

Abstract
Pharmacogenetics (PGx) seeks to enable selection of the right dose of the right drug for each patient to optimize therapeutic outcomes. Most PGx focuses on pharmacokinetics (PK), due to our relatively advanced understanding of the genes involved in PK and the causative effects of variants in those genes. Genetic variants can also affect pharmacodynamics (PD), but relatively few PGx-PD associations have been identified. This is partially due to a more limited understanding of the relevant genes and the consequences of genetic variation, but is also due in part to the potential confounding of PK variability in assessments of clinical outcomes that have a contribution from both PK and PD. For example, it is challenging to confirm the effect of mu opioid receptor (OPRM1) genetic variation on opioid response due to the contribution of CYP2D6 genotype to bioactivation of some opioid drugs (i.e., codeine and tramadol). The objectives of this mini-review are to describe several recent efforts to discover and validate PGx-PD that disentangle the influence of PK variability and propose potential approaches that could be used in future PGx-PD analyses. We use the effect of OPRM1 genetics on opioid response to illustrate how these analyses could be conducted and conclude by discussing how PGx-PD could be translated into clinical practice to improve therapeutic outcomes.
Pharmacogenetics of Pharmacodynamic Drug Response (PGx-PD)
The promise of pharmacogenetics (PGx) is the ability to select the right dose of the right drug for each patient. This idea acknowledges that each patient is unique, and optimal treatment should incorporate those factors that define the patient as a unique individual rather than assuming that the population mean or median sufficiently represents the patient1. Most of the work in PGx and personalized medicine has focused on pharmacokinetics (PK) as the phenotype of interest, including individualized dosing to achieve target drug exposures.
The focus on PGx of PK (PGx-PK) is due to several factors. PGx-PK builds upon substantial understanding of drug PK, including knowledge of the specific enzymes and transporters primarily responsible for the absorption, distribution, metabolism, and excretion of individual drugs. It also builds on substantial work to identify functional variants in the genes coding for these enzymes and transporters and the translation of genotypes to predicted activity phenotypes2. PK data are relatively easy to collect and measure, and provide a sensitive, quantitative phenotypic endpoint for PGx-PK analyses, with the caveat that often systemic PK is measured, and this may not accurately reflect PK at the target site. Clinical translation is relatively straightforward; adjustment of dosing reduces PK variability across PGx-PK groups, or substitution of an alternate agent with a different metabolic pathway may avoid inefficacy or toxicity. There are many examples of this approach, including recent guidelines for dosing tacrolimus based on CYP3A5 genotype3, or avoiding codeine in individuals with extreme CYP2D6 genetic phenotypes4, including poor or ultrarapid metabolizers.
Despite the potential for genetic variants to also affect drug sensitivity, or pharmacodynamics (PD), there are relatively few established PGx-PD associations. Genetic variants may affect the expression, function, occupancy, or activation of a drug target, among many other possible biological mechanisms5. The clinical consequence of PGx-PD is that a systemic exposure within the desired range may not necessarily elicit the desired response if genetic variation results in a drug target that is non-functional or not expressed to an appreciable extent (putting the patient at risk for an off-target or noxious on-target response). There are several reasons for the relative paucity of validated PGx-PD effects, including incomplete understanding of candidate PD genes, incomplete knowledge of the functional effects of variants within those genes, lack of well-phenotyped PD endpoints, variable efficacy endpoints for different indications of the same drug, and perhaps smaller effects from many PD genes and variants, similar to the genetics of complex diseases.
