

Abstract
Binge-eating disorder (BED) is the most common eating disorder, yet its genetic architecture remains largely unknown. Studying BED is challenging because it is often comorbid with obesity, a common and highly polygenic trait, and it is underdiagnosed in biobank datasets. To address this limitation, we apply a supervised machine learning approach (leveraging 822 cases) to estimate the probability of each individual having BED based on electronic medical records from the Million Veteran Program. We perform a genome-wide association study on individuals of African (n = 77,574) and European (n = 285,138) ancestry while controlling for body mass index to identify three independent loci near the HFE, MCHR2 and LRP11 genes and suggest APOE as a risk gene for BED. We identify shared heritability between BED and several neuropsychiatric traits, and implicate iron metabolism in the pathophysiology of BED. Overall, our findings provide insights into the genetics underlying BED and suggest directions for future translational research.
Introduction
BED is a common, heritable (41–57%) psychiatric disorder1,2 with lifetime prevalence estimated between 0.9 and 3%3,4. BED was a provisional diagnosis until the publication of the DSM 5 in 2014 when sufficient evidence was available to conclude that BED can be reliably discriminated from other eating disorders5, including anorexia nervosa or bulimia nervosa. Because BED was only recently added to the most widely used diagnostic classification systems, a sufficiently powered case-control cohort has not yet been recruited6 and few subjects can be identified with traditional approaches to electronic medical record (EMR)-driven genetic studies in existing genotyped cohorts. Thus, identification of genetic risk variants for BED has lagged behind similar efforts for other neuropsychiatric disorders. As a result, our understanding of the biological processes underlying the pathophysiology of BED is limited and the development of genetically based, targeted treatments for BED remains out of reach.
BED is epidemiologically associated with obesity, metabolic dysfunction, multiple psychiatric disorders and low overall well-being4,7. In studies where both cases and controls are categorized as overweight or obese, heritability of BED is higher than when BMI is not considered1,2,8. BED patients have a higher genetic liability for obesity when compared to healthy controls and those with other eating disorders9. While research distinguishing BED from obesity is limited, individuals with BED consistently consume more calories in laboratory meals than obese individuals without BED and demonstrate higher levels of psychiatric comorbidity, eating disorder psychopathology, distress and quality of life impairment5,10,11. Frequent binge eating is associated with poor metabolic outcomes after adjusting for body mass index (BMI)12,13 and those with BED are resistant to sustained weight-loss after interventions with known efficacy4,14. Successful treatment of BED does not necessarily result in metabolic improvement or significant weight change14,15 and approximately one third of individuals with current or recent BED do not have obesity3. Overall, BED is a serious disorder with psychiatric and metabolic components that is epidemiologically distinct from obesity1–5, 8–15.
Towards elucidating the genetic architecture of BED, we leveraged the Million Veteran Program (MVP)16 to develop and validate a supervised machine-learning algorithm based on clinically diagnosed BED cases to estimate each individual’s likelihood of having BED. By performing a bi-ancestral genome-wide association study (GWAS), we identified three genetic loci that are significantly associated with BED independent of BMI and we implicated a fourth gene through MAGMA gene analysis17. We validated our approach by performing polygenic risk score (PRS) association analysis in three independent cohorts: the Adolescent Brain and Cognitive Development Study (ABCD)18, the Philadelphia Neurodevelopmental Cohort (PNC)19 and the UK Biobank (UKBB)20. By performing genetic correlation analysis, we found that BED has considerable genetic overlap with several other neuropsychiatric phenotypes, including depression, bipolar disorder and attention-deficit/hyperactivity disorder. Finally, using gene-based and partitioned heritability analyses, we implicate iron metabolism in the pathophysiology of BED.
Results
Computational phenotyping approach
There are significant challenges in identifying individuals with BED using EMR: BED was not fully recognized by the American Psychiatric Association until 201421, BED remains underdiagnosed22,23 and the International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9-CM) does not have a BED-specific diagnostic code. Consequently, in large, longstanding EMRs such as the Veterans Health Administration’s MVP where diagnoses were coded in ICD-9-CM until 2015, most individuals with BED are likely either undiagnosed or diagnosed using codes corresponding to other or unspecified eating disorders (ambiguous codes). Using single ICD codes, we investigated the prevalence of eating disorder diagnoses (n = 4,266) within the MVP and found that BED (n = 851) was underrepresented relative to bulimia nervosa (n = 876) even though BED has a higher prevalence22 (Supplementary Table 1). After excluding those who had not been genotyped, there were less than 500 individuals with BED for any given genetic ancestry.
To identify individuals in the MVP with a high likelihood of having BED and allow for the performance of a well-powered GWAS, we developed a machine learning approach reliant upon individuals clinically diagnosed with BED. We first constructed a list of subjects reliably diagnosed with BED (n = 822) and a list of controls who had no eating disorder diagnoses (n = 766,705; Supplementary Table 2, Methods). To calculate a BED-score for each subject in the MVP, we built a LASSO logistic regression model across our BED plus controls cohort (n = 767,527; Supplementary Fig. 1). Our hypothesis-free model generated multiple predictors of BED (Top predictors: Fig. 1a, full list: Supplementary Table 3), many of which have known associations with BED (Supplementary Note). To reverse validate that our BED scores specifically predict BED rather than the other traits upon which they were built, we tested the association between BED score and a set of 1,752 phecodes plus the ICD-10 code for BED in a hold-out group comprising 10% of the cohort and found that BED scores predicted BED better than other disorders (Top codes: Fig. 1b, full list: Supplementary Table 4, Methods). The top 3 non-BED phecodes were related to BMI.
Fig. 1: Machine learning model to predict BED within the MVP.
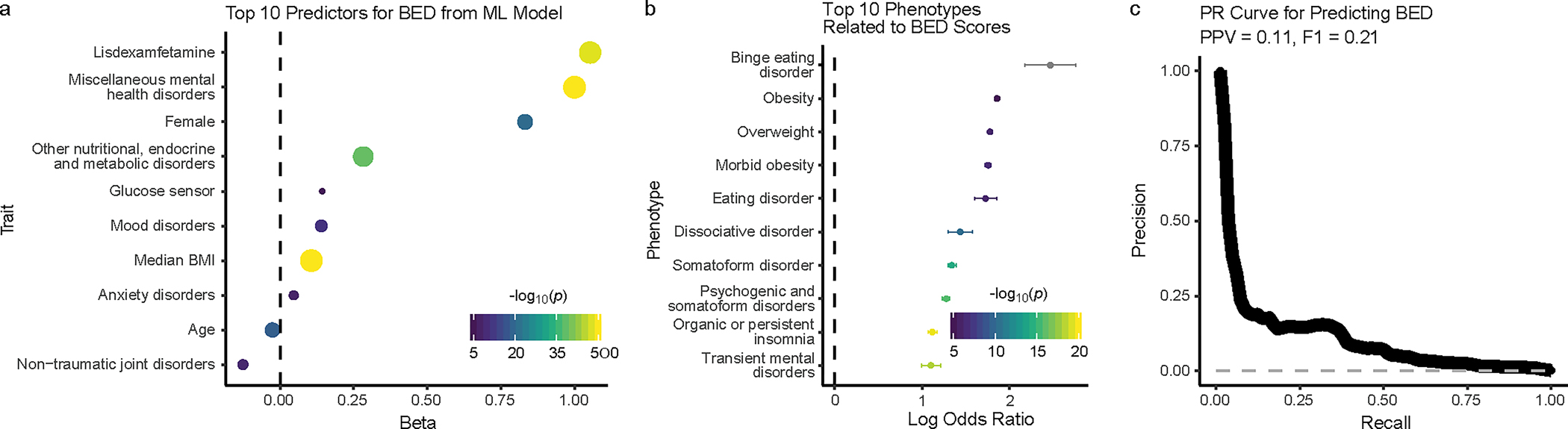
a, Top 10 predictors from the machine learning LASSO logistic regression model for predicting BED (y axis). To rank the predictors, uncorrected P values with a Wald Z-Test were computed from an analogous unpenalized logistic regression model. The strength of the statistical association (from the LASSO regression) is represented on the x axis as beta and in the size and color of the data points, corresponding to the negative log10 of uncorrected two-sided P value (−log10p). The dashed gray line is at 0 on the x axis. P values smaller than 10−50 are capped at that value. b, The ten phenotypes with the strongest association with our model-derived BED scores from an independent logistic regression on a hold-out set are shown on the y axis. The strength of the association is shown on the x axis as mean log odds ratio with the two-sided 95% confidence interval. The strength of the statistical association (comparing the prediction of BED vs. each phecode) is represented by the color of the data points, corresponding to the negative log10 of the uncorrected two-sided P value (−log10p) generated from a one-sided difference of means Z-test. As we tested whether the log odds ratio for BED is higher than the log odds ratio for the other phenotype, no test was performed on BED and it is colored gray. The dashed gray line is at 0 on the x axis. c, Precision recall (PR) curve (thick black line) for predicting BED in a stratified test set containing 10% of the data. The x axis shows the recall rate and the y axis shows the precision. Positive predictive value (PPV) is 0.11 with a phenotype prevalence of 0.001. F1 score is 21%. The dashed gray line represents chance performance.
We then assessed the predictive performance of our model against the hold-out group: the F1 score was 21% (Fig. 1c), the sensitivity-specificity area under the curve was 97.1% (Supplementary Fig. 2), and, while our prevalence of identified BED cases was only 0.1%, the average positive predictive value was 11.0% (Fig. 1c). Furthermore, when we assessed model performance on a test set that aligned more closely with real-world prevalence, the F1 score increased to 54% and the average positive predictive value increased to 50.5% (Supplementary Fig. 3). Assessment of model performance should be interpreted in the context of the probable presence of undiagnosed individuals. Thus, we leveraged the extensive information within the MVP EMR to classify undiagnosed but highly probable cases of BED from a smaller cohort of individuals with clinician-diagnosed BED using a hypothesis-free supervised model that relied on many factors previously associated with BED.
