

Abstract
Aminobenzotriazole (ABT) is commonly used as a non‐selective inhibitor of cytochrome P450 (CYP) enzymes to assign contributions of CYP versus non‐CYP pathways to the metabolism of new chemical entities. Despite widespread use, a systematic review of the drug–drug interaction (DDI) potential for ABT has not been published nor have the implications for using it in plated hepatocyte models for low clearance reaction phenotyping. The goal being to investigate the utility of ABT as a pan‐CYP inhibitor for reaction phenotyping of low clearance compounds by evaluating stability over the incubation period, inhibition potential against UGT and sulfotransferase enzymes, and interaction with nuclear receptors involved in the regulation of drug metabolizing enzymes and transporters. Induction potential for additional inhibitors used to ascribe fraction metabolism (f m), pathway including erythromycin, ketoconazole, azamulin, atipamezole, ZY12201, and quinidine was also investigated. ABT significantly inhibited the clearance of a non‐selective UGT substrate 4‐methylumbelliferone, with several UGTs shown to be inhibited using selective probe substrates in human hepatocytes and rUGTs. The inhibitors screened in the induction assay were shown to induce enzymes regulated through Aryl Hydrocarbon Receptor, Constitutive Androstane Receptor, and Pregnane X Receptor. Lastly, a case study identifying the mechanisms of a clinical DDI between Palbociclib and ARV‐471 is provided as an example of the potential consequences of using ABT to derive f m. This work demonstrates that ABT is not an ideal pan‐CYP inhibitor for reaction phenotyping of low clearance compounds and establishes a workflow that can be used to enable robust characterization of other prospective inhibitors.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
ABT is often used to delineate the contribution of CYPs versus non‐CYP pathways to metabolic clearance. However, its drug–drug interaction (DDI) potential has not been systematically investigated.
WHAT QUESTION DID THIS STUDY ADDRESS?
ABT inhibits several UGT isoforms and induces enzymes regulated through AhR, CAR, and PXR thereby complicating the interpretation of fraction metabolism through these pathways as highlighted using palbociclib as a case example.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
Palbociclib is reported to be a substrate of CYP3A and SULT2A1, however, the results here demonstrate the importance of glucuronidation in contributing to the observed clinical DDI with ARV‐471. This highlights the consequence of using ABT to define fraction metabolism and presents an opportunity for optimizing in vitro models for this purpose.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Given that ABT inhibits/induces multiple CYPs and UGTs fraction metabolism (f m) data generated using ABT should be interpreted with caution. If glucuronidation is determined to be a relevant metabolic pathway, ABT should not be used to assign f m for CYPs.
INTRODUCTION
Drug–drug interactions (DDIs) are a concern with polypharmacy as they can lead to toxicological observations or clinical failure. Therefore, assessing the potential for DDIs of new chemical entities (NCEs) is conducted both from the perspective of how the NCE exposure may be impacted (victim) as well as how the NCE may impact the exposure of co‐medicants (perpetrator). Over the past few decades, a significant understanding of CYP enzymes has made it possible to design NCEs that are less prone to CYP metabolism and demonstrate reduced clearance. 1 , 2 In molecules with low CYP metabolism, understanding the role of other drug metabolizing enzymes, such as UGTs, sulfotransferase (SULTs), aldehyde oxidase (AO), and carboxylesterase (CES), among others, is crucial. In vitro reaction phenotyping studies are usually conducted to identify the drug metabolizing enzymes involved in the metabolism of an NCE, with regulatory agencies recommending clinical DDI studies if a single drug metabolizing enzyme contributes to greater than 25% of total clearance. 3 , 4 In cases when more than one drug metabolizing enzyme is involved, it is valuable to determine the fraction metabolized (f m) by each enzyme in relation to total hepatic clearance to understand the potential for clinical variability and DDI. For moderate to high intrinsic clearance (Clint) drugs, f m can be calculated using relatively simple systems, like liver S9 with and without cofactors, such as NADPH for CYPs, UDPGA for UGTs, or PAPS for SULTs. 5 , 6 However, this method is not appropriate for low clearance drugs. 7
