

Abstract
Background:
DNA methylation causes silencing of tumor suppressor and differentiation-associated genes, being linked to chemoresistance. Previous studies demonstrated that hypomethylating agents (HMA) re-sensitize ovarian cancer (OC) to chemotherapy. NTX-301 is a highly potent and orally bioavailable HMA, in early clinical development.
Methods:
The anti-tumor effects of NTX-301 were studied in OC models by using cell viability, stemness and ferroptosis assays, RNA sequencing, lipidomic analyses and stimulated Raman spectroscopy.
Results:
OC cells (SKOV3, IC50=5.08nM; OVCAR5 IC50=3.66nM) were highly sensitive to NTX-301 compared to fallopian tube epithelial cells. NTX-301 downregulated expression of DNA methyltransferases (DNMTs)1–3 and induced transcriptomic reprogramming with 15,000 differentially expressed genes (DEGs, p<0.05). Among them, Gene Ontology enrichment analysis identified regulation of fatty acid biosynthesis and molecular functions related to aldehyde dehydrogenase (ALDH) and oxidoreductase, known features of cancer stem cells. Low dose NTX-301 reduced the ALDH(+) cell population and expression of stemness associated transcription factors. Stearoyl-Coenzyme A desaturase 1 (SCD), which regulates production of unsaturated fatty acids (UFAs), was among the top DEGs downregulated by NTX-301. NTX-301 treatment decreased levels of UFAs and increased oxidized lipids and this was blunted by deferoxamine, indicating cell death via ferroptosis. NTX-301 induced ferroptosis was rescued by oleic acid. In vivo, monotherapy with NTX-301 significantly inhibited OC and patient derived xenograft (PDX) growth (p<0.05). Decreased SCD levels and increased oxidized lipids were detected in NTX-301-treated xenografts.
Conclusions:
NTX-301 is active in OC models. Our findings point to a new mechanism by which epigenetic blockade disrupts lipid homeostasis and promotes cancer cell death.
Introduction:
Targeting genomic drivers in cancer has focused mainly on “drugging” mutated, amplified, or overexpressed oncogenes. Less success has been achieved attempting to target tumor suppressor genes (TSG), which are also critical to cancer initiation and progression. TSGs are lost in cancer through mutation, loss of heterozygosity, or epigenetic mechanisms. The latter are key regulators of transcription, have been implicated in silencing of TSGs and other genes, and include DNA methylation, post-transcriptional histone modifications and chromatin organization. Epigenetic modifications have been shown to act as inactivating “hits” during cancer initiation and/or progression, similar to genetic events (1). Given that the epigenome is involved in regulating a vast transcriptional program and is modifiable by druggable enzymes, such as DNA methyl transferases (DNMT) or histone modifiers, there has been considerable interest in developing and testing epigenome-targeting drugs as cancer agents. While early success was registered in hematological malignancies where several epigenome-directed drugs have been approved (2), solid tumors have proven to be more difficult to target (3, 4).
We and others have tested DNMT inhibitors in preclinical models of ovarian, lung, and gastrointestinal malignancies and showed that global hypomethylation induced by such agents leads to significant transcriptomic reprogramming and exerts potent anti-tumorigenic effects (5, 6). These findings were eventually translated into bedside interventions with several early-stage clinical trials completed and providing evidence of benefit in selected patients (4, 7–10). DNMT inhibitors are nucleoside analogues which after phosphorylation, are incorporated into DNA in place of cytosine during DNA replication. The DNMTs form a stable covalent bond with these faulty nucleosides and are depleted from the cellular/nuclear pool, leading to reduced ability to deposit new methyl groups on CpG sites in newly synthesized DNA (11). Some of the challenges encountered in clinical studies testing DNMT inhibitors in solid tumors included the short half-life (minutes) of these agents which ultimately affected incorporation of the nucleosides in tumor cell DNA, the poor intra-tumoral distribution of the drugs during the brief period of time they are active, and the low therapeutic index, leading to toxicity to normal tissue, in particular myelotoxicity (10, 12) that prevented opportunities for rational combinations with other cytotoxic agents. Continuing efforts to bring epigenome targeting to solid tumors, here we describe the effects of novel hypomethylating agent (HMA) 5-aza-4′-thio-2′-deoxycytidine (NTX-301), in preclinical models of ovarian cancer (OC).
NTX-301 is a derivative of deoxycytidine with a 4-thio modification that increases its chemical stability, rate of incorporation into DNA, and selectivity index compared to traditional HMAs, 5-azacitadine and decitabine (13). NTX-301 was recently shown to be highly active in acute myelogenous leukemia models (AML) including in high risk p53 mutant AML (14) and is bioavailable when administered orally, rendering it attractive for future clinical investigations.
OC is uniquely suited for epigenome targeting as a disease driven by loss of TSGs, rather than oncogenes. Loss of TP53 through mutation is a pathognomonic feature of high grade serous (HGS) tumors. Aside from mutations, epigenetic silencing through methylation of cytosine residues within cytosine-guanine (CpG) islands has been shown to repress expression of other TSGs in HGSOC, such as BRCA1 (15), p16 (16), PALB2, MLH1, RASSF1A, OPCML and others (15). Methylation profiles of primary and recurrent HGS tumors versus normal ovarian surface epithelial cells revealed thousands of differentially methylated CpGs and expression of DNMTs has been shown to correlate with progression free survival (17). DNA methylation and histone modifications have also been associated with emergence of chemo-resistance in OC (17, 18) and with modulation of anti-tumor immunity via silencing of tumor antigens and immunogenic endogenous retroviruses (19). As such, HMAs have been tested by us and others in women with recurrent OC as re-sensitizers to carboplatin (4) or as priming agents for immunotherapy (20, 21).
With promising, but not definitive results in the clinical arena for epigenome targeting drugs, there remains interest in exploring novel HMAs, which have increased potency, improved bioavailability and toxicity profile and are more suitable for combination treatments. Here we report that the 4-thio-decitabine derivative NTX-301 effectively depletes DNMT1, 3A and 3B, induces global hypomethylation and transcriptional reprogramming leading to inhibition of HGSOC cell proliferation and tumor growth in vivo. Mechanistically, among other gene sets, NTX-301 significantly altered the fatty acid metabolism pathway. A key enzyme in this pathway, the stearoyl co-A desaturase (SCD) which regulates the last step in lipogenesis, converting saturated fatty acids (SFAs) into unsaturated fatty acids (UFAs) was potently inhibited by NTX-301. This led to depletion of UFAs in cells and tumors treated with the HMA and cell death through ferroptosis. Our findings demonstrate a new mechanism by which DNMT targeting attacks cancer cells, linking the epigenome to regulation of lipid metabolism, which is a key metabolic dependency for HGSOC cells and tumors.
Materials and Methods:
Human specimens.
Deidentified ovarian tumors were collected and processed fresh from patients who provided “written informed consent for tissue banking”. This study was approved by Northwestern University IRB#: STU00202468) and follows the ethical principles promoted by the Declaration of Helsinki. Human tumor tissues and OC xenografts were enzymatically disassociated into single cell suspensions and cultured as previously described (6, 8). Tumor characteristics are listed in Supplemental Table S1.
Cell culture.
OVCAR5 cells were a generous gift from Dr. Marcus Peter, Northwestern University; OVCAR4 cells were from Dr. Mazhar Adli, Northwestern University; immortalized human fallopian tube luminal epithelial cells (FT190) were from Dr. R. Drapkin of University of Pennsylvania (22); COV362 cells were from Dr. Kenneth Nephew, Indiana University; NoEM cells were from Dr. S. Bulun, Northwestern University. SKOV3 cells were purchased from the American Type Culture Collection (ATCC). Cells were maintained in a 37°C incubator with 5% CO2. The media for culturing the cell lines are described in Supplemental Material. Cells lines used in the experiments were at low-passages and were tested negative for mycoplasma-free by Charles River Animal Diagnostic Services or by using Universal Mycoplasma Detection Kit (ATCC # 30–1012). Cell lines were authenticated by IDEXX BioAnalytics with short tandem repeat (STR) profiling.
Chemicals and reagents.
NTX-301 was provided by PinotBio. Carboplatin (Cat# C2538), oleic acid (Cat# O3008), dimethyl sulphoxide (DMSO, Cat# D2650), were from Sigma-Aldrich.
Cell viability, clonogenic, and spheroid formation assays
were performed as previously described (6, 23) (see Supplemental Material).
Aldefluor assay and flow cytometry.
Aldehyde dehydrogenase (ALDH) activity was measured by flow cytometry using an Aldefluor assay kit (Stemcell Technologies, Cat# 01700, Cambridge, MA, USA) following the manufacturer’s instructions and as described previously (23).
NTX-301 half maximal inhibitory concentration (IC50).
Five thousand OC cells, or OVCAR5 cells transfected with shRNA targeting SCD (shSCD) or shctrl vectors per well were seeded in 96-well plates and treated with different concentrations of NTX-301. NTX-301 was refreshed every day for 4 days and cell viability was measured with a CCK8 assay on day 5. IC50 values were calculated using GraphPad Prism (RRID: SCR_002798) software as described previously (23).
In vivo experiments.
Animal studies were conducted according to a protocol (# IS00017143) approved by the Institutional Animal Care and Use Committee (IACUC) of Northwestern University. 100,000 OVCAR5 cells were injected SQ into the right flank of female, 6–8 weeks old, athymic nude mice (Foxn1nu, Envigo). When tumors developed to measurable size (100mm3), mice were randomly allocated to the experimental treatments and then treated intraperitoneally (i.p.) with 100ul diluent (vehicle) or NTX-301 (0.5mg/kg or 1mg/kg, 5-day/week for 3 weeks). Patient Derived Xenografts (PDX) studies were approved by IACUC (protocol #IS00007992). Detailed protocols for evaluating the effects of NTX-301 on preventing OC tumor recurrence after chemotherapy in vivo, and on targeting OC PDX tumors are described in Supplemental Material.
Large-area hyperspectral SRS imaging.
