

Abstract
Background and Aims:
The thiazide-sensitive Na+-Cl− cotransporter NCC and the Cl−/HCO3− exchanger pendrin are expressed on apical membranes of distal cortical nephron segments and mediate salt absorption, with pendrin working in tandem with the epithelial Na+ channel (ENaC) and the Na+-dependent chloride/bicarbonate exchanger (NDCBE), whereas NCC is working by itself. A recent study showed that NCC and pendrin compensate for loss of each other under basal conditions, therefore masking the role that each plays in salt reabsorption. Carbonic anhydrase II (CAII, CA2 or CAR2) plays an important role in acid-base transport and salt reabsorption in the proximal convoluted tubule and acid-base transport in the collecting duct. Animals with CAII deletion show remodeling of intercalated cells along with the downregulation of pendrin. NCC KO mice on the other hand show significant upregulation of pendrin and ENaC. Neither model shows any significant salt wasting under baseline conditions. We hypothesized that the up-regulation of pendrin is essential for the prevention of salt wasting in NCC KO mice.
Methods and Results:
To test this hypothesis, we generated NCC/CAII double KO (dKO) mice by crossing mice with single deletion of NCC and CAII. The NCC/CAII dKO mice displayed significant downregulation of pendrin, along with polyuria and salt wasting. As a result, the dKO mice developed volume depletion, which was associated with the inability to concentrate urine.
Conclusions:
We conclude that the upregulation of pendrin is essential for the prevention of salt and water wasting in NCC deficient animals and its downregulation or inactivation will result in salt wasting, impaired water conservation and volume depletion in the setting of NCC inactivation or inhibition.
Keywords: Collecting duct, Intercalated cells, Acid base transport, Salt absorption
Introduction
The thiazide-sensitive Na+-Cl− cotransporter NCC and the Cl−/HCO3− exchanger pendrin (SLC26A4) are expressed on apical membranes of distal cortical nephron segments and mediate salt absorption, with pendrin working in tandem with the epithelial Na+ channel (ENaC) and the Na+-dependent chloride/bicarbonate exchanger (NDCBE), whereas NCC is working by itself [1–15]. Pendrin is expressed on the apical membrane of intercalated cells in the connecting tubule (CNT) and the cortical collecting duct (CCD) [6–9], whereas NCC is primarily expressed on the apical membrane of distal convoluted tubule (DCT) cells [2].
Single deletion of pendrin or NCC does not cause salt wasting or excessive diuresis under basal conditions [16–19]. Kidney functions, including sodium and chloride excretion, urine output, and blood urea nitrogen (BUN) levels in mutant mice are comparable to wild type animals. Both pendrin KO and NCC KO mice, however, show signs of volume depletion or develop hypotension during salt restriction [17,18]. These findings have led investigators to conclude that pendrin and NCC are predominantly active during salt depletion and/or in response to increased aldosterone levels, and their contribution to salt reabsorption at baseline conditions is minimal.
A recent study from our laboratory [20] showed that NCC and pendrin compensate for loss of the other under basal conditions; therefore masking the role that each plays in salt reabsorption. In these studies it was determined that mice with double knockout of pendrin and NCC develop significant salt and fluid wasting, along with volume depletion, nephrogenic diabetes insipidus and renal failure under baseline conditions.
Carbonic anhydrase II (CAII) plays an important role in acid-base transport and salt absorption in the proximal convoluted tubule and in acid-base transport in the collecting duct. Inhibition of CAII in the proximal tubule with the use of carbonic anhydrase inhibitors causes salt and bicarbonate wasting [21, 22]. The role of CAII in acid-base transport in the collecting duct is less well understood. Animals with CAII deletion show significant reduction in the number of B-intercalated cells along with the downregulation of pendrin expression [23, 24].
We hypothesized that the up-regulation of pendrin, along with ENaC activation, is essential for the prevention of salt wasting in NCC KO mice [11, 25]. As such, we hypothesized that the prevention of up-regulation of pendrin in NCC KO mice should result in salt wasting and volume depletion. To test this hypothesis, we generated NCC/CAII double KO (dKO) mice by crossing mice with single deletion of NCC and CAII.
Materials and Methods
Animal models
Details of generation of CAR2 (CAII) null mice and NCC null mice have been reported before [17,26,27]. NCC/CAII dKO mice were generated by crossing CAR2 null mice with NCC null mice. Wild type and mutant animals were housed and cared for in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Cincinnati. All animal handlers were IACUC-trained. Animals had access to food and water ad libitum, were housed in humidity, temperature, and light/dark controlled rooms, and were inspected daily. Animals were euthanized with the use of excess anesthetics (pentobarbital sodium) according to institutional guidelines and approved protocols.
Tail DNA genotyping
NCC KO genotyping:
NCC primers:
KO forward, 5’ AGG GTC AAG GGC ACG GTT GGC 3’; KO reverse, 5’ GGT AAA GGG AGC GGG TCC GAG G 3’; KO rev pk, 5’ GCA TGC TCC AGA CTG CCT TG 3’. PCR Conditions: 94 °C, 2 min, 1 cycle; 94 °C, 30 sec, 68 °C, 30 sec, 35 cycles; 68 °C, 3 min, 1 cycle; hold at 4 °C. The mutant band for the NCC KO is 188 bp and the wt band is 265 bp in size. rev pk refers to reverse primer complementary to the sequences in the phosphoglycerate kinase promoter, which drives the neo gene.
