

Abstract
BACKGROUND:
GWAS of schizophrenia demonstrated that variations in the non-coding regions are responsible for most of common variation heritability of the disease. It is hypothesized that these risk variants alter gene expression. Thus, studying alterations in gene expression in schizophrenia may provide a direct approach to understanding the etiology of the disease. In this study we use Cultured Neural progenitor cells derived from Olfactory Neuroepithelium (CNON) as a genetically unaltered cellular model to elucidate the neurodevelopmental aspects of schizophrenia.
METHODS:
We performed a gene expression study using RNA-Seq of CNON from 111 controls and 144 individuals with schizophrenia. Differentially expressed (DEX) genes were identified with DESeq2, using covariates to correct for sex, age, library batches and one surrogate variable component.
RESULTS:
80 genes were DEX (FDR<10%), showing enrichment in cell migration, cell adhesion, developmental process, synapse assembly, cell proliferation and related gene ontology categories. Cadherin and Wnt signaling pathways were positive in overrepresentation test, and, in addition, many genes are specifically involved in Wnt5A signaling. The DEX genes were modestly, but significantly, enriched in the genes overlapping SNPs with genome-wide significant association from the PGC GWAS of schizophrenia (PGC SCZ2). We also found substantial overlap with genes associated with other psychiatric disorders or brain development, enrichment in the same GO categories as genes with mutations de novo in schizophrenia, and studies of iPSC-derived neural progenitor cells.
CONCLUSIONS:
CNON cells are a good model of the neurodevelopmental aspects of schizophrenia and can be used to elucidate the etiology of the disorder.
Keywords: Schizophrenia, RNA-seq, cell culture, neural progenitors, olfactory neuroepithelium, Wnt5a signaling
Introduction.
Schizophrenia (SCZ) is a devastating psychiatric disorder with an estimated lifetime prevalence of 0.55% worldwide (1). With heritability of about 81% (2), genetics plays a critical role in the disease’s etiology, however the mechanism by which genetic variation contributes to the disease is unknown. Many direct and indirect pieces of evidence indicate that aberrations in brain development are major contributors (3–5). Post-mortem studies of the brains of adults with SCZ provide important information about functional changes associated with the disease, but it is highly unlikely that the gene expression profiles of differentiated cells can provide a full picture of changes in neurodevelopment. Different biological models, such as hiPSC-derived neural progenitor cells (6) and olfactory epithelium derived cells/tissue (7,8) have been suggested to study neurodevelopmental aberrations in schizophrenia. For this study, we have used Cultured Neural progenitor cells derived from Olfactory Neuroepithelium (CNON) of individuals with, and without, schizophrenia (9). These cells are not genetically modified neural progenitors. Here we present a study of transcriptome expression profiles using RNA-Seq (strand-specific, rRNA-depleted total RNA) in CNON lines derived from 144 SCZ and 111 control (CTL) individuals.
Materials and Methods
Recruitment and sample collection.
This study was approved by the University of Southern California and SUNY Downstate IRBs. We collected samples from 144 patients with DSM-IV criteria for schizophrenia (SCZ) and 111 controls. Most patients and control subjects were recruited from participants of the Genomic Psychiatry Cohort (GPC) study (1R01MH085548) (10), and a few patients were recruited through Los Angeles county/University of Southern California outpatient psychiatric clinic. Given the common variation overlap of SCZ and Major Depressive Disorder (MDD) (11), we excluded controls that endorsed either MDD probe question on the GPC screener (10), effectively removing individuals with a potential history of MDD.
Cell culture and transcriptome sequencing
We developed cell cultures from olfactory biopsies (12) as described previously (9) and in Supplemental Materials. RNA was purified from ~400,000 cells grown on 6 cm Petri dishes (~90% of confluence), using the Direct-Zol RNA MiniPrep kit (Zymo Research), according to manufacturer’s protocol. RNA libraries were prepared in batches of 24–48 samples with approximately equal numbers of cases and controls with TruSeq Stranded Total RNA LT Library preparation kits with Ribo-Zero Gold (Illumina) according to manufacturer’s protocol, using a Hamilton STARlet liquid handling robot to increase library preparation consistency. Equimolar pools of at least 4 libraries, containing both cases and controls, were constructed after quantification using the KAPA Library Quantification Kit (Kapa Biosystems), and sequenced together using HiSeq2000 DNA Sequencers (Illumina) with 100 bp single-end reads. On average, each sample was run in 3.93 lanes across 3.91 flow cells, to reduce potential channel and flow cell bias.
Mapping and assignment of reads to genes
We performed pre-mapping QC, read trimming, mapping and assignment to the sense strand of gene models (GenCode v22, max mismatch=6) using GT-FAR v12 (https://genomics.isi.edu/gtfar) (see Supplemental Materials for detail). The number of uniquely mapped reads uniquely assigned to each gene model was used as a proxy of gene expression in DEX gene analysis.
RT-qPCR
Reverse transcription quantitative Polymerase Chain Reaction (RT-qPCR) was performed in duplicates using the Biomark HD (Fluidigm) on a Flex Six Gene Expression IFC (Integrated Fluidic Circuit), according to manufacturer’s protocols and using the recommended reagents. Normalized relative expression (to ACTB) was calculated using the ΔΔCt method (13), and log-transformed expression values were analyzed by ANOVA controlling for sex.
Differential gene expression analysis
We performed main differential gene expression (DEX) analysis between SCZ and controls using DESeq2 v1.16.1 (14) in R v3.4.1, an algorithm assessing difference between mean gene expression in groups using a generalized linear model and assuming a negative binomial distribution of RNA-Seq reads. The analysis used covariates of sex, age, 4 library batches and 1 surrogate variable (SVA v3.24.4). Procedures used to arrive at this covariate set are further described in Supplemental Materials. DESeq2’s built-in normalization process was used.
The analysis was done on 23,920 expressed genes, defined as having on average at least 3.5 reads per sample, based on the density plot of log-transformed baseMean for all genes (Supplemental Figure S1). Resulting p-values were adjusted for multiple comparisons based on the Benjamini-Hochberg False Discovery Rate (FDR) using the p.adjust function in R. Transcripts Per Million transcripts (TPM) values were calculated by dividing the mean number of unnormalized reads uniquely mapped to a gene by the median transcript length (15) in Gencode 22 for that gene and normalizing the resulting values so they sum to one million (16). Hierarchical clustering analysis was done in R using the ‘hclust’ function where the distance is calculated as one minus the Pearson correlation coefficient and using average linkage.
Permutation analysis
To test that our findings are not due to random variation we performed two forms of permutation analysis. First, 1,000 analyses were run under the same conditions as the main analysis, except with labels for diagnosis randomly permuted for all samples. Second, 250 analyses each of random subsets in 4 different configurations were analyzed: random half of cases vs. other half of cases, and random half of controls vs. other half of controls (null comparisons); and, due to the difference in number of cases and controls, random half of cases vs. random same number of controls, and random half of controls vs. random same number of cases (case/control comparisons). The numbers of differentially expressed (DEX) genes in the permuted analyses were compared to the number seen in the original, unpermuted analysis by Wilcoxon test. The number of DEX genes in the null comparisons was compared to the number DEX in the case/control comparisons by Mann-Whitney test.
GWAS enrichment analysis
We identified GWAS variants’ p-values within a given gene based on the PGC SCZ2 dataset (https://www.med.unc.edu/pgc/results-and-downloads) and Gencode 22 annotation back-ported to human reference genome GRCh37 (hg19) to match coordinates used in GWAS. Fisher’s Exact Test was used to test for enrichment of genes co-localized with genome-wide significant (p<5×10−8) GWAS peaks and calculate estimated odds-ratio. Due to a very broad peak in the HLA region, genes from this region were removed from the analysis, as were genes from the Y chromosome.
WGCNA
Weighted Gene Correlation Network Analysis (WGCNA) (17) was performed for all expressed genes on residuals after correction for effects of three explicit batches as well as one surrogate variable, with a soft-threshold power of 5 to achieve approximate scale-free topology (SFT R2>0.85; truncated R2>0.95), and a minimum module size of 50 genes. Modules of coexpressed genes produced by the algorithm were tested for enrichment of DEX genes by Fisher’s exact test and for correlation of the eigengene with diagnosis (SCZ vs. control), after controlling for sex and age (linear model, no interactions); the p-value cutoff was adjusted for multiple comparisons. Gene set enrichment analysis was applied to modules that had a significant p-value on the aforementioned statistical tests.
Results
We collected samples of olfactory neuroepithelium and established CNON cell lines from 144 individuals with DSM-IV SCZ and 111 CTL (Supplemental Table S1) (9,12). Strand-specific RNA-Seq of total RNA was performed, resulting in an average of 23.38 million (7.1 – 106.7 million) uniquely mapped reads per sample, after exclusion of rRNA and mitochondrial genes. DESeq2 was used to normalize the read counts assigned to expressed genes and perform differential gene expression analysis.
Characterization of CNON cells.