Tissue blocks were cryopreserved in OCT (Optimal Cutting Temperature) at −80°C, sliced using a Leica CM1950 cryostat into 10 μm thick tissue layers, which were placed on glass slides and kept frozen until imaging. Multiplex Stimulated Raman Scattering (SRS) was performed by a femtosecond laser with two synchronized outputs beams (Insight DeepSee, Spectra-Physics, Santa Clara, CA) to measure fatty acid content, as previously described (24) (see Supplemental Material).
RNA extraction and quantitative RT-PCR analysis.
Total RNA was isolated with Trizol reagent (Invitrogen, Carlsbad, CA) and quantitative PCR was performed by using the iTaq Universal SYBR Green Supermix (Bio-Rad, Berkeley, California) on a 7900HT real-time PCR instrument (Applied Biosystems, Foster City, CA, see Supplemental Material). Primer sequences (Integrated DNA Technologies, USA) are in Suppl. Table S2.
ChIP-PCR.
ChIP was performed with anti-H3 (Active Motif, Cat# 39763), and anti-H3K27Ac (Abcam, Cat#4729), and mouse IgG antibodies (Santa Cruz, sc-2025), as previously described (see SM). Primer sequences are listed in Suppl. Table S3.
Lipidomics.
OVCAR-5 cells treated with DMSO or NTX-301 (100nM, 2-Day) were used for lipidomics analysis at the Bindley Bio-science Center, Purdue University, as described previously (25) (see SM). Statistical analysis was performed utilizing MetaboAnalystR 3.0 (12).
Cell transduction and transfection.
OVCAR5 cells were transduced with a lentiviral vector as described previously (25). The shRNAs targeted SCD (MilliporeSigma Cat#: SHCLNV) or were non-targeting (shctrl, MilliporeSigma Cat#: SHC001V). Selection of transfected cells with puromycin (2 μg/ml) (Gibco Cat#: A1113803) started 48 h after transduction and continued for 2 weeks. Gene knockdown was verified by western blot.
BODIPY staining for lipid peroxidation.
Intracellular lipid peroxidation was determined by using the lipid peroxidation sensor BODIPY 581/591 C11 (Thermo Fisher Scientific, Cat# D3861). as previously described (see Supplemental Material). Data were analyzed using FlowJo (RRID:SCR_008520) software.
Apoptosis Assay.
Cell apoptosis was analyzed using Annexin V staining (Thermal Fisher Scientific, USA, Cat# V13245, see Supplemental Material).
RNA sequencing (RNA-seq) and data analysis.
Total RNA was extracted with TRI Reagent (Sigma, Cat# T9424) and processed as previously described (25) (see Supplemental Material). The log2(fold-change) and P- value of total normalized counts and DEG counts were then analyzed by GSEA, Ingenuity Pathway Analysis (IPA) (RRID:SCR_008653) (QIAGEN). Data are deposited in the NCBI gene expression omnibus (GEO: GSE236985).
Genome-wide methylation analysis:
Total DNA was extracted using QIAmp DNA Mini Kit (Cat# 51304) according to the manufacturer’s instructions. Genome-wide methylation analysis were performed using the Epic array (Illumina; Northwestern University Sequencing Core). EPIC array data were processed and filtered with the following specifications. Probes with a >= 0.10 proportion of samples with an NA value were removed. The detection p-value threshold used was 0.01. Non-cg probes, probes aligning to multiple regions, and those in which the probed CpG falls in a known SNP region were removed. Missing beta values were imputed using ChAMP, using the K-nearest neighbors’ method (KNN) with k = 3 neighbors. Finally, beta values for the remaining 749,282 probes were normalized with BMIQ. Differentially methylated probes (DMPs) were identified using limma, which utilizes a linear model with empirical Bayes procedure to compute pairwise comparisons between the control and treatment groups. Data are deposited in the NCBI gene expression omnibus (GEO: GSE236984) (RRID:SCR_005012).
Western Blotting and immunohistochemistry (IHC)
were performed as previously described (see Supplemental Material). Primary antibody information is listed in the Suppl. Table S4.
Statistical Analysis.
Statistical analysis used the two-tailed Student’s t-test for two group comparisons or ANOVA (one-way/two-way) for >2 group comparisons, by using the GraphPad Prism software (RRID:SCR_002798). P values < 0.05 were considered significant.
Data Availability.
Sequencing and methylation data are deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (RRID:SCR_005012) data repository: GSE236985 and GSE236984. The analysis was performed by using publicly available software described in methods.
Results
OC cells are highly sensitive to NTX-301 treatment.
Cell viability assays determined the sensitivity and half maximum inhibitory concentration (IC50) to NTX-301 on a panel of OC cells, immortalized fallopian tube epithelial cells (FT-190) and normal endometrial stromal cells (NoEM) (26). Most OC cells (OVCAR5, IC50=3.66nM; OVCAR4, IC50=29.69nM; SKOV3, IC50=5.08nM) were highly sensitive to NTX-301 (p<0.05) compared to FT-190 (IC50=103.3nM) and NoEM cells (IC50=4.14mM) (Fig. 1A–C, Fig. S1A), but some showed similar IC50 (COV362, IC50=137nM) (Fig. S1B). Primary OC-derived cells also exhibited high sensitivity to NTX-301 (IC50~35nM) (Fig. 1B–C, Table S1). Intriguingly, low dose NTX-301 (10–50nM) significantly reduced the number of colonies formed by cell lines (OVCAR5 and OVCAR4)) and primary OC cells (P<0.05) (Fig. 1D–E; Fig. S1C–D). Next, to examine the effects of NTX-301 on the substrate enzymes (DNMTs), we treated OC cells with NTX-301 and the first generation HMA, decitabine (Fig. 1F–G; Fig. S1E–F). NTX-301 (100nM) significantly inhibited the expression of DNMTs (1–3), especially DNMT1 in OC cells (OVCAR5, Fig. 1F; SKOV3, Fig. 1G, OVCAR4 and COV362, Fig. S1E–F), to a higher degree compared with decitabine. Importantly, NTX-301 effectively inhibited DNMTs expression in primary OC-derived cells (Fig. 1H), confirming that it is a highly potent DNMT inhibitor.
Figure 1. OC cells are highly sensitive to NTX-301 treatment.
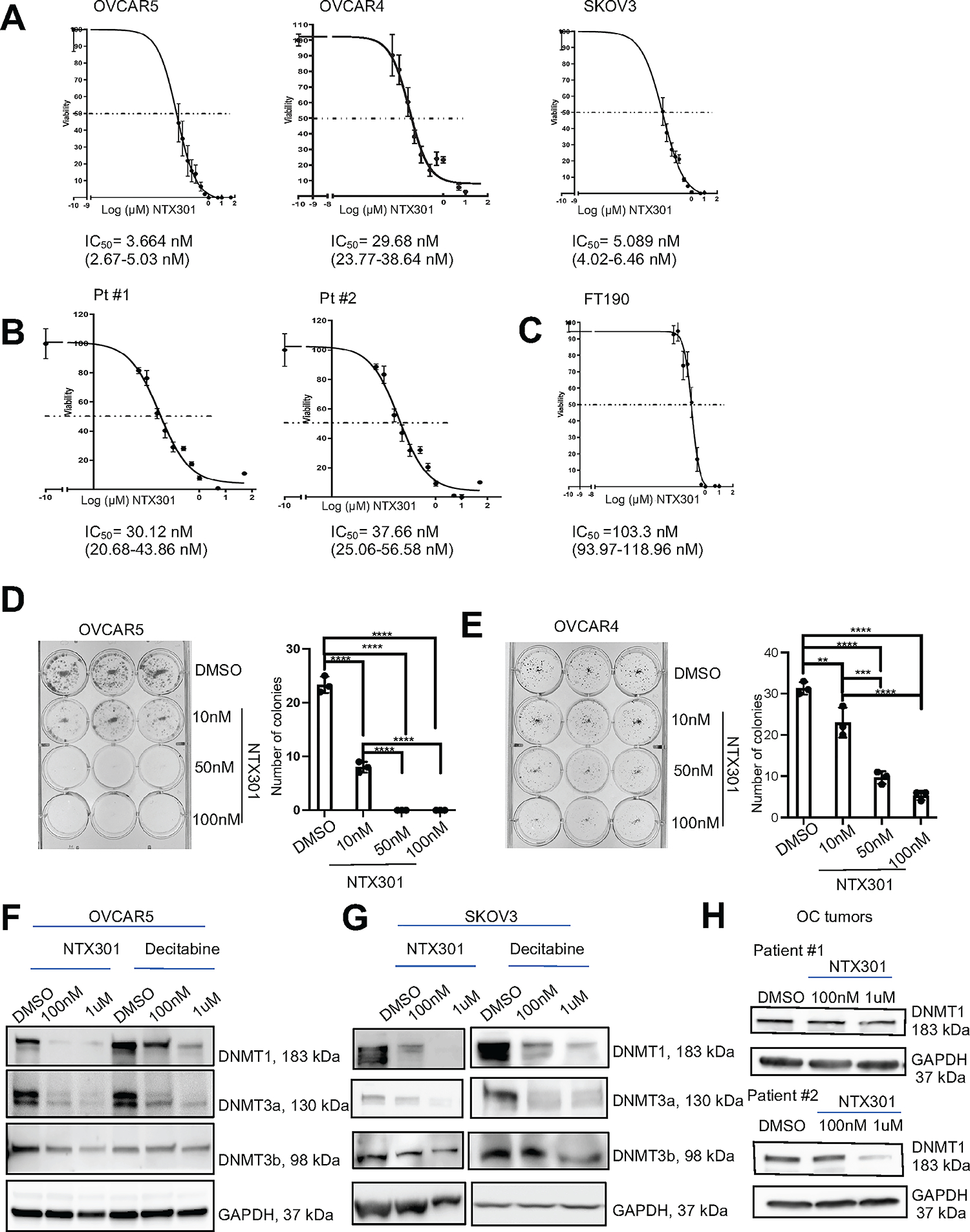
(A-C) Effects of NTX-301 on cell viability and estimation of IC50 values in OVCAR5, OVCAR4 and SKOV3 cells (A), OC cells derived from HGSOC tumors (B) and immortalized fallopian tube epithelial cells (FT-190) (C). Cells were treated daily with NTX-301 for 4 days, and cell viability was measured (CCK8 assay) on day 5. (D, E) Colony formation assay (left), and numbers of colonies (mean ± SD, n=3) (right) developed from 1,000 OVCAR5 (D) or OVCAR4 (E) cells treated with different doses of NTX-301 for 7 days. (F, G) Western blot of DNMT1, DNMT3a, DNMT3b, and GAPDH (loading control) in OVCAR5 (F) and SKOV3 (G) cells treated with DMSO, NTX-301(100nM, or 1uM), or decitabine (100nM or 1uM) for 48 hours (n=2 replicates). (H) Western blot of DNMT1 and GAPDH (loading control) in primary OC cells isolated from human HGSOC tumors and treated with DMSO or NTX-301 (100nM or 1uM) for 48 hours (n=2). For all comparisons: *p<0.05, **p<0.01, ***p<0.001.
