often involves positive or negative enrichment at the proteomic level, these techniques provide more accurate and sensitive results than traditional 2‐DE gel proteomics.
Finally, there are several other chemical biology techniques that have not yet been used for the purposes of antimalarial target deconvolution such as PROTACs. These methods, including PROTACs, could represent a new area of development for antimalarial research in terms of both therapeutics and target deconvolution.
ACKNOWLEDGMENTS
This work was supported by the National Health and Medical Research Council of Australia (Development Grants 2014427 and 2018883 to B.E.S. and A.F.C.; Ideas Grant 2001073 to W.N.), the Australian Cancer Research Foundation, the Victorian State Government Operational Infrastructure Support, and Australian Government NHMRC IRIISS. A.F.C. is a Howard Hughes International Scholar and an Australia Fellow of the NHMRC. B.E.S. is a Corin Centenary Fellow. Open access publishing facilitated by The University of Melbourne, as part of the Wiley ‐ The University of Melbourne agreement via the Council of Australian University Librarians.
Biographies
Brodie L. Bailey is a PhD graudate from the Chemical Biology and Infection & Immunity Divisions at the Walter and Eliza Hall Institute. Dr Bailey received an undergraduate degree in Medicinal Chemistry from the University of Auckland and as part of the degree researched the synthesis of HIF prolyl hydroxylase inhibitors for the treatment of anemia. Dr Bailey's PhD research focused on the development of a novel antimalarial series and the identification of its molecular target. Dr Bailey's interests include antimalarial drug discovery and emerging chemical biology techniques.
Dr William Nguyen is a mid‐career researcher with expertise in Medicinal Chemistry and early‐stage Drug Discovery. Nguyen is currently appointed as a Senior Research Fellow at the Walter and Eliza Hall Institute. Dr Nguyen obtained a PhD from Monash University researching Kv1.3 agonists in autoimmune diseases. Dr Nguyen held a postdoctoral position at Scripps Research Institute (Florida, USA) before joining the Walter and Eliza Hall Institute as a Research Fellow in 2015. Dr Nguyen's past research includes the development of orexin‐1 antagonists for the treatment of nicotine addiction, NEMO‐binding domain mimetics for inhibiting IKK/NF‐κB toward treating neurodegenerative disorders, and latency‐reversing agents for the treatment of HIV. Dr Nguyen's current research is focused on developing tool compounds to uncover dissect biological pathways and uncover new survival mechanisms of parasites that cause schistosomiasis and malaria.
Prof Alan F. Cowman AC FRS FAA FRSE FAHMS is Deputy Director at the Walter and Eliza Hall Institute of Medical Research (WEHI). Currently, he has a Senior Principal Research Fellowship from the National Health and Medical Research Council of Australia. Prof Alan Cowman was elected as a Fellow of the Royal Society (FRS) in 2011, the Australian Academy of Sciences (FAA) in 2009, and appointed a Companion of the Order of Australia (AC) in 2019. Prof Cowman is a recipient of the Glaxo Award for Advanced Research in Infectious Diseases, Gottschalk Medal for Medical Science and Biology from the Australian Academy of Sciences, Boehringer‐Mannheim Medal, Glaxo‐Wellcome Australia Medal, the Howard Taylor Ricketts Medal, the Victoria Prize from the Victorian Government and the Mahathir Science Prize from the Mahathir Science Award Foundation. Prof Cowman's research is focused on essential mechanisms that drive the translation and development of tools for the elimination and eradication of Malaria. Some of the key scientific contributions to the field include understanding effectors of antimalarial resistance, parasite‐host protein trafficking and mechanisms of P. falciparum parasite invasion into erythrocytes, and identification of vaccine candidates.
Dr Brad E. Sleebs is a medicinal chemist with extensive experience in early‐stage Drug Discovery and Chemical Biology. Dr Sleebs received his PhD from La Trobe University and joined The Walter and Eliza Hall Institute as a Research Officer in 2005. In 2018 Dr Sleebs was appointed as a Laboratory Head in the Chemical Biology Division at the Walter and Eliza Hall Institute. Dr Sleebs is a Fellow of the Royal Australian Chemical Institute, an Coirn Fellow, and is a recipient of the RACI Peter Andrews Award for Innovation in Medicinal Chemistry. Dr Sleebs' past research includes the development of anxiolytics and agents that target the BH3 family of proteins for the treatment of blood cancers. He directly contributed to collaborations with Bionomics Ltd that resulted in a Phase II clinical candidate for the treatment of anxiety and with Genentech and AbbVie developing a pre‐clinical candidate for targeting blood cancers. Dr Sleebs' current research focuses on developing small molecule probes to better understand biological processes that are essential to the survival of the malaria parasite and in collaboration with industry partners the development of novel antimalarial agents.
Bailey BL, Nguyen W, Cowman AF, Sleebs BE. Chemo‐proteomics in antimalarial target identification and engagement. Med Res Rev. 2023;43:2303‐2351. 10.1002/med.21975
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed.
REFERENCES
- 1. Tse EG, Korsik M, Todd MH. The past, present and future of anti‐malarial medicines. Malar J. 2019;18(1):93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. World Health Organization . World Malaria Report 2021. Geneva: World Health Organization; 2021. [Google Scholar]
- 3. Cibulskis RE, Alonso P, Aponte J, et al. Malaria: global progress 2000–2015 and future challenges. Infect Dis Poverty. 2016;5(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cui L, Mharakurwa S, Ndiaye D, Rathod PK, Rosenthal PJ. Antimalarial drug resistance: literature review and activities and findings of the ICEMR network. Am J Trop Med Hyg. 2015;93(3):57‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korenromp EL, Williams BG, Gouws E, Dye C, Snow RW. Measurement of trends in childhood malaria mortality in Africa: an assessment of progress toward targets based on verbal autopsy. Lancet Infect Dis. 2003;3(6):349‐358. [DOI] [PubMed] [Google Scholar]
- 6. Trape JF, Pison G, Preziosi MP, et al. Impact of chloroquine resistance on malaria mortality. C R Acad Sci III. 1998;321(8):689‐697. [DOI] [PubMed] [Google Scholar]
- 7. World Health Organization . Global Technical Strategy for Malaria. World Health Organization; 2015. [Google Scholar]
- 8. Laurens MB. RTS,S/AS01 vaccine (Mosquirix™): an overview. Hum Vaccines Immunother. 2020;16(3):480‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gamo F‐J, Sanz LM, Vidal J, et al. Thousands of chemical starting points for antimalarial lead identification. Nature. 2010;465(7296):305‐310. [DOI] [PubMed] [Google Scholar]
- 10. Guiguemde WA, Shelat AA, Bouck D, et al. Chemical genetics of Plasmodium falciparum . Nature. 2010;465(7296):311‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guiguemde WA, Shelat AA, Garcia‐Bustos JF, Diagana TT, Gamo F‐J, Guy RK. Global phenotypic screening for antimalarials. Chem Biol. 2012;19(1):116‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bermúdez M, Moreno‐Pérez DA, Arévalo‐Pinzón G, Curtidor H, Patarroyo MA. Plasmodium vivax in vitro continuous culture: the spoke in the wheel. Malar J. 2018;17(1):301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duffy S, Avery VM. Identification of inhibitors of Plasmodium falciparum gametocyte development. Malar J. 2013;12(1):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Roth A, Maher SP, Conway AJ, et al. A comprehensive model for assessment of liver stage therapies targeting Plasmodium vivax and Plasmodium falciparum . Nat Commun. 2018;9(1):1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Meister S, Plouffe DM, Kuhen KL, et al. Imaging of Plasmodium liver stages to drive next‐generation antimalarial drug discovery. Science. 2011;334(6061):1372‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schafer C, Dambrauskas N, Steel RW, et al. A recombinant antibody against Plasmodium vivax UIS4 for distinguishing replicating from dormant liver stages. Malar J. 2018;17(1):370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Maher S, Vantaux A, Cooper C, et al. A phenotypic screen for the liver stages of Plasmodium vivax . Bio‐protocol. 2021;11(23):e4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mullard A. Malaria Medicine box expands. Nat Rev Drug Discov. 2018;17(10):693‐695. [DOI] [PubMed] [Google Scholar]
