

Abstract
Hypoxic reprogramming of vasculature relies on genetic, epigenetic, and metabolic circuitry, but the control points are unknown. In pulmonary arterial hypertension (PAH), a disease driven by hypoxia inducible factor (HIF)–dependent vascular dysfunction, HIF-2α promoted expression of neighboring genes, long noncoding RNA (lncRNA) histone lysine N-methyltransferase 2E-antisense 1 (KMT2E-AS1) and histone lysine N-methyltransferase 2E (KMT2E). KMT2E-AS1 stabilized KMT2E protein to increase epigenetic histone 3 lysine 4 trimethylation (H3K4me3), driving HIF-2α–dependent metabolic and pathogenic endothelial activity. This lncRNA axis also increased HIF-2α expression across epigenetic, transcriptional, and posttranscriptional contexts, thus promoting a positive feedback loop to further augment HIF-2α activity. We identified a genetic association between rs73184087, a single-nucleotide variant (SNV) within a KMT2E intron, and disease risk in PAH discovery and replication patient cohorts and in a global meta-analysis. This SNV displayed allele (G)–specific association with HIF-2α, engaged in long-range chromatin interactions, and induced the lncRNA-K MT2E tandem in hypoxic (G/G) cells. In vivo, KMT2E-AS1 deficiency protected against PAH in mice, as did pharmacologic inhibition of histone methylation in rats. Conversely, forced lncRNA expression promoted more severe PH. Thus, the KMT2E-AS1/KMT2E pair orchestrates across convergent multi-ome landscapes to mediate HIF-2α pathobiology and represents a key clinical target in pulmonary hypertension.
INTRODUCTION
Cellular reprogramming by hypoxia relies on incompletely defined genetic, epigenetic, and metabolic circuitry. Such fundamental concepts are important for pulmonary hypertension (PH) and pulmonary arterial hypertension (PAH)—diseases of lung blood vessels linked to hypoxia and its master transcription factors, including hypoxia inducible factor α (HIF-α) (1). HIF-2α in pulmonary arterial endothelial cells (PAECs) is particularly important in promoting this disease (2). However, the broad heterogeneity of disease and hypoxia dependent molecular circuitry has bred confusion regarding the development of crucial endothelial pathophenotypes (3).
An integrated understanding of genetic, epigenetic, and metabolic landscapes in the hypoxic endothelium and pulmonary vasculature is lacking. HIF-dependent pathways regulate metabolic and mitochondrial programs that are dysregulated in hypoxic and diseased pulmonary vasculature in PH (4). Genome-w ide molecular profiling in PH (5) has revealed that epigenetic marks of the genome and associated histones are altered in hypoxia across various PH subtypes (6). Furthermore, G9a histone methyltransferase inhibitors targeting primarily histone H3 lysine 9 marks (H3K9) have been reported to improve experimental PAH (7). However, the roles of other hypoxia-driven histone methylation marks in PAH have been poorly described. Histone H3 lysine 4 trimethylation (H3K4me3) is enriched near promoters of activated genes and drives transcription (8). Although H3K4me3 is increased in hypoxia (9) and controlled by HIF-1α and HIF-2α (10), any pathogenic roles of H3K4me3 in orchestrating metabolic reprogramming in hypoxia are still unknown. Moreover, because hypoxia and HIF-2α constitute crucial triggers of the World Symposium on Pulmonary Hypertension (WSPH) group 1 PH (PAH) and group 3 PH (PH due to hypoxic lung disease), hypoxic regulation of H3K4me3 may exert control over key pathogenic pathways in these PH subtypes. Last, genomic insights are advancing regarding various associations between genetic variants with PAH risk, survival, and disease severity (11). However, because of the limited global number of patients with PAH, there are barriers to generating a comprehensive catalog of genetic variants causatively linked to disease initiation or progression.
Long noncoding RNAs (lncRNAs) can exert regulatory activity across genomic, epigenetic, and metabolic domains (12). lncRNAs are single stranded RNAs that affect cellular function by complexing with chromosomal DNA, RNAs, or proteins and may prevent microRNA binding to target mRNAs. lncRNAs are dysregulated in PH, and certain lncRNAs are controlled by PH triggers, such as hypoxia (13). Characterization of lncRNAs in pulmonary vascular cells has been limited (14), and functional data regarding their roles in PH are just emerging (15, 16). Yet, the majority of lncRNAs do not carry full sequence conservation in mammals, making it challenging to translate in vivo lncRNA biology between rodents and humans.
We hypothesized that specific lncRNAs serve as broad effectors of HIF dependent pathobiology, orchestrating epigenetic, metabolic, and genomic processes in health and disease. Here, combining insights into genetic epidemiology with molecular mechanism, we identified a lncRNA-protein pair, governed in part by an endogenous human single-nucleotide variant (SNV), which carries crucial epigenetic and metabolic functions in endothelial cells and controls PH in vivo.
RESULTS
Mouse lncRNA 5031425E22Rik and the human ortholog KMT2E-AS1 were up regulated in rodent and human types of PH
Using published RNA sequencing data (17), we identified differentially expressed lncRNAs in lungs of PAH mice induced by the small-molecule SU5416 and chronic hypoxia versus controls (fig. S1A). After filtering for length, conservation, and nonprotein coding status, we identified nine candidates, one of which was an up regulated lncRNA 5031425E22Rik (or E22) that mapped to a human ortholog histone lysine N-methyltransferase 2E gene-antisense 1 (KMT2E-AS1). Across mammals, this lncRNA gene sits on the antisense strand and adjacent (head-to-head) to the histone lysine N-methyltransferase 2E gene (KMT2E), a member of a regulatory family controlling H3K4me3 and chromatin remodeling (Fig. 1A) (18). This lncRNA does not carry obvious sequence homology with KMT2E. Nonetheless, given its conserved chromosomal location and partially conserved sequence with human KMT2E-AS1, we analyzed this lncRNA further. Inference from RNA sequencing reads indicated that this lncRNA is most abundantly expressed as a ~2-kb isoform. E22 and KMT2E-AS1 transcripts were up-regulated in pulmonary vascular tissue, particularly CD31-positive endothelial cells, in mouse and human cases of PH, accompanied by increased KMT2E (Fig. 1, B to E; fig. S1, B to E; and table S1). By immunofluorescence microscopy and fluorescence in situ hybridization (FISH), increased KMT2E-AS1 and KMT2E were also observed in CD31-positive endothelial cells of remodeled pulmonary arterioles of WSPH group 1 PAH and group 3 PH (Fig. 1, F and G, and table S1), in hypoxic mice (fig. S1F), and in PAH rat models (fig. S1, G and H). H3K4me3 was increased in both groups 1 and 3 PH (Fig. 1H), consistent with rodents with PH (fig. S1I).
Fig. 1. Human KMT2E-AS1 and neighboring KMT2E are up-regulated across in vivo and in vitro models of PH.
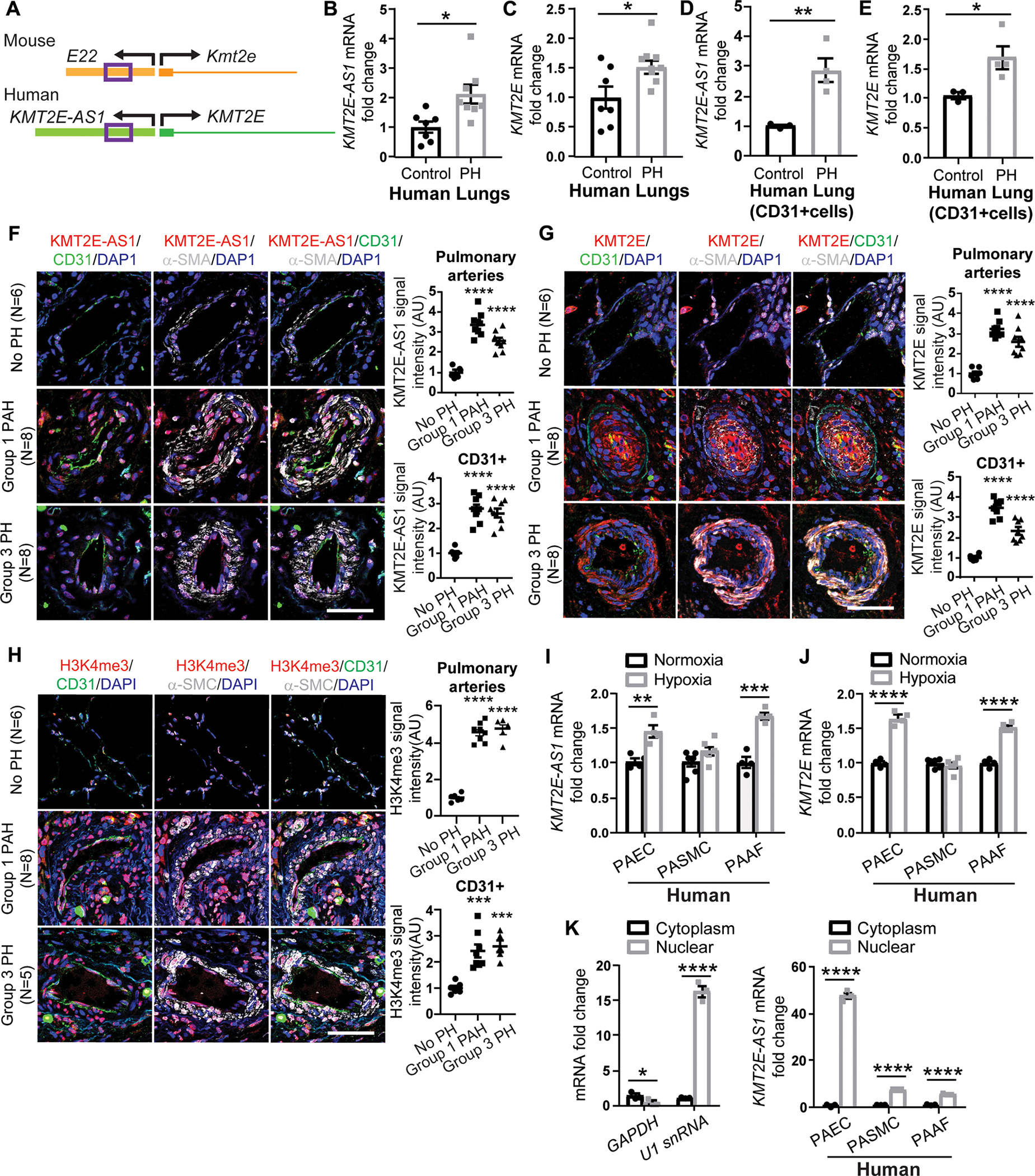
(A) Gene structure of mouse lncRNA 5031425E22Rik (E22) located adjacent to protein-coding gene Kmt2e is encoded on the opposite DNA strand and positioned in the opposite transcriptional direction. human lncRNA ortholog KMT2E-AS1 and neighboring gene KMT2E show similar genomic architecture; sequence conservation (purple box 500 bp region) is shown within mouse E22 and human KMT2E-AS1. (B to E) human lncRNA KMT2E-AS1 and KMT2E transcripts in lung tissue (B and C) (n = 7 to 10; *P < 0.05, unpaired Student’s t test; data represent the mean ± SEM) and cd31+ cells (d and e) (n = 3 or 4; *P < 0.05, **P < 0.01, unpaired Student’s t test; data represent the mean ± SEM) of patients with WSPH group 1 PAH (table S1). (F to H) representative FISH and immunofluorescence (IF) staining and quantifications of KMT2E-AS1 (red; F), KMT2e (red; G), and h3K4me3 (red; H) in cd31+ endothelium of human lung from individuals with group 1 and group 3 PH versus non-PH controls (n = 5 to 8; ***P < 0.001, ****P < 0.0001 versus no PH, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent the mean ± SEM). Scale bars, 50 μm. (I) KMT2E-AS1 transcript in cultured PAECs, adventitial fibroblasts (PAAFs), and PASMCs (n = 4 to 6; **P < 0.01, ***P < 0.001, unpaired Student’s t test; data represent the mean ± SEM). (J) KMT2E transcript in human PAECs, PASMCs, and PAAFs (n = 4 to 6; ****P < 0.0001, unpaired Student’s t test for PAECs and PAAFs, Mann-Whitney test for PASMCs; data represent mean ± SEM). (K) KMT2E-AS1 (right graph) in cytosolic and nucleic fractions of human cell types [glyceraldehyde phosphate dehydrogenase (GAPDH) and U1 small nuclear RNA (snRNA) served as cytoplasmic and nuclear controls, respectively; left graphs] (n = 3; *P < 0.05, ****P < 0.0001, unpaired Student’s t test; data represent mean ± SEM). DAPI, 4′,6-diamidino-2-phenylindole; AU, arbitrary units.
In cultured PAECs, hypoxic exposure drove similar up-regulation of KMT2E-AS1 and KMT2E (Fig. 1, I and J). Pulmonary arterial adventitial fibroblasts also displayed induction of this lncRNA-KMT2E pair, but other pulmonary vascular cell types, such as pulmonary artery smooth muscle cells (PASMCs), did not (Fig. 1, I and J). This specific induction, particularly in PAECs, led to subcellular localization of KMT2E-AS1 mainly to the nucleus (Fig. 1K). In mouse PAECs and fibroblasts, hypoxia up-regulated E22 with predominant localization in the nucleus (fig. S1, J to L). Other known inflammatory cytokine triggers of PH, including interleukin-1β (IL-1β) and IL-6, did not alter KMT2E-AS1 (fig. S1M) in PAECs, suggesting the central mechanism of hypoxia in controlling this lncRNA axis.
Hypoxic induction of KMT2E-AS1 and KMT2E was HIF-2α dependent
Previous chromatin immunoprecipitation sequencing (ChIP-Seq) identified interactions between HIF-2α and the KMT2E promoter (19). Correspondingly, by small interfering RNA (siRNA) knockdown of HIF 1α and HIF-2α, we found that HIF-2α knockdown prevented the increase of KMT2E-AS1 and KMT2E transcripts in hypoxic PAECs (Fig. 2, A and B). Conversely, lentiviral overexpression of a constitutively active HIF-2α gene (20) in normoxia promoted the increased expression of this lncRNA-KMT2E pair (Fig. 2, C and D). Thus, HIF-2α is both necessary and sufficient for the hypoxic up regulation of KMT2E-AS1 and the neighboring KMT2E in endothelial cells.