NTX-301 induced metabolic reprogramming in OC cells.
To explore the molecular mechanism by which NTX-301 inhibited OC cells, RNA sequencing was performed in OVCAR5 cells treated with low dose NTX-301 (10nM and 100nM respectively, 72 hours) vs DMSO. Treatment with low dose NTX-301 reset the transcriptome of OC cells in a dose dependent manner (Fig. 2A). Among NTX-301-induced 11,500 differentially expressed genes (DEGs) (100nM, adj. P<0.05), 5861 genes were significantly upregulated and 5595 were down-regulated, compared to DMSO (P<0.05) (Fig. 2A–B, Tables S5 –S6). Among them, several TSGs were significantly upregulated, including NKX2–5, BCL6B, MKRN3, PDCD1, CDH13, the TP53 targets BTG4 and PALM3, and others (Table S7). Gene Set Enrichment Analysis (GSEA) indicated that NTX-301 induced changes in OC transcriptomes towards pathways (Fig. 2C) related to metabolic reprogramming, including Fatty acid metabolism (Fig. 2D), Pyruvate metabolism, Propanoate metabolism, Ascorbate and aldarate metabolism, and other cancer related pathways, such as Cell cycle, transcriptional misregulation in cancer, JAK-STAT signaling, etc. (Fig. S2A). In addition, Gene Ontology (GO) analysis classified upregulated DEGs in pathways related to DNA repair, such as Fanconi anemia pathway and p53 signaling pathways (Fig. S2A); down-regulated DEGs in molecular functions related to aldehyde dehydrogenase (ALDH) and oxidoreductase activity (Fig. S2B), known features of cancer stem cells (CSCs). WikiPathways classified NTX-301 down-regulated DEGs in pathways related to metabolism, including cholesterol metabolism and fatty acid biosynthesis (Fig. 2E), suggesting a potential role of this HMA in promoting metabolic reprogramming in OC cells. We further examined gene sets related to “stemness” and lipid metabolism in NTX-301 treated OC cells. Clear differences including downregulation of ALDH1 isoforms and key lipid metabolism genes were observed in NTX-301 vs. DMSO treated OC cells (Fig. 2B, 2F–G), supporting that NTX-301 inhibits stemness features and lipid metabolism in this context.
Figure 2. NTX-301 induced transcriptome and methylome reprogramming in OC cells.
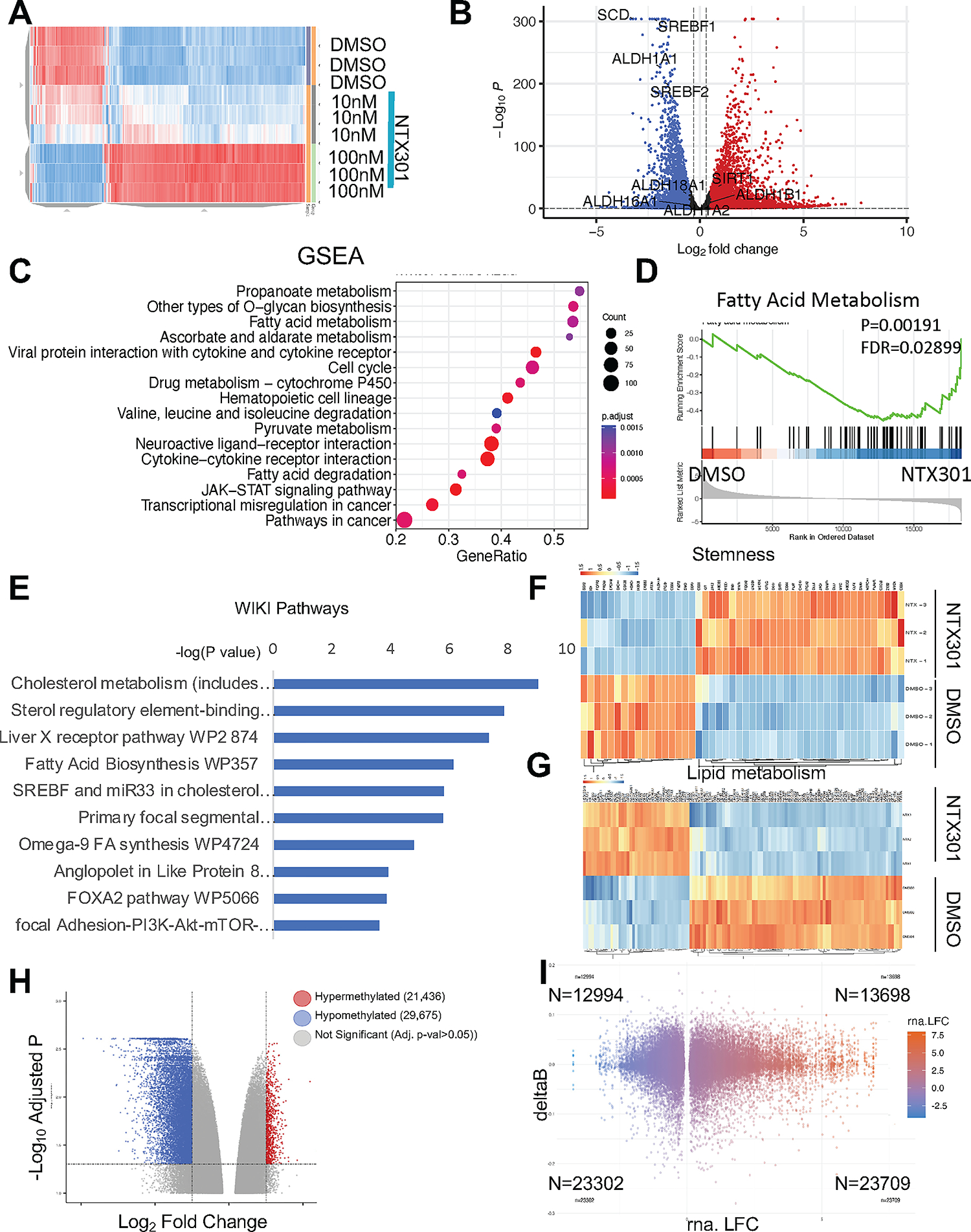
(A) Hierarchical clustering heatmap for DEGs (FDR<0.05) measured by RNAseq in OVCAR5 cells treated with NTX-301. Rows represent replicates (n=3/group). (B) Volcano plot of DEGs comparing OVCAR5 cells treated with NTX-301 100nM with DMSO. NTX-301-induced upregulated genes are shown in red and downregulated gene are shown in blue. (C) Dot plot of Gene Set Enrichment Analysis (GSEA) of DEG shows the top sixteen KEGG pathways in OVCAR5 cells treated with NTX-301 (100nM). The size of the circles represents the counts of DEGs within each term, and the color of the circles represents statistical significance. Gene ratio (x axis) is the relative number of DEGs per term. (D) GSEA enrichment plots for the KEGG “fatty acid metabolism” gene set using the gene expression profiles of OVCAR5 cells treated with NTX-301 (100nM) vs. DMSO (n=3). (E) WikiPathways analysis of downregulated DEGs in OVCAR5 OC cells treated with NTX-301 (100nM) vs. DMSO shows the top significantly enriched molecular pathways. Gene expression was measured by RNA-seq (n=3 replicates per group). (F, G) Hierarchical clustering heatmap for DEGs (FDR<0.05) stemness-related genes (F) (23) and lipid metabolism (G) in OVCAR5 cells treated with NTX-301 (100nM) vs. DMSO. Rows represent replicates (n=3/group). (H) Volcano plot of DMPs comparing OVCAR5 cells treated with NTX-301 vs control. Hypermethylated CpG probes are shown in red (positive delta Beta) and hypomethylated CpG probes are shown in blue (negative delta Beta). (I) Scatter plot shows overlapping DEGs associated with promoter-associated DMPs in response to treatment with NTX-301 (100nM, 3days; n = 3 replicates per group). A total of 23,709 genes were both upregulated and were associated with hypomethylated CpG sites (lower right quadrant), =whereas 23,302 genes were downregulated and were associated with hypomethylated CpGs (lower left quadrant).
To determine whether NTX-301 induced transcriptome changes are due to its hypomethylating effects, genome wide DNA methylation was profiled by using the Illumina Infinium Epic array. Compared to DMSO treated cells, NTX-301 induced global DNA methylation changes (Fig. 2H), 51,111 probes being differentially methylated between NTX-301 treated and the DMSO group (adjusted p-value < 0.05), with 29,675 of those probes being hypomethylated (delta beta value < 0) (Fig. S2C). The top differentially methylated probes (DMPs) are included in Supplemental Table S8 and S9. NTX-301 mediated DNA CpG Island demethylation was widely distributed, with ~ 15% events occurring in regulatory regions (TSS, 3’ UTR), relevant to transcription regulation (Fig. S2C). Integration of NTX-301 induced DNA methylome and transcriptome changes, revealed that the majority of hypomethylated genes were upregulated, including TSGs (Table S7), supporting that NTX-301 reversed the repressed expression of critical genes regulating stemness, cell survival, DNA repair and chemoresistance (Fig. 2I, Table S7).