- 19. Younis Y, Douelle F, Feng T‐S, et al. 3,5‐diaryl‐2‐aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J Med Chem. 2012;55(7):3479‐3487. [DOI] [PubMed] [Google Scholar]
- 20. Plouffe D, Brinker A, McNamara C, et al. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high‐throughput screen. Proc Natl Acad Sci USA. 2008;105(26):9059‐9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ashton TD, Devine SM, Möhrle JJ, et al. The development process for discovery and clinical advancement of modern antimalarials. J Med Chem. 2019;62(23):10526‐10562. [DOI] [PubMed] [Google Scholar]
- 22. Coteron JM, Marco M, Esquivias J, et al. Structure‐guided lead optimization of triazolopyrimidine‐ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54(15):5540‐5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baldwin J, Michnoff CH, Malmquist NA, et al. High‐throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280(23):21847‐21853. [DOI] [PubMed] [Google Scholar]
- 24. Swinney DC. Phenotypic vs. target‐based drug discovery for first‐in‐class medicines. Clin Pharm Ther. 2013;93(4):299‐301. [DOI] [PubMed] [Google Scholar]
- 25. Ashley EA, Phyo AP. Drugs in development for malaria. Drugs. 2018;78(9):861‐879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng W, Thorne N, McKew JC. Phenotypic screens as a renewed approach for drug discovery. Drug Discov Today. 2013;18(21‐22):1067‐1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Greenbaum D. Is chemical genetics the new frontier for malaria biology? Trends Pharmacol Sci. 2008;29(2):51‐56. [DOI] [PubMed] [Google Scholar]
- 28. Nzila A, Mwai L. In vitro selection of Plasmodium falciparum drug‐resistant parasite lines. J Antimicrob Chemother. 2010;65(3):390‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flannery EL, Fidock DA, Winzeler EA. Using genetic methods to define the targets of compounds with antimalarial activity. J Med Chem. 2013;56(20):7761‐7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Creek DJ, Barrett MP. Determination of antiprotozoal drug mechanisms by metabolomics approaches. Parasitology. 2014;141(1):83‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sexton AE, Doerig C, Creek DJ, Carvalho TG. Post‐genomic approaches to understanding malaria parasite biology: linking genes to biological functions. ACS Infect Dis. 2019;5(8):1269‐1278. [DOI] [PubMed] [Google Scholar]
- 32. Cowell AN, Winzeler EA. Advances in omics‐based methods to identify novel targets for malaria and other parasitic protozoan infections. Genome Med. 2019;11(1):63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Luth MR, Gupta P, Ottilie S, Winzeler EA. Using in vitro evolution and whole genome analysis to discover next generation targets for antimalarial drug discovery. ACS Infect Dis. 2018;4(3):301‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Murithi JM, Owen ES, Istvan ES, et al. Combining stage specificity and metabolomic profiling to advance antimalarial drug discovery. Cell Chem Biol. 2020;27(2):158‐171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Creek DJ, Chua HH, Cobbold SA, et al. Metabolomics‐based screening of the Malaria Box reveals both novel and established mechanisms of action. Antimicrob Agents Chemother. 2016;60(11):6650‐6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gunasekera AM, Myrick A, Roch KL, Winzeler E, Wirth DF. Plasmodium falciparum: Genome wide perturbations in transcript profiles among mixed stage cultures after chloroquine treatment. Exp Parasitol. 2007;117(1):87‐92. [DOI] [PubMed] [Google Scholar]
- 37. Sawada R, Iwata M, Tabei Y, Yamato H, Yamanishi Y. Predicting inhibitory and activatory drug targets by chemically and genetically perturbed transcriptome signatures. Sci Rep. 2018;8(1):156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Challis MP, Devine SM, Creek DJ. Current and emerging target identification methods for novel antimalarials. Int J Parasitol Drugs Drug Resist. 2022;20:135‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Carvalho LAR, Bernardes GJL. The impact of activity‐based protein profiling in malaria drug discovery. ChemMedChem. 2022;17:e202200174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lu K‐Y, Mansfield CR, Fitzgerald MC, Derbyshire ER. Chemoproteomics for Plasmodium parasite drug target discovery. ChemBioChem. 2021;22(16):2591‐2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Markus MB. Malaria: origin of the term “hypnozoite”. J Hist Biol. 2011;44(4):781‐786. [DOI] [PubMed] [Google Scholar]
- 42. Bunnik EM, Cook KB, Varoquaux N, et al. Changes in genome organization of parasite‐specific gene families during the Plasmodium transmission stages. Nat Commun. 2018;9(1):1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Burrows JN, Duparc S, Gutteridge WE, et al. New developments in anti‐malarial target candidate and product profiles. Malar J. 2017;16(1):26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Swearingen KE, Lindner SE. Plasmodium parasites viewed through proteomics. Trends Parasitol. 2018;34(11):945‐960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kennedy M, Fishbaugher ME, Vaughan AM, et al. A rapid and scalable density gradient purification method for Plasmodium sporozoites. Malar J. 2012;11:421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hollingdale MR, Leef JL, McCullough M, Beaudoin RL. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei from sporozoites. Science. 1981;213(4511):1021‐1022. [DOI] [PubMed] [Google Scholar]
- 47. Hollingdale MR, Leland P, Schwartz AL. In vitro cultivation of the exoerythrocytic stage of Plasmodium berghei in a hepatoma cell line. Am J Trop Med Hyg. 1983;32(4):682‐684. [DOI] [PubMed] [Google Scholar]
- 48. Calvo‐Calle JM, Moreno A, Eling WMC, Nardin EH. In vitro development of infectious liver stages of P. yoelii and P. berghei malaria in human cell lines. Exp Parasitol. 1994;79(3):362‐373. [DOI] [PubMed] [Google Scholar]
- 49. Long GW, Leath S, Schuman R, et al. Cultivation of the exoerythrocytic stage of Plasmodium berghei in primary cultures of mouse hepatocytes and continuous mouse cell lines. In Vitro Cell Dev Biol. 1989;25(9):857‐862. [DOI] [PubMed] [Google Scholar]
- 50. Mazier D, Beaudoin RL, Mellouk S, et al. Complete development of hepatic stages of Plasmodium falciparum in vitro. Science. 1985;227(4685):440‐442. [DOI] [PubMed] [Google Scholar]
- 51. Mazier D, Collins WE, Mellouk S, et al. Plasmodium ovale: in vitro development of hepatic stages. Exp Parasitol. 1987;64(3):393‐400. [DOI] [PubMed] [Google Scholar]
- 52. Mazier D, Landau I, Druilhe P, et al. Cultivation of the liver forms of Plasmodium vivax in human hepatocytes. Nature. 1984;307(5949):367‐369. [DOI] [PubMed] [Google Scholar]
- 53. Collins WE, Campbell CC, Hollingdale MR. In vitro culture of exoerythrocytic parasites of the North Korean strain of Plasmodium vivax in hepatoma cells. Am J Trop Med Hyg. 1986;35(2):275‐276. [DOI] [PubMed] [Google Scholar]
- 54. Hollingdale MR, Schwartz AL, Campbell CC, Collins WE. In vitro culture of two populations (dividing and nondividing) of exoerythrocytic parasites of Plasmodium vivax . Am J Trop Med Hyg. 1985;34(2):216‐222. [DOI] [PubMed] [Google Scholar]
- 55. Brewer TG, Leelaudomlipi S, Coleman RE, et al. Establishment of a human hepatocyte line that supports in vitro development of the exo‐erythrocytic stages of the malaria parasites Plasmodium falciparum and P. vivax . Am J Trop Med Hyg. 2006;74(5):708‐715. [PubMed] [Google Scholar]