Genetic architecture of BED independent of BMI
A strong genetic correlation has been reported between a BED-inclusive phenotype and BMI9. Therefore, we sought to examine the genetic underpinnings of BED while controlling for BMI by leveraging our inverse-rank normal transformed model-derived (MD) BED scores to perform ancestry-specific GWAS on the African ancestry (AFR; n = 77,574; Fig. 2a) and European ancestry (EUR; n = 285,138; Fig. 2a) populations within the MVP: AFR-MD-BED*BMI and EUR-MD-BED*BMI, respectively (Table 1, Supplementary Fig.4, Supplementary Table 5). From our EUR-MD-BED*BMI GWAS, we discovered two genome-wide significant loci, one within an exon of HFE (p = 1.88×10−9) and one near MCHR2 (p = 5.57×10−10; Fig. 2b, Table 1). For the SNP near MCHR2, since no other nearby SNPs were identified as suggested hits (p < 10−5), we performed regional analysis with the TOPMED24 imputation panel and uncovered additional SNPs in linkage disequilibrium (LD) below the suggestive p < 10−5 threshold (Supplementary Fig. 5, Methods). One of the genome-wide significant SNPs at the HFE locus, rs1800562, corresponds to the C282Y missense mutation with pathogenicity for hemochromatosis. Next, we utilized MAGMA17 to aggregate variants corresponding to protein coding genes and found an association with the EUR-MD-BED*BMI phenotype and APOE (p = 7.03×10−7). We calculated heritability using LD Score regression for the EUR-MD-BED*BMI GWAS to assess the amount of variation explained by an additive SNP model and attained an h2 of 2.14% (s.e. = 0.23%, p = 6.74×10−21). For the AFR-MD-BED*BMI GWAS, we achieved a h2 of 1.65% (s.e. = 0.72%, p = 1.10×10−2). However, limitations for estimating heritability for AFR ancestry are well-documented and the AFR heritability results should be interpreted cautiously25,26. No loci achieved genome-wide significance in the smaller AFR-MD-BED*BMI GWAS (Fig. 2a, Table 1).
Fig. 2: Bi-ancestral GWAS of BED.
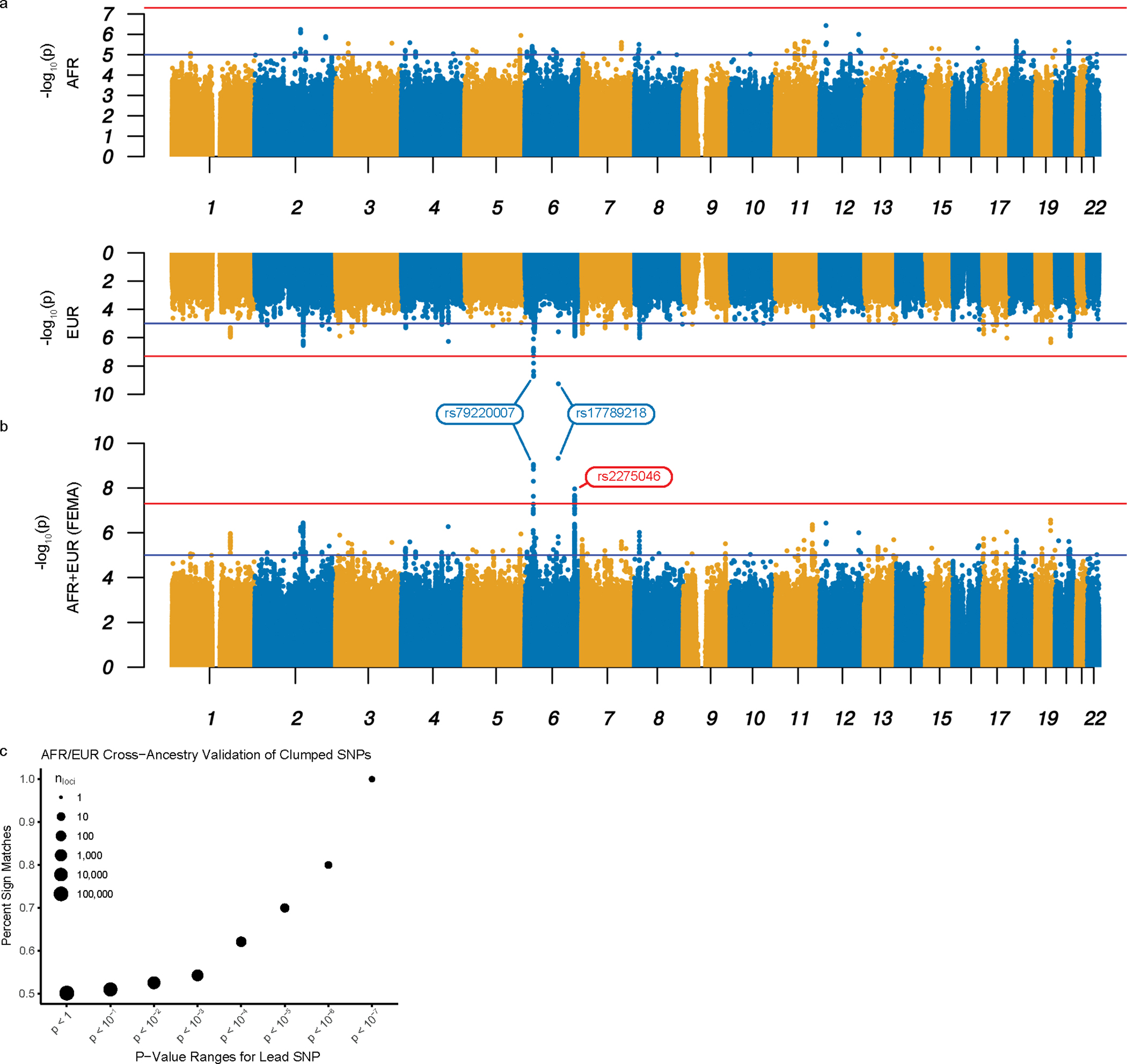
a-b, Miami plot for the AFR-MD-BED*BM (top) and EUR-MD-BED*BMI (bottom) GWAS (a); Manhattan plot for the FEMA-MD-BED*BMI GWAS (b). The x axis denotes the chromosome and position of the corresponding SNP. The strength of the SNP-phenotype association is on the y axis as the negative log10 of uncorrected two-sided P value (−log10p) generated from a two-sided T-test. The red lines represent genome-wide significance (p = 5.0×10−8). The blue lines represent the suggestive genome-wide association threshold (p = 1.0×10−5). Genome-wide significant hits shared by EUR and FEMA GWAS are labeled blue and were confirmed in the EUR replication cohort; the unique genome-wide significant hit in FEMA is labeled red and was not replicated in the EUR replication cohort. c, Sign test between the effect sizes of AFR-MD-BED*BMI and EUR-MD-BED*BMI with progressive restriction of the SNP inclusion threshold. The percentage of clumped SNPs with the same sign is shown on the y axis. The threshold below which lead SNPs were included in the correlation analysis is shown on the x axis as the uncorrected two-sided P value. The size of the point denotes the log10 count of the included loci.
Table 1.
Loci identified in MD-BED*BMI GWAS and MAGMA
| AFR-MD-BED*BMI | EUR-MD-BED*BMI | FEMA-MD-BED*BMI | EUR-MD-BED*BMI Replication | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNP | Chr | Position | Closest Gene | Ref. allele | Effect allele | EAF | Beta (s.e.) | p | EAF | Beta (s.e.) | p | p | Beta (s.e.) | p (one-sided) |
| rs79220007 | 6 | 26098474 | HFE | T | C | 0.01 | 0.023 (0.017) | 0.22 | 0.06 | 0.021 (0.004) | 1.9 × 10−9 | 8.8 × 10−10 | 0.009 (0.004) | 1.6 × 10−2 |
| rs17789218 | 6 | 100600097 | MCHR2 | T | C | 0.05 | 0.007 (0.009) | 0.41 | 0.25 | 0.013 (0.002) | 5.6 × 10−10 | 4.7 × 10−10 | 0.014 (0.002) | 1.9 × 10−8 |
| rs2275046 | 6 | 150157001 | LRP11 | A | G | 0.80 | 0.016 (0.005) | 8.3 × 10−4 | 0.36 | 0.009 (0.002) | 1.3 × 10−6 | 1.1 × 10−8 | 0.001 (0.002) | 0.32 |
| rs7412 | 19 | 45412079 | APOE | C | T | 0.11 | 0.008 (0.006) | 0.17 | 0.08 | 0.016 (0.003) | 4.4x 10−7 | 3.7 × 10−7 | 0.012 (0.004) | 1.3 × 10−3 |
Abbreviations: Chr: chromosome; EAF: effect allele frequency; s.e.: standard error; Ref.: reference. Uncorrected P values in the non-replication cohorts correspond to two-sided T-tests, whereas the uncorrected P values from the replication cohort analysis corresponds to a one-sided T-test.
We then attempted cross-ancestral replication of our genome-wide significant SNPs from the EUR-MD-BED*BMI GWAS, noting that this replication had limited power (Table 1, Supplementary Table 6, Methods). We did not replicate rs17789218 or rs79220007 in the AFR cohort (Table 1). Looking beyond the genome-wide significant loci, we performed the sign test for clumped SNPs between the EUR and AFR GWAS across the whole genome (Methods). We plotted the percentage of SNPs that share the same sign between the EUR and the AFR cohorts’ for lead SNPs using a series of P value thresholds as computed from our EUR-MD-BED*BMI GWAS (Fig. 2c). By limiting our analysis to SNPs with negative log10 P values below 5, we obtained 70% sign matched effects between the AFR- and EUR-MD-BED*BMI GWAS. When we further restricted our selection of SNPs to those with lower P values, we found progressively increasing percentages of matched signs up to 100% while selecting more restrictive values for the negative log10 P value threshold. As we progressively restricted our analysis to those SNPs with the strongest association to BED in the EUR-MD-BED*BMI GWAS, the increased percentage of sign-matched SNPs from the AFR- and the EUR- MD-BED*BMI GWAS suggest a common genomic signal for BED shared by the EUR and AFR ancestries.
To investigate further the cross-ancestral genetics of BED, we conducted fixed-effects meta-analysis27 from the summary statistics of the two ancestry-specific GWAS (FEMA-MD-BED*BMI; Fig. 2b, Table 1). In the FEMA-MD-BED*BMI, both of our lead SNPs from the genome-wide significant loci identified in the EUR-MD-BED*BMI GWAS and one additional locus located within an intron of LRP11, achieved genome-wide significance (Table 1). We also performed a Multi-Ancestry Meta-Analysis28 and found similar results (MAMA-MD-BED*BMI; Supplementary Fig. 6).