NTX-301 inhibits ALDH+ ovarian CSC population by targeting ALDH and oxidoreductase activity.
Based on observations that NTX-301 inhibited aldehyde dehydrogenases involved in oxidoreductase activity, we examined the impact of NTX-301 on stemness features in vitro and in vivo. Treatment of OVCAR5 and SKOV3 cells with low doses of NTX-301 reduced the ALDH(+) population (Fig. 3A, B) and inhibited cell renewal ability (spheroid formation assay, Fig. 3C, D) in a dose-dependent manner. Further, low dose NTX-301 (10nM) suppressed expression of stemness associated transcription factor (TFs) SOX2 (Fig. 3E–G) and stemness related gene, ALDH1A1 (Fig. 3E) in SKOV3 and OVCAR5 cells. As a control, DMNT1 expression was potently inhibited by NTX-301 (Fig. 3E).
Figure 3. NTX-301 inhibits ALDH+ ovarian CSC population.
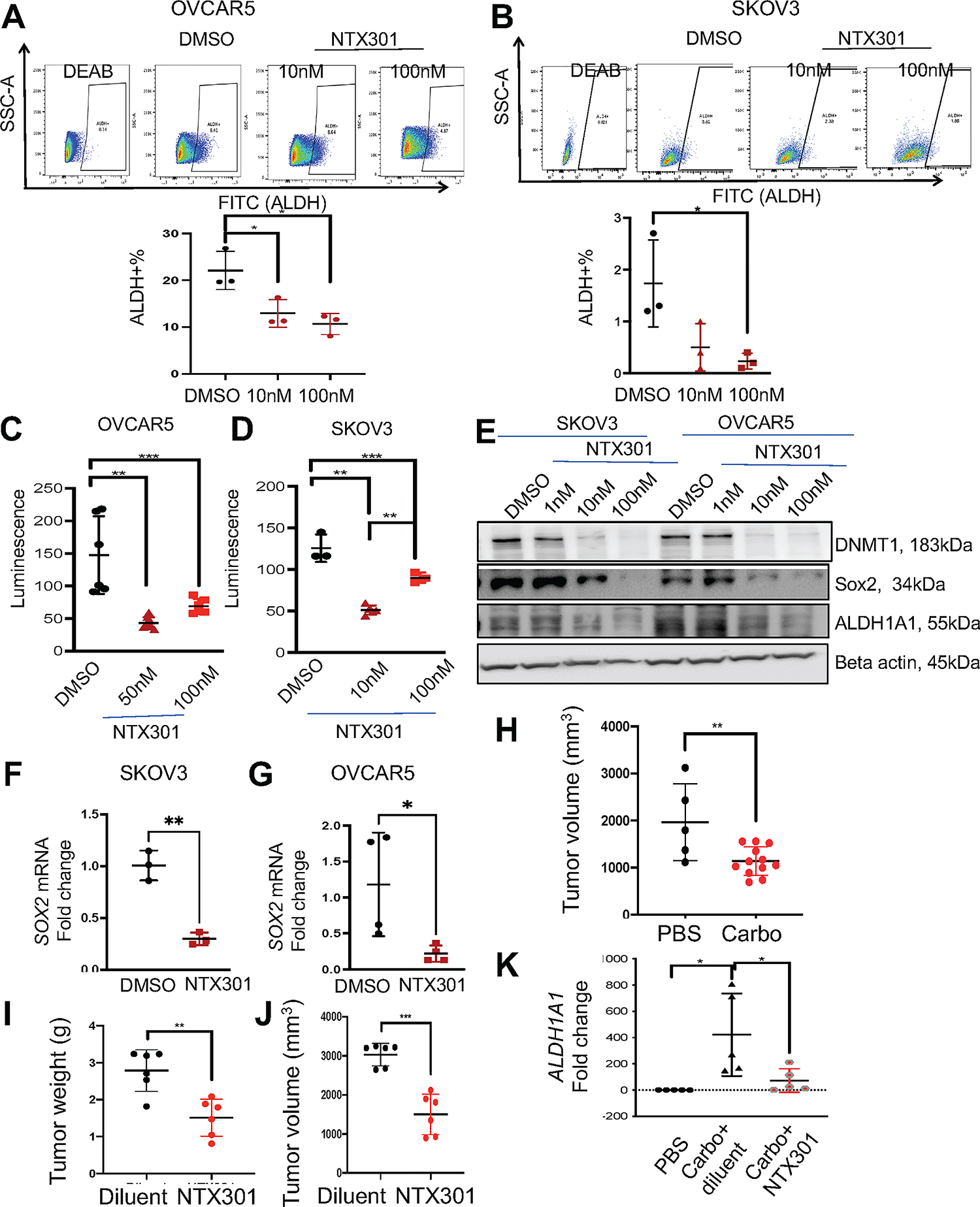
(A, B) Representative results of FACS side scatter analysis (top), and percentage (mean ± SD, n=3) (bottom) of ALDH(+) cells in OVCAR5 (A) and SKOV3 (B) OC cells treated with DMSO or NTX-301 (10nM and 100nM, 3 days). (C, D) Effects of NTX-301 (10nM and 100nM, 3days) on spheroid formation assessed by measuring cell viability in OVCAR5 (C) and SKOV3 (D) cells (n = 4–6 per dose). (E) Western blot measured protein levels of DNMT1, Sox2, ALDH1A1, and beta actin (loading control) in SKOV3 and OVCAR5 cells treated with DMSO or NTX-301 for 3 days (n=2). (F, G) mRNA expression levels of SOX2 measured by qRT-PCR (n=3–4) in SKOV3 (F) and OVCAR5 (G) cells treated with DMSO or NTX-301 (100nM, 3days). (H-K) Tumor bearing SQ xenografts were treated with PBS or carboplatin (40mg/kg weekly, 3-weeks) before being randomly assigned to receive NTX-301 (1.0 mg/kg) or diluent. Effects of carboplatin on tumor volume (H) and NTX-301 on tumor weight (I) and tumor volume (J) (n=6/group). (K) mRNA expression levels of ALDH1A1 measured by qRT-PCR in xenografts (n=5, 6) after treatment with diluent or NTX-301 as described in (H). Carbo, carboplatin. For all comparisons, *p<0.05, **p<0.01, ***p<0.001.
To assess the effects of NTX-301 on ovarian CSCs in vivo, we employed a xenograft model enriched in CSCs. It is known that platinum treatment induces enrichment of CSCs both in vitro and in vivo (23, 27). OC xenografts were developed through subcutaneous (SQ) implantation of OVCAR5 cells. Tumor bearing mice were treated with carboplatin (40mg/kg, ip weekly for 3 weeks). After platinum induced tumor regression, mice were treated with NTX-301 (1.0mg/kg i.p daily X 5 days per week) or diluent and residual tumors were assessed. As anticipated, carboplatin reduced tumor volumes (Fig. 3H). Subsequent treatment with NTX-301 significantly reduced recurrent xenograft weights (Fig. 3I) and volumes (Fig. 3J). mRNA expression levels of ALDH1A1, a surrogate marker for CSCs, were increased in platinum followed by diluent treated tumors, but suppressed in platinum followed by NTX-301 treated xenografts (Fig. 3K). These findings indicate that NTX-301 targeted ovarian CSCs and are consistent with the observed inhibitory effects of the agent at transcriptome level on oxidoreductase and ALDH activities which CSCs use to protect against oxidative stress (28).
NTX-301 induces ferroptosis by targeting SCD mediated lipid-oriented oxidative stress.
Given the observed inhibition of fatty acid biosynthesis and metabolism among transcriptomic changes induced by the HMA (Fig 2C–G), we explored its potential effects on metabolic reprograming. Stearoyl-Coenzyme A desaturase 1 (SCD), the key enzyme that converts saturated (SFAs) to unsaturated fatty acids (UFAs) was among the top downregulated DEGs (fold change 9.370±04, FDR<0.05). SCD is upregulated in cancer, including OC (25, 29) and its inhibition induces lipid oxidation and cell death through ferroptosis (30). We therefore examined whether NTX-301 altered lipid metabolism and induced ferroptosis by targeting SCD. NTX-301 downregulated SCD expression at mRNA (OVCAR5, Fig. S3A) and protein levels (OVCAR5, Fig. 4A; SKOV3. Fig. 4B, primary HGSOC cells, Fig. 4C) to a higher degree than decitabine (Fig. 4A–B). SCD expression was less affected by treatment with NTX-301 in non-malignant FT190 cells (Fig. S3B). We next assessed the abundance of SFAs and UFAs in OC cells in response to NTX-301 by mass spectrometry analysis and hyperspectral SRS (hSRS) techniques. NTX-301 induced a modest reduction in 16-C UFA (palmitoleic acid, Fig. 4E), significant reduction in 18-C UFAs (oleic and linoleic acid, Fig. 4G–H), but had no effects on abundance of 16-C and 18-C SFAs (palmitic and stearic acids, Fig. 4D and 4F). Furthermore, hSRS imaging (Fig. 4I–K) detected a significant reduction in the C-D bond signal corresponding to UFAs in OVCAR5 cells treated with NTX-301 compared with control cells. This indicates that NTX-301 significantly disrupted the lipid balance in OC cells by inhibiting SCD. To confirm, we assessed the sensitivity to NTX-301 after stable SCD knock down (KD) by stable transduction of shRNAs targeting the enzyme (shSCD; Fig S3C), as previously described (25). KD SCD reduced the sensitivity of OC cells to NTX-301 treatment compared with OC cells transfected with control shRNA (shCtrl) vector (~80-fold change in IC50, Fig. 4L), supporting that the effects of NTX-301 in OC cells are at least partly dependent upon SCD. Because SCD inhibition was shown to induce ferroptosis (31), oxidized lipid levels were measured by using C11-BODIPY staining in OC cells treated with NTX-301. NTX-301 caused increased levels of oxidized lipids compared to DMSO, and this was blunted by the iron chelator deferoxamine (DFOA, Fig. 4M–N), indicating that NTX-301 promoted cell death via ferroptosis. Additionally, Annexin V-FITC and propidium iodide (PI) staining assessed apoptosis. NTX301 (100nM and1μM) induced ~50–60% cell death, as assessed by trypan blue staining after 48- or 72-hours treatment. Among the dead cells, only 3–6% cells were apoptotic (Figs. S3D–I) and 4–6% were apoptotic and necrotic (Figs. S3D–I), suggesting that NTX-301 induced cell death mainly through ferroptosis. NTX-301 induced ferroptosis was rescued by addition of oleic acid (200μM), an UFA, into the culture medium (Fig. 4O–P), supporting that it was mediated through SCD depletion. Combined, the transcriptomic and lipidomic analyses coupled with hSRS imaging demonstrate that NTX-301 disrupted the lipid balance in OC cells, by targeting SCD to induce lipid oriented oxidative stress, leading to ferroptosis.