- 56. Tweedell RE, Tao D, Hamerly T, et al. The selection of a hepatocyte cell line susceptible to Plasmodium falciparum sporozoite invasion that is associated with expression of glypican‐3. Front Microbiol. 2019;10:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Pewkliang Y, Rungin S, Lerdpanyangam K, et al. A novel immortalized hepatocyte‐like cell line (imHC) supports in vitro liver stage development of the human malarial parasite Plasmodium vivax . Malar J. 2018;17(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Trager W, Jensen JB. Human malaria parasites in continuous culture. Science. 1976;193(4254):673‐675. [DOI] [PubMed] [Google Scholar]
- 59. Moon RW, Hall J, Rangkuti F, et al. Adaptation of the genetically tractable malaria pathogen Plasmodium knowlesi to continuous culture in human erythrocytes. Proc Natl Acad Sci USA. 2013;110(2):531‐536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferrer J, Rosal MD, Vidal JM, et al. Effect of the haematocrit layer geometry on Plasmodium falciparum static thin‐layer in vitro cultures. Malar J. 2008;7:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Noulin F, Borlon C, Van Den Abbeele J, D'Alessandro U, Erhart A. 1912‐2012: a century of research on Plasmodium vivax in vitro culture. Trends Parasitol. 2013;29(6):286‐294. [DOI] [PubMed] [Google Scholar]
- 62. Lambros C, Vanderberg JP. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol. 1979;65(3):418‐420. [PubMed] [Google Scholar]
- 63. Ribaut C, Berry A, Chevalley S, et al. Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malar J. 2008;7(1):45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Nillni EA, Londner MV, Spira DT. A simple method for separation of uninfected erythrocytes from those infected with Plasmodium berghei and for isolation of artificially released parasites. Z Parasitenkd Parasitol Res. 1980;64(3):279‐284. [DOI] [PubMed] [Google Scholar]
- 65. Tripathi AK, Mlambo G, Kanatani S, Sinnis P, Dimopoulos G. Plasmodium falciparum gametocyte culture and mosquito infection through artificial membrane feeding. J Vis Exp. 2020;(161). 10.3791/61426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ifediba T, Vanderberg JP. Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature. 1981;294(5839):364‐366. [DOI] [PubMed] [Google Scholar]
- 67. Armistead JS, Moraes Barros RR, Gibson TJ, et al. Infection of mosquitoes from in vitro cultivated Plasmodium knowlesi H strain. Int J Parasitol. 2018;48(8):601‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Delves MJ, Straschil U, Ruecker A, et al. Routine in vitro culture of P. Falciparum gametocytes to evaluate novel transmission‐blocking interventions. Nat Protoc. 2016;11(9):1668‐1680. [DOI] [PubMed] [Google Scholar]
- 69. Fivelman QL, McRobert L, Sharp S, et al. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Mol Biochem Parasitol. 2007;154(1):119‐123. [DOI] [PubMed] [Google Scholar]
- 70. Eksi S, Haile Y, Furuya T, Ma L, Su X, Williamson KC. Identification of a subtelomeric gene family expressed during the asexual‐sexual stage transition in Plasmodium falciparum . Mol Biochem Parasitol. 2005;143(1):90‐99. [DOI] [PubMed] [Google Scholar]
- 71. Brockelman CR. Conditions favoring gametocytogenesis in the continuous culture of Plasmodium falciparum . J Protozool. 1982;29(3):454‐458. [DOI] [PubMed] [Google Scholar]
- 72. Boltryk SD, Passecker A, Alder A, et al. CRISPR/Cas9‐engineered inducible gametocyte producer lines as a valuable tool for Plasmodium falciparum malaria transmission research. Nat Commun. 2021;12(1):4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Carter R, Beach RF. Gametogenesis in culture by gametocytes of Plasmodium falciparum . Nature. 1977;270(5634):240‐241. [DOI] [PubMed] [Google Scholar]
- 74. Janse CJ, Mons B, Rouwenhorst RJ, van der Klooster PFJ, Overdulve JP, van der Kaay HJ. In vitro formation of ookinetes and functional maturity of Plasmodium berghei gametocytes. Parasitology. 1985;91(1):19‐29. [DOI] [PubMed] [Google Scholar]
- 75. Sinden RE, Hartley RH, Winger L. The development of Plasmodium ookinetes in vitro: an ultrastructural study including a description of meiotic division. Parasitology. 1985;91(2):227‐244. [DOI] [PubMed] [Google Scholar]
- 76. Carter EH, Suhrbier A, Beckers PJA, Sinden RE. The in vitro cultivation of P. falciparum ookinetes, and their enrichment on Nycodenz density gradients. Parasitology. 1987;95:25‐30. [DOI] [PubMed] [Google Scholar]
- 77. Bounkeua V, Li F, Vinetz JM. In vitro generation of Plasmodium falciparum ookinetes. Am J Trop Med Hyg. 2010;83(6):1187‐1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Al‐Olayan EM, Beetsma AL, Butcher GA, Sinden RE, Hurd H. Complete development of mosquito phases of the malaria parasite in vitro. Science. 2002;295(5555):677‐679. [DOI] [PubMed] [Google Scholar]
- 79. Yeo AET, Rieckmann KH. Prolonged exposure of Plasmodium falciparum to ciprofloxacin increases anti‐malarial activity. J Parasitol. 1994;80(1):158‐160. [PubMed] [Google Scholar]
- 80. Yeo AET, Rieckmann KH. Increased antimalarial activity of azithromycin during prolonged exposure of Plasmodium falciparum in vitro. Int J Parasitol. 1995;25(4):531‐532. [DOI] [PubMed] [Google Scholar]
- 81. Divo AA, Geary TG, Jensen JB. Oxygen‐ and time‐dependent effects of antibiotics and selected mitochondrial inhibitors on Plasmodium falciparum in culture. Antimicrob Agents Chemother. 1985;27(1):21‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Lamarque M, Tastet C, Poncet J, et al. Food vacuole proteome of the malarial parasite Plasmodium falciparum . Proteom Clin Appl. 2008;2(9):1361‐1374. [DOI] [PubMed] [Google Scholar]
- 83. Sam‐Yellowe TY, Florens L, Wang T, et al. Proteome analysis of rhoptry‐enriched fractions isolated from Plasmodium merozoites. J Proteome Res. 2004;3(5):995‐1001. [DOI] [PubMed] [Google Scholar]
- 84. Lal K, Prieto JH, Bromley E, et al. Characterisation of Plasmodium invasive organelles; an ookinete microneme proteome. Proteomics. 2009;9(5):1142‐1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Oehring SC, Woodcroft BJ, Moes S, et al. Organellar proteomics reveals hundreds of novel nuclear proteins in the malaria parasite Plasmodium falciparum . Genome Biol. 2012;13(11):R108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Hata M, Sato S, Kita K. Method for the separation of mitochondria and apicoplast from the malaria parasite Plasmodium falciparum . Parasitol Int. 2019;69:99‐102. [DOI] [PubMed] [Google Scholar]
- 87. Christophers SR, Fulton JD. Experiments with isolated malaria parasites (Plasmodium Knowlesi) free from red cells. Ann Trop Med Parasitol. 1939;33(2):161‐170. [Google Scholar]
- 88. O'Cualain RD, Hyde JE, Sims PF. A protein‐centric approach for the identification of folate enzymes from the malarial parasite, Plasmodium falciparum, using OFFGEL™ solution‐based isoelectric focussing and mass spectrometry. Malar J. 2010;9:286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Sargeant T, Marti M, Caler E, et al. Lineage‐specific expansion of proteins exported to erythrocytes in malaria parasites. Genome Biol. 2006;7(2):R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Maier AG, Rug M, O'Neill MT, et al. Exported proteins required for virulence and rigidity of Plasmodium falciparum‐infected human erythrocytes. Cell. 2008;134(1):48‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Jonsdottir TK, Gabriela M, Crabb BS, F. de koning‐Ward T, Gilson PR. Defining the essential exportome of the Malaria parasite. Trends Parasitol. 2021;37(7):664‐675. [DOI] [PubMed] [Google Scholar]
- 92. Galmozzi A, Dominguez E, Cravatt BF, Saez E. Application of activity‐based protein profiling to study enzyme function in adipocytes. Meth Enzymol. 2014;538:151‐169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Dormán G, Flachner B, Hajdú I, András CD. Target Identification and Polypharmacology of Nutraceuticals. In: Elsevier Inc; 2016:263‐286. [Google Scholar]
- 94. Kawatani M, Osada H. Affinity‐based target identification for bioactive small molecules. MedChemComm. 2014;5(3):277‐287. [Google Scholar]
- 95. Gilbert IH. Drug discovery for neglected diseases: molecular target‐based and phenotypic approaches. J Med Chem. 2013;56(20):7719‐7726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Van Vleet TR, Liguori MJ, Lynch JJ III, Rao M, Warder S. Screening strategies and methods for better off‐target liability prediction and identification of small‐molecule pharmaceuticals. SLAS Discovery. 2019;24(1):1‐24. [DOI] [PubMed] [Google Scholar]