Genome-wide association studies (GWAS) have identified near monogenic PGx-PD associations of genes that were unlikely to have been selected for candidate genetic studies6. To date, these striking PGx-PD associations have been primarily observed in PK-independent outcomes, such as the associations for HLA genes with drug-induced hypersensitivity6 or CACNA1A/RYR1 with malignant hyperthermia7. Unlike these strongly penetrant genetic associations, most clinical outcomes are multifactorial, including a contribution from both PK and PD5, 8 (Figure 1). There has been limited success identifying these PGx-PD associations, partially due to the confounding effects of PK in the analysis. For example, there is evidence that genetic variation in the mu opioid receptor (OPRM1) is associated with response to opioid analgesics, but this association has been difficult to validate due to the confounding of variability in morphine systemic exposure.4
Figure 1:
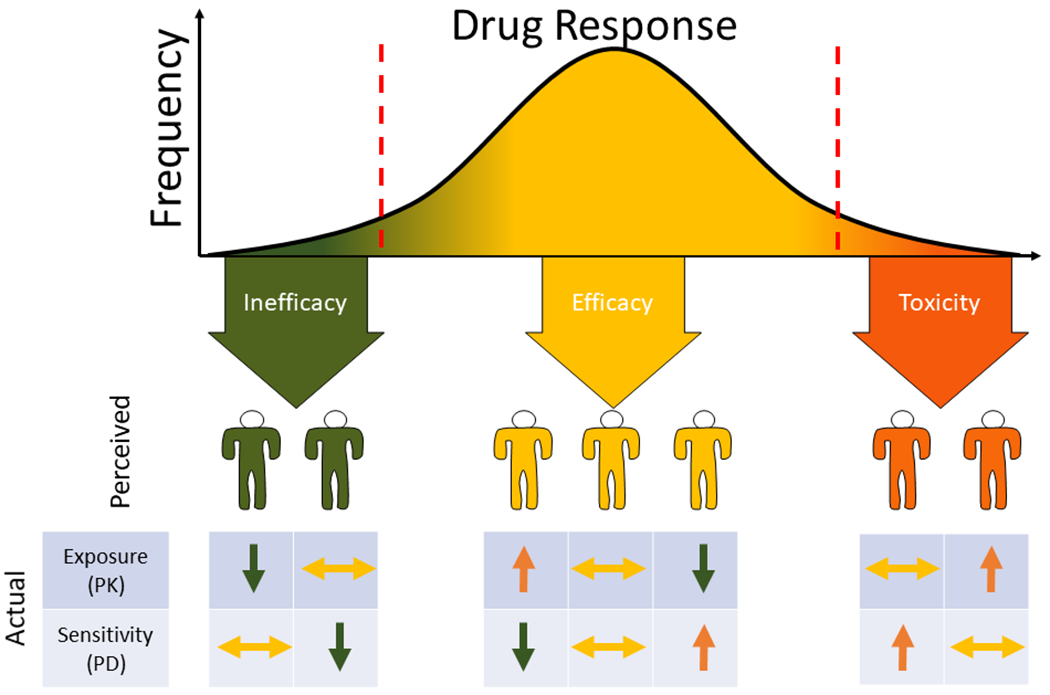
Drug response is a consequence of drug exposure (pharmacokinetics, PK) and sensitivity (pharmacodynamics, PD). A patient who has ineffective treatment could be due to inadequate exposure or reduced sensitivity (i.e., resistance). Similarly, a patient who experiences toxicity could be due to supra-therapeutic exposure or enhanced sensitivity. Patients who experience efficacy without toxicity could have normal exposure and sensitivity, or off-setting increased exposure and decreased sensitivity, or vice-versa, that produce a typical response.
The objective of this mini-review is to describe potential approaches to deconvolute the confounding effects of PK to isolate PGx-PD effects. We illustrate these approaches using recent efforts to identify PGx-PD and by returning to the example of the putative association of OPRM1 genetics on opioid response. We conclude by describing what is needed to advance PGx-PD research and integrate PGx-PD into individualized treatment to improve therapeutic outcomes.
Approaches to Identifying PGx-PD Associations
Drugs Dosed to Standardize Target Concentrations
For some drugs, particularly those with narrow therapeutic windows, clinical testing of drug concentrations is pursued, and dosing is adjusted to achieve a target concentration, commonly referred to as therapeutic drug monitoring (TDM). By adjusting the dose to achieve a target concentration, such as a maximum or minimum concentration (e.g., Cmax or Cmin), TDM can enhance efficacy and/or reduce toxicity. Tacrolimus, some antibiotics, and some anti-epileptics undergo TDM per routine. Effective use of TDM minimizes variability in exposure (at least as indicated by sampling of blood) and reduces the contribution of exposure variability in the analysis of treatment outcomes. This strategy was used to identify the PGx-PD association between variants in the KCNQ1 gene (encoding the pore-forming subunit of the voltage-gated potassium channel, KvLQT1) and new-onset post-transplant diabetes mellitus in patients treated with tacrolimus9.