CAII KO genotyping:
Oligo 4N (Normal Primer):
5’ CGC TAT TTT TTG AAG ATT GGA CCT GCC ACA C 3’; Oligo 4M (Mutant Primer): 5’ CGC TAT TTT TTG AAG ATT GGA CCT GCC ACA T 3’; Oligo 5R (Common Antisense Primer): 5’ TGG AAG CCA TTA TTT ACT CCG GGT AAA CTG 3’. PCR Conditions: 94 °C, 3 min, 1 cycle; 94 °C, 30 sec, 64 °C, 30 sec, 68 °C, 30 sec, 35 cycles; 68 °C, 3 min, 1 cycle; hold at 4 °C. The mutant band for CAII KO is 190 bp and the wt band is 190 bp in size.
Antibodies
Pendrin antibodies were generated in our laboratory as described (19). NHE3 antibodies were gifts from Dr. Alicia McDonough, University of Southern California. Antibodies against AQP2 (Santa Cruz Biothech.), Ser256p-AQP2 (Assay Biotech), Ser261p-AQP2 (Abnova) and renin (My Biosource) were used in these studies. Antibodies against ENaC (β and γ subunits) were purchased from StressMarq Biosciences Inc. (Victoria, BC Canada).
Western blot analysis
Membrane proteins were prepared from mouse kidneys as previously described [28–30]. Membrane proteins were size-fractionated by SDS/PAGE (40μg/lane) and transferred to nitrocellulose membrane. Western blot analyses were performed using anti-AQP2, anti-Ser256p-AQP2, anti-Ser261p-AQP2 and anti-ENaC antibodies. Appropriate horseradish peroxidase-conjugated IgGs (Thermo Scientific, Rockford, IL) were used as secondary antibodies. The bands were visualized by chemiluminescence method (Invitrogen, Carlsbad, CA) and captured on light-sensitive imaging film (Denville Scientific Inc, Metuchen, NJ).
Immunofluorescence labeling studies
Animals were euthanized with an overdose of pentobarbital sodium and perfused through the left ventricle with 0.9% saline followed by cold 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Kidneys were removed, cut in tissue blocks, and fixed in formaldehyde solution overnight at 4 °C. The tissue was either frozen on dry ice or fixed in paraffin, and 6 μm sections were cut with a cryostat and stored until used. Single-immunofluorescence labeling with pendrin antibodies was performed as described [28–30].
RNA isolation and Northern hybridization
Total cellular RNA was extracted from kidneys, according to established methods, quantitated spectrophotometrically and stored at −80 °C. Hybridization was performed according to established protocols [19].
Gene-specific DNA fragments were generated by RT-PCR and used as specific probes for Northern blot hybridization. For pendrin, a PCR fragment corresponding to nucleotides 1883-2217(NM_011867), was generated from mouse kidney. For renin, a PCR fragment corresponding to nucleotides 291-600(NM_031192) was generated from mouse kidney. Each Northern blot hybridization was performed on four independent samples from four different animals.
Urine and serum electrolytes analysis
Mice were housed in metabolic cages and had free access to rodent chow and water. Food intake, water intake, and urine volume were measured daily. Urine was collected under mineral oil. Urine chloride and sodium concentrations were measured using a digital chloridometer (HBI Haake Buchler Instruments, Inc.). Serum concentration of Na+, K+, Ca++, and HCO3− were measured using i-STATR-1 analyzer with i-STAT EG7+ cartridges (Abbott Laboratories, Abbott Park, IL).
Blood pressure monitoring
Systolic blood pressure in conscious mice was determined using a tail-cuff sphygmomanometer (Visitech BP2000; Visitech Systems, Apex, NC, USA). Measurements for each mouse represent mean value of three consecutive recordings performed in the last week of experiments. All experimental animals were preconditioned for blood pressure measurements as previously described [20]. For salt loading studies, animals were subjected to 280 mM NaCl added to their drinking water for 2 weeks. Blood pressure was recorded during the last 3 days of salt loading.
Statistical analysis
The results for chloride excretion, urine volume, urine osmolarity and blood pressure are presented as means ± SE. Statistical significance between experimental groups was determined by unpaired Student’s t- test or Anova and P<0.05 was considered significant.
Results
Expression of pendrin in kidneys of NCC KO or CAII KO mice
Figure 1a is a representative northern blot and shows the mRNA expression of pendrin in kidneys of wild type, NCC KO and CAII KO mice. As shown, the expression of pendrin is increased in NCC KO mice but decreased in CAII KO mice. Figure 1b depicts representative immunofluorescence labeling of pendrin in kidneys of NCC KO (middle panel) and CAII KO mice (right panel) vs. their wild type littermates (left panel). The results indicate that pendrin abundance is increased in NCC KO but decreased significantly in CAII KO mice and confirm the mRNA expression studies in Figure 1a and previous reports [11, 24].
Fig. 1.

Expression of pendrin in kidneys of NCC KO or CAII KO mice. a. Northern hybridization of pendrin mRNA. The RNA loading was determined by the intensity of 18S rRNA. b. Immunofluorescence labeling of pendrin. The expression of pendrin is significantly increased in kidneys of NCC KO mice. However, CAII KO mice show significant down-regulation of pendrin in their kidneys. G: Glomerulus.