- 97. Graves PR, Kwiek JJ, Fadden P, et al. Discovery of novel targets of quinoline drugs in the human purine binding proteome. Mol Pharmacol. 2002;62(6):1364‐1372. [DOI] [PubMed] [Google Scholar]
- 98. Zheng W, Li G, Li X. Affinity purification in target identification: the specificity challenge. Arch Pharmacal Res. 2015;38(9):1661‐1685. [DOI] [PubMed] [Google Scholar]
- 99. Dunham WH, Mullin M, Gingras A‐C. Affinity‐purification coupled to mass spectrometry: basic principles and strategies. Proteomics. 2012;12(10):1576‐1590. [DOI] [PubMed] [Google Scholar]
- 100. Cong F, Cheung AK, Huang SMA. Chemical genetics–based target identification in drug discovery. Annu Rev Pharmacol Toxicol. 2012;52(1):57‐78. [DOI] [PubMed] [Google Scholar]
- 101. Arrowsmith CH, Audia JE, Austin C, et al. The promise and peril of chemical probes. Nat Chem Biol. 2015;11(8):536‐541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chemical Probe Criteria for Classical Modulators and Bifunctional Degraders (e.g. PROTACs). 2021. https://www.chemicalprobes.org/chemical-probe-criteria-classical-modulators-and-bifunctional-degraders-eg-protacs
- 103. Chan JNY, Nislow C, Emili A. Recent advances and method development for drug target identification. Trends Pharmacol Sci. 2010;31(2):82‐88. [DOI] [PubMed] [Google Scholar]
- 104. Yeh E, DeRisi JL. Chemical rescue of malaria parasites lacking an apicoplast defines organelle function in blood‐stage Plasmodium falciparum . PLoS Biol. 2011;9(8):e1001138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Chen X, Wang Y, Ma N, et al. Target identification of natural medicine with chemical proteomics approach: probe synthesis, target fishing and protein identification. Signal Transduct Target Ther. 2020;5(1):72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Terai T, Maki E, Sugiyama S, et al. Rational development of caged‐biotin protein‐labeling agents and some applications in live cells. Chem Biol. 2011;18(10):1261‐1272. [DOI] [PubMed] [Google Scholar]
- 107. Buckie JW, Cook GMW. Specific isolation of surface glycoproteins from intact cells by biotinylated concanavalin A and immobilized streptavidin. Anal Biochem. 1986;156(2):463‐472. [DOI] [PubMed] [Google Scholar]
- 108. Taldone T, Rodina A, DaGama Gomes EM, et al. Synthesis and evaluation of cell‐permeable biotinylated PU‐H71 derivatives as tumor Hsp90 probes. Beilstein J Org Chem. 2013;9:544‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew Chem Int Ed. 2009;48(38):6974‐6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Nguyen SS, Prescher JA. Developing bioorthogonal probes to span a spectrum of reactivities. Nat Rev Chem. 2020;4(9):476‐489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kolb HC, Finn MG, Sharpless KB. Click chemistry: diverse chemical function from a few good reactions. Angew Chem Int Ed. 2001;40(11):2004‐2021. [DOI] [PubMed] [Google Scholar]
- 112. Chen X, Wu Y‐W. Selective chemical labeling of proteins. Org Biomol Chem. 2016;14(24):5417‐5439. [DOI] [PubMed] [Google Scholar]
- 113. Broichhagen J, Kilian N. Chemical biology tools to investigate malaria parasites. ChemBioChem. 2021;22(13):2219‐2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Agard NJ, Prescher JA, Bertozzi CR. A strain‐promoted [3 + 2] azide−alkyne cycloaddition for covalent modification of biomolecules in living systems. J Am Chem Soc. 2004;126(46):15046‐15047. [DOI] [PubMed] [Google Scholar]
- 115. Boger DL. Diels‐Alder reactions of heterocyclic aza dienes. Scope and applications. Chem Rev. 1986;86(5):781‐793. [Google Scholar]
- 116. Wright MH, Sieber SA. Chemical proteomics approaches for identifying the cellular targets of natural products. Nat Prod Rep. 2016;33(5):681‐708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Speers AE, Cravatt BF. A tandem orthogonal proteolysis strategy for high‐content chemical proteomics. J Am Chem Soc. 2005;127(28):10018‐10019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Sawa M, Hsu T‐L, Itoh T, et al. Glycoproteomic probes for fluorescent imaging of fucosylated glycans in vivo. Proc Natl Acad Sci USA. 2006;103(33):12371‐12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Seo SY, Corson TW. Small Molecule Target Identification Using Photo‐affinity Chromatography. Vol 622. Academic Press Inc; 2019:347‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Murale DP, Hong SC, Haque MM, Lee JS. Photo‐affinity labeling (PAL) in chemical proteomics: a handy tool to investigate protein‐protein interactions (PPIs). Proteome Sci. 2017;15(1):14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Dorman G, Prestwich GD. Benzophenone photophores in biochemistry. Biochemistry. 1994;33(19):5661‐5673. [DOI] [PubMed] [Google Scholar]
- 122. Kauer JC, Erickson‐Viitanen S, Wolfe HR, Degrado WF. p‐Benzoyl‐L‐phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin‐binding peptide. J Biol Chem. 1986;261(23):10695‐10700. [PubMed] [Google Scholar]
- 123. Smith E, Collins I. Photoaffinity labeling in target‐ and binding‐site identification. Future Med Chem. 2015;7(2):159‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Lapinsky DJ. Tandem photoaffinity labeling–bioorthogonal conjugation in medicinal chemistry. Bioorg Med Chem. 2012;20(21):6237‐6247. [DOI] [PubMed] [Google Scholar]
- 125. Dubinsky L, Krom BP, Meijler MM. Diazirine based photoaffinity labeling. Bioorg Med Chem. 2012;20(2):554‐570. [DOI] [PubMed] [Google Scholar]
- 126. Geurink PP, Prely LM, van der Marel GA, Bischoff R, Overkleeft HS. Photoaffinity labeling in activity‐based protein profiling. In: Sieber SA, ed. Activity‐Based Protein Profiling. Springer Berlin Heidelberg; 2012:85‐113. [DOI] [PubMed] [Google Scholar]
- 127. Brunner J. New photolabeling and crosslinking methods. Annu Rev Biochem. 1993;62(1):483‐514. [DOI] [PubMed] [Google Scholar]
- 128. Sakurai K, Ozawa S, Yamada R, Yasui T, Mizuno S. Comparison of the reactivity of carbohydrate photoaffinity probes with different photoreactive groups. ChemBioChem. 2014;15(10):1399‐1403. [DOI] [PubMed] [Google Scholar]
- 129. Mackinnon AL, Taunton J. Target identification by diazirine photo‐cross‐linking and click chemistry. Curr Protoc Chem Biol. 2009;1(1):55‐73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Foley M. Quinoline antimalarials: mechanisms of action and resistance and prospects for new agents. Pharmacol Ther. 1998;79(1):55‐87. [DOI] [PubMed] [Google Scholar]
- 131. Fu Y, Buryanovskyy L, Zhang Z. Quinone reductase 2 is a catechol quinone reductase. J Biol Chem. 2008;283(35):23829‐23835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Percário S, Moreira D, Gomes B, et al. Oxidative stress in malaria. Int J Mol Sci. 2012;13(12):16346‐16372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Chen Y, Koppaka V, Thompson DC, Vasiliou V. Focus on molecules: ALDH1A1: from lens and corneal crystallin to stem cell marker. Exp Eye Res. 2012;102:105‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Lindquist NG. Accumulation of drugs on melanin. Acta Radiol: Diagn. 1973;325:1‐92. [PubMed] [Google Scholar]
- 135. Rynes RI. Antimalarial drugs in the treatment of rheumatological diseases. Rheumatology. 1997;36(7):799‐805. [DOI] [PubMed] [Google Scholar]
- 136. Van Beek MJ, Piette WW. Antimalarials. Dermatol Clin. 2001;19(1):147‐160. [DOI] [PubMed] [Google Scholar]
- 137. Paquet T, Le Manach C, Cabrera DG, et al. Antimalarial efficacy of MMV390048, an inhibitor of Plasmodium phosphatidylinositol 4‐kinase. Sci Transl Med. 2017;9(387):eaad9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Arendse LB, Murithi JM, Qahash T, et al. The anticancer human mTOR inhibitor sapanisertib potently inhibits multiple Plasmodium kinases and life cycle stages. Sci Transl Med. 2022;14(667):eabo7219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Liu Q, Xu C, Kirubakaran S, et al. Characterization of Torin2, an ATP‐competitive inhibitor of mTOR, ATM and ATR. Cancer Res. 2013;73(8):2574‐2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Sun W, Tanaka TQ, Magle CT, et al. Chemical signatures and new drug targets for gametocytocidal drug development. Sci Rep. 2014;4:3743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bosch SS, Lunev S, Batista FA, et al. Molecular target validation of aspartate transcarbamoylase from Plasmodium falciparum by torin 2. ACS Infect Dis. 2020;6(5):986‐999. [DOI] [PubMed] [Google Scholar]