Fig. 2. KMT2E-AS1 interacts with KMT2E to enhance protein stability and increase histone 3 lysine 4 trimethylation (H3K-4me3).
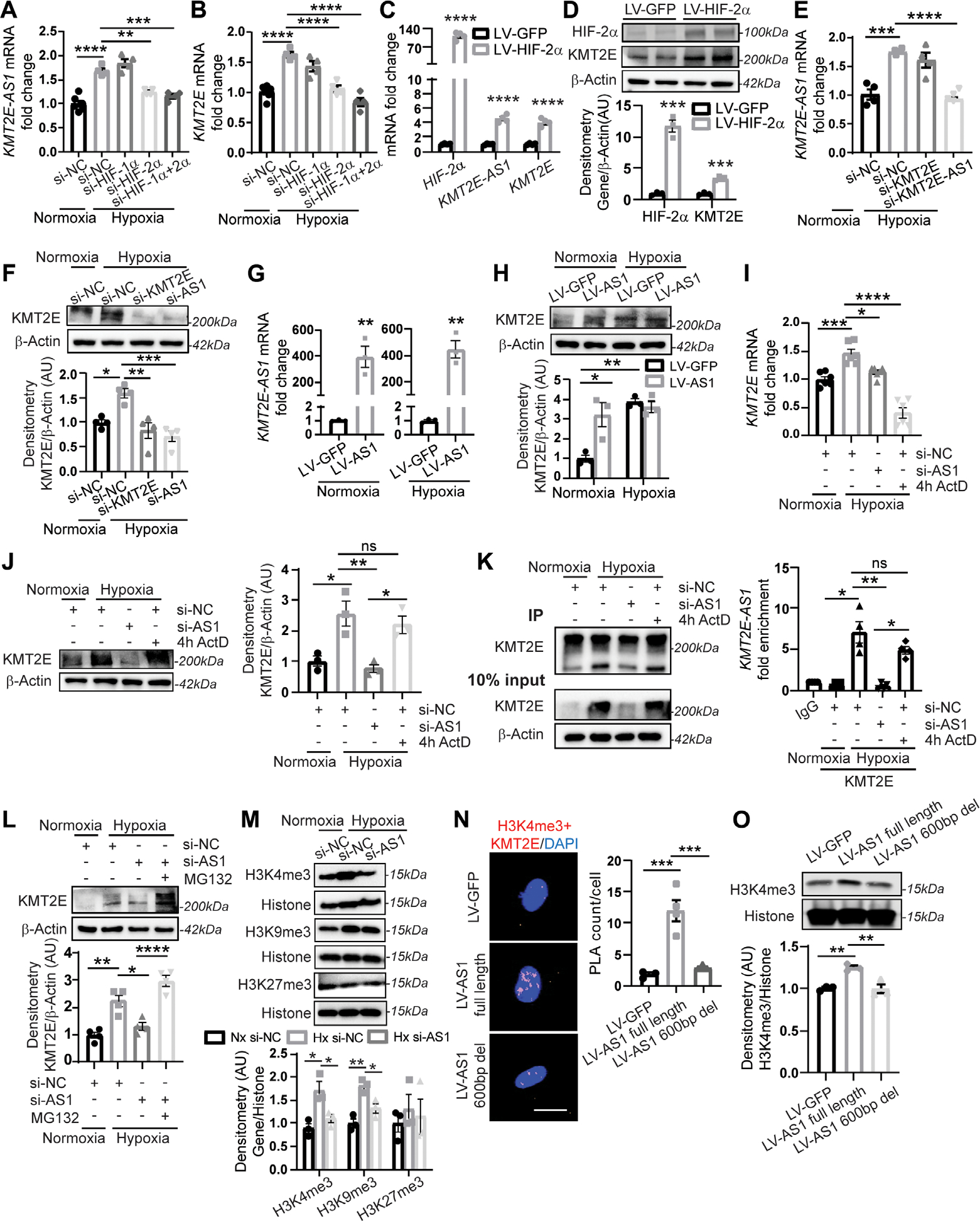
(A and B) KMT2E-AS1 (a) and KMT2E (B) transcripts in hypoxic human PAECs with knockdown (siRNA) of HIF-2α and/or HIF-1α versus normoxic scramble control (NC) (n = 4 to 6; **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (C) KMT2E-AS1, KMT2E, and HIF-2α transcripts in normoxic human PAECs with overexpression of a constitutively active HIF-2α (lV-HIF-2α versus LV-GFP control) (n = 4; ****P < 0.0001, unpaired Student’s t test; data represent mean ± SEM). (D) HIF-2α and KMT2e proteins in normoxic human PAECs transduced with lV-HIF-2α versus LV-GFP control (n = 3; ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM). (E) KMT2E-AS1 transcript in human PAECs under hypoxia and siRNA knockdown of KMT2E or KMT2E-AS1 (n = 4; ***P < 0.001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (F) KMT2e protein in human PAECs under hypoxia and knockdown of KMT2E or KMT2E-AS1 (n = 4; *P < 0.05, **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (G) KMT2E-AS1 transcript in human PAECs posttransduction with KMT2E-AS1 lentivirus (LV-AS1) versus GFP control (LV-GFP) under normoxia (left) and hypoxia (right) (n = 3; **P < 0.01, unpaired Student’s t test; data represent mean ± SEM). (H) KMT2e protein in human PAECs transduced as in (G) by (n = 3; *P < 0.05, **P < 0.01, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (I) KMT2E transcript in human PAECs under hypoxia and KMT2E-AS1 knockdown or 4 hour (4h) exposure to the transcriptional inhibitor actinomycin D (ActD) (n = 6; *P < 0.05, **P < 0.01, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (J) KMT2e protein in human PAECs after KMT2E-AS1 knockdown or actinomycin d (ActD) exposure under hypoxia (n = 3; *P < 0.05, **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). ns, not significant. (K) RNA immunoprecipitation (RIP)–qPCR [IP: KMT2e versus immunoglobulin G (IgG) negative control] of KMT2e protein in normoxia and hypoxia (left immunoblot). KMT2E-AS1 transcript in the IP fraction of human PAECs after KMT2E-AS1 knockdown or ActD exposure (4 hours) under hypoxia (right graph) (n = 4; *P < 0.05, **P < 0.01, Kruskal Wallis test followed by dunn’s post hoc analysis; data represent mean ± SEM). (L) KMT2e protein in human PAECs after KMT2E-AS1 knockdown or proteasomal inhibitor MG132 in hypoxia (n = 4; *P < 0.05, **P < 0.01, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (M) h3K4me3, h3K9me3, and h3K27me3 in human PAECs after hypoxia and KMT2E-AS1 knockdown (n = 3; *P < 0.05, **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis for H3K4me3 and H3K9me3, Kruskal-Wallis test followed by dunn’s post hoc analysis for H3K27me3; data represent mean ± SEM). (N) nuclear interaction of H3K4me3 marks and KMT2e protein, measured by proximity ligation assay (PLA; red, left images) and quantified by PLA counts per cell (right graph) in human PAECs after lentiviral expression of KMT2E-AS1 (full length) or KMT2E-AS1 deletion mutant (fig. S8) versus GFP control (n = 3 or 4; ***P < 0.001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). Scale bar, 20 μm. (O) H3K4me3 in human PAECs under lentiviral expression of KMT2E-AS1 (full length) or KMT2E-AS1 deletion mutant (n = 3; **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM).
KMT2E-AS1 interacted with KMT2E to enhance protein stability that increases histone methylation, including H3K4me3
Considering the conserved chromosomal neighboring location of KMT2E-AS1 and KMT2E, the functions of lncRNAs to complex with proteins to affect chromatin remodeling (12), and the known function of KMT2E in H3K4me3 (18), we hypothesized that KMT2E-AS1 carries epigenetic activity stemming from physical interactions with the KMT2E protein. We screened for and identified an effective siRNA targeting KMT2E-AS1. Accordingly, we found that siRNA knockdown of KMT2E-AS1 in hypoxia (Fig. 2E) down regulated KMT2E protein expression by immunoblot (Fig. 2F). Conversely, forced expression of KMT2E-AS1 in normoxia increased KMT2E protein (Fig. 2, G and H). Our results showed transcriptional control of this tandem pair by HIF-2α (Fig. 2, A to D). Yet, in hypoxia, short-term (4 hours) exposure to the transcriptional inhibitor actinomycin D reduced KMT2E transcript (Fig. 2I) but not protein expression (Fig. 2J), suggesting that KMT2E protein is stabilized in hypoxia. The in silico PRIdictor algorithm (21) predicted that KMT2E-AS1 transcript bound to the KMT2E protein (prediction score of 0.534, >0.5 threshold). To validate RNA-protein interaction, RNA-protein immunoprecipitation assay, using antibodies against KMT2E followed by reverse transcription quantitative polymerase chain reaction (RT-q PCR) of KMT2E-AS1, revealed that this lncRNA directly and specifically complexed with KMT2E under hypoxic conditions (Fig. 2K). Actinomycin D did not disrupt this physical interaction (P > 0.05), reflecting the stability of such RNA-protein complexes despite acute reductions in RNA transcript production. Pretreating with the proteasomal inhibitor MG132 reversed the effect of KMT2E-AS1 knockdown on KMT2E (Fig. 2L), indicating that the stabilized complex was responsible for protein expression.
To investigate the downstream consequences of KMT2E stabilization, we performed immunoblots specific for H3K4me3 in hypoxic PAECs. KMT2E-AS1 knockdown decreased H3K4me3 (Fig. 2M). Moreover, forced expression of KMT2E-AS1 increased the interaction of H3K4me3 with KMT2E by proximity ligation assay (Fig. 2N), whereas lentiviral delivery of a deletion mutant of this lncRNA missing a ~600-bp conserved sequence (fig. S8F) failed to increase this interaction. Correspondingly, forced expression of full length KMT2E-AS1, but not the deletion mutant lentivirus, increased downstream H3K4me3 as assessed by immunoblot (Fig. 2O). Thus, KMT2E-AS1 is a hypoxia-driven lncRNA that complexes with and stabilizes KMT2E protein to increase H3K4me3 histone tri-methylation.
We then determined whether other major histone lysine methylation marks that can be dependent on hypoxia (22), H3K9 and H3K27, were also controlled by this lncRNA-KMT2E tandem. In cultured PAECs, H3K9me3, but not H3K27, marks were increased by hypoxia and modestly reversed by KMT2E-AS1 knockdown (Fig. 2M), a finding consistent with previous studies showing that KMT2E deficiency can induce a histone demethylase, lysine-specific histone demethylase 1A (LSD1), that specifically reduces H3K9me3 (23, 24). Similarly, immunofluorescence staining of human group 1 PAH and group 3 PH demonstrated increased expression of H3K9me3 but not H3K-27me3 in diseased pulmonary arterioles (fig. S2, A to C). However, H3K4me3 reversal in PAECs by KMT2E-AS1 knockdown was more robust than the reversal of H3K9me3 (Fig. 2M), consistent with a primary role of this lncRNA axis in H3K4me3.
KMT2E-AS1 drove hypoxic metabolic reprogramming
Because KMT2E-AS1 acted in conjunction with KMT2E to regulate primarily epigenetic H3K4me3, we sought to define the landscape of transcriptional alterations under this lncRNA’s control during hypoxic endothelial reprogramming. In PAECs, siRNA knockdown of either KMT2E or KMT2E-AS1 (fig. S3, A and B) phenocopied each other by reversing the expression of 2480 genes that were altered by hypoxia (Fig. 3, A and B, and table S3). Gene set enrichment analysis (GSEA) (25) of these RNA sequencing results revealed that >50% of represented pathways were “hypoxia” and “metabolism” gene networks (Fig. 3A), consistent with the major activities of HIF-2α. Five hundred fifty four of 1285 reversed metabolism genes (Fig. 3B) were related to energy generation and the tricarboxylic acid cycle. To determine which of these changes were controlled directly by H3K4me3, H3K4me3 ChIP-Seq was performed in hypoxic versus normoxic PAECs (fig. S3C and table S4). The promoters of many of the top dysregulated genes in hypoxia and metabolism networks regulated by the lncRNA-KMT2E axis carried more H3K4me3 marks in hypoxia, suggesting a direct epigenetic mechanism by which the lncRNA axis modulates HIF-2α metabolic phenotypes (Fig. 3B, fig. S3C, and tables S3 and S4). Furthermore, not all H3K4me3-dependent genes in hypoxia were regulated by the lncRNA-KMT2E axis (fig. S3C), emphasizing an epigenetic specificity of this regulatory axis. By RNA sequencing and ChIP-Seq overlay (fig. S3B) and further independent ChIP-qPCR (Fig. 3C), we confirmed the lncRNA mir210hg as one of several KMT2E-AS1–dependent and H3K4me3-dependent genes.
Fig. 3. KMT2E-AS1 regulates a gene network driving hypoxic metabolic reprogramming.