Generation of NCC/CAII dKO mice
The NCC/CAII dKO mice were generated by crossing mice with single deletion of NCC or CAII with each other. Figure 2a shows tail genotyping demonstrating the generation of NCC/CAII dKO mice, as verified by the presence of mutant bands and the absence of wt bands. We next examined the expression of pendrin in kidneys of WT, NCC KO, CAII KO and NCC/CAII dKO mice. The results (Fig. 2b) demonstrate that pendrin expression in NCC/CAII dKO mice is significantlydecreased relative to that in NCC KO or WT mice (p<0.05, n=4) and resembles the pattern seen in CAII KO mice. Immunofluorescence confirmed the mRNA expression studies (data not shown). Figure 2c depicts body weights of 2 months old dKO, single KO, and WT mice. As shown, NCC/NCC dKO mice are significantly smaller than either single KO or WT animals. The average body weight was ~35% lower in dKO mice vs. WT or single KO mice (Fig. 2c) (p<0.05, n=4 in each group).
Fig. 2.
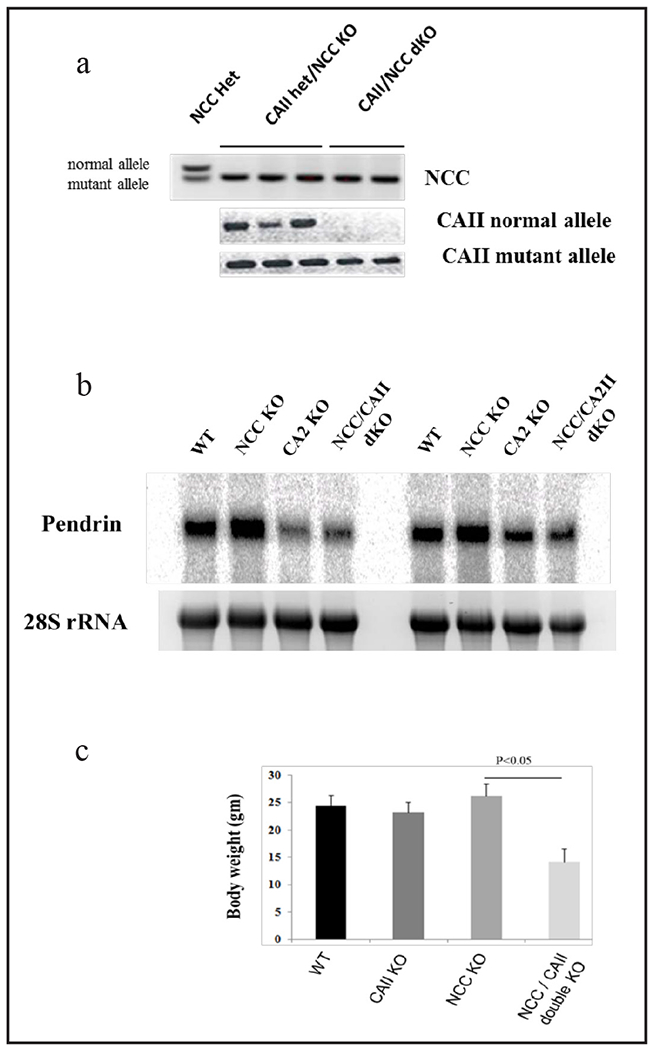
Generation and characterization of NCC/CAII dKO mice. a. Generation of NCC/CAII dKO mice. Generation of NCC/CAII dKO mice by crossing mice with single NCC or CAII deletions was confirmed by tail genotyping. b. Northern hybridization of pendrin in kidneys of NCC and CAII single KO and NCC/CAII dKO mice. c. Body weights in all genotypes. Results indicate that NCC/CAII dKO mice are significantly smaller than WT or NCC and CAII single KO mice (p<0.05, n=4 in each group).
NCC/CAII dKO mice have profound renal water and salt wasting
To assess the impact of single or double gene deletion on salt and water excretion, balanced studies were performed on all genotypes in metabolic cages. Figure 3 (a and b) shows the presence of polyuria (increased urine output) along with polydipsia (increased water intake) in NCC/CAII dKO mice. Water intake and urine output in NCC KO or CAII KO mice are depicted for comparison (Fig. 3a and b). As demonstrated, urine output and water intake are comparable in WT and NCC KO mice, moderately increased in CAII KO mice (p<0.05 for urine output, n=4) and significantly enhanced in NCC/CAII dKO mice (p<0.05, n=4).
Fig. 3.
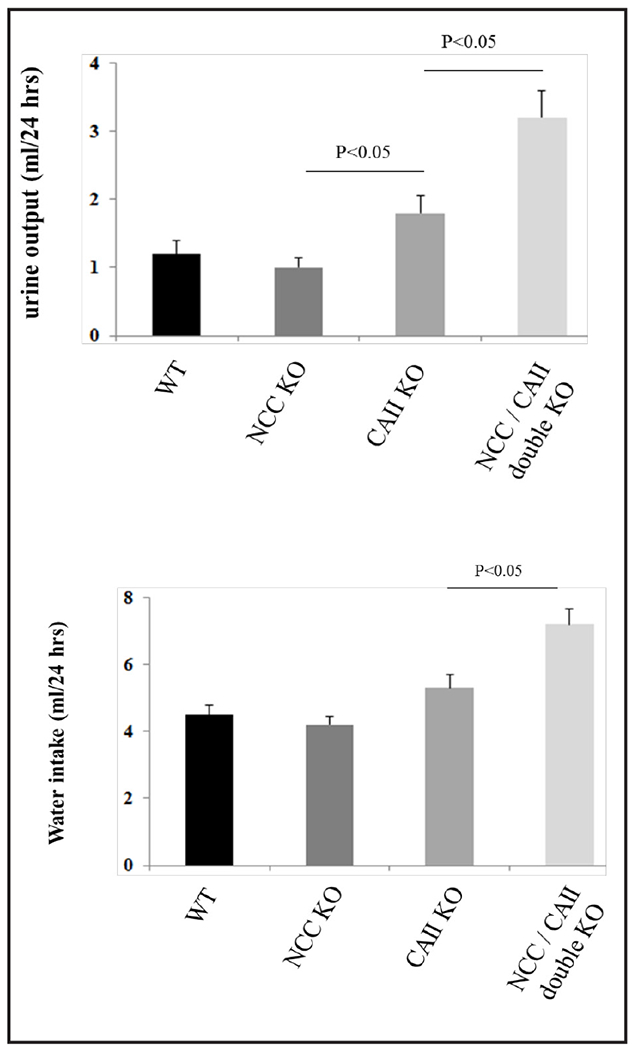
NCC/CAII dKO mice have increased urine output and water intake. a. NCC/CAII dKO mice exhibit polyuria (increased urine output); b. NCC/CAII dKO mice develop polydipsia (increased water intake).