- 142. Krishnan K, Ziniel P, Li H, et al. Torin 2 derivative, NCATS‐SM3710, has potent multistage antimalarial activity through inhibition of P. falciparum phosphatidylinositol 4‐kinase (Pf PI4KIIIβ). ACS Pharmacol Transl Sci. 2020;3(5):948‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Knockaert M, Gray N, Damiens E, et al. Intracellular targets of cyclin‐dependent kinase inhibitors: identification by affinity chromatography using immobilised inhibitors. Chem Biol. 2000;7(6):411‐422. [DOI] [PubMed] [Google Scholar]
- 144. Gray NS, Wodicka L, Thunnissen AMWH, et al. Exploiting chemical libraries, structure, and genomics in the search for kinase inhibitors. Science. 1998;281(5376):533‐538. [DOI] [PubMed] [Google Scholar]
- 145. Dai Y. Cyclin‐dependent kinase inhibitors. Curr Opin Pharmacol. 2003;3(4):362‐370. [DOI] [PubMed] [Google Scholar]
- 146. Chang YT, Gray NS, Rosania GR, et al. Synthesis and application of functionally diverse 2,6,9‐trisubstituted purine libraries as CDK inhibitors. Chem Biol. 1999;6(6):361‐375. [DOI] [PubMed] [Google Scholar]
- 147. Rachidi N, Taly JF, Durieu E, et al. Pharmacological assessment defines Leishmania donovani casein kinase 1 as a drug target and reveals important functions in parasite viability and intracellular infection. Antimicrob Agents Chemother. 2014;58(3):1501‐1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Allocco JJ, Donald R, Zhong T, et al. Inhibitors of casein kinase 1 block the growth of Leishmania major promastigotes in vitro . Int J Parasitol. 2006;36(12):1249‐1259. [DOI] [PubMed] [Google Scholar]
- 149. Durieu E, Prina E, Leclercq O, et al. From drug screening to target deconvolution: a target‐based drug discovery pipeline using Leishmania casein kinase 1 isoform 2 to identify compounds with antileishmanial activity. Antimicrob Agents Chemother. 2016;60(5):2822‐2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Kato N, Sakata T, Breton G, et al. Gene expression signatures and small‐molecule compounds link a protein kinase to Plasmodium falciparum motility. Nat Chem Biol. 2008;4(6):347‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Le Roch KG, Zhou Y, Blair PL, et al. Discovery of gene function by expression profiling of the malaria parasite life cycle. Science. 2003;301(5639):1503‐1508. [DOI] [PubMed] [Google Scholar]
- 152. Chapman TM, Osborne SA, Bouloc N, et al. Substituted imidazopyridazines are potent and selective inhibitors of Plasmodium falciparum calcium‐dependent protein kinase 1 (PfCDPK1). Bioorg Med Chem Lett. 2013;23(10):3064‐3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Ansell KH, Jones HM, Whalley D, et al. Biochemical and antiparasitic properties of inhibitors of the Plasmodium falciparum calcium‐dependent protein kinase PfCDPK1. Antimicrob Agents Chemother. 2014;58(10):6032‐6043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Green JL, Moon RW, Whalley D, et al. Imidazopyridazine inhibitors of Plasmodium falciparum calcium‐dependent protein kinase 1 also target cyclic GMP‐dependent protein kinase and heat shock protein 90 to kill the parasite at different stages of intracellular development. Antimicrob Agents Chemother. 2016;60(3):1464‐1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Banumathy G, Singh V, Pavithra SR, Tatu U. Heat shock protein 90 function is essential for Plasmodium falciparum growth in human erythrocytes. J Biol Chem. 2003;278(20):18336‐18345. [DOI] [PubMed] [Google Scholar]
- 156. Mellacheruvu D, Wright Z, Couzens AL, et al. The CRAPome: a contaminant repository for affinity purification‐mass spectrometry data. Nat Methods. 2013;10(8):730‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Pino P, Caldelari R, Mukherjee B, et al. A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress. Science. 2017;358(6362):522‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Nasamu AS, Glushakova S, Russo I, et al. Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion. Science. 2017;358(6362):518‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Favuzza P, de Lera Ruiz M, Thompson JK, et al. Dual plasmepsin‐targeting antimalarial agents disrupt multiple stages of the malaria parasite life cycle. Cell Host Microbe. 2020;27(4):642‐658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Hodder AN, Christensen J, Scally S, et al. Basis for drug selectivity of plasmepsin IX and X inhibition in Plasmodium falciparum and vivax . Structure. 2022;30:947‐961. [DOI] [PubMed] [Google Scholar]
- 161. Morita M, Sanai H, Hiramoto A, et al. Plasmodium falciparum endoplasmic reticulum‐resident calcium binding protein is a possible target of synthetic antimalarial endoperoxides, N‐89 and N‐251. J Proteome Res. 2012;11(12):5704‐5711. [DOI] [PubMed] [Google Scholar]
- 162. van Heusden GPH. 14‐3‐3 proteins: insights from genome‐wide studies in yeast. Genomics. 2009;94(5):287‐293. [DOI] [PubMed] [Google Scholar]
- 163. Fierro MA, Asady B, Brooks CF, et al. An endoplasmic reticulum CREC family protein regulates the egress proteolytic cascade in malaria parasites. mBio. 2020;11(1):e03078‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Wang J, Zhang C‐J, Chia WN, et al. Haem‐activated promiscuous targeting of artemisinin in Plasmodium falciparum . Nat Commun. 2015;6(1):10111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Ismail HM, Barton V, Phanchana M, et al. Artemisinin activity‐based probes identify multiple molecular targets within the asexual stage of the malaria parasites Plasmodium falciparum 3D7. Proc Natl Acad Sci USA. 2016;113(8):2080‐2085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166. Robert A, Cazelles J, Meunier B. Characterization of the alkylation product of heme by the antimalarial drug artemisinin. Angew Chem Int Ed. 2001;40(10):1954‐1957. [DOI] [PubMed] [Google Scholar]
- 167. Liu Y, Wong VK‐W, Ko BC‐B, Wong M‐K, Che C‐M. Synthesis and cytotoxicity studies of artemisinin derivatives containing lipophilic alkyl carbon chains. Org Lett. 2005;7(8):1561‐1564. [DOI] [PubMed] [Google Scholar]
- 168. Bousejra‐El Garah F, Pitié M, Vendier L, Meunier B, Robert A. Alkylating ability of artemisinin after Cu(I)‐induced activation. J Biol Inorg Chem. 2009;14(4):601‐610. [DOI] [PubMed] [Google Scholar]
- 169. Kehr S, Jortzik E, Delahunty C, Yates JR 3rd, Rahlfs S, Becker K. Protein S‐glutathionylation in malaria parasites. Antioxid Redox Signal. 2011;15(11):2855‐2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Stocks PA, Bray PG, Barton VE, et al. Evidence for a common non‐heme chelatable‐iron‐dependent activation mechanism for semisynthetic and synthetic endoperoxide antimalarial drugs. Angew Chem Int Ed. 2007;46(33):6278‐6283. [DOI] [PubMed] [Google Scholar]
- 171. Klonis N, Crespo‐Ortiz MP, Bottova I. Artemisinin activity against Plasmodium falciparum requires hemoglobin uptake and digestion. Proc Natl Acad Sci USA. 2011;108(28):11405‐11410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Bakar NA, Klonis N, Hanssen E, Chan C, Tilley L. Digestive‐vacuole genesis and endocytic processes in the early intraerythrocytic stages of Plasmodium falciparum . J Cell Sci. 2010;123:441‐450. [DOI] [PubMed] [Google Scholar]
- 173. Jourdan J, Walz A, Matile H, et al. Stochastic protein alkylation by antimalarial peroxides. ACS Infect Dis. 2019;5(12):2067‐2075. [DOI] [PubMed] [Google Scholar]
- 174. Ismail HM, Barton VE, Panchana M, et al. A click chemistry‐based proteomic approach reveals that 1,2,4‐trioxolane and artemisinin antimalarials share a common protein alkylation profile. Angew Chem Int Ed. 2016;55(22):6401‐6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Uhlemann A‐C, Wittlin S, Matile H, Bustamante LY, Krishna S. Mechanism of antimalarial action of the synthetic trioxolane RBX11160 (OZ277). Antimicrob Agents Chemother. 2007;51(2):667‐672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Hartwig CL, Lauterwasser EMW, Mahajan SS, Hoke JM, Cooper RA, Renslo AR. Investigating the antimalarial action of 1,2,4‐trioxolanes with fluorescent chemical probes. J Med Chem. 2011;54(23):8207‐8213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Schulze CJ, Navarro G, Ebert D, DeRisi J, Linington RG. Salinipostins A–K, long‐chain bicyclic phosphotriesters as a potent and selective antimalarial chemotype. J Org Chem. 2015;80(3):1312‐1320. [DOI] [PubMed] [Google Scholar]