There are several limitations to this approach. One is that the TDM metric used to individualize dosing (i.e., Cmin) is unlikely to capture all individual variability in PK and may not be the primary determinant of the outcome of interest. For example, for PD outcomes such as nephrotoxicity or efficacy, Cmax or systemic exposure defined as area under the concentration-time curve (AUC) may be more relevant than Cmin (trough), but these data may not be available. Additionally, the steepness of the concentration-response slope may be an important consideration. It is also required that the TDM measurements are available for a time relevant to the PD outcome. For example, a GWAS of tacrolimus-exposed individuals was unable to include drug levels as a covariate in the analysis of nephrotoxicity because data were not available at the same time as renal function measurements12.
Since TDM is only used for specific drugs, the use of clinically obtained drug concentrations is relevant to a limited number of medications. There is evidence that TDM may be beneficial for a larger number of medications, even including broad therapeutic index drugs. A CYP2D6 genotype-stratified PK study of atomoxetine revealed a 50-fold range in systemic exposure in patients receiving standard dosing, and suggested that even maximal recommended dosing would fail to achieve target exposure in a substantial proportion of CYP2D6 normal metabolizers (NMs)10. A recently published CPIC guideline recommends checking the atomoxetine concentration in patients exhibiting inadequate clinical response to inform subsequent therapeutic decisions11. Expanding TDM to more agents could have direct clinical benefit while improving the identification of PGx-PD associations.
For the example of OPRM1, since TDM is not used to guide codeine dosing, codeine or morphine metabolite data are not readily available to investigate PGx-PD effects for OPRM1. A concentration-controlled clinical trial13, in which patients are randomly assigned to receive personalized codeine dosing to achieve one of several pre-specified morphine exposure levels, could be a possible alternate source of data that is similar to a TDM situation, with which PGx-PD analyses could be conducted without the confounding of PK.
Investigate PGx-PD Associations by Adjusting for PK
For drugs that do not undergo TDM, there are several analytical approaches to reduce the contribution of PK variability to investigate PGx-PD associations. One straightforward approach that does not require measured drug concentrations is to conduct the PGx-PD analysis within a PGx-PK stratum. For instance, investigating the association of OPRM1 genotype with analgesic response to CYP2D6 substrates, such as codeine or tramadol, in only CYP2D6 normal metabolizers. This approach will reduce the contribution of PK variability but will not eliminate it completely as CYP2D6 activity can vary several-fold within individuals with the same genotype14. An extension of this approach is to conduct the analysis within each CYP2D6 metabolic phenotype strata or adjust for metabolic phenotype.
When concentrations measurements are available, the simplest approach is to adjust for measured drug concentrations in the analysis. For example, nephrotoxicity due to vancomycin is partially determined by vancomycin trough concentrations. In order to identify PGx-PD associations, a GWAS of nephrotoxicity adjusted for vancomycin trough concentrations, which enabled identification of a variant near the GJA1 gene (encoding connexin43)15. Applying this approach OPRM1, an analysis of the association of OPRM1 genotype with analgesia that adjusted for measured morphine concentrations during codeine treatment could substantially reduce the contribution of PK variability.
Demonstrate PK-outcome Association Stratified by PGx-PD
PK-outcomes associations can be detected using standard statistical approaches such as regression models. However, these associations can be confounded by PGx-PD effects, which can lead to an inability to detect the PK-outcome association in pooled patients. If the PK-outcome association is revealed by stratifying the cohort by the PGx-PD genotype, this provides evidence that the genotype is contributing to the outcome of interest. Importantly, this type of stratified analysis can accommodate PGx-PD effects that invert the PK-outcome association (Figure 2), as has been recently reported for paroxetine and genotype of the paroxetine drug target SLC6A416. In patients with genetic variants associated with low SLC6A4 expression, patients with lower plasma concentrations had better clinical improvement than patients with higher concentrations, potentially due to target saturation and off-target effects. In patients with variants associated with high SLC6A4 expression, the response improved with higher blood concentration. This inverse association within each genotype group would be difficult to detect using any of the other strategies proposed in this commentary. In terms of OPRM1, at least one study has analyzed the association between opioid concentration and analgesic response within OPRM1 genotype groups17.
