

Abstract
Metabolomics samples like human urine or serum contain upwards of a few thousand metabolites, but individual analytical techniques can only characterize a few hundred metabolites at best. The uncertainty in metabolite identification commonly encountered in untargeted metabolomics adds to this low coverage problem. A multiplatform (multiple analytical techniques) approach can improve upon the number of metabolites reliably detected and correctly assigned. This can be further improved by applying synergistic sample preparation along with the use of combinatorial or sequential non-destructive and destructive techniques. Similarly, peak detection and metabolite identification strategies that employ multiple probabilistic approaches have led to better annotation decisions. Applying these techniques also addresses the issues of reproducibility found in single platform methods. Nevertheless, the analysis of large data sets from disparate analytical techniques presents unique challenges. While the general data processing workflow is similar across multiple platforms, many software packages are only fully capable of processing data types from a single analytical instrument. Traditional statistical methods such as principal component analysis were not designed to handle multiple, distinct data sets. Instead, multivariate analysis requires multiblock or other model types for understanding the contribution from multiple instruments. This review summarizes the advantages, limitations, and recent achievements of a multiplatform approach to untargeted metabolomics.
Keywords: nuclear magnetic resonance, mass spectrometry, metabolomics, multiplatform, metabolome coverage, metabolite assignment
Graphical Abstract
Introduction
Metabolomics is commonly employed to answer a fundamental question about a biological system: what is changing or different between two or more states of the system, such as between a disease and healthy control.[1] In this manner, metabolomics is hypothesis generating and will routinely inform follow-up or alternative investigations.[2] Metabolomics has been used across a variety of scientific endeavors that includes investigations involving the environment,[3] nutrition,[4] functional genomics,[5] toxicology,[6] and various aspects of human health and drug discovery.[7] A rapidly expanding application of metabolomics to issues of human health includes the discovery of biomarkers to diagnose and treat various diseases.[8] In this capacity, metabolomics has been applied to numerous human diseases where it is now quite common for a few dozen replicate studies to populate the scientific literature.[9] A disease state disrupts or distorts the normal metabolic homeostasis in a healthy individual, where detecting these metabolic changes provides valuable insights about the disease.[10] Complete coverage of the metabolome is thus needed to fully understand or conceptualize disease development and progression; and to gain insights into the roles that metabolism plays in human health.[11] Unfortunately, achieving the total coverage of the metabolome is a daunting if not currently impossible task due to the size and chemical complexity of the metabolome and the inherent limitations of analytical techniques.[12] Most single platform techniques typically identify a few hundred metabolites at best.[12c, 13] With the metabolome surpassing 217,000 compounds, there is room for improvement.[14] One approach to partly addressing this challenge is to utilize multiple, complementary analytical techniques. A multiplatform approach to metabolomics can improve the overall coverage of the metabolome.[12a, 15]
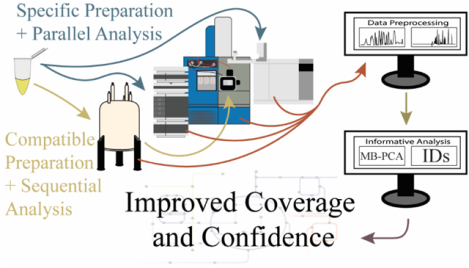
Untargeted metabolomics has been dominated by the application of a single analytical platform, such as nuclear magnetic resonance (NMR), gas chromatography-mass spectrometry (GC-MS), or liquid chromatography-mass spectrometry (LC-MS).[16] Each of these techniques present unique strengths and limitations. Overall, NMR and MS detect different chemical classes of metabolites with minimal overlap and are thus highly complementary in their application to metabolomics.[17] Combining NMR and MS in a metabolomics study provides a clear advantage to expanding the coverage of the metabolome. A multiplatform approach to metabolomics is summarized in Figure 1 and highlights a typical workflow that includes sample preparation, spectra acquisition, data analysis, and metabolite identification.
Figure 1.
Flow diagram for an untargeted multiplatform metabolomics study that describes the methods for improving the coverage of the metabolome and confidence in metabolite identifications. The metabolome can be extracted from cells, tissue, or biofluids. The sample preparation protocol is either compatible for both instruments or specific to one instrument. Sample preparation is then followed by the multiplatform data collection that encompasses any combination of two or more instruments. The data collection can occur in parallel or sequentially. Data processing is subdivided into preprocessing and informative analysis steps, which includes statistics and metabolite identifications. A variety of commercial or free academic software is available for platform specific analysis of a single type of data. There are limited software packages capable of processing multiple data types.
The general concept of a multiplatform approach to metabolomics is not well cited or described in the scientific literature. In fact, there are some disparities surrounding the simple definition and scope of the method. For example, combining multiple instruments is not the only feature that defines multiplatform metabolomics. The use of different sample preparation protocols, computational software, and statistical methods are also important features of a multiplatform approach. A query of current publications in PubMed demonstrates this definitional problem. As shown in Figure 2, a “multiplatform” query yielded a significantly lower result than a query with both the terms “NMR” and “MS”. In either case, both searches demonstrate an upward trend. While NMR and MS represent the most common platforms for untargeted metabolomics, there is a growing number of studies utilizing other analytical techniques (e.g., FTIR, TLC/GC-FID, LC-UV) in multiplatform analysis.[12b, 12c, 18] The actual scale and popularity of multiplatform metabolomics is, thus, difficult to define.
Figure 2.
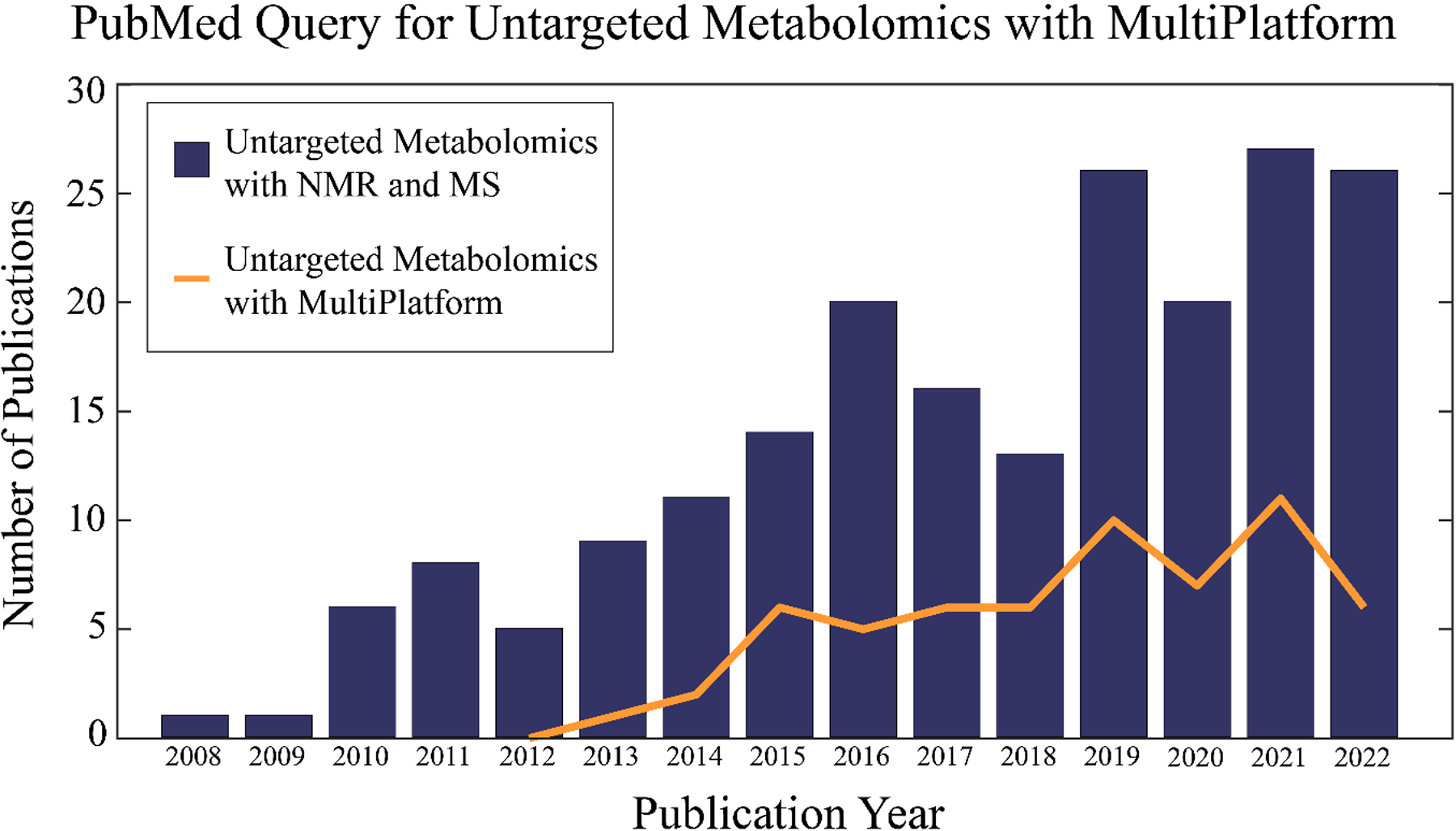
A bar graph of the number of articles in PubMed per year identified by the query: ((untargeted) OR (non-targeted)) AND ((metabolomics) OR (metabonomics)) AND ((nuclear magnetic resonance) AND (mass spectrometry)). A line plot of the number of articles in PubMed per year identified by the query: ((untargeted) OR (non-targeted)) AND ((metabolomics) OR (metabonomics)) AND (multiplatform). The chart extends up to October 26, 2022 with no hits found before 2008. The first query found a total of 203 papers and the second query identified a total of 60 manuscripts.
Besides the choice of analytical technique, the sample preparation protocol is likely the most critical step of a metabolomics study.[19] Optimized methods for metabolome extractions from a variety of biological samples have typically been devised based on the specific study question and analytical platform being utilized.[20] A multiplatform approach necessitates specific methods for a single sample preparation that involves any number of analytical techniques combined into a single study.[16d, 21] A readily available approach would simply employ existing sample preparation protocols that are used in parallel.[17a] Of course, this parallel approach has two serious limitations. First, it requires a duplicate set of biological samples, which may not be practical or possible; and is also clearly inefficient. More importantly, parallel sample preparation means each analytical method is characterizing the metabolome of a distinct set of samples. While these biological samples may be similar, they are clearly not identical, which may lead to a higher biological variance or a poor correlation between analytical methods. One unique challenge that differs from single platform strategies is the potential combination of destructive and non-destructive analytical techniques,[22] which may require a sequential approach to sample preparation and analysis. A sequential method will efficiently use each sample but at the expense of an increase in time or a slower throughput. Instead, the optimization of a combined sample preparation protocol that utilizes an identical set of biological samples is required to achieve the true benefits of a multiplatform approach. The compatibility of sample preparation methods and a comparison of instrument performance has been previously described by Beltran et al.[23]
Data processing and statistical analysis is another area of the multiplatform concept that represents a challenge. A large variety of processing software, statistical toolkits, and complex computational models are available to the scientific community and have been extensively used in metabolomics.[24] While software is readily accessible for processing single platform data types, few software packages are available for the complete processing of multiple data types from a variety of instrumentation.[24f] Not only do NMR and MS instrumentation have different file types, but different vendors have unique and proprietary data formats.[25] Multiple data formats make the preprocessing step burdensome in terms of time, programming, and computer resources. Currently, there is no immediate solution to this problem except to have program capability for preprocessing multiple data types.
The presence of two or more data sets produced by multiplatform metabolomics investigation is another unique challenge in data analysis. Simply put, the data sets need to combine in an unbiased manner by addressing fundamental differences in the structure of each individual data set, like variations in both the number and the dynamic range of the spectral features. An assortment of univariate and multivariate statistical models is available for the analysis of metabolomics where spectra alignment, normalization, and scaling are some of the key preprocessing steps.[24a, 26] Correctly preprocessing data sets from multiple analytical techniques is critical to obtaining a reliable statistical model and the accurate identification of the changes in the metabolome. An example of analyzing multiplatform data is multiblock statistical models such as multiblock PCA (MB-PCA),[27] which allows for the direct incorporation of multiplatform data into a single statistical model.
Multiplatform metabolomics has been utilized by several groups to expand the coverage of the metabolome and to improve our confidence in metabolite identification.[13, 27–28] With this greater coverage and higher confidence level, metabolomics has the potential to provide greater insights into a wider range of biological questions. Unfortunately, researchers are often sample limited, which can make a multiplatform analysis difficult to accomplish. There are other barriers that hinder the broad adoption of a multiplatform approach to metabolomics such as cost, lower throughput, lack of expertise, and complex data analysis, among others. However, these problems can often be overcome by proper experimental design and choice of analytical methods, by the optimization of sample preparation protocols, and the growing availability of software toolkits.[22] This review discusses both the advantages and limitations of a multiplatform approach to metabolomics and highlights some of the recent advancement. Specifically, we discuss the confidence and coverage of metabolite identification, the reproducibility of sample preparation, and the data analysis associated with multiplatform metabolomics.
A Multiplatform Approach Addresses Single Platform Limitations.
Multiple analytical techniques can be utilized or combined to expand the coverage of the metabolome.[16d, 21] The choice of analytical technique is dependent on several factors such as complementarity, sensitivity to specific chemical classes, added confidence in metabolite identifications, and improvements to experimental design. For example, NMR is a highly reproducible, high-throughput, non-destructive, and readily quantifiable technique. It provides redundant spectral features to validate metabolite identification, requires little to no sample preparation, and is not dependent on chromatography. Despite these strengths, NMR suffers from poor sensitivity compared to MS methods, and is generally limited to detecting the most abundant (≥ 1 μM) metabolites. Conversely, MS has a higher sensitivity (nM), and a higher resolution (~103–104) and dynamic range (~103–104) relative to NMR. But MS only detects metabolites that are readily ionized and it is dependent on chromatography to separate metabolites of similar mass. Further, GC-MS requires volatile or derivatized samples, while LC-MS suffers from matrix effects such as ion suppression.[29] MS tends to have a lower throughput than one-dimensional (1D) 1H NMR because of the additional chromatography time (~30–60 m) and the need to run the experiment in both positive and negative modes. Unless MS/MS or MSe is also used, metabolite identification may be ambiguous since it is limited to matching an exact mass and maybe a retention time. LC-MS and GC-MS are collectively challenged by instrument stability and batch variability,[30] and suffer from peak shifting and variable peak intensities due to matrix composition and other instrumental factors.[31] Similarly, NMR chemical shifts and peak shapes are sensitive to subtle changes in pH, temperature, ionic strength, and mixture composition.[32] While a common occurrence, NMR spectral changes due to variable sample conditions are easily recognizable. Nevertheless, these experimental uncertainties still diminish the reliability and accuracy of metabolite identifications using either NMR or MS. A multiplatform approach may resolve these concerns by providing redundant and complementary experimental results that may be combined to confirm a metabolite’s assignment.[33]
While LC-MS, 1D 1H NMR, and GC-MS are the most common analytical techniques used for untargeted metabolomics, other analytical methods have also been utilized. Direct injection mass spectrometry (DI-MS),[33d] which utilizes electrospray ionization (ESI) or matrix assisted laser desorption ionization (MALDI),[34] avoids the problems caused by chromatography but may encounter a higher occurrence of ion suppression.[35] Similarly, while debated, ion mobility coupled with mass spectrometry (IMS) potentially reduces the need for chromatography by including an additional ion separation based on the shape and size of the molecule (collision cross-section (CCS)).[36] In this regard, IMS has the potential to eliminate problems with both ion competition and chromatography while still providing a means to separate individual metabolites. Inductively coupled plasma mass spectrometry (ICP-MS) can provide elemental analysis and detect the presence of salts, metals, and metalloids that would be difficult by other methods.[37] Mass spectrometry-based metabolomics imaging provides spatial distribution of metabolites, but it has low throughput and requires large tissue or cell samples because of the relatively low resolution (50 to 200 μm).[38] MS imaging also creates large data sets, easily a terabyte or more per sample, which makes statistical analysis using standard software packages impractical.
Spectroscopy methods are highly desirable approaches for metabolomics due to the non-destructive, speed, and low cost of these techniques. Fourier transform infrared spectroscopy (FT-IR),[18a] and liquid chromatography coupled with spectroscopy methods such as infrared (LC-IR) and ultraviolet-visible spectroscopy (LC-UV) have also been used for metabolomics.[12c] Similarly, two-dimensional (2D) correlation IR and Raman have garnered some recent interest.[39] Also, IR and UV spectroscopy provide unique methods relative to NMR and MS for detecting and identifying metabolites. IR and UV rely on the absorbance of specific molecular vibrations or chromophores, respectively. Of course, the spectral resolution is limited given the high similarity in absorbance between different functional groups, the lack of neighbor-effects, and inherently broad peaks.
Another analytical technique that has seen recent success in identifying new metabolites is LC-MS-solid phase extraction (SPE)-NMR. Two notable advantages of this approach include the ability to: 1) easily remove and replace the solvent, and 2) concentrate low abundant metabolites for NMR analysis. Thus, HPLC-MS-SPE-NMR enables the simultaneous sequential analysis of a single sample by both NMR and MS and allows for a direct comparison between the two acquired spectra.[40] A recent perspective suggested cryogenic electron microscopy could be used alongside HPLC-MS-SPE-NMR to further improve the accuracy of structural identification.[41]
A Multiplatform Approach Improves Metabolome Coverage.