The RNA-Seq data in this study are consistent with our previous observations using Affymetrix Human Exon 1.0 ST arrays (9) that CNON lines are neural progenitors (Supplemental table S2). In order to determine the period of human brain development the CNON lines most resemble, we compared RNA-Seq data from CNON to 647 poly-A RNA-Seq 76 bp datasets from post-mortem human brain samples across 41 individuals, 26 brain regions and 10 developmental stages (from “Early Fetal” to “Middle Adulthood”) (www.brainspan.org, (18)). To mitigate the differences in RNA-Seq methodology, we transformed the gene expression values of each CNON sample using the coordinates described by the principal components of the BrainSpan data. The first principle component of the BrainSpan data roughly corresponds to developmental age and separates the pre- and post-natal samples (Supplemental Figure S2). The CNON samples form a tight cluster within the prenatal samples, particularly those from the mid fetal period (weeks 13–24, second trimester).
Genes differentially expressed in neural progenitor cells in SCZ
Using our analysis model (see Supplemental Materials for details), 80 genes were DEX between SCZ and CTL at a false discovery rate (FDR) of 10%, corresponding to a maximum p-value of 3.3×10−4 (Table 1; complete gene list in Supplemental Table S3). The average fold change was 1.8 (range 1.08 to 9.09) (Figure 1) at expression levels of 0.07 to 552 TPM.
Table 1. Genes significantly DEX between SCZ and Control at FDR < 0.1.
To facilitate the comparison of expression of each gene, normalized read counts were transformed to transcripts per million transcripts (TPM) (16), using the median transcript length (15) in Gencode release 22 for each gene as gene length. Gene symbols and gene types are taken from Gencode release 27 where available. Notes: 1) Gene lies under genome-wide significant PGC SCZ2 GWAS peak (17); 2) De novo non-silent mutations in the gene has been identified in patients with SCZ; 3) De novo missense mutation(s) in the gene has been identified in patients with autism spectrum disorder; 4) CNVs were identified in multiple patients with SCZ, 5) gene found significantly associated with psychiatric disorder or related traits (other than PGC SCZ2 GWAS).
| Ensembl ID | Gene Symbol | Gene Type | TPM | Log2 Fold-Change | FDR | Notes |
|---|---|---|---|---|---|---|
| ENSG00000278099 | U1 | snRNA | 4.84 | 3.184 | 9.77×10−10 | |
| ENSG00000170627 | GTSF1 | protein_coding | 0.362 | 2.162 | 5.50×10−07 | |
| ENSG00000149564 | ESAM | protein_coding | 0.347 | −1.423 | 3.79×10−06 | 1, 2, 5 |
| ENSG00000115461 | IGFBP5 | protein_coding | 552 | 1.045 | 5.54×10−04 | |
| ENSG00000124749 | COL21A1 | protein_coding | 0.926 | 1.184 | 2.31×10−03 | |
| ENSG00000180447 | GAS1 | protein_coding | 24.3 | 0.842 | 2.47×10−03 | |
| ENSG00000157766 | ACAN | protein_coding | 0.217 | −1.232 | 5.69×10−03 | |
| ENSG00000164532 | TBX20 | protein_coding | 0.115 | −1.509 | 5.77×10−03 | |
| ENSG00000135914 | HTR2B | protein_coding | 2.74 | 1.169 | 5.77×10−03 | |
| ENSG00000008517 | IL32 | protein_coding | 2.33 | 1.044 | 5.77×10−03 | |
| ENSG00000165197 | VEGFD | protein_coding | 0.614 | 1.125 | 6.66×10−03 | |
| ENSG00000141469 | SLC14A1 | protein_coding | 7.48 | −1.679 | 6.66×10−03 | |
| ENSG00000260604 | AL590004.4 | lincRNA | 0.602 | 0.813 | 9.83×10−03 | |
| ENSG00000114251 | WNT5A | protein_coding | 186 | 0.521 | 9.83×10−03 | |
| ENSG00000184731 | FAM110C | protein_coding | 0.591 | −1.003 | 2.71×10−02 | 5 |
| ENSG00000162434 | JAK1 | protein_coding | 81.7 | 0.180 | 2.98×10−02 | 3 |
| ENSG00000143631 | FLG | protein_coding | 0.127 | −1.248 | 3.41×10−02 | 3 |
| ENSG00000133048 | CHI3L1 | protein_coding | 1.036 | 1.513 | 3.49×10−02 | |
| ENSG00000118689 | FOXO3 | protein_coding | 9.45 | 0.207 | 3.49×10−02 | 1, 5 |
| ENSG00000226237 | GAS1RR | lincRNA | 0.719 | 0.503 | 3.49×10−02 | |
| ENSG00000172156 | CCL11 | protein_coding | 1.45 | 1.426 | 3.67×10−02 | |
| ENSG00000230552 | AC092162.2 | lincRNA | 0.460 | 0.707 | 4.12×10−02 | |
| ENSG00000203648 | AC007618.1 | processed_pseudogene | 0.662 | 0.522 | 4.53×10−02 | |
| ENSG00000076706 | MCAM | protein_coding | 15.52 | −1.012 | 4.53×10−02 | 5, 3 |
| ENSG00000109819 | PPARGC1A | protein_coding | 2.09 | 0.715 | 4.53×10−02 | |
| ENSG00000188227 | ZNF793 | protein_coding | 8.96 | 0.244 | 4.61×10−02 | |
| ENSG00000145819 | ARHGAP26 | protein_coding | 16.85 | 0.324 | 4.61×10−02 | 3 |
| ENSG00000120324 | PCDHB10 | protein_coding | 0.096 | 0.914 | 4.64×10−02 | |
| ENSG00000135250 | SRPK2 | protein_coding | 63.78 | 0.107 | 4.91×10−02 | 1, 5 |
| ENSG00000233476 | EEF1A1P6 | processed_pseudogene | 0.550 | 0.382 | 4.91×10−02 | |
| ENSG00000112852 | PCDHB2 | protein_coding | 1.22 | 0.647 | 4.91×10−02 | |
| ENSG00000189058 | APOD | protein_coding | 3.91 | 0.885 | 4.91×10−02 | |
| ENSG00000120337 | TNFSF18 | protein_coding | 3.05 | 1.216 | 4.91×10−02 | |
| ENSG00000197181 | PIWIL2 | protein_coding | 0.520 | −0.718 | 4.91×10−02 | |
| ENSG00000279118 | AC093535.2 | TEC | 7.38 | 0.454 | 4.91×10−02 | |
| ENSG00000277232 | GTSE1-AS1 | lincRNA | 0.858 | −0.423 | 4.91×10−02 | |
| ENSG00000235531 | MSC-AS1 | antisense_RNA | 36.55 | 0.343 | 5.50×10−02 | |
| ENSG00000170921 | TANC2 | protein_coding | 19.16 | 0.213 | 5.71×10−02 | 2,5 |
| ENSG00000154721 | JAM2 | protein_coding | 7.324 | 0.712 | 5.92×10−02 | |
| ENSG00000137558 | PI15 | protein_coding | 0.147 | 1.091 | 5.92×10−02 | 2 |
| ENSG00000124107 | SLPI | protein_coding | 1.241 | −2.265 | 5.92×10−02 | |
| ENSG00000109158 | GABRA4 | protein_coding | 0.362 | 0.483 | 5.92×10−02 | |
| ENSG00000154229 | PRKCA | protein_coding | 89.5 | 0.191 | 5.92×10−02 | 3 |
| ENSG00000113205 | PCDHB3 | protein_coding | 0.136 | 0.846 | 5.92×10−02 | 5 |
| ENSG00000255583 | AC084357.3 | transcribed_unprocessed_pseudogene | 0.819 | −0.824 | 5.92×10−02 | |
| ENSG00000183690 | EFHC2 | protein_coding | 0.162 | 0.908 | 5.92×10−02 | |
| ENSG00000176896 | TCEANC | protein_coding | 1.374 | 0.226 | 5.92×10−02 | |
| ENSG00000021645 | NRXN3 | protein_coding | 0.615 | −0.797 | 6.20×10−02 | |
| ENSG00000258071 | ARL2BPP2 | processed_pseudogene | 2.53 | 0.300 | 6.42×10−02 | |
| ENSG00000104903 | LYL1 | protein_coding | 0.485 | −0.502 | 6.93×10−02 | 5 |
| ENSG00000169122 | FAM110B | protein_coding | 17.59 | 0.387 | 7.28×10−02 | |
| ENSG00000245680 | ZNF585B | protein_coding | 30.62 | 0.194 | 7.28×10−02 | |
| ENSG00000169306 | IL1RAPL1 | protein_coding | 0.074 | −0.899 | 7.28×10−02 | 3, 4 |
| ENSG00000068305 | MEF2A | protein_coding | 24.27 | 0.212 | 7.81×10−02 | |
| ENSG00000234155 | LINC02535 | lincRNA | 0.797 | −0.376 | 7.93×10−02 | |
| ENSG00000272674 | PCDHB16 | protein_coding | 0.648 | 0.610 | 7.93×10−02 | 3 |
| ENSG00000111371 | SLC38A1 | protein_coding | 137 | 0.273 | 7.93×10−02 | |
| ENSG00000236535 | RC3H1-IT1 | sense intronic | 2.202 | 0.334 | 7.93×10−02 | |
| ENSG00000164265 | SCGB3A2 | protein_coding | 5.455 | 0.395 | 7.93×10−02 | |
| ENSG00000163815 | CLEC3B | protein_coding | 6.303 | 0.691 | 7.93×10−02 | |
| ENSG00000223361 | FTH1P10 | transcribed_processed_pseudogene | 0.732 | −0.906 | 8.33×10−02 | |
| ENSG00000139508 | SLC46A3 | protein_coding | 4.206 | 0.302 | 8.33×10−02 | |
| ENSG00000173947 | PIFO | protein_coding | 0.142 | 0.517 | 8.52×10−02 | |
| ENSG00000146453 | PNLDC1 | protein_coding | 0.181 | 1.274 | 8.52×10−02 | |
| ENSG00000182240 | BACE2 | protein_coding | 32.35 | 0.304 | 8.58×10−02 | |
| ENSG00000113269 | RNF130 | protein_coding | 24.13 | 0.173 | 8.78×10−02 | |
| ENSG00000139926 | FRMD6 | protein_coding | 284 | 0.263 | 8.81×10−02 | |
| ENSG00000260522 | AC106785.1 | processed_pseudogene | 2.283 | 0.385 | 8.83×10−02 | |
| ENSG00000279250 | AC022919.1 | TEC | 1.262 | 0.419 | 9.29×10−02 | |
| ENSG00000226261 | AC064836.1 | processed_pseudogene | 3.389 | −0.519 | 9.88×10−02 | |
| ENSG00000189067 | LITAF | protein_coding | 452 | 0.250 | 9.88×10−02 | |
| ENSG00000164244 | PRRC1 | protein_coding | 34.65 | 0.131 | 9.88×10−02 | |
| ENSG00000272769 | AC097532.2 | lincRNA | 0.237 | 0.476 | 9.88×10−02 | |
| ENSG00000174697 | LEP | protein_coding | 1.893 | 1.342 | 9.88×10−02 | |
| ENSG00000157216 | SSBP3 | protein_coding | 4.987 | 0.264 | 9.88×10−02 | 2 |
| ENSG00000261824 | LINC00662 | lincRNA | 21.58 | 0.205 | 9.88×10−02 | |
| ENSG00000106459 | NRF1 | protein_coding | 8.061 | −0.142 | 9.88×10−02 | |
| ENSG00000162383 | SLC1A7 | protein_coding | 0.142 | 0.828 | 9.88×10−02 | |
| ENSG00000228495 | LINC01013 | lincRNA | 1.26 | 0.921 | 9.90×10−02 | |
| ENSG00000197928 | ZNF677 | protein coding | 20.20 | 0.171 | 9.92×10−02 |
Figure 1. Volcano plot for SCZ vs. CTL DEX comparison.