Long‐term coculture systems, such as Hμrel and HepatoPac have emerged as invaluable tools for the prediction of human pharmacokinetics for low Clint compounds, 8 , 9 , 10 , 11 with recent examples where their utility for reaction phenotyping shows promise. 7 , 12 , 13 In these models, NCEs may be incubated with various inhibitors over longer durations, enabling DDI assessment and reaction phenotyping at a more relevant timescale. However, complete DDI assessment for several commonly used inhibitors, including ABT, azamulin, ketoconazole, erythromycin, quinidine, ZY12201, and atipamezole, is still lacking. Whereas specific inhibitors for CYPs are well‐characterized and widely used, enzymes including UGTs lack specific inhibitors. 14 In such cases, pan‐CYP inhibitors like ABT have been used to inhibit CYP metabolism and calculate f mUGT from f total. This method can theoretically be used for f m characterization of low Clint compounds, as long‐term coculture systems can be treated with pan‐CYP inhibitors followed by f m characterization. 15
A concentration of 1 mM ABT is often used to inhibit CYPs for in vitro DDI studies. 16 , 17 , 18 , 19 , 20 Although it has been reported that ABT can inhibit and induce activity of non‐CYP enzymes, only limited data have been published, and the impact of ABT on UGTs remains unreported. 21 , 22 , 23 , 24 In this series of experiments, the inhibition potential for ABT toward seven major UGT and eight major SULT isoforms was determined using recombinant enzymes and suspension hepatocytes. In addition, the induction potential for ABT and potential alternative inhibitors toward nuclear receptor pathways involved in the regulation of drug metabolizing enzymes was evaluated using long‐term Hμrel coculture or relative induction score (RIS)‐qualified sandwich cultured hepatocytes (Table S1).
Finally, the impact of using ABT as a nonselective CYP inhibitor on the interpretation of f m is demonstrated by using palbociclib as a case study. Palbociclib is a small molecule inhibitor of cyclin dependent kinases 4 and 6 used to treat cancers as a monotherapy or in combination with other drugs. 25 Co‐administration of palbociclib with ARV‐471, a selective PROteolysis TArgeting Chimera (PROTAC) protein degrader targeting estrogen receptor to treat breast cancer, 26 increased palbociclib exposure by ~50%, leading to higher incidence of neutropenia (https://endpts.com/arvinas‐reports‐delay‐for‐pfizer‐partnered‐protein‐degradation‐program/). ABT coupled with the hepatocyte relay assay was used for reaction phenotyping of palbociclib and assignment of CYP versus non‐CYP pathways (new drug application [NDA] 207103). Mechanistic investigations into the DDI between palbociclib and ARV‐471 were conducted to determine the mechanisms behind unexpected increases in palbociclib exposure. Our data demonstrates that ABT inhibits multiple UGTs and therefore its use to determine f m by CYPs can lead to underestimation of non‐CYP metabolism and in turn miss potential DDIs.
MATERIALS AND METHODS
Chemicals and reagents
ABT (Sigma‐Aldrich), palbociclib sulfate (TRC), uridine 5′‐diphosphoglucuronic acid, UDPGA (Sigma‐Aldrich), MgCl2 (Sigma‐Aldrich), Alamethicin (Sigma‐Aldrich), dimethyl sulfoxide, DMSO (Sigma‐Aldrich), ACN (liquid chromatography‐mass spectrometry [LC–MS] grade; Fisher Chemical), ACN (Optima LC/MS grade, Fisher Chemical) formic acid (Fisher Chemical), potassium phosphate (Sigma‐Aldrich) 5,5‐diethyl‐1,3‐diphenyl‐2‐iminobarbituric acid (“39:an”; Sigma‐Aldrich), D‐Saccharolactone (Sigma‐Aldrich), ultra‐pure water (Millipore), recombinant UGT supersomes (UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15; Corning), β‐estradiol (Sigma‐Aldrich), chenodeoxycholic acid (CDCA; Sigma‐Aldrich), trifluoperazine (TFP; Sigma‐Aldrich), serotonin (Sigma‐Aldrich), propofol (Sigma‐Aldrich), zidovudine (Sigma‐Aldrich), oxazepam (Sigma‐Aldrich), raloxifene (Sigma‐Aldrich), dehydroepiandrosterone (DHEA; Sigma‐Aldrich), DHEA‐sulfate (Sigma‐Aldrich), 4‐methylumbelliferone (4‐MU; Sigma‐Aldrich), palbociclib sulfate (Toronto Research Chemicals); recombinant sulfotransferases (SULT1A1, SULT1A3, SULT1B1, SULT1C2, SULT1C4, SULT1E1, SULT2A1, and SULT2B1; R&D Systems), palbociclib, and ARV‐471 were synthesized by AZ chemistry.