Figure 4. NTX-301 induces ferroptosis in OC cells by targeting SCD-mediated lipid balance.
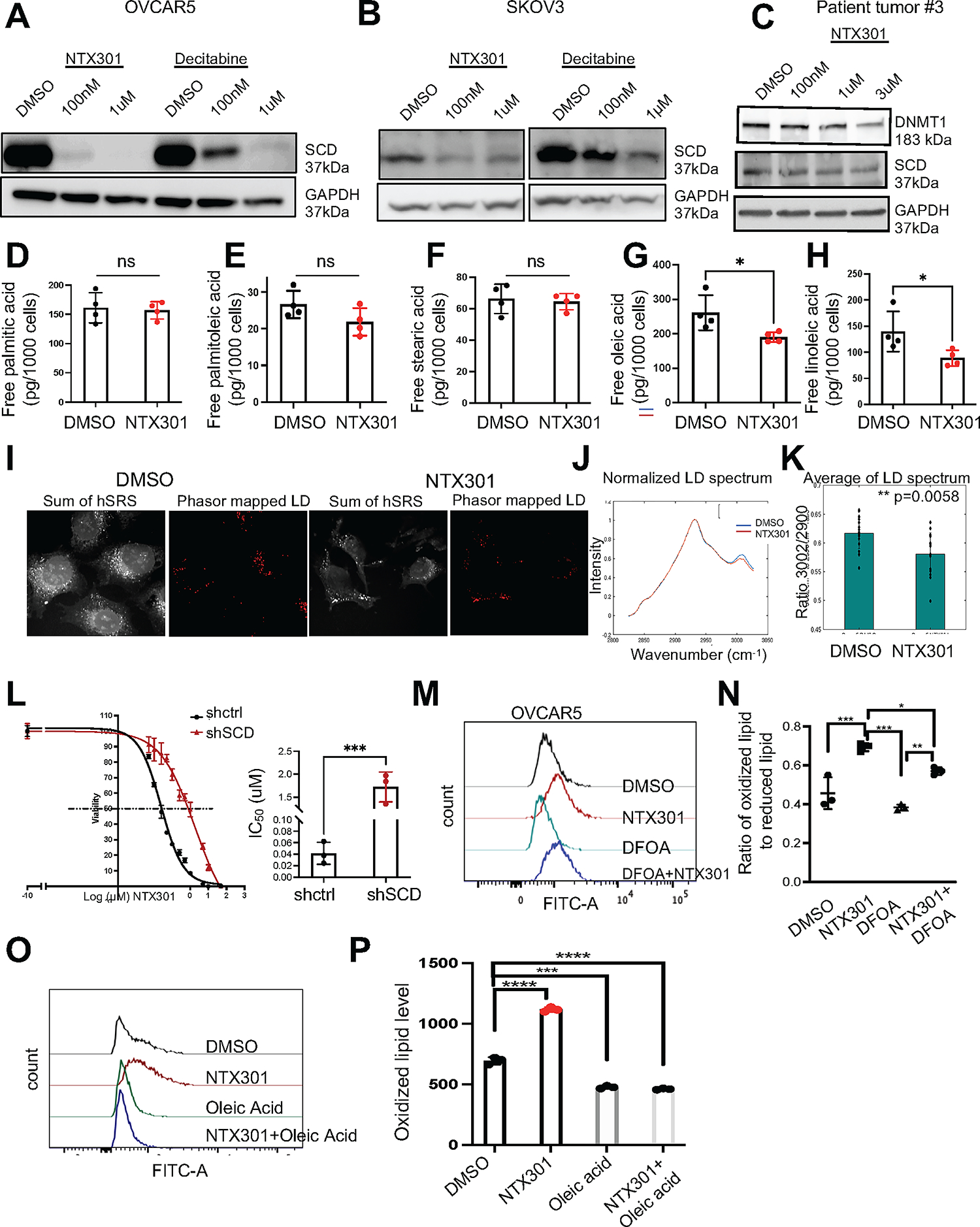
(A, B) Western blot measured SCD and GAPDH (loading control) protein levels in OVCAR5 (A) and SKOV3 (B) OC cells treated with DMSO, NTX-301 or decitabine for 2 days (n=2). (C) Western blot of DNMT1 and SCD in cells isolated from HGSOC tumors and treated with DMSO or NTX-301. (D-H) Lipidomics analysis measured amounts (means ± SD, n=4) of palmitic acid (16:0) (D) palmitoleic acid (16:1) (E) stearic acid (18:0) (F) oleic acid (18:1) (G), and linoleic acid (18:1) (H) in OVCAR5 cells treated with DMSO or NTX-301(100nM, 2days). (I) Representative hSRS images of DMSO- and NTX-301 (100nM, 2days)-treated OVCAR5 cells show lipid droplets (LD) phasor maps. Scale bars: 20 μm. (J) Average hSRS LD spectra of samples described in (I) and normalized to 2900 cm−1. (K) hSRS peak ratio at 3002 cm−1 to 2900 cm−1. Each data point represents one field of view on a plate. Bars indicate means ± SD. (L) Cell survival curves (left) and quantification of IC50 (right) of OVCAR5 transfected with shctrl or shSCD vectors in response to NTX-301 treatment. (M) Representative histograms and (N) mean (± SD, n = 3) fluorescence intensity of BODIPY 581/591-C11 in OVCAR5 OC cells treated for 2 days with DMSO, NTX-301 (500nM), DFOA (800nM) and NTX-301 plus DFOA. (O) Representative histograms and (P) mean (± SD, n = 3) fluorescence intensities (right) of BODIPY 581/591-C11 show effects of oleic acid (200μM, 2days) on rescuing lipid peroxidation levels in OVCAR5 cells treated with DMSO or NTX-301 (100nM, 2days, n=3). For all comparisons, *p<0.05, **p<0.01, ***p<0.001, ***p<0.0001.
NTX-301-induced SIRT1 re-expression causes epigenetic repression of SCD.
To understand how SCD was repressed in response to this HMA, we examined potential direct effects of NTX-301 on transcriptional regulators of SCD. SCD expression is highly dependent on epigenetic regulation, especially histone acetylation, with H2K27ac enrichment in the promoter region of the gene being known to activate its transcription (32). H3K27ac peak analysis from available chromatin immunoprecipitation (ChIP) sequencing databases demonstrates conserved H3K27ac deposition in the promoter region of the SCD gene at ~1kb from the transcription start site (TSS) in OVCAR5 cells (33) and in other cell lines profiled by ENCODE (GM12878, and HEK293T) (34, 35) (Fig. 5A). ChIP-qPCR using an antibody against H3K27Ac and primers flanking the observed binding region (hg38_dna range=chr10:100346479–100346745) confirmed H3K27Ac at the SCD promoter and decreased deposition of this active mark in NTX-301 treated cells (Fig. 5B, Fig. S4A). Interestingly, NTX-301 and decitabine induced global reduction in total H3K27ac levels compared to DMSO (Fig. 5C and Fig. S4B). As histone deacetylases (HDACs) regulate H3K27Ac levels, we examined expression levels of key HDACs in NTX-301 treated cells. SIRT1 and HDAC9 were significantly upregulated by NTX-301, while other HDACs were inhibited (Fig. S4C–E). SIRT1 is an NAD+-dependent class III HDAC which has been shown to regulate SCD (36). Quantitative RT-PCR confirmed increased SIRT1 mRNA expression levels in OVCAR5 and primary HGSOC cells treated with NTX-301 (Fig. 5D and E). To determine whether upregulation of SIRT1 by NTX-301 was dependent on reversal of DNA methylation, we examined the effects of the drug on regulatory CpG sites in the SIRT1 gene. Three CpG sites were found to be significantly hypomethylated in response to NTX-301, including a 5’-UTR probe (D Beta value of −16%) and two probes located in the gene body (D Beta values of −11% and −9%, respectively, Fig. 5F). Collectively, these results support that NTX-301 induced upregulation of SIRT1, which suppressed SCD expression in OC cells by decreasing H3K27Ac at its promoter.
Figure 5. Inhibitory effects of NTX-301 on SCD in OC cells.