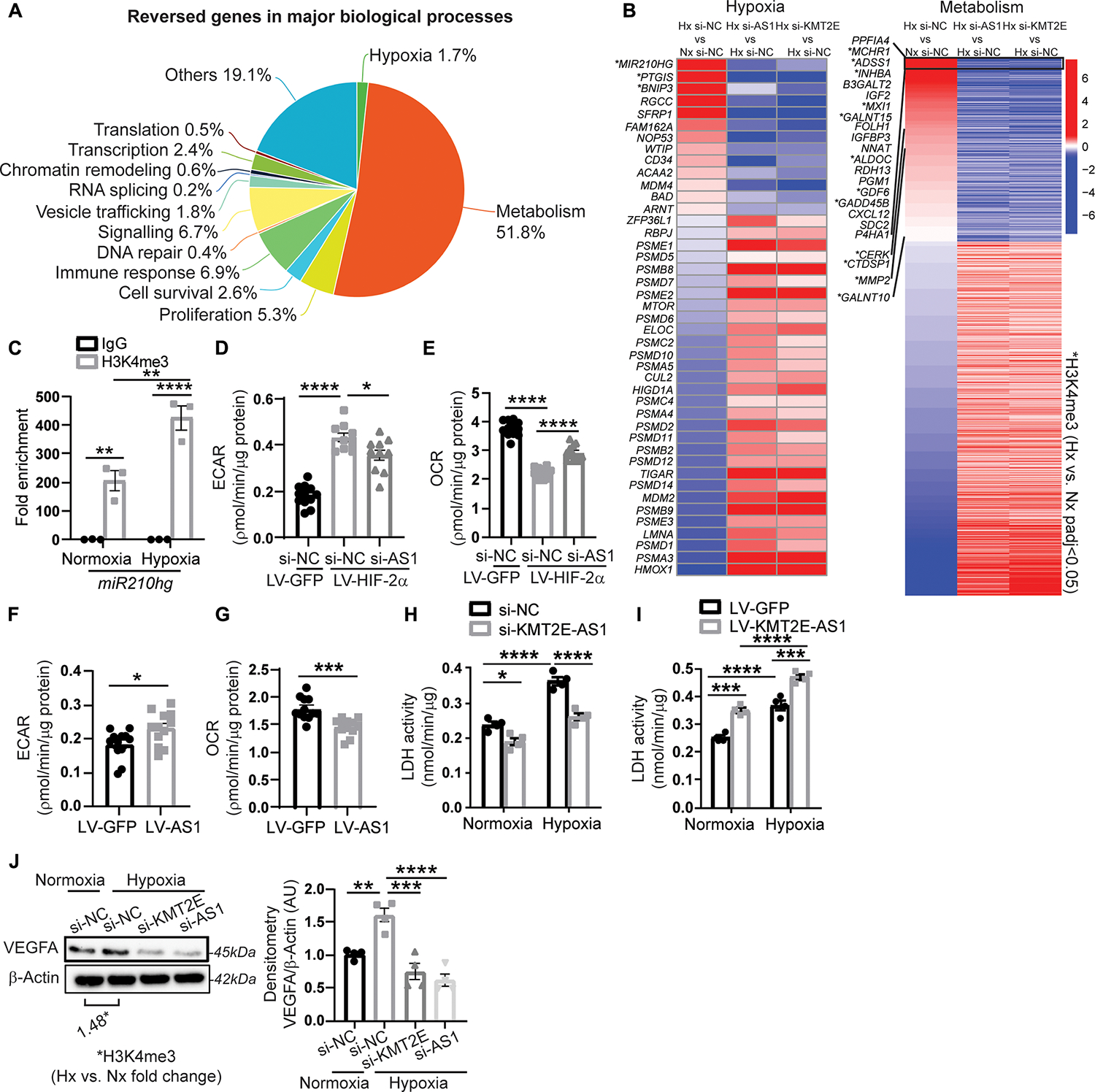
(A) RNA sequencing of human PAECs revealed that 2480 genes are altered by hypoxia and reversed by either KMT2E-AS1 or KMT2E in hypoxia. GSEA reveals the major biological processes represented by these reversed genes. (B) heatmaps display genes in hypoxia and metabolism networks that are altered by hypoxia (leftmost column) and are reversed by KMT2E-AS1 (middle column) and KMT2E (rightmost column) knockdown in hypoxia. Adjusted P < 0.05 for each gene shown. H3K4me3 ChIP-Seq was also performed in hypoxic versus normoxic PAECs. A subcohort of these genes displays increased h3K4me3 marks in hypoxia by co-analyzing these ChIP-Seq and RNA sequencing data (* indicates methylated genes with adjusted P < 0.05). (C) ChIP qPCR of h3K4me3 binding at the promoter site of the lncRNA miR210hg (n = 3; **P < 0.01, ****P < 0.0001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (D and E) extracellular acidification rate (ECAR) (d) and baseline OCR (E) of human PAECs after KMT2E-AS1 knockdown and HIF-2α overexpression (n = 10 to 12; *P < 0.05, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (F and G) ECAR (F) and baseline OCR (G) after KMT2E-AS1 overexpression (n = 12; *P < 0.05, ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM). (H and I) LDH enzymatic activity of human PAECs after KMT2E-AS1 knockdown (H) and KMT2E-AS1 overexpression (I), a representative measure of glycolysis (n = 4; *P < 0.05, ***P < 0.001, ****P < 0.0001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (J) VEGF abundance in hypoxic PAECs after knockdown of KMT2E and KMT2E-AS1 (n = 4; **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM), overlayed with ChIP-seq of h3K4me3 at the VEGFA gene in hypoxia versus normoxia.
Consistent with KMT2E-AS1’s control over HIF-dependent metabolism, KMT2E-AS1 knockdown mitigated the hypoxic induction of extracellular acidification rate (ECAR), a representative measure of glycolysis, as well as concomitant lactate dehydrogenase (LDH) enzymatic activity (Fig. 3, D and H); forced KMT2E-AS1 expression increased ECAR and LDH activity (Fig. 3, F and I). Moreover, in cultured PAECs, KMT2E-AS1 knockdown increased baseline oxygen consumption rate (OCR) (Fig. 3E), whereas forced KMT2E-AS1 decreased OCR (Fig. 3G). Furthermore, vascular endothelial growth factor (VEGFA) was up-regulated by hypoxia, consistent with increased H3K4me3 at its gene locus (Fig. 3J). Both KMT2E-AS1 and KMT2E knockdown decreased VEGFA expression. Thus, these data demonstrated that KMT2E-AS1 regulates a gene network that decreases oxidative metabolism, increases glycolysis, and controls hypoxic PAEC adaptation.
KMT2E-AS1 induced HIF via increasing HIF transcription and controlling HIF degradation
In addition to the ability of KMT2E-AS1 to stabilize KMT2E protein, knockdown of KMT2E-AS1 reduced KMT2E transcript (Fig. 2I and fig. S3A). Guided by the identification of “hypoxic” response pathway genes regulated by this lncRNA axis that control HIF transcription (miR210hg) and HIF protein degradation (ELOC) (Fig. 3B), we determined whether HIF-2α–dependent KMT2E-AS1 reciprocally regulates HIF-2α expression. siRNA knockdown of KMT2E-AS1 or KMT2E in hypoxic PAECs decreased HIF-2α expression (Fig. 4A), whereas forced expression of this lncRNA increased HIF-2α in normoxia and hypoxia (Fig. 4B). The hypoxia dependent miR210hg gene was induced by this lncRNA axis (Fig. 3B and fig. S3B). miR210hg is a lncRNA that directly binds the 5′ untranslated region of HIF transcripts (26), leading to increased translation and increased HIF protein expression. Moreover, for both RNA and protein, KMT2E-AS1 knockdown increased elongin C (ELOC), a crucial component of the Von Hippel Lindau complex that controls HIF-2α protein stability (Fig. 4, C and D), indicating that the lncRNA augments HIF-2α expression via modulation of the hydroxylase-ubiquitin-proteasome pathway. The proteasomal inhibitor MG132 reversed the down regulation of HIF-2α by lncRNA knockdown (Fig. 4E). Thus, hypoxic induction of KMT2E-AS1 and KMT2E participates in a positive feedback loop to increase HIF-2α expression via multiple epigenetic, transcriptional, and posttranscriptional mechanisms to activate HIF-2α transcription and translation as well as to alter HIF-2α protein degradation.
Fig. 4. KMT2E-AS1 induces HIF-2α activation and prevents HIF-2α degradation in driving endothelial pathophenotypes.
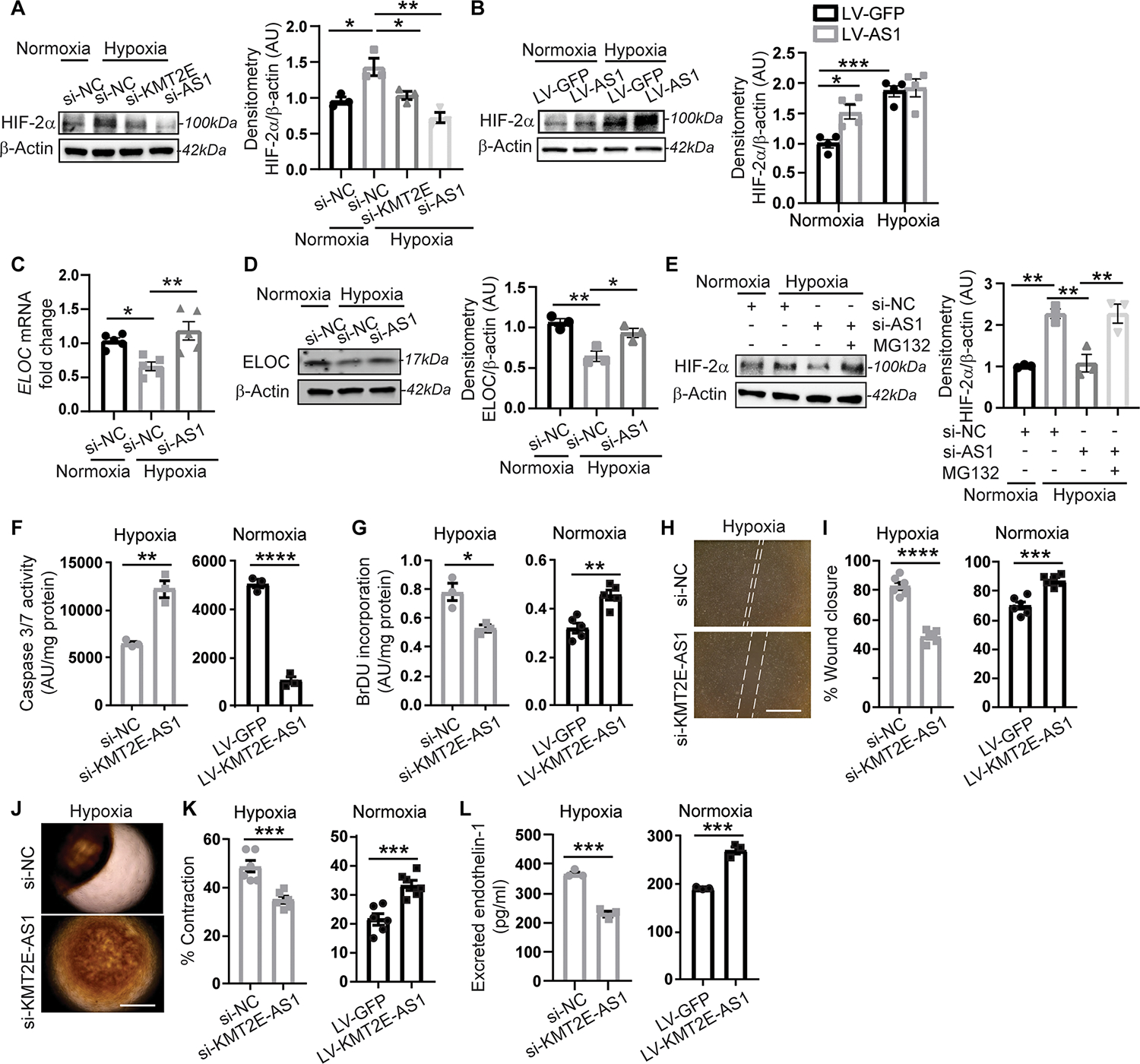
(A and B) HIF-2α protein in hypoxic human PAECs after knockdown of KMT2E-AS1 (a) and overexpression of KMT2E-AS1 (B) (n = 3 or 4; *P < 0.05, **P < 0.01, ***P < 0.001, one-way (A) or two-way (B) ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM. (C and D) ELOC RNA (c) and protein (D) expression in human PAECs after hypoxia and KMT2E-AS1 knockdown (Fig. 3B) (n = 3 to 5; *P < 0.05, **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (E) HIF-2α protein in human PAECs after KMT2E-AS1 knockdown and MG132 treatment (n = 3; **P < 0.01, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (F) apoptotic caspase 3/7 activity in human PAECs after KMT2E-AS1 knockdown under hypoxia (left) and KMT2E-AS1 overexpression under normoxia (right) (n = 3; **P < 0.01, ****P < 0.0001, unpaired Student’s t test; data represent mean ± SEM). (G) BrdU proliferative potential after KMT2E-AS1 knockdown in hypoxia (left) and forced KMT2E-AS1 expression in normoxia (right) (n = 3 to 5; *P < 0.05, **P < 0.01, unpaired Student’s t test; data represent mean ± SEM). (H and I) Scratch wound healing assay (h) measures migration of human PAECs after KMT2E-AS1 knockdown under hypoxia (left, I) and KMT2E-AS1 overexpression in normoxia (right, I) (n = 6; ***P < 0.001, ****P < 0.0001, unpaired Student’s t test; data represent mean ± SEM). (J and K) human PASMC contraction in gel matrix (J) with conditioned media from PAECs after knockdown of KMT2E-AS1 under hypoxia (K, left graph) and after forced expression of KMT2E-AS1 under normoxia (K, right graph) (n = 6; ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM). (L) endothelin-1 in conditioned media from human PAECs after KMT2E knockdown under hypoxia (left) and forced KMT2E expression in normoxia (right) by enzyme linked immunosorbent assay (n = 3; ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM).
KMT2E-AS1 promoted endothelial pathophenotypes of PH
KMT2E-AS1 drove endothelial pathophenotypes linked to HIF biology and PH. Knockdown of KMT2E-AS1 increased PAEC apoptotic potential in hypoxia, whereas forced expression of KMT2E-AS1 via lentiviral transduction decreased apoptosis in normoxia (Fig. 4F). KMT2E-AS1 knockdown decreased PAEC proliferation in hypoxia, whereas forced expression increased proliferation in normoxia as observed by 5 bromo-2′-deoxyuridine (BrdU) incorporation (Fig. 4G). Thus, KMT2E-AS1 is necessary and sufficient to promote PAEC proliferation. Consistent with these alterations in cell survival and proliferative capacity, KMT2E-AS1 knockdown decreased PAEC migration in hypoxia, whereas forced expression increased such activity by wound closure assay in vitro (Fig. 4, H and I). Similarly, by quantifying gel contraction as a surrogate of smooth muscle cell contraction when exposed to PAEC conditioned media, we found that KMT2E-AS1 knockdown in hypoxic PAECs generated conditioned media that decreased contraction seen in hypoxia, whereas forced expression of KMT2E-AS1 in PAECs increased contraction in normoxia (Fig. 4, J and K). Consistent with these alterations in vasomotor activity, KMT2E-AS1 knockdown decreased secreted endothelin-1 (EDN1) in hypoxia, whereas forced expression of KMT2E-AS1 increased EDN1 in normoxia (Fig. 4L). Via sequences different from that of the above siRNA, antisense oligonucleotide gapmers targeting KMT2E-AS1 phenocopied siRNA silencing of KMT2E-AS1 with knockdown of KMT2E-AS1 and downstream VEGFA and EDN1 expression (fig. S4, A to C). We found that KMT2E-AS1 inhibited PAEC apoptosis and enhanced proliferation, migration, and vasomotor tone, consistent with a role that promotes endothelial dysregulation and PAH.