Stability of ABT and inhibition of 4‐MU metabolism in Hμrel coculture
Mixed gender, five donor pooled hepatocytes in Hμrel co‐culture 96‐well plates were received from the vendor as “ready to use” (Table S2). The media was changed, and cells were acclimatized in an incubator set at 37°C, 5% CO2, for 24 h before initiation of any experiments. Following the acclimation period, the cells were treated with ABT, and samples were removed at designated timepoints to monitor for depletion of ABT.
The nonselective UGT substrate, 4‐MU, was incubated with Hμrel and stromal only cells in the presence or absence of varying concentrations of ABT (0.125, 0.25, 0.5, 0.75, and 1 mM), and half‐maximal inhibitory concentration (IC50) was determined.
Evaluation of the impact of ABT on the activity of UGTs
Isoform‐specific substrates β‐estradiol, CDCA, TFP, serotonin, propofol, zidovudine, and oxazepam were incubated at concentrations five times lower than the reported K m values so that IC50 values would approximate K i. Recombinant UGT 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 were purchased from Corning. Incubations were performed at 0.1 or 0.2 mg/mL for 30 min as described. 27 Inhibition of UGTs using specific probe substrates was also performed in mixed gender pooled cryopreserved human hepatocytes from BioIVT lot: MTJ.
Formation of glucuronide metabolites was monitored using liquid chromatography tandem mass spectrometry. IC50 values were corrected to free values using in silico approaches previously described. 28
Evaluation of the impact of ABT on activity of SULTS
Raloxifene was used as a substrate for the evaluation of SULT inhibition by ABT. Recombinant SULTs SULT1A1, SULT1A3, SULT1B1, SULT1C2, SULT1C4, SULT1E1, SULT2A1, and SULT2B1 were incubated at 20 μg/mL with or without 1 mM ABT in presence of PAPS as cofactor. Loss of raloxifene parent was monitored using high resolution mass spectrometry to determine the inhibition potential of ABT on each SULT isoform.
Investigations into the induction potential of ABT and other commonly used inhibitors towards drug metabolizing enzymes
Mixed gender, five donor pooled hepatocytes in Hμrel co‐culture 96‐well plates were received from the vendor as “ready to use.” The media was changed, and cells were acclimatized in an incubator set at 37°C, 5% CO2, for 24 h before initiation of any experiments. The impact of ABT, erythromycin, azamulin, and positive controls for induction (omeprazole, phenobarbital, and rifampin) was determined using Hμrel. For alternative potential inhibitors, the induction assessment was conducted using a single female donor HH1103 (In Vitro ADMET Laboratories, LLC), which has been qualified for RIS quantitative predictions. HH1103 was recovered and plated according to the vendor recommendations (Table S2). Test articles were prepared in DMSO and added to the culture at selected concentrations for 48 h with a media change at 24 h. Fold changes in mRNA were determined using TaqMan RT‐PCR (reverse transcriptase polymerase chain reaction), and induction parameters were derived where possible. Positive controls for induction were included. CYP1A2, CYP2B6, and CYP3A4 mRNA levels changes were used as sensitive markers of Aryl Hydrocarbon Receptor (AhR), Constitutive Androstane Receptor (CAR), and Pregnane X Receptor (PXR) activation. In addition, for Hμrel samples, induction of UGT1A1, UGT1A6, CYP2C8, and CYP2C9 was also evaluated.
Evaluation of the impact of ARV‐471 on intrinsic clearance of palbociclib using Hμrel
Mixed gender, five donor pooled hepatocytes in Hμrel co‐culture 96‐well plates were received from the vendor as “ready to use.” The media was changed, and cells were acclimatized in an incubator set at 37°C, 5% CO2, for 24 h before initiation of any experiments. Incubation media was prepared by adding equivalent volumes of maintenance media and probing media and contained 2.5% fetal bovine serum (FBS). Palbociclib was incubated at a total concentration of 0.045 μM, which was set to result in an equal unbound concentration as the observed unbound peak plasma concentration at steady‐state following a 125 mg dose (0.038 μM). ARV‐471 was solubilized in 90% DMSO containing 10% 1N HCL (hydrogen chloride). Varying concentrations of ARV‐471 were added to incubation media (0, 0.3, 1, 3, 10, and 30 μM), and the final solvent concentration was 0.4%. Samples were removed at designated timepoints by adding an equivalent volume of quench containing 10 nM IS (39:an) directly to the well. The wells were scraped, and the intrinsic clearance was determined by generating PAR of analyte to IS using high resolution mass spectrometry (HRMS).