After completing our initial analysis, we obtained an additional release of genetic data from the MVP and replicated rs79220007 (β = 0.009, s.e. = 0.004, p = 1.6×10−2), rs17789218 (β = 0.01, s.e. = 0.002, p = 1.9×10−8) and rs7412 (β = 0.01, s.e. = 0.004, p = 1.3×10−3) but not rs2275046 (β = 0.001, s.e. = 0.002, p = 0.32; Supplementary Note). We further validated our EUR-MD-BED*BMI by using the sign test for clumped SNPs on this cohort (Supplementary Fig. 7).
We attempted fine-mapping29 to better identify causal/effect variants; however, our findings were limited by the extended LD blocks within which the index SNPs for the LRP11 and HFE loci reside, the latter falling within the major histocompatibility complex (MHC) locus. Fine-mapping of the locus near MCHR2 was constrained by the limited coverage quality of this region by the MVP genotyping arrays (Supplementary Table 7).
GWAS validation
To further validate our results between the EUR and the AFR cohorts, we performed additional internal and external validations using only the EUR ancestry cohort due to inadequate numbers of AFR ancestry subjects in the validation cohorts. As internal validation, we tested the relationship between our primary GWAS methodology (MD-BED), BMI and an alternative ICD-based BED phenotype (ICD-BED) comprised of individuals reliably diagnosed with BED and/or diagnosed with an ambiguous eating disorder and with a high probability of having BED based on a second machine learning algorithm designed to discriminate BED from other eating disorders (Methods).
We computed the genetic correlation across five BED-related GWAS on EUR individuals in the MVP (Supplementary Table 5): Model-Derived BED (EUR-MD-BED; n = 285,138), Model-Derived BED adjusted for BMI (EUR-MD-BED*BMI; n = 285,138), ICD-based BED (EUR-ICD-BED; n = 549 cases, n = 284,648 controls; Supplementary Fig. 8a), ICD-based BED adjusted for BMI (EUR-ICD-BED*BMI; n = 549 cases, n = 284,648 controls; Supplementary Fig. 8b) and BMI (EUR-BMI; n = 291,593). We observed reasonably high correlations between the EUR-ICD-BED GWAS and the other GWAS, with genetic correlation rg ranging from 0.48 to 0.86 (Fig. 3a). We note that the EUR-MD-BED*BMI GWAS achieved high genetic correlation (rg = 0.85, s.e. = 0.36) with the EUR-ICD-BED*BMI GWAS. In contrast, when we did not adjust our MD-BED phenotype for BMI, we observed that EUR-MD-BED GWAS achieved greater genetic correlation with BMI (rg = 0.93, s.e. = 0.01) than it did with any of the BED GWAS (rg = 0.71, s.e. = 0.22 with EUR-ICD-BED). These results confirm that the two approaches to classification of BED identify common genetic factors, but suggest that without adjustment, our machine learning approach may be inflated by the influence of BMI-associated genetic determinants.
Fig. 3: Validation of the MD-BED phenotype.
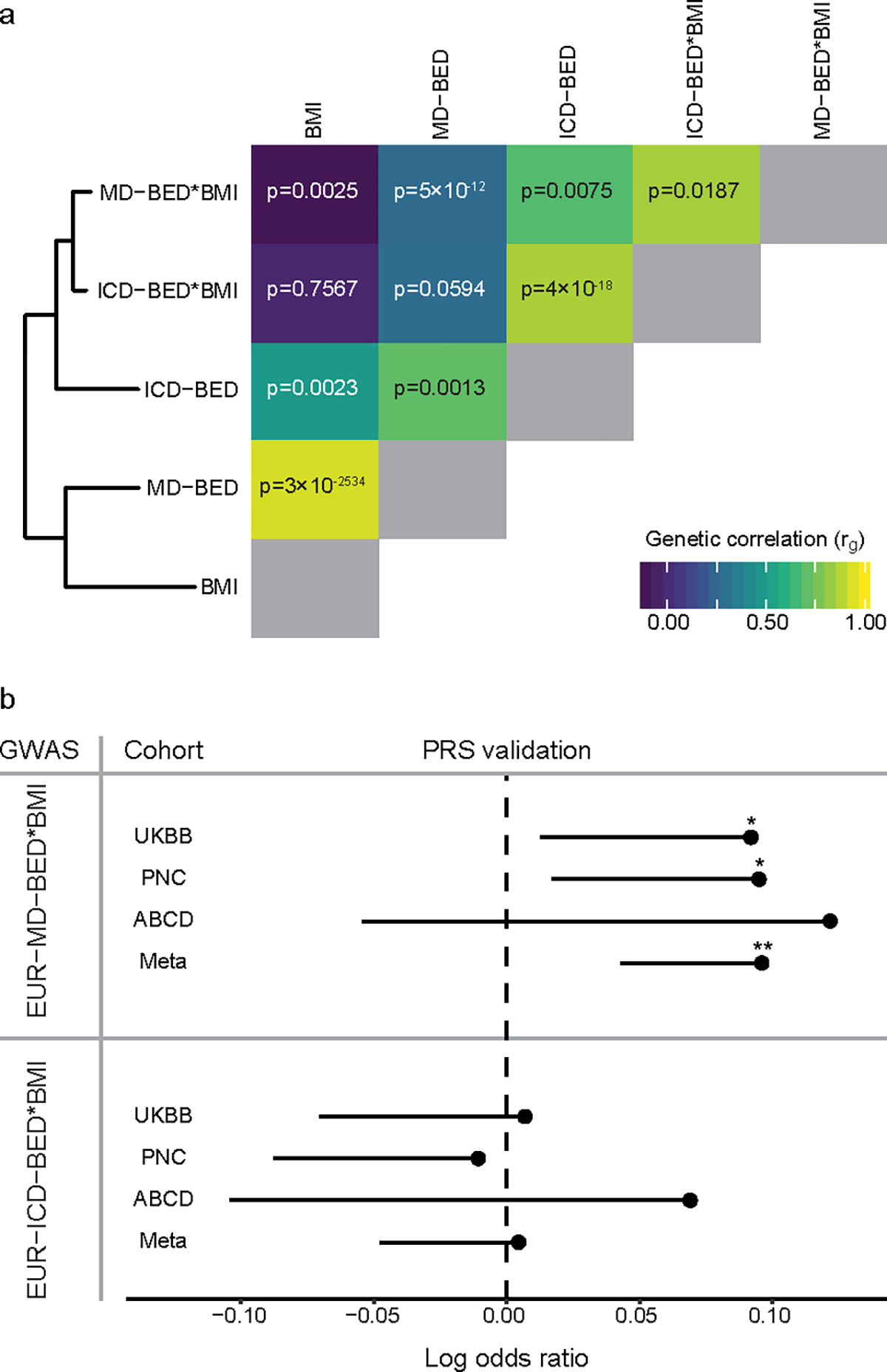
a, On the left is a hierarchical clustering of five EUR BED-related phenotypes. On the right is a heat map of the genetic correlation matrix. The diagonal genetic correlation entries in gray represent a correlation of 1 between each GWAS and itself. Genetic correlation values for each comparison are shown on the heat map. b, PRS validation of EUR-MD-BED*BMI and EUR-ICD-BED*BMI GWAS with UKBB (cases = 461), PNC (cases = 531), ABCD (cases = 94) and a meta-analysis of those cohorts. The MVP (vertical) and external (horizontal) cohorts are shown on the y axis. The mean log odds ratio for the PRS predictor is shown on the x axis. Confidence intervals are one-sided standard errors and uncorrected P values are generated using a one-sided Wald Z-test. *p < 0.05. **p < 0.01. P values for validating the MD-BED*BMI PRS are: UKBB, p = 0.03; PNC, p = 0.02; ABCD, p = 0.13; Meta, p = 0.001. P values for validating the BED-ICD PRS are: UKBB, p = 0.44; PNC, p = 0.59; ABCD, p = 0.26; Meta, p = 0.44.
While the genetic correlations between the EUR-MD-BED*BMI GWAS and the two ICD-BED GWAS were high, the ICD-based approach identified different genome-wide significant loci (Supplementary Table 8, Supplementary Note). However, heritability of the EUR-ICD-BED GWAS was only nominally significant (h2 = 22.3%–29.5%, p = 0.05) and heritability of the EUR-ICD-BED*BMI GWAS was not statistically significant (h2 = 16.9%–22.4%, p = 0.11).
We also validated our EUR-MD-BED*BMI approach using three external cohorts with BED-inclusive phenotypes by leveraging our GWAS summary statistics to compute PRS (see Supplementary Table 9 and Supplementary Note for phenotype definitions and cohort details). Our EUR-MD-BED*BMI PRS significantly predicted BED in the UKBB (p = 0.03; Fig. 3b) and in the PNC (p = 0.02; Fig. 3b), but did not reach significance in the ABCD cohort (p = 0.13, Fig. 3b). However, the effect sizes were similar in all validation sets (log odds ratios: 0.09–0.12) and the inverse-variance weighted meta-analysis across the UKBB, ABCD and PNC was robust (p = 1.39×10−3; Fig. 3b). The EUR-ICD-BED*BMI PRS failed to validate in any of the individual cohorts or through meta-analysis (Fig. 3b) in the setting of its small sample size. We then validated rs17789218, located near MCHR2, using an inverse-variance weighted meta-analysis of the case-control, BMI-adjusted GWAS from ABCD, PNC and UKBB after adjusting for cross-study heterogeneity30 and applying a power weighted P value approach to computing the false discovery rate31 (FDR; ABCD-PNC-UKBB-BED*BMI; q < 0.05; Supplementary Tables 10–12, Methods, see Supplementary Note for discussion of power to replicate other alleles in these cohorts).