KMT2E SNV rs73184087 (G) allele was associated with WSPH group 1 PAH risk
Genetic control of HIF-2α activity can be facilitated by SNVs (27) that alter transcription factor binding sites. SNVs alter HIF binding sites even outside canonical promoter regions (27) with consequent disruptions of long-range genomic interactions with active promoter sites. Thus, we screened for such SNVs relevant to this lncRNA-K MT2E locus within a previously reported WSPH group 1 PAH discovery cohort (“PAH Biobank”) of European-descent (N = 694) individuals versus individuals without disease (N = 1560) (28) (table S5). Among the 883 genotyped and imputed SNVs (with minor allele frequency > 1%) within and flanking (+/−200 kb) the KMT2E-AS1 KMT2E tandem locus, we prioritized 59 SNVs (table S6) with predicted HIF-2α binding to one of either the minor or major alleles using position weight matrices (PWMs) derived from HIF-2α ChIP-Seq (19). Among these SNVs, by Firth’s penalized logistic regression, we observed a significant association for rs73184087 (GRCh38:7:105087911) with the risk for developing PAH, with the G allele conferring an adjusted odds ratio (OR) of 1.86 [95% confidence interval (CI):1.30–2.67; P = 7.40 × 10−4] in the discovery cohort (Fig. 5A). Linkage disequilibrium pattern analysis revealed that there was no strong association of SNV rs73184087 with other neighboring SNVs (fig. S5). PWM scoring predicted more robust HIF-2α binding to the effect allele (G) of this SNV (log odds score 10.01, P < 10−8) versus the non-effect allele (A) (log odds score 4.56). We then replicated the SNV association with disease risk in a second, independent PAH cohort of participants of European descent from the University of Pittsburgh Medical Center (UPMC cohort, table S7, N = 96 cases versus N = 401 non-P AH individuals, adjusted OR 2.53 [95% CI:1.25–5.13]; P = 0.01). We also replicated the association of rs73184087 in the All of Us (dataset v6) cohort, using International Classification of Diseases 10 (ICD10) codes to identify patients with PAH (European-descent) and other filters (such as pulmonary vasodilator medication use) to increase confidence in diagnostic accuracy (table S8, N = 52 cases versus N = 11,821 controls, adjusted OR 2.44 [95% CI: (1.25, 4.79)]; P = 0.01). Last, we replicated the association of this SNV in a global meta-analysis of five cohorts (N = 2181 cases versus N = 10,060 controls; total N = 12,241) including the PAH Biobank, UPMC cohort, and three additional European cohorts from a prior study (OR = 1.30 [95% CI: 1.08–1.56], P = 0.005). On the basis of the association with disease risk, this SNV was further characterized by functional validation.
Fig. 5. G allele of KMT2E SNV rs73184087 binds HIF-2α to control the KMT2E-AS1/KMT2E pair.
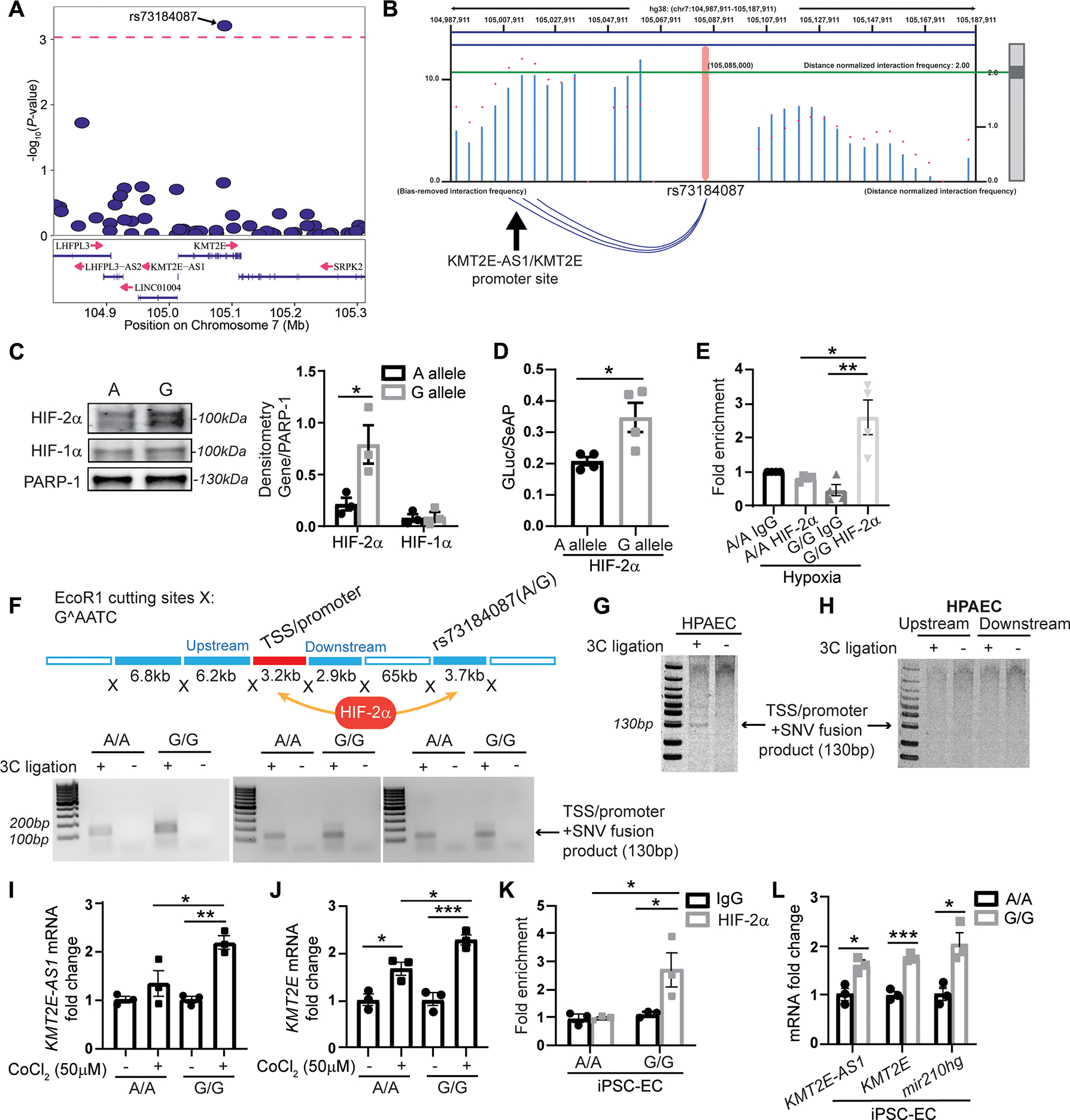
(A) among 883 genotyped and imputed SNVs in the PAH discovery cohort (table S5) within and flanking (+/−200 kb) the lncRNA KMT2e locus, SNVs (table S6) are displayed with predicted HIF-2α binding to either the minor or major SNV allele. independent effective SNV test count was calculated at 53.84 (46). Of those, SNV rs73184087 ranks the highest and meets the P value threshold of 0.00093 (as indicated by the dashed line on the plot). (B) high throughput chromatin conformation capture (HI-C) in lung tissue (29) displays long range interactions between SNV rs73184087 and the transcription start site/promoter region of KMT2E-AS1/KMT2e (as indicated by the blue arcs below the graph). A distance-normalized frequency (magenta dots) greater than the threshold of 2.0 by default (green line) defines a significant interaction with a SNV. (C) SNV binding of HIF-2α and HIF-1α is compared across the SNV a versus G allele (n = 3; *P < 0.05, unpaired Student’s t test; data represent mean ± SEM). (D) Luciferase activity in protein lysates of human embryonic kidney 293T cells transfected with a constitutively active HIF-2α plasmid and a luciferase reporter plasmid carrying the lncRNA-KMT2E promoter and SNV rs73184087 (A versus G allele) (n = 4; *P < 0.05, unpaired Student’s t test; data represent mean ± SEM). Luciferase activity is normalized to constitutively secreted alkaline phosphatase (GLuc/SeAP). (E) ChIP qPCR of HIF-2α binding to SNV in hypoxic transformed lymphocytes from patients with WSPH group 1 PAH carrying SNV rs73184087 (G/G) versus (A/A) genotypes (table S2) (n = 4; *P < 0.05, **P < 0.01, Kruskal-Wallis test followed by dunn’s post hoc analysis; data represent mean ± SEM). (F) In a chromatin conformation capture (3c) assay (top diagram) with three pairs of matched transformed lymphocytes (A/A versus G/G) from patients with WSPH group 1 PAH, PCR is used to detect transcription start site (TSS)/promoter + SNV fusion products indicative of an interaction between SNV rs73184087 and the lncRNA KMT2E promoter. (G and H) detection of a lncRNA-KMT2E promoter and SNV fusion product (G) by 3c assay in human PAECs versus detection of negative control ligation products representing SNV interactions upstream or downstream of the promoter (h). (I and J) KMT2E-AS1 (I) and KMT2E (J) transcripts in transformed lymphocytes carrying A/A or G/G genotype with HIF-α induction by cobalt chloride (50 μM) by RT-qPCR (n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (K) ChIP-qPCR of HIF-2α binding with SNV in inducible pluripotent stem cell differentiated endothelial cells (IPSC-ECS) carrying G/G versus A/A genotype (n = 3; *P < 0.05, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (L) KMT2E-AS1, KMT2E, and miR210hg transcripts in IPSC-ECS carrying A/A versus G/G genotype by RT-qPCR (n = 3; *P < 0.05, ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM).
SNV rs73184087 displayed allele specific binding to HIF-2α and long range interaction with the shared lncRNA KMT2E promoter
SNV rs73184087 is located at a KMT2E intronic site 75 kb downstream of the lncRNA-K MT2E shared promoter. On the basis prior capture Hi C mapping in lung tissue (29), we found a long range genomic interaction between this SNV and the shared promoter (Fig. 5B), consistent with the notion that SNV-bound HIF-2α can readily gain access to the promoter for transcriptional activation. In hypoxic PAEC extracts, oligonucleotides carrying the risk allele SNV rs73184087 (G) displayed preferential and increased binding to HIF-2α, but not HIF-1α, as compared with the major allele (A) (Fig. 5C). Confirming the functionality of such binding, placement of SNV rs73184087 downstream of a luciferase reporter gene demonstrated increased reporter gene expression with the risk (G) allele versus ancestral (A) allele in the presence of constitutive HIF-2α expression (Fig. 5D). In transformed lymphocytes from patients with WSPH group 1 PAH carrying SNV rs73184087 (G/G) versus (A/A) genotypes (table S2), ChIP-qPCR via pulldown of HIF-2α demonstrated enrichment of binding to the (G/G) versus (A/A) SNV in hypoxia (Fig. 5E). To confirm the long range interactions of this SNV regardless of its genotype with the shared promoter, using transformed PAH lymphocytes carrying SNV rs73184087 (G/G) or (A/A) genotypes, 3C assays revealed SNV promoter interaction driven by either the (G/G) or (A/A) genotype (Fig. 5F). A 3C assay using PAECs with the A/A genotype confirmed a specific interaction between the SNV and promoter (Fig. 5G) but not upstream or downstream of the TSS/promoter (Fig. 5H). Furthermore, under cobalt (II) chloride treatment where HIF-α is stabilized in normoxia (Fig. 5, I and J), lymphocytes with (G/G) genotype increased KMT2E-AS1 and KMT2E more robustly as compared with those with (A/A) genotype.
To determine whether such HIF-2α interaction is observed in endothelial cells, we generated inducible pluripotent stem cells (iPSCs) from these transformed lymphocyte lines (30) followed by redifferentiation (31) of those lines into endothelial cells, iPSC-ECs (fig. S6). Consistent with our findings in transformed lymphocytes, ChIP-qPCR in these iPSC-ECs revealed that the G/G locus displayed greater HIF-2α binding versus the A/A locus (Fig. 5K). This increased binding correlated with increased transcript expression of KMT2E-AS1, KMT2E, and downstream miR210hg (Fig. 5L). Together, these data define an intronic SNV rs73184087 (G) allele-specific mechanism by which HIF-2α controls the expression of the lncRNA-K MT2E pair, thus offering a mechanistic explanation underlying the enrichment of SNV rs73184087 (G) allele in WSPH group 1 PAH.
Mouse lncRNA 5031425E22Rik (E22) phenocopied KMT2E-AS1 via a 500 bp conserved sequence
We determined whether mouse lncRNA E22 carries similar activity as KMT2E-AS1 in PAECs. Hypoxia up regulated E22 and KMT2E in mouse PAECs (fig. S1, J and K), mirroring the regulation of KMT2E-AS1 in human PAECs (Fig. 1, I and J). Lentiviral forced expression of E22 (fig. S7A) drove consequent decreased oxygen consumption and increased glycolysis (fig. S7, B and C). As with KMT2E-AS1, this mouse lncRNA controlled similar endothelial pathophenotypes, including migration, contraction, and regulation of vasomotor effectors (fig. S7, D to F).
Computational predictions of secondary structures of E22 and KMT2E-AS1 revealed putative conserved similarities across these mouse and human isoforms (fig. S8A), indicating its importance in this lncRNA’s conserved functions. Yet, because of the genomic proximity of this region to the lncRNA KMT2E promoter (fig. S8B), it was possible that the chromosomal region encoding this sequence was also important in controlling canonical promoter function in cis rather than lncRNA function in trans, as reported for other lncRNAs (12). To clarify these roles, we used a deletion mutant analysis to map a sequence responsible for an lncRNA-dependent function, such as VEGFA induction. This approach identified a 550- to 600-bp region in the 5′ end of the lncRNA transcript, conserved in both KMT2E-AS1 and mouse E22 (fig. S8, C to K). This region corresponded to the same functional domain of KMT2E-AS1 that controlled the interaction of H3K4me3 with KMT2E and thus the expression of H3K4me3 (Fig. 2, N and O). Last, by reporter gene assay, deletion of this region did not affect canonical lncRNA-K MT2E promoter activity (fig. S8L), emphasizing the specific importance of this region for lncRNA activity. Therefore, these data demonstrated that mouse E22 and its conserved 550-b p domain can serve as a surrogate to define the in vitro and in vivo pathobiology of human KMT2E-AS1.