Evaluation of ARV‐471 as a direct or time‐dependent inhibitor of CYP3A or SULT2A1
Varying concentrations of ARV‐471 were incubated with recombinant SULT2A1, and the formation of dehydroepiandrosterone sulfate (DHEAS) was monitored. The IC50 value was determined. The formation of palbociclib sulfate was monitored over the incubation time‐course used in the Clint study to evaluate the impact of ARV‐471 on sulfate formation. The activity of 1'hydroxylation of midazolam and sulfation of DHEA were determined over the incubation time course as CYP3A and SULT2A1 are reported as the primary metabolic pathways for palbociclib. Maximal rate of metabolism concentrations of both substrates were used, and midazolam was incubated for 5 min, whereas DHEA was incubated for 15 min. In addition, the morphology, adenosine triphosphate content, lactate dehydrogenase, and MTT activity were determined at 10 and 30 μM as markers of cell health.
Biotransformation pathway of palbociclib in two long‐term co‐cultured human hepatocyte models
Significant metabolism of some substrates has been observed to occur in stromal only cells used in the Hμrel model (Figure S1 and data on file). Therefore, an additional co‐culture model containing all human cells, TruVivo, was used to clarify biotransformation pathways for palbociclib (donor demographics are located in Table S2). Metabolite identification was conducted for palbociclib by incubating 5 μM palbociclib with Hμrel or TruVivo co‐culture cells and feeder or stromal cells with and without 20 μM ARV‐471. Mixed gender, five donor pooled hepatocytes in Hμrel co‐culture 96‐well plates were received from the vendor as “ready to use.” The media was changed, and cells were acclimatized in an incubator set at 37°C, 5% CO2, for 24 h before initiation of any experiments. Incubation media was prepared by adding equivalent volumes of maintenance media and probing media and contained 2.5% FBS. The TruVivo model was prepared according to the vendor recommendations using a single female donor (2017942‐01). The TruVivo model was acclimated in an incubator set at 37°C, 5% CO2 for 9 days post recovery and seeding. Media was changed daily. Samples were removed at designated timepoints by quenching with an equal volume of ACN and metabolite profiling was conducted using HRMS followed by processing with the Oniro platform (Molecular Discovery, UK). Because the incubation concentration is much higher than clinically relevant, the Clint was also determined for comparison with the data derived at the anticipated clinical concentration.
RESULTS
Stability of ABT in Hμrel coculture and incubation with 4‐MU
ABT was incubated with Hμrel up to 72 h, and concentrations of ABT relative to 0 h were measured in media samples (Figure S1A). No significant loss of ABT was observed indicating ABT remained stable for 72 h in the presence of Hμrel co‐culture cells.
To determine if ABT has any inhibitory effect on UGT, a nonselective UGT substrate 29 4‐MU was incubated with various concentrations of ABT ranging from (0–1 mM). IC50 of 4‐MU inhibition was determined to be 0.661 mM (confidence interval 0.542–0.820) confirming the ability of ABT to inhibit UGTs (Figure S1C). Additionally, Significant formation of 4‐MU glucuronide was also observed in stromal only cells (Figure S1B).
Identifying UGT and SULT isoforms inhibited by ABT
Recombinant UGTs were incubated with isoform‐specific substrates in the presence of varying concentrations of ABT, and IC50 values were determined (Figure S2). UGT isoforms 1A1, 1A3, 1A4, 1A6, 1A9, 2B7, and 2B15 were inhibited by ABT (Table 1). In addition, suspended human hepatocytes were incubated with various isoform‐specific UGT substrates in the presence of ABT. Isoforms 1A3, 1A4, 1A9, 2B7, and 2B15 were inhibited by ABT (Table 1). UGT1A1 and 1A6 showed inhibition in recombinant UGT but was not inhibited in human hepatocytes.
TABLE 1.