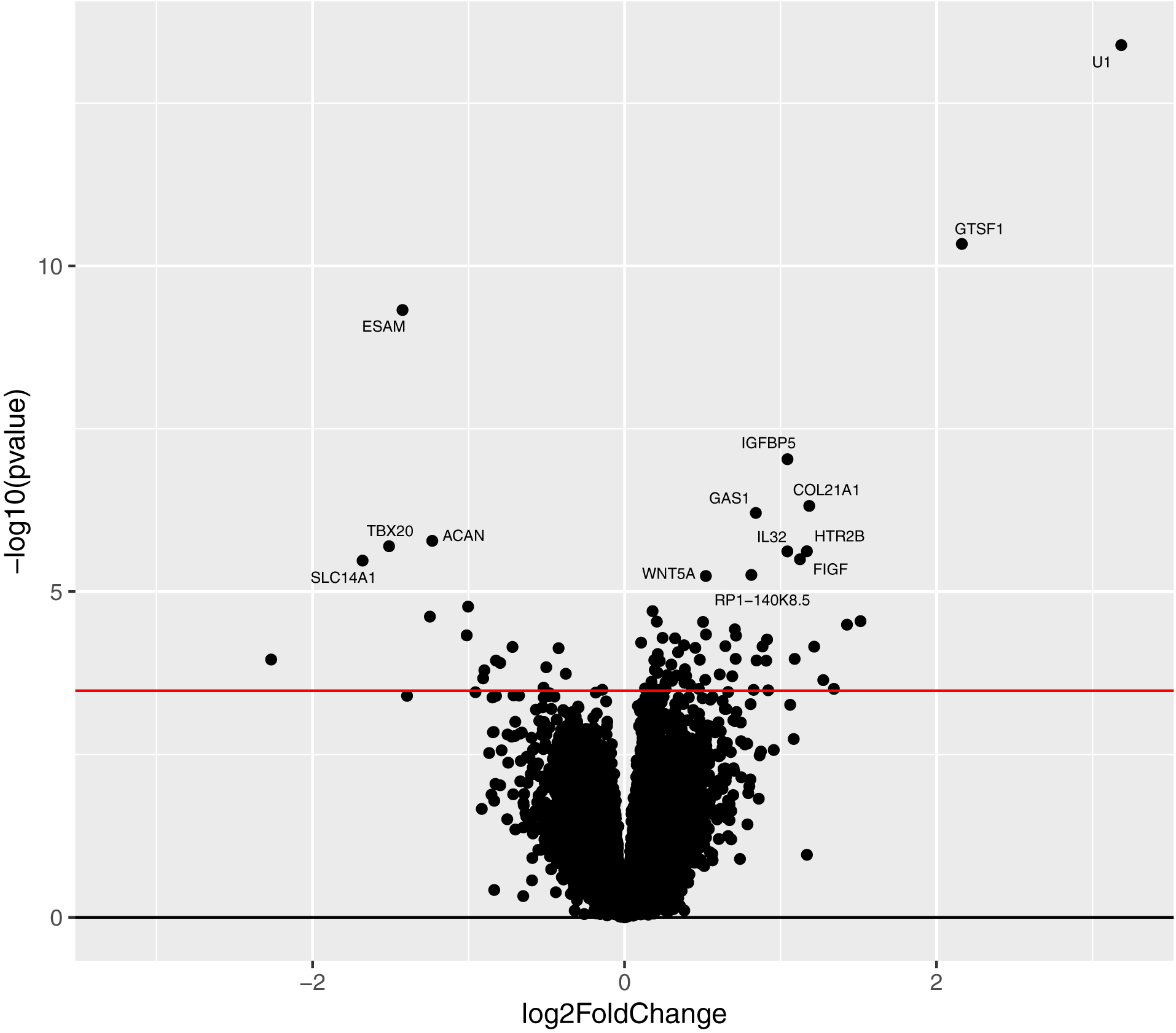
Genes with raw p < 10−5 are labeled. Red line indicates FDR < 10%.
To evaluate the accuracy of RNA-Seq gene quantification and DEX analysis, we compared results with RT-qPCR. For that purpose, we used a subset of 146 samples and performed DEX analysis on RNA-Seq data on only these samples. From this list of DEX genes we selected 5 genes (CCL8, HTR2B, PLAT, PPARGC1A, and VAV3) to include genes with fold change differences in both directions and spanning a range of gene expression levels. Expression of these genes and ACTB (used for normalization) was assessed by RT-qPCR on the same set of 146 samples. Expression data from RT-qPCR and RNA-Seq were highly correlated within each gene (mean r=0.75, range 0.63 – 0.90, all p-values <2×10−15) (Supplemental Figure S3A–E), and mean expression of all six genes had correlation r=0.943 (p=0.0047) (Supplemental Figure S3F). DEX of 4 of 5 genes was replicated, while one gene (PLAT) did not reach significance (p=0.15) but shows a trend in the same direction as that seen in RNA-Seq (Supplemental Figure S3GH).
Permutation analysis of differential expression
To assess the probability that DEX findings could be due to random statistical variation, we performed two forms of permutation analysis. In the first, we randomly permuted the diagnosis labels, but held all other factors constant. We found a median of 11 differentially expressed genes at FDR<10%, which is significantly lower (p<2×10−16, Wilcoxon signed rank test) than in the unpermuted data. In the second permutation analysis, where we permute subsets of the data to compare case-control comparisons to null comparisons (SCZ vs. SCZ and CTL vs. CTL) (see Methods for detail), the null comparisons resulted in significantly fewer differentially expressed genes at FDR<10% (median=4, mean=38.45), as compared to case/control comparisons (median=9, mean=152.98); p<3.6×10−15 by Mann-Whitney test (Supplemental Figure S4 and Supplemental Materials). These permutation analyses strongly suggest that the majority of genes found to be DEX are not likely to be false positives.
GWAS enrichment analysis
It is thought that causal variants in SCZ GWAS loci regulate expression of genes involved in the etiology of SCZ (19,20). Previous studies have shown that regulatory variants are likely to be localized near to or within the genes they directly regulate (21), we tested the hypothesis that DEX genes are more likely to be co-localized with genome-wide significant (p<5×10−8) variants from the PGC SCZ2 GWAS (22), excluding the HLA region of chromosome 6. Three DEX genes (ESAM, FOXO3, and SRPK2) were found to overlap independent genome-wide significant variants (Fisher’s exact test, odds-ratio=3.8, p=0.049) (Supplemental Figure S5). Two other genes just missed being overlapped with genome-wide significant peaks. DEX gene IL1RAPL1 is almost genome-wide significant (p=5.3×10−8), and AC007618.3 sits within an intron of CACNA1C, in which another intron contains one of the most significant GWAS signals (22). For comparison, genes that were DEX (FDR<10%) based on either sex or age were not found to be enriched for overlap with SCZ GWAS variants (p>0.05).