Figure 4 (a and b) depicts the chloride and sodium excretion rates in WT and NCC/CAII dKO mice. As indicated, NCC/CAII dKO mice display significant chloride and sodium wasting vs. WT mice (p<0.04 and p<0.05, respectively, n=4).
Fig. 4.

NCC/CAII dKO mice demonstrate salt wasting. NCC/CAII dKO mice exhibit salt wasting as depicted by increased sodium (a) and chloride (b) excretion vs. WT mice.
NCC/CAII dKO mice have severe volume depletion but are unable to concentrate their urine
To determine whether excessive salt and water wasting (Figs. 3 and 4) cause volume depletion in dKO mice, the expression levels of renin in kidneys of WT and mutant mice were examined. As shown in Figure 5a, renin expression levels are profoundly increased in kidneys of dKO mice, whereas they remain comparable in kidneys of single KOs and WT mice. An analysis of renin expression levels in WT and dKO mice demonstrates ~600% increase in kidneys of dKO mice (Fig. 5a, right panel is a densitometric analysis, p<0.0001, n=4).
Fig. 5.
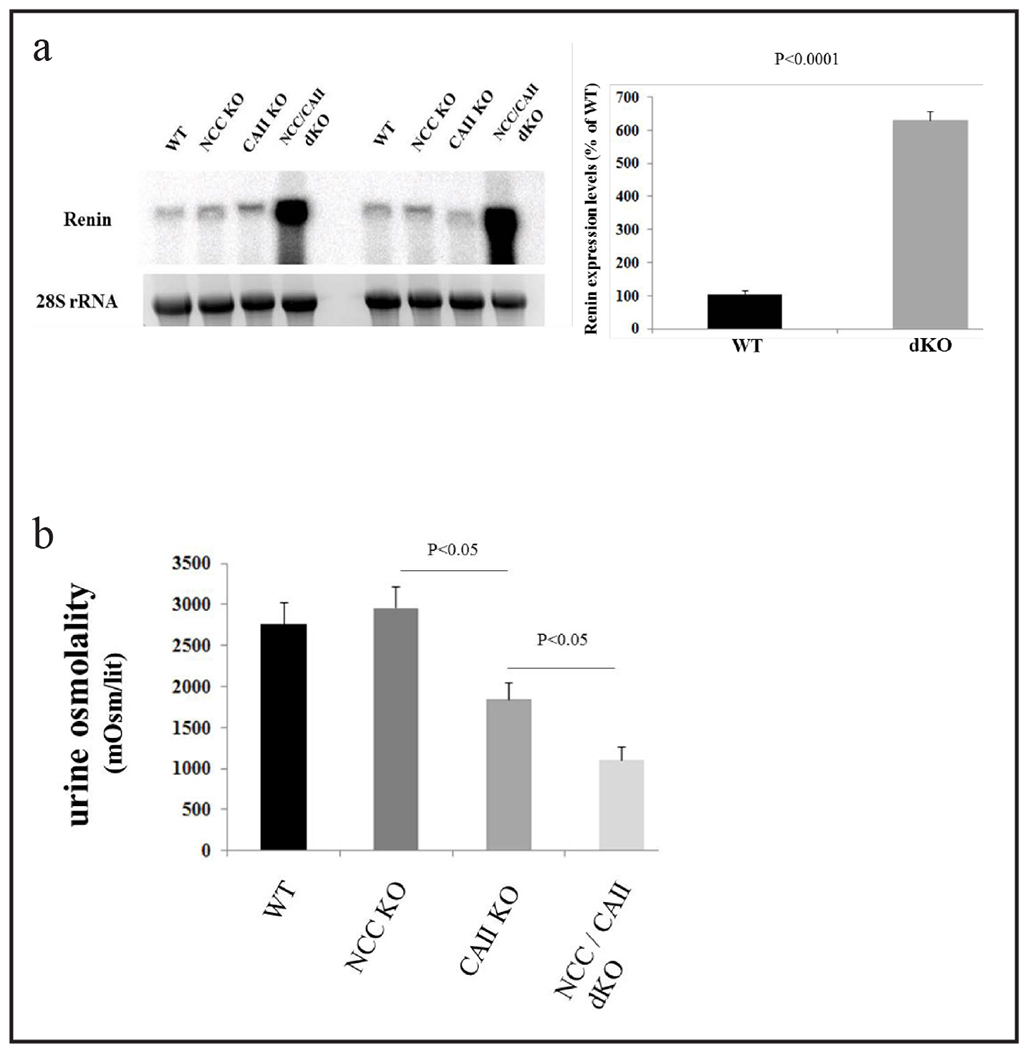
NCC/CAII dKO mice are volume depleted but have impaired urine concentrating ability. a. Northern hybridization of renin mRNA. Renin expression levels are profoundly increased in kidneys of NCC/CAII dKO mice, whereas they remain comparable in kidneys of NCC and CAII single KO and WT mice. b. NCC/CAII dKO mice have impaired urine concentrating ability. Urine osmolality is very low in NCC/CAII dKO mice vs. other genotypes.