Inhibition constants for ABT in suspension human hepatocytes (HHEPs) and recombinant UGTs (rUGTs).
| UGT isoform | Substrate | Substrate concentration (μM) | Ki,u (mM) | |
|---|---|---|---|---|
| HHEPs | rUGTs | |||
| 1A1 | β‐Estradiol | 3 | No inhibition | >20 a |
| 1A3 | CDCA | 2 | 6.03 | 1.90 |
| 1A4 | TFP | 1 | 2.52 | 1.54 |
| 1A6 | Serotonin | 1715 | No inhibition | 15.8 |
| 1A9 | Propofol | 10 | 4.58 | 1.79 |
| 2B7 | Zidovudine | 100 | 5.45 | 3.90 |
| 2B15 | Oxazepam | 10 | >20 b | 7.30 |
Up to 59% inhibition was observed in at least one of the triplicate incubations at 20 mM.
Up to 45% inhibition was observed in at least one of the triplicate incubations at 20 mM.
Inhibition of SULT isoforms 1A1, 1A3, 1B1, 1C2, 1C4, 1E1, 2A1, and 2B1 was performed using recombinant enzymes. No significant inhibition of any SULT isoform tested was observed when using raloxifene as a probe substrate with 1 mM ABT (data not shown).
Induction of CYPs and UGTs in plated sandwich cultured hepatocytes and Hμrel coculture by ABT
Plated sandwich cultured human hepatocytes and Hμrel co‐cultured hepatocytes were treated with several standard inhibitors for 48 h, and the mRNA fold changes relative to DMSO control were determined for CYP1A2, CYP2B6, and CYP3A4 as sensitive markers of AhR, CAR, and PXR, respectively. In Hμrel incubations, additional enzymes were also evaluated including CYP2C8, CYP2C9, UGT1A1, and UGT1A6 because they are reported to be regulated through the same nuclear receptor pathways. The mRNA levels were determined using TaqMan RT‐PCR and selective primer/probe pairs for the gene of interest. Induction parameters were derived where possible. Induction was considered to be positive when the following criteria were met, changes in mRNA levels were concentration‐dependent (by statistical analysis) and greater than twofold when compared with the vehicle control. No increases in UGT1A6 mRNA levels were observed. Positive controls for induction were included and demonstrated expected levels of induction, confirming the performance of the models (Figure 1d, Table S4). Half‐maximal effective concentration (EC50) and maximum effect (E max) are summarized in Table S3, and concentration response profiles are presented in Figure 1. ABT induced mRNA levels of CYP1A2, CYP2B6, CYP2C8, CYP3A4, and UGT1A1, suggesting that it acts as an agonist of AhR, CAR, and PXR (Figure 1a/e). Additional inhibitors were also evaluated and demonstrated varying impact on genes regulated through AhR, CAR, and PXR. Erythromycin induced CYP1A2, 2B6, and 3A (Figure 1b), azamulin, a potential alternative CYP3A inhibitor, induced CYP2B6, 2C8, 2C9, 3A4, and UGT1A1 (Figure 1c), ketoconazole and quinidine induced CYP1A2 and CYP3A4 (Figure 1f/h) but quinidine did not induce CYP2B6. CYP2B6 was not evaluated for ketoconazole given the low CYP3A4 response (E max = 2‐fold). Atipamezole used as a selective CYP3A inhibitor, induced CYP1A2, 2B6, and 3A4, sensitive enzymes regulated through AhR, CAR, and PXR, respectively (Figure 1g). ZY12201, recently reported as a pan‐CYP inhibitor, also induced enzymes regulated through AhR, CAR, and PXR (Figure 1i). 30
FIGURE 1.