Figure 2:
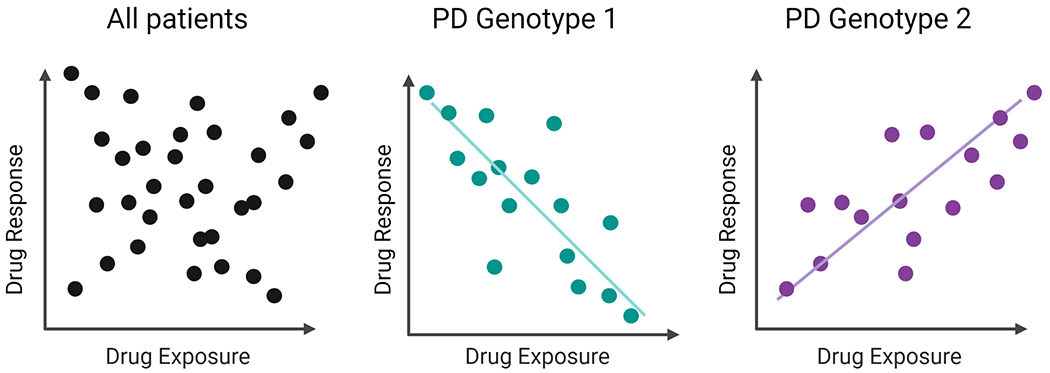
In the combined cohort there is no apparent association between drug exposure (PK) and response. However, when the cohort is stratified by pharmacogenetic variant that impacts pharmacodynamics (PGx-PD), there are inverse associations between exposure and response within each PGx-PD genotype, as per the example in the text of paroxetine and SLC6A4.
Introduce PGx-PD into PK-outcomes Model
Another somewhat related approach is to first establish the PK-outcomes model and then introduce the PGx-PD variable. Multivariable models retain only variables that explain residual variability in the outcome of interest. Including measured PK in a model accounts for the contribution of PK variability, and the residual variability will be predominantly contributed by PD (Figure 3). This approach was recently used in a PGx-PD analysis of paclitaxel-induced peripheral neuropathy. First, a model was created that included systemic paclitaxel PK and other clinical variables that were associated with the risk of neuropathy18. Then genes involved in hereditary neuropathy that had previously been reported to increase risk of paclitaxel-induced neuropathy were investigated, including variants in EPHA5. Introducing those variants into the neuropathy-prediction model demonstrated that these genotypes affect a patient’s neuropathy sensitivity after accounting for variability in cumulative paclitaxel exposure19. Importantly, a post-hoc analysis confirmed that this association for EPHA5 would not have been detected without including measured paclitaxel PK in the multivariable model. In the case of OPRM1, this approach would be attempted by first modeling the morphine-analgesia association and then adding OPRM1 genotype as a covariate in the model to see if it explains residual variability in the resulting analgesic effect.