The increase in urine output (Fig. 3) in the setting of volume depletion (Fig. 5a) suggests impaired urine concentrating ability in kidneys of NCC/CAII dKO mice. To examine this issue further, urine osmolality was measured in all genotypes. As demonstrated in Figure 5b, urine osmolality is reduced by ~70% in dKO mice vs. the other genotypes (p<0.05, n=4).
The low urine osmolality and increased urine output in the face of severe volume depletion suggest impaired water conservation by the kidney, which points to altered AQP2 trafficking. Figure 6a is a representative western blot and examines the expression levels of total AQP2 in microsomal proteins isolated from kidney medulla of all genotypes. As indicated, AQP2 abundance, when normalized for B-actin levels (Fig. 6a, bottom panel), is increased in kidneys of NCC KO, CAII KO and NCC/CAII dKO mice (p<0.05, n=4 for each group). Figure 6b (middle panel) is a representative western blot and examines the expression of membrane-targeted Ser256p-AQP2 in kidneys of various genotypes. As indicated, the abundance of Ser256p-AQP2 is increased in kidneys of NCC KO mice (p<0.05, n=4) but returns to normal levels in kidneys of CAII KO and NCC/CAII dKO mice (Fig. 6b, middle panel). The abundance of Ser261p-AQP2, which represents the intracellular/subapical/recycling pool, however, is significantly increased in kidneys of CAII KO and further increased in NCC/CAII dKO mice (Fig. 6b, top panel, p<0.05, n=4 for each group vs. wt). The loading equality for protein samples is shown by B-actin intensity. The protein levels for ENaC subunits (β and γ) were examined by western blot in WT, single KO and NCC/CAII dKO mice. As indicated, a representative blot in Fig. 6c shows that the abundance of both ENaC subunits, when normalized for B-actin levels, increased in NCC KO mice (p<0.05, n=4) but was not significantly different in NCC/CAII KO mice vs. WT mice (Fig. 6c). The expression of ENaC-β and ENaC-γ subunits increased by 80 and 150%, respectively in NCC KO mice (n=4, p<0.05). However, they increased by only 15 and 22%, respectively, in NCC/CAII dKO mice (n=4, p>0.05). The abundance of the Na+/H+ exchanger NHE3 did not change significantly in microsomal membranes isolated from kidney cortices of NCC/CAII dKO mice (data not shown).
Fig. 6.

Expression of salt and water transporters in NCC/CAII dKO mice. a. AQP2 abundance in kidneys of NCC/CAII dKO mice. Total AQP2 abundance is increased in kidneys of NCC KO, CAII KO and NCC/CAII dKO mice. b. Aberrant trafficking of AQP2 in kidneys of NCC/CAII dKO mice. NCC/CAII dKO mice show increased abundance of intracellular/recycling AQP2 (Ser261p-AQP2) and mildly reduced levels of apical membrane targeted AQP2 (Ser256p-AQp2). c. ENaC subunits (β and γ) abundance in WT, NCC and CAII single KO and NCC/CAII dKO mice.
Discussion
Pendrin, which was first identified by linkage analysis in patients with Pendred syndrome [31], is an anion exchanger which can function in Cl−/HCO3− exchange mode [6, 19]. It can also transport iodide [32]. Despite its role as a chloride absorbing transporter, there is little evidence of salt wasting in mice with the genetic deletion of pendrin and in humans with inactivating mutation of pendrin under baseline conditions. Similar to pendrin deficient mice, NCC null mice also do not demonstrate any noticeable salt wasting under baseline conditions. Both pendrin and NCC KO mice, however, show signs of volume depletion or develop hypotension in response to salt restriction [17, 18]. Both NCC and pendrin display adaptive regulation by excess aldosterone. These findings have led investigators to conclude that pendrin and NCC are predominantly active during salt depletion (and or in response to aldosterone), and their contribution to salt reabsorption under baseline conditions is minimal.
Pendrin expression increases significantly in kidneys of NCC KO mice [11]. This response parallels enhanced expression of the epithelial sodium channel (ENaC) in these animals [17]. Taken together, these results suggest that pendrin, working in tandem with ENaC, is activated and plays an important role in salt reabsorption in the distal nephron of NCC KO mice. In addition to ENaC, recent studies suggest that NDCBE can also collaborate with pendrin to reabsorb salt in the distal nephron [13]. Whether NDCBE activity is enhanced in kidneys of NCC KO mice remains unanswered at the present.
The most salient feature of the present studies is the profound fluid and electrolyte wasting in NCC/CAII dKO mice but not in NCC KO or CAII KO mice (Results). Given the upregulation of pendrin in kidneys of NCC KO mice [11] and its significant downregulation in kidneys of CAII null mice [24], these results strongly suggest that severe salt wasting in NCC/CAII dKO mice is mainly due to the prevention of upregulation of pendrin in the setting of NCC deficiency. The fluid and electrolyte loss resulted in severe volume depletion in NCC/CAII dKO mice, as verified by the profound upregulation of renin expression. This picture is completely distinct from that in mice with single deletion of NCC, which do not display any major electrolyte wasting under baseline conditions and display only mild (but statistically not significant) upregulation of renin expression in their kidneys (Results). Published reports indicate that CAII is a cytosolic enzyme expressed in almost all nephron segments except loop of Henle [33]. As for the salt reabsorption, there is no evidence that CAII deficiency causes any impairment in the proximal tubule or any other nephron segment. This is supported by normal renin expression and salt excretion in kidneys of CAII KO mice (Results).