Shared genetic architecture between BED and other traits
To investigate the genetic overlap between our model-derived BED scores and other traits, we computed the genetic correlation between the EUR-MD-BED*BMI GWAS and a curated set of 44 psychiatric disorders, behavioral phenotypes and health-related traits and contrasted it with the results of our EUR-BMI (Fig. 4; Supplementary Fig. 9). We found significant positive genetic correlations between EUR-MD-BED*BMI and lobar intracerebral hemorrhage (rg = 0.56, p = 2.02×10−6), depression (rg = 0.52, p = 2.46×10−45), cannabis use (rg = 0.44, p = 9.11×10−9), bipolar disorder (rg = 0.42, p = 7.63×10−21), neuroticism (rg = 0.38, p = 5.43×10−10), attention-deficit/hyperactivity disorder, (rg = 0.34, p = 1.26×10−7), externalizing32,33 (rg = 0.32, p = 9.40×10−12), schizophrenia (rg = 0.24, p = 2.94×10−9) and anorexia nervosa (rg = 0.21, p = 4.00×10−4), as well as a cross-psychiatric disorder GWAS (rg = 0.44, p = 1.58×10−11). The strength of the genetic correlation between these traits was greater with the EUR-MD-BED*BMI GWAS than with the EUR-BMI GWAS (Supplementary Fig. 9). We found significant negative genetic correlations between EUR-MD-BED*BMI and educational attainment (rg = −0.11, p = 0.001), intelligence quotient (rg = −0.12, p = 0.02), cognitive performance (rg = −0.13, p = 8.00×10−4) and subjective well-being (rg = −0.21, p = 0.006). We computed the genetic correlation between our EUR-MD-BED*BMI GWAS and 1,427 traits from the IEU Open GWAS project34 that met our quality control standards (Supplementary Table 13; Methods), and broadly confirmed the genetic association between BED and depression and neuroticism. Among other traits, we also found an association with risk taking (rg = 0.30, p = 1.92×10−11) and lifetime smoking (rg = 0.19, p = 1.05×10−5). To validate the negative genetic association between BED and cognitive functioning, we assessed the relationship between our EUR-MD-BED*BMI PRS and neurocognitive measures obtained in the UKBB35 and found strong negative associations across nearly all domains tested (Supplementary Fig. 10).
Fig. 4: Genetic correlation with other traits.
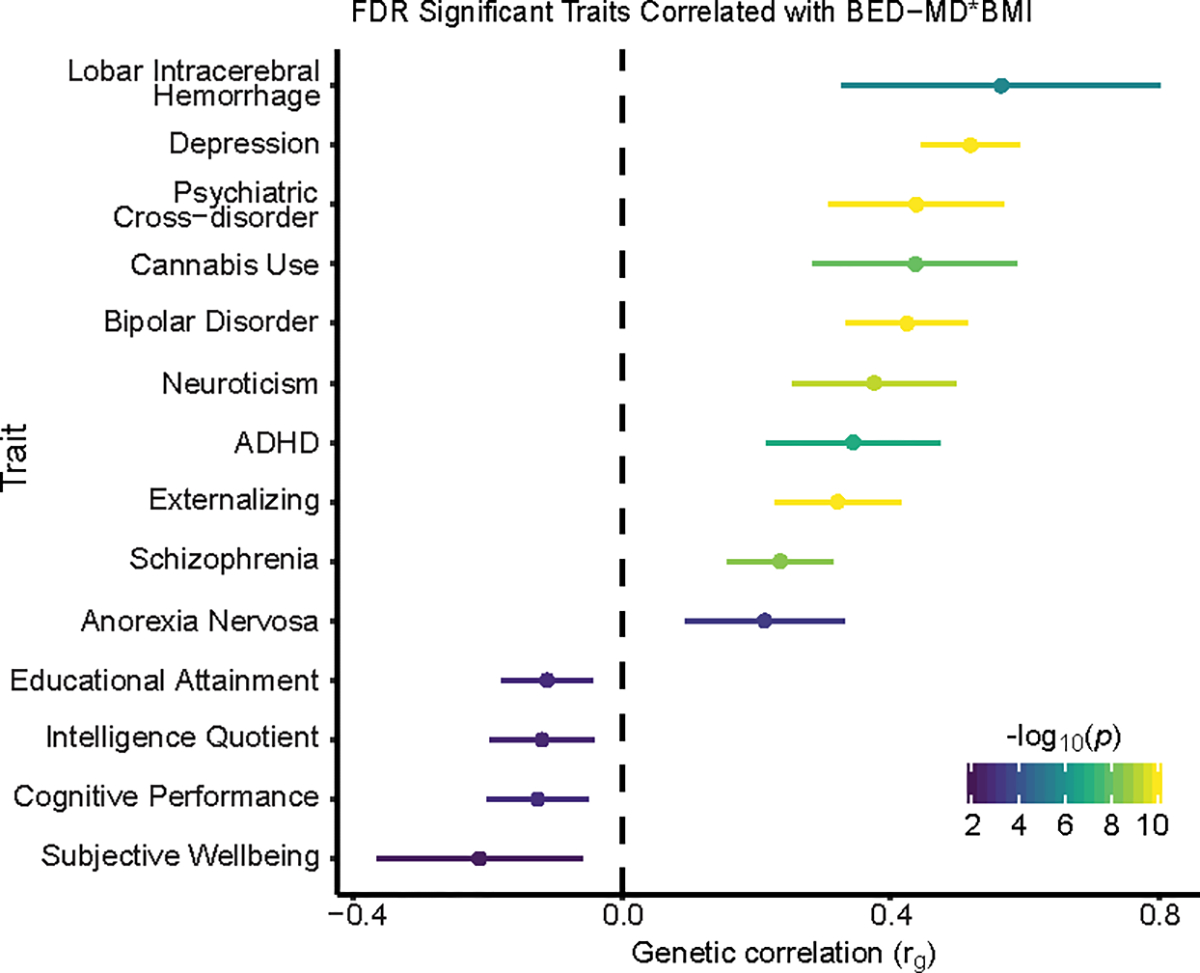
Traits with significant genetic correlation to EUR-MD-BED*BMI at the FDR significant threshold (q < 0.05) from our curated set of GWAS are ranked by rg on the y axis. The strength of the averaged genetic correlation is shown on the x axis as rg with the 95% confidence interval for each trait shown and through the color of the error bar corresponding to the uncorrected P value as generated from a two-sided Z-test when performing LD Score regression. P values smaller than 10−10 are capped at that value.
Pathways and cell types
We used FUMA, excluding the MHC to ensure that results were not driven by the high LD structure and gene density in this region, to identify enrichment in the overlap of gene sets from our EUR-MD-BED*BMI and a large GWAS database (Supplementary Fig. 11). Apart from enrichment for neuropsychiatric, obesity-related, autoimmune and cancer traits, we found enrichment for gene sets involved in heme metabolism and biosynthesis, and uric acid metabolism. Phenome-wide association studies (PheWAS) of the lead SNPs from the FEMA-MD-BED*BMI GWAS were also consistent with iron dysregulation through associations with disorders of iron metabolism and iron deficiency anemia (Supplementary Fig. 12).
To elucidate the role of iron and heme metabolism in BED, we assessed the relationship between the PRS from our EUR-MD-BED*BMI and EUR-BMI GWAS with both an iron deficiency and an iron overload phenotype derived from the UKBB (Fig. 5a). BED was very strongly positively correlated with iron overload (β = 0.66, p = 1.62×10−60) and negatively correlated with iron deficiency (β = −0.03, p = 0.01). Differently, BMI was positively correlated with iron deficiency (β = 0.05, p = 1.03×10−7) and was not significantly correlated with iron overload (β = −0.01, p = 0.73). We then found a positive correlation between BED scores and iron deficiency in the MVP EMR data (β = 0.27, p = 4.03×10−71) that persisted after adjustment for BMI (β = 0.32, s.e. = 0.02), in contrast to the negative correlation between the EUR-MD-BED*BMI PRS and iron deficiency in the UKBB, suggesting that the predictors for our BED scores are not driving the negative relationship between iron deficiency and the EUR-MD-BED*BMI PRS. To test for a putative causal relationship between BED scores and iron overload we performed Generalized Summary-data-based Mendelian Randomisation (GSMR)36, where we leveraged summary statistics from our EUR-MD-BED*BMI and a transferrin saturation GWAS37 based on an independent cohort comprising subjects from deCODE genetics and the INTERVAL study (Fig. 5b). Using GSMR, we confirmed a statistically robust relationship (β = 0.02, p = 5.45×10−6) that persisted after outlier removal (Supplementary Fig. 13; Methods), further supporting that transferrin saturation, a biomarker of whole-body iron stores, affects BED scores. After linking BED probability scores with iron overload, we hypothesized that if iron/heme metabolism is critical for BED we would also see independent downstream enrichment of EUR-MD-BED*BMI risk variants for regions of the genome regulated by heme. Thus, we tested the relationship between enrichment of BED risk variant-homologs and heme-regulated open chromatin regions from a murine erythroid cell β-estradiol stimulation model38. After correcting for multiple comparisons, we found significant enrichment of our BED risk variant-homologues in wild-type mice exposed to β-estradiol, an induced high heme state (FDR adjusted p = 0.03), but not in mutants with reduced heme expression (Fig. 5c), suggesting enrichment of BED risk variants in heme-regulated open chromatin regions.
Fig. 5: Iron overload in BED.
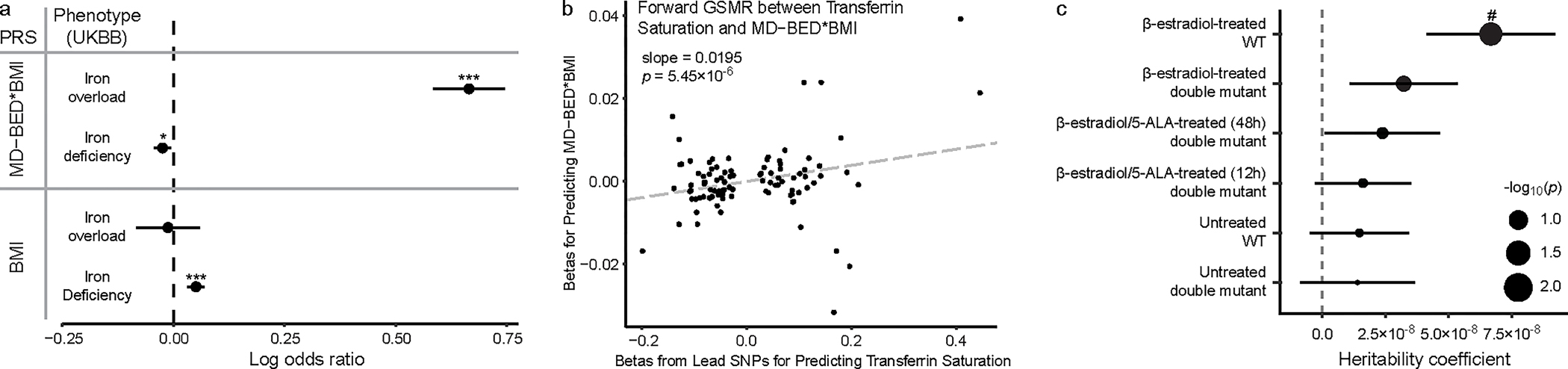
a, PRS associations between EUR-MD-BED*BMI and EUR-BMI GWAS and iron overload (n = 790 cases, n = 385,100 controls) and iron deficiency (n = 11,247 cases, n = 374,643 controls). PRS scores and iron phenotypes are on the y axis. The coefficients, as log odds ratios (mean ± s.e.m.), from the logistic regression for PRS predictors are on the x axis. EUR-MD-BED*BMI PRS predicts iron overload (p = 1.62×10−60) and iron deficiency (p = 0.01). EUR-BMI PRS predicts iron deficiency (p = 1.03×10−7) but not iron overload (p = 0.73). *p < 0.05. ***p < 0.001. b, Scatter plot with generalized linear regression from GSMR between lead SNPs from the transferrin saturation GWAS from deCODE and INTERVAL and EUR-MD-BED*BMI. Transferrin saturation lead SNP betas are on the x axis. EUR-MD-BED*BMI betas are on the y axis. P values from GSMR are from a two-sided Z-test. c, Enrichment of BED risk variant-homologs in open chromatin regions (OCR) in wild type (WT) and heme-deficient mutant murine erythroid cells treated with β-estradiol and/or 5-aminolevulinic acid hydrochloride (5-ALA) (β-estradiol-treated WT: n = 1,010,459 OCR, p = 0.005; β-estradiol-treated double-mutant: n = 1,263,093 OCR, p = 0.07; β-estradiol/5-ALA-treated (48hr) double mutant: n = 1,229,810 OCR, p = 0.15; β-estradiol/5-ALA-treated (12hr) double mutant: n = 1,229,810 OCR, p = 0.20; Untreated WT: n = 1,488,490 OCR, p = 0.23; Untreated double mutant: n = 1,001,591 OCR, p = 0.27). Cell lines are on the y axis. Heritability is on the x axis. Positive coefficients signify enriched heritability. Dot size reflects negative log10 of uncorrected P value (−log10p) from a two-sided LD Score regression Z-test. Error bars indicate standard errors from LD Score regression mean estimates. #p < 0.05 after FDR correction.