The index SNP of another GWAS-significant locus (rs56972983, chromosome 5) is an eQTL for PCDHB16, in GTEx v7 (aorta). Three other DEX protocadherin genes, PCDHB10, PCDHB2 and PCDHB3, also have eQTLs within a broader region of this GWAS peak. It is noteworthy that all 48 protocadherin genes transcribed in CNON from the alpha, beta and gamma clusters, are expressed at a higher level in SCZ.
Gene set enrichment, pathway and network analyses of DEX genes
Gene set enrichment analysis (GSEA) shows significant (corrected p<0.05) enrichment of the DEX genes in 47 GO terms from 10 categories (related groups of terms), including cell migration, cell adhesion, developmental process, cell proliferation, synapse assembly and PANTHER pathway analysis (23) shows over-representation of DEX genes involved in the cadherin and Wnt pathways (FDR=1.39% and 1.91% respectively). Hierarchical clustering of DEX genes further supports the finding of involvement of Wnt signaling (Figure 2). The most prominent cluster of correlated genes includes WNT5A (a Wnt ligand), LEP (encoding Leptin) and JAK1 (both are in a cytokine-induced WNT5A signaling loop), MCAM (encoding WNT5A receptor CD146), PRKCA (encoding protein kinase C, in the non-canonical Wnt/calcium pathways), PPARGC1A (activator of transcriptional factors, involved in canonical Wnt signaling) and FOXO3 (interact with β-catenin and alter the output of the canonical Wnt-signaling pathway) (Figure 3). Together, this grouping of DEX genes in the WNT5A signaling pathway implicates altered signaling through Wnt pathways in schizophrenia.
Figure 2. Heatmap and hierarchical clustering of DEX genes.
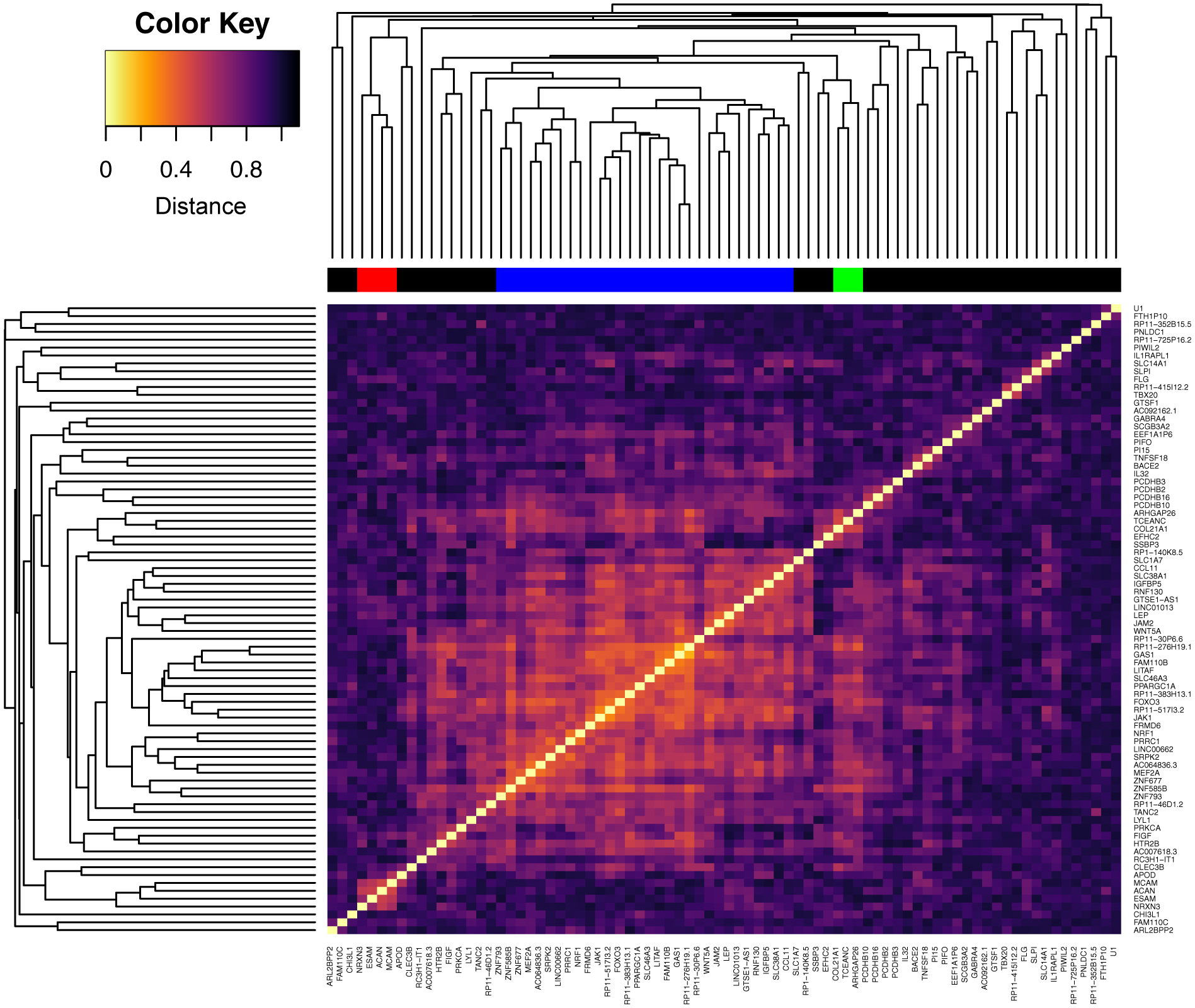
Clustering was performed using average linkage and a distance of one minus the absolute value of the Pearson correlation coefficient between the two genes. Lighter color indicates higher correlation. Genes were assigned to a group (colors in bar above heatmap) based on a cutoff at clustering distance of 0.6.
Figure 3. Wnt signaling branches involving genes with differential expression between SCZ and control groups.
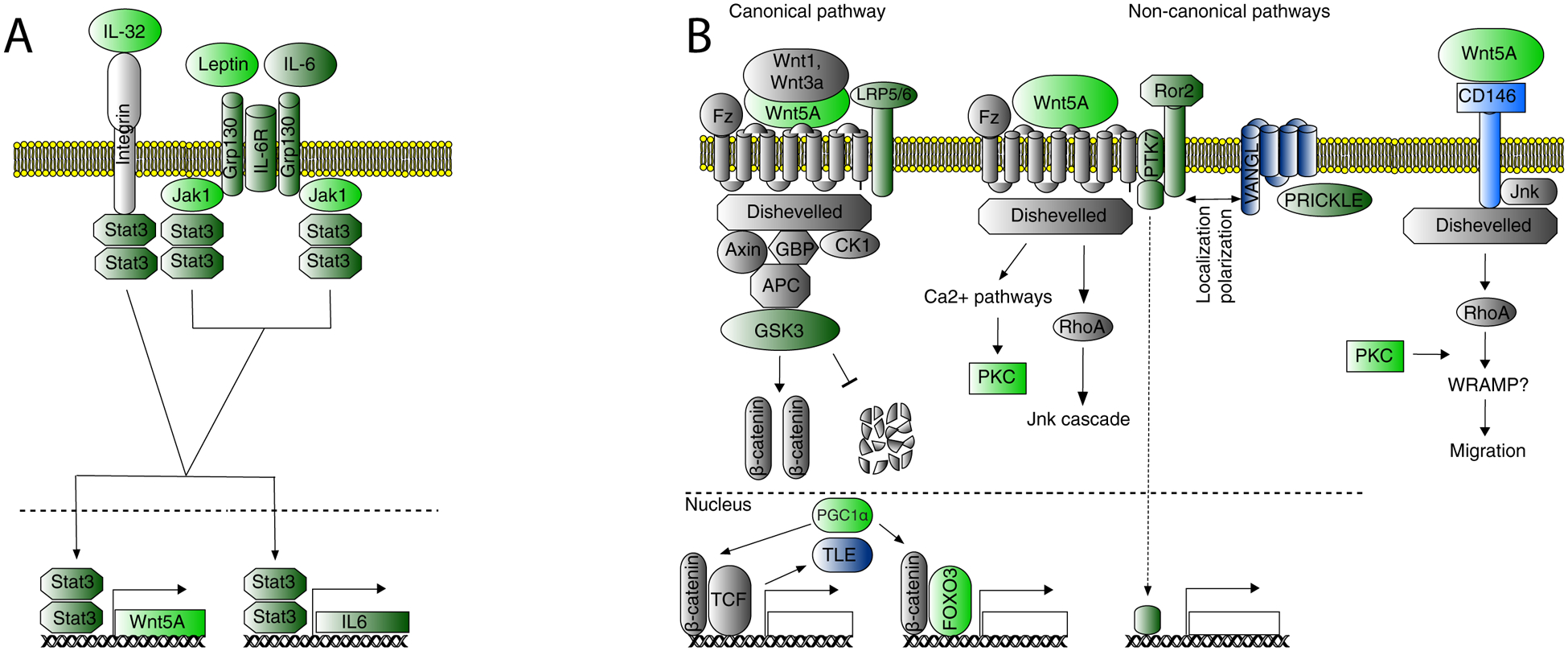
Light green: DEX (FDR<10%), over-expressed in SCZ; light blue: DEX (FDR<10%), over-expressed in SCZ; dark-green: over-expressed in SCZ, p<0.05 (nominally significant); dark-blue: under-expressed in SCZ, p<0.05; grey: not differentially expressed (p>0.05); empty box: unknown genes. (A) Regulation of WNT5A expression. WNT5A is regulated by JAK1, LEP, STAT3, IL6ST, and IL6, which are known as the STAT3-WNT5A signaling loop (25). All genes corresponding to these proteins have higher expression in SCZ group (p<0.05), the first two genes being DEX, pointing to specific mechanism of elevated WNT5A expression in SCZ. IL32, another DEX gene, may regulate IL6 production (26) and/or directly contribute to increased expression of WNT5A through activating STAT3. (B) Wnt signaling pathways. The information about involvement of WNT5A in induction of canonical signaling is conflicting, but it is generally agreed that it is mostly involved in non-canonical signaling. Three genes involved in canonical pathway show changes in expression in line with enhanced signaling. LRP5, co-receptor of fizzled, has higher expression in SCZ, while TLE, repressor of β-catenin target genes, has lower expression (p<0.05). GSK3B was previously implicated in bipolar disorder (27), another psychiatric disease which shares some genetic susceptibility with SCZ. GSK3B did not reach the level of transcriptome-wide significance in our study (#294 in rank of significance, p=0.002), but direction of expression change is in line with increased Wnt signaling. DEX gene FOXO3 encodes a transcription factor, which interferes with transcription factors of canonical Wnt signaling (28), is over-expressed in SCZ and its expression highly correlates with WNT5A expression in CNON. PPARG coactivator 1 alpha, also encoded by a DEX gene, alters expression of transcription factors involved in canonical Wnt signaling (29). Non-canonical WNT5A signaling pathways. Frizzled, Dishevelled, VANGL and PRICKLE are core proteins of the Planar Cell Polarity pathway, activated by WNT5A, with ROR2 (p<0.05) and PTK7 (p<0.05) being significant co-receptors (25,30). Complexes of Frizzled and Dishevelled are localized on the opposite side of cells from VANGL and PRICKLE, polarizing cells and playing a role in polarized movement (31). In CNON VANGL proteins are presented almost exclusively by under-expressed VANGL1 (p<0.05), while both PRICKLE1 and PRICKLE2 genes are expressed comparably, with PRICKLE2 being over-expressed in SCZ (p<0.05). MCAM, a DEX gene with lower expression in SCZ, encodes CD146, a strong WNT5A receptor for a different non-canonical signaling pathway that regulates cell migration (32). Over-expression of DEX gene protein kinase C could result from both Ca2+ and planar cell polarity (PCP) non-canonical Wnt signaling pathways.