The results of the present studies are in agreement with those in mice with double deletion of NCC and pendrin [20], which showed salt wasting, volume depletion and a sharp increase in BUN levels, consistent with pre-renal failure [20]. The volume depletion and kidney hypoperfusion improved significantly in response to salt repletion, strongly suggesting that salt wasting was the primary pathogenic event in NCC/pendrin dKO mice [20]. It is worth mentioning that CAII null mice have increased urine output (Fig. 3) but their salt excretion is not significantly different relative to WT or NCC KO mice.
In addition to enhanced expression of renin, the volume depletion resulting from salt wasting via kidney or gastrointestinal tract invariably stimulates arginine vasopressin (AVP) release from the pituitary gland [34]. Reports indicate that the water-absorbing channel in the kidney collecting duct (AQP2) is under the control of AVP and plays an essential role in water reabsorption in volume-depleted states [35–37]. It is known that AVP enhances the phosphorylation of AQP2 on serine residue 256, therefore increasing the surface expression and activity of AQP2 in collecting duct principal cells, resulting in urine concentration augmentation [38, 39]. It is known that the phosphorylation of serine 261, unlike the serine residue 256, results in the internalization and recycling of AQP2 in principal cells [38, 39]. Given the presence of severe volume depletion and elevated renin/AVP levels, the impaired urine concentrating ability in NCC/CAII dKO mice suggests defects in water absorption machinery in the collecting duct. Indeed, our studies support this conclusion by demonstrating enhanced expression of intracellularly-located, serine 261-phosphorylated AQP2 in the medullary collecting duct of NCC/CAII dKO mice compared to WT animals (Fig. 6). Our results provide a molecular basis for impaired urine concentrating ability in the volume-depleted, NCC/CAII dKO mice.
These are the first reports demonstrating aberrant AQP2 trafficking in kidneys of animals with salt wasting and severe volume depletion, despite the presence of elevated circulating AVP levels. We suggest that the increase in total AQP2 in kidneys of NCC/CAII dKO mice (Fig. 6a) is predominantly due to enhanced expression of Ser261p-AQP2, which is located intracellularly, and is therefore not involved with water reabsorption and urine concentration.
It is unlikely that altered AQP2 trafficking is only observed in NCC/CAII dKO and mice. Rather, we believe that any model of renal salt wasting that results in volume depletion can potentially cause aberrant AQP2 trafficking. It is important to note that volume depletion subsequent to gastrointestinal electrolyte loss or salt restriction does not impair urine concentration or cause abnormal AQP2 trafficking. In support of this statement, we find that Slc26a3 (DRA) null mice, which exhibit diarrhea subsequent to decreased absorption of salt in the intestine [40], develop volume depletion but are able to concentrate their urine appropriately and process AQP2 properly in the collecting duct (data not shown). Further, we find that mice with the double deletion of pendrin and NCC display aberrant trafficking of the AQP2 water channel in medullary collecting ducts in a manner very similar to that in NCC/CAII dKO mice (manuscript in preparation). Taken together, we propose that it is the salt wasting via kidney, which can potentially cause impaired trafficking of AQP2 and water conservation once volume depletion is ensued. While the signal(s) mediating the differential regulation of phosphorylated AQP2 isoforms in kidneys of NCC/CAII dKO mice is (are) not clear at the present, possibilities such as increased PGE2 (prostaglandin E2) and 20-HETE (hydroxyeicosatetraenoic Acid), which are known to antagonize the effect of AVP on the collecting duct should strongly be considered.
In conclusion, NCC/CAII dKO mice develop significant salt wasting subsequent to the prevention of upregulation of kidney pendrin in the setting of NCC deficiency. The salt wasting in NCC/CAII dKO mice is associated with impaired water conservation and aberrant AQP2 trafficking despite severe volume depletion. We propose that the targeted inhibition of NCC and pendrin provides a strong diuretic regimen for the treatment of fluid overload in patients with congestive heart failure, nephrotic syndrome, generalized edema, and chronic kidney disease. The diagram in Figure 7 depicts the salt and water wasting in NCC/CAII dKO mice. According to this scheme, while pendrin downregulation in CAII KO mice does not cause any significant salt wasting under basal condition due to compensatory upregulation of NCC (Fig. 7b) vs. WT mice (Fig. 7a), it causes significant salt wasting in the setting of NCC inactivation or inhibition (Fig. 7c).
Fig. 7.
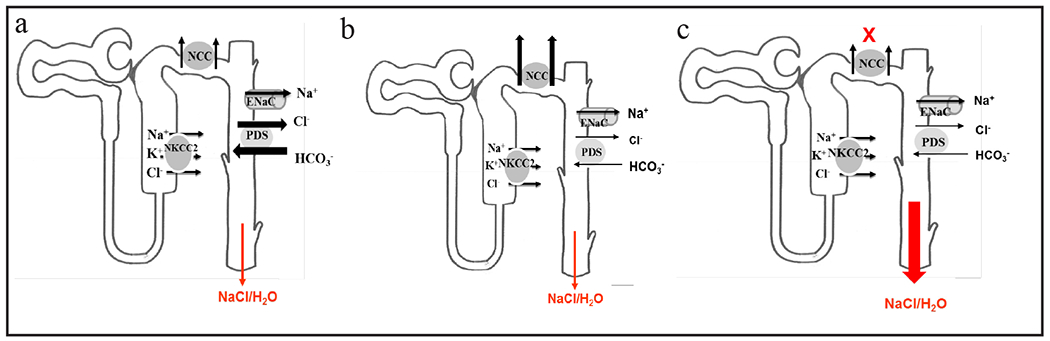
Schematic diagram depicting the synergic role of pendrin and NCC in salt and water absorption in the kidney distal nephron. The downregulation of pendrin in CAII null mice does not cause significant salt and water wasting due to compensatory activation of NCC (b) vs. WT mice (a). However, the downregulation of pendrin causes significant salt wasting in the setting of NCC inactivation (c). NCC downregulation per se does not cause significant salt wasting due to the compensatory upregulation of pendrin and the ENaC [20].