Based on our gene set enrichment findings, we also attempted to explore the relationship between uric acid metabolism and BED. We found genetic associations with urate and gout; however, in contrast to iron/heme metabolism dysregulation, observed BMI heavily confounded these associations (Supplementary Table 14, Supplementary Note).
Finally, to identify cell types in which the genetic drivers of BED are likely to have an effect, we performed partitioned heritability analysis for our EUR-MD-BED*BMI GWAS with two chromatin accessibility atlases39,40 and found nominal enrichment across several neural lineages including from limbic system neurons, inhibitory neurons, astrocytes, enteric neurons and enteric glia (Supplementary Fig. 14, Supplementary Note). These results point to a potential pleiotropic effect of the genetic drivers across neural tissues and suggest shared dysfunction across the central and enteric nervous systems.
Discussion
We report the first GWAS of a model-derived BED phenotype from 362,712 individuals in the MVP, including individuals with AFR and EUR ancestries, and the identification of three loci that have a genome-wide significant association with BED independent of BMI; two of these loci are replicated in a later tranche of EUR genotypes in MVP not included in the discovery GWAS. We further implicate APOE in BED through MAGMA and subsequent replication of the association for its lead SNP. To power our study, we developed a machine learning approach to identify the probability of individuals having BED across the MVP based on clinician-diagnosed cases. We leveraged the diversity of the MVP to conduct separate GWAS in AFR and EUR cohorts and perform both cross-cohort and meta-analytic validation of our results. Our machine learning approach was effective in performing a GWAS on a common, yet underdiagnosed disorder in a very large cohort. With the limited number of identified cases, our algorithm-based approach shared high genetic correlation with our more traditional case-control GWAS (rg = 0.85) and was well-powered for SNP heritability estimation (p = 6.74×10−21) and PRS validation across external cohorts (p = 1.39×10−3). In contrast, the case-control GWAS did not achieve significant heritability (p = 0.11) or validate in external cohorts via PRS association (p = 0.44).
We found significant PRS association between our MD-BED phenotype built on clinically diagnosed veterans and gender-balanced civilian cohorts. Of these three cohorts, only the ABCD used DSM-based criteria for BED diagnosis; however, it contains only 94 cases as the cohort is substantially younger than the typical age of onset of BED18. The PNC cohort was also younger than the mean age of onset of BED19 and the closest available phenotype captured a much larger population than would be expected to have BED. Despite these limitations, our PRS significantly predicted BED in two of the three civilian cohorts (PNC and UKBB) and through meta-analysis. Consequently, we provide strong evidence for the external validity of our GWAS approach, even in the presence of demographic confounders.
A recent UKBB study confirmed the expected partial genetic association between BMI and over-eating/binge-eating in a EUR-only cohort but was not powered to detect associations with individual loci, was not based on diagnosed BED and did not examine the BMI-independent genetic contributions to over-eating/binge-eating9. However, their phenotype was similar to the UKBB phenotype that we used as one of the validations of our approach. As expected, we found a high-degree of genetic correlation between BED and BMI using both our machine learning and ICD-based approaches.
While there is substantial epidemiological and genetic overlap between BED and obesity, most obese individuals do not have BED and many people with BED are not obese3. Therefore, we examined the genetics of BED while adjusting for BMI. When comparing our EUR-MD-BED*BMI PRS to a curated set of behavioral and psychiatric traits, we revealed genetic correlations between BED and depression, bipolar disorder and attention-deficit/hyperactivity disorder, all common comorbidities of BED7, along with anorexia nervosa. We also identified a strong association with intracerebral lobar hemorrhage. This risk may be mediated through shared associated factors including hypertension and through APOE41. Using a large GWAS repository34, we confirmed the association with depression and neuroticism and also identified genetic correlation with traits associated with impulsivity. We note that some of the traits with genetic correlation to our BED phenotype, e.g., depression, are components of our predictor model, contributing to the correlation. Depression is genetically correlated with the over-eating/binge-eating phenotype in the UKBB which is not reliant on a predictor model including depression9. Together, these cross-disorder genetic commonalities provide biological validation of the epidemiological evidence that the BED subpopulation within obesity is a nexus for enhanced psychopathology.
Examining the individual genes, pathways and cell-types implicated in BED provides further insight into the biology underlying BED and evidence against the theory that BED is merely an associated feature of the co-occurrence of obesity and general psychopathology42. Several lines of evidence implicate dysfunctional iron metabolism, heme signaling and resultant iron overload as a driver of BED. We identified multiple SNPs with significant association to BED within the HFE gene, including rs1800562, a missense variant pathogenic for hemochromatosis43. We identified iron and heme-related phenotypes using both FUMA and PheWAS. Our FUMA analysis identified iron dysregulation even while excluding the region of the MHC containing HFE. We confirmed that BED is genetically associated with iron overload through a PRS association study in the UKBB, transferrin saturation via GSMR in an independent cohort, and further implicated heme metabolism by demonstrating that BED risk variants are enriched for heme-induced open chromatin regions. Heme metabolism has been implicated in insulin resistance44 and could partially mediate the association between BED and metabolic dysfunction. Iron deficiency is implicated in pica, another eating disorder in which individuals repetitively eat non-nutritive, nonfood substances45. This contrasts with BED in which we implicate iron overload and where patients repetitively engage in consumption of highly nutritive food substances. Iron load may be a partial driver of consumptions on a spectrum from non-nutritive to binge eating through an undiscovered pathway. Iron overload has also been associated with cocaine use disorder46, which shares several key features with BED including loss-of-control and impulsivity and animal models have shown an association between binge eating and cocaine craving47. Peripheral iron excess may lead to iron accumulation in the basal ganglia, causing oxidative damage and dysfunction of reward circuitry46.
Conclusive identification of SNP-gene associations was limited by indeterminate fine-mapping. A preponderance of evidence implicating iron overload in BED suggests that the locus associated with rs79220007 corresponds to HFE, and we were able to replicate this SNP in an independent MVP cohort. We implicated APOE through MAGMA and, while rs7412, the lead SNP located within APOE did not reach genome-wide significance in our primary GWAS, its replication suggests this association may persist with a larger sample. This association along with the observation that individuals with BED and obesity have an unfavorable lipid profile compared to those with obesity and not BED48, suggests that lipid signaling abnormalities may be associated with BED. rs17789218, also replicated in our MVP replication cohort, and in a meta-analysis of the UKBB, PNC and ABCD cohorts, is located near MCHR2. MCHR2 encodes a G protein-coupled receptor for melanin-concentrating hormone, a conserved cyclic peptide that plays an important role in energy and glucose metabolism49 and regulates impulsive eating50. We did not replicate rs2275046 (identified in our cross-ancestry analysis) in our EUR MVP replication cohort.
In conclusion, we report the first GWAS investigating BED; we implicate and replicate two loci, one additional gene and iron metabolism in the pathophysiology of BED independent of BMI. We demonstrate that BED is a complex, metabolic-psychiatric disorder by inculpating both neural tissues and peripheral metabolic pathways known to influence brain function. Through identification of disrupted iron metabolism, we find an actionable target for future translational research.
Methods
Participants
The protocol from this study was approved by the Department of Veterans Affairs (VA) Central Institutional Review Board. The MVP cohort has been previously described16,51,52. Analysis of MVP data was conducted using electronic medical data v20.1 comprising 819,417 patients with demographic information. Of the 819,417 patients, release 3 of the genomics data consists of 459,777 patients with diverse genetic ancestries. Ancestry was assigned using the HARE program53 with the 1000 Genomes project54 as a reference panel. Confirmatory analysis was performed on release 4 of the MVP (n = 135,069), the UKBB35 (n = 386,085), ABCD18,55 (n = 4,659) and the PNC19 (n = 4,861), all previously described (Supplementary Table 9).
Phenotyping
We developed a series of eating disorder phenotype definitions from data available from the subjects’ associated EMR available through the MVP. For eating disorder prevalence estimates by diagnostic code within the MVP, we counted subjects that had at least 1 instance of any individual eating disorder coded in either ICD-9-CM or ICD-10-CM (Supplementary Table 1).
We built our BED machine learning model (MD-BED) by leveraging available diagnostic codes, medication prescriptions, BMI measurements and demographic data. The set of reliably diagnosed BED subjects (Supplementary Table 2) comprised those with either two ICD-10-CM BED codes (F50.81) or a single BED code and no other specific eating disorder codes. The set of control subjects comprised all MVP subjects with available EMR data, excluding those with any specific eating disorder diagnosis code and those who self-reported having an eating disorder on the enrollment questionnaire (“Do you have an eating disorder?”).