WGCNA analysis
WGCNA identified 23 gene expression modules containing 78.0% of expressed genes (18,636 genes). Module size ranged from 3,934 to 64 genes. Only module #3, containing 32 DEX genes (out of 2,675 genes in the module), showed significant enrichment of DEX genes (Fisher’s exact test; OR=3.6; raw p=4.0×10−11). This enrichment was driven by genes that were in the main cluster of DEX genes seen in the hierarchical clustering (Figure 2), including WNT5A, GAS1, and FOXO3. Based on GSEA, this module shows extremely significant enrichment for GO terms “cell cycle process”, “chromosome segregation”, and “DNA replication” (corrected p<5×10−18 for all), among others. Additionally, the module eigengene showed a significant correlation with SCZ status (p=0.004).
Hierarchical clustering of samples based on the expression of genes in WGCNA module #3 shows a clear separation into two groups, an effect further supported by examination of the heatmap (Figure 4A). The same separation is apparent when SCZ and CTL samples are examined separately (Figure 4BC). The smaller subgroup shows an enrichment of SCZ samples as compared to controls (Fisher’s exact test, p=3.6×10−05, OR=4.76).
Figure 4. Heatmaps of sample-sample correlation based on genes from WGCNA module #3.
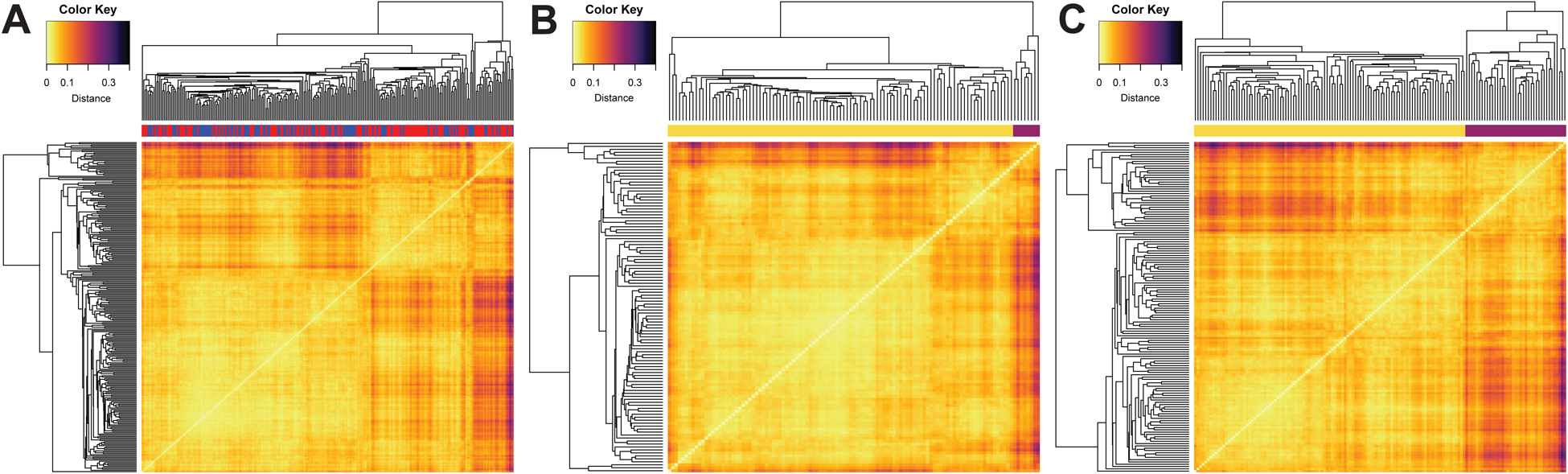
Clustering was performed using average linkage and a distance of one minus the Pearson correlation coefficient between the two samples. Lighter color indicates higher correlation. (A) Heatmap for all samples. Color bar at top indicates SCZ (red) or CTL (blue) status. (B) Heatmap for only CTL samples. (C) Heatmap for only SCZ samples. Color bar at top indicates membership in a SCZ-enriched subset of samples (purple) or the main group of samples (yellow).
Convergence with other genetic studies
Although SCZ GWAS is considered the most general and direct way to identify causative common genetic variants, consideration of other related phenotypes, and other types of genetic studies, such as de novo mutations, mutations segregating with psychiatric disorders in multigeneration families, and gene expression studies in relevant models also provide information about genes likely involved in SCZ. We found that many genes identified in these studies are also DEX in our study. FOXO3, SRPK2, BACE2, GABRA4, RC3H1, NRXN3, TANC2 and LYL1 are genome-wide significant in DEPICT-based association in GWAS meta-analysis of intelligence and associated traits (32). SNP within PRKCA is significant in GWAS of neuroticisms (33). Four DEX genes have been reported to have de novo non-silent mutations in individuals with SCZ (ESAM, PI15, SSBP3, TANC2) (34,35), seven have been reported to have de novo missense mutation(s) in patients with autism spectrum disorder (JAK1, IL1RAPL1, MCAM, FLG, PRKCA, PCDHB16, TANC2) (36–39), copy number variations of IL1RAPL1 have been identified in multiple patients with SCZ (40) and point mutations in the same gene have been observed in individuals with mental retardation (41). An exonic deletion in NRXN3 was found to segregate with neurodevelopmental and neuropsychiatric conditions in a three-generation Chinese family (42). Moreover, among 19 genes most correlated with WNT5A (R>0.6) nine have de novo mutations in either SCZ, autism spectrum disorder or developmental disorder. In total, 18 of the 80 genes overlap with previously published results from genome-wide studies of psychiatric disorders, and some of them have supported evidence from several independent studies (ESAM, FOXO3, SRPK2, IL1RAPL1, TANC2, PRKCA, NRXN3).
The largest transcriptome study of SCZ was performed by the CommonMind Consortium (CMC) using post-mortem adult dorsolateral prefrontal cortex (258 SCZ vs. 279 CTL) (43), which has a very different pattern of gene expression than CNON or the fetal brain (Supplemental Figure S2, and (18)). Only two genes showed significant differences after correcting for multiple comparisons in both studies (FAM110C and CLEC3B) and the differences were in opposite directions in both cases. However, correlation of test statistics for genes expressed in both studies was highly significant (r=0.174; p<2.2×10−16, n=14924 genes); this correlation increased to r=0.42 (p<2.2×10−16, n=453 genes) on the set of genes that were nominally significant in both studies.