Figure 3:
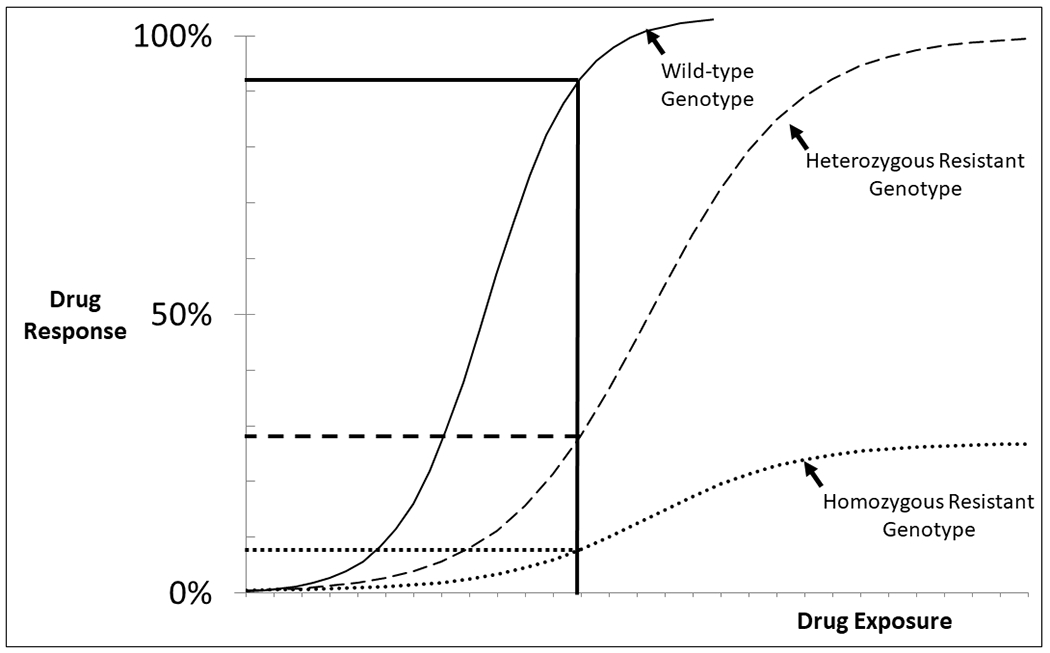
Pharmacogenetics of pharmacodynamics (PGx-PD) affects the patient’s drug response at a given exposure level. In this example, at a given drug exposure (solid vertical line) a patient with wild-type PGx-PD would have near complete drug response (solid horizontal line). At that same exposure, a patient carrying a single resistance allele would have a small response (dashed horizontal line) and a patient with homozygous resistant genotype would have almost no drug response (dotted horizontal line). The corollary is that achieving the same drug response in patients with different PGx-PD genotypes requires different drug exposures.
Incorporation of Genetics in Pharmacometric Models
The previously described multivariable statistical approaches are empirical, simpler approaches for investigators who do not have expertise in population pharmacokinetic-pharmacodynamic (popPKPD) modeling. The ideal methods to investigate PGx-PD effects are likely to develop popPKPD or possibly physiologically-based PKPD (PBPKPD) models. PopPKPD models20 are typically used to understand the relationship between drug concentration and PD response by accounting for the variability in PK and PD parameters from covariates. Traditionally, these covariates are clinical variables that affect PK or PD parameters, though it has become increasingly common to investigate PGx factors affecting PK parameters (i.e., drug clearance). Similarly, genetic factors of PD response could be explored in a popPKPD model to understand variability in PD parameters, including maximal drug effect (Emax) or potency (EC50).This approach was recently used within a study of the effectiveness of buprenorphine for reducing illicit opioid use21. The base PKPD model identified the buprenorphine EC50 for successful opioid abstinence. One of the covariates associated with this EC50 was the rs678849 genotype of the delta-opioid receptor (OPRD1). A very similar approach could be used for OPRM1 by building a popPKPD model that relates morphine exposure to analgesia, and then investigating the OPRM1 genotype as a covariate on the analgesic response parameters Emax or EC50.
On the other hand, PBPKPD models22 provide a mechanistic representation of the drug in the biological system by explicitly considering the organs and tissues to estimate drug concentrations within each tissue. Thus, PBPKPD models can potentially link target site concentrations to the PD response and any PGx factors affecting the PD response, for instance the binding of the drug to its target site, could be modeled. In the OPRM1 example, the PBPKPD model could be built in which the concentrations of morphine in the brain could be estimated, followed by modeling of the binding of morphine to OPRM1 and then investigating the effects of OPRM1 genotype on this binding and the resulting analgesic effect.