Acknowledgements
These studies were supported by a Merit Review award from the Department of Veterans Affairs, funds from the Center on Genetics of Transport and Epithelial Biology at the University of Cincinnati, an NIH award (R56DK62809) and by grants from Dialysis Clinic Inc. (to M.S).
References
- 1.Ellison DH: The thiazide-sensitive na-cl cotransporter and human disease: reemergence of an old player. J Am Soc Nephrol 2003;14:538–540. [DOI] [PubMed] [Google Scholar]
- 2.Câmpean V, Kricke J, Ellison D, Luft FC, Bachmann S: Localization of thiazide sensitive N+-Cl− cotransport and associated gene products in mouse DCT. Am J Physiol Renal Physiol 2001;281F1028-1035. [DOI] [PubMed] [Google Scholar]
- 3.Weinstein AM: A mathematical model of rat distal convoluted tubule. I. Cotransporter function in early DCT. Am J Physiol Renal Physiol 2005;289:F699–720. [DOI] [PubMed] [Google Scholar]
- 4.Eladari D, Chambrey R, Peti-Peterdi J: A new look at electrolyte transport in the distal tubule. Annu Rev Physiol 2012;74:325–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Glover M, Zuber AM, O’Shaughnessy KM: Hypertension, dietary salt intake, and the role of the thiazide-sensitive sodium chloride transporter NCCT. Cardiovasc Ther 2011;29:68–76. [DOI] [PubMed] [Google Scholar]
- 6.Soleimani M, Greeley T, Petrovic S, Wang Z, Amlal H, Kopp P, Burnham CE: Pendrin: an apical Cl−/HCO3− exchanger in the kidney cortex. Am J Physiol Renal Physiol 2001;280:F356–F364. [DOI] [PubMed] [Google Scholar]
- 7.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED: Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 2001;98:4221–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Verlander JW, Hassell KA, Royaux IE, Glapion DM, Wang ME, Everett LA, Green ED, Wall SM: Deoxycorticosterone upregulates PDS (Slc26a4) in mouse kidney: role of pendrin in mineralocorticoid-induced hypertension. Hypertension 2003;42:356–362. [DOI] [PubMed] [Google Scholar]
- 9.Petrovic S, Wang Z, Ma L, Soleimani M: Regulation of the apical Cl−/HCO3− exchanger pendrin in rat cortical collecting duct in metabolic acidosis. Am J Physiol Renal Physiol 2003;284:F103–112. [DOI] [PubMed] [Google Scholar]
- 10.Quentin F, Chambrey R, Trinh-Trang-Tan MM, Fysekidis M, Cambillau M, Paillard M, Aronson PS, Eladari D: The Cl−/HCO3− exchanger pendrin in the rat kidney is regulated in response to chronic alterations in chloride balance. Am J Physiol Renal Physiol 2004;287:F1179–1188. [DOI] [PubMed] [Google Scholar]
- 11.Vallet M, Picard N, Loffing-Cueni D, Fysekidis M, Bloch-Faure M, Deschênes G, Breton S, Meneton P, Loffing J, Aronson PS, Chambrey R, Eladari D: Pendrin regulation in mouse kidney primarily is chloride-dependent. J Am Soc Nephrol 2006;17:2153–2163. [DOI] [PubMed] [Google Scholar]
- 12.Sharma AK, Rigby AC, Alper SL: STAS domain structure and function. Cell Physiol Biochem 2011;28:407–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D: The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 2010;120:1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozansky DJ, Cornwall T, Subramanya AR, Rogers S, Yang YF, David LL, Zhu X, Yang CL, Ellison DH: Aldosterone mediates activation of the thiazide-sensitive Na-Cl cotransporter through an SGK1 and WNK4 signaling pathway. J Clin Invest 2009;119:2601–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang CL, Zhu X, Ellison DH: The thiazide-sensitive Na-Cl cotransporter is regulated by a WNK kinase signaling complex. J Clin Invest 2007;117:3403–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schultheis PJ, Lorenz JN, Meneton P, Nieman ML, Riddle TM, Flagella M, Duffy JJ, Doetschman T, Miller ML, Shull GE: Phenotype resembling Gitelman’s syndrome in mice lacking the apical Na+–Cl− cotransporter of the distal convoluted tubule. J Biol Chem 1998;273:29150–29155. [DOI] [PubMed] [Google Scholar]
- 17.Loffing J, Vallon V, Loffing-Cueni D, Aregger F, Richter K, Pietri L, Bloch-Faure M, Hoenderop JG, Shull GE, Meneton P, Kaissling B: Altered renal distal tubule structure and renal Na+ and Ca2+ handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol 2004;15:2276–2288. [DOI] [PubMed] [Google Scholar]
- 18.Wall SM, Kim YH, Stanley L, Glapion DM, Everett LA, Green ED, Verlander JW: NaCl restriction upregulates renal Slc26a4 through subcellular redistribution: role in Cl- conservation. Hypertension 2004;44:982–987. [DOI] [PubMed] [Google Scholar]