Overexpression of pulmonary vascular mouse lncRNA 5031425E22Rik worsened PH
To investigate whether forced expression of mouse E22 in pulmonary vasculature could promote these epigenetic and metabolic alterations in PAECs and thus cause PH, mice received orotracheal administration of a recombinant adeno associated virus (serotype 6) carrying the 5031425E22Rik transgene (AAV6-E22 ) 4 weeks before a 3 week exposure to normoxia or hypoxia (Fig. 6A). AAV was chosen for long-term transgene delivery, and serotype 6 was selective for optimal endothelial delivery based on in vitro infection studies of PAECs, as previously described (32). Compared with AAV6-GFP–treated littermate mice, mice that were administered AAV6-E22 demonstrated a selective up-regulation of E22 in pulmonary vascular CD31-positive endothelial cells, as assessed by in situ stain (fig. S9A and Fig. 6B). AAV6-E22 increased KMT2E expression, H3K4me3, and H3K9me3 (but not H3K27me3), and HIF-dependent and vasoconstrictive gene expression with greatest consistency in normoxia and across most of the same indices in hypoxia (Fig. 6, C to F, and figs. S9, A to C, and S10, A and B). AAV6 E22 increased the presence of proliferation markers (Ki67+) in CD31+ (endothelial) pulmonary arteriolar cells in comparison with AAV6-GFP mice, most robustly in normoxia and with a further trend in hypoxia (Fig. 6, G and H). As a result, in both normoxia and hypoxia, AAV6-E22 increased pulmonary arteriolar muscularization but less robustly vascular thickness (Fig. 6, I and J), whereas forced lncRNA expression increased right ventricular systolic pressure (RVSP) (Fig. 6K) and right ventricular remodeling (RV to body weight ratio) (Fig. 6L) primarily in hypoxia. Delivery did not affect heart rate, systemic mean arterial pressure, or left ventricular (LV) function (fig. S9, D to H). Thus, the mouse lncRNA E22 alone was sufficient to promote epigenetic functions and metabolic reprogramming and to induce histologic and hemodynamic indices of PH. However, the effects of forced E22 expression in isolation were not equally robust across all indices. In that regard, H3K4me3, H3K9me3, and histologic muscularization all displayed higher signals with hypoxic exposure alone compared with normoxic AAV6-E22 delivery (Fig. 6, E, F, and J), potentially indicating the importance of the hypoxic milieu and precise endogenous and tandem transcriptional elevation of KMT2E to drive the full extent of disease.
Fig. 6. Pulmonary vascular delivery of an AAV6-E22 transgene promotes PH in mice.
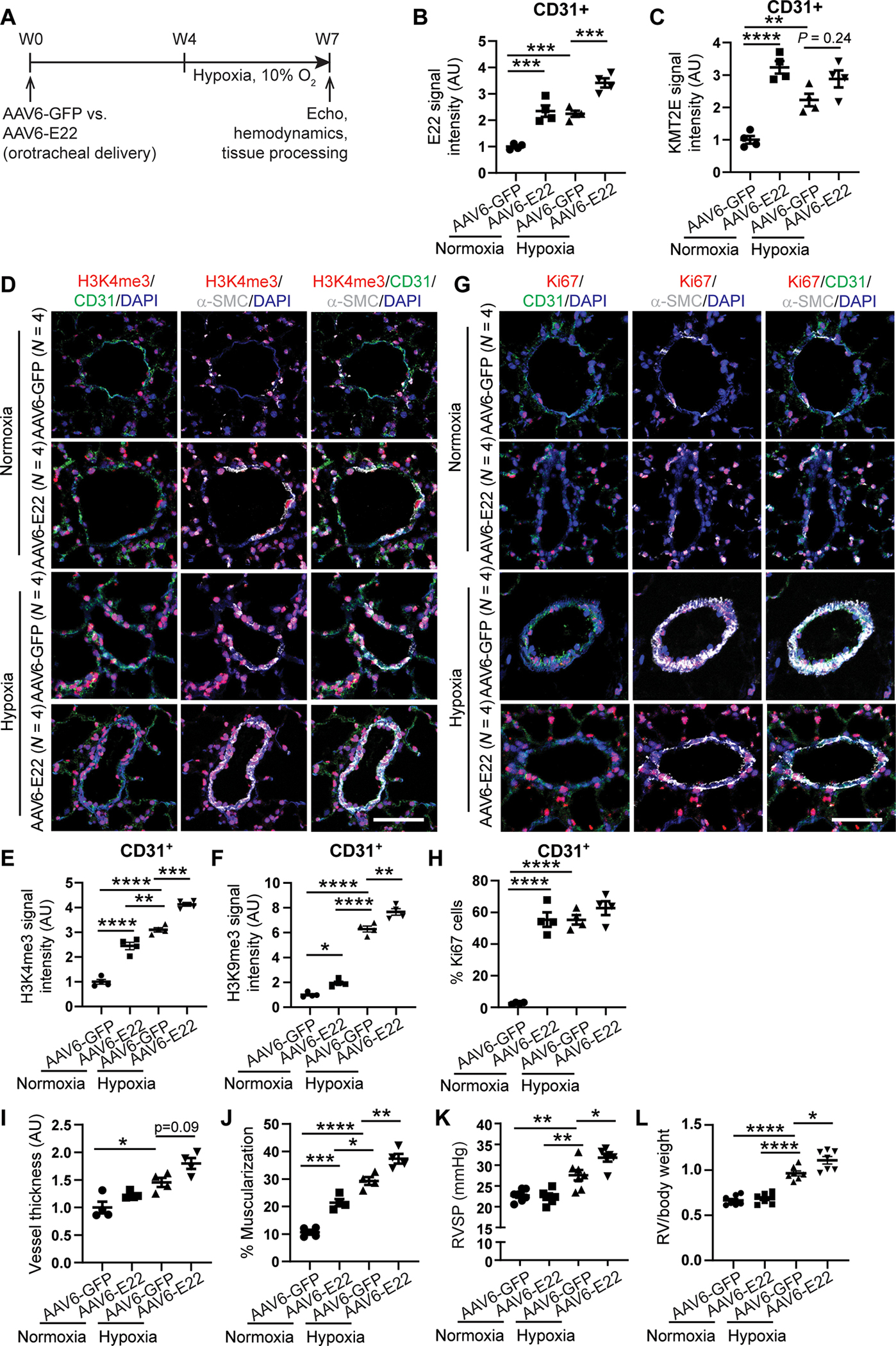
(A) experimental design for an AAV serotype 6 (AAV6) carrying either GFP or E22 transgene delivered orotracheally to wild type c57Bl6 mice 4 weeks before exposure to 3 weeks of chronic hypoxia. (B and C) E22 (B) and KMT2e (c) expression in mouse AAV6-GFP or AAV6-E22 mouse lung CD31+ endothelial cells by FISH and IF staining (n = 4; ***P < 0.001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (D to H) representative IF images for H3K4me3 (red; d) and Ki67 proliferation marker stains (red; G) in AAV6-E22 versus AAV6-GFP mouse lungs. IF quantifications in pulmonary CD31+ vascular endothelium of H3K4me3 (E), H3K9me3 (F), and Ki67 (H) expression (n = 4; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). Scale bars, 50 μm. (I and J) Vessel thickness (I) and muscularization (J) for normoxic and hypoxic AAV6 E22 versus AAV6 GFP mouse lungs as indicated by α-S MA staining (white; d, G) (n = 4; *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (K and L) right ventricular systolic pressure (RVSP, K) and RV/body weight mass index (l) in AAV6-E22 versus AAV6-GFP mice (n = 6 or 7; *P < 0.05, **P < 0.01, ****P < 0.0001, two-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM).
Mice with a deletion in the conserved 5031425E22Rik/KMT2E-AS1 sequence were protected from PH
To investigate the full pathobiologic activity of this lncRNA KMT2E tandem, we determined whether E22 was necessary to drive PAH when both the lncRNA and KMT2E are up-regulated. We therefore used CRISPR-Cas9 technology to generate a mouse genetically deficient specifically in the conserved 550-bp sequence in E22 responsible for the control of PAEC activity (Fig. 7A). These knockout mice displayed decreased active E22 and KMT2E in CD31 positive pulmonary vascular endothelial cells (Fig. 7B and fig. S11A), consistent with knockdown of KMT2E-AS1 in human PAECs (Fig. 2, E and F). Consequently, under conditions of hypoxic PH to model group 3 PH, in both lung tissue and CD31-positive PAECs, H3K4me3 and H3K9me3, but not H3K27me3, were down-regulated in knockout mice as compared with wild-type littermate controls (Fig. 7, C to E, and fig. S10, C and D). This was accompanied by downstream reduction of VEGFA and EDN1 (fig. S11, B and C), consistent with our studies of cultured PAECs (Figs. 3J and 4L and figs. S7F and S8, E, G, I, and K) and control of VEGFA by H3K4me3 in hypoxia (Fig. 3J). As in cultured cells, in situ staining of PAECs displayed lower expression of the proliferation marker Ki67 (Fig. 7, F and G). Knockout mice were protected from histologic and hemodynamic manifestations of hypoxic PH, including demonstrating decreased pulmonary remodeling/muscularization, RVSP, and RV/body weight index (Fig. 7, H to K) but without other differences in heart rate, blood pressure, or echocardiographic measures of LV function (fig. S11, D to H). To address the possibility of off-target CRISPR-Cas9 editing, a separate mouse line using alternative guide primer pairing was generated with a smaller conserved sequence deletion (300 bp). These mice exhibited similar reductions of E22 and RVSP (fig. S11, I to K).
Fig. 7. E22 knockout mice display decreased KMT2E and H3K4me3 along with disease improvement in mouse models of WSPH groups 1 and 3 PH.
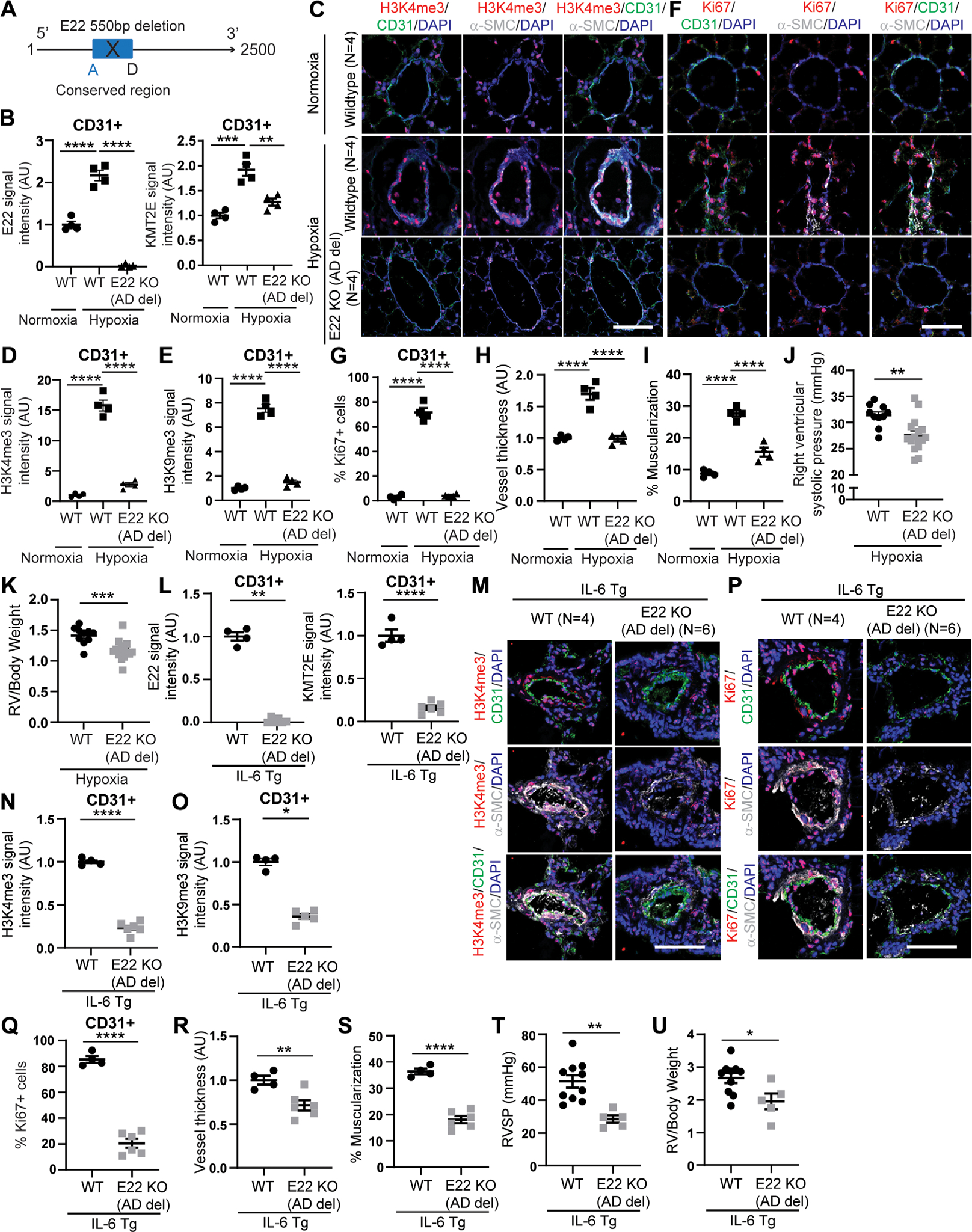
(A) CRISPR-Cas9 edited mice deficient in a conserved 500-bp sequence (denoted a and d) shared between human KMT2E-AS1 and mouse E22. (B) FISH and IF staining for full-length E22 and KMT2e expression in CD31+ lung endothelial cells of hypoxic E22 knockout (KO) mice with ad deletion versus wild-type (WT) controls (n = 4; **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (C to G) representative IF images of H3K4me3 (red; C) and Ki67 (red; F) in hypoxic E22 (KO) mice versus wild-type (WT) controls. IF quantifications in CD31+ PAECs (green) for H3K4me3 (D), H3K9me3 (E), and Ki67 (G) expression (n = 4; ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). Scale bars, 50 μm. (H to I) Vessel thickness (H) and muscularization (I) for E22 ad KO mice versus WT controls as indicated by α-SMA staining (white; C and F) (n = 4; ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (J and K) RVSP (J) and RV remodeling (RV/body weight ratio, K) in hypoxic E22 KO mice versus WT controls (n = 10 to 16; **P < 0.01, ***P < 0.001, unpaired Student’s t test; data represent mean ± SEM). (L) E22 and KMT2e expression in lung cd31+ endothelium of interleukin 6 transgenic (IL-6 TG) mice crossed onto E22 KO (ad deletion) mice by FISH and IF staining (n = 4 to 6; **P < 0.01, ****P < 0.0001, Mann-Whitney test for E22, unpaired Student’s t test for KMT2e; data represent mean ± SEM). (M to Q) representative IF images of h3K4me3 (red; M) and Ki67 (red; P). IF quantifications of h3K4me3 (N), h3K9me3 (O), and Ki67 (Q) in hypoxic IL-6 TG E22 KO mouse lung endothelium versus hypoxic IL-6 TG PAH mice [n = 4 to 6; *P < 0.05, ****P < 0.0001, unpaired Student’s t test for h3K4em3 (N) and Ki67 (Q), Mann-Whitney test for H3K9me3 (o); data represent mean ± SEM]. Scale bars, 50 μm. (R and S) Vessel thickness (R) and muscularization (S) for IL-6 TG E22 KO mice versus WT controls (n = 4 to 6; **P < 0.01, ****P < 0.0001, unpaired Student’s t test; data represent mean ± SEM). (T and U) RVSP (T) and RV remodeling (U) in IL-6 TG e22 KO mice versus controls (n = 5 to 10; *P < 0.05, **P < 0.01, unpaired Student’s t test; data represent mean ± SEM).