Clinical Translation and Recommendations for Future Work
Integrating PGx-PD into Precision Treatment
As mentioned earlier, the clinical translation of PGx-PK by adjusting doses according to PGx-PK genotype to standardize drug concentration is relatively straightforward. In this sense, “individualized treatment” means stratifying dosing so all patients achieve the same exposure. PGx-PD is somewhat more complex to translate into clinical practice since it implies that the optimal exposure level for each patient is distinct. The appropriate dosing patients with each PD-PGx genotype is a function of the direction of effect (i.e., sensitive vs. resistant) of that genotype and the relevant clinical outcome (i.e., efficacy and/or toxicity) (Figure 4). Patients who are “sensitive” to the therapeutic effects of a drug may achieve greater benefit at typical levels of exposure; this may enable downward titration of the exposure to reduce risk of toxicity (or maintaining typical exposure to enhance efficacy without increasing toxicity). Patients who are “sensitive” to drug toxicity cannot tolerate typical exposure and require reduced exposure, which may reduce efficacy. Toxicity “resistant” patients can tolerate higher exposure, which could allow for upward titration of exposure to enhance efficacy or maintaining exposure to maintain efficacy, potentially with less toxicity. Finally, patients “resistant” to therapeutic effects will require higher exposure or may not be able to achieve therapeutic response at any tolerable exposure level. The sensitivity/resistance to therapeutic effects and toxicity may be linked, in which case proper titration could yield the typical balance of efficacy and toxicity, or may be independent, based on the biologic mechanism. Importantly, these situations demonstrate the complexity of translating PGx-PD into clinical practice by individualizing dosing so each patient achieves the exposure that optimizes their clinical outcomes.
Figure 4:
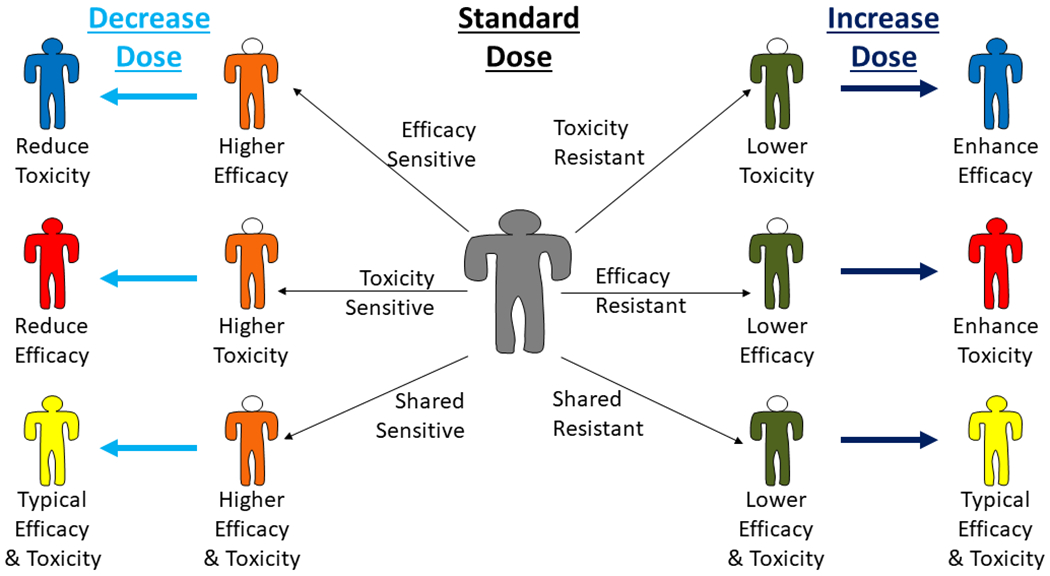
Integrating pharmacogenetics of pharmacodynamics (PGx-PD) into clinical practice requires adjusting dosing so that patients achieve the exposure that is consistent with their optimal treatment outcomes. Patients can be “sensitive” (orange bodies) or “resistant” (dark green bodies) to efficacy and/or toxicity. In each case, treating that patient with standard dosing will result in higher or lower efficacy and/or toxicity than is typical. Depending on the PGx-PD genotype, a dose decrease or increase could result in superior (blue), inferior (red), or similar (yellow) treatment outcomes.