We also compared our results with data from studies of iPSC-derived neural progenitor cells, which are most similar to the first trimester samples in BrainSpan (44). We found three DEX genes, all changed in the same direction, in common with a study based on microarray expression profiling of 4 SCZ and 4 CTL individuals (ARHGAP26, NRXN3, LRRC61) (44), and there was a significant positive correlation in z statistics testing for differential expression between SCZ and CTL groups for genes with publicly available data (Pearson’s r=0.117, p=0.025, n=367 genes). This work was extended using RNA-Seq and two additional controls (4SCZ/6CTL) (45) and shared 5 genes with our DEX list (GAS1, WNT5A, BACE2, FRMD6, PRKCA); all genes except PRKCA, had the same direction of effect. As before, the overall comparison of test statistics with our study was significant (r=0.15, p=4.26×10−5, n=724 genes). This latter study implicated Wnt signaling in schizophrenia, which agrees with our findings of involvement of WNT5A in etiology of the disease.
However, the largest SCZ study using iPSC-derived NPCs (10SCZ/9CTL) from the same group is significantly negatively correlated in test statistics with our data for all genes (r=−0.08, p<2.2×10−16, n=15,862). The results of this latest study are also significantly negatively correlated with two previous studies from the same group (r=−0.33, p=7.87×10−12, n=412 with microarray study and r=−0.19, p=1.13×10−6, n=654 with RNA-seq study).
Discussion
Schizophrenia is a complex genetic disorder that originates during fetal development, typically manifests symptoms in adolescence and early adulthood, and persists throughout adult life. While post-mortem brain transcriptome studies assess changes in gene expression of differentiated neuronal and glial cells in the adult brain, we developed a genetically unmodified cell-based system, CNON, to study the neurodevelopmental component of the disorder. These cells, developed from olfactory neuroepithelium, are neural progenitors, and express a transcriptome most similar to the mid-fetal period of the brain (Supplemental Figure S2), a time of increased risk for the development of schizophrenia (46,47). We examined differences in mean gene expression between SCZ and CTL groups, but other approaches for finding biologically important differences are also possible (48). We identified 80 DEX genes at FDR<10% and found an overrepresentation of genes annotated with gene ontology terms related to processes of cell proliferation, migration and differentiation, all fundamental aspects of neurodevelopment.
We looked for convergence of our transcriptome data analysis with results of other types of genomic or transcriptomic studies of SCZ or related psychiatric diseases that were performed on a comprehensive, genome-wide or transcriptome-wide basis. GWAS is considered the most general way to identify causative genomic loci associated with common variants. The main mechanistic explanation for involvement of GWAS loci in disease etiology is through altering gene expression (19,20). However, the specific causal SNPs are generally not known, and neither are the genes which they regulate, or the developmental stage or type of cells where the regulation is important for the development of the disease. Despite these complexities, our study shows a significant agreement between our DEX gene list and genome-wide significant loci in the PGC SCZ2 GWAS (odds-ratio=3.8, p=0.049). Other genomic approaches, such as identification of de novo non-synonymous mutations in psychiatric disorders, also provide independent evidence for involvement of some DEX genes with SCZ or neurodevelopment in general, suggesting that alterations in expression of some genes and changes in gene function could result in similar phenotype. Finally, a transcriptome study of iPSC-derived NPCs, a similar cellular model of SCZ, agrees with our conclusion of involvement of Wnt signaling in SCZ etiology (45).
Analysis of the DEX genes provides insight into the neurodevelopmental processes altered in SCZ. These genes appear to function in a number of biological pathways and functions, with the largest group being involved in Wnt signaling in general, and WNT5A signaling in particular.
Most DEX genes in this group (co-expressed with DEX gene WNT5A), as observed in both the hierarchical clustering of DEX genes (Figure 2) and the WGCNA analysis (Figure 4), have known functions in Wnt signaling. The Wnt signaling pathway was also found to be over-represented in the PANTHER pathway analysis.
Wnt signaling is one of the most versatile signaling mechanisms involved in regulation of different cellular and organismal functions, including development of organs and tissues, balance between cell proliferation and differentiation, cell migration, and stem and progenitor cell maintenance. These functions are critical for proper brain development. Our finding of a perturbation in Wnt signaling is consistent with previous studies (reviewed in (49–51)). As presented above, Brennand and colleagues found a gene expression signal of altered Wnt signaling, using both microarrays (6) and RNA-Seq (45). Genomic disruption of Disc1 (disrupted in schizophrenia 1), which segregates with psychiatric disorders including schizophrenia, results in an increased level of canonical Wnt signaling in neural progenitor cells (52). Gene expression in blood cells shows alterations in Wnt signaling in SCZ and BD, and plasma level of dickkopf 1 and sclerostin, known inhibitors of Wnt signaling, are decreased in patients (53).
In CNON lines, WNT5A has the highest expression of Wnt ligand genes (TPM=185.5) and is DEX (FDR=1.9%, SCZ>CTL). Transcription of WNT5A is regulated by a cytokine-induced signaling loop, which includes DEX genes JAK1 (FDR=3%, SCZ>CTL) and Leptin (LEP, FDR=9.9%, SCZ>CTL) (Figure 3A) (24). Additionally, IL-32 (FDR=0.58%, SCZ>CTL) has been shown to increase the expression of IL-6, another ligand of the same loop (25), and may increase WNT5A through IL-6 or via STAT3. Lastly, PI15 (FDR=0.059, SCZ>CTL) has been shown to induce WNT5A (54). Two DEX genes encode proteins in the downstream canonical signaling pathway of WNT5A; PGC1-alpha (FDR=4.5%, SCZ>CTL) and FOXO3 (FDR=3.5%, SCZ>CTL), the latter is a product of DEX gene that lies under a PGC SCZ2 GWAS peak (Figure 3B). WNT5a is also involved in multiple interrelated non-canonical Wnt signaling pathways. There are two DEX genes whose products are involved in non-canonical Wnt signaling: MCAM (FDR=4.5%, CTL>SCZ), encoding cell surface glycoprotein CD146, and PRKCA (FDR=5.9%, SCZ>CTL), encoding protein kinase C alpha (PKC). CD146 is a high affinity receptor for WNT5A (31) and in conjunction with PKC regulates localized membrane retraction, establishing directionality of locomotion (31,55).
The DEX gene with the highest correlation with WNT5A is GAS1, growth arrest-specific protein 1. Transcription of this gene is induced by several Wnt ligands (56). Among the functions of GAS1 is attenuation of SHH signaling (56), one of key pathways in neurodevelopment. GAS1 is known to regulate the proliferation of the external germinal layer and Bergmann glia, influencing the size of the cerebellum (57), which has been observed to be altered in SCZ (58).
Analysis of individuals by hierarchical clustering and heatmap analysis for WGCNA module #3, which showed a significant enrichment of DEX genes, identifies a group of individuals with abnormal expression of genes involved either in WNT5A regulation or downstream Wnt signaling. This subset is significantly enriched for individuals with SCZ and may represent a molecular subtype.
In summary, our results show that DEX analysis of CNON cells produces biologically meaningful results, demonstrates convergence with other genome- and transcriptome-wide studies of SCZ and related traits, and provides insight into specific mechanisms of developmental aspects of the disease. We also show that CNON are a good cellular model to study developmental aspects of brain disorders. Further studies using this model will improve the mechanistic view of SCZ etiology with finer detail. CNON cells are derived from living individuals, providing numerous opportunities for personalized medicine at a substantially lower cost than the development of iPSCs. Cell lines provide additional opportunities to test hypotheses using molecular tools such as CRISPR, siRNA and miRNA knock-down. These technologies can be combined with our ongoing epigenetic studies (59) and functional tests for proliferation, migration, cell adhesion, and to evaluate cellular phenotypes in 2D and 3D cultures.
Supplementary Material
Table 2: Gene Ontology enrichment results based on 80 DEX genes (FDR < 10%).
GSEA done by g:Profiler (23) using the ordered query option that takes into account which genes are more significant. Hierarchical filtering was applied to produce only the most significant term per parent term. Complete table of significant terms is presented in Supplemental Table S4. P-values were corrected for multiple comparisons by algorithm g:SCS, the default option in g:Profiler.