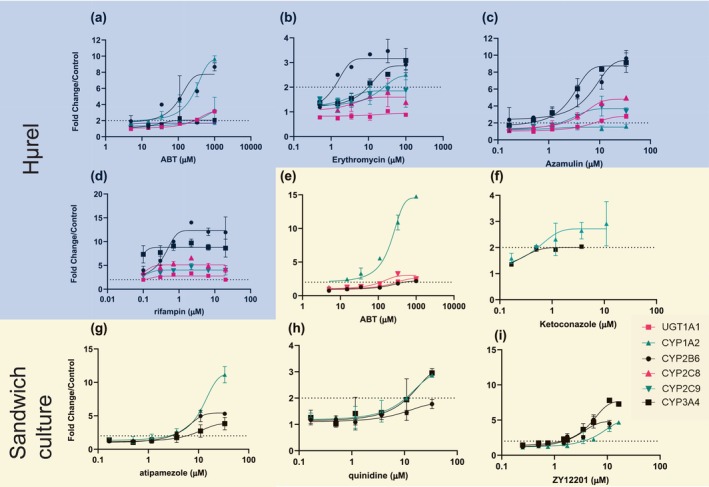
Evaluation of mRNA level changes for UGT1A1 (pink squares), CYP1A2 (teal triangles), CYP2B6 (black circles), CYP2C8 (pink triangles), CYP2C9 (teal upside down triangles), or CYP3A4 (black squares), UGT1A6 (data not shown) following administration of ABT, erythromycin, azamulin, or rifampin (a, b, c, d, respectively) to Hμrel co‐culture model or ABT, ketoconazole, atipamezole, quinidine, or ZY12201 to sandwich cultured hepatocytes (e, f, g, h, i, respectively). Note studies conducted with Hμrel (blue background) looked at all mRNA end points, whereas studies conducted in sandwich cultured hepatocytes (yellow background) focused on sensitive markers of AhR, CAR, and PXR activation (CYP1A2, CYP2B6, and CYP3A4, respectively). Each data point is representative of two with error, dashed line represents twofold change from vehicle control. Induction potential for ABT and other commonly used inhibitors in the Hμrel co‐culture model (a, b, c, d) or sandwich cultured human hepatocytes (e, f, g, i) Dashed line represents twofold increase in mRNA levels.
Evaluation of the impact of ARV‐471 on the Clint of palbociclib using Hμrel
Incubation of a clinically relevant concentration of palbociclib (0.038 μM unbound) with increasing concentrations of ARV‐471 showed concentration dependent inhibition of palbociclib Clint by ARV‐471 that could recapitulate the observed clinical effect (50% increase in palbociclib exposure at ~3 μM ARV‐471; Figure 2a,b).
FIGURE 2.
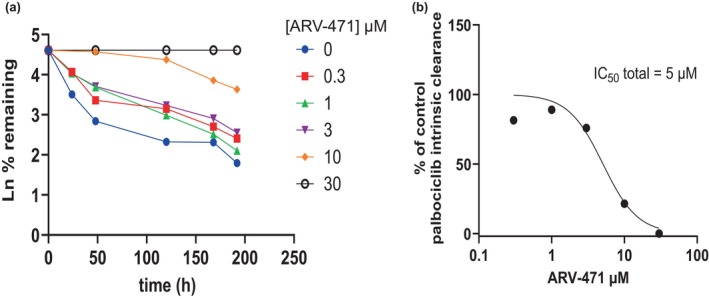
Impact of ARV‐471 on palbociclib intrinsic clearance (a) mean of two, derivation of ARV‐471 inhibition parameters (b) mean of two. Impact of ARV‐471 on palbociclib intrinsic clearance (a), derivation of ARV‐471 inhibition parameters (b).
Evaluation of ARV‐471 as a direct or time‐dependent inhibitor of CYP3A or SULT2A1
Administration of ARV‐471 resulted in concentration dependent inhibition of SULT2A1 as measured by DHEAS formation, in both recombinant enzyme (Figure 3a) and the Hμrel model (Figure 3b), albeit at much higher concentrations than would be clinically relevant (IC50 > 35 μM). Additionally, there was an apparent activation of palbociclib sulfation by ARV‐471 observed over the time course (Figure 2c). In contrast, there was no direct inhibition of CYP3A activity perpetrated by ARV‐471 (Figure 4a). However, there was a concentration and time‐dependent increase in midazolam activity with induction parameters of 3.8‐fold and 0.25 μM determined for E max and EC50 of ARV‐471, respectively (Figure 4b).
FIGURE 3.
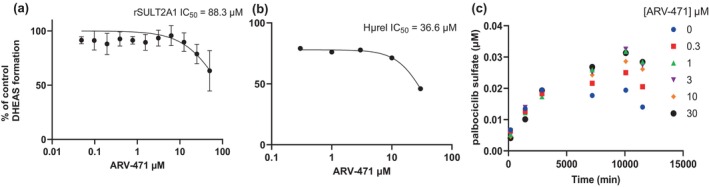
Impact of ARV‐471 on DHEAS formation in recombinant SULT2A1 mean of three (a), Hμrel (b) mean of three, and the formation of palbociclib sulfate in Hμrel (c) mean of two. Impact of ARV‐471 on DHEAS formation in recombinant SULT2A1 (a), Hμrel (b), and the formation of palbociclib sulfate in Hμrel (c).