We trained a machine learning model to calculate a BED probability score across the MVP utilizing the cohort of reliably diagnosed BED cases described above (n = 822). We first mapped ICD diagnoses to Clinical Classification Software codes and computed log counts for diagnoses for each subject. Next, we computed 25th, 50th and 75th percentile BMI measures using height and weight values from each record. We removed records with nonsensical height, weight and BMI values, where we required BMI to range from 9 to 150 kg/m2, median height to range from 35 to 95 inches, and weight entries to range from 40 to 1400 pounds. We further removed patients with less than two valid, distinct BMI measurements. Of the 767,527 patients that passed these filters, we constructed a 90%–10% split of our data to train our model. Due to the large number of medication ingredients (3,456) available for analysis, we used a LASSO regression model (using the cv.glmnet function with parameters alpha = 1, family = binomial and nfolds = 10) prior to the creation of the final model to reduce the number of the medication ingredients included. For this step, we took a random sample of 90,000 control subjects to identify the relevant medications, while retaining all of our cases from the training set. To compute the area deprivation index for patients with missing ZIP codes, we imputed the median value. Subjects without any prescriptions or diagnoses were filtered out. We constructed our final MD-BED model by incorporating log counts of medications at the ingredient level, demographic variables at enrollment (self-reported sex, age, race, and ethnicity, and ZIP code-associated deprivation index), log counts for diagnoses and BMI (as calculated above) as model predictors and trained it on the remaining subjects.
To develop a ranking system for variables included in our model and identify a set of top predictors of BED, we developed an analogous, unpenalized logistic regression model. Ranking of top variables was based on P values from this analogous unpenalized logistic regression model, which was trained on all of the 767,527 patients, and used only the variables that were retained in the LASSO regression model used to calculate BED scores. The ranking of top variables was only performed to identify important predictors from the model and did not impact downstream analyses.
To test whether our BED scores predict BED better than other phenotypes, we ran an independent logistic regression analysis for a set of 1,752 phecodes plus the ICD-10-CM code for BED (1,753 phenotypes) leveraging the same test set, that was excluded when training our LASSO regression model to predict BED. We compared the log odds ratio of the BED score to each phenotype (Top phenotypes: Fig. 1b, full list: Supplementary Table 4). Furthermore, we applied the same test to assess the relationship between BED scores and iron deficiency anemia within the MVP.
To construct our MD-BED*BMI model, after assessing the performance of the LASSO logistic regression model for various parameter choices through cross-validation, we initially retained both the model with the lowest binomial deviance (Min) as well as the simplest model that had a mean binomial deviance within one standard deviation of the best performing model (1SE) to avoid overfitting. We then completed GWAS (as described below) on both models adjusting for BMI and retained the model with mean binomial deviance within one standard deviation of the best performing mode (MD-BED*BMI) as it outperformed the model with the lowest binomial deviance (MD-Min-BED*BMI) in SNP heritability (2.14% (s.e. = 0.23%) compared to 1.65% (s.e. = 0.22%)) and did not differ on predictive performance (Supplementary Fig. 15).
For all GWAS utilizing BMI (i.e., EUR-BMI), participant BMI quartile was calculated as above and all participants with distinct and valid BMI measurements at the 25th and 75th percentile were retained.
For privacy reasons, the MVP places a restriction on case-control GWAS requiring at least 500 cases for the summary statistics to be shared with the scientific community for reproducibility, and there are less than 500 individuals of a single ancestry with both an ICD-10-CM BED code and available genetic information. Therefore, to identify enough BED-probable subjects to perform an ICD-based case-control GWAS for the EUR-ICD-BED phenotype, we developed a second machine learning classifier to identify probable BED cases from within the subpopulation of all patients with eating disorder diagnoses. To generate the ICD-BED score, we followed the same procedure as above with cases defined as all reliably diagnosed BED cases. We again built a LASSO regression model, this time training it on the entire cohort of reliably diagnosed BED subjects and training it to distinguish them from within the subpopulation of all subjects with any eating disorder diagnosis. Cases for the GWAS were then then defined as the set of reliably diagnosed subjects with BED as above plus subjects with at least one ambiguous eating disorder code (307.50, 307.59, F50.89, F50.9) and a within-eating disorders BED score greater than the 25th percentile of reliably diagnosed BED cases (n = 549). Controls were defined as all remaining subjects excluding those with any eating disorder diagnosis code and those who self-reported having an eating disorder as above (n = 284,648).
For the UKBB, inclusion and exclusion was based on the Mental Health Questionnaire and associated ICD-9 and ICD-10 codes. Subjects reporting a “diagnosis of psychological over-eating or binge-eating” were included as cases. Those with a self-reported or ICD coded diagnosis of anorexia nervosa or bulimia nervosa were excluded from both the case and control groups. All remaining subjects were retained as controls. For ABCD (data release 4.0), case-control definitions were based on the K-SADS. Cases were subjects who received a K-SADS diagnosis of BED and controls were subjects who did not receive a K-SADS diagnosis for either BED or bulimia nervosa. A diagnosis of anorexia nervosa could not be made from the available data from ABCD as it could not be determined whether an individual met all criteria or had been formally diagnosed. For the PNC, case-control definitions were based on a modified version of the K-SADS (third study accession, dbGaP phs000607.v3.p2). Cases were those who answered yes to EAT007 (lifetime binge eating) and no to EAT008 (lifetime purging after a binge-eating episode). Subjects were excluded from the control group if they answered yes to EAT007 or EAT008 or screened positive for an anorexia nervosa-like phenotype (yes to EAT001, EAT002 & EAT003 AND either 1) male 2) no to EAT004 3) no to EAT005 or 4) yes to EAT004, EAT005 and EAT006).
In the UKBB, the gout, iron overload, and iron deficiency phenotypes were defined by the presence of at least one relevant phecode or self-reported diagnosis code. Hospital inpatient ICD-9 and ICD-10 codes were converted to phecodes using previously established methods56. Gout phenotype cases included individuals with phecodes 274.1 (gout) or 274.11 (gouty arthropathy), as well as self-reported gout at the baseline assessment. Iron overload phenotype cases included individuals with phecodes 275.1 (disorders of iron metabolism) or 277.1 (disorders of porphyrin metabolism). Iron deficiency phenotype cases included individuals with phecodes 262 (mineral deficiency NEC) or 280.1 (iron deficiency anemias, unspecified or not due to blood loss), as well as self-reported iron deficiency anemia at baseline.
Genotyping, quality control and imputation
Genotyping, quality control and imputation in MVP is handled by a dedicated data team. Data from release 3 of the MVP used in this study includes genotyping data from 455,789 individuals; DNA was extracted from whole blood (which was collected during enrollment to the MVP) and genotyping was performed with the MVP 1.0 Genotyping array16. Ancestry as determined by HARE (Harmonized Ancestry and Race/Ethnicity) analysis53. Prephasing was performed using EAGLE v257 and genotypes were imputed using Minimac v3 with the 1000 Genomes Project phase 3, version 5 reference panel58. Relatedness between MVP participants was inferred using kinship coefficient calculated using software KING59. Related individuals are removed using a kinship coefficient cut off >= 0.0884. Principal component analysis to generate ancestral principal components was performed using EIGENSOFT v.660,61. The replication MVP dataset comprises the set of 135,069 European ancestry individuals from the 4th data release of the MVP who were not included in the 3rd data release. This dataset became available to us after the initial analysis was completed and was thus used only as a replication cohort. For the follow-up analysis with rs17889218, we used the TOPMED imputation to identify regional variants in LD with the lead SNP that passed the threshold for suggestive significance (p < 10−5).
Genome-wide association studies
GWAS analysis was conducted employing either logistic regression for binary traits (case-control definition of BED from the ICD model) and linear regression for continuous traits (BMI, MD-BED scores) using PLINK 2.0. We conducted separate analyses for AFR and EUR genetic ancestries and included age, sex, record density and the top 10 principal components to adjust for potential confounders. SNPs with minor allele frequency less than 0.5% or an effective minor allele count < 30 were removed from the analysis per MVP regulation. SNPs with a HWE P value less than 5×10−8 or an imputation r2 < 0.4 were removed from the analysis. To assess whether a SNP was genome-wide significant, we used the standard multiple-testing correction threshold, p < 5×10-8. To satisfy the model assumptions of performing linear regression, we applied an inverse-rank normal transformation of the MD-BED scores to ensure the prediction errors follow an approximately normal distribution for all model derived GWAS. To compute power adjusting for winner’s curse, we estimated adjusted effect sizes from the corrected P values in the discovery cohort (release 3 of the MVP) and computed power using the estimated standard error in the replication cohort62.
Enrichment Analyses
We utilized FUMA (v1.3.7)63 and its implementation of MAGMA (v1.0.8)17 to determine genes and gene sets that are linked to the MD BED phenotype. We used a Bonferroni-corrected P value threshold of 2.684×10−6 to account for multiple testing of 18,626 protein coding genes using the 10k UKBB European reference panel (release 2b) to account for LD. In addition, we considered 54 tissue types from GTEx V8 to assess for gene enrichment across different tissues. We excluded genes from the extended MHC region starting at 25.7Mb in GENE2FUNC. We also provide results with less stringent default parameters for excluding annotations from the MHC region, between MOG and COL11A2.
LD Score Regression, Heritability and Genetic Correlation
We estimated SNP heritability and genetic correlation using the LDSC package26,64. Heritability for case-control phenotypes was computed on the liability scale for a range of plausible population prevalences, while heritability for continuous phenotypes was computed on the observed scale. To compute the heritability and genetic correlation estimates, the 1000 Genomes Project - European Project was used to construct the reference panel. For genetic correlation analysis with external GWAS, we first curated a list of 44 GWAS of interest. To supply a more comprehensive, hypothesis-free approach, we computed genetic correlation between EUR-MD-BED*BMI and 1,427 (of 2,150) traits from the IEU Open GWAS project34 after removal of 723 traits that did not have statistically robust estimates for heritability (Z < 2).
Meta-Analysis
We used the inverse-variance weighting method to meta-analyze both the AFR and EUR MD-BED*BMI GWAS and the log odds ratio for the PRS on the external cohorts (UKBB, ABCD and PNC). To account for the LD structure and the possibility of different effect sizes between different populations, we also employed Multi-ancestry Meta-analysis (MAMA)28 using the 1000 Genomes Project as a reference panel. For meta-analysis with MAMA, we filtered out SNPs with minor allele frequencies that were smaller than 1% to ensure our sample size from the reference panel was large enough to attain sufficiently accurate LD Scores. MAMA did not yield any additional hits and as such was not further discussed in the main paper.