To model angioproliferative group 1 PAH, the same mice carrying the 550-b p deletion were crossed with the pulmonary interleukin-6 transgenic (IL-6 TG) mouse (33) and exposed to chronic hypoxia. We observed decreased E22 and KMT2E (Fig. 7L and fig. S12A), H3K4me3 and H3K9me3 (but not H3K27me3) (Fig. 7, M to O, and fig. S10, E and F), VEGFA and EDN1 (fig. S12, B and C), and Ki67 (Fig. 7, P and Q) in CD31 positive pulmonary vascular endothelium. This resulted in a reduction of vascular remodeling, RVSP, and RV/body weight ratio (Fig. 7, R to U) but no differences in heart rate, blood pressure, or LV function (fig. S12, D to G). These findings demonstrated that this E22 lncRNA is necessary for promoting experimental groups 1 and 3 PH in vivo via epigenetic control of endothelial proliferation.
The histone lysine methyltransferase inhibitor chaetocin ameliorated PAH in vivo
To determine whether histone lysine methylation, as regulated by the lncRNA-K MT2E pair (Figs. 2, 6, and 7 and fig. S10), promotes PAH, pharmacologic inhibition of histone lysine methyltransferase activity across H3K4me3 and H3K9me3 was pursued with the small-molecule chaetocin (34) in a rat model of PAH (SU5416-hypoxia). A disease reversal protocol was used, where PAH was allowed to develop over 3 weeks of hypoxia before rats were removed from hypoxia and serial intraperitoneal administration of chaetocin was performed over two subsequent weeks of normoxia (Fig. 8A). In these PAH rats, elevated expression of lncRNA rat homolog of E22 (Fig. 8B) was observed and was not altered by chaetocin. Vascular KMT2E was modestly reduced by chaetocin (Fig. 8C and fig. S13A). However, chaetocin decreased downstream H3K4me3 and H3K9me3 (Fig. 8, D to F, and fig. S13, B to D), VEGFA (fig. S13, E and F), EDN1 (fig. S13, G and H), as well as Ki67 (Fig. 8, G and H) in CD31-positive pulmonary vascular endothelial cells. Chaetocin reduced vascular remodeling, RVSP, and RV to body weight ratio (Fig. 8, I to L) without alterations in heart rate, blood pressure, or LV function (fig. S13, I to M). When considering these comprehensive gain and loss-of function analyses via both pharmacologic and genetic means, we conclude that the mouse E22 and human KMT2E-AS1 carry conserved functions as epigenetic mediators of hypoxic metabolic reprogramming, crucial to pulmonary vascular proliferation, remodeling, and overt PH in vivo.
Fig. 8. The histone lysine methyltransferase inhibitor chaetocin alleviates PAH in a disease-reversal dosing protocol for SU5416 hypoxic rats.
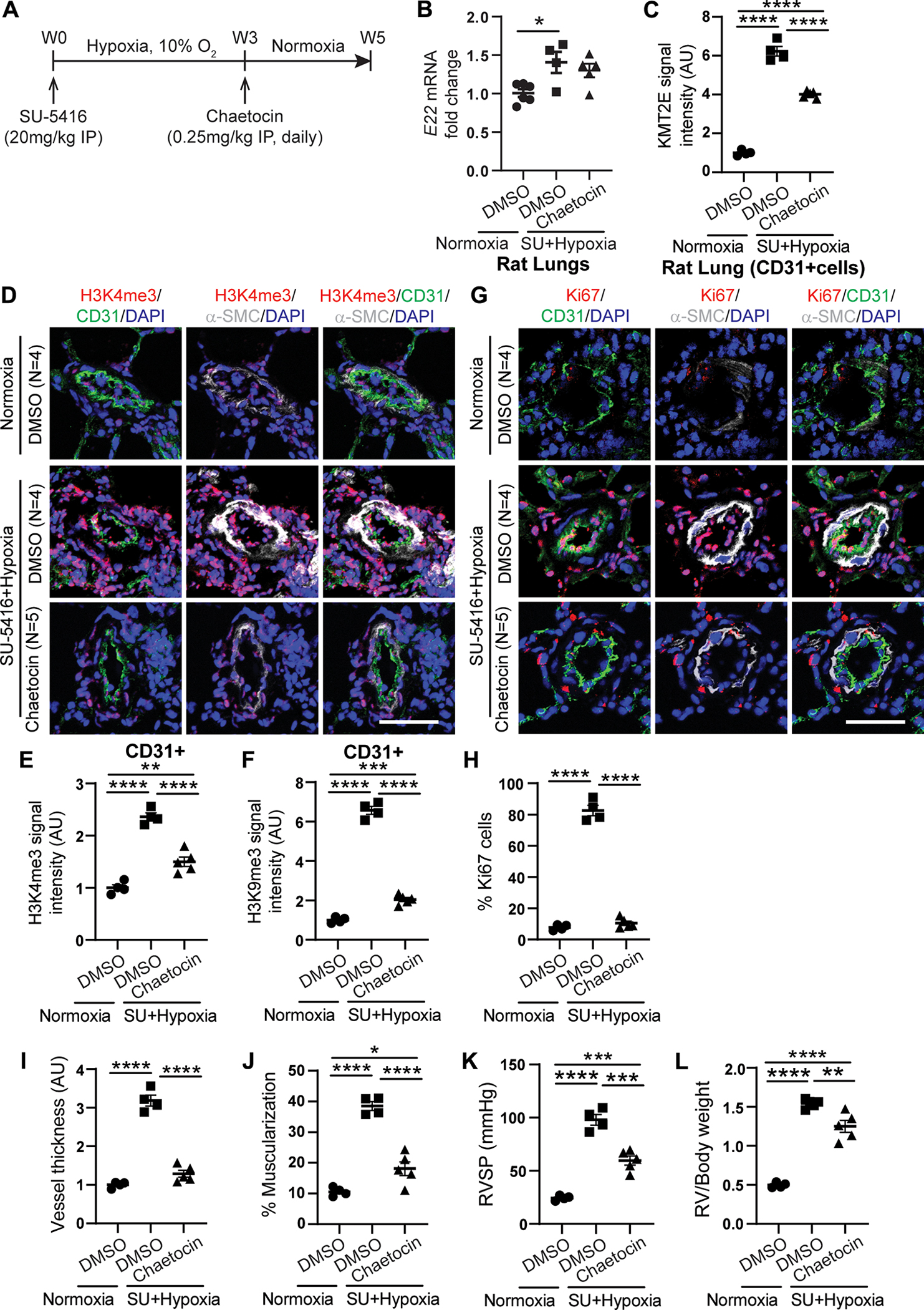
(A) experimental design whereby Sprague-Dawley rats were dosed with SU5416 (SU) and exposed to chronic hypoxia for 3 weeks to generate PAH. Subsequently, PAH rats were dosed with chaetocin versus dimethyl sulfoxide (DMSO) vehicle control by intraperitoneal (IP) injection daily for 2 weeks in normoxia. (B) E22 transcript in lungs of DMSO- and chaetocin-treated SU + hypoxic rats compared with normoxic controls (n = 4 to 6; *P < 0.05, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (C) IF staining quantification of KMT2E expression in CD31+ lung endothelial cells of DMSO and chaetocin-treated SU + hypoxic rats versus normoxic controls (n = 4 or 5; ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (D to H) representative IF images of H3K4me3 (red; D) and Ki67 (red; G). IF quantifications for h3K4me3 (E), H3K9me3 (F), Ki67 (h) in CD31+ PAECs (green) in SU5415 + hypoxic PAH rats versus controls (n = 4 or 5; **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). Scale bars, 50 μm. (I and J) Vascular thickness (I) and muscularization (J) for chaetocin treated SU5415 hypoxic PAH rats versus controls as indicated by α-SMA stain (white, d and e) (n = 4 or 5; *P < 0.05, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM). (K and L) RVSP (K) and RV remodeling (RV/body weight ratio, l) in chaetocin-treated SU5415-hypoxic PAH rats versus controls (n = 4 or 5; **P < 0.01, ***P < 0.001, ****P < 0.0001, one-way ANOVA followed by Bonferroni’s post hoc analysis; data represent mean ± SEM).
DISCUSSION
Here, we defined the KMT2E-AS1/KMT2E pair as a lynchpin of HIF-2α–dependent endothelial pathophenotypes for promoting PH, encompassing robust and convergent genetic, epigenetic, and metabolic processes in this complex disease (fig. S14). These findings established epigenetic, metabolic, and RNA-based paradigms that control HIF-2α–dependent physiology and endothelial reprogramming. Furthermore, by describing the mechanistic underpinnings of large scale epidemiologic and genetic associations, this work offers insight into the genomic functionality of SNVs contributing to clinical manifestations of PH. As a result, these findings introduce the potential of this lncRNA KMT2E pair as diagnostic markers as well as the lncRNA-K MT2E and downstream H3K4me3 as therapeutic targets.
Our work describes an lncRNA-dependent mechanism by which HIF-2α controls endothelial pathobiology and PH, with implications for other ischemic and hypoxic diseases (1). Although HIF-2α is canonically considered a transcription factor that binds promoter elements directly, our findings add to the emerging roles of genomic architecture, epigenetics (10), and context-specific features (35) that modulate HIF’s functions. This is particularly relevant for HIF-specific control of endothelial metabolism in ischemia and hypoxia (36). Our work more specifically identifies poorly defined regulatory paradigms that govern HIF-2α activity, including allele specific intronic SNV binding, long-range chromatin interactions, and lncRNA-dependent modulation of epigenetic histone methylation. In the case of SNV rs73184087, HIF-2α complexing to this locus occurs independently of a canonical HIF response element. This suggests that other factors mediate the interaction of this SNV with HIF-2α and potentially aid additional long range genomic interactions. Whether HIF-2α uses such strategies to exert genome-wide effects (19) and whether other SNVs may control such binding and chromatin contact (27) in additional conditions is not known. Overall, our findings should guide further understanding of context-specific alterations of local hypoxic or HIF-dependent cellular states, such as those described for senescent cells in PH (31) and beyond.
The connected biology between this lncRNA and KMT2E offers guidance in functional understanding of human lncRNAs. Defining the pathobiologic actions of lncRNAs is challenging because of the lack of full sequence conservation among most human lncRNAs. In this study, partial conservation of lncRNA sequence and secondary structure (fig. S8A) coupled with tandem gene chromosomal location guided our investigation. It is possible that other head-to-head chromosomal arrangements among multiple lncRNA-protein coding gene pairs also predict lncRNA activity based on the protein coding gene’s binding or functions. The connected biology of this gene tandem may underlie their cell type specificity and promoter-specific epigenetic activity (fig. S3C). For example, on the basis of our findings that endothelial cells, but not smooth muscle cells, displayed robust KMT2E-AS1 up-regulation (Fig. 1, I and J), the context dependence of KMT2E function may be critically dependent on this lncRNA partner. KMT2E also controls diverse biological processes (37), across hematopoiesis, cancer stem cell renewal, spermatogenesis, autism spectrum disorder (38), and atherogenesis (39). Thus, similar to its binding partner, KMT2E-AS1 may also carry broad biologic actions in cancer as well as neurologic and cardiovascular conditions. Future studies should also explore whether this lncRNA integrates into a broader network of hypoxia-relevant noncoding RNAs—a process that could be aided by computational algorithms applied to pulmonary vascular biology (40).
The identification of a noncoding SNV rs73184087 as a pathogenic mediator of HIF-2α’s epigenetic activity establishes a fundamental paradigm of a positive feedback loop that augments and sustains hypoxic reprogramming in PH. Although carrying a modest predicted effect on differential HIF-2α binding, SNV rs73184087 was the sole significant variant associated with PAH risk (P = 0.00074), perhaps reflecting the importance of genomic location and context in addition to binding. Other predicted HIF binding sites exist in the enhancer regions of the lncRNA-K MT2E gene locus (19), and these may be active, independent of SNV rs73184087 genotypes. Thus, such genetic control does not define a simple “on-off” switch for hypoxic reprogramming. Rather, our findings demonstrate that the presence of the G allele at rs73184087 augments an existing ability of HIF-2α to promote transcription of the lncRNA-K MT2E tandem, with more robust induction of downstream phenotypes. Such tunable activity could explain cell type differences, with the actions of the G allele at rs73184087 observed in normoxia in iPSC-ECs but primarily in hypoxia in transformed lymphocytes. These differences may stem from the fact that endothelium can carry augmented expression of HIF-2α even in normoxia and in PAH disease states (1). Thus, a more obvious display of the effects of the G/G SNP genotype could be seen in endothelium without an exogenous hypoxic trigger.