- 178. Yoo E, Schulze CJ, Stokes BH, et al. The antimalarial natural product Salinipostin A identifies essential α/β serine hydrolases involved in lipid metabolism in P. falciparum parasites. Cell Chem Biol. 2020;27(2):143‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Zhang M, Wang C, Otto TD, et al. Uncovering the essential genes of the human malaria parasite Plasmodium falciparum by saturation mutagenesis. Science. 2018;360(6388):eaap7847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180. Farazi TA, Waksman G, Gordon JI. The biology and enzymology of protein N‐myristoylation. J Biol Chem. 2001;276(43):39501‐39504. [DOI] [PubMed] [Google Scholar]
- 181. Frearson JA, Brand S, McElroy SP, et al. N‐myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature. 2010;464(7289):728‐732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ebiike H, Masubuchi M, Liu P, et al. Design and synthesis of novel benzofurans as a new class of antifungal agents targeting fungal N‐myristoyltransferase. Bioorg Med Chem Lett. 2002;12(4):607‐610. [DOI] [PubMed] [Google Scholar]
- 183. Wright MH, Clough B, Rackham MD, et al. Validation of N‐myristoyltransferase as an antimalarial drug target using an integrated chemical biology approach. Nat Chem. 2013;6(2):112‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184. Brunner R, Aissaoui H, Boss C, et al. Identification of a new chemical class of antimalarials. J Infect Dis. 2012;206(5):735‐743. [DOI] [PubMed] [Google Scholar]
- 185. Brunner R, Ng CL, Aissaoui H, et al. UV‐triggered affinity capture identifies interactions between the Plasmodium falciparum multidrug resistance protein 1 (PfMDR1) and antimalarial agents in live parasitized cells. J Biol Chem. 2013;288(31):22576‐22583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Sharma N, Kashif M, Singh V, et al. Novel antiplasmodial compounds leveraged with multistage potency against the parasite Plasmodium falciparum: in vitro and in vivo evaluations and pharmacokinetic studies. J Med Chem. 2021;64(12):8666‐8683. [DOI] [PubMed] [Google Scholar]
- 187. Richardson LW, Ashton TD, Dans MG, et al. Substrate peptidomimetic inhibitors of P. falciparum plasmepsin X with potent antimalarial activity. ChemMedChem. 2022;17(18):e202200306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Liu K, Shi H, Xiao H, et al. Functional profiling, identification, and inhibition of plasmepsins in intraerythrocytic malaria parasites. Angew Chem Int Ed. 2009;48(44):8293‐8297. [DOI] [PubMed] [Google Scholar]
- 189. Omara‐Opyene AL, Moura PA, Sulsona CR, et al. Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J Biol Chem. 2004;279(52):54088‐54096. [DOI] [PubMed] [Google Scholar]
- 190. Boddey JA, Hodder AN, Günther S, et al. An aspartyl protease directs malaria effector proteins to the host cell. Nature. 2010;463(7281):627‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191. Boonyalai N, Collins CR, Hackett F, Withers‐Martinez C, Blackman MJ. Essentiality of Plasmodium falciparum plasmepsin V. PLoS One. 2018;13(12):e0207621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Marapana DS, Wilson DW, Zuccala ES, et al. Malaria parasite signal peptide peptidase is an ER‐resident protease required for growth but not for invasion. Traffic. 2012;13(11):1457‐1465. [DOI] [PubMed] [Google Scholar]
- 193. Harbut MB, Patel BA, Yeung BKS. Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc Natl Acad Sci. 2012;109(52):21486‐21491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194. Déchamps S, Shastri S, Wengelnik K, Vial HJ. Glycerophospholipid acquisition in Plasmodium—a puzzling assembly of biosynthetic pathways. Int J Parasitol. 2010;40(12):1347‐1365. [DOI] [PubMed] [Google Scholar]
- 195. Calas M, Cordina G, Bompart J, et al. Antimalarial activity of molecules interfering with Plasmodium falciparum phospholipid metabolism. structure−activity relationship analysis. J Med Chem. 1997;40(22):3557‐3566. [DOI] [PubMed] [Google Scholar]
- 196. Vial HJ, Wein S, Farenc C. Prodrugs of bisthiazolium salts are orally potent antimalarials. Proc Natl Acad Sci. 2004;101(43):15458‐15463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Held J, Supan C, Salazar CLO, et al. Safety and efficacy of the choline analogue SAR97276 for malaria treatment: results of two phase 2, open‐label, multicenter trials in African patients. Malar J. 2017;16(1):188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Wein S, Maynadier M, Bordat Y, et al. Transport and pharmacodynamics of albitiazolium, an antimalarial drug candidate. Br J Pharmacol. 2012;166(8):2263‐2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199. Penarete‐Vargas DM, Boisson A, Urbach S, et al. A chemical proteomics approach for the search of pharmacological targets of the antimalarial clinical candidate albitiazolium in Plasmodium falciparum using photocrosslinking and click chemistry. PLoS One. 2014;9(12):e113918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Kubicek S, O'Sullivan RJ, August EM, et al. Reversal of H3K9me2 by a small‐molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25(3):473‐481. [DOI] [PubMed] [Google Scholar]
- 201. Vedadi M, Barsyte‐Lovejoy D, Liu F, et al. A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat Chem Biol. 2011;7(8):566‐574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Liu F, Barsyte‐Lovejoy D, Li F, et al. Discovery of an in vivo chemical probe of the lysine methyltransferases G9a and GLP. J Med Chem. 2013;56(21):8931‐8942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203. Xiong Y, Li F, Babault N, et al. Structure‐activity relationship studies of G9a‐like protein (GLP) inhibitors. Bioorg Med Chem. 2017;25(16):4414‐4423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204. Ma A, Yu W, Xiong Y, Butler KV, Brown PJ, Jin J. Structure‐activity relationship studies of SETD8 inhibitors. MedChemComm. 2014;5(12):1892‐1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205. Curry E, Green I, Chapman‐Rothe N, et al. Dual EZH2 and EHMT2 histone methyltransferase inhibition increases biological efficacy in breast cancer cells. Clin Epigenetics. 2015;7(1):84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Gilson PR, Tan C, Jarman KE, et al. Optimization of 2‐anilino 4‐amino substituted quinazolines into potent antimalarial agents with oral in vivo activity. J Med Chem. 2017;60(3):1171‐1188. [DOI] [PubMed] [Google Scholar]
- 207. Mizukawa Y, Ikegami‐Kawai M, Horiuchi M, et al. Quest for a potent antimalarial drug lead: synthesis and evaluation of 6,7‐dimethoxyquinazoline‐2,4‐diamines. Bioorg Med Chem. 2021;33:116018. [DOI] [PubMed] [Google Scholar]
- 208. Sundriyal S, Malmquist NA, Caron J, et al. Development of diaminoquinazoline histone lysine methyltransferase inhibitors as potent blood‐stage antimalarial compounds. ChemMedChem. 2014;9(10):2360‐2373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209. Sundriyal S, Chen PB, Lubin AS, et al. Histone lysine methyltransferase structure activity relationships that allow for segregation of G9a inhibition and anti‐Plasmodium activity. MedChemComm. 2017;8(5):1069‐1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Chen PB, Ding S, Zanghì G, et al. Plasmodium falciparum PfSET7: enzymatic characterization and cellular localization of a novel protein methyltransferase in sporozoite, liver and erythrocytic stage parasites. Sci Rep. 2016;6:21802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211. Lubin AS, Rueda‐Zubiaurre A, Matthews H, et al. Development of a photo‐cross‐linkable diaminoquinazoline inhibitor for target identification in Plasmodium falciparum . ACS Infect Dis. 2018;4(4):523‐530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212. Richard D, Bartfai R, Volz J, et al. A genome‐wide chromatin‐associated nuclear peroxiredoxin from the malaria parasite Plasmodium falciparum . J Biol Chem. 2011;286(13):11746‐11755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213. Gill J, Yogavel M, Kumar A, et al. Crystal structure of malaria parasite nucleosome assembly protein. J Biol Chem. 2009;284(15):10076‐10087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214. Muralidharan V, Oksman A, Pal P, Lindquist S, Goldberg DE. Plasmodium falciparum heat shock protein 110 stabilizes the asparagine repeat‐rich parasite proteome during malarial fevers. Nat Commun. 2012;3:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215. Dziekan JM, Wirjanata G, Dai L, et al. Cellular thermal shift assay for the identification of drug–target interactions in the Plasmodium falciparum proteome. Nat Protoc. 2020;15(6):1881‐1921. [DOI] [PubMed] [Google Scholar]