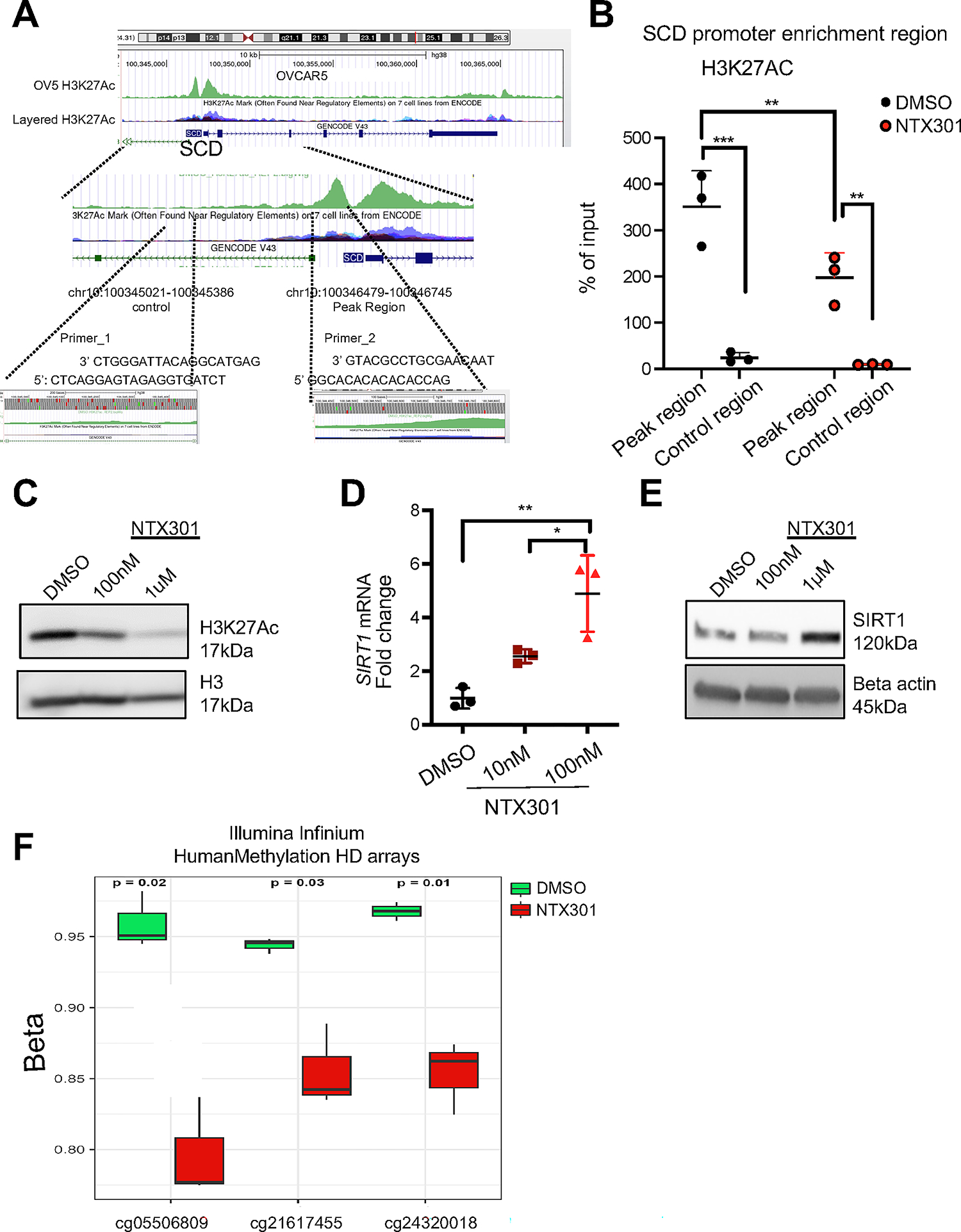
(A) UCSC Genome Browser on Human GRCh38/hg38 tracks of H3K27Ac peaks in the SCD promoter in OVCAR5 cells (33) and other cell lines previously recorded in the ENCODE dataset (GM12878, and HEK293T) (34, 35). The H3K27Ac binding motif along with the position of primer sequences (Primer_2) used for q-PCR are shown. Amplification of a sequence 1 kb downstream was used as a control (Primer_1). (B) ChIP-PCR shows H3K27Ac enrichment in the SCD gene (Primer_2) in OVCAR5 cells treated with DMSO or NTX-301 (100nM, 2days, n=3). Enrichment of H3K27Ac on a sequence 1 kb downstream was used as a control (Primer_1). Results are means ± SD, which was analyzed by two-way ANOVA with multiple comparisons. For all comparisons, *p<0.05; **p<0.01; ***p<0.001. (C) Western blot of H3K27Ac and H3 in OVCAR5 OC cells treated with DMSO or NTX-301 for 2 days. (D, E) mRNA expression levels (mean fold-change ± SD) of SIRT1 measured by qRT-PCR (D) and western blot of SIRT1 and beta actin (loading control) (E) in OVCAR5 cells treated with DMSO or NTX-301 for 2 days (n=3 replicates). (F) DNA methylation levels for 3 significantly demethylated CpG sites (p-adj< 0.05) in the SIRT1 gene as measured with an Illumina EPIC array in OVCAR5 cells treated with DMSO or NTX-301 (100nM, 3 days; n = 3 replicates per group).
NTX-301 monotherapy inhibits xenograft growth in vivo by targeting lipid metabolism.
To test the effects of NTX-301 in vivo, we used SQ OVCAR5 xenografts. Tumor bearing mice were treated with diluent, and two doses of NTX-301 (0.5mg/kg, or 1mg/kg SQ daily) for three weeks (Fig. S5A). Mice tolerated NTX-301 at both dose levels, with modest changes in body weight (Fig. S5B). Monotherapy with NTX-301 significantly inhibited xenograft growth (p<0.05, Fig. S5C), decreased tumor sizes (Fig. 6A–B) and tumor weights (Fig. 6C) at both dose levels. To determine whether NTX-301 induced ferroptosis, we measured oxidized lipid levels in cells dissociated from NTX-301 or diluent treated tumors. Compared to diluent, NTX-301 potently induced oxidized lipids in xenografts (Fig. 6D–E). In addition, significant reduction of DNMT1 (Fig. 6F) and SCD (Fig. 6G) were observed in NTX-301-treated xenografts compared with control tumors. To determine whether SCD inhibition led to alterations in tumor lipid content, we used hSRS microscopy and measured the ratio between the peak at 3002 cm−1 corresponding to C=C-H vibration from unsaturated lipids and the SRS peak at 2900 cm−1 corresponding to other C-H bonds in fatty acids. After normalization, the C=C-H peak (3002 cm−1) was lower in the NTX-301 treated tumors, supporting that treatment with this HMA caused decreased levels of UFAs (p=0.04, Fig. 6H–J).
Figure 6. NTX-301 inhibits OC tumor growth by targeting lipid metabolism.
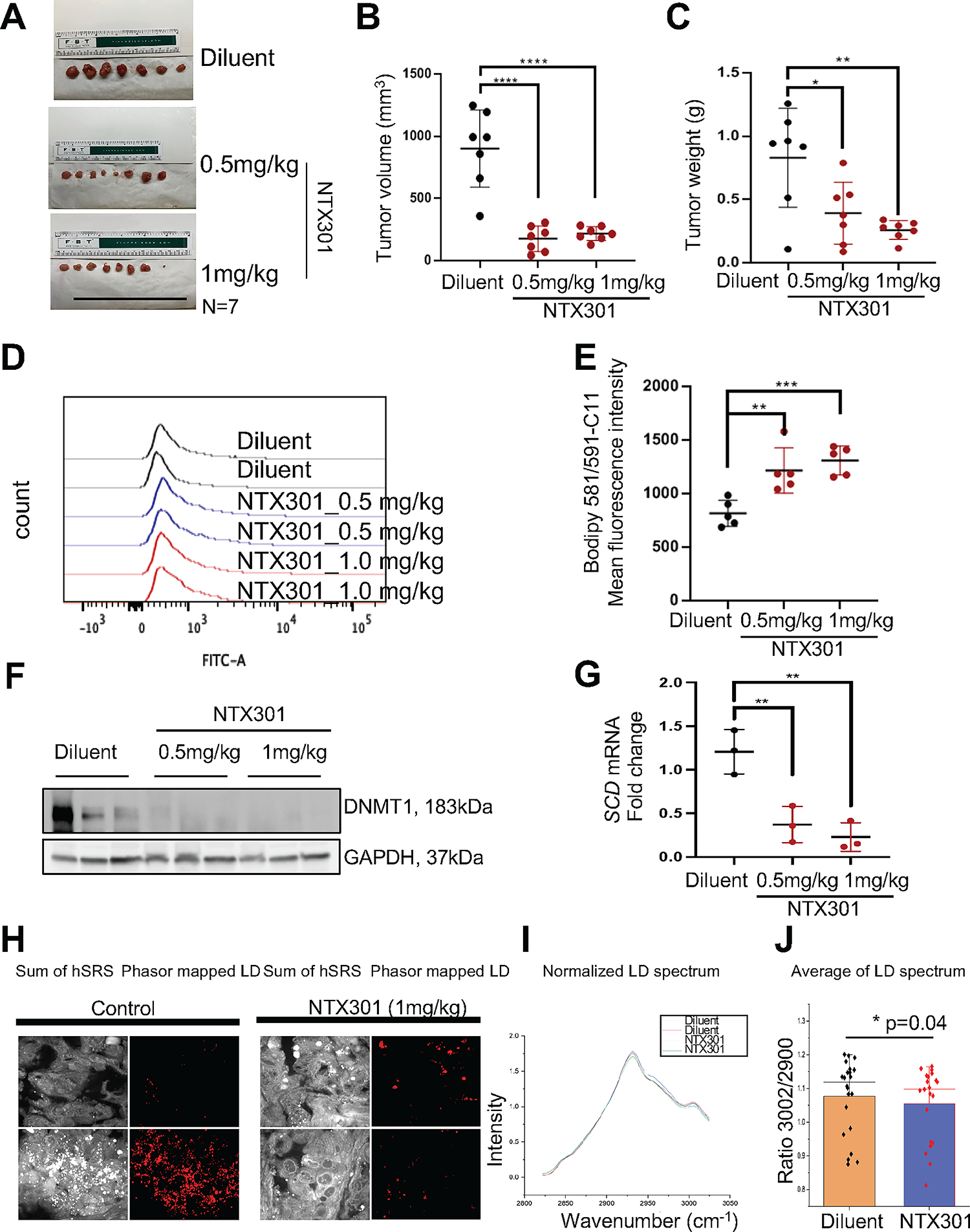
(A-C) Images of harvested SQ xenografts (A) and means ± SD of tumor volumes (B) and tumor weights (C) from mice treated with diluent or NTX-301. (D, E) Representative histograms (D) and mean (± SD, n = 5) fluorescence intensity of BODIPY 581/591-C11 (E) in cells obtained from OVCAR5 xenografts in the experiment described in (A). (F) Protein levels of DNMT1 and GAPDH (loading control) measured by western blotting in xenograft tumors (n = 3 per group) from the experiment described in (A). (G) mRNA expression levels of SCD in xenograft tumors collected from indicated groups and measured by qRT-PCR (n = 3 per group). (H) Representative hSRS images of xenograft sections from mice treated with diluent or NTX-301 (1mg/ml), and corresponding lipid droplets phasor maps. Scale bars: 20 μm. (I) Average hSRS spectra of sections of xenografts from mice treated with diluent or NTX-301 (1mg/ml), and normalized to 2900 cm−1. (J) Box-scatter plot of the ratio of hSRS peaks at 3002 cm−1 and 2900 cm−1. Each data point represents one field of view in a tissue section, bars indicate means ± SD, p=0.04. *p<0.05; **p<0.01; ***p<0.001.