| GO Term Name | GO Term ID | Corrected p-value | DEX Genes |
|---|---|---|---|
| Angiogenesis | GO:0001525 | 3.53E-06 | 11 genes |
| Cell migration | GO:0016477 | 9.48E-06 | 15 genes |
| Cell adhesion | GO:0007155 | 0.000125 | 16 genes |
| Synapse assembly | GO:0007416 | 0.00031 | 7 genes: IL1RAPL1, NRXN3, PCDHB2, PCDHB3, PCDHB10, PCDHB16, WNT5A |
| Positive regulation of endothelial cell proliferation | GO:0001938 | 0.000864 | 5 genes: CCL11, HTR2B, PRKCA, VEGFD, WNT5A |
| Calcium-dependent cell-cell adhesion via plasma membrane cell adhesion molecules | GO:0016339 | 0.00283 | 4 genes: PCDHB2, PCDHB3, PCDHB10, PCDHB16 |
| Regulation of cell proliferation | GO:0042127 | 0.00291 | 12 genes |
| Striated muscle hypertrophy | GO:0014897 | 0.0094 | 5 genes: HTR2B, IGFBP5, LEP, MEF2A, PRKCA |
| Regulation of lipoprotein lipid oxidation | GO:0060587 | 0.0425 | 2 genes: APOD, LEP |
| Mammary gland morphogenesis | GO:0060443 | 0.05 | 3 genes: CCL11, IGFBP5, WNT5A |
KEY RESOURCES TABLE
| Resource Type | Specific Reagent or Resource | Source or Reference | Identifiers | Additional Information |
|---|---|---|---|---|
| Add additional rows as needed for each resource type | Include species and sex when applicable. | Include name of manufacturer, company, repository, individual, or research lab. Include PMID or DOI for references; use “this paper” if new. | Include catalog numbers, stock numbers, database IDs or accession numbers, and/or RRIDs. RRIDs are highly encouraged; search for RRIDs at https://scicrunch.org/resources. | Include any additional information or notes if necessary. |
| Biological Sample | patients with schizophrenia and control group | This paper | ||
| Cell Line | 255 Cultured Neural progenitor cells derived from Olfactory Neuroepithelium | This paper | primary | |
| Chemical Compound or Drug | Matrigel basement membrane | BD Bioscience | cat. 354234 | |
| Chemical Compound or Drug | Coon’s medium | Sigma | cat. F6636–10L | |
| Chemical Compound or Drug | Dispase | BD Bioscience | cat. 354235 | |
| Chemical Compound or Drug | Leibovitz’s L-15 Medium (1X), liquid | Invitrogen | 11415–064 | |
| Chemical Compound or Drug | Bovine Pituitary Extract | Gibco | 13028–014 | |
| Chemical Compound or Drug | antibiotic–antimycotic | Gibco | 15240–096 | |
| Chemical Compound or Drug | Human transferrin | Gibco | 0030124SA | |
| Chemical Compound or Drug | Sodium Selenite | Sigma | S9133–1MG | |
| Chemical Compound or Drug | Nutrient Mixture F-12 Ham, Coon's Modification | Sigma | F6636–10L | |
| Chemical Compound or Drug | hydrocortisone | Sigma | H0135 | |
| Chemical Compound or Drug | Thyroxine | Sigma | T0397 | |
| Chemical Compound or Drug | antibiotic–antimycotic | Gibco | 15240–096 | |
| Chemical Compound or Drug | Endothelial Cell Growth Supplement | Millipore | 02–102 | |
| Chemical Compound or Drug | Fetal Bovine Serum USDA | KSE Scientific | N/A | |
| Commercial Assay Or Kit | TruSeq Stranded Total RNA LT Library preparation kits with Ribo-Zero Gold | Illumina, Inc. | cat. RS-122–2301 and RS-122–2302 | |
| Commercial Assay Or Kit | Direct-Zol RNA MiniPrep kit | Zymo Research | R2051 | |
| Commercial Assay Or Kit | KAPA Library Quantification Kit | Kapa Biosystems | KK4824 | |
| Deposited Data; Public Database | BrainSpan database | www.brainspan.org | DOI: 10.1126/science.aat7615 | |
| Deposited Data; Public Database | https://www.gencodegenes.org/human/release_22.html | v22 | ||
| Genetic Reagent | ||||
| Organism/Strain | ||||
| Recombinant DNA | ||||
| Sequence-Based Reagent | ||||
| Software; Algorithm | HTSeq Python package | https://doi.org/10.1093/bioinformatics/btp120; https://doi.org/10.1093/bioinformatics/btu638 | RRID:SCR_005514 | |
| Software; Algorithm | DESeq2 | http://www.bioconductor.org/packages/release/bioc/html/DESeq2.html | v1.16.1 | doi: 10.1186/s13059-014-0550-8 |
| Software; Algorithm | GT-FAR | https://genomics.isi.edu/gtfar | v12 | |
| Software; Algorithm | SVA (R package) | available at: http://www.bioconductor.org | v3.24.4 | doi: 10.1093/bioinformatics/bts034 |
| Software; Algorithm | WGCNA (R package) | available at: http://www.bioconductor.org | https://horvath.genetics.ucla.edu/html/CoexpressionNetwork/Rpackages/WGCNA/#cranInstall |
Acknowledgments
This study was supported by a NARSAD Young Investigator Award and a NIMH grant MH086874 to OVE and NIMH grant MH086873 to JAK. Previous version of this manuscript is available at BioRxiv (doi: https://doi.org/10.1101/209197)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosures
The authors reported no biomedical financial interests or potential conflicts of interest
Conflict of interest.
The authors declare no conflict of interest.
References
- 1.Goldner EM, Hsu L, Waraich P, Somers JM. Prevalence and incidence studies of schizophrenic disorders: a systematic review of the literature. Can J Psychiatry [Internet]. 2002. Nov [cited 2015 May 30];47(9):833–43. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12500753 [DOI] [PubMed] [Google Scholar]
- 2.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a Complex Trait. Arch Gen Psychiatry. 2003;60:1187–92. [DOI] [PubMed] [Google Scholar]
- 3.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44(7):660–9. [DOI] [PubMed] [Google Scholar]
- 4.Lewis DA, Levitt P. Schizophrenia as a disorder of neurodevelopment. Annu Rev Neurosci [Internet]. 2002. Jan [cited 2015 Mar 19];25:409–32. Available from: http://www.ncbi.nlm.nih.gov/pubmed/12052915 [DOI] [PubMed] [Google Scholar]
- 5.Raedler TJ, Knabel MB, Weinberger DR. Schizophrenia as a developmental disorder of the cerebral cortex. Curr Opin Neurobiol. 1998;8:157–61. [DOI] [PubMed] [Google Scholar]
- 6.Brennand KJ, Simone a, Jou J, Gelboin-Burkhart C, Tran N, Sangar S, et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature [Internet]. 2011;473(7346):221–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21490598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cascella NG, Takaki M, Lin S, Sawa A. Neurodevelopmental involvement in schizophrenia: the olfactory epithelium as an alternative model for research. J Neurochem. 2007;102(3):587–94. [DOI] [PubMed] [Google Scholar]
- 8.Lavoie J, Sawa A, Ishizuka K. Application of olfactory tissue and its neural progenitors to schizophrenia and psychiatric research. Curr Opin Psychiatry. 2017;30(3):176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Evgrafov OV, Wrobel BB, Kang X, Simpson G, Malaspina D, Knowles J a. Olfactory neuroepithelium-derived neural progenitor cells as a model system for investigating the molecular mechanisms of neuropsychiatric disorders. Psychiatr Genet [Internet]. 2011. Oct [cited 2011 Dec 20];21(5):217–28. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21451437 [DOI] [PubMed] [Google Scholar]
- 10.Pato MT, Sobell JL, Medeiros H, Abbott C, Sklar BM, Buckley PF, et al. The genomic psychiatry cohort: Partners in discovery. Am J Med Genet Part B Neuropsychiatr Genet. 2013;162(4):306–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Consortium C-DG of the PG. Genetic relationship between five psychiatric disorders estimated from genome-wide SNPs. 2015;27(3):320–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wrobel BB, Mazza JM, Evgrafov OV., Knowles JA. Assessing the efficacy of endoscopic office olfactory biopsy sites to produce neural progenitor cell cultures for the study of neuropsychiatric disorders. Int Forum Allergy Rhinol. 2013;3(2):133–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karlen Y, McNair A, Perseguers S, Mazza C, Mermod N. Statistical significance of quantitative PCR. BMC Bioinformatics [Internet]. 2007;8:131. Available from: http://www.ncbi.nlm.nih.gov/pubmed/17445280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol [Internet]. 2014. Dec 5 [cited 2014 Dec 23];15(12):550. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4302049&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li H-D. GTFtools: a Python package for analyzing various modes of gene models. bioRxiv. 2018;300. [Google Scholar]
- 16.Wagner GP, Kin K, Lynch VJ. Measurement of mRNA abundance using RNA-seq data: RPKM measure is inconsistent among samples. Theory Biosci [Internet]. 2012. Aug 8 [cited 2012 Oct 29];131(4):281–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22872506 [DOI] [PubMed] [Google Scholar]
- 17.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics [Internet]. 2008. Jan [cited 2011 Jun 13];9:559. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2631488&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li M, Santpere G, Imamura Kawasawa Y, Evgrafov OV, Gulden FO, Pochareddy S, et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science [Internet]. 2018;362(6420):eaat7615. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30545854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallagher MD, Chen-Plotkin AS. The Post-GWAS Era: From Association to Function. Am J Hum Genet [Internet]. 2018;102(5):717–30. Available from: 10.1016/j.ajhg.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tak YG, Farnham PJ. Making sense of GWAS: using epigenomics and genome engineering to understand the functional relevance of SNPs in non-coding regions of the human genome. Epigenetics Chromatin [Internet]. 2015;8(1):57. Available from: http://epigeneticsandchromatin.biomedcentral.com/articles/10.1186/s13072-015-0050-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauberg ME, Zhang W, Giambartolomei C, Franzén O, Morris DL, Vyse TJ, et al. Large-Scale Identification of Common Trait and Disease Variants Affecting Gene Expression. Am J Hum Genet. 2017;100(6):885–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ripke S, Neale BM, Corvin A, Walters JTR, Farh K-H, Holmans P a., et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature [Internet]. 2014;511:421–7. Available from: http://www.nature.com/doifinder/10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mi H, Poudel S, Muruganujan A, Casagrande JT, Thomas PD. PANTHER version 10: Expanded protein families and functions, and analysis tools. Nucleic Acids Res. 2016;44(D1):D336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katoh M, Katoh M. STAT3-induced WNT5A signaling loop in embryonic stem cells, adult normal tissues, chronic persistent inflammation, rheumatoid arthritis and cancer (Review)..pdf. Int J Mol Med. 2007;19:273–8. [PubMed] [Google Scholar]