- 216. Martinez Molina D, Nordlund P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu Rev Pharmacol Toxicol. 2016;56(1):141‐161. [DOI] [PubMed] [Google Scholar]
- 217. Reinhard FBM, Eberhard D, Werner T, et al. Thermal proteome profiling monitors ligand interactions with cellular membrane proteins. Nat Methods. 2015;12(12):1129‐1131. [DOI] [PubMed] [Google Scholar]
- 218. Jafari R, Almqvist H, Axelsson H, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9(9):2100‐2122. [DOI] [PubMed] [Google Scholar]
- 219. Franken H, Mathieson T, Childs D, et al. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat Protoc. 2015;10(10):1567‐1593. [DOI] [PubMed] [Google Scholar]
- 220. Friman T. Mass spectrometry‐based Cellular Thermal Shift Assay (CETSA®) for target deconvolution in phenotypic drug discovery. Bioorg Med Chem. 2020;28(1):115174. [DOI] [PubMed] [Google Scholar]
- 221. Amos B, Aurrecoechea C, Barba M, et al. VEuPathDB: the eukaryotic pathogen, vector and host bioinformatics resource center. Nucleic Acids Res. 2022;50(D1):D898‐D911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222. Kawatkar A, Schefter M, Hermansson N‐O, et al. CETSA beyond soluble targets: a broad application to multipass transmembrane proteins. ACS Chem Biol. 2019;14(9):1913‐1920. [DOI] [PubMed] [Google Scholar]
- 223. Dziekan JM, Yu H, Chen D, et al. Identifying purine nucleoside phosphorylase as the target of quinine using cellular thermal shift assay. Sci Transl Med. 2019;11(473):eaau3174. [DOI] [PubMed] [Google Scholar]
- 224. Dai L, Prabhu N, Yu LY, Bacanu S, Ramos AD, Nordlund P. Horizontal cell biology: monitoring global changes of protein interaction states with the proteome‐wide cellular thermal shift assay (CETSA). Annu Rev Biochem. 2019;88(1):383‐408. [DOI] [PubMed] [Google Scholar]
- 225. Anderson A. Targeting DHFR in parasitic protozoa. Drug Discov Today. 2005;10(2):121‐128. [DOI] [PubMed] [Google Scholar]
- 226. Kerr ID, Lee JH, Pandey KC, et al. Structures of falcipain‐2 and falcipain‐3 bound to small molecule inhibitors: implications for substrate specificity. J Med Chem. 2009;52(3):852‐857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227. Shenai BR, Sijwali PS, Singh A, Rosenthal PJ. Characterization of native and recombinant falcipain‐2, a principal trophozoite cysteine protease and essential hemoglobinase of Plasmodium falciparum . J Biol Chem. 2000;275(37):29000‐29010. [DOI] [PubMed] [Google Scholar]
- 228. Xie SC, Dogovski C, Hanssen E, et al. Haemoglobin degradation underpins the sensitivity of early ring stage Plasmodium falciparum to artemisinins. J Cell Sci. 2016;129(2):406‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Wong W, Bai XC, Sleebs BE, et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat Microbiol. 2017;2:17031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Nguyen W, Hodder AN, de Lezongard RB, et al. Enhanced antimalarial activity of plasmepsin V inhibitors by modification of the P(2) position of PEXEL peptidomimetics. Eur J Med Chem. 2018;154:182‐198. [DOI] [PubMed] [Google Scholar]
- 231. Pai MY, Lomenick B, Hwang H, et al. Drug affinity responsive target stability (DARTS) for small‐molecule target identification. Methods Mol Biol. 2015;1263:287‐298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232. Park C, Marqusee S. Pulse proteolysis: a simple method for quantitative determination of protein stability and ligand binding. Nat Methods. 2005;2(3):207‐212. [DOI] [PubMed] [Google Scholar]
- 233. Lomenick B, Jung G, Wohlschlegel JA, Huang J. Target identification using drug affinity responsive target stability (DARTS). Curr Protoc Chem Biol. 2011;3(4):163‐180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234. Lomenick B, Hao R, Jonai N, et al. Target identification using drug affinity responsive target stability (DARTS). Proc Natl Acad Sci USA. 2009;106(51):21984‐21989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235. Park C, Zhou S, Gilmore J, Marqusee S. Energetics‐based protein profiling on a proteomic scale: identification of proteins resistant to proteolysis. J Mol Biol. 2007;368(5):1426‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236. West GM, Tang L, Fitzgerald MC. Thermodynamic analysis of protein stability and ligand binding using a chemical modification‐ and mass spectrometry‐based strategy. Anal Chem. 2008;80(11):4175‐4185. [DOI] [PubMed] [Google Scholar]
- 237. Lomenick B, Olsen RW, Huang J. Identification of direct protein targets of small molecules. ACS Chem Biol. 2011;6(1):34‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238. Tan CSH, Go KD, Bisteau X, et al. Thermal proximity coaggregation for system‐wide profiling of protein complex dynamics in cells. Science. 2018;359(6380):1170‐1177. [DOI] [PubMed] [Google Scholar]
- 239. Lu K‐Y, Quan B, Sylvester K, Srivastava T, Fitzgerald MC, Derbyshire ER. Plasmodium chaperonin TRiC/CCT identified as a target of the antihistamine clemastine using parallel chemoproteomic strategy. Proc Natl Acad Sci USA. 2020;117(11):5810‐5817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240. Derbyshire ER, Prudêncio M, Mota MM, Clardy J. Liver‐stage malaria parasites vulnerable to diverse chemical scaffolds. Proc Natl Acad Sci USA. 2012;109(22):8511‐8516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241. Gao Y, Thomas JO, Chow RL, Lee GH, Cowan NJ. A cytoplasmic chaperonin that catalyzes β‐actin folding. Cell. 1992;69(6):1043‐1050. [DOI] [PubMed] [Google Scholar]
- 242. Yaffe MB, Farr GW, Miklos D, Horwich AL, Sternlicht ML, Sternlicht H. TCP1 complex is a molecular chaperone in tubulin biogenesis. Nature. 1992;358(6383):245‐248. [DOI] [PubMed] [Google Scholar]
- 243. Trinh HV, Grossmann J, Gehrig P, et al. iTRAQ‐based and label‐free proteomics approaches for studies of human adenovirus infections. Int J Proteom. 2013;2013:1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244. Ziegler S, Pries V, Hedberg C, Waldmann H. Target identification for small bioactive molecules: finding the needle in the haystack. Angew Chem Int Ed. 2013;52(10):2744‐2792. [DOI] [PubMed] [Google Scholar]
- 245. Zhang H, Yan W, Aebersold R. Chemical probes and tandem mass spectrometry: A strategy for the quantitative analysis of proteomes and subproteomes. Curr Opin Chem Biol. 2004;8(1):66‐75. [DOI] [PubMed] [Google Scholar]
- 246. Gygi SP, Rist B, Gerber SA, Turecek F, Gelb MH, Aebersold R. Quantitative analysis of complex protein mixtures using isotope‐coded affinity tags. Nat Biotechnol. 1999;17(10):994‐999. [DOI] [PubMed] [Google Scholar]
- 247. Nirmalan N, Sims PFG, Hyde JE. Quantitative proteomics of the human malaria parasite Plasmodium falciparum and its application to studies of development and inhibition. Mol Microbiol. 2004;52(4):1187‐1199. [DOI] [PubMed] [Google Scholar]
- 248. Sims PF, Hyde JE. Proteomics of the human malaria parasite Plasmodium falciparum . Expert Rev Proteom. 2006;3(1):87‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249. Smolka MB, Zhou H, Purkayastha S, Aebersold R. Optimization of the isotope‐coded affinity tag‐labeling procedure for quantitative proteome analysis. Anal Biochem. 2001;297(1):25‐31. [DOI] [PubMed] [Google Scholar]
- 250. Thompson A, Schäfer J, Kuhn K, et al. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal Chem. 2003;75(8):1895‐1904. [DOI] [PubMed] [Google Scholar]