NTX-301 monotherapy inhibited patient derived xenograft (PDX) growth by targeting SCD.
Further, NTX-301 effects were tested in a platinum tolerant PDX model generated from a HGSOC tumor implanted under the ovarian bursa of NSG mice (37) and subsequently treated with carboplatin weekly for 6 weeks. PDX material was subsequently implanted SQ into NSG mice. Once tumors’ sizes reached ~150 mm3, mice were treated with diluent or NTX-301 (1mg/kg, 5-day/week, 4 weeks). NTX-301 treatment was well tolerated, with no significant changes in body weights being detected (Fig. S6A). NTX-301 significantly inhibited PDX growth (Fig. S6B), tumor volumes (Fig. S6C–D) and tumor weights (Fig. S6E). Histological examination and staining for PAX8 and vimentin demonstrated that PDX maintained the histological characteristics of the original HGSOC tumor, with strong PAX8 and P53 expression and absent vimentin staining (Fig. S6F). Importantly, NTX-301 treated PDXs displayed increased levels of oxidized lipids as measured through C11 BODIPY staining compared to control tumors (Fig. S6G–H). These data support that NTX-301 suppressed tumor growth and induced ferroptosis by targeting SCD in vivo.
Discussion:
Our fundings support that the 4-thio-decitabine derivative NTX-301 elicits potent hypomethylating effects which led to tumor inhibition in HGSOC models. We identify metabolic reprogramming induced by NTX-301 reflected in depletion of UFAs, as a new mechanism triggered by this HMA, leading to ferroptosis in cancer cells and tumors. Our results have several implications.
First, the robust anti-tumor activity of NTX-301 in several OC models provide a solid foundation for considering clinical testing of this new HMA. Interestingly, the agent was active both in established ovarian xenografts, as well as in residual tumors following platinum. These results support the possibility of testing NTX-301 as a maintenance strategy after optimal response to standard chemotherapy. This is particularly important, as it has been proposed that resistant CSCs are enriched in tumors residual after platinum (6, 27) and that platinum-resistant cells can be eradicated through induction of ferroptosis (23). HMAs have been tested as resensitizers to platinum in several clinical trials targeting patients with recurrent OC with mixed results. A phase I/II trial tested repetitive low doses of decitabine prior to platinum in women with platinum-resistant OC and reported a response rate of 35% and progression free survival (PFS) of 10.2 months (7). A subsequent randomized, multicenter, phase 2 trial evaluated guadecitabine and carboplatin against established second-line treatment in women with platinum-resistant HGSOC (12). Patients treated with the HMA combination had a higher PFS rate at 6 months compared with standard of care treated subjects, however, the median PFS was not statistically different between arms, leading to diminished enthusiasm for continued clinical development. Coupled with the favorable pharmacokinetic properties and oral bioavailability of NTX-301, the data presented here demonstrating potent activity in vitro and in vivo support revisiting the potential clinical utility of HMAs, particularly in a setting of minimal residual disease after chemical tumor debulking with platinum.
Second, NTX-301 targeted the ALDH+ CSCs population, inhibiting their tumor initiation capacity, consistent with our and others’ previous studies (6). Epigenetic modifications including altered DNA methylation and histone modifications play important roles in regulating gene expression and maintaining the stemness phenotype in both normal or malignant stem cells (38). We have previously shown that ovarian CSCs harbor methylation traits linked to stemness and targetable by HMAs (6, 27). Guadecitabine, a potent DNMT inhibitor, inhibited the CSC population by inducing expression of differentiation associated genes. Similar inhibitory effects are described here for NTX-301. Other epigenetic modifiers, such as bromodomain and extra-terminal (BET) (39), EZH2 (40) and disruptor of telomeric silencing-1–like (Dot1L) inhibitors (41) were shown to inhibit CSCs. The small molecule inhibitor (JQ1) blocked ALDH expression by directly affecting a super-enhancer element controlled by BRD4 in the ALDH1A1 regulatory region (39). Inhibition of the histone methyltransferase EZH2 suppressed Disabled Homolog 2-Interacting Protein (DAB2IP) required for supporting CSC maintenance (40). DAB2IP is silenced by EZH2-mediated H3K27 trimethylation and regulates stemness-associated pathways, particularly WNT/C-JUN signaling. Combined EZH2 and WNT/C-JUN inhibitors suppressed CSCs in vitro and tumor growth in vivo (40). We identified DOT1L, as a key regulator of self-renewal and tumor initiation capacity of ovarian CSCs (41). EPZ-5676 inhibited DOT1L’s methyltransferase activity and targeted ovarian CSCs by blocking stemness associated Wnt/β-catenin signaling (41). These results support that such epigenetic dependencies, including DNA methylation alterations, could be targeted to eliminate CSCs and prevent tumor recurrence.
Third, we show that NTX-301 causes depletion of UFAs, through suppression of the desaturase SCD. The enzyme catalyzes transformation of SFAs into UFAs, specifically, stearic (18:0) and palmitic acid (16:0), into their Δ9-monounsaturated counterparts, oleic (18:1) and palmitoleic acid (16:1) (30). UFAs play critical roles in regulating cell membrane integrity and fluidity, cellular signaling and protection from oxidative stress (30). In the absence of UFAs, saturated lipids undergo peroxidation which damages cellular membranes and causes cell death through ferroptosis. Therefore, maintaining the balance between UFAs and SFAs reduces susceptibility to ferroptosis and promotes the survival or normal and malignant cells (30, 42). High levels of SCD have been reported in many tumors, including HGSOC (30, 42, 43). We recently showed that by mediating the dynamics between SFAs and UFAs, SCD controlled cancer cell survival and tumor progression (25). SCD blockade by a small molecule was shown to induce ferroptosis by depleting the lipophilic antioxidant coenzyme Q10 (30). Here, we demonstrate that NTX-301 targeted lipid metabolism by inhibiting SCD, reducing cellular levels of UFAs, and ultimately promoting lipid peroxidation and cell death. Importantly, NTX-301 induced ferroptosis was rescued by addition of the SCD’s product, oleic acid, confirming that depletion of the enzyme was a key contributor. SCD downregulation induced by NTX-301 was related to hypomethylation and re-expression of the deacetylase SIRT1, causing loss of the active H3K27Ac histone mark on the SCD promoter. We cannot exclude other mechanisms, such as direct effects of NTX-301 on SCD gene body demethylation (44) contributing to decreased SCD expression during treatment with this HMA.
As anticipated for an agent with global transcriptomic effects, other pathways were also impacted, including genes networks related to DNA repair, such as Fanconi anemia pathway and p53 signaling pathways. A recent report examining the effects of NTX-301 in a panel of leukemia cell lines identified among gene sets upregulated by this HMA, gene sets related to DNA repair, p53 and immune activation (45), similar to findings in our models. That integrated analysis found SREBF1/2 and mTORC1/2 as upstream regulators of downregulated genes in leukemia cells (45). Interestingly, SCD, the target identified in our model is known to be regulated by SREBF1 (46), supporting the idea that shared pathways are affected by NTX-301 across different cancer models. In all, we demonstrate that a novel HMA depleted cellular levels of UFAs in OC cells and tumors by targeting SCD, which induced ferroptosis. Our findings identify a new mechanism by which DNMT blockade attacks cancer cells, linking the epigenome to regulation of lipid metabolism, a key dependency of HGSOC cells and tumors.
Supplementary Material
Statement of translational relevance:
Development of novel agents to target recurrent ovarian cancer (OC) is critically needed. Epigenetic alterations, including DNA methylation of tumor suppressor genes, have been linked to platinum resistance in OC and hypomethylating agents (HMAs) have shown efficacy in previous clinical trials. NTX-301 is a novel, highly potent and orally bioavailable HMA. Here we demonstrate that it is highly active in OC cell lines, platinum-resistant xenografts, and patient derived xenografts. We identify blockade of lipid synthesis through inhibition of stearoyl CoA desaturase as a novel mechanism through which NTX-301 targets platinum resistant OC cells, inducing ferroptosis. Our data support further clinical testing of NTX-301 in women with ovarian cancer.
Acknowledgements:
Funding and drug for this study was contributed by PinotBio to D. Matei, the National Cancer Institute R01 CA224275 to D. Matei and J.X. Cheng, and the US Department of Veterans Affairs IO1BX000792 to D. Matei. Flow cytometry analyses were performed in the Northwestern University Flow Cytometry Core Facility supported by the Cancer Center Support Grant NCI CA060553. PDX experiments were performed in the Center for Developmental Therapeutics (CDT) partly supported by Cancer Center Support Grant NCI CA060553.
Footnotes
Conflict of Interest: PinotBio provided drug and funding to support part of the study.