Fine-mapping
For each of our loci, where we have a genome-wide significant hit and the minor allele frequency is sufficiently large (> 1%), we performed fine-mapping to identify candidate causal variants using SUSIE with the UKBB as a reference panel. We ran SUSIE29,65 both including and excluding outlying SNPs flagged by the algorithm, where we specified the upper bound for the number of causal variants to be 5 and the window to span 1 megabase pairs.
Phenome-Wide Association Studies
PheWAS were conducted leveraging EUR subjects in the MVP (n = 296,407). We then filtered imputed genotypes by minor allele frequency (> 0.01), variant level missingness (< 0.02) and imputation r2 (> 0.9). Phenotypes were derived by aggregating ICD Codes in the EMR data using the categorizations provided in the Phecode Map v1.266; phenotypes with less than 500 cases were removed from the analysis. We then performed a logistic regression, adjusted for sex, age and top 10 ancestry principal components, to assess the presence of an association between the flagged SNP and the phenotype. We assessed significance using a Bonferroni corrected two-sided P value at the 0.05 level.
Polygenic Risk Scores and Validation
The Polygenic Risk Score-Continuous Shrinkage (PRS-CS)67 method was used to compute PRS for the external cohorts from the GWAS summary statistics derived from the MVP cohort. For further details for PRS validation and quality control processes for the ABCD, PNC, and UKBB cohorts, we refer the reader to the Supplementary Note.
Concordance of EUR and AFR GWAS hits
To assess the similarity between the AFR- and EUR- MD-BED*BMI GWAS, we clumped lead SNPs in Plink68 using the European 1000 Genomes reference panel. In particular, we required that all SNPs in the same clump must have an r2 of at least 0.2 with the lead SNP and be at most 250 kbp away from the lead SNP. We then computed the Spearman correlation between the linear regression coefficients for the respective GWAS using different P value filters, (e.g., p < 10−4) as described in the main text.
MVP-Derived GWAS SNP Replication in Additional Cohorts
To estimate the appropriate adjusted effect size for computing power, as cross-study heterogeneity (e.g., phenotypic misclassification, comparing a GWAS from a continuous phenotype (MD-BED*BMI GWAS) to a binary phenotype (ABCD-PNC-UKBB-BED*BMI GWAS)) can result in diminished effect sizes for all SNPs in the GWAS, we leveraged the PheMED tool30. Our uncorrected effect sizes came from the replication cohort of the MD-BED*BMI GWAS from the MVP. Since we had heterogeneous power for detecting true effects across different tests of the genome-wide significant SNPs from the MD-BED*BMI GWAS, we leveraged a weighted P value approach to compute the FDR, where we more heavily weight tests based on their power31. For further details about SNP replication on the ABCD, PNC and UKBB cohorts for the model derived and ICD GWAS, we refer the reader to the Supplementary Note.
Generalised Summary-data-based Mendelian Randomisation
We leveraged the GCTA software tool69 to perform GSMR using the European participants from the 1000 Genomes Project as a reference panel with the default parameters. Since GSMR requires at least 10 genome wide significant loci, we could only conduct forward GSMR to assess if a given phenotype (transferrin saturation, baseline urate levels) influences our model derived BED phenotype, and not backward GSMR as our MD-BED GWAS lacks the requisite number of genome-wide significant loci. We used available summary statistics for the transferrin saturation37 and urate level70 phenotypes. As part of GSMR, pleiotropic SNPs were filtered out using the HEIDI-outlier method (with the default threshold of p = 0.01 for the primary analysis and p = 0.05 for secondary analysis presented in Supplementary Fig. 13) before assessing the correlation of lead SNPs from the summary statistics for transferrin saturation phenotype with the model derived BED phenotype.
Partitioned Heritability
We examined an overlap of common genetic variants of BED and open chromatin from (i) wild-type and heme-deficient mutant murine erythroid cells treated with β-estradiol and/or 5-aminolevulinic acid hydrochloride (5-ALA)38, (ii) a sciATAC-seq3 study of human fetal cell types, (iii) a scATAC-seq study across six human adult brain regions40 using an LD Score partitioned heritability approach71. We used LD Scores with a baseline model of general genomic annotation (such as conserved regions and coding regions) that corrects for the general genetic context of tested sets of open chromatin regions. All regions of open chromatin were extended by 500 base pairs in either direction. The broad MHC-region (chr6:25–35MB) was excluded due to its extensive and complex LD structure, but otherwise default parameters were used for the algorithm. In the case of mouse cell lines, we merged all peaks of open chromatin for all replicates belonging to the same type of experiment (i.e., untreated / β-estradiol treated wild-type, untreated / β-estradiol mutant and untreated / β-estradiol treated double mutant) and converted mice genome coordinates of resulting peak sets to human genome coordinates using liftOver (https://genome.ucsc.edu/cgi-bin/hgLiftOver).
Supplementary Material
Acknowledgements
This research is based on data from the Million Veteran Program, Office of Research and Development, Veterans Health Administration, and was supported by award #MVP006. This publication does not represent the views of the Department of Veteran Affairs or the United States Government. This study was also supported by the National Institutes of Health (NIH), Bethesda, MD under award numbers T32MH087004 (K.T.), T32MH096679 (T.G.), T32MH122394 (A.M.), K08MH122911 (G.V.), R01MH125246, R01AG067025, U01MH116442, R01MH109677 (P.R.), and by the Veterans Affairs Merit grants BX002395 and BX004189 (P.R.). This study has also been funded in part by the Brain & Behavior Research Foundation via the 2020 NARSAD Young Investigator Grant #29350 (G.V.). We thank Shing Wan Choi and Paul F. O’Reilly for their guidance and expertise in utilizing data from the UK Biobank. We thank the participants in the UK Biobank and the scientists involved in the construction of this resource. This research has been conducted using the UK Biobank Resource under application 18177 (P.F.O.). This work was supported, in part, through the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. Data used in the preparation of this article were obtained from the Adolescent Brain Cognitive Development (ABCD) Study (https://abcdstudy.org), held in the NIMH Data Archive (NDA). This is a multisite, longitudinal study designed to recruit more than 10,000 children aged 9–10 and follow them over 10 years into early adulthood. The ABCD Study is supported by the National Institutes of Health and additional federal partners under award numbers U01DA041022, U01DA041028, U01DA041048, U01DA041089, U01DA041106, U01DA041117, U01DA041120, U01DA041134, U01DA041148, U01DA041156, U01DA041174, U24DA041123, U24DA041147, U01DA041093, and U01DA041025. A full list of supporters is available at https://abcdstudy.org/federal-partners.html. A listing of participating sites and a complete listing of the study investigators can be found at https://abcdstudy.org/scientists/workgroups/. ABCD consortium investigators designed and implemented the study and/or provided data but did not necessarily participate in analysis or writing of this report. This manuscript reflects the views of the authors and may not reflect the opinions or views of the NIH or ABCD consortium investigators. The ABCD data repository grows and changes over time. The ABCD data used in this report came from DOI 10.15154/1527728. DOIs can be found at https://nda.nih.gov/study.html?id=1661. Support for data collection for the PNC, acquired through dbGaP (accession no. phs000607, v3.p2), was provided by grant RC2MH089983 awarded to R. Gur and RC2MH089924 was awarded to H. Hakonarson. Participants were recruited and genotyped through the Center for Applied Genomics (CAG) at The Children’s Hospital in Philadelphia (CHOP). Phenotypic data collection occurred at the CAG/CHOP and at the Brain Behavior Laboratory, University of Pennsylvania.
Footnotes
Competing Interests Statement
Dr. Hildebrandt is a scientific advisory board member of Noom, Inc. and Drs. Hildebrandt and Sysko receive funding from and have equity in Noom, Inc. (non-publicly traded company). Dr. Sysko receives royalties from Wolters Kluwer Health.
Code Availability
EIGENSOFT v6 https://github.com/dreichlab/eig
FUMAv1.3.7: https://fuma.ctglab.nl/
GCTA v.1.93.2: https://yanglab.westlake.edu.cn/software/gcta/#Overview
KINGv2.0: https://www.chen.kingrelatedness.com
LD Score Regression v1.0.1: https://github.com/bulik/ldsc
liftOver v1.2.0: https://genome.ucsc.edu/cgi-bin/hgLiftOver
Minimac v3: https://genome.sph.umich.edu/wiki/Minimac3
Multi-Ancestry Meta Analysis: https://github.com/JonJala/mama
PheMED: https://github.com/DiseaseNeuroGenomics/PheMED
PRS-CS: https://github.com/getian107/PRScs
susieR as implemented in echolocatoR72: https://github.com/RajLabMSSM/echolocatoR
Data Availability
BED GWAS summary statistics from the Million Veteran Program data will become available on dbGaP (accession number phs001672).