- 25.Lin X, Yang L, Wang G, Zi F, Yan H, Guo X, et al. Interleukin-32a promotes the proliferation of multiple myeloma cells by inducing production of IL-6 in bone marrow stromal cells. Oncotarget [Internet]. 2017;8(54):92841–54. Available from: http://www.oncotarget.com/fulltext/21611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Luykx JJ, Boks MPM, Terwindt APR, Bakker S, Kahn RS, Ophoff RA. The involvement of GSK3β in bipolar disorder: Integrating evidence from multiple types of genetic studies. Eur Neuropsychopharmacol [Internet]. 2010;20(6):357–68. Available from: 10.1016/j.euroneuro.2010.02.008 [DOI] [PubMed] [Google Scholar]
- 27.Hoogeboom D, Essers MAG, Polderman PE, Voets E, Smits LMM, Burgering BMT. Interaction of FOXO with β-catenin inhibits β-catenin/T cell factor activity. J Biol Chem. 2008;283(14):9224–30. [DOI] [PubMed] [Google Scholar]
- 28.Monteagudo S, Cornelis FMF, Aznar-Lopez C, Yibmantasiri P, Guns LA, Carmeliet P, et al. DOT1L safeguards cartilage homeostasis and protects against osteoarthritis. Nat Commun. 2017;8(May). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez S, Scerbo P, Giordano M, Daulat AM, Lhoumeau AC, Thomé V, et al. The PTK7 and ROR2 protein receptors interact in the vertebrate WNT/Planar cell polarity (PCP) pathway. J Biol Chem. 2015;290(51):30562–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davey CF, Moens CB. Planar cell polarity in moving cells: think globally, act locally. Development [Internet]. 2017;144(2):187–200. Available from: http://dev.biologists.org/lookup/doi/10.1242/dev.122804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ye Z, Zhang C, Tu T, Sun M, Liu D, Lu D, et al. Wnt5a uses CD146 as a receptor to regulate cell motility and convergent extension. Nat Commun. 2013;4. [DOI] [PubMed] [Google Scholar]
- 32.Lee JJ, Wedow R, Okbay A, Kong E, Maghzian O, Zacher M, et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat Genet. 2018;50(8):1112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kichaev G, Bhatia G, Loh P-RR, Gazal S, Burch K, Freund MK, et al. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. Am J Hum Genet [Internet]. 2019;104(1):65–75. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30595370%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6323418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature [Internet]. 2014;506(7487):179–84. Available from: http://www.nature.com.myaccess.library.utoronto.ca/nature/journal/v506/n7487/full/nature12929.html [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu B, Ionita-Laza I, Roos JL, Boone B, Woodrick S, Sun Y, et al. De novo gene mutations highlight patterns of genetic and neural complexity in schizophrenia. Nat Genet [Internet]. 2012. Dec [cited 2016 Feb 3];44(12):1365–9. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3556813&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De Novo Gene Disruptions in Children on the Autistic Spectrum. Neuron [Internet]. 2012;74(2):285–99. Available from: 10.1016/j.neuron.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012;485(7397):246–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012;485(7397):242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruzzo EK, Pérez-Cano L, Jung JY, Wang L kai, Kashef-Haghighi D, Hartl C, et al. Inherited and De Novo Genetic Risk for Autism Impacts Shared Networks. Cell. 2019;178(4):850–866.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Melhem N, Middleton F, McFadden K, Klei L, Faraone SV, Vinogradov S, et al. Copy number variants for schizophrenia and related psychotic disorders in Oceanic Palau: risk and transmission in extended pedigrees. Biol Psychiatry [Internet]. 2011. Dec 15 [cited 2016 Feb 21];70(12):1115–21. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3224197&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montani C, Gritti L, Beretta S, Verpelli C, Sala C. The Synaptic and Neuronal Functions of the X-Linked Intellectual Disability Protein Interleukin-1 Receptor Accessory Protein Like 1 (IL1RAPL1). Dev Neurobiol. 2019;79(1):85–95. [DOI] [PubMed] [Google Scholar]
- 42.Yuan H, Wang Q, Liu Y, Yang W, He Y, Gusella JF, et al. A rare exonic NRXN3 deletion segregating with neurodevelopmental and neuropsychiatric conditions in a three-generation Chinese family. Am J Med Genet Part B Neuropsychiatr Genet [Internet]. 2018. Aug 4 [cited 2018 Aug 7]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/30076746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci [Internet]. 2016;19(11):1442–53. Available from: http://www.nature.com/doifinder/10.1038/nn.4399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brennand K, Savas JN, Kim Y, Tran N, Simone A, Hashimoto-Torii K, et al. Phenotypic differences in hiPSC NPCs derived from patients with schizophrenia. Mol Psychiatry [Internet]. 2015;20(3):361–8. Available from: 10.1038/mp.2014.22\nhttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4182344&tool=pmcentrez&rendertype=abstract\nhttp://www.nature.com/doifinder/10.1038/mp.2014.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Topol A, Zhu S, Tran N, Simone A, Fang G, Brennand KJ. Altered WNT Signaling in Human Induced Pluripotent Stem Cell Neural Progenitor Cells Derived from Four Schizophrenia Patients. Biol Psychiatry. 2015;29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boksa P Maternal infection during pregnancy and schizophrenia. J Psychiatry Neurosci. 2008;33(3):183–5. [PMC free article] [PubMed] [Google Scholar]
- 47.Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: A systematic review of population-based studies. Psychol Med. 2013;43(2):239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Igolkina AA, Armoskus C, Newman JRB, Evgrafov OV, McIntyre LM, Nuzhdin SV, et al. Analysis of gene expression variance in schizophrenia using structural equation modeling. Front Mol Neurosci. 2018;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Panaccione I, Napoletano F, Forte AM, Kotzalidis GD, Del Casale A, Rapinesi C, et al. Neurodevelopment in schizophrenia: the role of the wnt pathways. Curr Neuropharmacol [Internet]. 2013;11(5):535–58. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3763761&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mulligan KA, Cheyette BNR. Neurodevelopmental Perspectives on Wnt Signaling in Psychiatry. Mol Neuropsychiatry [Internet]. 2016;2:219–46. Available from: https://www.karger.com/Article/Pdf/453266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Singh K An emerging role for Wnt and GSK3 signaling pathways in schizophrenia. Clin Genet [Internet]. 2013. Jun [cited 2017 Jul 14];83(6):511–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23379509 [DOI] [PubMed] [Google Scholar]
- 52.Srikanth P, Han K, Callahan DG, Makovkina E, Muratore CR, Lalli MA, et al. Genomic DISC1 Disruption in hiPSCs Alters Wnt Signaling and Neural Cell Fate. Cell Rep [Internet]. 2015;12(9):1414–29. Available from: 10.1016/j.celrep.2015.07.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hoseth EZ, Krull F, Dieset I, Mørch RH, Hope S, Gardsjord ES, et al. Exploring the Wnt signaling pathway in schizophrenia and bipolar disorder. Transl Psychiatry. 2018;8(1):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nimmagadda S, Buchtová M, Fu K, Geetha-Loganathan P, Hosseini-Farahabadi S, Trachtenberg AJ, et al. Identification and functional analysis of novel facial patterning genes in the duplicated beak chicken embryo. Dev Biol [Internet]. 2015;407(2):275–88. Available from: 10.1016/j.ydbio.2015.09.007 [DOI] [PubMed] [Google Scholar]
- 55.Witze ES, Litman ES, Argast GM, Moon RT, Ahn NG. Wnt5a Control of Cell Polarity and Directional Movement by Polarized Redistribution of Adhesion Receptors. Science (80- ) [Internet]. 2008. Apr 18;320(5874):365–9. Available from: http://www.sciencemag.org/cgi/doi/10.1126/science.1151250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee CS, Buttitta L, Fan C-M. Evidence that the WNT-inducible growth arrest-specific gene 1 encodes an antagonist of sonic hedgehog signaling in the somite. Proc Natl Acad Sci [Internet]. 2001;98(20):11347–52. Available from: http://www.pnas.org/cgi/doi/10.1073/pnas.201418298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Y, May NR, Fan C-M. Growth Arrest Specific Gene 1 Is a Positive Growth Regulator for the Cerebellum. Dev Biol. 2001;236(1):30–45. [DOI] [PubMed] [Google Scholar]
- 58.Moberget T, Doan NT, Alnæs D, Kaufmann T, Córdova-Palomera A, Lagerberg TV, et al. Cerebellar volume and cerebellocerebral structural covariance in schizophrenia: a multisite mega-analysis of 983 patients and 1349 healthy controls. Mol Psychiatry [Internet]. 2017. May 16 [cited 2017 Sep 29]; Available from: http://www.ncbi.nlm.nih.gov/pubmed/28507318 [DOI] [PubMed] [Google Scholar]
- 59.Spitsyna VN, Farnham PJ, Guo Y, Knowles JA, Lay FD, Rhie SK, et al. Using 3D epigenomic maps of primary olfactory neuronal cells from living individuals to understand gene regulation. Sci Adv. 2018;4(12):eaav8550. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.