Conclusion and Future Directions for Research and Practice
We have described several approaches for reducing confounding by PK to assist with identifying PGx-PD effects for multi-factorial clinical outcomes. Other challenges mentioned earlier, such as the limited understanding of the genes responsible for PD effects and the consequences of genetic variation in those genes and the lack of well-phenotyped PD endpoints, require additional consideration and investigation but are beyond the scope of this mini-review. In addition to a general recognition of the challenges with PGx-PD, there are several other initiatives that would improve our ability to conduct the analyses described within this mini-review. First, a greater effort is needed to collect samples for PK analysis, as this is the most direct way to account for PK variability in PGx-PD studies. One highly efficient potential approach is to collect scavenged samples, which reduces the cost and some of the regulatory issues around PK sampling and is especially beneficial in patients who are difficult to sample including neonates, children, and the elderly23. PGx-PD analyses would also benefit from development of more precise biomarkers defining clinically relevant outcomes. These analyses also require further development of modeling approaches that integrate PGx-PD analyses, perhaps including simulation approaches to determine the optimal exposure and necessary dosing for patients based on PD genotype. Finally, clinical translational researchers will likely need to develop prospective study designs to demonstrate the clinical utility of individualized treatment based on PGx-PD effects24. One possible study design would be a variation of concentration-controlled clinical trials, PGx-PD stratified studies, in which participants are genotyped for the drug target of interest. In such a study design a pharmacometric model is used to individualize drug doses to achieve a target exposure (Cmax or AUC); inadequate therapeutic response at the initial exposure level can be followed with an increase in exposure and re-assessment of therapeutic response, allowing for exposure-response relationships to be established for each drug target genotype. This approach is analogous to genetics-stratified dose escalation studies that have been used to validate PGx-PK effects in oncology25. A more concerted effort to discover and validate PGx-PD effects will someday usher in a new era of personalized treatment in which patients are dosed to achieve their personalized target concentration, improving therapeutic outcomes, and realizing the promise of PGx.
Footnotes
Conflict of Interest Statement: All other authors declared no competing interests for this work.
References:
- 1.Leeder JS. Who Believes They Are “Just Average”: Informing the Treatment of Individual Patients Using Population Data. Clin Pharmacol Ther. 2019;106:939–941. doi: 910.1002/cpt.1612. Epub 2019 Sep 1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Caudle KE, Sangkuhl K, Whirl-Carrillo M, et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci. 2020;13:116–124. doi: 110.1111/cts.12692. Epub 12019 Oct 12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birdwell KA, Decker B, Barbarino JM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guidelines for CYP3A5 Genotype and Tacrolimus Dosing. Clin Pharmacol Ther. 2015;98:19–24. doi: 10.1002/cpt.1113. Epub 2015 Jun 1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Crews KR, Monte AA, Huddart R, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for CYP2D6, OPRM1, and COMT Genotypes and Select Opioid Therapy. Clin Pharmacol Ther. 2021;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hertz DL, McLeod HL. Using pharmacogene polymorphism panels to detect germline pharmacodynamic markers in oncology. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014;20:2530–2540. [DOI] [PubMed] [Google Scholar]
- 6.Daly AK. Using genome-wide association studies to identify genes important in serious adverse drug reactions. Annual Review of Pharmacology and Toxicology. 2012;52:21–35. [DOI] [PubMed] [Google Scholar]
- 7.Gonsalves SG, Dirksen RT, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for the Use of Potent Volatile Anesthetic Agents and Succinylcholine in the Context of RYR1 or CACNA1S Genotypes. Clin Pharmacol Ther. 2019;105:1338–1344. doi: 1310.1002/cpt.1319. Epub 2019 Jan 1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McLaughlin MJ, Wagner J, Shakhnovich V, Carleton B, Leeder JS. Considerations for Implementing Precision Therapeutics for Children. Clin Transl Sci. 2019;12:140–150. doi: 110.1111/cts.12607. Epub 12019 Jan 12625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tavira B, Coto E, Díaz-Corte C, et al. KCNQ1 gene variants and risk of new-onset diabetes in tacrolimus-treated renal-transplanted patients. Clin Transplant. 2011;25:E284–291. doi: 210.1111/j.1399-0012.2011.01417.x. Epub 02011 Mar 01411. [DOI] [PubMed] [Google Scholar]