For the external validation sets for the Partitioned Heritability Analysis: open chromatin regions from a murine erythroid cell beta-estradiol stimulation model (GEO GSE114996), open chromatin atlas of adult human brains (GEO GSE147672), open chromatin atlas of developing human organs (https://descartes.brotmanbaty.org/bbi/human-chromatin-during-development/)
For GWAS summary statistics for genetic correlation analyses, we refer the reader to the IEU Open GWAS Project: https://gwas.mrcieu.ac.uk/
References
- 1.Mitchell KS et al. Binge eating disorder: a symptom-level investigation of genetic and environmental influences on liability. Psychol. Med. 40, 1899–1906 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichborn-Kjennerud T, Bulik CM, Tambs K & Harris JR Genetic and environmental influences on binge eating in the absence of compensatory behaviors: a population-based twin study. Int. J. Eat. Disord. 36, 307–314 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Udo T & Grilo CM Prevalence and Correlates of DSM-5-Defined Eating Disorders in a Nationally Representative Sample of U.S. Adults. Biol. Psychiatry 84, 345–354 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brownley KA et al. Binge-Eating Disorder in Adults: A Systematic Review and Meta-analysis. Ann. Intern. Med. 165, 409–420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wonderlich SA, Gordon KH, Mitchell JE, Crosby RD & Engel SG The validity and clinical utility of binge eating disorder. Int. J. Eat. Disord. 42, 687–705 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Bulik CM et al. The Binge Eating Genetics Initiative (BEGIN): study protocol. BMC Psychiatry 20, 307 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Javaras KN et al. Co-occurrence of binge eating disorder with psychiatric and medical disorders. J. Clin. Psychiatry 69, 266–273 (2008). [DOI] [PubMed] [Google Scholar]
- 8.Javaras KN et al. Familiality and heritability of binge eating disorder: results of a case-control family study and a twin study. Int. J. Eat. Disord. 41, 174–179 (2008). [DOI] [PubMed] [Google Scholar]
- 9.Hübel C et al. One size does not fit all. Genomics differentiates among anorexia nervosa, bulimia nervosa, and binge-eating disorder. Int. J. Eat. Disord 54, 785–793 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guss JL, Kissileff HR, Devlin MJ, Zimmerli E & Walsh BT Binge size increases with body mass index in women with binge-eating disorder. Obes. Res. 10, 1021–1029 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Anderson DA, Williamson DA, Johnson WG & Grieve CO Validity of test meals for determining binge eating. Eat. Behav 2, 105–112 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Kenardy J et al. Disordered eating behaviours in women with Type 2 diabetes mellitus. Eat. Behav. 2, 183–192 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Hudson JI et al. Longitudinal study of the diagnosis of components of the metabolic syndrome in individuals with binge-eating disorder. Am. J. Clin. Nutr. 91, 1568–1573 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hilbert A et al. Meta-analysis on the long-term effectiveness of psychological and medical treatments for binge-eating disorder. Int. J. Eat. Disord 53, 1353–1376 (2020). [DOI] [PubMed] [Google Scholar]
- 15.Peat CM et al. Comparative Effectiveness of Treatments for Binge-Eating Disorder: Systematic Review and Network Meta-Analysis. Eur. Eat. Disord. Rev. 25, 317–328 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Gaziano JM et al. Million Veteran Program: a mega-biobank to study genetic influences on health and disease. J. Clin. Epidemiol 70, 214–223 (2016). [DOI] [PubMed] [Google Scholar]
- 17.de Leeuw CA, Mooij JM, Heskes T & Posthuma D MAGMA: generalized gene-set analysis of GWAS data. PLoS Comput. Biol. 11, e1004219 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volkow ND et al. The conception of the ABCD study: From substance use to a broad NIH collaboration. Dev. Cogn. Neurosci. 32, 4–7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Satterthwaite TD et al. The Philadelphia Neurodevelopmental Cohort: A publicly available resource for the study of normal and abnormal brain development in youth. Neuroimage 124, 1115–1119 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ollier W, Sprosen T & Peakman T UK Biobank: from concept to reality. Pharmacogenomics 6, 639–646 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Diagnostic and statistical manual of mental disorders (DSM-5) 5th edn (American Psychiatric Association Publishing, Washington, DC, 2013). [Google Scholar]
- 22.Kessler RC et al. The prevalence and correlates of binge eating disorder in the World Health Organization World Mental Health Surveys. Biol. Psychiatry 73, 904–914 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sonneville KR & Lipson SK Disparities in eating disorder diagnosis and treatment according to weight status, race/ethnicity, socioeconomic background, and sex among college students. Int. J. Eat. Disord. 51, 518–526 (2018). [DOI] [PubMed] [Google Scholar]
- 24.Taliun D et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. Nature 590, 290–299 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Polimanti R et al. Leveraging genome-wide data to investigate differences between opioid use vs. opioid dependence in 41,176 individuals from the Psychiatric Genomics Consortium. Mol. Psychiatry 25, 1673–1687 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bulik-Sullivan B et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet. 47, 291–295 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Willer CJ, Li Y & Abecasis GR METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Turley P et al. Multi-Ancestry Meta-Analysis yields novel genetic discoveries and ancestry-specific associations. BioRxiv (2021) doi: 10.1101/2021.04.23.441003. [DOI] [Google Scholar]
- 29.Zou Y, Carbonetto P, Wang G & Stephens M Fine-mapping from summary data with the “Sum of Single Effects” model. PLoS Genet. 18, e1010299 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Burstein D et al. Detecting and adjusting for hidden biases due to phenotype misclassification in genome-wide association studies. medRxiv (2023) doi: 10.1101/2023.01.17.23284670. [DOI] [Google Scholar]
- 31.Genovese CR, Roeder K & Wasserman L False discovery control with p-value weighting. Biometrika 93, 509–524. [Google Scholar]
- 32.Karlsson Linnér R et al. Multivariate analysis of 1.5 million people identifies genetic associations with traits related to self-regulation and addiction. Nat. Neurosci. 24, 1367–1376 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams C et al. Facilitating the Application of Externalizing Summary Statistics in Behavioral and Biomedical Research.
- 34.Elsworth B et al. The MRC IEU OpenGWAS data infrastructure. BioRxiv (2020) doi: 10.1101/2020.08.10.244293. [DOI] [Google Scholar]
- 35.Sudlow C et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhu Z et al. Causal associations between risk factors and common diseases inferred from GWAS summary data. Nat. Commun. 9, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bell S et al. A genome-wide meta-analysis yields 46 new loci associating with biomarkers of iron homeostasis. Commun. Biol. 4, 156 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tanimura N et al. GATA/Heme Multi-omics Reveals a Trace Metal-Dependent Cellular Differentiation Mechanism. Dev. Cell 46, 581–594.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Domcke S et al. A human cell atlas of fetal chromatin accessibility. Science 370, eaba7612 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Corces MR et al. Single-cell epigenomic analyses implicate candidate causal variants at inherited risk loci for Alzheimer’s and Parkinson’s diseases. Nat. Genet. 52, 1158–1168 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.An SJ, Kim TJ & Yoon B-W Epidemiology, risk factors, and clinical features of intracerebral hemorrhage: an update. J. Stroke 19, 3–10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stunkard AJ & Allison KC Binge eating disorder: disorder or marker? Int. J. Eat. Disord 34 Suppl, S107–16 (2003). [DOI] [PubMed] [Google Scholar]
- 43.Hinckley JD et al. Quantitative trait locus linkage analysis in a large Amish pedigree identifies novel candidate loci for erythrocyte traits. Mol. Genet. Genomic Med. 1, 131–141 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galmozzi A et al. PGRMC2 is an intracellular haem chaperone critical for adipocyte function. Nature 576, 138–142 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Borgna-Pignatti C & Zanella S Pica as a manifestation of iron deficiency. Expert Rev. Hematol 9, 1075–1080 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Ersche KD et al. Disrupted iron regulation in the brain and periphery in cocaine addiction. Transl. Psychiatry 7, e1040 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Barnea R et al. Trait and state binge eating predispose towards cocaine craving. Addict. Biol 22, 163–171 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Succurro E et al. Obese patients with a binge eating disorder have an unfavorable metabolic and inflammatory profile. Medicine (Baltimore) 94, e2098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Al-Massadi O et al. Multifaceted actions of melanin-concentrating hormone on mammalian energy homeostasis. Nat. Rev. Endocrinol. 17, 745–755 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Noble EE et al. Hypothalamus-hippocampus circuitry regulates impulsivity via melanin-concentrating hormone. Nat. Commun. 10, 4923 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods-only references
- 51.Harrington KM et al. Gender differences in demographic and health characteristics of the million veteran program cohort. Women’s Health Issues 29 Suppl 1, S56–S66 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gelernter J et al. Genome-wide association study of post-traumatic stress disorder reexperiencing symptoms in >165,000 US veterans. Nat. Neurosci. 22, 1394–1401 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fang H et al. Harmonizing Genetic Ancestry and Self-identified Race/Ethnicity in Genome-wide Association Studies. Am. J. Hum. Genet. 105, 763–772 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.1000 Genomes Project Consortium et al. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56–65 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karcher NR & Barch DM The ABCD study: understanding the development of risk for mental and physical health outcomes. Neuropsychopharmacology 46, 131–142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wu P et al. Mapping ICD-10 and ICD-10-CM Codes to Phecodes: Workflow Development and Initial Evaluation. JMIR Med. Inform. 7, e14325 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Loh P-R et al. Reference-based phasing using the Haplotype Reference Consortium panel. Nat. Genet. 48, 1443–1448 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.1000 Genomes Project Consortium et al. A global reference for human genetic variation. Nature 526, 68–74 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Manichaikul A et al. Robust relationship inference in genome-wide association studies. Bioinformatics 26, 2867–2873 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Price AL et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909 (2006). [DOI] [PubMed] [Google Scholar]
- 61.Patterson N, Price AL & Reich D Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bigdeli TB et al. A simple yet accurate correction for winner’s curse can predict signals discovered in much larger genome scans. Bioinformatics 32, 2598–2603 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Watanabe K, Taskesen E, van Bochoven A & Posthuma D Functional mapping and annotation of genetic associations with FUMA. Nat. Commun. 8, 1826 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bulik-Sullivan B et al. An atlas of genetic correlations across human diseases and traits. Nat. Genet. 47, 1236–1241 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang G, Sarkar A, Carbonetto P & Stephens M A simple new approach to variable selection in regression, with application to genetic fine mapping. J. Royal Statistical Soc. B (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Denny JC et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat. Biotechnol. 31, 1102–1110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ge T, Chen C-Y, Ni Y, Feng Y-CA & Smoller JW Polygenic prediction via Bayesian regression and continuous shrinkage priors. Nat. Commun. 10, 1776 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Purcell S et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang J, Lee SH, Goddard ME & Visscher PM GCTA: a tool for genome-wide complex trait analysis. Am. J. Hum. Genet. 88, 76–82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Churchhouse C et al. Rapid GWAS of thousands of phenotypes for 337,000 samples in the UK Biobank — Neale lab. http://www.nealelab.is/blog/2017/7/19/rapid-gwas-of-thousands-of-phenotypes-for-337000-samples-in-the-uk-biobank.
- 71.Finucane HK et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat. Genet. 47, 1228–1235 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schilder BM, Humphrey J, & Raj T echolocatoR: an automated end-to-end statistical and functional genomic fine-mapping pipeline, Bioinformatics. 38.2, 536–539 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
BED GWAS summary statistics from the Million Veteran Program data will become available on dbGaP (accession number phs001672).
For the external validation sets for the Partitioned Heritability Analysis: open chromatin regions from a murine erythroid cell beta-estradiol stimulation model (GEO GSE114996), open chromatin atlas of adult human brains (GEO GSE147672), open chromatin atlas of developing human organs (https://descartes.brotmanbaty.org/bbi/human-chromatin-during-development/)
For GWAS summary statistics for genetic correlation analyses, we refer the reader to the IEU Open GWAS Project: https://gwas.mrcieu.ac.uk/