- 251. Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine‐reactive isobaric tagging reagents. Mol Cell Proteom. 2004;3(12):1154‐1169. [DOI] [PubMed] [Google Scholar]
- 252. Dayon L, Hainard A, Licker V, et al. Relative quantification of proteins in human cerebrospinal fluids by MS/MS using 6‐plex isobaric tags. Anal Chem. 2008;80(8):2921‐2931. [DOI] [PubMed] [Google Scholar]
- 253. Choe L, D'Ascenzo M, Relkin NR, et al. 8‐plex quantitation of changes in cerebrospinal fluid protein expression in subjects undergoing intravenous immunoglobulin treatment for Alzheimer's disease. Proteomics. 2007;7(20):3651‐3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254. McAlister GC, Huttlin EL, Haas W, et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal Chem. 2012;84(17):7469‐7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255. Stopfer LE, Mesfin JM, Joughin BA, Lauffenburger DA, White FM. Multiplexed relative and absolute quantitative immunopeptidomics reveals MHC I repertoire alterations induced by CDK4/6 inhibition. Nat Commun. 2020;11(1):2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256. Bai B, Tan H, Pagala VR, et al. Deep profiling of proteome and phosphoproteome by isobaric labeling, extensive liquid chromatography, and mass spectrometry. Methods Enzymol. 2017;585:377‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Briolant S, Almeras L, Belghazi M, et al. Plasmodium falciparum proteome changes in response to doxycycline treatment. Malar J. 2010;9(1):141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258. Chen X, Wei S, Ji Y, Guo X, Yang F. Quantitative proteomics using SILAC: principles, applications, and developments. Proteomics. 2015;15(18):3175‐3192. [DOI] [PubMed] [Google Scholar]
- 259. Ong SE, Blagoev B, Kratchmarova I, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteom. 2002;1(5):376‐386. [DOI] [PubMed] [Google Scholar]
- 260. Ong S‐E, Mann M. A practical recipe for stable isotope labeling by amino acids in cell culture (SILAC). Nat Protoc. 2006;1(6):2650‐2660. [DOI] [PubMed] [Google Scholar]
- 261. Sherman IW. Transport of amino acids and nucleic acid precursors in malarial parasites. Bull World Health Organ. 1977;55(2‐3):211‐225. [PMC free article] [PubMed] [Google Scholar]
- 262. Prieto JH, Koncarevic S, Park SK, Yates J 3rd, Becker K. Large‐scale differential proteome analysis in Plasmodium falciparum under drug treatment. PLoS One. 2008;3(12):e4098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263. Overmyer KA, Tyanova S, Hebert AS, Westphall MS, Cox J, Coon JJ. Multiplexed proteome analysis with neutron‐encoded stable isotope labeling in cells and mice. Nat Protoc. 2018;13(2):293‐306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264. Neilson KA, Ali NA, Muralidharan S, et al. Less label, more free: approaches in label‐free quantitative mass spectrometry. Proteomics. 2011;11(4):535‐553. [DOI] [PubMed] [Google Scholar]
- 265. Liu H, Sadygov RG, Yates JR. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal Chem. 2004;76(14):4193‐4201. [DOI] [PubMed] [Google Scholar]
- 266. Drabovich AP, Pavlou MP, Batruch I, Diamandis EP. Chapter 2 – Proteomic and mass spectrometry technologies for biomarker discovery. In: Issaq HJ, Veenstra TD, eds. Proteomic and Metabolomic Approaches to Biomarker Discovery. Academic Press; 2013:17‐37. [Google Scholar]
- 267. Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal Bioanal Chem. 2007;389(4):1017‐1031. [DOI] [PubMed] [Google Scholar]
- 268. Zybailov B, Mosley AL, Sardiu ME, Coleman MK, Florens L, Washburn MP. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae . J Proteome Res. 2006;5(9):2339‐2347. [DOI] [PubMed] [Google Scholar]
- 269. Zhang Y, Wen Z, Washburn MP, Florens L. Refinements to label free proteome quantitation: how to deal with peptides shared by multiple proteins. Anal Chem. 2010;82(6):2272‐2281. [DOI] [PubMed] [Google Scholar]
- 270. Griffin NM, Yu J, Long F, et al. Label‐free, normalized quantification of complex mass spectrometry data for proteomic analysis. Nat Biotechnol. 2010;28(1):83‐89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271. Ishihama Y, Oda Y, Tabata T, et al. Exponentially modified protein abundance index (emPAI) for estimation of absolute protein amount in proteomics by the number of sequenced peptides per protein. Mol Cell Proteom. 2005;4(9):1265‐1272. [DOI] [PubMed] [Google Scholar]
- 272. Podwojski K, Eisenacher M, Kohl M, et al. Peek a peak: a glance at statistics for quantitative label‐free proteomics. Expert Rev Proteom. 2010;7(2):249‐261. [DOI] [PubMed] [Google Scholar]
- 273. Dost B, Bandeira N, Li X, Shen Z, Briggs SP, Bafna V. Accurate mass spectrometry based protein quantification via shared peptides. J Comput Biol. 2012;19(4):337‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274. Perkins DN, Pappin DJC, Creasy DM, Cottrell JS. Probability‐based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20(18):3551‐3567. [DOI] [PubMed] [Google Scholar]
- 275. Bondarenko PV, Chelius D, Shaler TA. Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed‐phase liquid chromatography−tandem mass spectrometry. Anal Chem. 2002;74(18):4741‐4749. [DOI] [PubMed] [Google Scholar]
- 276. Chelius D, Bondarenko PV. Quantitative profiling of proteins in complex mixtures using liquid chromatography and mass spectrometry. J Proteome Res. 2002;1(4):317‐323. [DOI] [PubMed] [Google Scholar]
- 277. Schwanhäusser B, Busse D, Li N, et al. Global quantification of mammalian gene expression control. Nature. 2011;473(7347):337‐342. [DOI] [PubMed] [Google Scholar]
- 278. Shin JB, Krey JF, Hassan A, et al. Molecular architecture of the chick vestibular hair bundle. Nat Neurosci. 2013;16(3):365‐374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279. Wiśniewski JR, Ostasiewicz P, Duś K, Zielińska DF, Gnad F, Mann M. Extensive quantitative remodeling of the proteome between normal colon tissue and adenocarcinoma. Mol Syst Biol. 2012;8:611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280. Silva JC, Gorenstein MV, Li GZ, Vissers JPC, Geromanos SJ. Absolute quantification of proteins by LCMSE. Mol Cell Proteom. 2006;5(1):144‐156. [DOI] [PubMed] [Google Scholar]
- 281. Listgarten J, Emili A. Statistical and computational methods for comparative proteomic profiling using liquid chromatography‐tandem mass spectrometry. Mol Cell Proteom. 2005;4(4):419‐434. [DOI] [PubMed] [Google Scholar]
- 282. Nesvizhskii AI. CRAPome: Contaminant Repository for Affinity Purification. 2022. Accessed May 5, 2022. https://reprint-apms.org/?q=reprint-home
- 283. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proc Natl Acad Sci USA. 2001;98(15):8554‐8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284. Nowak RP, Jones LH. Target validation using PROTACs: applying the four pillars framework. SLAS Discov. 2021;26(4):474‐483. [DOI] [PubMed] [Google Scholar]
- 285. Li Y, Meng Q, Wang P, et al. Identification of PDE6D as a potential target of sorafenib via PROTAC technology. bioRxiv. 2020. [Google Scholar]
- 286. Chessum NEA, Sharp SY, Caldwell JJ, et al. Demonstrating in‐cell target engagement using a pirin protein degradation probe (CCT367766). J Med Chem. 2018;61(3):918‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287. Ng CL, Fidock DA, Bogyo M. Protein degradation systems as antimalarial therapeutic targets. Trends Parasitol. 2017;33(9):731‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Hamilton MJ, Lee M, Le Roch KG. The ubiquitin system: an essential component to unlocking the secrets of malaria parasite biology. Mol BioSyst. 2014;10(4):715‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289. Krishnan KM, Williamson KC. The proteasome as a target to combat malaria: hits and misses. Transl Res. 2018;198:40‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Grohmann C, Marapana DS, Ebert G. Targeted protein degradation at the host–pathogen interface. Mol Microbiol. 2021;117:760‐681. [DOI] [PubMed] [Google Scholar]
- 291. Maple HJ, Clayden N, Baron A, Stacey C, Felix R. Developing degraders: principles and perspectives on design and chemical space. MedChemComm. 2019;10(10):1755‐1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed.