

Abstract
PARP inhibitor (PARPi)-resistant BRCA mutant (BRCAm) high-grade serous ovarian cancer (HGSOC) represents a new clinical challenge with unmet therapeutic needs. Here, we performed a quantitative high-throughput drug combination screen that identified the combination of an ATR inhibitor (ATRi) and an AKT inhibitor (AKTi) as an effective treatment strategy for both PARPi-sensitive and PARPi-resistant BRCAm HGSOC. The ATRi and AKTi combination induced DNA damage and R-loop-mediated replication stress (RS). Mechanistically, the kinase domain of AKT1 directly interacted with DHX9 and facilitated recruitment of DHX9 to R-loops. AKTi increased ATRi-induced R-loop-mediated RS by mitigating recruitment of DHX9 to R-loops. Moreover, DHX9 was upregulated in tumors from PARPi-resistant BRCAm HGSOC patients, and high co-expression of DHX9 and AKT1 correlated with worse survival. Together, this study reveals an interaction between AKT1 and DHX9 that facilitates R-loop resolution and identifies combining ATRi and AKTi as a rational treatment strategy for BRCAm HGSOC irrespective of PARPi resistance status.
Introduction
High-grade serous ovarian cancer (HGSOC), the most common subtype of ovarian cancer, remains the second leading cause of gynecologic cancer death worldwide (1). The disease presents at late stages and approximately 80% of patients recur after initial surgical debulking and platinum-based chemotherapy (2). HGSOC is also characterized by genomic instability because almost all cases exhibit p53 dysfunction, causing loss of G1/S cell cycle checkpoints and premature entry into S phase, thus preventing optimal DNA replication and DNA repair (3).
Successful introduction of PARP inhibitors (PARPis) has led to new therapeutic opportunities in ovarian cancer, particularly for BRCA mutant (BRCAm) HGSOC (4). However, most patients eventually discontinue treatments due to the emergence of resistance, highlighting the unmet need for a more effective therapy (4). Several PARPi resistance mechanisms have been identified, including BRCA-dependent (e.g., reversion mutations of BRCA) or BRCA-independent homologous recombination (HR) restoration (e.g., loss of negative regulators of HR repair, e.g., 53BP1 or REV7), increased replication fork stability, and upregulation of survival pathways or drug efflux activity (5). Of those, increased ATR signaling pathway is important for optimal DNA replication and DNA repair (6). Also, increased protein levels of ATR/CHK1 pathway are found in acquired PARPi-resistant and platinum-resistant BRCAm HGSOC cell lines and patient-derived xenografts models (7–9), indicating its critical role in drug resistance.
Increasingly, there is recognition that replication stress is a main source of genomic instability and may be a crucial vulnerability of drug-resistant HGSOC cells (10). Replication stress is traditionally defined as the slowing or stalling of replication fork progression and/or DNA synthesis, but is an evolving concept including any events blocking optimal DNA replication processes (11). Among numerous sources of replication stress, aberrant R-loops are a major cause of replication stress (12). R-loops, which are the intermediates of transcription, consist of a DNA:RNA hybrid and a displaced single-stranded DNA (ssDNA)(13). R-loops occur naturally during mitochondrial DNA replication and transcription-coupled recombination, and play vital roles in regulating gene expression, DNA replication, and histone modifications (13). Under normal conditions, ribonuclease H (RNase H)(14) and several DNA/RNA helicases such as DHX9 (15), senataxin (16), and aquarius (17), unwind and remove aberrant R-loops, thus promote the replication fork restart and HR repair to preserve genome integrity (13). However, pathological R-loop accumulation, either due to excessive formation or impaired resolution, can compromise genomic integrity by exposing ssDNA to nicks and other types of damage, which thereby impair transcription, leading to high levels of replication stress and consequent DNA damage (13).
Multiple studies have identified ATR as a key regulator of the R-loop response. ATR is essential for R-loop resolution and fork restart in concert with the endonuclease MUS81-EME1 (18), thereby protecting cells from R-loop-mediated replication stress and consequent DNA damage (19,20). This recent observation becomes important as a potential therapeutic opportunity with ATR inhibitor (ATRi) for PARPi-resistant HGSOC because aberrant R-loops induce ATR pathway activation and increased reliance on ATR for R-loop resolution. However, while targeting ATR is a scientifically rational approach for PARPi-resistant HGSOC, only modest clinical activity has been reported with ATRi monotherapy in previously PARPi-treated patients. For instance, berzosertib (M6620) monotherapy yielded a response rate of 6% in patients with advanced or relapsed solid tumors who progressed on PARPi (21), highlighting the complexity of PARPi resistance in the clinic and the urgent need for developing combination treatment strategies as well as identifying reliable biomarkers for patient selection.
Hence, we conducted an unbiased high-throughput drug combination screening to identify the novel combination with ATRi using PARPi-resistant BRCAm HGSOC cells. In that screen, several compounds targeting phosphoinositide 3-kinase (PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway were found to be synergistic with ATRi. In this study, we aimed to determine whether pharmacologic inhibition of ATR and AKT pathways affects R-loop dynamics as a novel mechanism of action and investigated how these R-loop regulators function in concert in HGSOC preclinical models. Here, we report that dual inhibition of ATR and AKT as a novel therapeutic strategy for both PARPi-sensitive and PARPi-resistant BRCAm HGSOCs. The combination therapy induces apoptosis, DNA damage, and lethal replication stress in BRCA2 mutant (BRCA2m) HGSOC in vitro and in vivo models. Mechanistically, AKT1 directly binds on the site of R-loops, and its activation prevents R-loop formation. AKT1 also physically interacts with DHX9 on R-loops, and their interaction is highly dependent on the kinase activity of AKT1. Collectively, our data provide novel insights into how AKT1 responds to R-loop formation in concert with DHX9 to prevent pathological R-loop accumulation and consequent lethal replication stress, establishing a new role for AKT in R-loops response. More importantly, our findings support the clinical development of ATRi and AKT inhibitor (AKTi) combination for HGSOC patients who progressed on PARPi.
Materials and Methods
Cell lines
PEO1 (BRCA2 mutation 5193C>G) was purchased from Sigma-Aldrich (#10032308-1VL, Saint Louis, MO, USA) and UWB1.289 (BRCA1 mutation 2594delC) was obtained from the American Type Culture Collection (ATCC; #CRL-2945, Manassas, VA, USA). OVCAR3 (BRCA wild-type [BRCAwt]) and OVCAR8 (BRCA1 promoter methylation) were received from NCI-60 collection at the NCI Frederick (Frederick, MD, USA). In addition, we have used two PARPi-resistant PEO1 variants, PEO1/OlaR (22) and PEO1/OlaJR (23), which harbor different mechanisms of PARPi resistance as previously described (23). Briefly, PEO1/OlaJR restores HR repair by a heterozygous BRCA2 reversion mutation in addition to increased drug efflux activity (23). In contrast, PEO1/OlaR, a gift from Dr. Bitler lab, retains its BRCA2 mutation while acquiring PARPi resistance through multiple mechanisms such as adoption of a mesenchymal-like phenotype, activation of ATR/CHK1 pathway, and suppression of the EZH2/MUS81 pathway, etc. (22). PARPi-resistant UWB/OlaR cells were generated in-house from parental UWB1.289 by exposure to olaparib from 0.4 μM to 20 μM for 10 months. PARPi-resistant cell lines were routinely maintained at 5 μM (PEO1/OlaR), 20 μM (UWB/OlaR and PEO1/OlaJR) of olaparib. Cells were cultured without olaparib for at least three days before being used for in vitro experiments. PEO1, UWB1.289, OVCAR3 and OVCAR8 cells were grown in RPMI1640 medium supplemented with (+) L-glutamine, 10% fetal bovine serum, 0.01 mg/mL insulin, and 1% penicillin/streptomycin. All cell lines were routinely tested for Mycoplasma using MycoAlert ® Mycoplasma Detection Kit (LT-07-318, Lonza, Rockville, MD, USA).
Chemical preparation
For in vitro assays, PARPi olaparib (#S1060), ATRi ceralasertib (#S7693), AKTi capivasertib (#S8019) were purchased from Selleck Chemicals (Houston, TX, USA). ATRi M4344 (#HY-136270) and AKTi M2698 (#HY-100501) were from MedCahemExpress (Monmouth Junction, NJ, USA). Caspase Inhibitor Z-VAD-FMK (#G7231, 20 mM in DMSO) was purchased from Promega (Madison, WI, USA). Hydroxyurea (HU; #H8627) was from Sigma-Aldrich. 100 mM of olaparib, capivasertib and HU as well as 10 mM of ceralasertib, M4344 and M2698 were prepared as stocks in dimethyl sulfoxide (DMSO; #S-002-M, Sigma-Aldrich). All drugs were stored in aliquots at −80°C until use. We used a consistent drug ratio of 1:10 within the clinically achievable concentrations of < 0.5 μM for ceralasertib and < 5 μM for capivasertib, for the 5-day XTT assay. For mechanistic studies conducted over a shorter duration of 6-72 hours, we used 1 μM ceralasertib and 10 μM capivasertib to achieve a therapeutic effect.
For in vivo studies, ceralasertib (AstraZeneca, Cambridge, United Kingdom) was prepared in the solution containing 10% DMSO and 40% propylene glycol (#P4347, Sigma-Aldrich) and 50% de-ionized sterile water. Capivasertib (AstraZeneca) was dissolved in 10% DMSO and 25% 2-hydroxy-propyl-β-cyclodextrin pH 5.0 (#332593, Sigma-Aldrich). Olaparib (#V300, InvivoChem, Libertyvile, IL, USA) was formulated in PBS containing 10% DMSO and 10% (w/v) 2-hydroxy-propyl-β-cyclodextrin.
High-throughput drug combination screening
High-throughput drug combination screening was performed as previously reported (24). Briefly, we first screened two PARPi-resistant (PEO1/OlaR and PEO1/OlaJR) against 2,450 combinations with ATRi using a 96-hour cell proliferation assay with an ATP-based readout CellTiter-Glo (#G7570, Promega) to determine activity and synergy of compounds in a dose-response manner. Each 6 × 6 matrix was scored by the sum of excess over highest single agent (ExcessHSA) for evidence of synergistic (ExcessHSA score < − 20), additive (− 20 ≤ ExcessHSA score ≤ 20), or antagonistic (ExcessHSA score > 20) effects, and the average ExcessHSA score of each compound was ranked accordingly. We deprioritized chemotherapy combinations with ATRi due to overlapping bone marrow toxicity for potential human clinical trials development. 10 × 10 matrix screens of the combination of PI3K/AKT pathway inhibitors with ATRi were conducted for confirmation. CellTiter-Glo added after 96 hours of compound incubation to inform on cell viability as described by the manufacturers. Luminescence signal was measured for 6 × 6 and 10 × 10 matrix screening using a Pherastar (BMG Labtech, Ortenberg, Germany) or a ViewLux (Perkin-Elmer, Waltham, MA, USA) reader, respectively.
Gene set enrichment analysis (GSEA) and protein association networks
RNA sequencing (RNAseq) data were obtained from fresh frozen tumor biopsy samples from 14 BRCAm PARPi-resistant HGSOC patients enrolled in the clinical trial at the National Cancer Institute (ClinicalTrials.gov, NCT02203513) as detailed before (25). Differential gene expression and Z-score normalization across genes were analyzed using DESeq2 (26). The gene list of PI3K/AKT pathway was downloaded from GSEA (KEGG dataset https://www.gsea-msigdb.org/gsea/msigdb/index.jsp). The list of R-loop regulators from immunoprecipitation of S9.6 (27) or syntenic DNA:RNA hybrids (28) (IP-MS) and proximity ligation (Prox-MS) methods (29,30) as well as known R-loop regulators (12,20) is provided in Supplementary Table S1. Protein association networks for the R-loop regulators were constructed using the STRING database version 11.5 (https://string-db.org).
Cell proliferation assays: XTT and colony-forming assays
Cell growth was evaluated for the drug response of ATRi and AKTi. For the short-term cell survival, XTT assay (#X6493, Thermo Fisher Scientific, Pittsburg, PA, USA) was performed. 2,000 cells/well were seeded in 96-well plates and treated with drugs for 5 days. Plates were measured by Synergy™ HTX Multi-Mode Microplate Reader with Gen5™ software (BioTek Instruments, Winooski, VT, USA). IC50 values were calculated using GraphPad Prism version 9.4.1 (GraphPad Software, Inc., La Jolla, CA, USA). The CI values were evaluated by CompuSyn software (ComboSyn, Inc., Paramus, NJ, USA). CI values < 1 indicate synergism while values > 1 indicate antagonism (31).
Colony-forming assay was used to determine the long-term effect of ATRi and/or AKTi treatment on cell survival. 5,000 cells were seeded in 24-well plates and treated with ATRi and/or AKTi. Media and drugs were changed every three days for 12 days. Fixed colonies were stained with 0.01% (w/v) crystal violet in PBS. Colony images were scanned and quantification of colony area percentage using Image J (NIH, Bethesda, MD, USA).
Immunoblotting
Cells were collected for protein extraction and subjected to immunoblotting. Blots were visualized using the Licor Odyssey Imaging System. Cleaved PARP (c-PARP, #5625), AKT1 (#2938), phospho-S6 ribosomal protein Ser235/Ser236 (#2211), phospho-CHK1 Ser345 (#2348), GAPDH (#5174), ECL goat anti-mouse IgG HRP (#7076) and ECL goat anti-rabbit IgG HRP (#7074) antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). DHX9 (#A300-855A) antibody was purchased from Bethyl Laboratories, Inc. (Montgomery, TX, USA).
Immunofluorescence staining
Cells were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.25% Triton-X 100, and blocked with 1% bovine serum albumin. For DNA damage endpoints, the cells with > 5 γH2AX foci were considered as γH2AX positive. Separately, cells were incubated with Alexa Fluor® 488 anti-H2A.X Phospho (Ser139) antibody (#613406, BioLegend, San Diego, CA) and anti-pRPA2 (S4/S8) (#ab87277, abcam, Cambridge, MA) to examine replication stress. Double-positive γH2AX and pRPA2 (S4/S8) cells (γH2AX+/pRPA+) were cells under replication stress. The signal intensity per nucleus were quantified using ImageJ.
Alkaline comet assay
DNA fragmentations were analyzed by alkaline comet assay according to the manufacturer’s instruction (Trevigen). The mean tail moment from three independent experiments (each experiment scored at least 100 cells/treatment) was calculated as an index of DNA damage by using CometScore Pro (TriTek Corporation, Sumerduck, VA, USA). Apoptosis inhibitor Z-VAD-FMK was used to exclude apoptosis-driven DNA fragmentation when conducting alkaline comet assays.
DNA fiber assay
DNA fiber assays were conducted to assess replication fork progression as described (32). Briefly, cells were treated with or without treatments for 24 hours and then labeled with 100 μM 5-chloro-2′-deoxyuridine (CldU; #C6891, Sigma-Aldrich) and 200 μM 5-Iodo-2’-deoxyuridine (IdU; #I7125, Sigma-Aldrich) for 20 minutes each. CldU- and IdU-labeled DNA fibers were stained with mouse anti-IdU (1:250; #NBP2-44056, Novus Biological, Centennial, CO) and rat anti-CldU (1:200; #NB500-169, Novus Biological) antibodies, respectively. Anti-rat Alexa Fluor 488 (1:250; #A-11006, Thermo Fisher Scientific) and anti-mouse Alexa 594 (1:250; #A-11005, Thermo Fisher Scientific) were used as secondary antibodies. Images were captured using a Zeiss LSM 780 confocal microscope. Fiber length was measured using ImageJ software. Replicating 1 μm roughly corresponds to 2.59 kb (33).
In vitro R-loop detection using dot-blot
Total nucleic acid was extracted by a standard SDS/Proteinase K digestion, followed by phenol/chloroform extraction and ethanol precipitation. The indicated amount of purified nucleic acid was applied to the Hybond N+/Positive nylon membrane (#RPN303B, GE Healthcare, Chicago, IL, USA) assembled in the Bio-Dot Apparatus (#1706545, Bio-Rad, Hercules, CA, USA). The membranes were then UV cross-linked (0.12 J/m2) and blocked with 5% milk/TBST (0.1% Tween 20) for 1 hour at room temperature. The level of R-loops was detected using the mouse monoclonal S9.6 antibody (#ENH001, Kerafast, Inc., Boston, MA, USA). Samples were treated with RNase H (#M0297L, New England BioLabs, Inc., Ipswich, MA) for 3 hours at 37°C to digest DNA:RNA hybrid as a negative control.
DNA-RNA immunoprecipitation (DRIP)
DRIP was performed as described (34). Briefly, DNA:RNA hybrids were immunoprecipitated using the S9.6 antibody (#ENH001, Kerafast, Inc.) from gently extracted and enzymatically digested DNA. In vitro RNase H digestion was used as a negative control. For each DRIP, 4 μg of genomic DNA was incubated with 10 μg of S9.6 antibody in the binding buffer (10 mM NaPO4 [pH 7.0], 140 mM NaCl, and 0.05% Triton X-100) and bound to Protein G Dynabeads (#10003D, Thermo Fisher Scientific). Control IP was performed using 10 μg of anti-mouse IgG antibody. Precipitated DNA was resuspended in 100 μl of distilled water, and 2 μl was used for qPCR analysis.
Chromatin immunoprecipitation (ChIP)
Cells were transfected with siRNAs against AKT1 for 72 hours. Cells were crosslinked with 3% formaldehyde and processed for ChIP using Pierce Agarose ChIP Kit (#26156, Thermo Fisher Scientific) according to manufacturer’s instructions. Anti-DHX9 (#A300-855A, Bethyl Laboratories, Inc) and anti-rabbit IgG antibody was used to immunoprecipitate chromatin.
Real-time PCR (qPCR)
mRNA expression levels of R-loop regulators were validated by qPCR. Total RNA from the cells was isolated using a RNeasy™ Mini kit (Qiagen, Germantown, MD). cDNA was synthesized using an iScript Advanced cDNA Synthesis Kit (#1725037, Bio-Rad, Hercules, CA) according to manufacturer’s instructions. Primers specific for DHX9, AQR, SUPT16H, SETX, TRDMT1, ARID1A, ILF3, PDCD11, DDX21, SIRT7 and endogenous control GAPDH were purchased from Bio-Rad (#10025636). qPCR was performed using SsoAdvanced Universal SYBR Green Supermix (#1725270, Bio-Rad) on a Bio-Rad CFX Opus 96 Real-Time PCR System and analyzed using CFX Maestro software.
Primers pairs for DRIP-qPCR (34) including R-loop-positive CALM3 (forward 5’-GAGGAATTGTGGCGTTGACT-3’, reverse 5’-AGAGTGGCCAAATGAGCAGT-3’) and RPL13A genes (forward 5’-AGGTGCCTTGCTCACAGAGT-3’, reverse 5’-GGTTGCATTGCCCTCATTAC-3’) as well as R-loop-negative SNRPN gene (forward 5’-GCCAAATGAGTGAGGATGGT-3’, reverse 5’-TCCTCTCTGCCTGACTCCAT-3’) were purchased from Bio-Rad. DRIP-qPCR analysis in the R-loop-positive CALM3 and RPL13A was presented as the percentage of input. R-loop-negative SNRPN gene was used as a negative control.
Primer pairs specific for DHX9 ChIP-qPCR analysis including promoter (5’-CCGAAAGTTGCCTTTTATGGC-3’, reverse 5’-CAAAGGCGAGGCTCTGTGC-3’), intron 1 (forward 5’-CGGGGTCTTTGTCTGAGC-3’, reverse 5’-CAGTTAGCGCCCAAAGGAC-3’) and pause (forward 5’-CCGAAAGTTGCCTTTTATGGC-3’, reverse 5’-CAAAGGCGAGGCTCTGTGC-3’), and 5’ pause (forward 5’-TTACCCAGAGTGCAGGTGTG-3’, reverse 5’-CCCCAATAAGCAGGAACAGA-3’) of β actin gene (35) were purchased from Bio-Rad. DHX9 ChIP-qPCR analysis of different genomic regions of β actin was presented as the percentage of input fold change that normalized with siControl group.
Proximity ligation assay (PLA)
The PLA was performed to detect the interactions between DHX9 and R-loops, AKT1 and R-loops or proliferating cell nuclear antigen (PCNA) and RNA polymerase II (RNAPII) at distances < 40 nm in situ following standard protocol as described (36). For R-loop interactions, cells were first transfected with or without ppyCAG_RNaseH1_WT (#111906, addgene, Watertown, MA, USA) for 48 hours. We re-seeded the transfected cells for further ATRi and/or AKTi treatments or wild-type AKT1 WT or AKT1 K179M transfection for another 48 hours. Cells were then fixed with 4% PFA, pretreated with RNase T1 (#EN0541, Thermo Fisher Scientific) and ShortCut® RNase III (#M0245S, New England BioLabs, Inc.) for 1 hour and applied to Duolink™ In Situ Red Starter Kit Mouse/Rabbit (#DUO92101, Sigma) according to manufacturer’s instructions. S9.6 (#ENH001, Kerafast, Inc.), DHX9 (#A300-855A, Bethyl Laboratories, Inc.) and AKT1 (#2938, Cell Signaling Technology) antibodies were used to examine the recruitment of DHX9 or AKT1 to the R-loops while PCNA (#ab18197, abcam) and RNAPII (#2629, Cell Signaling Technology) antibodies were used to detect TRCs. Average PLA foci were calculated by examining at least 50 cells per treatment in three different experiments. The number of foci per nucleus was determined using the ImageJ macro, Foci-analyzer.
Co-immunoprecipitation (co-IP) assay
Cell lysates were incubated with antibodies overnight at 4°C, followed by incubation with Protein A/G polyacrylamide beads (#53133, Thermo Fisher Scientific) for 2 hours. The beads were washed three times, and bound proteins were eluted with SDS-PAGE loading buffer. Proteins were separated on an SDS-PAGE gel and analyzed by immunoblotting. HA-Tag (#3724), rabbit IgG control (#3900), ECL goat anti-mouse IgG HRP (#7076) and ECL goat anti-rabbit IgG HRP (#7074) antibodies were from Cell Signaling Technology. GFP (#sc-9996) and AKT1 (#sc-5298) were from Santa Cruz Biotechnology (Dallas, TX, USA) and DHX9 antibody (#A300-855A) was purchased from Bethyl Laboratories, Inc.
siRNA transfections
As previously described (23), the cells were transfected with siRNA against AKT1 (#L-003000-00-0005), DHX9 (#L-009950-00-0005) or non-targeting control siRNA (#D-001810-01-05) for 48 hours using DharmaFECT 1 siRNA Transfection Reagent (#T-2001-01) purchased from Horizon Discovery, Saint Louis, MO, USA.
Plasmid transfections
To determine the interaction sites between DHX9 and AKT1, the cDNA clones of wild-type and serial deletion mutants AKT1 and DHX9 were transfected into cells. pCDNA3-HA-Akt1 (#73408), pCDNA3-HA-Akt1-K179M (#73409), pCDNA3-HA-Akt1-aa1-149 (#73410) and pCDNA3-HA-Akt1-aa1-408 (#73412) were from addgene. GFP-DHX9, GFP-DHX9 RBD (amino acids 1-262), GFP-DHX9 RGG (amino acids 1064-1270), and GFP-DHX9 helicase domain (amino acids 255-1077) were gifts from Dr. Yang (37). Cells were seeded and transfected with the above plasmids or empty vector at a 3:1 FuGENE® transfection teagent: DNA ratio according to the manufacturer’s instruction (#E2311, Promega).
Live imaging of AKT1 and DHX9 on R-loops
To determine the potential direct interaction of AKT1 and DHX9 in R-loop resolution, pEGFP-RNASEH1 (GFP-RNase H1, #108699, addgene), pmCherry-Akt1 (#86631, addgene) and full-length DHX9 cDNA clone (pTagBFP2-DHX9, VectorBuilder Inc., Chicago, IL, USA) were transfected into cells using FuGENE® transfection reagent for 72 hours. Cells were re-seeded onto an 8-well high glass bottom chamber slide (#80807, ibidi USA, Inc.). Live imaging was taken by Nikon SoRa Spinning Disk (Nikon Instruments Inc., Melville, NY, USA).
Animal study
All animal procedures reported in this study that were performed by NCI-CCR affiliated staff were approved by the NCI Animal Care and Use (ACU) committee and in accordance with federal regulatory requirements and standards. All components of the intramural NIH ACU program are accredited by Association for Assessment and Accreditation of Laboratory Animal Care International (AAALAC). Subcutaneous xenograft mouse models were used to evaluate the efficacy of ATRi ceralasertib and AKTi capivasertib in in vivo. 5×106 cells were suspended in 50 μl of cold PBS and mixed with 50 μl of Matrigel. A total of 100 μl cell mixture was subcutaneously injected to 6-week-old female NSG mouse (NCI, Frederick, MD, USA). The volume of tumor was measured once per week according to the formula V = ½ (length × width2). When tumors reached 50 mm3, mice were randomized into 5 groups (n=4/group). Mice received vehicle or 50 mg/kg ceralasertib orally once daily for 5 days, followed by 2 days of no treatment for a total of 4 weeks. For combination studies, 2 study arms were added, and mice received 130 mg/kg capivasertib orally twice daily for 4 days, followed by 3 days of no treatment with or without 50 mg/kg ceralasertib orally once daily for 5 days, followed by 2 days of no treatment for 4 weeks. Mice received 100 mg/kg olaparib orally once daily for 5 days, followed by 2 days of no treatment as a positive control group (8,9). All mice were sacrificed at the end of treatments.
Intraperitoneal injection mouse models were used to evaluate the metastasis and survival rate in mice with ATRi ceralasertib and/or AKTi capivasertib treatment. 5×106 cells were suspended in 100 μl of PBS and were injected to 6-week-old female NSG mouse intraperitoneally. After 2-3 weeks, mice were randomized into 4 groups (n=10/group). Mice received vehicle or 50 mg/kg ceralasertib orally once daily for 5 days, followed by 2 days of no treatment for a total of 4 weeks. For combination studies, 2 study arms were added, and mice received 130 mg/kg capivasertib orally twice daily for 4 days, followed by 3 days of no treatment with or without 50 mg/kg ceralasertib orally once daily for 5 days, followed by 2 days of no treatment. Disease progression was monitored based on overall health until a predetermined endpoint was reached. Survival time reflects the time required for the animals to reach any endpoints, including tumor ulceration, weight loss exceeding 15%, weight gain exceeding 5g, anorexia, and/or diarrhea.
Immunohistochemistry (IHC) staining
Tumor tissue sections were deparaffinized in xylene and rehydrated in graded alcohols, preincubated with 3% H2O2, boiled in 10 mM citrate buffer and blocked with goat serum. Afterwards, sections were incubated with primary antibodies against R-loops (#ENH001, Kerafast, Inc.), pRPA32 (S4/S8) (#ab87277, abcam) and γH2AX (Ser139) (#9718, Cell Signaling Technology) at 1:100 dilution for overnight incubation, respectively. Sections were labeled by avidin-biotin-peroxidase complex using VECTASTAIN® ABC-HRP Kit (peroxidase) (rabbit IgG #LS-J1010 and mouse IgG #LS-J1011, LSBio, Seattle, WA, USA) and followed by diaminobenzidine development using ImmPACT DAB substrate (#SK-4105, Vector Laboratories, CA, USA) according to manufacturer’s instructions. The slides were counterstained with hematoxylin stain solution (#MHS16, Sigma-Aldrich) and mounted in Eukitt Mounting Medium (#15322, Electron Microscopy Sciences, Hatfield, PA, USA). Images were captured using an ECLIPSE Ts2 microscope (Nikon Instruments Inc.). Image J software with the IHC profiler plugin was used to analyze the percentage of positive staining area as previously described (38).
Genetic alteration databases of HGSOC patients
Genetic alterations including mutations, structural variants, and copy number changes along with mRNA expression profiling of HGSOC patients from The Cancer Genome Atlas (TCGA) database were downloaded from cBioPortal (https://www.cbioportal.org/) and GEPIA (http://gepia.cancer-pku.cn/). The prognostic value of AKT1, ATR, DHX9, and other R-loop regulators was evaluated using Kaplan-Meier Plotter (http://www.kmplot.com), which contains mRNA expression data and survival information of HGSOC patients. Tumors with only serous histology and TP53 mutation (indicating HGSOC) were included for this analysis. To analyze the PFS of HGSOC patients, patient samples were split into two groups by median expression (high versus low expression) and assessed by a Kaplan-Meier survival plot, with the hazard ratio with 95% confidence intervals and log rank p value.
Statistical analysis
All experiments were performed at least in triplicate. Data were analyzed using one-way ANOVA test and shown as mean ± SEM. The overall survival of mice bearing tumors was analyzed using Kaplan–Meier curves applying log-rank (Mantel-Cox) test. All differences were considered statistically significant if P < 0.05. All statistical analyses were done using GraphPad Prism version 9.4.1.
Data availability
Raw RNAseq data from PARPi-resistant BRCAm HGSOC tumors are available in Gene Expression Omnibus (GEO) at GSE252610. The genomic alteration of HGSOC analyzed in this study was obtained from cBioPortal at https://www.cbioportal.org/ and GEPIA at http://gepia.cancer-pku.cn/. The mRNA expression data and survival data of AKT1, ATR, DHX9, and other R-loop regulators analyzed in this study were obtained from Kaplan-Meier Plotter [ovarian cancer] at http://www.kmplot.com. All summary data are available in the article or uploaded as Supplementary Data, and all raw data are available upon request from the corresponding author.
Results
High-throughput drug combination screening identifies PI3K/AKT pathway inhibitors for combination with ATR blockade in PARPi-resistant BRCA2m HGSOC cells
First, we performed a high-throughput drug combination screen using two acquired PARPi-resistant BRCA2m HGSOC cell lines (PEO1/OlaR [no BRCA2m reversion mutation] (22) and PEO1/OlaJR [BRCA2 heterozygous reversion mutation] (23)) (Fig. 1A) to identify the drug candidates for combination with ATRi. Ceralasertib (AZD6738) was selected as a lead ATRi compound for the screens against the Mechanism Interrogation Plate Library (MIPE 5.0)(39) because ceralasertib is currently in the late stage of clinical development for recurrent HGSOC (40,41). Overall, 11.6% of these compounds (284/2,450) showed synergistic effect (ExcessHSA < −20) with ceralasertib and all compounds were ranked based on average ExcessHSA scores (Fig. 1A and Supplementary Table S2). Among the top 100 candidates, the most obvious trend was towards compounds targeting the cell cycle checkpoint (10.2%, 29/284), e.g., the CHK1 inhibitor AZD-7762 (rank 2) and the WEE1 inhibitor adavosertib (rank 12). It was also notable that PI3K/AKT pathway related drugs (7.0%, 20/284) such as a PI3K inhibitor CUDC-907 (rank 84), AKTi triciribine (rank 98) and the mTOR inhibitor ridaforolimus (rank 18) act synergistically with ATRi.
Figure 1. PI3K/AKT pathway inhibitors acting synergistically with an ATRi in PARPi-resistant BRCA2m HGSOC cells.
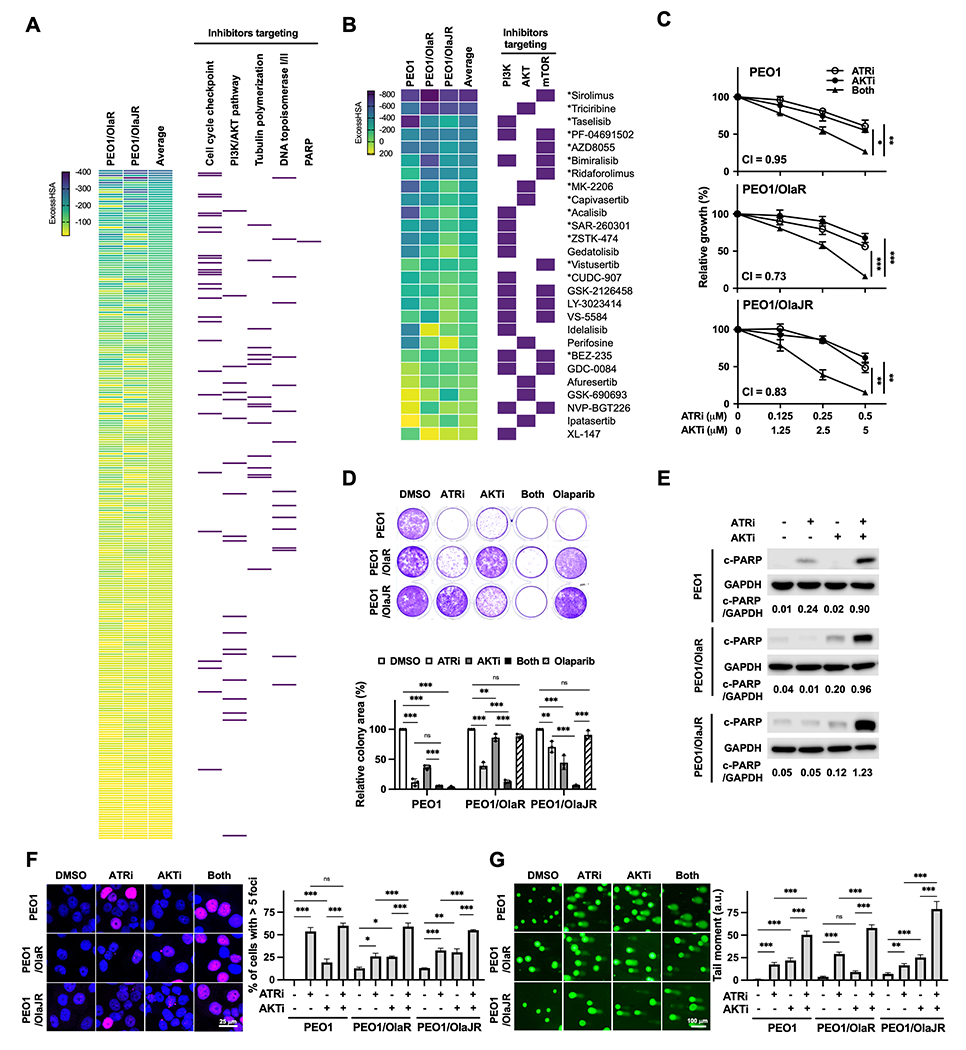
A, Hierarchal view of 6 × 6 initial drug combination screening in PARPi-resistant (PEO1/OlaR and PEO1/OlaJR) BRCA2m HGSOC cells. Drugs that were found to be synergistic with ATRi ceralasertib (ExcessHSA < −20) were ranked using the average ExcessHSA values; more negative ExcessHSA values indicate greater potency. Purple rectangles on the right highlight the agents from key mechanistic classes, including inhibitors targeting cell cycle checkpoint, PI3K/AKT pathway, topoisomerase I/II, tubulin polymerization, and PARP. B, 10 × 10 matrix screening of PI3K/AKT pathway inhibitors and ATRi ceralasertib in PARPi-sensitive (PEO1) and PARPi-resistant (PEO1/OlaR and PEO1/OlaJR) BRCA2m HGSOC cells. Purple rectangles on the right highlight drugs targeting PI3K isoform, AKT, and mTOR. * Indicates the synergy seen in all cell lines. C, Cell growth was assessed using XTT assays (n = 4). Cells were treated with ATRi ceralasertib and/or AKTi capivasertib at indicated doses for 5 days. Combination index (CI) values <1 indicate synergism. D, Long-term survival was evaluated by colony-forming assays (n = 3). Cells were treated with ATRi (0.5 μM) and/or AKTi (5 μM) and grown for 12 days. E-G, Cells were treated with ATRi (1 μM) and AKTi (10 μM) for 48 hours. E, Cell apoptosis effect was assessed by immunoblotting of cleaved PARP (c-PARP). GAPDH was used as a loading control. Densitometric values of c-PARP relative to GAPDH are shown. F-G, DNA damage was examined by immunofluorescence staining for γH2AX foci (n = 3) (F) and alkaline comet assay (n = 3) (G). F, Representative images of γH2AX foci (pink) and nuclei (DAPI, blue) (left). The percentage of cells with > 5 γH2AX foci representing cells with DNA damage is plotted (right). G, Representative images of comet assays are shown (left). The tail moment, including the product of the tail length and the fraction of total DNA, is plotted (right). Data from C, D, F, G were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
For the subsequent experiments, we prioritized the inhibitors of PI3K/AKT pathway for ATRi combination given that PI3K/AKT pathway is frequently upregulated in HGSOC, and its activation is associated with aggressive phenotypes and platinum and/or PARPi resistance (42,43). Consistently, there was increased expression of PI3K/AKT pathway genes in RNAseq data from PARPi-resistant BRCAm HGSOC patients tumors (Supplementary Fig. S1A). Based on the findings from the initial 6 x 6 matrix drug combination screen and our interest of clinical trial development, a more detailed analysis was conducted with the 10 x 10 matrix screens. We shortlisted 15 drugs targeting the PI3K/AKT pathway that exhibited consistent synergistic effects with ATRi in all HGSOC cell lines (indicated as * in Fig. 1B and Supplementary Table S3). Notably, none of these cell lines harbored the PIK3CA mutation (23).
AKT inhibition shows synergistic cytotoxic effects with ATRi in drug-resistant HGSOC cells
We selected ATRi ceralasertib and AKTi capivasertib for further in vitro and in vivo studies. Capivasertib was chosen because it showed strong synergism with ATRi in all cell lines (mean ExcessHSA −308.10, Supplementary Table S3) and in the late stage of clinical investigation for women’s cancers (44). Cell growth assays were performed to validate the cytotoxic effects of ATRi and AKTi in PARPi-resistant BRCA2m HGSOC cells along with platinum-resistant OVCAR3 (BRCAwt) and OVCAR8 (BRCA1 promoter methylation) HGSOC cells. Using clinically attainable concentrations of both drugs (ceralasertib ~0.98 μM (41,45) and capivasertib ~4.45 μM (46)), the combination resulted in synergistic cytotoxic effects (combination index [CI] < 1) in both PARPi-resistant and PARPi-sensitive BRCA2m PEO1 cells (Fig. 1C). Combination treatment also significantly reduced colony-forming ability of PARPi-resistant HGSOC cells (Fig. 1D). We noted PARPi-resistant PEO1/OlaR exhibited a greater sensitivity to ATRi alone likely due to its elevated ATR/CHK1 activation (23) while PARPi-resistant PEO1/OlaJR showed a greater sensitivity to AKTi monotherapy possibly due to its increased AKT activation (23) (Fig. 1C–D). The on-target effects of ATRi and/or AKTi were confirmed by immunoblotting, showing reduced phosphorylated CHK1 (pCHK1 Ser345) and phosphorylated S6 ribosomal protein (pS6 Ser235/Ser236, Supplementary Fig. S1B), consistent with previous reports (47,48). Additionally, cell survival assays with another ATRi (M4344) and AKTi (M2698) for combination exhibited reduced cell growth (Supplementary Fig. S1C), indicating the synergistic cytotoxicity of ATRi and AKTi was unlikely due to drug-specific or off-target effects. The combination also significantly reduced cell growth compared to each drug alone in platinum-resistant HGSOC cell lines (Supplementary Fig. S2A–B), suggesting broad applicability of the ATRi and AKTi combination in drug-resistant HGSOC regardless of HR repair proficiency status.
We examined apoptosis and DNA damage endpoints to understand the mechanisms behind the increased cytotoxicity induced by the combination. As shown in Fig. 1E and Supplementary Fig. S2C, the 48-hour combination treatment resulted in greater apoptosis as evidenced by increased cleaved PARP (c-PARP), compared to ATRi or AKTi alone. The combination also increased the percentage of cells with > 5 γH2AX foci (Fig. 1F and Supplementary Fig. S2D) and the mean alkaline comet tail moment (Fig. 1G and Supplementary Fig. S2E) in all cell lines compared to each drug alone. Since both ATR/CHK1 and PI3K/AKT pathways are critical for the survival of drug-resistant cells, we speculate that ATRi and AKTi combination would have broad therapeutic potential in various drug-resistant HGSOC subtypes, as multiple resistance mechanisms often co-exist in the clinical setting.
AKTi augments ATRi-induced DNA damage and replication stress
Replication protein A (RPA) was analyzed to determine if the observed increase in DNA damage was linked to replication stress given that RPA protects ssDNA at replication forks from degradation, thus used as a marker of stalled replication forks (49). We found a higher population of double positive cells for phosphorylated RPA (pRPA) and γH2AX with the combination of ATRi and AKTi compared to cells treated with ATRi or AKTi alone, especially in PARPi-resistant and platinum-resistant HGSOC cells (Fig. 2A and Supplementary Fig. S2F). Furthermore, DNA fiber assays revealed a significant reduction in replication fork speed indicating stalled replication forks with combination therapy compared to each monotherapy, in both PARPi-sensitive and -resistant PEO1 cells (Fig. 2B). These results suggest that dual inhibition of ATR and AKT can induce greater cell death by increasing DNA damage and lethal replication stress in various HGSOC cell lines, including those with PARPi or platinum resistance as well as drug-sensitive HGSOC cell lines. We focused on PARPi-resistant HGSOC cells for subsequent mechanistic investigations as they represent an important population with an unmet medical need.
Figure 2. AKTi enhances ATRi-induced DNA damage and R-loop accumulation.
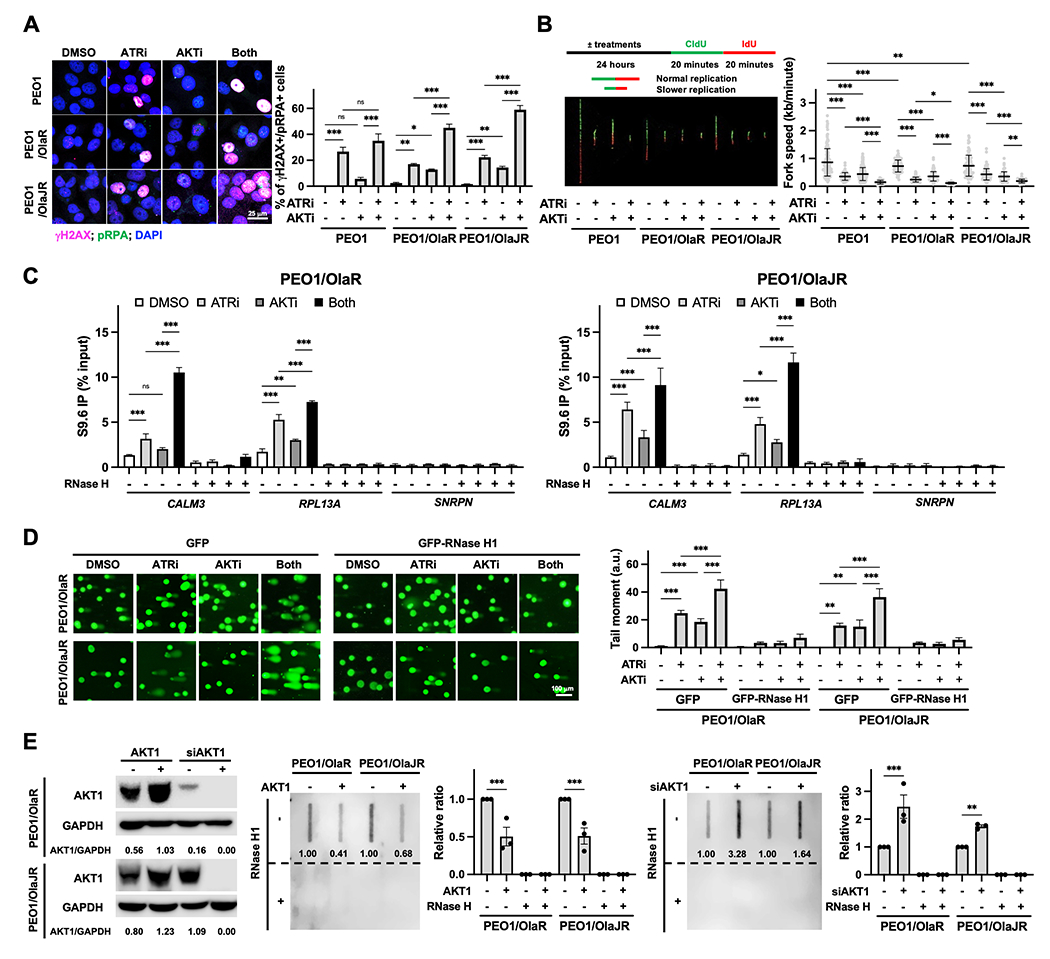
A, Immunofluorescence staining of γH2AX (pink; DNA damage marker) and pRPA (green; stalled replication fork marker) were performed to examine replication stress (n = 3). Cells were treated with ATRi ceralasertib (1 μM) and AKTi capivasertib (10 μM) for 48 hours. Representative images are shown (left). The percentage of double-positive cells, indicative of replication stress, is plotted (right). B, DNA fiber assays were performed to assess replication fork dynamics (n = 3). The schematics (left, top) and representative fibers (left, bottom) are shown. Average replication fork speeds in each group are shown (right). C, DRIP was conducted using the S9.6 monoclonal antibody in PARPi-resistant cells treated with ATRi and/or AKTi. RNase H treatment was used as a negative control. The qPCR analysis of R-loop positive foci of CALM3 and RPL13A genes and R-loop negative SNRPN gene (n = 3) was plotted using the percentage of input. D, Alkaline comet assay (n = 3) in cells treatment with ATRi ceralasertib (1 μM) and/or AKTi capivasertib (10 μM) in the presence of 20 μM pan-caspase inhibitor Z-VAD-FMK for 72 hours with GFP or GFP-RNase H1 overexpression. Representative immunofluorescence images of the comet tail are shown (left). The tail moment is plotted (right). E, Effect of AKT1 overexpression or knockdown on R-loop formation (n = 3). Cells were transfected with siAKT1 (25 nM) or HA-tagged AKT1 (2 mg) for 72 hours. The overexpression or knockdown efficiency of AKT1 was assessed by immunoblotting and GAPDH was used as a loading control. Densitometric values of AKT1 relative to GAPDH are shown (left). Dot-blot analysis of R-loops using S9.6 antibody in genomic DNA from transfected cells (right). Ratios relative to control group are shown. Bottom panels show the same analysis using genomic DNA after RNase H1 treatment were used as negative control. Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
Increased DNA damages are associated with aberrant R-loops
It is increasingly recognized that pathological R-loop accumulation is a major source of replication stress and consequently activates ATR kinase for R-loop unwinding and DNA repair (12,20). While multiple studies have identified that ATR plays a critical role in preventing aberrant R-loops by decreasing the nuclease activity of MUS81-EME1 (18) or increasing the nuclear localization of DDX19 (50), how R-loops contributes to the ATRi alone or ATRi-based combination therapy effects is still not fully understood. Hence, we performed DNA-RNA immunoprecipitation (DRIP)-qPCR experiments using the S9.6 antibody (34) to address this question. Gene loci where R-loops have been previously shown to occur during normal transcription were selected for analysis, including RPL13A and CALM3 genes (51,52). While ATRi alone was expected to induce aberrant R-loops, it is notable that AKTi monotherapy also resulted in increased R-loops (Fig. 2C). Notably, we observed greater R-loop levels within these genes in cells treated with the combination compared to monotherapy (Fig. 2C) while increased R-loops were diminished by RNase H treatment (Fig. 2C). SNRPN negative control locus was similar to the background resulting from RNase H treatment (Fig. 2C). These data suggest that pharmacologic inhibition of ATR and AKT induces R-loop accumulation in PARPi-resistant HGSOC cells.
We also conducted a set of experiments using ectopic expression of RNase H1 which resolves R-loops (14) to determine whether R-loop accumulation induced by the combination treatment ultimately leads to DNA damage. Accordingly, DNA damages by combination therapy were remarkably decreased by RNase H1 overexpression (Fig. 2D). Of note, AKTi-induced DNA damage was also significantly reduced by RNase H1 overexpression (Fig. 2D), suggesting that DNA damage by AKTi monotherapy was associated with R-loop formation. These results indicate that lethal replication stress with ATRi and AKTi combination is associated with R-loop–mediated DNA damage, opening the possibility of AKT’s direct role for R-loop resolution.
We then knocked down AKT1 using AKT1 siRNAs and performed a dot-blot assay to evaluate the levels of R-loops. Depletion of AKT1 significantly induced higher levels of R-loops relative to the basal level, whereas overexpression of AKT1 resolved R-loop formation (Fig. 2E). Again, the increased R-loop accumulation was diminished by RNase H1 overexpression (Fig. 2E). We also conducted cell growth assays in cells with RNase H1 overexpression to determine whether the combination-induced cell growth inhibition was due to R-loop accumulation. RNase H1 overexpression partially rescued the reduced cell growth caused by AKTi and ATRi (Supplementary Fig. S3A–B). Collectively, our findings reveal a possible direct involvement of AKT1 in R-loop–mediated replication stress response and the novel mechanisms of action of AKTi and ATRi combination in PARPi-resistant HGSOC cells.
R-loop resolution genes are upregulated in PARPi-resistant BRCAm HGSOC patients
Although R-loop dynamics have been extensively studied in cell line models (12,20), the potential of R-loop resolution–related genes as biomarkers of replication stress in clinical settings remains unknown. We speculated that PARPi-resistant BRCAm HGSOC tumors might upregulate R-loop resolution genes to circumvent lethal replication stress and exhibit higher tolerance to PARPi. To test this idea, we first identified candidate R-loop resolution genes in the context of HGSOC. We referred to the published literature that used various cancer models (12,20,27–30) and generated a consensus list of 66 genes (Fig. 3A and Supplementary Table S1).
Figure 3. R-loop resolution genes are upregulated in PARPi-resistant BRCAm HGSOC patients.
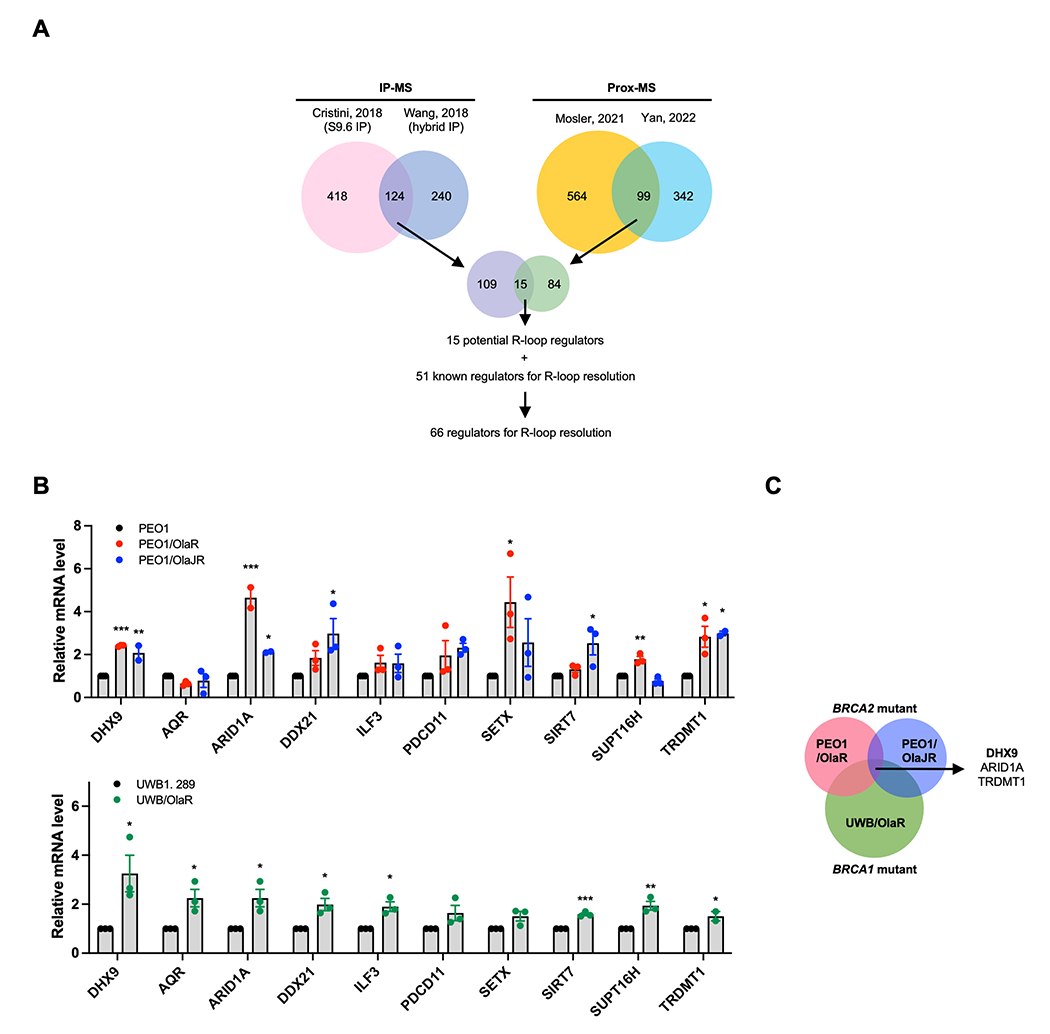
A, Scheme for generating the list of R-loop regulators identified by published proteomic studies. B, mRNA expression levels of R-loop resolution genes in PARPi-sensitive (PEO1, UWB1.289) and PARPi-resistant HGSOC cell lines (PEO1/OlaR, PEO1/OlaJR, UWB/OlaR) were analyzed by qPCR (n = 3). C, Venn diagram showing the overlap of upregulated R-loop regulators in PARPi-resistant BRCAm HGSOC cells. Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
To further validate the clinical relevance of these upregulated genes, we cross-referenced TCGA and Kaplan-Meier plotter ovarian cancer databases. In the public datasets, 10 genes with high expression were associated with poor progression-free survival (PFS) and possible resistance to platinum drugs (Supplementary Table S4). Moreover, alterations (e.g., mutations, amplification, or mRNA levels) of those genes were relatively frequent (>10%) in HGSOC patients (Supplementary Table S4). In our RNAseq data from PARPi-resistant BRCAm HGSOC tumors, these R-loop resolution genes were highly expressed in about two thirds of tumor samples (9/14) (Supplementary Fig. S4A). Also, the STRING database exhibited possible interaction of ATR and AKT1 pathways with those 10 genes, except TRDMT1 (Supplementary Fig. S4B).
In addition, we examined their mRNA expression levels in three PARPi-resistant BRCAm HGSOC cell lines for subsequent mechanistic investigations. Our findings indicated consistent upregulation of DHX9, ARID1A, and TRDMT1 in all PARPi-resistant BRCAm cell lines as compared to their parental counterparts (Fig. 3B–C). We focused on the DNA/RNA helicase DHX9 for further mechanistic studies, as it is a known substrate of ATR (53) and indirectly activates AKT by enhancing the activity of PI3K regulatory subunit p85 for cell survival (54) but its interaction with AKT for R-loop resolution is not well understood.
AKT1 regulates the interaction between R-loop and DHX9
We first investigated whether AKT1 affects the colocalization of DHX9 and R-loops by using PLA. While ATRi was expected to mitigate the recruitment of DHX9 (53), AKTi alone also decreased the nuclear DHX9-R-loop PLA foci (Fig. 4A), suggesting AKT independently modulates the interaction between DHX9 and R-loops. The addition of ATRi further reduced the DHX9-R-loop PLA foci (Fig. 4A). We also silenced AKT1 using AKT1 siRNAs to evaluate the biological role of AKT1 in R-loop dynamics. Depletion of AKT1 significantly mitigated the DHX9-R-loops PLA signals compared to those transfected with control siRNAs (Fig. 4B), supporting the notion of AKT1’s involvement in regulating the interaction between DHX9 and R-loops.
Figure 4. AKTi augments ATRi-induced R-loop accumulation by decreasing DHX9 recruitment to R-loops.
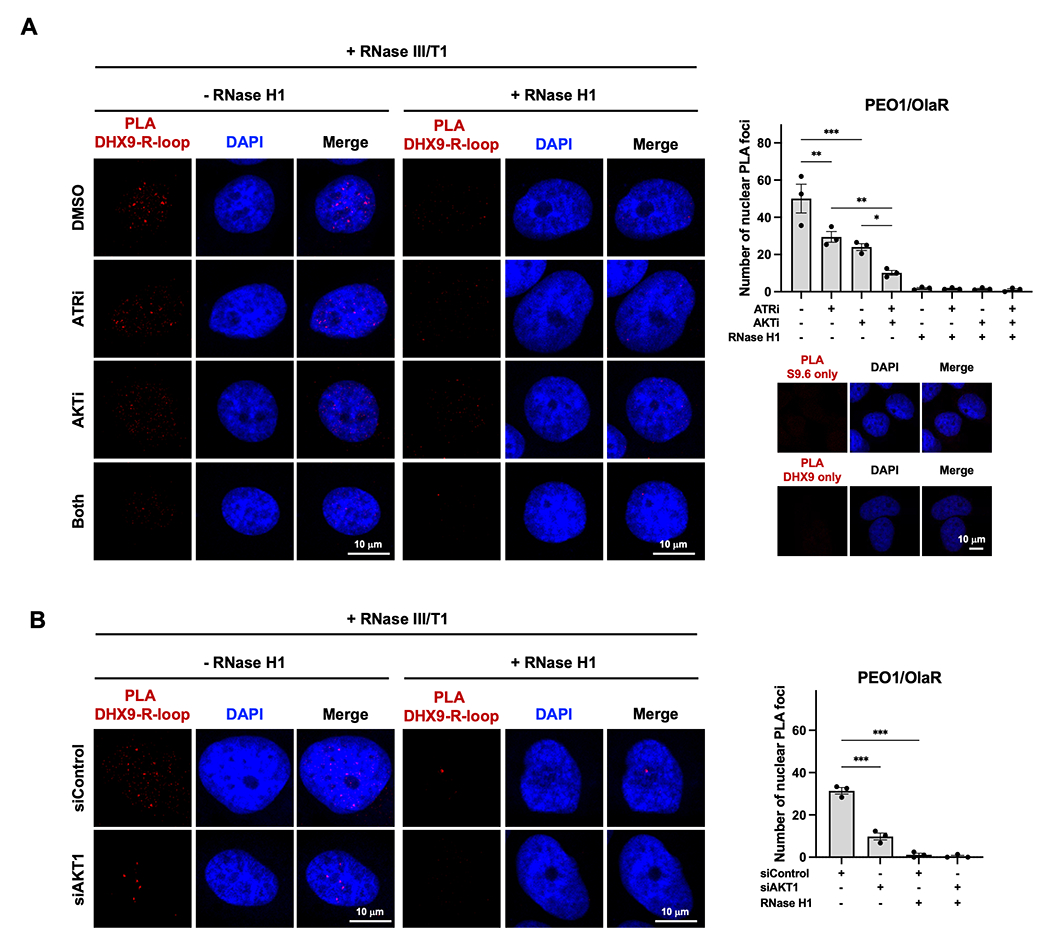
A-B, PARPi-resistant PEO1/OlaR cells were transfected with (+) or without (−) RNase H1. A, PLA was conducted to detect the interactions between R-loops and DHX9 in cells treated with ATRi ceralasertib (1 μM) and/or AKTi capivasertib (10 μM) for 48 hours (n = 3). Representative immunofluorescence images of DHX9-R-loops PLA foci (red) and DAPI (blue) in cells pretreated with RNase III/T1 treatment are shown (left). Number of PLA nuclear foci was plotted (right top). Representative immunofluorescence images of DHX9 or S9.6 antibody only PLA are shown (right bottom). B, Representative immunofluorescence images of DHX9-R-loops PLA foci (red) and DAPI (blue) in cells transfected with siRNAs against AKT1 or scramble control for 48 hours and RNase III/T1 pretreatment (left). Number of PLA nuclear foci was counted and plotted (right) (n = 3). Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant.
AKT1 directly interacts with DHX9 for R-loop resolution
We next questioned how AKT1 affects the recruitment of DHX9 to the R-loops. Evidence has shown that DNA repair proteins such as BRCA1 (55) and DNA-PK (56) directly interact with DHX9 and facilitate its recruitment to the R-loops for R-loop resolution and R-loop–associated DNA damage repair. We thus hypothesized that AKT1 may also bind to DHX9 to assist its recruitment to the R-loops. In support of this idea, immunofluorescence staining exhibited endogenous DHX9 colocalized with AKT1 on R-loops in PARPi-resistant HGSOC cells (Supplementary Fig. S5A, white dotted circles 1-3). Co-IP assay also verified that endogenous DHX9 physically bound to AKT1 in PARPi-resistant HGSOC cells (Fig. 5A), indicating a direct interaction between DHX9 and AKT1.
Figure 5. Endogenous AKT1 physically interacts with DHX9.
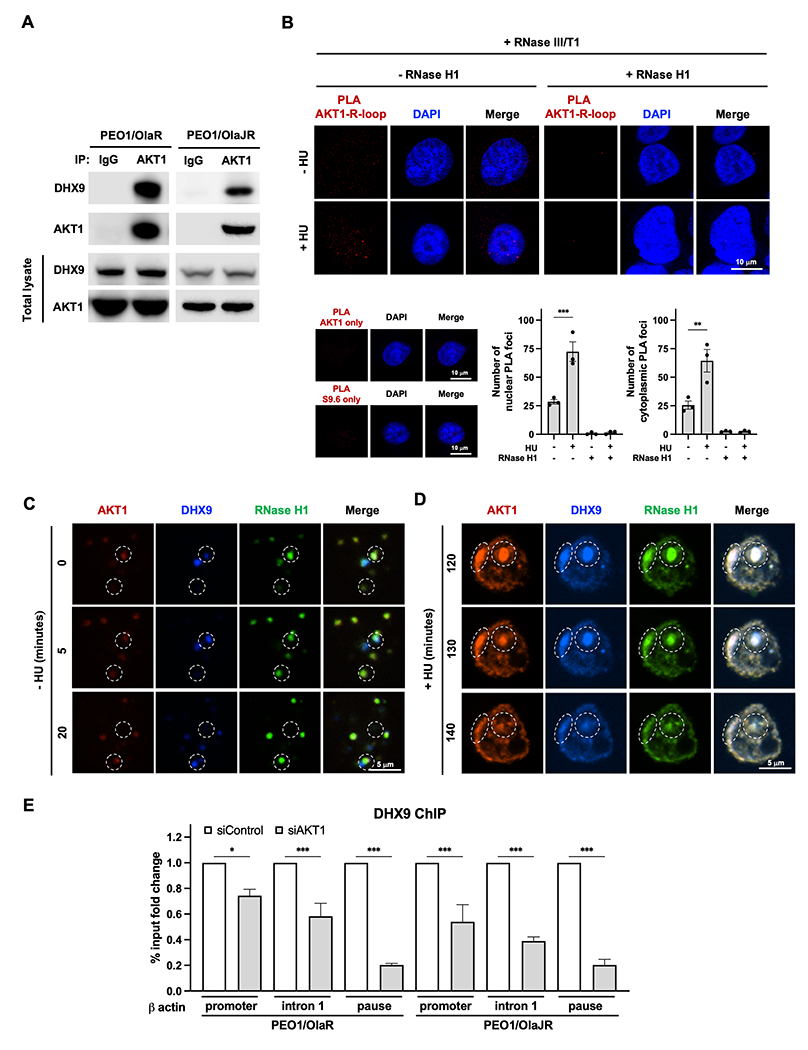
A, Co-IP was performed to study the interaction between endogenous DHX9 and AKT1 in PARPi-resistant BRCA2m HGSOC cells (n = 3). Bound AKT1 or DHX9 proteins were analyzed by immunoblotting. B, PLA was conducted to detect the interactions between R-loops and AKT1 under replication stress (n = 3). Representative immunofluorescence images of AKT1-R-loops PLA foci (red) and DAPI (blue) in cells treated with (+) or without (−) 4 mM HU treatment for 2 hours (toxic replication stress condition) with (+) or without (−) RNase H treatment are shown (left). Number of PLA nuclear foci was plotted (bottom). Data are shown as mean ± SEM. **, P < 0.01. C-D, AKT1 couples with DHX9 to resolve R-loops in PARPi-resistant BRCA2m HGSOC cells (n = 3). Representative images of AKT1 (red), and DHX9 (blue) and RNase H1 (green) in PEO1/OlaR cells (C) without (−) or (D) with (+) 4 mM HU treatment for 2 hours (toxic replication stress condition) captured from live cell imaging. In the same time frame (20 minutes), AKT1 showed up immediately when R-loops were displayed and cleared R-loops with DHX9 within 20 minutes (C, white dotted circle, captured from Supplementary Movie S1), while AKT1 and DHX9 and RNase H1 formed significantly larger foci in the nucleus of PARPi-resistant cells with HU treatment (D, white dotted circles, captured from Supplementary Movie S2). E, DHX9 ChIP in PARPi-resistant cells transfected siRNAs against AKT1 (siAKT1) or scramble control (siControl). ChIP-qPCR analysis of DHX9 recruitment to different R-loop-prone genomic regions of β actin was conducted. Data are presented as % input fold change compared to siControl group (n = 3). Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; ***, P < 0.001.
AKT1 has not been previously reported as part of the R-loop interactome, we therefore conducted a PLA analysis to investigate the direct association between AKT1 and R-loops under replication stress. Our findings suggest AKT1’s direct involvement in R-loop resolution under replication stress, as evidenced by the increased colocalization of AKT1 with both nuclear and cytoplasmic R-loops (Fig. 5B), which are caused by aberrant R-loop processing (57). To further visualize the interaction between AKT1 and DHX9 on R-loops, we co-transfected the mCherry-AKT1, BFP-DHX9, and GFP-RNase H1 and performed fluorescence live cell imaging. In live cell imaging, AKT1 appeared first when RNase H1 and DHX9 were displayed, and their signals rapidly disappeared at the same location after 20 minutes without any treatment (Supplementary Movie S1 and Fig. 5C, white dot circle). Also, PARPi-resistant HGSOC cells were treated with 4 mM of HU for 2 hours to induce high levels of replication stress as HU is known to deplete deoxynucleoside triphosphate pools (58). Live cell imaging showed that AKT1, RNase H1 and DHX9 formed significantly larger foci in the nucleus of PARPi-resistant cells with HU treatment, which decreased after 20 minutes (Supplementary Movie S2 and Fig. 5D), suggesting AKT1 prevents excessive R-loop accumulation in concert with RNase H1 and DHX9 in the cells under toxic replication stress. This finding supports our notion that AKT1’s interaction with R-loop regulators is crucial in removing of aberrant R-loops under both physiologic and toxic replication stress conditions.
We also evaluated the role of AKT1 on recruitment of DHX9 to genomic sites prone to R-loops to assess the importance of their interactions in resolving R-loops. β-actin was used as a tested gene because of its known R-loop enrichment in promoter-proximal and termination regions (35), and DHX9 enrichment in promoter-proximal regions (27). ChIP-qPCR results demonstrated a significant decrease in recruitment of DHX9 to R-loop-prone genomic regions when AKT1 is silenced (Fig. 5E), supporting a direct role of AKT1 in DHX9 recruitment to R-loops.