The concept of metabolome coverage refers to detecting all the known or reported metabolites for a biological sample in an established database such as the human metabolomics database (HMDB, https://hmdb.ca/),[14] serum metabolome database (SMDB, https://serummetabolome.ca/),[12b] or the urine metabolome database (UMDB, https://urinemetabolome.ca/metabolites).[12c] The serum metabolome database is comprised of over 4500 metabolites. Thus, a study that successfully identified 100 serum metabolites would only have an approximate coverage of 2.2%. The use of multiple complementary analytical techniques would be expected to improve upon this low coverage. For example, a multiplatform approach that combined HPLC-MS, GC-MS, capillary electrophoresis mass spectrometry (CE-MS), and 1H NMR was shown to improve the coverage of metabolites in breast milk by a factor of 6 to 7.[13] The increased coverage can be attributed to the unique chemical sensitivity of the four distinct platforms used in the study and the two sample preparation methods comprising a Folch and a single-phase methanol:methyl tert-butyl ether extraction. The two sample extraction protocols were needed to collect metabolites from breast milk with distinct solubility profiles. A flow diagram shown in Figure 3A summarizes the multiple sample preparation and analysis methods used in this study. Specifically, 1D 1H NMR was used to analyze both the polar and non-polar phases of the Folch extraction in either deuterated water or deuterated chloroform. CE-MS was used to analyze the polar phase of the Folch extraction and reverse-phase (C18) UPLC-MS utilized the non-polar phase. The single-phase methanol:methyl tert-butyl ether extraction was used by both GC-MS and (C8) HPLC-MS. A total of 639 unique metabolites were detected consisting of 63 metabolites observed by 1D 1H NMR, 23 metabolites by CE-MS, 105 metabolites by UPLC-MS, 392 metabolites by HPLC-MS, and 56 metabolites by GC-MS. The authors reported relative standard deviation (RSD) between biological replicates with NMR recognized as the most reproducible with an RSD of 6.4% and CE-MS with the highest RSD of 24.2%.
Figure 3.
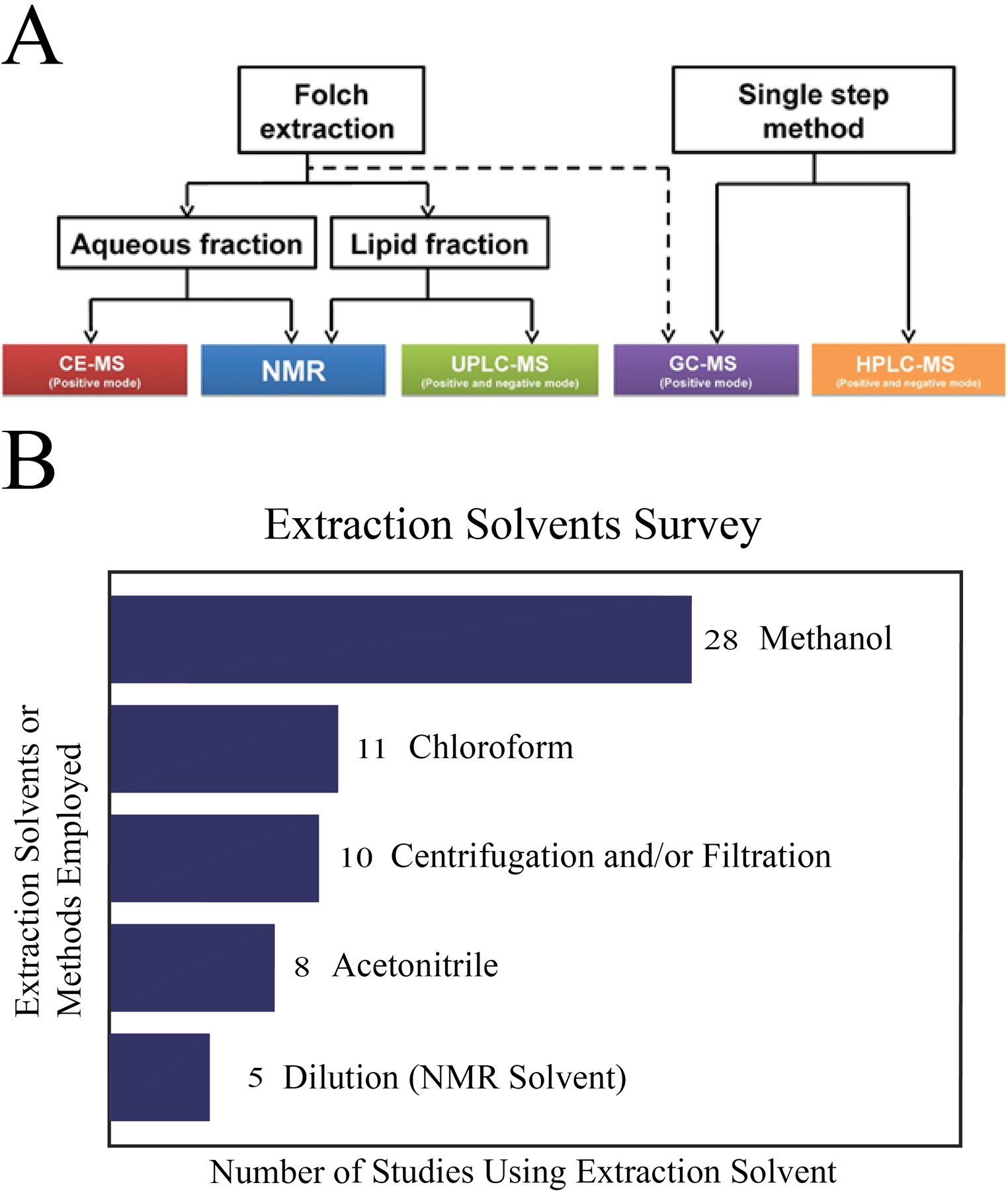
Details of multiple extractions. (A) Workflow displaying the different metabonomic techniques selected to analyze the aqueous and lipid fraction of breast milk extractions. Reprinted with permission from Andreas, N.J., Hyde, M.J., Gomez-Romero, M., Lopez-Gonzalvez, M.A., Villaseñor, A., Wijeyesekera, A., Barbas, C., Modi, N., Holmes, E. and Garcia-Perez, I. (2015), Multiplatform characterization of dynamic changes in breast milk during lactation. ELECTROPHORESIS, 36: 2269–2285. Copyright 2015 Wiley-VCH GmbH, Weinheim. (B) Survey of 43 multiplatform untargeted metabolomics papers. A bar graph summarizing the number and type of solvents or methods used to extract metabolites from a biological sample.
A study by Bouatra et al. provides another example of increasing the coverage of the metabolome by using a multiplatform approach.[12c] Human urine samples from 22 healthy individuals were analyzed by six analytical methods. GC-MS relied on four different solvent extraction and derivatization protocols that targeted distinct metabolite classes corresponding to polar, organic acids, volatiles, and bile acids. A total of 445 urine metabolites were identified with each individual analytical technique identifying the following number of metabolites: 209 by NMR, 179 by GC-MS, 127 by DI-MS, 40 by ICP-MS and 10 by HPLC. Approximately 17% of the 2651 metabolites within the urine metabolome database (UMDB) were detected by the combination of these six analytical techniques.[12c] In comparison, the best metabolome coverage achieved by any single platform was only 0.15% to 8%. At a minimum, a multiplatform approach doubled the coverage of the urine metabolome.
Chaby et al. sent NIST SRM 1950 - metabolites in human plasma, control biometric matched plasma purchased from bioIVT, and plasma samples from patients suffering from post-traumatic stress disorder (PTSD) to five commercial metabolomics vendors that consisted of Biocrates, HMT, Lipotype, Metabolon, and Nightingale.[42] The vendors used a combination of targeted and untargeted metabolomics and lipidomics, and a range of analytical techniques that included LC-MS, GC-MS, CE-MS, and NMR. The overlap in the coverage of the metabolome between the five vendors was extremely low where specific percentages were difficult to assess (Table 1). A minimum of two vendors were required to report an entire metabolite class. Notably, several chemical classes had only a few metabolites identified across all vendors. The coverage of PTSD associated metabolites ranged from 16 to 70 percent. Metabolon had the highest coverage of metabolites associated with PTSD, but this was attributed to the fact that only Metabolon was previously used in a large-scale PTSD study. NMR (i.e., Nightingale) had the lowest coefficient of variance (CV) but also the lowest coverage. The largest number of metabolites detected by any vendor was 950. Lipid classes across all platforms had the highest CV. In general, CVs ranged from 0.9 to 63.2% with an accuracy of 0.6 to 99.1% to the NIST standard plasma samples. Clearly, the low coverage and high variability in precision and accuracy across the five analytical platforms makes a strong argument for the critical need for a multiplatform approach especially regarding identifying reliable metabolite biomarkers for disease diagnosis and prognosis.
Table 1:
Summary of NIST and PTSD Metabolites Detected by Vendors[42]
| Vendor | NIST Metabolitesa | Average Errorb | Error Rangec | PTSD Biomarkersd | Coveragee | Unique Metabolitesf | Percent Changeg |
|---|---|---|---|---|---|---|---|
| Biocrates | 24 | −19.29% | (−99.49% : 18.92%) | 22 | 29% | 0 | −16% |
| HMT | 13 | −16.74% | (−38.5% : 1.97%) | 48 | 63% | 3 | 11% |
| Lipotype | 1 | −40.33% | N/A | 24 | 32% | 4 | 35% |
| Metabolon | N/A | N/A | N/A | 53 | 70% | 4 | 61% |
| Nightingale | 12 | −1.03% | (−19.63% : 15.25%) | 12 | 16% | 0 | 0% |
| Average | 12.5 | −19.35% | (−49.49% : 12.05%) | 31.8 | 42% | - | - |
total number of metabolites detected by the vendor that matches known metabolites in the standard NIST plasma sample. The total number of known metabolites in the NIST plasma sample is 26.
average error in the quantification of the detected NIST metabolites.
error range in the quantification of the detected NIST metabolites.
N/A - not applicable; metabolon did not submit an analysis of the standard NIST plasma sample
total number of metabolites detected by the vendor that matches literature defined PTSD metabolite biomarkers. The total number of PTSD metabolite biomarkers identified from scientific literature is 76.
percentage of metabolites detected by the vendor that matches literature defined PTSD metabolite biomarkers.
the number of PTSD metabolite biomarkers detected by the vendor that was not detected by any other vendor.
percent change in the number of metabolites detected between two identical replicate sets of PTSD plasma samples.
Our laboratory has also made significant contributions to describing the complementarity of NMR and MS to expanding the coverage of the metabolome. For example, we utilized two dimensional (2D) 1H-13C HSQC NMR combined with GC-MS to characterize chlamydomonas reinhardtii cells treated with lipid accumulation modulators (WD30030 and WD10784).[33c] The combination of NMR and GC-MS was essential to achieve an extensive coverage of the key metabolic pathways affected by the addition of WD30030 and WD10784 (Figure 4). Of the 47 key metabolites affected by these compounds, 14 metabolites were uniquely identified by NMR, 16 metabolites were uniquely identified by GC−MS, and 17 metabolites were identified by both NMR and GC−MS. Despite separate extraction techniques and parallel analytical analysis, the commonly identified metabolites yielded a reasonable correlation (r2 0.55) in relative metabolite concentrations. In a separate study, we utilized 1D 1H NMR and DI-MS to characterize the metabolome of human dopaminergic neurons following treatments with known toxins related to Parkinson’s disease.[32b, 43] We noted that splitting the cell lysate extraction products (90:10) between the two analytical platforms was an effective high throughput method for metabolomics. More importantly, an MB-PCA model of the combined NMR and DI-MS data sets yielded dramatically better results than an analysis of either independent data set. Distinct sets of metabolites were identified by both NMR and DI-MS that yielded a consensus model of the impact of paraquat on dopaminergic neurons, which indicated that paraquat hijacked the pentose phosphate pathway resulting in an increase in oxidative stress and cell death. The complete metabolic response to paraquat would not have been possible if only NMR or MS was used alone, instead of the multiplatform approach.
Figure 4.
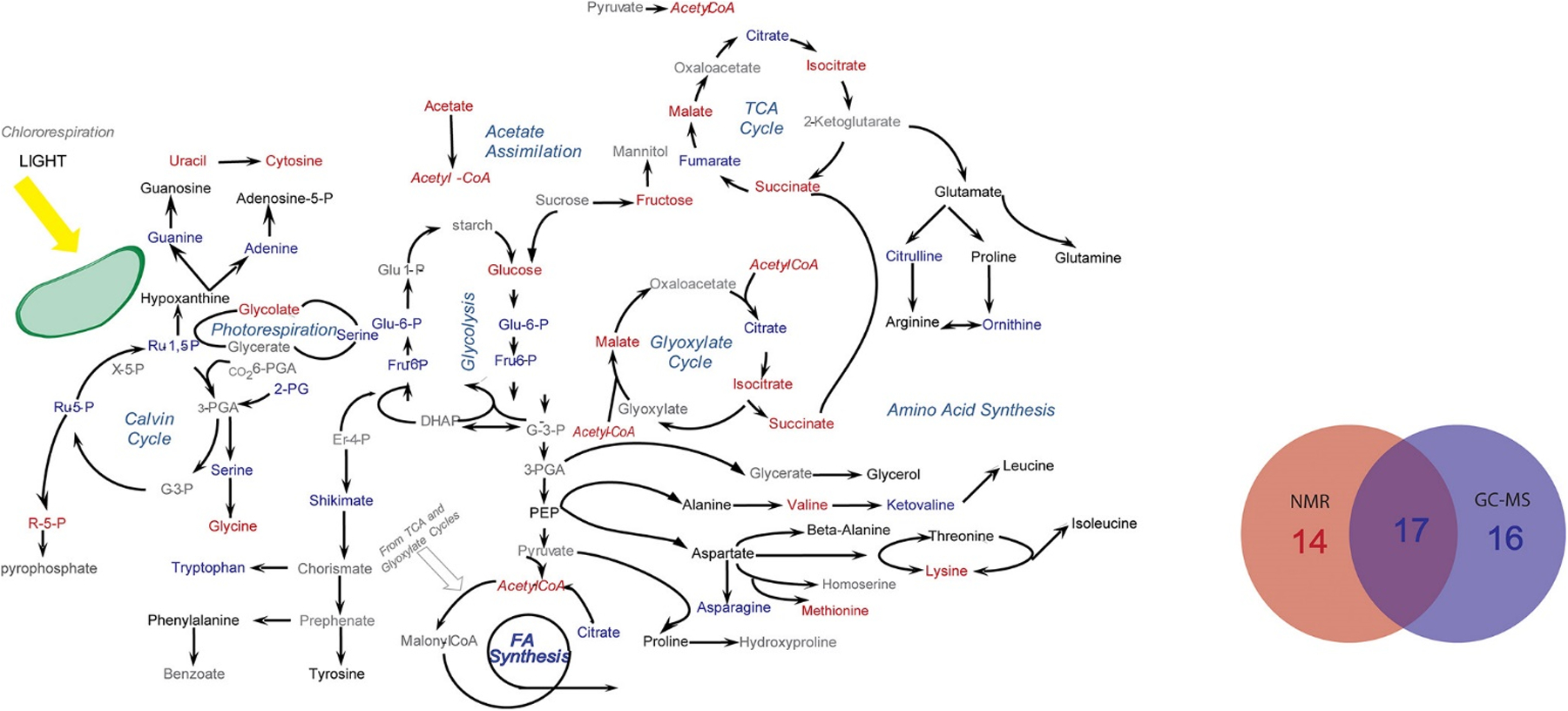
Metabolic pathway summarizing the coverage of the C. reinhardtii metabolome (metabolites of interest) from the combined application of NMR and GC–MS. Metabolites that were only identified by NMR are colored blue. Metabolites that were only identified by GC–MS are colored red. Metabolites identified by both methods are colored black, and metabolites that are not identified are colored gray. The embedded Venn diagram identifies the total number of metabolites of interest within these metabolic pathways that were identified either by NMR, by GC–MS, or by both techniques. Reprinted with permission from Bhinderwala, F.; Wase, N.; DiRusso, C.; Powers, R., Combining Mass Spectrometry and NMR Improves Metabolite Detection and Annotation. Analytical Chemistry 2018, 17 (11), 4017–4022. Copyright 2018 American Chemical Society.
A multiplatform approach is not restricted to just combining distinct analytical methods like NMR and MS. We and others have also demonstrated the possibility of improving coverage of the metabolome with NMR by detecting multiple nuclei like carbon, nitrogen and phosphorus in addition to 1D 1H NMR.[44] These heteronuclear 2D NMR experiments combined with isotope labeling (or 100% natural abundance for 31P) significantly expand our abilities for detecting additional nitrogen-containing and phosphorus-containing metabolites. The large number of nitrogen- (>30% of metabolome) and phosphorus- (~36% of metabolome) containing metabolites offers an opportunity to greatly expand the coverage of the metabolome. We also demonstrated how modest manipulation of sample conditions (low pH and temperature) permits the sequential collection of 2D 1H-31P, 15N and 13C HSQC and/or HSQC-TOCSY spectra on the same NMR metabolomics sample.[44f] In addition to detecting naturally occurring nitrogen-containing and phosphorus-containing metabolites, 15N and 31P NMR can also be leveraged to detect specific classes of molecules by using chemoselective isotope tags. For example, DeSilva et al. used 2-chloro-4,4,5,5-tetramethyldioxaphospholane to label lipids containing a hydroxyl, aldehyde or carboxyl group with phosphorus that could be detected with 31P NMR experiments.[44d] Similarly, Gowda et al. tagged carboxyl groups with 15N-ethanolamine and amino groups with 13C-formic acid. The 13C or 15N tagged metabolites were detected using either a 2D 1H-13C HSQC or a 2D 1H-15N HSQC experiment, respectively.[44b]
Optimizing Sample Preparation for a Multiplatform Approach.