- 10.Brown JT, Abdel-Rahman SM, van Haandel L, Gaedigk A, Lin YS, Leeder JS. Single dose, CYP2D6 genotype-stratified pharmacokinetic study of atomoxetine in children with ADHD. Clin Pharmacol Ther. 2016;99:642–650. doi: 610.1002/cpt.1319. Epub 2016 Jan 1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown JT, Bishop JR, Sangkuhl K, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Cytochrome P450 (CYP)2D6 Genotype and Atomoxetine Therapy. Clin Pharmacol Ther. 2019;106:94–102. doi: 110.1002/cpt.1409. Epub 2019 Apr 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oetting WS, Wu B, Schladt DP, et al. Genetic Variants Associated With Immunosuppressant Pharmacokinetics and Adverse Effects in the DeKAF Genomics Genome-wide Association Studies. Transplantation. 2019;103:1131–1139. doi: 1110.1097/TP.0000000000002625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Peck C. The randomized concentration-controlled clinical trial (CCR) : an informationrich alternative to the randomized placebo controlled clinical trial (PCT). Clin. Pharmacol. Ther 1990;47:148. [Google Scholar]
- 14.Gaedigk A, Dinh JC, Jeong H, Prasad B, Leeder JS. Ten Years’ Experience with the CYP2D6 Activity Score: A Perspective on Future Investigations to Improve Clinical Predictions for Precision Therapeutics. J Pers Med. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Van Driest SL, McGregor TL, Velez Edwards DR, et al. Genome-Wide Association Study of Serum Creatinine Levels during Vancomycin Therapy. PLoS One. 2015;10:e0127791. doi: 0127710.0121371/journal.pone.0127791. eCollection 0122015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tomita T, Yasui-Furukori N, Nakagami T, et al. The influence of 5-HTTLPR genotype on the association between the plasma concentration and therapeutic effect of paroxetine in patients with major depressive disorder. PLoS One. 2014;9:e98099. doi: 98010.91371/journal.pone.0098099. eCollection 0092014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boswell MV, Stauble ME, Loyd GE, et al. The role of hydromorphone and OPRM1 in postoperative pain relief with hydrocodone. Pain Physician. 2013;16:E227–235. [PubMed] [Google Scholar]
- 18.Hertz DL, Kidwell KM, Vangipuram K, et al. Paclitaxel Plasma Concentration After the First Infusion Predicts Treatment-Limiting Peripheral Neuropathy. Clin Cancer Res. 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marcath LA, Kidwell KM, Vangipuram K, et al. Genetic variation in EPHA contributes to sensitivity to paclitaxel-induced peripheral neuropathy. Br J Clin Pharmacol. 2020;86:880–890. doi: 810.1111/bcp.14192. Epub 12020 Feb 14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Upton RN, Mould DR. Basic concepts in population modeling, simulation, and model-based drug development: part 3-introduction to pharmacodynamic modeling methods. CPT Pharmacometrics Syst Pharmacol. 2014;3:e88. doi: 10.1038/psp.2013.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ngaimisi E, Gopalakrishnan M, Ivaturi V, Zhang W, Young M, Laffont CM. Exposure-response analyses to support dosing recommendations for RBP-6000 buprenorphine montly formulation in subjects with opioid use disorder. Journal of Pharmacokinetics and Pharmacodynamics. 2017;44:S50. [Google Scholar]
- 22.Kuepfer L, Niederalt C, Wendl T, et al. Applied Concepts in PBPK Modeling: How to Build a PBPK/PD Model. CPT Pharmacometrics Syst Pharmacol. 2016;5:516–531. doi: 510.1002/psp1004.12134. Epub 12016 Oct 12119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Driest SL, Marshall MD, Hachey B, et al. Pragmatic pharmacology: population pharmacokinetic analysis of fentanyl using remnant samples from children after cardiac surgery. Br J Clin Pharmacol. 2016;81:1165–1174. doi: 1110.1111/bcp.12903. Epub 12016 Apr 12915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diouf B, Evans WE. Pharmacogenomics of Vincristine-Induced Peripheral Neuropathy: Progress Continues. Clin Pharmacol Ther. 2019;105:315–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sharma MR, Joshi SS, Karrison TG, et al. A UGT1A1 genotype-guided dosing study of modified FOLFIRINOX in previously untreated patients with advanced gastrointestinal malignancies. Cancer. 2019;125:1629–1636. [DOI] [PubMed] [Google Scholar]