AKT1 resolves aberrant R-loops by physically binding to DHX9 via its kinase domain
To identify the specific binding sites between AKT1 and DHX9, cells were transfected with serial deletion mutants of AKT1 and DHX9. Of note, AKT1 is a 480 amino acid protein that contains an N-terminal pleckstrin homology (PH) domain followed by a catalytic kinase domain and culminates in a regulatory C-terminal disordered tail (REG)(59) (Fig. 6A, top). Co-IP with the deletion mutants of AKT1 demonstrated that AKT1 interacts with DHX9 mainly through its kinase domain (Fig. 6A, bottom). Moreover, cells transfected with kinase-dead AKT1 mutant (K179M) revealed significantly weaker binding activity with DHX9 compared to those with wild-type AKT1 (WT) (Fig. 6A, bottom), indicating that the interaction between AKT1 and DHX9 is highly dependent on the kinase activity of AKT1.
Figure 6. AKT1 resolves R-loop formation by directly interacting with DHX9 via its kinase domain.
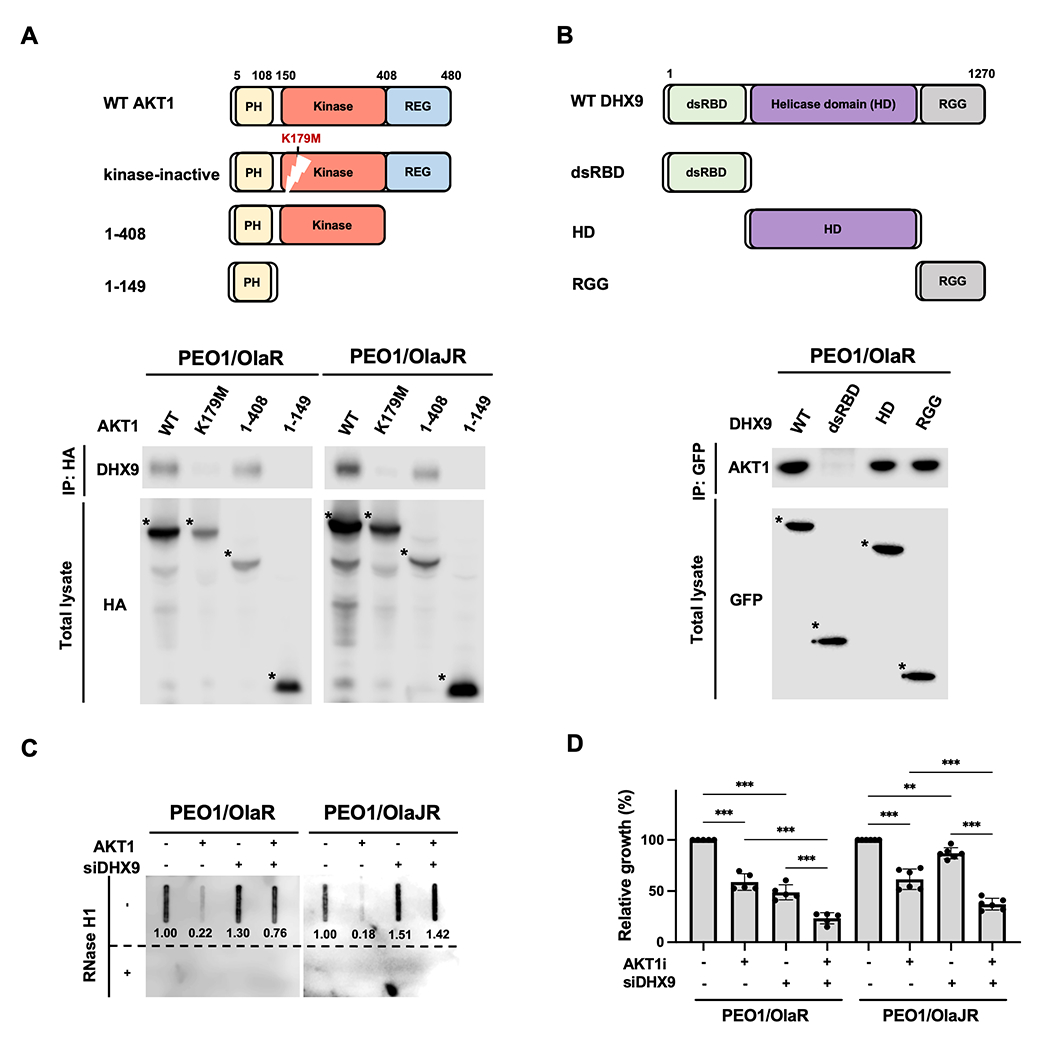
A-B, Schematic representations of AKT1, DHX9 and their serial-deletion mutants (top). Co-IP was performed to study the interaction between DHX9 and AKT1 (bottom). PARPi-resistant HGSOC cells were transfected with (A) HA-tagged full-length or deletion mutants of AKT1 or (B) GFP-tagged full-length or deletion mutants of DHX9. Cell lysates were collected for performing a co-IP assay (n = 3). Bound AKT1 or DHX9 proteins were analyzed by immunoblotting. The full-length or deletion mutants of AKT1 and DHX9 were detected (indicated as *). C, Dot-blot analysis of R-loops. Genomic DNAs were collected from PARPi-resistant cells transfected with siDHX9 and/or GFP-AKT1 (AKT1) for 72 hours. Ratios relative to control group are shown. Bottom panels show the same analysis using genomic DNAs after RNase H1 treatment as control. D, PARPi-resistant HGSOC cells were transfected with siRNAs against DHX9 or scramble controls for 48 hours and then treated with AKTi for another 48 hours. Cell growth was measured by XTT assay (n = 5). Data were analyzed using one-way ANOVA test and shown as mean ± SEM. **, P < 0.01; ***, P < 0.001.
We depleted endogenous AKT1 using AKT1-targeting siRNAs and reintroduced a kinase-dead AKT1 mutant into PARPi-resistant cells to examine the effect of AKT1 kinase activity on the recruitment of DHX9 to R-loops and R-loop resolution (Supplementary Fig. S5B). With reintroduction of the kinase-dead AKT1 mutant, there was a substantial increase in R-loop levels within RPL13A and CALM3 genes (Supplementary Fig. S5C) and a significant decrease in DHX9-R-loop PLA foci (Supplementary Fig. S5D), similar to the effects observed with AKT1 silencing. Conversely, reintroduction of AKT1 WT reduced R-loop levels and rescued the interaction between DHX9 and R-loops that were affected by AKT1 knockdown at baseline (Supplementary Fig. S5C–D).
Next, we expressed three truncation sites of DHX9 to identify the interaction domains of DHX9 with AKT1. These truncation sites of DHX9 include the double-stranded N-terminal RNA-binding domain (dsRBD), the helicase domain, and the C-terminal fragment containing nuclear localization/export signals and repeated arginine and glycine-glycine (RGG) residues (Fig. 6B, top). We noted that the helicase domain and RGG residues interact with endogenous AKT1 in PARPi-resistant HGSOC cells (Fig. 6B, bottom). It is possible that the helicase domain of DHX9 may be the main binding site for AKT1, as AKT1 did not affect DHX9’s subcellular localization (Fig. 4A), suggesting AKT1 could regulate DHX9’s helicase activity without affecting its subcellular localization for R-loop resolution. Also, it has been shown that RGG complements DHX9 helicase activity by regulating its interactions with DNA and RNA substrates (60).
To determine whether DHX9 is critical for AKT1-mediated R-loop resolution, PARPi-resistant cells were transfected with siRNA against DHX9 for 24 hours, followed by transfection with AKT1-overexpressing vector for another 48 hours. Using dot-blot analysis, we observed that while formation of R-loops was reduced upon the ectopic expression of AKT1, it increased after DHX9 depletion (Fig. 6C). Moreover, silencing DHX9 increased AKTi-induced cell growth inhibition (Fig. 6D) while overexpressing DHX9 partially rescued cell growth (Supplementary Fig. S3A–B). These data indicate that DHX9 is essential for AKT1-mediated R-loop resolution and the interaction between AKT1 and DHX9 is highly dependent on AKT1 kinase activity.
Increased R-loop accumulation is associated with transcription-replication conflicts
Transcription-replication conflicts (TRCs) are also the main sources of R-loop-induced replication stress and DNA damage (61). To evaluate whether blocking AKT and ATR signaling also affects TRCs, we used the PLA to measure TRCs. The interaction between PCNA and RNAPII was evaluated by PLA given that they are temporarily displaced from the replication forks to prevent replication stress during TRCs (61). Also, it has been shown that DHX9 interacts with PCNA and RNAPII to prevent TRCs (61). PLA signals for RNAPII-PCNA foci increased after treatment with either AKTi or ATRi alone and increased more significantly with the combination treatment (Supplementary Fig. S6). This finding suggests that the combination of AKTi and ATRi enhances TRCs, leading to further R-loop-induced replication stress. Overall, our data support the idea that pharmacologic inhibition of AKT and ATR signaling can exacerbate TRCs and R-loop-mediated replication stress in PARPi-resistant BRCAm HGSOC cells.
Combination treatment of ATRi and AKTi reduces tumor growth and prolongs survival in PARPi-resistant HGSOC animal models
For the future development of human clinical trials, we evaluated the therapeutic efficacy of the combination in in vivo HGSOC models. Immunodeficient NOD-SCID gamma (NSG) mice were used to develop subcutaneous and intraperitoneal xenograft models to monitor tumor growth, metastasis, and survival rate. PARPi-sensitive (PEO1) and PARPi-resistant (PEO1/OlaR and PEO1/OlaJR) cells were injected subcutaneously or intraperitoneally into NSG mice. The combination treatment resulted in near-complete tumor regression (Fig. 7A) without significant weight loss (Fig. 7B) compared to monotherapy in two PARPi-resistant models. Olaparib alone temporarily suppressed the tumor growth in PARPi-sensitive PEO1 models after 1-week treatment, but this activity was not maintained after few weeks (Supplementary Fig. S7A–B). Importantly, the combination of ATRi and AKTi resulted in > 3-fold increased median overall survival compared with control (24-29.5 days vs. 82.5-102.5 days, P < 0.001), ATRi (31.5-34 days vs. 82.5-102.5 days, P < 0.001), and AKTi alone (32-32.5 days vs. 82.5-102.5 days, P < 0.001) in PARPi-resistant HGSOC models and ~2-fold increased median overall survival in PEO1 models (Fig. 7C and Supplementary Fig. S7C).
Figure 7. ATRi and AKTi combination reduces tumor growth and prolongs survival, and high co-expression of DHX9 and AKT1 is associated with poor survival in HGSOC patients.
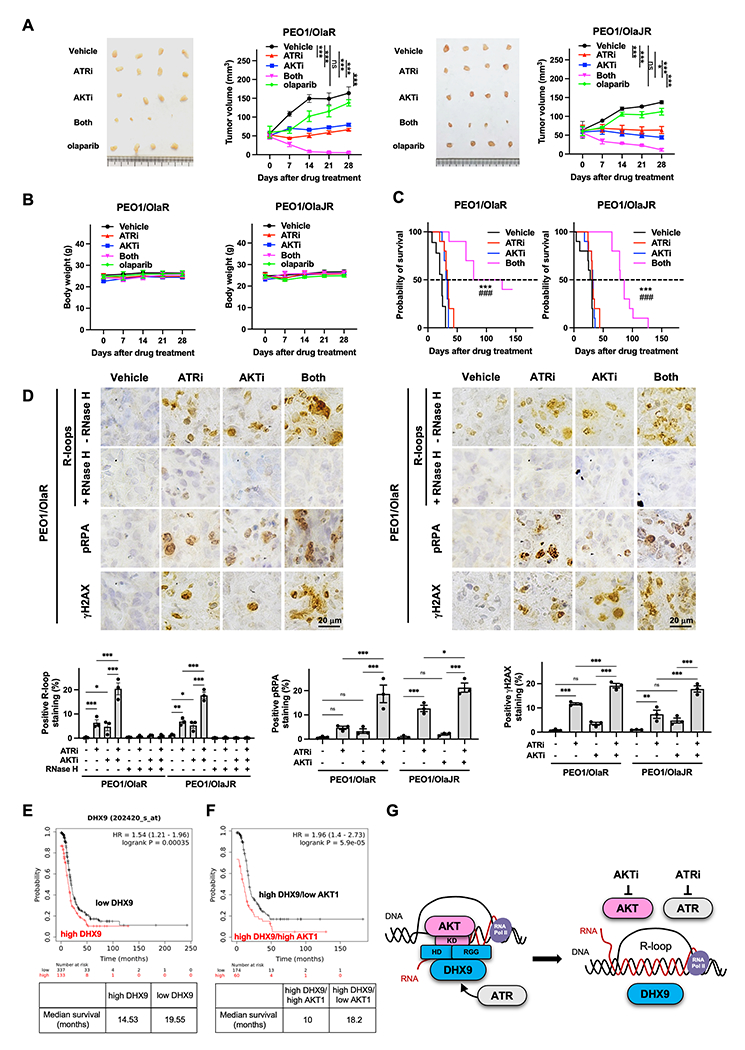
A-B, Tumor growth was measured using subcutaneous xenograft models (n = 4/group). Mice received vehicle, 130 mg/kg capivasertib, 50 mg/kg ceralasertib or 100 mg/kg olaparib. For combination, mice received 130 mg/kg capivasertib with or without 50 mg/kg ceralasertib. The tumor volume (A) and body weight (B) are plotted. Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. C, Overall survival was studied using intraperitoneal injection models. Mice received vehicle, 130 mg/kg capivasertib and/or 50 mg/kg ceralasertib. Survival is shown by Kaplan–Meier curve using the Mantel-Cox log-rank test. Data are shown as mean ± SEM. *, ATRi versus both; #, AKTi versus both; ***, ###, P < 0.001. D, Representative IHC images of R-loops, pRPA, and γH2AX (upper). The percentage of IHC positive staining area of nuclear R-loops, pRPA, and γH2AX are plotted (n = 3, bottom). Data were analyzed using one-way ANOVA test and shown as mean ± SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not significant. E-F, Prognostic value of DHX9 and AKT1 was obtained from Kaplan–Meier plotter (http://kmplot.com/analysis/) [ovarian cancer] database. E, HGSOC tumors with high (n = 337) and low DHX9 levels (n = 133) were divided using auto-select best cutoff on the website. F, HGSOC with high DHX9 levels were further divided into high- versus low-expression groups based on the median expression of AKT1 using multiple genes analysis. The progression-free survival of patients with HGSOC was analyzed by Kaplan–Meier plotter website, and the hazard ratios with 95% confidence intervals and log-rank P values were calculated. G, Proposed model of AKT1-dependent DHX9 function on R-loop resolution. While ATR is an important regulator of DNA replication and R-loop resolution, AKT1 also plays an essential role in R-loop resolution by directly recruiting DHX9 to R-loops through AKT1 kinase domain’s interaction with DHX9 helicase domain and RGG box. Hence, combined inhibition of AKT and ATR creates lethal replication stress by inducing aberrant R-loops in PARPi-resistant HGSOC.