This positive feedback loop that augments HIF-2α expression offers a mechanistic explanation for the association between rs73184087 and the risk of PAH. The integral relationship of SNV rs73184087 with HIF-2α biology may also portend this SNV’s role in other hypoxic or ischemic conditions. In contrast, given the complex regulation of H3K4me3, this SNV may carry a variety of contrasting activities in such HIF-dependent conditions. Given the importance of histone methylation in a variety of chronic human diseases (41), well-phenotyped control cohorts without pre-existing chronic disease will be essential for appropriate interpretation of any further targeted SNV rs73184087 association studies. It is notable that the SNV rs73184087 risk allele (G) is known to carry a much lower prevalence in African, Asian, and Latin cohorts (42). Future work should prioritize determining whether variations of this allele drive ethnic differences in risk or outcome of PAH.
Last, the epigenetic action of KMT2E-AS1/KMT2E in hypoxic endothelial metabolism clarifies the pathogenic role of histone methylation in PH and beyond. It also establishes a platform to advance epigenetic and RNA-based diagnostics and therapeutics. Unlike other SET domain–containing methyltransferase family (MLL or KMT2) members, KMT2E promotes H3K4me3 indirectly (43) in a cell-specific manner. Whereas the KMT2E-KMT2E-AS1 axis primarily controls hypoxic-driven H3K4me3, this axis also modulates H3K9me3, consistent with a therapeutic role of G9a histone methyltransferase inhibitors in PAH (7). Our genetic and pharmacologic data in vivo present an attractive opportunity for pursuing additive or synergistic therapy, with oligonucleotide inhibition of KMT2E-AS1 coupled with pharmacologic inhibition of both H3K4me3 and H3K9me3. More precise targeting of specific mediators of HIF-2α activity rather than HIF-2α itself may carry higher efficacy for improvement of PAH and may better address the risks of broad endothelial HIF-2α inhibition in the lung (44). Beyond oligonucleotide technology targeting KMT2E-AS1 directly, improvement of experimental PAH with chaetocin (Fig. 8) offers a foundation for optimizing small-molecule inhibitors that can target across both sets of H3K4me3 and H3K9me3 marks. Because KMT2E and downstream histone methylation carry pleiotropic activity beyond the vasculature and lung, simultaneous, lung specific, and controlled release of drugs would be ideal, as shown with recent inhaled microparticle drug delivery in PH (45).
Our study has several limitations. Although multiple independent PAH versus control cohorts were used to establish SNV associations with PAH risk, the ability to match clinical characteristics completely across all PAH groups was not possible. Although the PAH Biobank discovery cohort and the UPMC validation cohort were adjudicated by expert physicians to ensure clinical accuracy, phenotypic information in the All of Us dataset relied on ICD codes and could be prone to error. This study also did not fully define the epigenetic pleiotropy of KMT2E-AS1, setting the stage for future work to analyze the link between this lncRNA and other KMT2 factors besides KMT2E. Moreover, the full context of how this lncRNA behaves in hypoxia was not defined. KMT2E-AS1 may require both the endogenous hypoxic milieu and precise transcriptional elevation of KMT2E, as suggested by our data showing that the pathogenic effects of forced lncRNA expression in isolation (Fig. 6) were not as consistently robust as PH improvement after lncRNA knockout (Fig. 7). In summary, by coupling genetic epidemiology with mechanistic exploration, we have identified the KMT2E-AS1/KMT2E pair as a lynchpin of hypoxic, genetic, and metabolic endothelial reprogramming for promoting PH. These findings carry broad implications for understanding the fundamental molecular hierarchy driving HIF and hypoxic signaling and endothelial pathobiology. This work offers a roadmap toward more effective diagnostic and therapeutic opportunities focused on RNA-based and epigenetic platforms.
MATERIALS AND METHODS
Study design
This study was designed to investigate the role of lncRNA in pathogenetic development of PH and explore the potential of RNA-based and epigenetic platforms for early diagnosis and therapeutic opportunities. This objective was addressed by (i) identifying and determining the function of KMT2E-AS1/KMT2E lncRNA-protein complex in driving endothelial dysfunction via H3K4 trimethylation, (ii) identifying and determining functional SNV that mediates pathogenic epigenetic activity in orchestrating hypoxic reprogramming in PH, and (iii) determining the protective effects with knockdown of KMT2E-AS1/KMT2E and inhibition of H3K4me3 in hypoxic PH rodent models, revealing new prospective epigenetic and RNA-based diagnostics and therapeutics in PH.
Cell culture experiments were performed at least three times and at least in triplicate for each replicate. The number of animals in each group was calculated to measure at least a 20% difference between the means of experimental and control groups with a power of 80% and SD of 10%. The number of unique patient samples for this study was determined primarily by clinical availability. Animals sharing same sex, same genotype, and similar body weight were generated and randomly assigned to different experimental groups. No animals were excluded from analyses. Investigators performing hemodynamic data collection and histologic analysis were blinded to the groups.
All animal experiments were approved by the University of Pittsburgh School of Medicine Division of Laboratory Animal Resources (DLAR). All experimental procedures involving the use of human tissue and plasMA and the study of invasive and noninvasive hemodynamics were approved by institutional review boards at the University of Pittsburgh, Brigham and Women’s Hospital, and Boston Children’s Hospital (studies 19070274, 990835, 19050364, 19020233, 19050026). Ethical approval for this study and informed consent conformed to the standards of the Declaration of Helsinki.
Human patients
Informed consent was obtained for right heart catheterization and tissue sampling. For tissue samples derived from WSPH group 3 PH, diagnosis was made by an expert physician based on the criteria of having an elevated mean pulmonary arterial pressure (mPAP) > 20 mmHg by right heart catheterization with PH deemed to be driven by hypoxic lung disease. For tissue samples derived from WSPH group 1 PAH, diagnosis was made by an expert physician based on the criteria of having an elevated mPAP > 20 mmHg, pulmonary capillary wedge pressure ≤ 15 mm Hg, and pulmonary vascular resistance ≥ 3 Wood units by right heart catheterization. For PAH cases, the expert physician adjudicated the diagnosis by ruling out left heart disease, hypoxic lung disease, or chronic thromboembolic disease. For OCT fresh frozen lung samples, human specimens were collected from unused or discarded surgical samples from participants diagnosed with PH (40). Nondiseased human lung specimens were described previously (40).
Statistical analysis
Numerical quantifications for in vitro experiments using cultured cells or in situ quantifications of transcript expression and physiologic experiments using rodents or human reagents represent mean ± SEM. Immunoblot images are representative of experiments that have been repeated at least three times and quantified using ImageJ. Micrographs are representative of experiments in each relevant cohort. Normality of data distribution was determined by Shapiro Wilk test. Means of two sample groups were compared by an unpaired two tailed Student’s t test for normally distributed data, whereas Mann Whitney U nonparametric testing was used for non-normally distributed data. For comparisons among different groups, one- or two-w ay analysis of variance (ANOVA) tests followed by Bonferroni’s post hoc analysis or Kruskal Wallis tests (where appropriate for non-normally distributed data) were performed. A P value less than 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We thank a. Handen (University of Pittsburgh) for input on sequencing analyses; Sanofi for gapmer oligonucleotides; B. McMahon (University of Pittsburgh Small Animal Ultrasonography Core) for rodent echocardiogram measurements; S. Gingras (University of Pittsburgh) for generation of the lncRNA knockout mouse; and J. Park (University of Pennsylvania) for scientific advice. We thank the NIHR Bioresource–Rare Diseases Consortium, UK PAH Cohort Study Consortium, and the US PAH Biobank Consortium as reported in (28) for data access. The All of Us Research Program is supported by the National Institutes of Health, Office of the Director: Regional Medical Centers: 1 OT2 OD026549; 1 OT2 OD026554; 1 OT2 OD026557; 1 OT2 OD026556; 1 OT2 OD026550; 1 OT2 OD 026552; 1 OT2 OD026553; 1 OT2 OD026548; 1 OT2 OD026551; 1 OT2 OD026555; IAA #: AOD 16037; Federally Qualified health centers: HHSN 263201600085U; data and research center: 5 U2C Od023196; Biobank: 1 U24 OD023121; The Participant center: U24 OD023176; Participant Technology Systems Center: 1 U24 OD023163; Communications and Engagement: 3 OT2 OD023205; 3 OT2 OD023206; and Community Partners: 1 OT2 OD025277; 3 OT2 OD025315; 1 OT2 OD025337; 1 oT2 OD025276. In addition, the All of Us Research Program would not be possible without the partnership of its participants.
Funding:
This work was supported by NIH grants R01 HL124021 to S.Y.C., HL 122596 to S.Y.C., R00 HL133473 to H.W., HL105333 to W.C.N. and M.W.P.; HL137927, HL147148, and HL089856 to M.H.C.; HL141601 to A.B.; and HL143185 to V.N.P.; HL129964 to N.J.K.; AHA grant 18EIA33900027 to S.Y.C.; Plan Cancer 2018 grant 18CN045 to C.L. and B.M.; the French National Research Agency ANR-18-CE14-0025, ANR-21-CE44-0036, and ANR-20-CE14-0006 to T.B.; the French National Cancer Institute INCA–PLBIO 21–094 to T.B.; the BHF Intermediate Basic Science Research fellowship FS/15/59/31839 to C.J.R.; Academy of Medical Sciences Springboard fellowship SBF004\1095 to C.J.R.; and the NSF DBI-1942143 to J.W. The UK National Cohort of Idiopathic and Heritable PAH was supported by the NIHR BioResource, the BHF (SP/12/12/29836), and the UK Medical Research Council (MR/K020919/1). The Advanced High Resolution Rodent Ultrasound Imaging System provided by the University of Pittsburgh Small Animal Ultrasonography Core was supported by NIH S10OD023684.
Footnotes
Competing interests: S.Y.C. has served as a consultant for Merck and United Therapeutics. S.Y.C. is a director, officer, and shareholder in Synhale Therapeutics. M.R.W. has served as a consultant for Novartis, Accelerant, MSD, Janssen, MorphogenIX, Chiesi, and BenevolentAI. S.Y.C. has held research grants from United Therapeutics and Bayer. M.H.C. has received grant support from Bayer. S.Y.C., S.K., and T.B. have filed patent applications regarding the targeting of metabolism in PH (Patent: Compositions and Methods for Treating Pulmonary Vascular Disease, US10925869B2). M.R.W. has filed a patent application regarding ZIP12 antibodies (ZIP12 ANTIBODY, WO2022064216A1). The other authors declare that they have no competing interests.
Data and materials availability: All data associated with this study are present in the paper or the Supplementary Materials. RNA sequencing and ChIP-seq data have been uploaded to the NCBI Gene Expression Omnibus (GEO) repository (GSE232799). De-identified human/patient data are listed in supplementary tables. All original code used to process sequencing data has been uploaded to Zenodo (DOI:10.5281/zenodo.10096872). All newly generated cell lines, plasmids, or mouse lines will be shared upon request. Human and rodent samples and data from Cincinnati Children’s Hospital Medical Center and Brigham and Women’s Hospital shared with the University of Pittsburgh are covered by material transfer agreements.