References
- 1.Jones PA, and Baylin SB. The epigenomics of cancer. Cell. 2007;128(4):683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kantarjian H, Issa JP, Rosenfeld CS, Bennett JM, Albitar M, DiPersio J, et al. Decitabine improves patient outcomes in myelodysplastic syndromes: results of a phase III randomized study. Cancer. 2006;106(8):1794–803. [DOI] [PubMed] [Google Scholar]
- 3.Matei D, and Nephew KP. Epigenetic Attire in Ovarian Cancer: The Emperor’s New Clothes. Cancer Res. 2020;80(18):3775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oza AM, Matulonis UA, Alvarez Secord A, Nemunaitis J, Roman LD, Blagden SP, et al. A Randomized Phase II Trial of Epigenetic Priming with Guadecitabine and Carboplatin in Platinum-resistant, Recurrent Ovarian Cancer. Clin Cancer Res. 2020;26(5):1009–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fang F, Munck J, Tang J, Taverna P, Wang Y, Miller DF, et al. The novel, small-molecule DNA methylation inhibitor SGI-110 as an ovarian cancer chemosensitizer. Clin Cancer Res. 2014;20(24):6504–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Cardenas H, Fang F, Condello S, Taverna P, Segar M, et al. Epigenetic targeting of ovarian cancer stem cells. Cancer Res. 2014;74(17):4922–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matei D, Fang F, Shen C, Schilder J, Arnold A, Zeng Y, et al. Epigenetic resensitization to platinum in ovarian cancer. Cancer Research. 2012;72(9):2197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang F, Cardenas H, Huang H, Jiang G, Perkins SM, Zhang C, et al. Genomic and Epigenomic Signatures in Ovarian Cancer Associated with Resensitization to Platinum Drugs. Cancer Res. 2018;78(3):631–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azad NS, El-Khoueiry A, Yin J, Oberg AL, Flynn P, Adkins D, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat; a phase 2 consortium/stand Up 2 cancer study. Oncotarget. 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee V, Wang JS, Zahurak ML, Gootjes EC, Verheul HMW, Parkinson RM, et al. A phase I trial of a guadecitabine (SGI-110) and irinotecan in metastatic colorectal cancer patients previously exposed to irinotecan. Clin Cancer Res. 2018. [DOI] [PubMed] [Google Scholar]
- 11.Santi DV, Norment A, and Garrett CE. Covalent Bond Formation between a DNA-Cytosine Methyltransferase and DNA Containing 5-Azacytosine. P Natl Acad Sci-Biol. 1984;81(22):6993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Matei D, Ghamande S, Roman L, Alvarez Secord A, Nemunaitis J, Markham MJ, et al. A Phase I Clinical Trial of Guadecitabine and Carboplatin in Platinum-Resistant, Recurrent Ovarian Cancer: Clinical, Pharmacokinetic, and Pharmacodynamic Analyses. Clin Cancer Res. 2018;24(10):2285–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thottassery JV, Sambandam V, Allan PW, Maddry JA, Maxuitenko YY, Tiwari K, et al. Novel DNA methyltransferase-1 (DNMT1) depleting anticancer nucleosides, 4’-thio-2’-deoxycytidine and 5-aza-4’-thio-2’-deoxycytidine. Cancer Chemother Pharmacol. 2014;74(2):291–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lim B, Yoo D, Chun Y, Go A, Cho KJ, Choi D, et al. The preclinical efficacy of the novel hypomethylating agent NTX-301 as a monotherapy and in combination with venetoclax in acute myeloid leukemia. Blood Cancer J. 2022;12(4):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swisher EM, Gonzalez RM, Taniguchi T, Garcia RL, Walsh T, Goff BA, and Welcsh P. Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas. Mol Cancer. 2009;8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Makarla PB, Saboorian MH, Ashfaq R, Toyooka KO, Toyooka S, Minna JD, et al. Promoter hypermethylation profile of ovarian epithelial neoplasms. Clin Cancer Res. 2005;11(15):5365–9. [DOI] [PubMed] [Google Scholar]
- 17.Cardenas H, Fang F, Jiang G, Perkins SM, Zhang C, Emerson RE, et al. Methylomic Signatures of High Grade Serous Ovarian Cancer. Epigenetics. 2021;16(11):1201–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeller C, Dai W, Steele NL, Siddiq A, Walley AJ, Wilhelm-Benartzi CS, et al. Candidate DNA methylation drivers of acquired cisplatin resistance in ovarian cancer identified by methylome and expression profiling. Oncogene. 2012;31(42):4567–76. [DOI] [PubMed] [Google Scholar]
- 19.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell. 2015;162(5):974–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen S, Xie P, Cowan M, Huang H, Cardenas H, Keathley R, et al. Epigenetic priming enhances antitumor immunity in platinum-resistant ovarian cancer. J Clin Invest. 2022;132(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiappinelli KB, Zahnow CA, Ahuja N, and Baylin SB. Combining Epigenetic and Immunotherapy to Combat Cancer. Cancer Res. 2016;76(7):1683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perets R, Wyant GA, Muto KW, Bijron JG, Poole BB, Chin KT, et al. Transformation of the fallopian tube secretory epithelium leads to high-grade serous ovarian cancer in Brca;Tp53;Pten models. Cancer Cell. 2013;24(6):751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Y, Zhao G, Condello S, Huang H, Cardenas H, Tanner EJ, et al. Frizzled-7 Identifies Platinum-Tolerant Ovarian Cancer Cells Susceptible to Ferroptosis. Cancer Res. 2021;81(2):384–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tan Y, Li J, Zhao G, Huang KC, Cardenas H, Wang Y, et al. Metabolic reprogramming from glycolysis to fatty acid uptake and beta-oxidation in platinum-resistant cancer cells. Nat Commun. 2022;13(1):4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao G, Tan Y, Cardenas H, Vayngart D, Wang Y, Huang H, et al. Ovarian cancer cell fate regulation by the dynamics between saturated and unsaturated fatty acids. Proc Natl Acad Sci U S A. 2022;119(41):e2203480119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monsivais D, Dyson MT, Yin P, Navarro A, Coon JS, Pavone ME, and Bulun SE. Estrogen receptor β regulates endometriotic cell survival through serum and glucocorticoid-regulated kinase activation. Fertil Steril. 2016;105(5):1266–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Zong X, Mitra S, Mitra AK, Matei D, and Nephew KP. IL-6 mediates platinum-induced enrichment of ovarian cancer stem cells. JCI Insight. 2018;3(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nwani NG, Condello S, Wang Y, Swetzig WM, Barber E, Hurley T, and Matei D. A Novel ALDH1A1 Inhibitor Targets Cells with Stem Cell Characteristics in Ovarian Cancer. Cancers (Basel). 2019;11(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roongta UV, Pabalan JG, Wang XY, Ryseck RP, Fargnoli J, Henley BJ, et al. Cancer Cell Dependence on Unsaturated Fatty Acids Implicates Stearoyl-CoA Desaturase as a Target for Cancer Therapy. Mol Cancer Res. 2011;9(11):1551–61. [DOI] [PubMed] [Google Scholar]
- 30.Tesfay L, Paul BT, Konstorum A, Deng ZY, Cox AO, Lee J, et al. Stearoyl-CoA Desaturase 1 Protects Ovarian Cancer Cells from Ferroptotic Cell Death. Cancer Research. 2019;79(20):5355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol. 2016;12(7):497-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan GH, Luna A, Wang HP, Bozorgui B, Li XB, Sanchez M, et al. BET inhibition induces vulnerability to MCL1 targeting through upregulation of fatty acid synthesis pathway in breast cancer. Cell Rep. 2022;40(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gopi LK, and Kidder BL. Integrative pan cancer analysis reveals epigenomic variation in cancer type and cell specific chromatin domains. Nature Communications. 2021;12(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis C, Doyle F, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, et al. The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46(D1):D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qiang L, Kon N, Zhao W, Jiang L, Knight CM, Welch C, et al. Hepatic SirT1-Dependent Gain of Function of Stearoyl-CoA Desaturase-1 Conveys Dysmetabolic and Tumor Progression Functions. Cell Rep. 2015;11(11):1797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong R, Qiang W, Guo H, Xu X, Kim JJ, Mazar A, et al. Histologic and molecular analysis of patient derived xenografts of high-grade serous ovarian carcinoma. J Hematol Oncol. 2016;9(1):92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinberg AP, Koldobskiy MA, and Gondor A. Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat Rev Genet. 2016;17(5):284–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yokoyama Y, Zhu H, Lee JH, Kossenkov AV, Wu SY, Wickramasinghe JM, et al. BET Inhibitors Suppress ALDH Activity by Targeting ALDH1A1 Super-Enhancer in Ovarian Cancer. Cancer Res. 2016;76(21):6320–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zong X, Wang W, Ozes A, Fang F, Sandusky GE, and Nephew KP. EZH2-Mediated Downregulation of the Tumor Suppressor DAB2IP Maintains Ovarian Cancer Stem Cells. Cancer Res. 2020;80(20):4371–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y, Wang Y, Valdivia A, Huang H, and Matei D. DOT1 L Regulates Ovarian Cancer Stem Cells by Activating beta-catenin Signaling. Mol Cancer Res. 2023;21(2):140–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YQ, Wang YN, Zhao GY, Orsulic S, and Matei D. Metabolic dependencies and targets in ovarian cancer. Pharmacol Therapeut. 2023;245. [DOI] [PubMed] [Google Scholar]
- 43.Igal RA. Stearoyl CoA desaturase-1: New insights into a central regulator of cancer metabolism. Bba-Mol Cell Biol L. 2016;1861(12):1865–80. [DOI] [PubMed] [Google Scholar]
- 44.Yang X, Han H, De Carvalho DD, Lay FD, Jones PA, and Liang G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell. 2014;26(4):577–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lim B, Yoo D, Chun Y, Go A, Kim JY, Lee HY, et al. Integrative Analyses Reveal the Anticancer Mechanisms and Sensitivity Markers of the Next-Generation Hypomethylating Agent NTX-301. Cancers (Basel). 2023;15(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rodriguez-Cruz M, Sanchez Gonzalez R, Sanchez Garcia AM, and Lopez-Alarcon M. Coexisting role of fasting or feeding and dietary lipids in the control of gene expression of enzymes involved in the synthesis of saturated, monounsaturated and polyunsaturated fatty acids. Gene. 2012;496(1):28–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing and methylation data are deposited in the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) (RRID:SCR_005012) data repository: GSE236985 and GSE236984. The analysis was performed by using publicly available software described in methods.