We also assessed R-loop accumulation-related replication stress and DNA damage in vivo using IHC. Consistent with in vitro findings, the levels of γH2AX, pRPA, and R-loops were significantly elevated in tumors treated with combination compared to monotherapy, while the increased R-loops were significantly mitigated upon RNase H treatment (Fig. 7D and Supplementary Fig. S7D). These findings demonstrate the therapeutic potential of the combination treatment of ATRi and AKTi possibly via modulating R-loop dynamics and replication stress in vivo.
High co-expression of DHX9 and AKT1 is associated with worse survival in HGSOC patients
Lastly, we studied the correlations of mRNA levels of DHX9, AKT1 and ATR with the survival of HGSOC patients using the public Kaplan-Meier Plotter ovarian cancer database to evaluate these genes as a potential biomarker to predict the response for combination therapy. The survival analysis exhibited worse progression-free survival (PFS) in HGSOC with high expression of DHX9 compared with those with low DHX9 (14.53 vs. 19.55 months, P < 0.001) (Fig. 7E). Moreover, HGSOC with high DHX9/high AKT1 had worse PFS compared with those with high DHX9/low AKT1 (10 vs. 18.2 months, P < 0.001) (Fig. 7F), and the high levels of DHX9 were associated with elevated expressions of AKT1 in HGSOC tumors (Supplementary Fig. S7E). Of note, there was no difference of PFS between patients with high DHX9/high ATR and those with high DHX9/low ATR (Supplementary Fig. S7F, left) although we found that the high levels of DHX9 are also associated with high ATR expressions in HGSOC tumors (Supplementary Fig. S7F, right). Together, these data suggest that high co-expression of DHX9 and AKT may represent a subset of ovarian cancers with high replication stress that may benefit from combination treatment.
Discussion
Drug-resistant HGSOC, especially BRCAm HGSOC with acquired resistance to PARPi is an emerging patient population with unmet medical needs (5). Although strong preclinical evidence supports the use of ATR/CHK1 pathway blockade in PARPi-resistant BRCAm HGSOC (7–9), findings from earlier clinical trials with ATRi monotherapy have been disappointing (21). We therefore conducted a combination drug screening to address this challenge, which identified the synergistic effects between AKT blockade and ATRi in both PARPi-sensitive and PARPi-resistant BRCAm HGSOC cells. While some preclinical studies revealed that ATRi and PARPi combination is synergistic in PARPi-resistant HGSOC (8,9), our drug screen did not exhibit such synergy with multiple PARPis (e.g., olaparib, rucaparib and niraparib) except talazoparib which is known to cause greater PARP-DNA trapping activity than most PARPis (62). We prioritized this novel combination of AKTi and ATRi for further preclinical studies and possible development of the human clinical trials. In the current study, we identified the novel mechanisms of action of ATRi and AKTi combination in PARPi-resistant BRCAm HGSOC cells. Specifically, AKT plays a critical and direct role in the resolution of R-loops by binding to DHX9 and pharmacologic inhibition of AKT augments ATRi-induced R-loop-mediated replication stress and DNA damage (Fig. 7G).
There is growing evidence that aberrant R-loops lead to genomic instability and replication stress (13,61). Recent studies demonstrated that key DNA damage repair and replication proteins e.g., ATR, RNase H, DNA-RNA helicases, and topoisomerase 1/topoisomerase binding protein 1 (Top1/TopBP1) are involved in preventing aberrant R-loop formation and removing R-loops (12). Also, studies reported that cancer cells harboring high levels of R-loops particularly rely on the ATR/CHK1 pathway activation for survival (18,50). ATR protects replication forks from being cleaved by MUS81 via inhibiting EME1, thus preventing further DSBs induced by R-loop accumulation, as shown in HeLa cells (18). Though a high activity of ATR at baseline may suggest increased endogenous replication stress (8,9), we acknowledge that preconditions for the ATRi and AKTi combination response is complex. As such, in PARPi-resistant BRCAm HGSOC cells, we uncovered that pathological R-loop formation can be induced by AKTi not only by ATRi, suggesting other survival pathways also contribute to the dynamics of R-loop resolution.
AKT is a master regulator in maintaining genomic stability by involving DNA replication, DSB repair, and cell cycle regulation (63). For instance, AKT inhibits TopBP1, which leads to increased E2F1 transcription activity and DNA synthesis, ultimately promoting cell survival and preventing apoptosis (64). AKT also involves DSB repair process by inhibiting the recruitment of DNA end resection factors, e.g., RPA and RAD51 and stimulating DNA-PKcs autophosphorylation for initiating non-homologous end joining repair (63). Although these data may suggest that AKT activation is necessary for cancer cells’ survive against R-loop–associated replication stress, it remained elusive whether AKT directly involves R-loop regulation. Therefore, it is noteworthy that our data provide the first evidence of AKT1’s direct role in R-loop resolution by binding to DHX9 through its kinase domain for the recruitment of DHX9 to R-loops. Furthermore, our data suggest that DHX9 binds to AKT1 predominantly through its helicase domain.
There are conflicting reports for DHX9 as a promoter of R-loop formation (65,66). However, most studies (15,27,37,56,67,68) demonstrated DHX9’s function as resolving R-loops and a conflicting ability of DHX9 may depend on cellular context and binding partners. Specifically, DHX9 facilitates R-loop resolution by directly interacting with RNF168 in BRCA1/2 deficient tumor cells (68). Another study showed that loss of DHX9 or PARP1 independently increases R-loop accumulation in HeLa cells treated with camptothecin (27). In contrast, DHX9 promotes the R-loop accumulation in U2OS osteoblastoma cells lacking RNA splicing factors SFPQ and SF3B3 (65). However, it is worth noting that loss of SFPQ (0.32%, 1/316) or SF3B3 (1.27%, 4/316) is a rare event in HGSOC (Supplementary Fig. S8) thus it is possible that DHX9 likely supports R-loop resolution rather than R-loop formation in HGSOC.
DHX9 involves the early stage of HR by facilitating the end resection of damaged DNA, generating a 3’ ssDNA substrate that promotes HR through RAD51 recombinase (53). Other helicases, such as DDX1 (69) and senataxin (16), also enhance HR repair and reduce R-loops at DSBs. Thus, targeting of R-loop resolution proteins, e.g., DHX9 blockade (70) may be a potential strategy to overcome PARPi resistance, requiring further investigation. Similarly, Boros-Olah et al. reported that R-loop-related genes, including DHX9, may predict the response of drugs targeting of PI3K/AKT and cell cycle pathways, examined in >1000 cell lines, including 30 BRCAwt and 4 BRCAm ovarian cancer cell lines (71). Also, it has been reported that Dhx9 activates the ERK pathway in mice CD8+ T cells (72), serving as a compensatory survival pathway in the absence of PI3K/AKT signaling activation (73). Consistent with this, we observed an additive cytotoxic effect with DHX9 knockdown and AKTi, indicating that depleting DHX9 may impede compensatory survival pathway induction, thereby further inhibiting cell growth.
Multiple factors are involved in R-loop-mediated replication stress and transcription homeostasis. During S phase, transcription and replication machineries need to access the same template which can lead to collision in certain situations and/or at specific genomic regions (61). Also, pausing, stalling, and backtracking of transcribing RNA polymerases further increase the chance of TRC and replication fork stalling (61). As such, TRCs are the main sources of R-loop-induced replication stress and DNA damage. Here, we identified a novel function of AKT and found that PARPi-resistant HGSOC cells also depend on AKT1 to ensure efficient transcription during S-phase and thus prevents R-loop-dependent TRCs. Our finding is in line with previous reports. Peng et al. reported on Akt1/2 double-knockout causes severe growth retardation and defective transcription cascade in DBA/1lacJ mice (74). Also, AKT1-knockout cells resulted in impaired DNA synthesis leading to high sensitivity to PARPi (75). Our proposed mechanism thus supports the notion that ATRi/AKTi combination synergy exists not only by mitigating cell’s ability of R-loop-mediated replication stress but also by increased TRCs.
In summary, our study demonstrates the potential therapeutic benefit of combining ATRi and AKTi in drug-resistant HGSOC models. Moreover, this combination treatment is well-tolerated in murine models. Our findings also shed light on the mechanisms behind the observed effects, indicating that the ATRi and AKTi combination promotes lethal replication stress and DNA damage by increasing R-loop-mediated replication stress. Additionally, we reveal that AKT1 interacts with DHX9, playing a direct role in R-loop resolution. Collectively, our research provides compelling evidence for the ATRi and AKTi-based clinical trials, facilitating the development of new treatment options and biomarkers, and ultimately improving clinical outcomes for women with recurrent HGSOC.
Supplementary Material
Significance:
Inhibition of the AKT and ATR pathways cooperatively induces R-loop-associated replication stress in high-grade serous ovarian cancer, providing rationale to support the clinical development of AKT and ATR inhibitor combinations.
Acknowledgements
The authors thank Dr. Seth Steinberg for assistance in statistical analysis. This research was fully funded by the intramural research program of the NIH, National Cancer Institute (NCI), Center for Cancer Research (CCR; ZIA BC011525 [J.L.]), USA. The drug combination screening work was supported by the intramural research programs of the National Center for Advancing Translational Sciences and the CCR of NCI (FY21-NCI-01 [J.L., T.H., K.C.C.]), USA. AstraZeneca (AZ) supplied ceralasertib and capivasertib to the NCI CCR under a Material Transfer Agreement between AZ and NCI.
Footnotes
Conflict of interest: All authors declare no competing interests.