REFERENCES AND NOTES
- 1.Pullamsetti SS, Mamazhakypov A, Weissmann N, Seeger W, Savai R, Hypoxia-inducible factor signaling in pulmonary hypertension. J. Clin. Invest. 130, 5638–5651 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dai Z, Li M, Wharton J, Zhu MM, Zhao YY, Prolyl-4 hydroxylase 2 (PHD2) deficiency in endothelial cells and hematopoietic cells induces obliterative vascular remodeling and severe pulmonary arterial hypertension in mice and humans through hypoxia-inducible factor 2α. Circulation 133, 2447–2458 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Michelakis ED, Spatio-temporal diversity of apoptosis within the vascular wall in pulmonary arterial hypertension: heterogeneous BMP signaling may have therapeutic implications. Circ Res. 98, 172–175 (2006). [DOI] [PubMed] [Google Scholar]
- 4.culley MK, chan SY, Mitochondrial metabolism in pulmonary hypertension: Beyond mountains there are mountains. J. Clin. Invest. 128, 3704–3715 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hemnes AR, Using Omics to Understand and Treat Pulmonary Vascular Disease. Front. Med. 5, 157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benincasa G, DeMeo DL, Glass K, Silverman EK, Napoli C, Epigenetics and pulmonary diseases in the horizon of precision medicine: a review. Eur. Respir. J. 57, 2003406 (2021). [DOI] [PubMed] [Google Scholar]
- 7.Awada C, Bourgeois A, Lemay SE, Grobs Y, Yokokawa T, Breuils-Bonnet S, Martineau S, Krishna V, Potus F, Jeyaseelan J, Provencher S, Bonnet S, o. Boucherat, G9a/GLP targeting ameliorates pulmonary vascular remodeling in pulmonary arterial hypertension. Am. J. Respir. Cell Mol. Biol. 68, 537–550 (2023). [DOI] [PubMed] [Google Scholar]
- 8.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL, Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. U.S.A. 99, 8695–8700 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Batie M, Frost J, Frost M, Wilson JW, Schofield P, Rocha S, Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Ortmann BM, Burrows N, Lobb IT, ARNAiz E, Wit N, Bailey PSJ, Jordon l. h., Lombardi O, Penalver A, Mccaffrey J, Seear R, Mole DR, Ratcliffe PJ, Maxwell PH, Nathan JA, The HIF complex recruits the histone methyltransferase SET1B to activate specific hypoxia inducible genes. Nat. Genet. 53, 1022–1035 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, Soubrier F, Trembath RC, Loyd JE, Genetics and genomics of pulmonary arterial hypertension. Eur. Respir. J. 53, 1801899 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Statello L, Guo CJ, Chen LL, Huarte M, Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell. Biol. 22, 96–118 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sun M, Kraus WL, From discovery to function: The expanding roles of long non coding RNAs in physiology and disease. EndOCR. Rev. 36, 25–64 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zahid KR, Raza U, Chen J, Raj UJ, Gou D, Pathobiology of pulmonary artery hypertension: role of long non-coding RNAs. Cardiovasc. Res. 116, 1937–1947 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Zehendner CM, Valasarajan C, Werner A, Boeckel JN, Bischoff FC, John D, Weirick T, Glaser SF, Rossbach O, Jae N, demolli S, Khassafi F, Yuan K, De Jesus Perez VA, Michalik KM, Chen W, Seeger W, Guenther A, Wasnick RM, Uchida S, Zeiher AM, Dimmeler S, Pullamsetti SS, Long noncoding RNA TYKRIL plays a role in pulmonary hypertension via the p53 mediated regulation of PdGFRβ. Am. J. Respir. Crit. Care Med. 202, 1445–1457 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Omura J, Habbout K, Shimauchi T, Wu WH, Breuils Bonnet S, Tremblay E, Martineau S, Nadeau V, Gagnon K, Mazoyer F, Perron J, Potus F, Lin J. h., Zafar h., Kiely d. G., Lawrie A, Archer S. l., Paulin R, Provencher S, Boucherat O, Bonnet S, identification of long noncoding RNA h19 as a new biomarker and therapeutic target in right ventricular failure in pulmonary arterial hypertension. Circulation 142, 1464–1484 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Bertero T, cotrill KA, Lu Y, Haeger CM, Dieffenbach P, Annis S, Hale A, Bhat B, Kaimal V, Zhang YY, Graham BB, Kumar R, Saggar R, Saggar R, Wallace WD, Ross DJ, Black SM, Fratz S, Fineman JR, Vargas SO, Haley KJ, Waxman AB, Chau BN, Fredenburgh LE, Chan SY, Matrix remodeling promotes pulmonary hypertension through feedback mechanoactivation of the YAP/TAZ-miR-130/301 circuit. Cell Rep. 13, 1016–1032 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rao RC, dou Y, Hijacked in cancer: The KMT2 (Mll) family of methyltransferases. Nat. Rev. Cancer 15, 334–346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schodel J, oikonomopoulos S, ragoussis J, Pugh CW, ratcliffe PJ, Mole DR, High resolution genome-wide mapping of HIF-binding sites by ChIP-seq. Blood 117, e207–e217 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kondo K, Kim WY, lechpammer M, KaelinJr WG., inhibition of HIF2alpha is sufficient to suppress pVHL-defective tumor growth. PLoS Biol. 1, e83 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tuvshinjargal N, Lee W, Park B, Han K, Pridictor: Protein-RNA interaction predictor. Biosystems 139, 17–22 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Kim J, Lee h., Yi SJ, Kim K, Gene regulation by histone-modifying enzymes under hypoxic conditions: a focus on histone methylation and acetylation. Exp. Mol. Med. 54, 878–889 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sebastian S, Sreenivas P, Sambasivan R, cheedipudi S, Kandalla P, Pavlath GK, dhawan J, MLL5, a trithorax homolog, indirectly regulates H3K4 methylation, represses cyclin a2 expression, and promotes myogenic differentiation. Proc. Natl. Acad. Sci. U.S.A. 106, 4719–4724 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pereira Martins DA, Weinhauser I, Coelho-Silva JL, Franca-Neto PL, Almeida LY, Bianco TM, Silva CL, Franca RF, Traina F, Rego EM, Schuringa JJ, Lucena-Araujo AR, MLL5 improves ATRA driven differentiation and promotes xenotransplant engraftment in acute promyelocytic leukemia model. Cell Death Dis. 12, 371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Subramanian A, Tamayo P, Mootha V, Mukherjee S, Ebert B, Gillette M, Paulovich A, Pomeroy S, Golub T, Lander E, Mesirov J, Gene set enrichment analysis: a knowledge-based approach for interpreting genome wide expression profiles. Proc. Nat. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du Y, Wei N, Ma R, Jiang S. h., Song D, Long noncoding RNA MIR210hG promotes the Warburg effect and tumor growth by enhancing HIF-1α translation in triple-negative breast cancer. Front. Oncol. 10, 580176 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ortiz-Barahona A, Villar D, Pescador N, Amigo J, del Peso L, Genome wide identification of hypoxia inducible factor binding sites and target genes by a probabilistic model integrating transcription profiling data and in silico binding site prediction. Nucleic Acids Res. 38, 2332–2345 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.rhodes CJ, Batai K, Bleda M, Haimel M, Southgate L, Germain M, Pauciulo MW, Hadinnapola C, Aman J, Girerd B, Aarora J. Knight, Hanscombe KB, Karnes J. h., Kaakinen M, Gall H, Ulrich A, Harbaum L, Cebola I, Ferrer J, Lutz K, Swietlik EM, Ahmad F, Amouyel P, Archer SL, Argula R, Austin ED, Badesch D, Bakshi S, Barnett C, Benza R, Bhatt N, Bogaard HJ, Burger CD, chakinala M, church C, Coghlan JG, Condliffe R, Corris PA, Danesino C, Debette S, Elliott CG, Elwing J, Eyries M, Fortin T, Franke A, Frantz RP, Frost A, Garcia JGN, Ghio S, Ghofrani H-A, Gibbs JSR, Harley J, He H, Hill NS, Hirsch R, Houweling AC, Howard LS, Ivy D, Kiely DG, Klinger J, Kovacs G, Lahm T, Laudes M, Machado RD, Mac Kenzie Ross RV, Marsolo K, Martin l. J., Moledina S, Montani D, Nathan SD, Newnham M, Olschewski A, Olschewski H, Oudiz RJ, Ouwehand WH, Peacock AJ, Pepke-Zaba J, rehman Z, Robbins I, roden DM, rosenzweig EB, Saydain G, Scelsi L, Schilz R, Seeger W, Shaffer CM, Simms RW, Simon M, Sitbon O, Suntharalingam J, Tang H, Tchourbanov AY, Thenappan T, Torres F, Toshner MR, Treacy CM, noordegraaf AV, Waisfisz Q, Walsworth AK, Walter RE, Wharton J, White RJ, Wilt J, Wort SJ, Yung D, lawrie A, humbert M, Soubrier F, Trégouët D-A, Prokopenko L, Kittles R, Gräf S, nichols WC, Trembath RC, Desai AA, Morrell NW, Wilkins MR, Genetic determinants of risk in pulmonary arterial hypertension: inteRNAtional genome-wide association studies and meta-analysis. Lancet. Respir. Med. 7, 227–238 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jung I, Schmitt A, Diao Y, Lee AJ, Liu T, Yang D, Tan C, Eom J, Chan M, Chee S, chiang Z, Kim C, Masliah E, Barr CL, Li B, Kuan S, Kim D, Ren B, A compendium of promoter centered long-range chromatin interactions in the human genome. Nat. Genet. 51, 1442–1449 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fujimori K, Tezuka T, Ishiura H, Mitsui J, Doi K, Yoshimura J, Tada H, Matsumoto T, Isoda M, Hashimoto R, Hattori N, Takahashi T, Morishita S, Tsuji S, Akamatsu W, Okano H, Modeling neurological diseases with induced pluripotent cells reprogrammed from immortalized lymphoblastoid cell lines. Mol. Brain 9, 88 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Culley MK, Zhao J, Tai YY, Tang Y, Perk D, Negi V, Yu Q, Woodcock CC, Handen A, Speyer G, Kim S, Lai YC, Satoh T, Watson AM, Aaraj YA, SEMbrat J, Rojas M, Goncharov D, Goncharova EA, Khan OF, anderson DG, dahlman JE, Gurkar AU, Lafyatis R, Fayyaz AU, Redfield MM, Gladwin MT, Rabinovitch M, Gu M, Bertero T, Chan SY, Frataxin deficiency promotes endothelial senescence in pulmonary hypertension. J. Clin. Invest. 131, e136459 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yu Q, Tai YY, Tang Y, Zhao J, Negi V, Culley MK, Pilli J, Sun W, Brugger K, Mayr J, Saggar R, Saggar R, Wallace WD, ross DJ, Waxman AB, Wendell SG, Mullett SJ, SEMbrat J, Rojas M, Khan OF, Dahlman JE, Sugahara M, Kagiyama N, Satoh T, Zhang M, Feng N, Gorcsan iii J, Vargas SO, Haley KJ, Kumar R, Graham BB, Langer R, Anderson DG, Wang B, Shiva S, Bertero T, Chan SY, Bola (Bola Family Member 3) deficiency controls endothelial Metabolism and Glycine homeostasis in Pulmonary hypertension. Circulation 139, 2238–2255 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Steiner MK, Syrkina o. l., Kolliputi N, Mark EJ, Hales CA, Waxman AB, Interleukin-6 overexpression induces pulmonary hypertension. Circ. Res. 104, 236–244 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greiner D, Bonaldi T, Eskeland R, Roemer E, Lmhof A, Identification of a specific inhibitor of the histone methyltransferase SU(VAR)3–9. Nat. Chem. Biol. 1, 143–145 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Platt J. l., Salama R, Smythies J, Choudhry H, Davies JO, Hughes JR, Ratcliffe PJ, Mole DR, Capture-C reveals preformed chromatin interactions between HIF-binding sites and distant promoters. EMBO Rep. 17, 1410–1421 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Falkenberg KD, Rohlenova K, Luo Y, Carmeliet P, The metabolic engine of endothelial cells. Nat. Metab. 1, 937–946 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Zhang X, Novera W, Zhang Y, Deng LW, MLL5 (KMT2E): Structure, function, and clinical relevance. Cell. Mol. Life. Sci. 74, 2333–2344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dong S, Walker MF, carriero NJ, Dicola M, Willsey AJ, Ye AY, Waqar Z, Gonzalez LE, Overton JD, Frahm S, Keaney JF 3rd, Teran NA, Dea J, Mandell JD, Hus Bal V, Sullivan CA, dilullo NM, Khalil RO, Gockley J, Yuksel Z, Sertel SM, Ercan-Sencicek AG, Gupta AR, Mane SM, Sheldon M, Brooks AI, Roeder K, Devlin B, State MW, Wei L, Sanders SJ, De novo insertions and deletions of predominantly pateRNAl origin are associated with autism spectrum disorder. Cell Rep. 9, 16–23 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bjorkegren J. l., Hagg S, Talukdar HA, Foroughi asl H, Jain RK, Cedergren C, Shang MM, Rossignoli A, Takolander R, Melander O, hamsten A, Michoel T, Skogsberg J, PlasMA cholesterol-induced lesion networks activated before regression of early, mature, and advanced atherosclerosis. PLoS Genet. 10, e1004201 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Negi V, Yang J, Speyer G, Pulgarin A, Handen A, Zhao J, Tai YY, Tang Y, Culley MK, Yu Q, Forsythe P, Gorelova A, Watson AM, Al Aaraj Y, Satoh T, Sharifi-Sanjani M, Rajaratnam A, SEMbrat J, Provencher S, Yin X, Vargas SO, Rojas M, Bonnet S, Torrino S, Wagner BK, Schreiber SL, dai M, Bertero T, Al Ghouleh I, Kim S, Chan SY, Computational repurposing of therapeutic small molecules from cancer to pulmonary hypertension. Sci. Adv. 7, eabh3794 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sen P, Shah PP, nativio R, Berger SL, Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phan L, Jin Y, Zhang H, Qiang W, Shekhtman E, Shao D, revoe D, Villamarin R, Ivanchenko E, Kimura M, Wang ZY, Hao L, Sharapova N, Bihan M, Sturcke A, Lee M, Popova N, Wu W, Bastiani C, Ward M, holmes JB, lyoshin V, Kaur K, Moyer E, Felolo M, Kattman BL, AlFa: Allele Frequency aggregator. National Center for Biotechnology Information, U.S. national library of Medicine; (2020). [Google Scholar]
- 43.Zhou P, Wang Z, Yuan X, Zhou C, Liu L, Wan X, Zhang F, Ding X, Wang C, Xiong S, Wang Z, Yuan J, li Q, Zhang Y, Mixed lineage leukemia 5 (MLL5) protein regulates cell cycle progression and E2F1 responsive gene expression via association with host cell factor 1 (hcF 1). J. Biol. Chem. 288, 17532–17543 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pasupneti S, Tian W, Tu AB, dahms P, Granucci E, Gandjeva A, Xiang M, Butcher EC, SEMenza GL, Tuder RM, Jiang X, Nicolls MR, Endothelial HIF-2α as a key endogenous mediator preventing emphySEMa. Am. J. Respir. Crit. Care. Med. 202, 983–995 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.A. P. acharya, [Google Scholar]
- Tang Y, Bertero T, Tai YY, Harvey LD, Woodcock CC, Sun W, Pineda R, Mitash N, Konigshoff M, little SR, Chan SY, Simultaneous pharmacologic inhibition of Yes-associated protein 1 and glutaminase 1 via inhaled poly(lactic-co-glycolic) acid encapsulated microparticles improves pulmonary hypertension. J. Am. Heart Assoc. 10, e019091 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nyholt DR, A simple correction for multiple testing for single nucleotide polymorphisms in linkage disequilibrium with each other. Am. J. Hum. Genet. 74, 765–769 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhao Y, Wu D, Jiang D, Zhang X, Wu T, Cui J, Qian M, Zhao J, Oesterreich S, Sun W, Finkel T, Li G, a sequential methodology for the rapid identification and characterization of breast cancer associated functional SnPs. Nat. Commun. 11, 3340 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.dobin A, Davis CA, Schlesinger F, drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STar: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Love MI, huber W, Anders S, Moderated estimation of fold change and dispersion for RNA seq data with DESeq2. Genome. Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuleshov MV, Jones MR, Rouillard AD, FeRNAndez NF, duan Q, Wang Z, Koplev S, Jenkins S. l., Jagodnik KM, Lachmann A, Mcdermott MG, Monteiro CD, Gundersen GW, Ma’ayan A, Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic. Acids. Res. 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MacS). Genome. Biol. 9, r137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ross-innes CS, Stark R, Teschendorff AE, holmes KA, Ali HR, dunning MJ, Brown GD, Gojis O, Ellis IO, Green a. r., Ali S, chin SF, Palmieri C, Caldas C, Carroll JS, Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 481, 389–393 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bailey TL, Williams N, Misleh C, Li WW, MeMe: Discovering and analyzing DNA and protein sequence motifs. Nucleic. Acids. Res. 34, W369–W373 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.cock PJ, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJ, Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Touzet H, Varre JS, Efficient and accurate P-value computation for Position Weight Matrices. Algorithms Mol. Biol. 2, 15 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hafeez N, Kirillova A, Yue Y, Rao RJ, Kelly NJ, El Khoury W, Al Aaraj Y, Tai YY, Handen A, Tang Y, Jiang D, Wu T, Zhang Y, Mcnamara D, Kudryashova TV, Goncharova EA, Goncharov D, Bertero T, Nouraie M, Li G, Sun W, Chan SY, Single nucleotide polymorphism rs9277336 controls the nuclear alpha actinin 4-human leukocyte antigen-DPA1 axis and pulmonary endothelial pathophenotypes in pulmonary arterial hypertension. J. Am. Heart Assoc. 12, e027894 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pelletier S, Gingras S, Green DR, Mouse genome engineering via CRISPR-Cas9 for study of immune function. Immunity 42, 18–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ, Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.