- 19.Amlal H, Petrovic S, Xu J, Wang Z, Sun X, Barone S, Soleimani M: Deletion of the anion exchanger Slc26a4 (pendrin) decreases apical Cl-/HCO3-exchanger activity and impairs bicarbonate secretion in kidney collecting duct. Am J Physiol Cell Physiol 2010;299:C33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Soleimani M, Barone S, Xu J, Shull GE, Siddiqui F, Zahedi K, Amlal H: Double Knockout of Pendrin and Na-Cl Cotransporter (NCC) Causes Severe Salt Wasting, Volume Depletion and Renal Failure. Proc Nat Acad Sci USA 2012;109:23368–23373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Purkerson JM, Schwartz GJ: The role of carbonic anhydrases in renal physiology. Kidney Int 2007;71:103–115. [DOI] [PubMed] [Google Scholar]
- 22.Nicoletta JA, Schwartz GJ: Distal renal tubular acidosis. Curr Opin Pediatr 2004;16:194–198. [DOI] [PubMed] [Google Scholar]
- 23.Breton S, Alper SL, Gluck SL, Sly WS, Barker JE, Brown D: Depletion of intercalated cells from collecting ducts of carbonic anhydrase II-deficient (CAR2 null) mice. Am J Physiol 1995;269:F761–74. [DOI] [PubMed] [Google Scholar]
- 24.Sun X, Soleimani M, Petrovic S: Decreased expression of Slc26a4 (Pendrin) and Slc26a7 in the kidneys of carbonic anhydrase II-deficient mice. Cell Physiol Biochem 2008;21:95–108. [DOI] [PubMed] [Google Scholar]
- 25.Brooks HL, Sorensen AM, Terris J, Schultheis PJ, Lorenz JN, Shull GE, Knepper MA: Profiling of renal tubule Na+ transporter abundances in NHE3 and NCC null mice using targeted proteomics. J Physiol 2001;530:359–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lewis SE, Erickson RP, Barnett LB, Venta PJ, Tashian RE: N-ethyl-N-nitrosourea-induced null mutation at the mouse Car-2 locus: an animal model for human carbonic anhydrase II deficiency syndrome. Proc Natl Acad Sci U S A 1988;85:1962–1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spicer SS, Lewis SE, Tashian RE, Schulte BA: Mice carrying a CAR-2 null allele lack carbonic anhydrase II immunohistochemically and show vascular calcification. Am J Pathol 1989;134:947–954. [PMC free article] [PubMed] [Google Scholar]
- 28.Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, Riederer B, Bonhagen J, Arend LJ, Amlal H, Seidler U, Soleimani M: Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem 2009;284:29470–29479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, Wang Z, Alper SL, Forte JG, Shull GE, Ehrenfeld J, Seidler U, Soleimani M: Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci U S A 2008;105;17955–17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu J, Barone S, Li H, Holiday S, Zahedi K, Soleimani M: Slc26a11, a chloride transporter, localizes with the vacuolar H+-ATPase of A-intercalated cells of the kidney. Kidney Int 2011;80:926–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED: Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat Genet 1997;17:411–422. [DOI] [PubMed] [Google Scholar]
- 32.Scott DA, Wang R, Kreman TM, Sheffield VC, Karniski LP: The Pendred syndrome gene encodes a chloride-iodide transport protein. Nat Genet 1999;21:440–443. [DOI] [PubMed] [Google Scholar]
- 33.Purkerson JM, Schwartz GJ: The role of carbonic anhydrases in renal physiology. Kidney Int 2007;71:103–115. [DOI] [PubMed] [Google Scholar]
- 34.Boone M, Deen PM: Physiology and pathophysiology of the vasopressin-regulated renal water reabsorption. Pflugers Arch 2008;456:1005–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Promeneur D, Kwon TH, Frøkiaer J, Knepper MA, Nielsen S: Vasopressin V(2)-receptor-dependent regulation of AQP2 expression in Brattleboro rats. Am J Physiol Renal Physiol 2000;279:F370–382. [DOI] [PubMed] [Google Scholar]
- 36.Knepper MA, Verbalis JG, Nielsen S: Role of aquaporins in water balance disorders. Curr Opin Nephrol Hypertens 1997;6:367–371. [DOI] [PubMed] [Google Scholar]
- 37.Inoue T, Terris J, Ecelbarger CA, Chou CL, Nielsen S, Knepper MA: Vasopressin regulates apical targeting of aquaporin-2 but not of UT1 urea transporter in renal collecting duct. Am J Physiol 1999;276:F559–566. [DOI] [PubMed] [Google Scholar]
- 38.Moeller HB, MacAulay N, Knepper MA, Fenton RA: Role of multiple phosphorylation sites in the COOH-terminal tail of aquaporin-2 for water transport: evidence against channel gating. Am J Physiol Renal Physiol 2009;296:F649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xie L, Hoffert JD, Chou CL, Yu MJ, Pisitkun T, Knepper MA, Fenton RA: Quantitative analysis of aquaporin-2 phosphorylation. Am J Physiol Renal Physiol 2010;298:F1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schweinfest CW, Spyropoulos DD, Henderson KW, Kim JH, Chapman JM, Barone S, Worrell RT, Wang Z, Soleimani M: Slc26a3 (dra)-deficient mice display chloride-losing diarrhea, enhanced colonic proliferation, and distinct up-regulation of ion transporters in the colon. J Biol Chem 2006;281:37962–37971. [DOI] [PubMed] [Google Scholar]