Most metabolomics sample preparation strategies are platform and research question specific, and address individual instrument needs to optimize data quality. Comparing the metabolome profile between two samples prepared with different extraction methods is likely to incur inconsistencies and a higher variance. Thus, a sample preparation protocol for a combined multiplatform study needs to achieve a sample condition that is compatible with two or more instruments. For instance, NMR requires high metabolite concentrations in a deuterated solvent while MS requires a volatile or easily ionizable solvent. It is also important to consider the goals of the study and to select an extraction method that was tailored to optimize the detection of the desired metabolites. For example, maintaining oxidation state is key for distinguishing between metabolites such as nicotinamide adenine dinucleotide (NADH) and NAD+.[45] Alternatively, an aqueous extraction method would have a poor retention of hydrophobic lipids that requires an organic solvent. Recovering all the metabolites from a biological sample may require multiple extraction steps, a large volume of solvent, and a subsequent concentration step to achieve the necessary sensitivity. The extraction protocol also needs to quench all enzymatic activity to avoid biologically irrelevant perturbations to the metabolome from simply processing the samples. The sample should also contain a reduced matrix free of non-metabolite biomolecules and salts, where a number of metabolome extraction methods have been introduced for different matrices.[19] Multiple platforms require or allow different compositions, for instance: NMR is mostly unaffected by non-volatile salts, but greatly affected by changes in pH. MS is mostly unaffected by pH, but greatly affected by non-volatile salts.[31a, 32b] Many sample preparation methods reported in the scientific literature likely need to be adapted for a multiplatform approach.[19, 46]
A survey of sample preparation methods used by 43 different multiplatform metabolomics projects is summarized in Figure 3B. An average of 1.7 extraction methods were employed per study. Thus, it was more common to utilize multiple extraction methods specific to each analytical platform than a single uniform extraction protocol. A larger number of extractions techniques were likely required to improve the coverage of the metabolome, enhance the spectral quality, and improve the outcome of the data analysis. The survey results also indicate that methanol was the most popular choice for extraction solvent with chloroform being a second choice.[12c, 13, 17–18, 22–24, 28, 33a, 33b, 47] Simply, methanol was commonly utilized to extract polar metabolites and chloroform/acetonitrile was used for nonpolar metabolites. The Folch method was another common approach that used both methanol and chloroform to simultaneously extract polar and nonpolar metabolites from the same biological sample. Centrifugation and filtration methods were also routinely employed to pellet cell and tissue debris and to separate proteins and enzymes from the metabolome based on molecular weight differences. Acid extraction has also been used to improve enzyme quenching and to preserve the oxidation state of specific metabolites.[47c, 47j, 47q] Some studies using NMR to characterize the metabolome of urine and serum samples completely avoided an extraction step and only used dilution into a buffer.[47b, 47p, 47q, 47x, 47aa] Similarly, solid state NMR can be used for global tissue profiling without a necessary extraction step.[47z] Thus, multiple factors need to be considered when selecting and optimizing an extraction method for use across a set of analytical platforms.
Not surprisingly, Ye et al. found that different quenching and extraction procedures yielded significantly different metabolite profiles of Escherichia coli when using UPLC combined with time of flight mass spectrometry (TOF-MS).[46d] They compared four extractions protocols consisting of cold 80% methanol/water (−20°C), boiling 80% methanol/water (80°C), boiling 75% ethanol/water (95°C), and cold 20:20:20:40 acetonitrile/methanol/ethanol/water (−20°C); and four enzyme quenching protocols corresponding to liquid nitrogen, 60% methanol/water, 60% methanol/ethylene glycol, and 45% methanol/ethylene glycol. They characterized the following properties for the different quenching and extraction procedures: (1) enzyme quenching efficiency, (2) cryoprotectant capabilities, (3) stability of energy metabolites, and (4) the relative abundance of all detectable metabolites.
Enzyme quenching efficiency was assessed by comparing the normalized peak intensities of a representative set of 14 metabolites and a hierarchal clustering of a heatmap containing the normalized (i.e., standard normal variate) relative abundance of all detected metabolites. The use of 45% methanol/ethylene glycol as a quenching solvent produced a greater abundance of metabolites compared to the other enzyme quenching protocols. Specifically, approximately 77% of the high-abundance metabolites were attributed to 45% methanol/ethylene glycol. Conversely, liquid nitrogen had the lowest number of abundant metabolites, which was attributed to cell leakage. To further evaluate the cryoprotectant capabilities of the quenching solvents, the authors evaluated membrane integrity with confocal laser scanning microscopy and cell staining with SYTO 9 and propidium iodide (PI). PI-only stains cells with a broken membrane so the ratio of the number of SYTO 9 to PI-stained cells indicates the relative amount of cell membrane damage. Again, 45% methanol/ethylene glycol exhibited the least amount of membrane damage while liquid nitrogen had the highest. Maintaining an intact membrane while quenching enzyme activity allows for a distinction between intracellular and extracellular metabolites.
Similarly, maintaining the proper proportions of energy metabolites is particularly challenging due to the low inherent stability of these molecules.[48] The relative ratios of AMP, ADP and ATP determines the energy charge (EC) of a cell as defined by:
| (1) |
where EC typically ranges between 0.7 and 0.95.[49] 60% methanol/water, 60% methanol/ethylene glycol, and 45% methanol/ethylene glycol had a similar effectiveness in keeping EC between 0.8 and 0.9 while liquid nitrogen was below 0.8.
A comparison of the four metabolome extractions protocols resulted in four unique metabolic profiles as evident by their distinct clustering in a PCA scores plot. A heatmap containing the normalized relative abundance of all detected metabolites with hierarchical clustering analysis yielded the same outcome. Notably, the extraction protocol consisting of boiling 75% ethanol/water was grouped separately and exhibited the highest number of abundant metabolites. Of the four solvents, boiling 75% ethanol/water was shown to be the most reproducible with 145 metabolites having an RSD of 20% or less. This was a significant improvement over the 125, 120, and 95 metabolites with RSDs of 20% or less from cold 20:20:20:40 acetonitrile/methanol/ethanol/water, cold 80% methanol/water and boiling 80% methanol/water, respectively. Overall, Ye et al. demonstrated the broad diversity in metabolome coverage that results from the choice of quenching and extraction procedures for a single cell type. Clearly, the optimization of a sample preparation protocol becomes more complex with the inclusion of multiple analytical platforms while also considering the wide range of biofluids, tissue samples, and cell types encountered in a metabolomics study.
Chamberlain et al. studied the impact of matrix effects and ion efficiency in untargeted MS-based metabolomics and observed large quantitative changes in analytes depending on the type of biological sample.[29] Multiple isotope labeled standards such as creatine-D3, leucine-D10, and caffeine-D3 were added at a uniform concentration of 10 μg/mL to plasma, serum, and urine samples and then quantified using UHPLC-MS. Despite the uniform concentrations, significant variations in peak areas were observed between the different sample types and between the three metabolites. For example, leucine-D10 showed an 18.5% increase in peak area in urine relative to plasma and serum while creatine-D3 (125.8%) and caffeine-D3 (14.8%) showed a decrease. Similarly, leucine-D10 had a peak area that ranged from approximately 1×108 −1.25×108 compared to 4.7×107-6.0×107 for caffeine-D3 and 1.75×107-4.5×107 for creatine-D3. A similar outcome was observed for metabolites from a uniformly 13C-labeled yeast extract solution spiked into plasma, serum, and urine. Sample dependent matrix effects biased the ion efficiency of specific metabolites, even those in the same class.
An ion suppression effect of a solvent, acetonitrile, has also been observed that negatively affected several metabolites and resulted in reduced signal.[35a] These compounds included peroxides acetone, cyclohexanone, and cyclopentanone. This effect was also seen for vitamins including 25-hydroxyvitamin true positives.[50] The ion suppression effect of solvents are poorly studied and possibly wide spread. Thus, the type of biological sample and the choice of LC-MS solvents both play important roles in optimizing the sample preparation to reduce bias and maximize the number of metabolites detected by MS in a multiplatform approach.
Gowda et al. examined the effect of different extractions techniques on 1D 1H NMR spectra and noted the impact of proteins on metabolite peak intensities.[51] A simple methanol extraction protocol to remove these biomolecules from the serum by precipitation significantly improved the overall quality of the NMR spectra and the detection of serum metabolites compared to direct analysis or ultrafiltration. While ultrafiltration also produced well resolved peaks, precipitating the protein released bound metabolites and improved the accuracy of detecting some metabolites that included 2-oxoisocaproate, 2-hydroxyisovalerate, benzoate, and tryptophan.
Simplifying the overall extraction protocol by minimizing the number of steps is a valuable goal to reduce errors, improve reproducibility, and maintain a correlation between multiple analytical techniques.[52] Performing similar sample preparation steps for each analytical platform can reduce variability and improve the confidence in results. A true comparison between multiple analytical methods is achieved by using a single sample. In this approach, the workflow would proceed from a non-destructive spectroscopy (e.g., NMR) to destructive MS.[23, 47y, 47z] Conversely, the metabolome profile can be generated by two different analytical methods by characterizing two separate samples prepared with distinct protocols. This approach is likely to incur inconsistencies and a higher variance between the two analytical techniques, but it will also maximize the quality of the data obtained by each technique since the sample preparation protocol has been optimized per instrument. Between these two extremes is a compromise in which one sample is split between two or more platforms.[23] In all cases, there is a transition point where the sample needs to be transferred into a condition amenable to the analysis by each analytical instrument. The workflow is either parallel (i.e., two separate samples and conditions), sequential (i.e., one sample transferred into a second condition),[22] or forked (i.e., one sample split into two conditions).
A Multiplatform Approach Improves the Confidence in Metabolite Identification.
The confidence in a metabolite assignment is a major issue in untargeted metabolomics due to the complexity of a biological sample and our limited knowledge of the chemical composition of the metabolome.[53] While the concept of metabolome coverage deals with the absolute number of discoverable metabolites, the confidence or reliability in a metabolite identification is not as simple to define. The low reproducibility in metabolite assignments between replicate metabolomics studies is a growing concern.[54] This reliability is related to both the uncertainty in peak detection and the number of redundant and unique spectral features attributed to a metabolite.[55] Systematic biases, such as peak shifts in NMR or ion suppression in MS, negatively impact the confidence in a metabolite assignment and are inherent to the instrumental platform employed. Confidently assigning metabolites from heterogeneous mixtures is a complex task, but the use of multiple analytical techniques can reduce these ambiguities and remove uncertainties by providing multiple confirmatory spectral information.
In organic synthesis, there is a requirement for the exact mass, MS/MS fragmentation pattern, and multiple NMR experiments to confirm a new structure.[41] This is contrary to the current standard held by the Metabolomics Standards Initiative (MSI), which only requires two orthogonal techniques for reporting a metabolite identification.[56] This is an important distinction as structure elucidation isn’t required by MSI to identify a metabolite, which has allowed for LC-MS alone to be portrayed as two orthogonal techniques. The comparison of unknown metabolites to pure standards is a relatively effective approach to confirming a metabolite’s identity. Unfortunately, the number of contaminants and the presence of overlapping metabolites in a complex spectrum is high enough to increase the uncertainty in identification.[30] Also, pure standards are not commercially available for all metabolites. The level of uncertainty in untargeted metabolomics is well-beyond the errors of detecting a single analyte, and likely grows exponentially with the number of metabolites in a complex mixture. Reaching a similar level of confidence that is routinely achieved for organic structure identification would logically require more analytical approaches.
Repeated identification of a metabolite by multiple analytical techniques can provide greater confidence than a single method that a correct metabolite assignment has occurred. It can also improve the overall accuracy in the quantification of the metabolite. The Venn Diagram shown in Figure 5A summarizes the mouse lung metabolites identified by the three analytical platforms, HPLC-MS, CE-MS, and GC-MS. Only 7 metabolites were identified by all three platforms, where 10, 13, and 14 metabolites were shared among any two platforms.[57] A total of 38, 55 and 1081 metabolites were detected by a single platform. These results demonstrate an interesting and unsurprising trend; the number of identified metabolites decreases as the number of analytic methods detecting these metabolites increases. This trend also appears to parallel the likely increase in confidence with the 7 metabolites identified by the three platforms having the highest overall confidence (Figure 5B).
Figure 5.
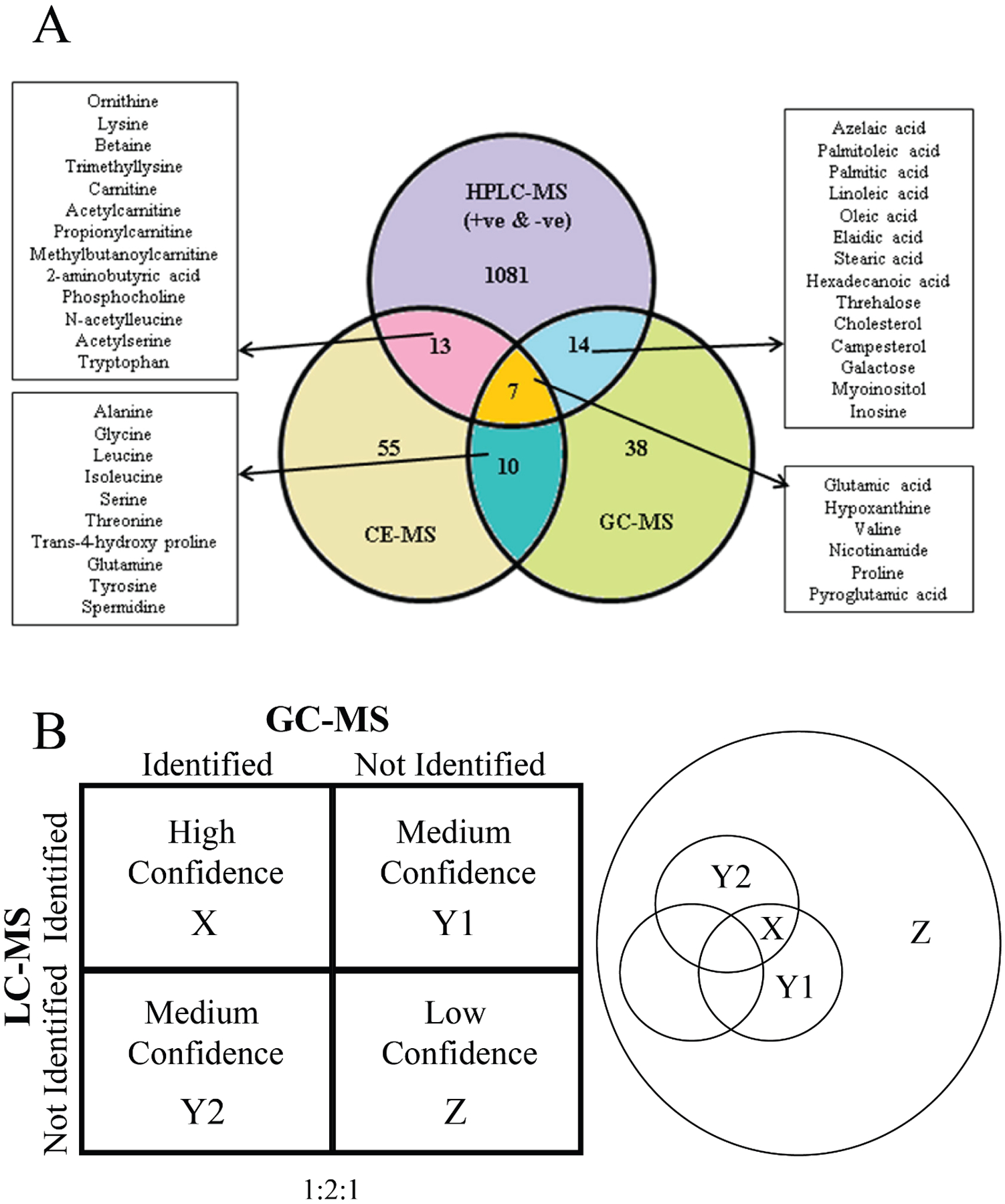
(A) Venn diagram presenting data for identified features detected in all 6 replicates for mouse lung pool compared between the three analytical platforms (LC–MS, GC/MS, and CE–MS). Reprinted with permission from Naz, S.; García, A.; Barbas, C., Multiplatform Analytical Methodology for Metabolic Fingerprinting of Lung Tissue. Analytical Chemistry 2013, 85 (22), 10941–10948. Copyright 2013 American Chemical Society. (B) (left) Confidence array based on the number of instrumental platforms used to identify a metabolite in a complex biological sample. (right) Venn diagram depicting the group of hypothetical metabolites detected by two (X), one (Y1, Y2), or no (Z) analytical instruments.