References
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, Bray F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209–49 [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DK, Alvarez RD, Backes FJ, Bakkum-Gamez JN, Barroilhet L, Behbakht K, et al. NCCN Guidelines(R) Insights: Ovarian Cancer, Version 3.2022. J Natl Compr Canc Netw 2022;20:972–80 [DOI] [PubMed] [Google Scholar]
- 3.Gralewska P, Gajek A, Marczak A, Rogalska A. Participation of the ATR/CHK1 pathway in replicative stress targeted therapy of high-grade ovarian cancer. J Hematol Oncol 2020;13:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dias MP, Moser SC, Ganesan S, Jonkers J. Understanding and overcoming resistance to PARP inhibitors in cancer therapy. Nat Rev Clin Oncol 2021;18:773–91 [DOI] [PubMed] [Google Scholar]
- 5.Gupta N, Huang T, Horibata S, Lee J. Cell cycle checkpoints and beyond: Exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor–resistant cancer. Pharmacol Res 2022;178:106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Veneziani AC, Scott C, Wakefield MJ, Tinker AV, Lheureux S. Fighting resistance: post-PARP inhibitor treatment strategies in ovarian cancer. Ther Adv Med Oncol 2023;15:17588359231157644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes Dev 2017;31:318–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim H, Xu H, George E, Hallberg D, Kumar S, Jagannathan V, et al. Combining PARP with ATR inhibition overcomes PARP inhibitor and platinum resistance in ovarian cancer models. Nat Commun 2020;11:3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim H, George E, Ragland R, Rafail S, Zhang R, Krepler C, et al. Targeting the ATR/CHK1 Axis with PARP Inhibition Results in Tumor Regression in BRCA-Mutant Ovarian Cancer Models. Clin Cancer Res 2017;23:3097–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pillay N, Tighe A, Nelson L, Littler S, Coulson-Gilmer C, Bah N, et al. DNA Replication Vulnerabilities Render Ovarian Cancer Cells Sensitive to Poly(ADP-Ribose) Glycohydrolase Inhibitors. Cancer Cell 2019;35:519–33 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nat Cell Biol 2014;16:2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petermann E, Lan L, Zou L. Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat Rev Mol Cell Biol 2022;23:521–40 [DOI] [PubMed] [Google Scholar]
- 13.Garcia-Muse T, Aguilera A. R Loops: From Physiological to Pathological Roles. Cell 2019;179:604–18 [DOI] [PubMed] [Google Scholar]
- 14.Arora R, Lee Y, Wischnewski H, Brun CM, Schwarz T, Azzalin CM. RNaseH1 regulates TERRA-telomeric DNA hybrids and telomere maintenance in ALT tumour cells. Nat Commun 2014;5:5220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chakraborty P, Grosse F. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst) 2011;10:654–65 [DOI] [PubMed] [Google Scholar]
- 16.Cohen S, Puget N, Lin YL, Clouaire T, Aguirrebengoa M, Rocher V, et al. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat Commun 2018;9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sollier J, Stork CT, Garcia-Rubio ML, Paulsen RD, Aguilera A, Cimprich KA. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol Cell 2014;56:777–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matos DA, Zhang JM, Ouyang J, Nguyen HD, Genois MM, Zou L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol Cell 2020;77:514–27 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Simoneau A, Zou L. An extending ATR-CHK1 circuitry: the replication stress response and beyond. Curr Opin Genet Dev 2021;71:92–8 [DOI] [PubMed] [Google Scholar]
- 20.Brickner JR, Garzon JL, Cimprich KA. Walking a tightrope: The complex balancing act of R-loops in genome stability. Mol Cell 2022;82:2267–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yap TA, O’Carrigan B, Penney MS, Lim JS, Brown JS, de Miguel Luken MJ, et al. Phase I Trial of First-in-Class ATR Inhibitor M6620 (VX-970) as Monotherapy or in Combination With Carboplatin in Patients With Advanced Solid Tumors. J Clin Oncol 2020;38:3195–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto TM, McMellen A, Watson ZL, Aguilera J, Ferguson R, Nurmemmedov E, et al. Activation of Wnt signaling promotes olaparib resistant ovarian cancer. Mol Carcinog 2019;58:1770–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang TT, Burkett SS, Tandon M, Yamamoto TM, Gupta N, Bitler BG, et al. Distinct roles of treatment schemes and BRCA2 on the restoration of homologous recombination DNA repair and PARP inhibitor resistance in ovarian cancer. Oncogene 2022;41:5020–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang TT, Brill E, Nair JR, Zhang X, Wilson KM, Chen L, et al. Targeting the PI3K/mTOR Pathway Augments CHK1 Inhibitor-Induced Replication Stress and Antitumor Activity in High-Grade Serous Ovarian Cancer. Cancer Res 2020;80:5380–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta N, Huang TT, Nair JR, An D, Zurcher G, Lampert EJ, et al. BLM overexpression as a predictive biomarker for CHK1 inhibitor response in PARP inhibitor-resistant BRCA-mutant ovarian cancer. Sci Transl Med 2023;15:eadd7872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cristini A, Groh M, Kristiansen MS, Gromak N. RNA/DNA Hybrid Interactome Identifies DXH9 as a Molecular Player in Transcriptional Termination and R-Loop-Associated DNA Damage. Cell Rep 2018;23:1891–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang IX, Grunseich C, Fox J, Burdick J, Zhu Z, Ravazian N, et al. Human proteins that interact with RNA/DNA hybrids. Genome Res 2018;28:1405–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mosler T, Conte F, Longo GMC, Mikicic I, Kreim N, Mockel MM, et al. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat Commun 2021;12:7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan Q, Wulfridge P, Doherty J, Fernandez-Luna JL, Real PJ, Tang HY, Sarma K. Proximity labeling identifies a repertoire of site-specific R-loop modulators. Nat Commun 2022;13:53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 2006;58:621–81 [DOI] [PubMed] [Google Scholar]
- 32.Nair J, Huang TT, Murai J, Haynes B, Steeg PS, Pommier Y, Lee JM. Resistance to the CHK1 inhibitor prexasertib involves functionally distinct CHK1 activities in BRCA wild-type ovarian cancer. Oncogene 2020;39:5520–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J Cell Biol 1998;140:1285–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sanz LA, Chedin F. High-resolution, strand-specific R-loop mapping via S9.6-based DNA-RNA immunoprecipitation and high-throughput sequencing. Nat Protoc 2019;14:1734–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skourti-Stathaki K, Proudfoot NJ, Gromak N. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol Cell 2011;42:794–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Alagia A, Ketley RF, Gullerova M. Proximity Ligation Assay for Detection R-loop detection of R-Loop Complexes upon DNA Damage. In: Aguilera A, Ruzov A, editors. R-Loops : Methods and Protocols. New York, NY: Springer US; 2022. p 289–303. [DOI] [PubMed] [Google Scholar]
- 37.Yuan W, Al-Hadid Q, Wang Z, Shen L, Cho H, Wu X, Yang Y. TDRD3 promotes DHX9 chromatin recruitment and R-loop resolution. Nucleic Acids Res 2021;49:8573–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Varghese F, Bukhari AB, Malhotra R, De A. IHC Profiler: an open source plugin for the quantitative evaluation and automated scoring of immunohistochemistry images of human tissue samples. PLoS One 2014;9:e96801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lin GL, Wilson KM, Ceribelli M, Stanton BZ, Woo PJ, Kreimer S, et al. Therapeutic strategies for diffuse midline glioma from high-throughput combination drug screening. Sci Transl Med 2019;11:eaaw0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mahdi H, Hafez N, Doroshow D, Sohal D, Keedy V, Do KT, et al. Ceralasertib-Mediated ATR Inhibition Combined With Olaparib in Advanced Cancers Harboring DNA Damage Response and Repair Alterations (Olaparib Combinations). JCO Precis Oncol 2021;5:PO.20.00439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah PD, Wethington SL, Pagan C, Latif N, Tanyi J, Martin LP, et al. Combination ATR and PARP Inhibitor (CAPRI): A phase 2 study of ceralasertib plus olaparib in patients with recurrent, platinum-resistant epithelial ovarian cancer. Gynecol Oncol 2021;163:246–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Burdett NL, Willis MO, Alsop K, Hunt AL, Pandey A, Hamilton PT, et al. Multiomic analysis of homologous recombination-deficient end-stage high-grade serous ovarian cancer. Nat Genet 2023;55:437–50 [DOI] [PubMed] [Google Scholar]
- 43.Swisher EM, Kwan TT, Oza AM, Tinker AV, Ray-Coquard I, Oaknin A, et al. Molecular and clinical determinants of response and resistance to rucaparib for recurrent ovarian cancer treatment in ARIEL2 (Parts 1 and 2). Nat Commun 2021;12:2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Howell SJ, Casbard A, Carucci M, Ingarfield K, Butler R, Morgan S, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol 2022;23:851–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yap TA, Krebs MG, Postel-Vinay S, El-Khouiery A, Soria JC, Lopez J, et al. Ceralasertib (AZD6738), an Oral ATR Kinase Inhibitor, in Combination with Carboplatin in Patients with Advanced Solid Tumors: A Phase I Study. Clin Cancer Res 2021;27:5213–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tamura K, Hashimoto J, Tanabe Y, Kodaira M, Yonemori K, Seto T, et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother Pharmacol 2016;77:787–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dillon MT, Barker HE, Pedersen M, Hafsi H, Bhide SA, Newbold KL, et al. Radiosensitization by the ATR Inhibitor AZD6738 through Generation of Acentric Micronuclei. Mol Cancer Ther 2017;16:25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davies BR, Greenwood H, Dudley P, Crafter C, Yu DH, Zhang J, et al. Preclinical pharmacology of AZD5363, an inhibitor of AKT: pharmacodynamics, antitumor activity, and correlation of monotherapy activity with genetic background. Mol Cancer Ther 2012;11:873–87 [DOI] [PubMed] [Google Scholar]
- 49.Lyu K, Kumagai A, Dunphy WG. RPA-coated single-stranded DNA promotes the ETAA1-dependent activation of ATR. Cell Cycle 2019;18:898–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hodroj D, Recolin B, Serhal K, Martinez S, Tsanov N, Abou Merhi R, Maiorano D. An ATR-dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J 2017;36:1182–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ginno PA, Lim YW, Lott PL, Korf I, Chedin F. GC skew at the 5’ and 3’ ends of human genes links R-loop formation to epigenetic regulation and transcription termination. Genome Res 2013;23:1590–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li X, Manley JL. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005;122:365–78 [DOI] [PubMed] [Google Scholar]
- 53.Chakraborty P, Hiom K. DHX9-dependent recruitment of BRCA1 to RNA promotes DNA end resection in homologous recombination. Nat Commun 2021;12:4126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koirala P, Huang J, Ho TT, Wu F, Ding X, Mo YY. LncRNA AK023948 is a positive regulator of AKT. Nat Commun 2017;8:14422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Anderson SF, Schlegel BP, Nakajima T, Wolpin ES, Parvin JD. BRCA1 protein is linked to the RNA polymerase II holoenzyme complex via RNA helicase A. Nat Genet 1998;19:254–6 [DOI] [PubMed] [Google Scholar]
- 56.Mischo HE, Hemmerich P, Grosse F, Zhang S. Actinomycin D induces histone gamma-H2AX foci and complex formation of gamma-H2AX with Ku70 and nuclear DNA helicase II. J Biol Chem 2005;280:9586–94 [DOI] [PubMed] [Google Scholar]
- 57.Crossley MP, Song C, Bocek MJ, Choi JH, Kousorous J, Sathirachinda A, et al. R-loop-derived cytoplasmic RNA-DNA hybrids activate an immune response. Nature 2023;613:187–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T. Hydroxyurea-stalled replication forks become progressively inactivated and require two different RAD51-mediated pathways for restart and repair. Mol Cell 2010;37:492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chu N, Viennet T, Bae H, Salguero A, Boeszoermenyi A, Arthanari H, Cole PA. The structural determinants of PH domain-mediated regulation of Akt revealed by segmental labeling. Elife 2020;9:e59151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xing L, Zhao X, Niu M, Kleiman L. Helicase associated 2 domain is essential for helicase activity of RNA helicase A. Biochim Biophys Acta 2014;1844:1757–64 [DOI] [PubMed] [Google Scholar]
- 61.Rinaldi C, Pizzul P, Longhese MP, Bonetti D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front Cell Dev Biol 2020;8:618157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther 2014;13:433–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu N, Lao Y, Zhang Y, Gillespie DA. Akt: a double-edged sword in cell proliferation and genome stability. J Oncol 2012;2012:951724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu K, Graves JD, Scott JD, Li R, Lin WC. Akt switches TopBP1 function from checkpoint activation to transcriptional regulation through phosphoserine binding-mediated oligomerization. Mol Cell Biol 2013;33:4685–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chakraborty P, Huang JTJ, Hiom K. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat Commun 2018;9:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dou P, Li Y, Sun H, Xie W, Zhang X, Zhang X, et al. C1orf109L binding DHX9 promotes DNA damage depended on the R-loop accumulation and enhances camptothecin chemosensitivity. Cell Prolif 2020;53:e12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Nakajima T, Uchida C, Anderson SF, Lee CG, Hurwitz J, Parvin JD, Montminy M. RNA helicase A mediates association of CBP with RNA polymerase II. Cell 1997;90:1107–12 [DOI] [PubMed] [Google Scholar]
- 68.Patel PS, Abraham KJ, Guturi KKN, Halaby MJ, Khan Z, Palomero L, et al. RNF168 regulates R-loop resolution and genomic stability in BRCA1/2-deficient tumors. J Clin Invest 2021;131:e140105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li L, Germain DR, Poon HY, Hildebrandt MR, Monckton EA, McDonald D, et al. DEAD Box 1 Facilitates Removal of RNA and Homologous Recombination at DNA Double-Strand Breaks. Mol Cell Biol 2016;36:2794–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Castro J, Daniels MH, Lu C, Brennan D, Gotur D, Lee Y-T, et al. Abstract 1136: Targeting DHX9 inhibition as a novel therapeutic modality in microsatellite instable colorectal cancer. Cancer Research 2023;83:1136- [Google Scholar]
- 71.Boros-Olah B, Dobos N, Hornyak L, Szabo Z, Karanyi Z, Halmos G, et al. Drugging the R-loop interactome: RNA-DNA hybrid binding proteins as targets for cancer therapy. DNA Repair (Amst) 2019;84:102642. [DOI] [PubMed] [Google Scholar]
- 72.Jiao A, Sun C, Wang X, Lei L, Liu H, Li W, et al. DExD/H-box helicase 9 intrinsically controls CD8(+) T cell-mediated antiviral response through noncanonical mechanisms. Sci Adv 2022;8:eabk2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mendoza MC, Er EE, Blenis J. The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation. Trends Biochem Sci 2011;36:320–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Peng XD, Xu PZ, Chen ML, Hahn-Windgassen A, Skeen J, Jacobs J, et al. Dwarfism, impaired skin development, skeletal muscle atrophy, delayed bone development, and impeded adipogenesis in mice lacking Akt1 and Akt2. Genes Dev 2003;17:1352–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mohammadian Gol T, Rodemann HP, Dittmann K. Depletion of Akt1 and Akt2 Impairs the Repair of Radiation-Induced DNA Double Strand Breaks via Homologous Recombination. Int J Mol Sci 2019;20:6316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNAseq data from PARPi-resistant BRCAm HGSOC tumors are available in Gene Expression Omnibus (GEO) at GSE252610. The genomic alteration of HGSOC analyzed in this study was obtained from cBioPortal at https://www.cbioportal.org/ and GEPIA at http://gepia.cancer-pku.cn/. The mRNA expression data and survival data of AKT1, ATR, DHX9, and other R-loop regulators analyzed in this study were obtained from Kaplan-Meier Plotter [ovarian cancer] at http://www.kmplot.com. All summary data are available in the article or uploaded as Supplementary Data, and all raw data are available upon request from the corresponding author.