As shown in Figure 5A, there were dozens to hundreds of metabolites that were only identified by one platform, which accounts for most of the identified metabolites. While the accuracy in the assignment of these metabolites has the lowest relative confidence, the repeated identification of other metabolites by multiple techniques will also limit the misidentification of these metabolites or improve their assignment confidence. The confidently assigned metabolites limit the possible chemical space available for assigning the remaining spectral features. Simply, if glucose has already been confidently assigned, it can no longer be misassigned to other spectral features. While the entire composition of any metabolome is currently unknown, there are well-established sets of metabolites (i.e., central carbon metabolism) and chemical classes (i.e., amino acids) that are commonly expected to be detected. If these commonly detected metabolites are “left” for the single platforms, the confidence in the assignment again increases. Thus, another reason for utilizing a multiplatform approach in untargeted metabolomics is the increase in the confidence of all metabolite assignments.
Software Approaches to Improve Confidence in Metabolite Identification.
An uncertainty or a lack of confidence is inherent in metabolite assignments.[55] An uncertainty in a metabolite assignment typically arises from an overlap in analytical signals and/or an ambiguity in reference spectral data. Simply, an exact mass or a single NMR resonance is shared by several compounds. Furthermore, the exact chemical composition of any biological sample or metabolome is currently unknown, but it is likely over a 100,000 compounds.[58] Thus, there is ample opportunity to misassign a spectral feature that corresponds to a currently unknown metabolite that resembles a known compound. An uncertainty in a metabolite assignment may also result from the software platform utilized to make the assignments.
Garcia et al. assessed the reliability of metabolite assignments by comparing different MS instruments and found a poor overlap in identifications.[47k] The metabolomes from butterhead and romaine lettuce were compared using an Agilent 6550 iFunnel Q-TOF LC/MS and a Waters Vion IMS QTOF. First, both data sets were analyzed with the Waters Progenesis QI software (case I). The analysis was repeated (case II) where the data set from the Waters instrument was processed with the Waters Progenesis QI software and the data set from the Agilent instrument was processed with the Agilent Mass Profinder software. Notably, the metabolites identified from the lettuce samples were dependent on both the processing software and the instrument. Only 26 metabolites (case I) were in common when Progenesis QI was used to analyze both the Waters (656 total metabolites) and Agilent (397 total metabolites) data sets. The situation improved when data sets were analyzed by the individual vendor software. A total of 101 metabolites (case II) were in common when Progenesis QI was used to analyze the Waters (581 total metabolites) data set and Mass Profinder was used to analyze the Agilent (372 total metabolites) data sets. Comparing the metabolites in common from Case I (26 metabolites) to Case II (101 metabolites) identified only 13 consistently identified metabolites. Overall, the processing software had a larger impact on the consistency of the outcome than the choice of instrument platform.
A database search of reference spectral data is the fundamental means by which metabolite assignments are accomplished.[59] The confidence in these metabolite assignments is dependent on the quality of the spectral matches and, of course, the presence of the metabolite in the database. In general, for NMR, a database search matches the chemical shifts, peak intensities, and peak pattern from the experimental spectra to the entire set of reference spectra where the best match minimizes these differences. Similarly, a MS query seeks to match exact masses, retention times, and fragmentation patterns. Thus, the accuracy of any assignment is directly proportional to the number of these matching spectral features, where matching more spectral features is generally better. Again, a multiplatform approach to metabolomics addresses this need by providing more spectral features to match against a database. Combining NMR and MS or MS/MS spectral data is a common means of obtaining a list of high confidence metabolite assignments. These methods are based on standard structure elucidation-based strategies.[47e, 60]
For example, the SUMMIT strategy uses direct injection high-resolution MS (e.g., Q-TOF, Orbitrap, FT-ICR) to assemble a list of exact masses for a metabolomics sample.[60] An experimental 1D 1H NMR and/or 2D 1H-13C HSQC spectra are also collected on the same metabolomics sample. The exact masses are converted to a molecular formula that are then used to calculate all possible chemical structures using the ChemSpider database (http://www.chemspider.com/).[61] A 1D 1H NMR and/or 2D 1H-13C HSQC spectra are calculated for each predicted structure using Mnova software (https://mestrelab.com/). The experimental NMR spectra are compared against all the predicted NMR structures to find the best matches and annotate the metabolomics sample. Boiteau et al expanded on the SUMMIT approach (i.e., NMR/MS2) to incorporate tandem mass spectrometry (MS/MS) to also identify unknown metabolites.[47e] Again, experimental MS/MS and 2D NMR spectra are compared against predicted spectra for every possible molecule consistent with the exact mass.
In addition to combining NMR and MS data, it is also possible to combine multiple NMR experiments to verify metabolite identifications. The COLMAR database (https://spin.ccic.osu.edu/index.php/colmar) allows for the identification of metabolites from either 1D 1H, 1D 13C, 2D 1H-13C HSQC or 2D 1H-1H-TOCSY spectra.[62] Notably, COLMAR also allows for queries using multiple 2D NMR spectra comprising 2D 1H-13C HSQC, 2D 1H-1H-TOCSYand 2D 1H-13C HSQC-TOCSY.[63] Recently, we demonstrated how a 2D 1H-13C HMBC experiment could similarly be combined with a 2D 1H-13C HSQC and 2D 1H-13C HSQC-TOCSY spectra to improve the accuracy of metabolite assignments. Interestingly, a 2D 13C-13C covariance matrix can be calculated from the experimental 2D 1H-13C HMBC spectrum. This HMBC covariance spectrum provides a complete and unique 13C-13C connectivity map for each spin system in the complex mixture which would be impractical to obtain experimentally. These 13C-13C connectivity maps can be used in the same way as 2D NMR spectra are used in database queries to make metabolite assignments. As discussed previously, using lesser common nuclei such as 15N or 31P in the NMR experiment would also improve confidence in metabolite assignments.[44e, 44f]
A Multiplatform Approach Addresses the Reproducibility Challenge of a Metabolomics Study.
Like all scientific endeavors, the reproducibility of a metabolomics study is a fundamental necessity.[28b, 47l] Similarly, methods employed by metabolomics should provide repeatable outcomes regardless of the instrument or research group, and the results should remain consistent over the course of months to years. Assessing the reproducibility of a metabolomics study presents a unique challenge since biological samples are typically limited, especially for clinical studies. Further, since biological samples are inherently variable, even obtaining a duplicate set of biological samples may likely lead to a large variance that will mask the true reproducibility of the study. A multiplatform approach can address this issue by obtaining replicate data sets on the same set of biological samples.
Martin et al. assessed the reproducibility of metabolomics by employing a multiplatform approach.[53b] Specifically, the same set of metabolomics samples were independently analyzed using five NMR and eleven LC-MS instruments. Notably, the NMR instruments were not all at the same field strength, and there was a larger variance in the configuration of the LC-MS instruments, different vendors, columns, ESI conditions, and mass analyzers (TOF, QTOF, orbitrap). The metabolomics samples consisted of human urine samples with or without the addition of 32 amino acid standards; and plasma from rats feed a diet with or without a vitamin D supplement. There was also an outlier purposely added to the group of human urine samples.
All the methods were able to discriminate between the spiked and non-spiked urine samples, but no method could differentiate between the two rat diets. To evaluate reproducibility, all the data sets were added to individual statistical blocks and a correlation (i.e., RV coefficients) was calculated between each pair of blocks. The data sets were identified to be highly convergent in spectral characteristics with RV coefficients ranging from 64 to 91%. The spectral information within either the NMR or LC-MS groups or between the NMR and LC-MS groups were highly convergent. The LC-MS methods had a lower average RV coefficient for the plasma samples relative to the urine samples, which was attributed to a matrix effect. The pairwise RV coefficients were used to construct a correlation network (Figure 6). While the overall network map shows the expected tight clustering, it also indicates the presence of subgroups based on instrument type or platform. There is a clear separation between NMR (N) and LC-MS platforms. There is also a subtle split between MS instruments, quadrupole (Q), orbitrap (O), and time of flight (T).
Figure 6.
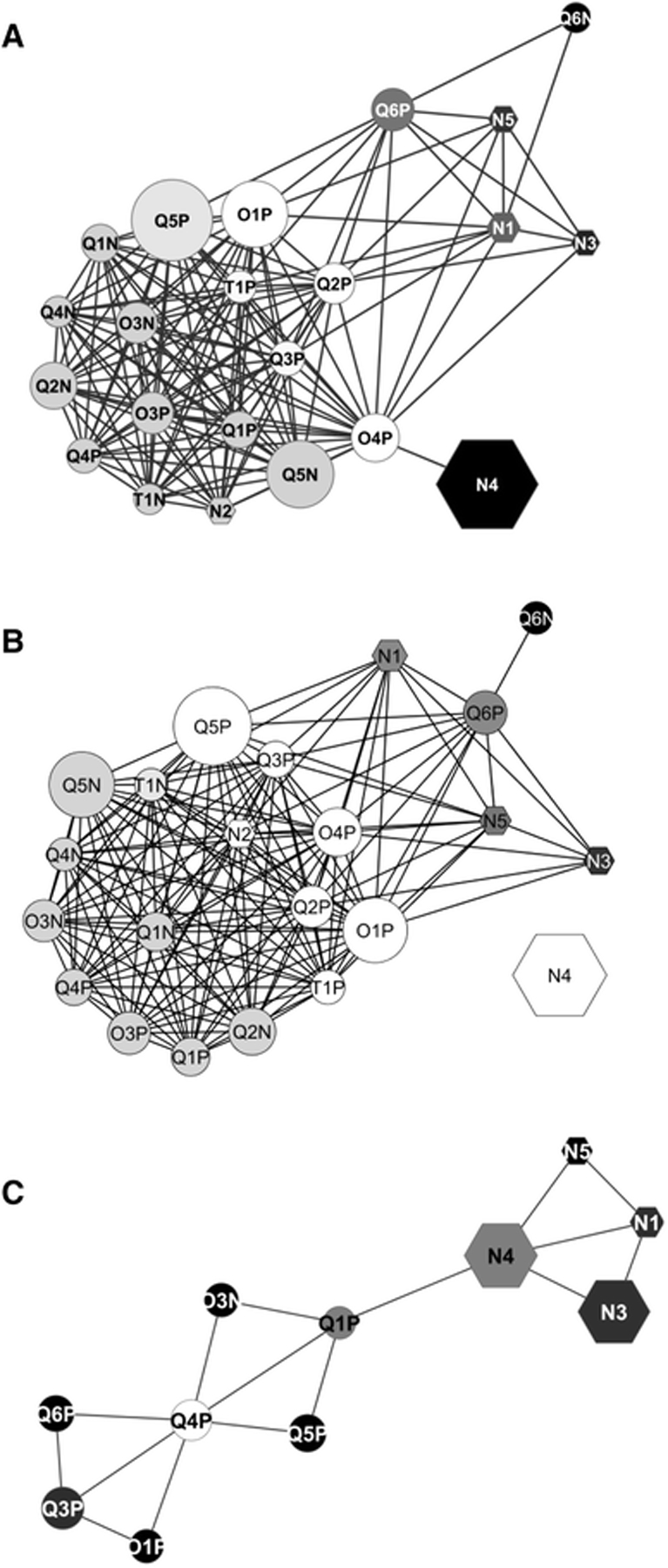
Correlations networks calculated from the pair-wise RV coefficients matrix from Test #1 (a) with spiked and non-spiked samples or with native urine samples only (b) and from Test #2 (c). Node labelling: N NMR platforms, Q QTOF mass spectrometer, O orbitrap mass spectrometer, T TOF mass spectrometer. The P or N appended to the mass spectrometer identifier number denotes positive or negative ionization mode, respectively. Node shapes: hexagon for nuclear magnetic resonance platforms, ellipse for mass spectrometers. The node size is proportional to the number of features retained by each instrument. The node colour from black to white indicates an increasing node degree (number of edges per node). The edges represent the RV coefficient values, with cut off values ≥0.791 in Test #1 and ≥0.708 in Test #2). At this cut off level, O3P was excluded from the Test #2 network (b) Martin, JC., Maillot, M., Mazerolles, G. et al. Can we trust untargeted metabolomics? Results of the metabo-ring initiative, a large-scale, multi-instrument inter-laboratory study. Metabolomics 11, 807–821 (2015). Copyright 2014 Springer Nature.
Both NMR and LC-MS were able to identify the urine outlier, a woman in mid-pregnancy, but the discriminating metabolites were different. LC-MS identified estroprogestative hormone derivatives and related steroid hormone derivatives while NMR identified alanine, threonine, lactate, and glycine. To further clarify, the LC-MS and NMR data sets achieved the same metabotypes but not necessarily at the feature and/or metabolite level. Nevertheless, Martin et al. demonstrated a high-level of reproducibility that was not dependent on instrument type, configuration, or data processing. While the focus of the study was to assess reproducibility, it also illustrated the inherent value of combining multiple analytical platforms for a metabolomics study since the NMR and LC-MS platforms covered distinct aspects of the metabolome (Figure 6).
One factor that may negatively impact reproducibility is sample matrix complexity. An example is the changes in urine content based on diet, physical activity, and other factors.[12c] This variability can impede sample processing differently across analytical platforms.[64] As discussed above, the study of PTSD patients by Chaby et al included an assessment of the reproducibility of the metabolomics experiments.[42] Along with the inconsistent metabolite coverage, each platform exhibited different levels of batch variations (Table 1). Identical replicate sets of PTSD serum samples were sent to the five vendors approximately 14 weeks apart. The percentage change in the number of PTSD biomarkers detected across the two batches ranged from −15% to 61%. Similarly, Martin et al. observed a significant and uniform sample-dependent decrease in the RV coefficients for the LC-MS platforms; going from an average of 0.87 for the human urine samples to an average of 0.74 for the rat plasma samples. Multiple studies have examined the effects of extractions protocols on sample matrices.[19, 46d, 65] Thus, applying an appropriate extraction method to minimize or eliminate variabilities due to matrix effects would ensure a multiplatform approach maintains an acceptable level of reproducibility across the different analytical platforms.
We recently explored the reproducibility of metabolomics platforms by conducting a meta-analysis of 24 pancreatic ductal adenocarcinoma (PDAC) metabolomics studies from the scientific literature.[54b] The majority of these studies relied on a single analytical platform, either NMR or LC-MS, to identify potential biomarkers for PDAC. Only one study used both NMR and LC-MS. The 24 metabolomics studies identified a total of 655 unique metabolites as potential biomarkers for PDAC where 87% of these metabolites were identified by a single study (Figure 7). Less than 1% of the 655 metabolites were detected in seven or more studies. More concerning was the fact that the direction of the metabolite fold change (FC) across the 24 PDAC metabolomics studies was inconsistent. Only 38% of the metabolites identified in at least five studies exhibited a consistent FC across all studies. There was no similarity in the set of identified metabolites based on analytical technique, cohort size, p-value, or FC cutoff. Recall, the analysis of serum from PTSD patients by five commercial metabolomics vendors also yielded a low consistency (16–70% coverage) in detecting literature defined PTSD metabolite biomarkers (Table 1).[42] The low reproducibility across multiple metabolomics studies does not appear to be disease dependent, but is likely a generic problem.
Figure 7.
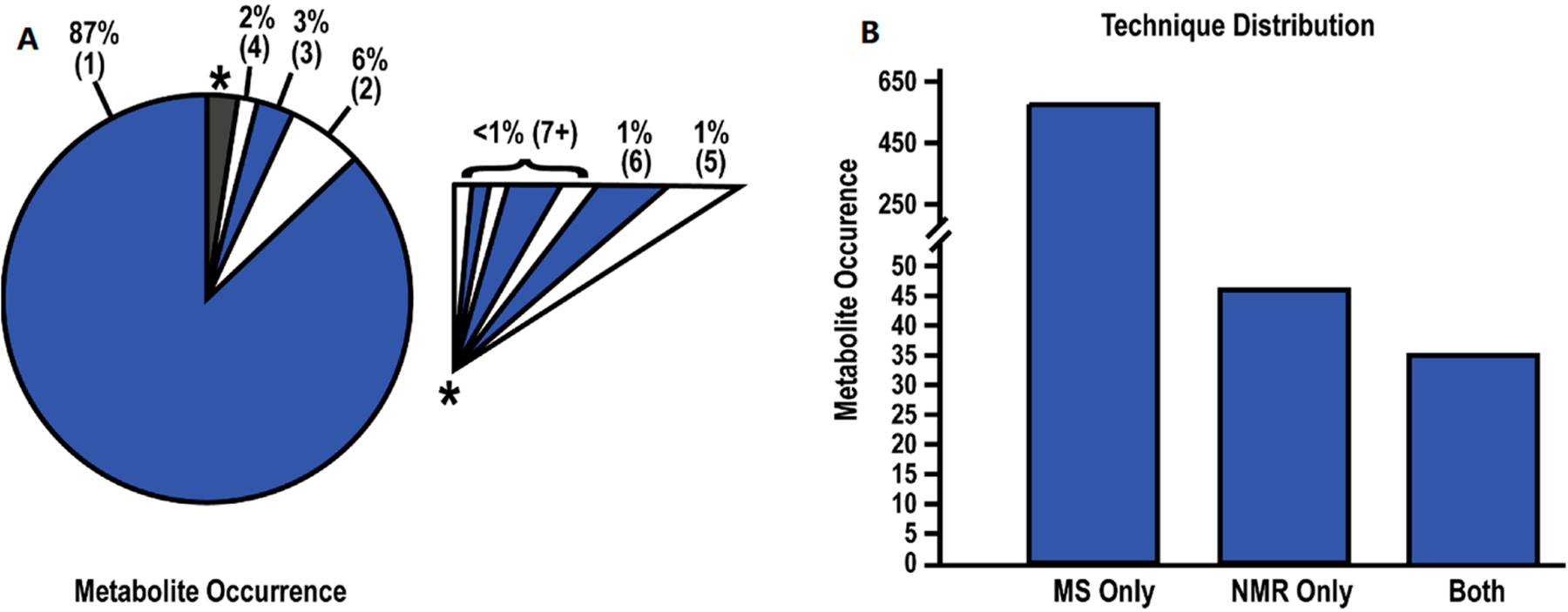
Summary of Metabolite Data. (A) Pie chart depicting the rate of metabolite occurrence across the 24 studies. Numbers within parentheses indicate the number of studies identifying the metabolites. An expanded view of the grey-starred slice of the pie chart is shown as an insert. (B) Bar chart depicting the number of metabolites identified by MS, NMR, or both techniques. Reprinted with permissions set under the CC BY 4.0 open access license (https://creativecommons.org/licenses/by/4.0/legalcode) from Roth, H.E.; Powers, R. Meta-Analysis Reveals Both the Promises and the Challenges of Clinical Metabolomics. Cancers 2022, 14, 3992. https://doi.org/10.3390/cancers14163992. Copyright 2022 MDPI.
Overall, study design and experimental parameters such as time of sample preprocessing, length of storage, sample preparation protocols, patient demographics, and bioinformatic analyses were likely major contributors to the low reproducibility of the PDAC and PTSD metabolomics studies. Of course, the fact that most studies used only a single analytical platform may also explain the high false positive rate which furthers the argument for a multiplatform approach to metabolomics.
Other routine factors may beneficially impact reproducibility. The intrinsic consistency of a high throughput and automated platform is likely to improve reproducibility in addition to reducing cost and increasing the speed of analysis. Simplifying the overall experimental protocol when combining two or more single platform methods is another easy and important path to improving reproducibility. Reducing the number of steps, especially the extraction procedure, will eliminate additional sources of error, bias, and sample variability. It will also increase throughput. Emphasizing quantitative NMR and MS methods of analysis for untargeted metabolomics may also improve reproducibility. Quantitation and targeted metabolomics requires creating standard curves and optimizing experimental conditions and parameters for each instrumental platform.[52b, 53a, 66] It is also necessary to incorporate quality controls and internal standards in the analysis. Finally, reliable quantitation requires routine calibrations and validation of instrument performance. Adopting these protocols into a standard untargeted metabolomics study using a multiplatform approach will likely improve the reproducibility of results.[28a, 47b, 64a, 67] Simply, it is more challenging to maintain two or more analytical platforms operating at the same high level of performance and rigor without synchronized quality control and quality assurance routines.
Processing and Analysis of Multiplatform Data sets.
Combining multiple platforms is a power tool to increase the quantity and quality of data available to compare complex systems. This increase in data generally is linked to an increased number of identifiable metabolites, but the increase in the amount of data is also difficult to manage. Further, merging inherently distinct data sets from different instrument types is an added challenge. There are a variety of file formats to contend with in addition to inconsistent data sizes, variable reference frames, differences in dynamic ranges, and a wide-distribution in the number of spectral features. Thus, a simple concatenation of two or more data sets is invalid since these differences would be the primary discriminating factors in any statistical model. There are also data specific preprocessing steps that need to be addressed. For example, NMR data are routinely phased, Fourier transformed, zero-filled and apodized. Similarly, MS data requires centroiding, deisotoping, imputing, and feature selection. Even preprocessing steps like spectral alignments that are conceptually similar require data-specific algorithms. Thus, most metabolomics software developed to date have been platform specific.[59]
Several software packages for metabolomics have been previously described in detail.[24f, 68] Many instrument vendors such as Agilent, Bruker, and Waters provide basic and advanced data analysis tools such as Mass Profinder, MetaboScape, and Progenesis QI, respectively. In addition, there is a growing collection of freely available metabolomics software applications that can be used for data preprocessing, processing, and statistical analysis.[69] Most of the freely available metabolomics software applications are for single platform data analysis and utilize R (www.r-project.org). Some examples include MetaboAnalyst and XCMS.[70] WebSpecmine is an R based package with an intuitive web site interface that can be used for the analysis of multiple data types including MS, NMR, IR, and UV-Vis.[71] MS-Dial is a Microsoft Windows program that includes all of the standard preprocessing capabilities for MS data but it is also capable of deconvolution of data-independent MS/MS spectra for metabolite identification and quantification.[72] Similarly, the commercial program Chenomx (www.chenomx.com) is specifically used to identify and quantify metabolites from 1D 1H NMR spectra. MVAPACK has been developed in the Octave (www.octave.org) environment for the full data analysis of NMR spectra.[73] MVAPACK can input standard NMR and text file formats and has been shown to simultaneously process multiple data sets from a multiplatform metabolomics study.[33d, 43, 74] The ability to use a single processing package for multiple platform data types would allow for a simplified data processing pipeline that would facilitate standardization and the development and adoption of best practices. This would likely lead to an overall improvement in the quality and scientific impact of metabolomics studies and provides further support for a multiplatform approach to metabolomics.
A major factor that will facilitate progress in multiplatform untargeted metabolomics is advancements in data processing methods and software.[75] Data interpretation methods can be exceptionally complex when interconnecting data from multiple analytical platforms. In fact, the proliferation of metabolomics software packages can be attributed to the multitude of analytical platforms in use, the complexity of untargeted metabolomics data, the diversity of algorithms required for data processing (i.e., from automated peak picking to advanced statistical modeling), and the need to address the individual requirements of each unique type of sample and data set.[24f, 68, 76] An identification-based analysis is commonly employed, which addresses both this processing complexity and the current lack of software for a multiplatform approach.[13, 77]
An identification-based approach requires spectra from each individual platform to be separately processed with available software. The data processing pipeline terminates at a common endpoint, typically a list of identified metabolites with an associated quantification.[15] The lists are then merged for any follow-up analysis. This can be an arduous task and ensuring consistency in processing parameters and analysis across multiple data processing pipelines can be difficult. Additionally, the automated assignment of metabolites routinely requires manual intervention and significant supervision to avoid false positives and true positives.[30, 78] Of course, overlapping metabolite identifications from two or more platforms will increase the confidence of any of these metabolite assignments. A primary problem with an identification-based approach is the routine occurrence of contradictory results. For example, one platform detects an increase in a metabolite while the other detects a decrease or there are large discrepancies in the magnitude of the metabolite’s fold change. Resolving these contradictions may not be straightforward. Possible solutions may involve a manual interrogation of the individual spectra to assess and choose the highest quality data, a comparison of the statistical significance and choosing the best likely outcome, a separate association experiment,[40, 79] or elimination of the metabolites from any further analysis. Alternatively, since NMR is generally more quantitative and reproducible relative to untargeted MS metabolomics studies, a contradiction may be resolved by simply choosing the NMR results.
Another fundamental challenge associated with a multiplatform approach is data size. In general, a study can quickly grow to a very large scale with increasing numbers of replicates, cohorts, groups, time points, and experimental conditions. A data set can easily consist of 1000s or more spectra. Of course, the situation simply multiplies with the application of two or more analytical platforms. The overall size of the data set can be daunting, where the physical storage of the data is a concern especially since GC-MS and LC-MS generate hundreds of megabytes to gigabytes of data per run.[80] A similar issue arises during data processing and the corresponding high demand for memory, and the impracticality of placing an entire large metabolomics data set in the memory of a single computer. Thus, a Big Data framework such as the MetHoS platform (www.methos.cebitec.uni-bielefeld.de) may become necessary. MetHoS distributes the data across a cluster of computers using cloud computing, which enables parallel processing and parallel data storage.[81]
A further confounding problem is the simple fact that data collection will likely occur over days or even months given the large number of samples and the use of multiple platforms. The resulting variability between different batches and instruments presents a unique challenge to data processing.[82] Again, the issue becomes more pronounced for a multiplatform approach, and highlights the importance of spectral alignment and normalization. Another important effect that can contribute to unintentional variation is injection order.[83] Injection order effects are typically managed by randomizing sample order, but very long runs can still cause injection bias.
Many methods have been developed to counteract batch effects by statistical methods.[83–84] Deng et al. categorized these methods into five groups: internal standard based (1), quality control metabolite based (2), quality control sample based (3), location-scale (4), and matrix factorization (5) methods.[84d] The introduced method uses a generalized additive model to determine group association of batches. This was described as:
| (2) |
where y is the batch effect component, x is the injection order, β0 is the intercept, f(x) is the smooth term, and ε is the error term. For example, the WaveICA algorithm was developed to remove batch effects in untargeted metabolomics studies using LC-MS by using a discrete wavelet transform (WT).[84d, 85] WaveICA deconvolutes the low-frequency intensity drift due to batch order from the high-frequency trends in the biological samples. The low frequency WT is then removed from the data set. WaveICA 2.0 was demonstrated using a cohort of 568 LC-MS plasma spectra collected from 497 colorectal cancer and 71 chronic enteritis patients. The LC-MS dataset was preprocessed using XCMS yielding a final dataset of 6,461 metabolite peaks and 74 quality control (QC) samples divided in 3 batches. A QC sample was created by combining a small aliquot from each of the 568 plasma samples. The removal of the batch effects were visualized by a comparison of the scaled intensities versus injection order plots and by the PCA score plots. The application of the WaveICA 2.0 algorithm clearly removed the three separate clusters in the PCA scores plot and the general downward trend in the batch order scaled intensities. Similarly, the average distance between the QC samples was also decreased. WaveICA 2.0 also resulted in an overall tighter cluster of PCA scores, the identification of more peaks that differentiated the two groups, and a greater classification accuracy using the area under a ROC curve (AUC). Groupings of QC samples by PCA allowed for visual comparison of the WaveICA 2.0 method with similar rank as two other statistical methods, QC-RLSC and QC-SVRC. The WaveICA 2.0 method was the only method able to increase the number of differential peaks, which doubled the number from the original data.
Karaman et al. demonstrated a preprocessing workflow for a large 8000 cohort data set of 1D 1H NMR spectra using QC samples.[86] QC samples were a combination of standard commercially available human serum samples and a pooled 50 μL aliquot of serum from each cohort participating in the study. 1D 1H NMR spectra were collected for each cohort and QC sample and combined into a single data matrix. The spectra were aligned with the Recursive Segment-wise Peak Alignment (RSPA) method.[87] Spectral regions corresponding to water, methanol and other interferents were removed, the spectra were normalized with probabilistic quotient normalization (PQN), and spectral outliers were removed based on Hotelling’s T2 values in a PCA scores plot. After this standard preprocessing, the authors separately applied a phase and cohort adjustment using a mean-centering operation to each of the six phase/cohort batches:
| (3) |
where is the pth intensity of the ith spectrum in the Mth batch, and NM is the number of samples in the batch. A PCA model using this preprocessing scheme removed all batch variations between the cohorts and QC samples.
Multivariate statistical analysis plays an important role in untargeted metabolomics to differentiate between groups and to aid in the identification of metabolic changes.[15, 26a, 88] PCA, partial least squares or projection to latent structures (PLS), and orthogonal projection to latent structures (OPLS) are commonly used in the analysis of a metabolomics data set derived from a single analytical platform. In fact, these statistical techniques were not designed for multiple data sets and a simple concatenation of data would likely lead to erroneous outcomes due to differences in the number and dynamic range of features. An alternative form of multivariate statistical analysis known as multiblock (MB) is a data integration method that can simultaneously use data from multiple data sources.[24a, 24d, 27, 33d, 89]
MB-PCA, MB-PLS, and MB-OPLS generate a single consensus statistical model from two or more metabolomics data sets.[24d, 89b] In this regard, a multiblock model can identify the dominant pattern of spectral features that are common across the entire integrated data set. The structure and design of a multiblock PCA is shown in Figure 8. In a single platform PCA approach, the data structure is two-dimensional where each column represents a spectral feature or metabolite, and each row is a biological replicate. The data matrix (Figure 8C) becomes three dimensional where the third axis represents the different instruments and/or sample types. Each data set derived from the individual instruments in a multiplatform approach occupies a separate block in the multiblock PCA (Figure 8B). As described in Figure 8C, a PCA model is generated for each block where the resulting loadings (p) are normalized to back calculate scores (t) for each block. All the scores are then combined into a global matrix T that is used to calculate a global PCA model and the resultant global weights and global score vector. Tanabe et al. compared the performance of PCA and MB-PCA using CE-MS, LC-MS (HILIC) and LC-MS (RPLC) metabolomics data sets collected from heart, kidney and liver tissues from ZDF and SD rats.[27] The MB-PCA score plot showed improved group separation relative to the PCA score plot (Figure 9). We observed a similar improvement with a MB-PCA model of 1D 1H NMR and DI-MS data sets produced from cell lysates of human dopaminergic neurons treated with environmental toxins (Figure 10).[33d] As these two examples illustrate, the MB matrix can be composed of either the raw spectra or quantified metabolites.
Figure 8.
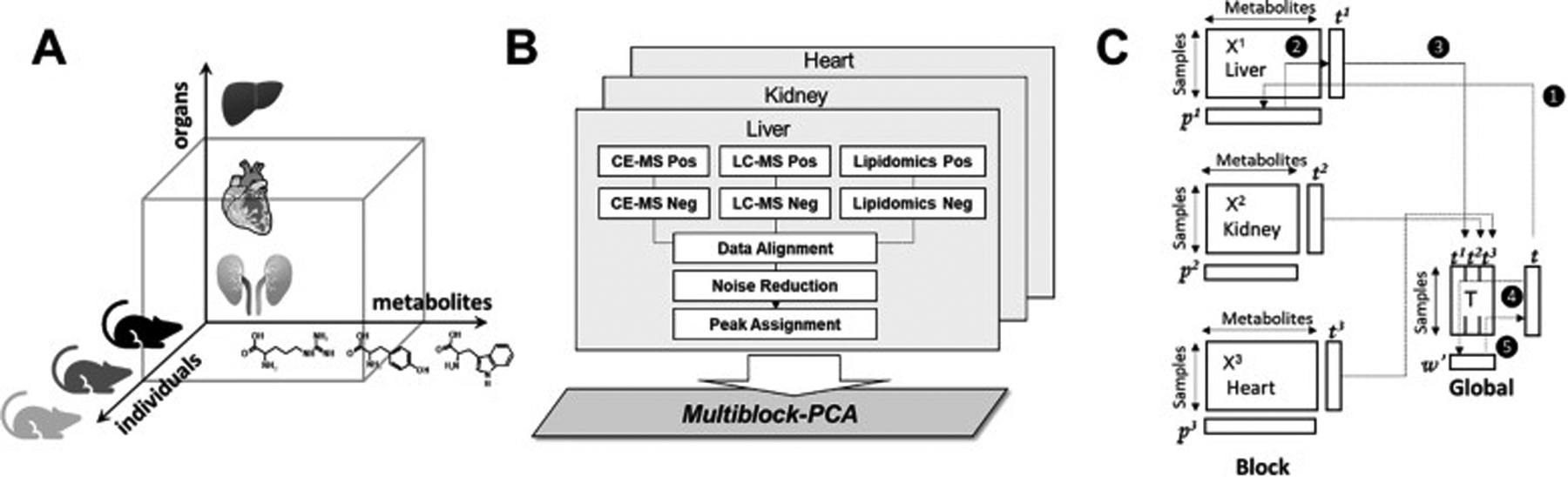
Multiblock metabolomics scheme. (A) Multiblock metabolomics requires a three-dimensional data structure, metabolites, individual samples, and organs. (B) Metabolomics data obtained from CE/MS and LC/MS (hilic and lipid modes) were merged into one data table. After noise reduction, peaks were identified based on the matched m/z values and normalized retention times of the corresponding standard compounds. This process was repeated for the heart, kidney, and liver, and three data matrices were integrated using multiblock PCA. (C) Multiblock PCA architecture: ❶All blocks of X1,2,3 were regressed by an arbitrary global score t to obtain the block loadings p1,2,3. ❷The block scores t1,2,3 were calculated with the normalized block loadings p1,2,3 using the following equation: tb = Xb pb where b = 1, 2, 3. ❸All block scores were combined to a global score matrix T. ❹The global score matrix T was regressed by the global score vector t, resulting in the global weights. ❺ Global weights were normalized to length one and a new global score vector t was then calculated. Reprinted with permission from K. Tanabe, C. Hayashi, T. Katahira, K. Sasaki, K. Igami, Multiblock metabolomics: An approach to elucidate whole-body metabolism with multiblock principal component analysis. Computational and Structural Biotechnology Journal 2021, 19, 1956–1965. Copyright 2021 Elsevier.
Figure 9.
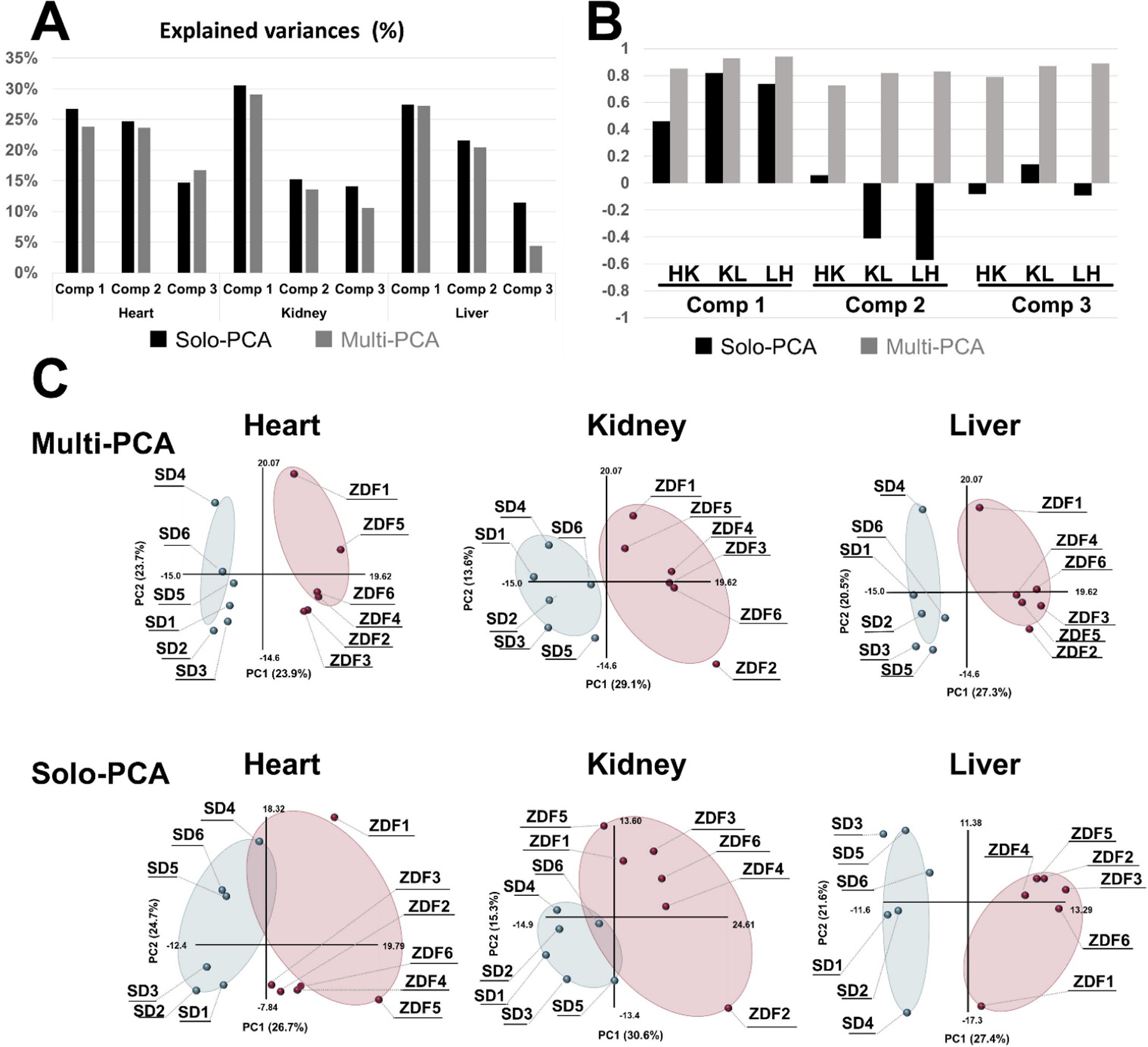
Comparison of multiblock PCA and solo PCA. Multiblock and solo-PCA were performed with the metabolomic data of the heart, kidney, and liver. (A) The explained variances (%) are indicated by black (solo) and gray (multiblock) bars for the first three components. (B) The cos θ values of the t block scores in the solo and multiblock PCAs are indicated by black and gray bars, respectively. HK: cos θ between heart and kidney, KL: cos θ between kidney and liver, LH: cos θ between liver and heart. (C) The tb block scores of the first and second components are plotted for the multiblock and solo PCAs for the three organs. Six SD rats and six ZDF rats are plotted as light blue and red solid circles in the scatter plots. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.) Reprinted with permission from K. Tanabe, C. Hayashi, T. Katahira, K. Sasaki, K. Igami, Multiblock metabolomics: An approach to elucidate whole-body metabolism with multiblock principal component analysis. Computational and Structural Biotechnology Journal 2021, 19, 1956–1965. Copyright 2021 Elsevier.
Figure 10.
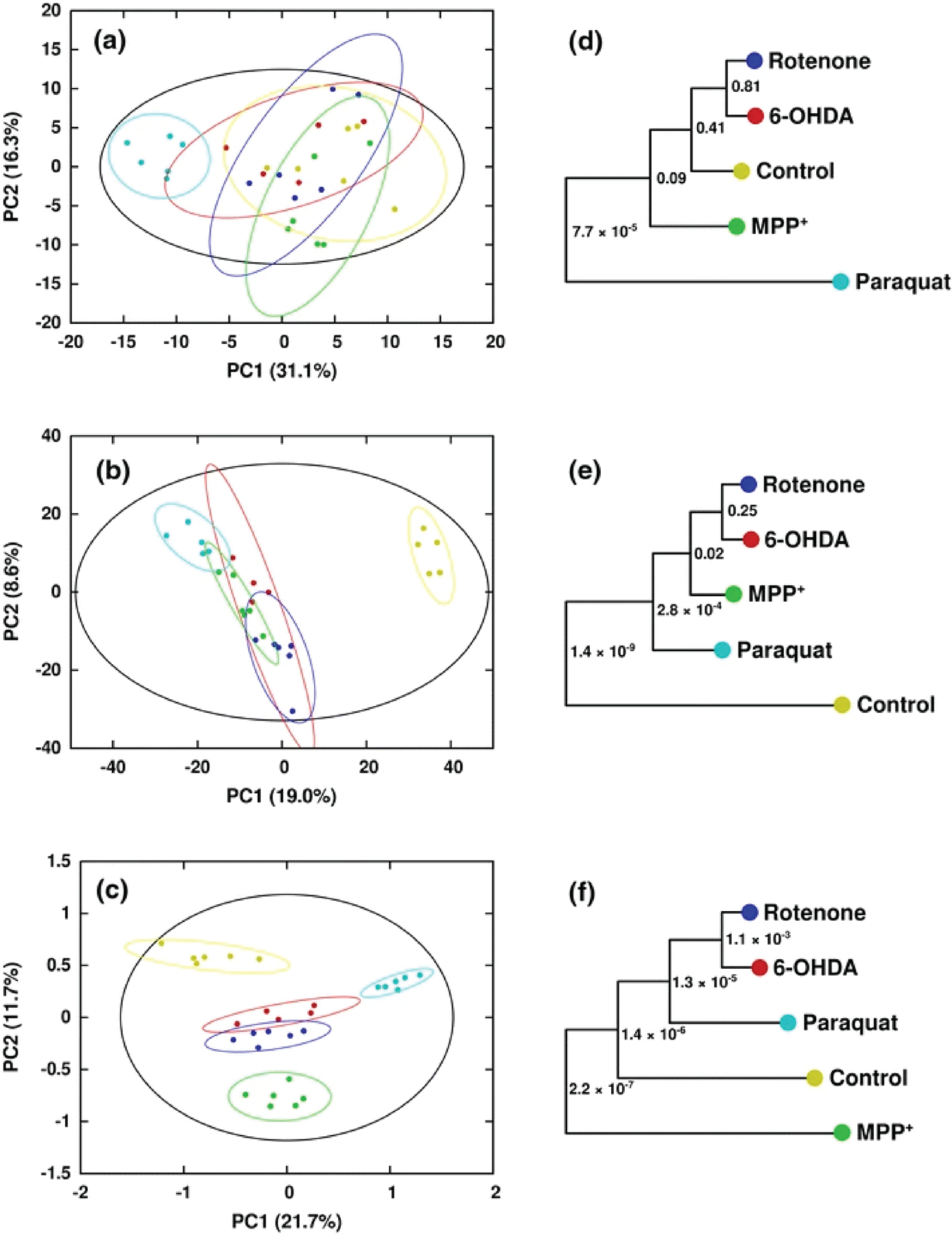
Scores generated from (a) PCA of 1H NMR in vacuo, (b) PCA of DI-ESI–MS in vacuo, and (c) MB-PCA of 1H NMR and DI-ESI–MS. Separations between classes are greatly increased upon combination of the two data sets via MB-PCA. Symbols designate the following classes: Control (yellow circle), Rotenone (blue circle), 6-OHDA (red circle), MPP+ (green circle), and Paraquat (turquoise colour circle). Corresponding dendrograms are shown in (d–f). The statistical significance of each node in the dendrogram is indicated by a p-value (Worley et al. 2013). Reprinted with permission from Marshall, D.D., Lei, S., Worley, B. et al. Combining DI-ESI–MS and NMR data sets for metabolic profiling. Metabolomics 11, 391–402 (2015). Copyright 2015 Springer Nature.
Conclusion and Perspectives
While multiplatform untargeted metabolomics is not a new concept, it is significantly underutilized despite the gain in information content and the improved coverage of the metabolome. Despite these advantages, a single analytical platform approach still dominates the metabolomics field, which may be a contributing factor to an abundance of tenuous metabolomics research studies populating the scientific literature. It may also explain the low reproducibility seen across multiple clinical studies aimed at identifying disease biomarkers.[42, 54b] MS methods have faced a growing concern over reproducibility and the accuracy of metabolite identifications.[47k] Conversely, the fact that a multiplatform approach to metabolomics has not been widely adopted is likely due to several practical considerations: (1) lack of expertise, (2) limited sample availability, (3) cost and time, (4) diminished throughput and concerns with sample degradation, (5) insufficient personnel to conduct multiple experiments, (6) inadequate software, and (7) the absence of well-established protocols and best practices.
Notwithstanding and as we have demonstrated throughout this review, a multiplatform approach provides several important advantages. It can deliver multi-fold increases in the coverage of the metabolome, it can significantly improve the confidence in metabolite assignments, it can simplify and potentially automate the assignment process, it can guide the development of best practices, and contribute to the optimization of experimental protocols. Overall, these contributions are likely to improve the accuracy and reproducibility of metabolomics studies, which in turn will increase the scientific value and insights derived from an untargeted metabolomics study.
NMR and MS have individually made enhancements in hardware, software, and other methodologies to improve metabolomics, but a parallel and separate application of these platforms to metabolomics is suboptimal. There is still a need for methodological developments to effectively integrate NMR and MS into a single metabolomics pipeline to take full advantage of a multiplatform approach and to maximize its utility. For example, single sample workflows have the potential of using precious samples more efficiently.[22–23, 33d, 47z] However, these workflows encounter difficulties when matching the sample conditions needed for both NMR and MS, such as buffer choice, derivatization needs, dilution effects and solvent-induced ion suppression. Nevertheless, the potential to enhance the capabilities and utility of multiplatform metabolomics makes this an area of current interest. The cost of untargeted metabolomics is still a large concern, where limiting instrument run times may be a means to improve performance and efficiency.[90] In this regard, automation has seen limited usage in metabolomics but it is clearly a valuable avenue to explore with great potential to improve throughput and reduce costs.[91] A combined approach to data analysis has further improved the information content produced by untargeted metabolomics. The use of multiblock models and other integrated data analysis tools have shown promise. Some software tools are already capable of streamlining the processing of multiplatform data such as MVAPACK and WebSpecmine, while others are in development.[71, 73] There are still issues to be resolved to make the combined analysis of multiple metabolomics data sets routine and widely accessible.
Untargeted metabolomics is a multifaceted technique routinely employed for the deconvolution of complex biological systems to answer important and fundamental scientific questions. The complexity of these systems dictates the need for efficiency and rigor that the multiplatform approach brings to metabolomics. The ability to increase the coverage of the metabolome and to improve our confidence in metabolite identifications is a highly desirable outcome. The ongoing developments in methods and workflows for streamlining a multiplatform analysis are highly promising and are expected to facilitate the widespread adoption of a multiplatform approach by the metabolomics field. This, in turn, will only improve the overall quality of metabolomics data and its scientific impact. The use of multiplatform techniques to study complex metabolic systems has the potential to improve our understanding of various issues related to the environment, nutrition, functional genomics, toxicology, and various aspects of human health and drug discovery.
Acknowledgments
We would like to thank Alexandra A. Crook and Darcy Cochran for their assistance in preparing the manuscript. This work was supported in part by funding from the National Institutes of Health (R01 AI148160, NIAID), and the Nebraska Center for Integrated Biomolecular Communication (NIH NIGMS P20 GM113126).
Footnotes
The authors declare no competing financial interest.
References
- [1].Johnson CH, Ivanisevic J, Siuzdak G, Nature Reviews Molecular Cell Biology 2016, 17, 451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Di Minno A, Gelzo M, Stornaiuolo M, Ruoppolo M, Castaldo G, Nutrition, Metabolism and Cardiovascular Diseases 2021, 31, 1645–1652. [DOI] [PubMed] [Google Scholar]
- [3].a Katam R, Lin C, Grant K, Katam CS, Chen S, International Journal of Molecular Sciences 2022, 23, 6985; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jin L, Godri Pollitt KJ, Liew Z, Rosen Vollmar AK, Vasiliou V, Johnson CH, Zhang Y, Current Environmental Health Reports 2021, 8, 7–22; [DOI] [PubMed] [Google Scholar]; c Wei S, Wei Y, Gong Y, Chen Y, Cui J, Li L, Yan H, Yu Y, Lin X, Li G, Yi L, Metabolomics 2022, 18, 35. [DOI] [PubMed] [Google Scholar]
- [4].a Lacalle-Bergeron L, Izquierdo-Sandoval D, Sancho JV, López FJ, Hernández F, Portolés T, TrAC Trends in Analytical Chemistry 2021, 135, 116161; [Google Scholar]; b Jacobs DM, van den Berg MA, Hall RD, Current Opinion in Biotechnology 2021, 70, 23–28. [DOI] [PubMed] [Google Scholar]
- [5].a Duarte NC, Becker SA, Jamshidi N, Thiele I, Mo ML, Vo TD, Srivas R, Palsson BØ, Proceedings of the National Academy of Sciences 2007, 104, 1777–1782; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Graham E, Lee J, Price M, Tarailo-Graovac M, Matthews A, Engelke U, Tang J, Kluijtmans LAJ, Wevers RA, Wasserman WW, van Karnebeek CDM, Mostafavi S, Journal of Inherited Metabolic Disease 2018, 41, 435–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a Tsunoda SM, Gonzales C, Jarmusch AK, Momper JD, Ma JD, Clinical Pharmacokinetics 2021, 60, 971–984; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Magny R, Auzeil N, Lefrère B, Mégarbane B, Houzé P, Labat L, Metabolites 2022, 12, 665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].a Kaspy MS, Semnani-Azad Z, Malik VS, Jenkins DJA, Hanley AJ, PROTEOMICS 2022, 22, 2100388; [DOI] [PubMed] [Google Scholar]; b Kumar A, Misra BB, PROTEOMICS 2019, 19, 1900042; [DOI] [PubMed] [Google Scholar]; c Powers R, Journal of Medicinal Chemistry 2014, 57, 5860–5870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a Trezzi J-P, Vlassis N, Hiller K, in Advances in Cancer Biomarkers: From biochemistry to clinic for a critical revision (Ed.: Scatena R), Springer; Netherlands, Dordrecht, 2015, pp. 41–57; [Google Scholar]; b Castelli FA, Rosati G, Moguet C, Fuentes C, Marrugo-Ramírez J, Lefebvre T, Volland H, Merkoçi A, Simon S, Fenaille F, Junot C, Analytical and Bioanalytical Chemistry 2022, 414, 759–789; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Chen C-J, Lee D-Y, Yu J, Lin Y-N, Lin T-M, Mass Spectrometry Reviews 2022, n/a, e21785; [Google Scholar]; d Kwon EH, Tennagels S, Gold R, Gerwert K, Beyer L, Tönges L, Biomolecules 2022, 12, 329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a Paul B, Lewinska M, Andersen JB, JHEP Reports 2022, 4, 100479; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Piras C, Noto A, Ibba L, Deidda M, Fanos V, Muntoni S, Leoni VP, Atzori L, Metabolites 2021, 11, 694; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Beyoğlu D, Idle JR, Metabolites 2020, 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Jans JJM, Broeks MH, Verhoeven-Duif NM, Current Opinion in Systems Biology 2022, 29, 100409. [Google Scholar]
- [11].Hollywood K, Brison DR, Goodacre R, PROTEOMICS 2006, 6, 4716–4723. [DOI] [PubMed] [Google Scholar]
- [12].a Ten-Doménech I, Ramos-Garcia V, Piñeiro-Ramos JD, Gormaz M, Parra-Llorca A, Vento M, Kuligowski J, Quintás G, Metabolites 2020, 10, 43; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Psychogios N, Hau DD, Peng J, Guo AC, Mandal R, Bouatra S, Sinelnikov I, Krishnamurthy R, Eisner R, Gautam B, Young N, Xia J, Knox C, Dong E, Huang P, Hollander Z, Pedersen TL, Smith SR, Bamforth F, Greiner R, McManus B, Newman JW, Goodfriend T, Wishart DS, PLOS ONE 2011, 6, e16957; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bouatra S, Aziat F, Mandal R, Guo AC, Wilson MR, Knox C, Bjorndahl TC, Krishnamurthy R, Saleem F, Liu P, Dame ZT, Poelzer J, Huynh J, Yallou FS, Psychogios N, Dong E, Bogumil R, Roehring C, Wishart DS, PLOS ONE 2013, 8, e73076; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gertsman I, Barshop BA, Journal of Inherited Metabolic Disease 2018, 41, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Andreas NJ, Hyde MJ, Gomez-Romero M, Lopez-Gonzalvez MA, Villaseñor A, Wijeyesekera A, Barbas C, Modi N, Holmes E, Garcia-Perez I, ELECTROPHORESIS 2015, 36, 2269–2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Wishart DS, Guo A, Oler E, Wang F, Anjum A, Peters H, Dizon R, Sayeeda Z, Tian S, Lee Brian L., Berjanskii M, Mah R, Yamamoto M, Jovel J, Torres-Calzada C, Hiebert-Giesbrecht M, Lui Vicki W., Varshavi D, Varshavi D, Allen D, Arndt D, Khetarpal N, Sivakumaran A, Harford K, Sanford S, Yee K, Cao X, Budinski Z, Liigand J, Zhang L, Zheng J, Mandal R, Karu N, Dambrova M, Schiöth Helgi B., Greiner R, Gautam V, Nucleic Acids Research 2021, 50, D622–D631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Marshall DD, Powers R, Progress in Nuclear Magnetic Resonance Spectroscopy 2017, 100, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a Alonso A, Marsal S, Julia A, Frontiers in Bioengineering and Biotechnology 2015; [DOI] [PMC free article] [PubMed]; b Fuhrer T, Zamboni N, Current Opinion in Biotechnology 2015, 31, 73–78; [DOI] [PubMed] [Google Scholar]; c Wishart DS, Cheng LL, Copié V, Edison AS, Eghbalnia HR, Hoch JC, Gouveia GJ, Pathmasiri W, Powers R, Schock TB, Sumner LW, Uchimiya M, Metabolites 2022, 12, 678; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Segers K, Declerck S, Mangelings D, Heyden YV, Eeckhaut AV, Bioanalysis 2019, 11, 2297–2318; [DOI] [PubMed] [Google Scholar]; e Nagana Gowda GA, Raftery D, in Cancer Metabolomics: Methods and Applications (Ed.: Hu S), Springer International Publishing, Cham, 2021, pp. 19–37. [Google Scholar]
- [17].a Paiva C, Amaral A, Rodriguez M, Canyellas N, Correig X, Ballescà JL, Ramalho-Santos J, Oliva R, Andrology 2015, 3, 496–505; [DOI] [PubMed] [Google Scholar]; b Lima AR, Carvalho M, Aveiro SS, Melo T, Domingues MR, Macedo-Silva C, Coimbra N, Jerónimo C, Henrique R, Bastos M. d. L., Guedes de Pinho P, Pinto J, Journal of Proteome Research 2022, 21, 727–739. [DOI] [PubMed] [Google Scholar]
- [18].a Patra B, Meena R, Rosalin R, Singh M, Paulraj R, Ekka RK, Pradhan SN, Applied Biochemistry and Biotechnology 2022; [DOI] [PubMed]; b Umar AH, Ratnadewi D, Rafi M, Sulistyaningsih YC, Metabolites 2021, 11, 42; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lebanov L, Chung Lam S, Tedone L, Sostaric T, Smith JA, Ghiasvand A, Paull B, Journal of Chromatography B 2021, 1179, 122861. [DOI] [PubMed] [Google Scholar]
- [19].Mushtaq MY, Choi YH, Verpoorte R, Wilson EG, Phytochemical Analysis 2014, 25, 291–306. [DOI] [PubMed] [Google Scholar]
- [20].Williams R, Lenz EM, Wilson AJ, Granger J, Wilson ID, Major H, Stumpf C, Plumb R, Molecular BioSystems 2006, 2, 174–183. [DOI] [PubMed] [Google Scholar]
- [21].Dunn WB, Bailey NJC, Johnson HE, Analyst 2005, 130, 606–625. [DOI] [PubMed] [Google Scholar]
- [22].Walker LR, Hoyt DW, Walker II SM, Ward JK, Nicora CD, Bingol K, Magnetic Resonance in Chemistry 2016, 54, 998–1003. [DOI] [PubMed] [Google Scholar]
- [23].Beltran A, Suarez M, Rodríguez MA, Vinaixa M, Samino S, Arola L, Correig X, Yanes O, Analytical Chemistry 2012, 84, 5838–5844. [DOI] [PubMed] [Google Scholar]
- [24].a Beniddir MA, Le Moyec L, Triba MN, Longeon A, Deville A, Blond A, Pham VC, de Voogd NJ, Bourguet-Kondracki M-L, Analytical and Bioanalytical Chemistry 2022, 414, 5929–5942; [DOI] [PubMed] [Google Scholar]; b Pathmasiri W, Kay K, McRitchie S, Sumner S, in Computational Methods and Data Analysis for Metabolomics (Ed.: Li S), Springer US, New York, NY, 2020, pp. 61–97; [DOI] [PubMed] [Google Scholar]; c Wishart DS, Briefings in Bioinformatics 2007, 8, 279–293; [DOI] [PubMed] [Google Scholar]; d Worley B, Powers R, Chemometrics and Intelligent Laboratory Systems 2015, 149, 33–39; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Zhang D, Zhang M, in NMR-Based Metabolomics: Methods and Protocols (Eds.: Gowda GAN, Raftery D), Springer New York, New York, NY, 2019, pp. 471–482; [Google Scholar]; f Misra BB, Metabolomics 2021, 17, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Rauh D, Blankenburg C, Fischer TG, Jung N, Kuhn S, Schatzschneider U, Schulze T, Neumann S, Pure and Applied Chemistry 2022, 94, 725–736. [Google Scholar]
- [26].a Worley B, Powers R, Current Metabolomics 2013, 1, 92–107; [DOI] [PMC free article] [PubMed] [Google Scholar]; b van den Berg RA, Hoefsloot HCJ, Westerhuis JA, Smilde AK, van der Werf MJ, BMC Genomics 2006, 7, 142; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Vu T, Riekeberg E, Qiu Y, Powers R, Metabolomics 2018, 14, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tanabe K, Hayashi C, Katahira T, Sasaki K, Igami K, Computational and Structural Biotechnology Journal 2021, 19, 1956–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].a Comte B, Monnerie S, Brandolini-Bunlon M, Canlet C, Castelli F, Chu-Van E, Colsch B, Fenaille F, Joly C, Jourdan F, Lenuzza N, Lyan B, Martin J-F, Migné C, Morais JA, Pétéra M, Poupin N, Vinson F, Thevenot E, Junot C, Gaudreau P, Pujos-Guillot E, eBioMedicine 2021, 69, 103440; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Gonzalez-Riano C, Tapia-González S, Perea G, González-Arias C, DeFelipe J, Barbas C, Molecular Neurobiology 2021, 58, 3224–3237. [DOI] [PubMed] [Google Scholar]
- [29].Chamberlain CA, Rubio VY, Garrett TJ, Metabolomics 2019, 15, 135. [DOI] [PubMed] [Google Scholar]
- [30].Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, McLean JA, Journal of the American Society for Mass Spectrometry 2016, 27, 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].a Annesley TM, Clinical Chemistry 2003, 49, 1041–1044; [DOI] [PubMed] [Google Scholar]; b Lu W, Su X, Klein MS, Lewis IA, Fiehn O, Rabinowitz JD, Annual Review of Biochemistry 2017, 86, 277–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].a Dona AC, Kyriakides M, Scott F, Shephard EA, Varshavi D, Veselkov K, Everett JR, Computational and Structural Biotechnology Journal 2016, 14, 135–153; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Bhinderwala F, Roth HE, Noel H, Feng D, Powers R, Journal of Magnetic Resonance 2022, 345, 107335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].a Kordalewska M, Wawrzyniak R, Jacyna J, Godzień J, López Gonzálves Á, Raczak-Gutknecht J, Markuszewski M, Gutknecht P, Matuszewski M, Siebert J, Barbas C, Markuszewski MJ, Biochemistry and Biophysics Reports 2022, 31, 101318; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhuang J, Yang X, Zheng Q, Li K, Cai L, Yu H, Lv J, Bai K, Cao Q, Li P, Yang H, Wang J, Lu Q, Metabolites 2022, 12, 558; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Bhinderwala F, Wase N, DiRusso C, Powers R, Journal of Proteome Research 2018, 17, 4017–4022; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Marshall DD, Lei S, Worley B, Huang Y, Garcia-Garcia A, Franco R, Dodds ED, Powers R, Metabolomics 2015, 11, 391–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].a Maia M, Figueiredo A, Cordeiro C, Sousa Silva M, Mass Spectrometry Reviews, n/a; [DOI] [PubMed] [Google Scholar]; b Ding Y, Pei C, Shu W, Wan J, Chemistry – An Asian Journal 2022, 17, e202101310; [DOI] [PubMed] [Google Scholar]; c Jiang X, Chen X, Chen Z, Yu J, Lou H, Wu J, Journal of Proteome Research 2021, 20, 4346–4356. [DOI] [PubMed] [Google Scholar]
- [35].a Colizza K, Mahoney KE, Yevdokimov AV, Smith JL, Oxley JC, Journal of The American Society for Mass Spectrometry 2016, 27, 1796–1804; [DOI] [PubMed] [Google Scholar]; b Alseekh S, Aharoni A, Brotman Y, Contrepois K, D’Auria J, Ewald J, Ewald JC, Fraser PD, Giavalisco P, Hall RD, Heinemann M, Link H, Luo J, Neumann S, Nielsen J, Perez de Souza L, Saito K, Sauer U, Schroeder FC, Schuster S, Siuzdak G, Skirycz A, Sumner LW, Snyder MP, Tang H, Tohge T, Wang Y, Wen W, Wu S, Xu G, Zamboni N, Fernie AR, Nature Methods 2021, 18, 747–756; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Cajka T, Fiehn O, Analytical Chemistry 2016, 88, 524–545; [DOI] [PubMed] [Google Scholar]; d Börnsen KO, in Mass Spectrometry of Proteins and Peptides: Mass Spectrometry of Proteins and Peptides (Ed.: Chapman JR), Humana Press, Totowa, NJ, 2000, pp. 387–404. [Google Scholar]
- [36].Chen X, Yin Y, Luo M, Zhou Z, Cai Y, Zhu Z-J, Analytica Chimica Acta 2022, 1210, 339886. [DOI] [PubMed] [Google Scholar]
- [37].Henry R, Cassel T, in The Handbook of Metabolomics (Eds.: Fan TW-M, Lane AN, Higashi RM), Humana Press, Totowa, NJ, 2012, pp. 99–125. [Google Scholar]
- [38].a Zhong AB, Muti IH, Eyles SJ, Vachet RW, Sikora KN, Bobst CE, Calligaris D, Stopka SA, Agar JN, Wu C-L, Mino-Kenudson MA, Agar NYR, Christiani DC, Kaltashov IA, Cheng LL, Frontiers in Molecular Biosciences 2022, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zemaitis KJ, Izydorczak AM, Thompson AC, Wood TD, Metabolites 2021, 11, 253; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhao C, Cai Z, Mass Spectrometry Reviews 2022, 41, 469–487. [DOI] [PubMed] [Google Scholar]
- [39].a Mello C, Crotti AEM, Vessecchi R, Cunha WR, Journal of Molecular Structure 2006, 799, 141–145; [Google Scholar]; b Noda I, Applied Spectroscopy 1990, 44, 550–561. [Google Scholar]
- [40].Agnolet S, Jaroszewski JW, Verpoorte R, Staerk D, Metabolomics 2010, 6, 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ghosh R, Bu G, Nannenga BL, Sumner LW, Frontiers in Molecular Biosciences 2021, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Chaby LE, Lasseter HC, Contrepois K, Salek RM, Turck CW, Thompson A, Vaughan T, Haas M, Jeromin A, Metabolites 2021, 11, 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Lei S, Zavala-Flores L, Garcia-Garcia A, Nandakumar R, Huang Y, Madayiputhiya N, Stanton RC, Dodds ED, Powers R, Franco R, ACS Chem Biol 2014, 9, 2032–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].a Cox N, Millard P, Charlier C, Lippens G, Analytical Chemistry 2021, 93, 4818–4824; [DOI] [PubMed] [Google Scholar]; b Gowda GAN, Tayyari F, Ye T, Suryani Y, Wei S, Shanaiah N, Raftery D, Analytical Chemistry 2010, 82, 8983–8990; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Ye T, Mo H, Shanaiah N, Gowda GAN, Zhang S, Raftery D, Analytical Chemistry 2009, 81, 4882–4888; [DOI] [PMC free article] [PubMed] [Google Scholar]; d DeSilva MA, Shanaiah N, Nagana Gowda GA, Rosa-Pérez K, Hanson BA, Raftery D, Magnetic Resonance in Chemistry 2009, 47, S74–S80; [DOI] [PMC free article] [PubMed] [Google Scholar]; e Bhinderwala F, Lonergan S, Woods J, Zhou C, Fey PD, Powers R, Analytical Chemistry 2018, 90, 4521–4528; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Bhinderwala F, Evans P, Jones K, Laws BR, Smith TG, Morton M, Powers R, Analytical Chemistry 2020, 92, 9536–9545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].a Nagana Gowda GA, Pascua V, Raftery D, Analytical Chemistry 2021, 93, 14844–14850; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagana Gowda GA, Raftery D, Analytical Chemistry 2017, 89, 4620–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a Hajjaj H, Blanc PJ, Goma G, François J, FEMS Microbiology Letters 1998, 164, 195–200; [Google Scholar]; b Faijes M, Mars AE, Smid EJ, Microbial Cell Factories 2007, 6, 27; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Grata E, Boccard J, Guillarme D, Glauser G, Carrupt P-A, Farmer EE, Wolfender J-L, Rudaz S, Journal of Chromatography B 2008, 871, 261–270; [DOI] [PubMed] [Google Scholar]; d Ye D, Li X, Wang C, Liu S, Zhao L, Du J, Xu J, Li J, Tian L, Xia X, Microbiology Spectrum 2021, 9, e00625–00621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].a Antunes AC, Acunha T. d. S., Perin EC, Rombaldi CV, Galli V, Chaves FC, Journal of the Science of Food and Agriculture 2019, 99, 6973–6980; [DOI] [PubMed] [Google Scholar]; b Bearden DW, Sheen DA, Simón-Manso Y, Benner BA, Rocha WFC, Blonder N, Lippa KA, Beger RD, Schnackenberg LK, Sun J, Mehta KY, Cheema AK, Gu H, Marupaka R, Nagana Gowda GA, Raftery D, Metabolites 2019, 9, 270; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Beltran A, Samino S, Yanes O, in Mass Spectrometry in Metabolomics: Methods and Protocols (Ed.: Raftery D), Springer New York, New York, NY, 2014, pp. 75–80; [Google Scholar]; d Bilbrey EA, Williamson K, Hatzakis E, Miller DD, Fresnedo-Ramírez J, Cooperstone JL, New Phytologist 2021, 232, 1944–1958; [DOI] [PubMed] [Google Scholar]; e Boiteau RM, Hoyt DW, Nicora CD, Kinmonth-Schultz HA, Ward JK, Bingol K, Metabolites 2018, 8, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Bub A, Kriebel A, Dörr C, Bandt S, Rist M, Roth A, Hummel E, Kulling S, Hoffmann I, Watzl B, JMIR Res Protoc 2016, 5, e146; [DOI] [PMC free article] [PubMed] [Google Scholar]; g Cambeiro-Pérez N, González-Gómez X, González-Barreiro C, Pérez-Gregorio MR, Fernandes I, Mateus N, de Freitas V, Sánchez B, Martínez-Carballo E, Molecules 2021, 26, 787; [DOI] [PMC free article] [PubMed] [Google Scholar]; h de Falco B, Grauso L, Fiore A, Bonanomi G, Lanzotti V, Phytochemical Analysis 2022, 33, 696–709; [DOI] [PubMed] [Google Scholar]; i Diémé B, Lefèvre A, Nadal-Desbarats L, Galineau L, Madji Hounoum B, Montigny F, Blasco H, Andres CR, Emond P, Mavel S, Journal of Pharmaceutical and Biomedical Analysis 2017, 142, 270–278; [DOI] [PubMed] [Google Scholar]; j Du S, Chen Y, Liu X, Zhang Z, Jiang Y, Zhou Y, Zhang H, Li Q, XuemeiWang Y Wang, Feng R, Journal of Proteomics 2022, 252, 104394; [DOI] [PubMed] [Google Scholar]; k García CJ, Yang X, Huang D, Tomás-Barberán FA, Metabolomics 2020, 16, 85; [DOI] [PubMed] [Google Scholar]; l Gonzalez-Riano C, Saiz J, Barbas C, Bergareche A, Huerta JM, Ardanaz E, Konjevod M, Mondragon E, Erro ME, Chirlaque MD, Abilleira E, Goñi-Irigoyen F, Amiano P, npj Parkinson’s Disease 2021, 7, 73; [DOI] [PMC free article] [PubMed] [Google Scholar]; m Hu Y, Zhang L, Wang H, Xu S, Mujeeb A, Nie G, Tang H, Wang Y, Metabolomics 2017, 13, 49; [Google Scholar]; n Li B, Hui F, Yuan Z, Shang Q, Shuai G, Bao Y, Chen Y, Journal of Functional Foods 2021, 78, 104355; [Google Scholar]; o Li X, Sarma SJ, Sumner LW, Jones AD, Last RL, Journal of Agricultural and Food Chemistry 2022, 70, 8010–8023; [DOI] [PMC free article] [PubMed] [Google Scholar]; p Mack C, Wefers D, Schuster P, Weinert CH, Egert B, Bliedung S, Trierweiler B, Muhle-Goll C, Bunzel M, Luy B, Kulling SE, Postharvest Biology and Technology 2017, 125, 65–76; [Google Scholar]; q Perales-Chorda C, Obeso D, Twomey L, Rojas-Benedicto A, Puchades-Carrasco L, Roca M, Pineda-Lucena A, Laguna JJ, Barbas C, Esteban V, Martí-Garrido J, Ibañez-Echevarria E, López-Salgueiro R, Barber D, Villaseñor A, Fernández de Rojas D. Hernández, Clinical & Experimental Allergy 2021, 51, 1295–1309; [DOI] [PubMed] [Google Scholar]; r Pérez-Míguez R, Castro-Puyana M, Sánchez-López E, Plaza M, Marina ML, Molecules 2020, 25, 887; [DOI] [PMC free article] [PubMed] [Google Scholar]; s Sobolev AP, Mannina L, Capitani D, Sanzò G, Ingallina C, Botta B, Fornarini S, Crestoni ME, Chiavarino B, Carradori S, Locatelli M, Giusti AM, Simonetti G, Vinci G, Preti R, Toniolo C, Reverberi M, Scarpari M, Parroni A, Abete L, Natella F, Di Sotto A, Food Chemistry 2018, 255, 120–131; [DOI] [PubMed] [Google Scholar]; t Zhu D, Kebede B, Chen G, McComb K, Frew R, Food Control 2020, 116, 107017; [Google Scholar]; u Münger LH, Trimigno A, Picone G, Freiburghaus C, Pimentel G, Burton KJ, Pralong FP, Vionnet N, Capozzi F, Badertscher R, Vergères G, Journal of Proteome Research 2017, 16, 3321–3335; [DOI] [PubMed] [Google Scholar]; v Peron G, Uddin J, Stocchero M, Mammi S, Schievano E, Dall’Acqua S, Journal of Pharmaceutical and Biomedical Analysis 2017, 140, 62–70; [DOI] [PubMed] [Google Scholar]; w Rubić I, Burchmore R, Weidt S, Regnault C, Kuleš J, Barić Rafaj R, Mašek T, Horvatić A, Crnogaj M, Eckersall PD, Novak P, Mrljak V, International Journal of Molecular Sciences 2022, 23, 1575; [DOI] [PMC free article] [PubMed] [Google Scholar]; x Sitole LJ, Tugizimana F, Meyer D, Journal of Pharmaceutical and Biomedical Analysis 2019, 176, 112796; [DOI] [PubMed] [Google Scholar]; y Torres S, Samino S, Ràfols P, Martins-Green M, Correig X, Ramírez N, Environment International 2021, 146, 106242; [DOI] [PubMed] [Google Scholar]; z van der Walt G, Lindeque JZ, Mason S, Louw R, Metabolites 2021, 11, 658; [DOI] [PMC free article] [PubMed] [Google Scholar]; aa Vanhaverbeke C, Touboul D, Elie N, Prévost M, Meunier C, Michelland S, Cunin V, Ma L, Vermijlen D, Delporte C, Pochet S, Le Gouellec A, Sève M, Van Antwerpen P, Souard F, Scientific Reports 2021, 11, 14205; [DOI] [PMC free article] [PubMed] [Google Scholar]; ab Venter L, Loots DT, Mienie LJ, Jansen van Rensburg PJ, Mason S, Vosloo A, Lindeque JZ, Metabolomics 2018, 14, 49; [DOI] [PubMed] [Google Scholar]; ac Xuan Q, Ouyang Y, Wang Y, Wu L, Li H, Luo Y, Zhao X, Feng D, Qin W, Hu C, Zhou L, Liu X, Zou H, Cai C, Wu J, Jia W, Xu G, Advanced Science 2020, 7, 2001714; [DOI] [PMC free article] [PubMed] [Google Scholar]; ad Yan X, Zhao M, Zou W, Tian P, Sun L, Wang M, Zhao C, Biomedical Chromatography 2021, 35, e5120; [DOI] [PubMed] [Google Scholar]; ae Yanibada B, Hohenester U, Pétéra M, Canlet C, Durand S, Jourdan F, Ferlay A, Morgavi DP, Boudra H, Journal of Dairy Science 2021, 104, 12553–12566. [DOI] [PubMed] [Google Scholar]
- [48].Gil A, Siegel D, Permentier H, Reijngoud DJ, Dekker F, Bischoff R, Electrophoresis 2015, 36, 2156–2169. [DOI] [PubMed] [Google Scholar]
- [49].a Atkinson DE, Walton GM, Journal of Biological Chemistry 1967, 242, 3239–3241; [PubMed] [Google Scholar]; b De la Fuente IM, Cortés JM, Valero E, Desroches M, Rodrigues S, Malaina I, Martínez L, PLOS ONE 2014, 9, e108676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Le J, Yuan T-F, Zhang Y, Wang S-T, Li Y, Journal of Lipid Research 2018, 59, 1783–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gowda GAN, Raftery D, Analytical Chemistry 2014, 86, 5433–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].a Malm L, Palm E, Souihi A, Plassmann M, Liigand J, Kruve A, Molecules 2021, 26, 3524; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Crook AA, Powers R, Molecules 2020, 25, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].a Xiao JF, Zhou B, Ressom HW, TrAC Trends in Analytical Chemistry 2012, 32, 1–14; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Martin J-C, Maillot M, Mazerolles G, Verdu A, Lyan B, Migné C, Defoort C, Canlet C, Junot C, Guillou C, Manach C, Jabob D, Bouveresse DJ-R, Paris E, Pujos-Guillot E, Jourdan F, Giacomoni F, Courant F, Favé G, Le Gall G, Chassaigne H, Tabet J-C, Martin J-F, Antignac J-P, Shintu L, Defernez M, Philo M, Alexandre-Gouaubau M-C, Amiot-Carlin M-J, Bossis M, Triba MN, Stojilkovic N, Banzet N, Molinié R, Bott R, Goulitquer S, Caldarelli S, Rutledge DN, Metabolomics 2015, 11, 807–821; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Creek DJ, Dunn WB, Fiehn O, Griffin JL, Hall RD, Lei Z, Mistrik R, Neumann S, Schymanski EL, Sumner LW, Trengove R, Wolfender J-L, Metabolomics 2014, 10, 350–353. [Google Scholar]
- [54].a Reveglia P, Paolillo C, Ferretti G, De Carlo A, Angiolillo A, Nasso R, Caputo M, Matrone C, Di Costanzo A, Corso G, Metabolomics 2021, 17, 78; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Roth HE, Powers R, Cancers 2022, 14, 3992; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Windley HR, Starrs D, Stalenberg E, Rothman JM, Ganzhorn JU, Foley WJ, American Journal of Primatology 2022, 84, e23397. [DOI] [PubMed] [Google Scholar]
- [55].Moseley HNB, Computational and Structural Biotechnology Journal 2013, 4, e201301006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sumner LW, Amberg A, Barrett D, Beale MH, Beger R, Daykin CA, Fan TWM, Fiehn O, Goodacre R, Griffin JL, Hankemeier T, Hardy N, Harnly J, Higashi R, Kopka J, Lane AN, Lindon JC, Marriott P, Nicholls AW, Reily MD, Thaden JJ, Viant MR, Metabolomics 2007, 3, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Naz S, García A, Barbas C, Analytical Chemistry 2013, 85, 10941–10948. [DOI] [PubMed] [Google Scholar]
- [58].Markley JL, Bruschweiler R, Edison AS, Eghbalnia HR, Powers R, Raftery D, Wishart DS, Curr. Opin. Biotechnol 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].a Ellinger JJ, Chylla RA, Ulrich EL, Markley JL, Curr Metabolomics 2013, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Vinaixa M, Schymanski EL, Neumann S, Navarro M, Salek RM, Yanes O, TrAC Trends in Analytical Chemistry 2016, 78, 23–35. [Google Scholar]
- [60].a Leggett A, Wang C, Li D-W, Somogyi A, Bruschweiler-Li L, Brüschweiler R, in Methods in Enzymology, Vol. 615 (Ed.: Wand AJ), Academic Press, 2019, pp. 407–422; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang C, He L, Li D-W, Bruschweiler-Li L, Marshall AG, Brüschweiler R, Journal of Proteome Research 2017, 16, 3774–3786; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Wang C, Zhang B, Timári I, Somogyi Á, Li D-W, Adcox HE, Gunn JS, Bruschweiler-Li L, Brüschweiler R, Analytical Chemistry 2019, 91, 15686–15693; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Bingol K, Bruschweiler-Li L, Yu C, Somogyi A, Zhang F, Brüschweiler R, Analytical Chemistry 2015, 87, 3864–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Pence HE, Williams A, J. Chem. Educ 2010, 87, 1123–1124. [Google Scholar]
- [62].Robinette SL, Zhang F, Brüschweiler-Li L, Brüschweiler R, Analytical Chemistry 2008, 80, 3606–3611. [DOI] [PubMed] [Google Scholar]
- [63].Bingol K, Li D-W, Zhang B, Brüschweiler R, Analytical Chemistry 2016, 88, 12411–12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].a Dudzik D, Barbas-Bernardos C, García A, Barbas C, Journal of Pharmaceutical and Biomedical Analysis 2018, 147, 149–173; [DOI] [PubMed] [Google Scholar]; b Gil RB, Lehmann R, Schmitt-Kopplin P, Heinzmann SS, Analytical and Bioanalytical Chemistry 2016, 408, 4683–4691. [DOI] [PubMed] [Google Scholar]
- [65].a Bögl T, Mlynek F, Himmelsbach M, Buchberger W, Talanta 2022, 236, 122849; [DOI] [PubMed] [Google Scholar]; b Liu Z, Wang P, Liu Z, Wei C, Li Y, Liu L, Journal of Separation Science 2021, 44, 3450–3461. [DOI] [PubMed] [Google Scholar]
- [66].Koek MM, Jellema RH, van der Greef J, Tas AC, Hankemeier T, Metabolomics 2011, 7, 307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Kirwan JA, Gika H, Beger RD, Bearden D, Dunn WB, Goodacre R, Theodoridis G, Witting M, Yu L-R, Wilson ID, the metabolomics Quality A, Quality Control C, Metabolomics 2022, 18, 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].a Misra BB, Mohapatra S, ELECTROPHORESIS 2019, 40, 227–246; [DOI] [PubMed] [Google Scholar]; b Misra BB, van der Hooft JJJ, ELECTROPHORESIS 2016, 37, 86–110. [DOI] [PubMed] [Google Scholar]
- [69].Spicer R, Salek RM, Moreno P, Cañueto D, Steinbeck C, Metabolomics 2017, 13, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].a Pang Z, Zhou G, Ewald J, Chang L, Hacariz O, Basu N, Xia J, Nature Protocols 2022, 17, 1735–1761; [DOI] [PubMed] [Google Scholar]; b Huan T, Forsberg EM, Rinehart D, Johnson CH, Ivanisevic J, Benton HP, Fang M, Aisporna A, Hilmers B, Poole FL, Thorgersen MP, Adams MWW, Krantz G, Fields MW, Robbins PD, Niedernhofer LJ, Ideker T, Majumder EL, Wall JD, Rattray NJW, Goodacre R, Lairson LL, Siuzdak G, Nature Methods 2017, 14, 461–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cardoso S, Afonso T, Maraschin M, Rocha M, Metabolites 2019, 9, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tsugawa H, Cajka T, Kind T, Ma Y, Higgins B, Ikeda K, Kanazawa M, VanderGheynst J, Fiehn O, Arita M, Nature Methods 2015, 12, 523–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Worley B, Powers R, ACS Chemical Biology 2014, 9, 1138–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].a Crook AA, Zamora-Olivares D, Bhinderwala F, Woods J, Winkler M, Rivera S, Shannon CE, Wagner HR, Zhuang DL, Lynch JE, Berryhill NR, Runnebaum RC, Anslyn EV, Powers R, Food Chemistry 2021, 354, 129531; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sakallioglu IT, Tripp B, Kubik J, Casey CA, Thomes P, Powers R, in Biology, Vol. 12, 2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Smilde AK, Westerhuis JA, Hoefsloot HCJ, Bijlsma S, Rubingh CM, Vis DJ, Jellema RH, Pijl H, Roelfsema F, van der Greef J, Metabolomics 2010, 6, 3–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].O’Shea K, Misra BB, Metabolomics 2020, 16, 36. [DOI] [PubMed] [Google Scholar]
- [77].O’Connell TM, Logsdon DL, Payne RM, Molecular Genetics and Metabolism 2022, 136, 306–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Vu T, Xu Y, Qiu Y, Powers R, Biostatistics 2022, 24, 140–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Tayyari F, Gowda GAN, Gu H, Raftery D, Analytical Chemistry 2013, 85, 8715–8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Guo J, Yu H, Xing S, Huan T, Chemical Communications 2022, 58, 9979–9990. [DOI] [PubMed] [Google Scholar]
- [81].Tzanakis K, Nattkemper TW, Niehaus K, Albaum SP, BMC Bioinformatics 2022, 23, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Han W, Li L, Mass Spectrometry Reviews 2022, 41, 421–442. [DOI] [PubMed] [Google Scholar]
- [83].Yue Y, Bao X, Jiang J, Li J, Journal of Chromatography B 2022, 1212, 123513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].a Niu J, Xu W, Wei D, Qian K, Wang Q, Analytical Chemistry 2022, 94, 8937–8946; [DOI] [PubMed] [Google Scholar]; b Niu J, Yang J, Guo Y, Qian K, Wang Q, BMC Bioinformatics 2022, 23, 270; [DOI] [PMC free article] [PubMed] [Google Scholar]; c Shaver AO, Garcia BM, Gouveia GJ, Morse AM, Liu Z, Asef CK, Borges RM, Leach FE, Andersen EC, Amster IJ, Fernández FM, Edison AS, McIntyre LM, Frontiers in Molecular Biosciences 2022, 9; [DOI] [PMC free article] [PubMed] [Google Scholar]; d Deng K, Zhao F, Rong Z, Cao L, Zhang L, Li K, Hou Y, Zhu Z-J, Metabolomics 2021, 17, 87; [DOI] [PubMed] [Google Scholar]; e Randall DW, Kieswich J, Swann J, McCafferty K, Thiemermann C, Curtis M, Hoyles L, Yaqoob MM, Microbiome 2019, 7, 127; [DOI] [PMC free article] [PubMed] [Google Scholar]; f Fages A, Pontoizeau C, Jobard E, Lévy P, Bartosch B, Elena-Herrmann B, Analytical and Bioanalytical Chemistry 2013, 405, 8819–8827; [DOI] [PubMed] [Google Scholar]; g Bongaerts M, Bonte R, Demirdas S, Jacobs EH, Oussoren E, van der Ploeg AT, Wagenmakers MAEM, Hofstra RMW, Blom HJ, Reinders MJT, Ruijter GJG, Metabolites 2021, 11, 8; [DOI] [PMC free article] [PubMed] [Google Scholar]; h Dunn WB, Broadhurst D, Begley P, Zelena E, Francis-McIntyre S, Anderson N, Brown M, Knowles JD, Halsall A, Haselden JN, Nicholls AW, Wilson ID, Kell DB, Goodacre R, C. The Human Serum Metabolome, Nature Protocols 2011, 6, 1060–1083; [DOI] [PubMed] [Google Scholar]; i Kuligowski J, Sánchez-Illana Á, Sanjuán-Herráez D, Vento M, Quintás G, Analyst 2015, 140, 7810–7817. [DOI] [PubMed] [Google Scholar]
- [85].Deng K, Zhang F, Tan Q, Huang Y, Song W, Rong Z, Zhu Z-J, Li K, Li Z, Analytica Chimica Acta 2019, 1061, 60–69. [DOI] [PubMed] [Google Scholar]
- [86].Karaman I, Ferreira DLS, Boulangé CL, Kaluarachchi MR, Herrington D, Dona AC, Castagné R, Moayyeri A, Lehne B, Loh M, de Vries PS, Dehghan A, Franco OH, Hofman A, Evangelou E, Tzoulaki I, Elliott P, Lindon JC, Ebbels TMD, Journal of Proteome Research 2016, 15, 4188–4194. [DOI] [PubMed] [Google Scholar]
- [87].Veselkov KA, Lindon JC, Ebbels TMD, Crockford D, Volynkin VV, Holmes E, Davies DB, Nicholson JK, Analytical Chemistry 2009, 81, 56–66. [DOI] [PubMed] [Google Scholar]
- [88].Hervé MR, Nicolè F, Lê Cao K-A, Journal of Chemical Ecology 2018, 44, 215–234. [DOI] [PubMed] [Google Scholar]
- [89].a Mehl F, Marti G, Merle P, Delort E, Baroux L, Sommer H, Wolfender J-L, Rudaz S, Boccard J, Flavour and Fragrance Journal 2015, 30, 131–138; [Google Scholar]; b Xu Y, Goodacre R, Metabolomics 2012, 8, 37–51; [Google Scholar]; c Brandolini-Bunlon M, Pétéra M, Gaudreau P, Comte B, Bougeard S, Pujos-Guillot E, Metabolomics 2019, 15, 134. [DOI] [PubMed] [Google Scholar]
- [90].Sakallioglu IT, Maroli AS, Leite ADL, Powers R, Journal of Chromatography A 2022, 1662, 462739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Malinowska JM, Palosaari T, Sund J, Carpi D, Lloyd GR, Weber RJM, Whelan M, Viant MR, Metabolites 2022, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]