FIGURE 4.
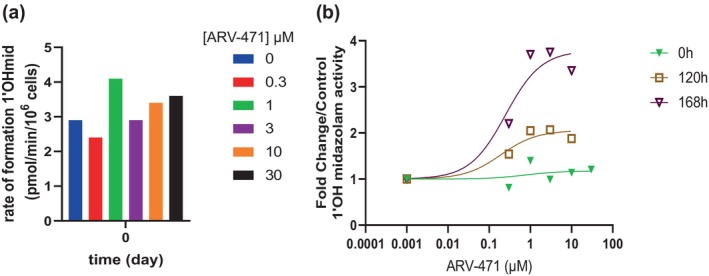
Impact of ARV‐471 on CYP3A activity as measured by 1'Ohmidazolam, direct inhibition (a) mean of three, time‐dependent inhibition (b) mean of three. The 0 h, 120 h, and 168 h refer to the time of exposure to the test article prior to determining the enzyme activity Impact of ARV‐471 on CYP3A activity as measured by 1'Ohmidazolam, direct inhibition (a), time‐dependent inhibition (b).
Biotransformation pathway of palbociclib in human hepatocytes
Palbociclib was incubated with two long‐term human hepatocyte models, Hμrel and TruVivo, at 5 μM in the presence and absence of 20 μM ARV‐471. In addition, the supporting cells were also incubated under the same conditions. The Clint was reduced at 5 μM when compared with 0.045 μM, suggesting saturation of metabolic clearance (Figure S3). Metabolite profiling demonstrated that both systems produced similar metabolites and that palbociclib metabolism was mediated through glucuronidation, sulfation, hydrolysis, ketone reduction, acetylation, and oxidation (Figure 5, Table S5). The supporting cell incubations showed the formation of select metabolites, including oxidation, ketone reduction, acetylation, and sulfation, albeit at much lower levels than was observed with the co‐culture models. In both models, the glucuronide metabolism was completely inhibited by ARV‐471 and accounted for up to 50% inhibition of metabolic clearance based on MS ionization and summing recovered metabolite area ratios in relation to remaining palbociclib (Table S6).
FIGURE 5.
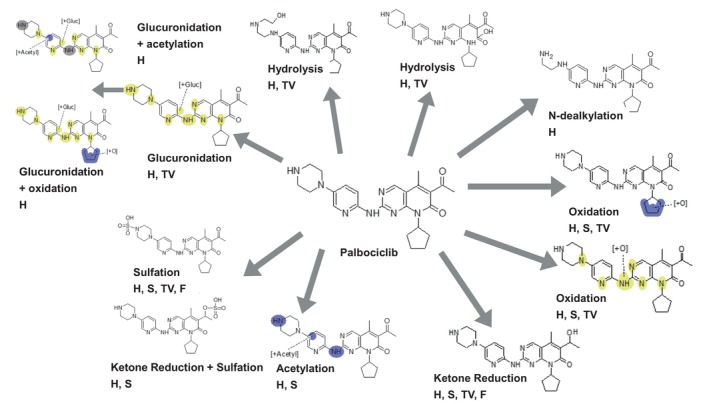
Metabolic scheme for palbociclib in co‐culture human hepatocyte models (H = Hμrel, S = stromal cells in H, TV = TruVivo, F = feeder cells in TV) pooled samples from three replicates, over a timecourse on two separate occasions.
DISCUSSION
ABT is routinely used to delineate CYP‐mediated metabolism from non‐CYP metabolism as it is reported to be a pan‐CYP inhibitor. 16 , 31 The ability of ABT to inhibit or induce other enzymes has not been comprehensively investigated, thus this series of studies aimed to evaluate the overall suitability of using ABT to inform fraction metabolism. Data from these studies indicate that ABT demonstrates complex DDIs, including inhibition and induction of several non‐CYP enzymes involved in hepatic clearance. This observation leads to some uncertainty when utilizing ABT in reaction phenotyping and the assessment of potential risks for clinical DDI. In addition, several other potential inhibitors were evaluated as inducers of drug metabolizing enzymes and like ABT showed varying magnitude of induction, thus complicating their utility to ascribe f m for these pathways in long‐term plated cell models and highlighting a need for further optimization of in vitro tools for this effort. A workflow, based on the work conducted here, can be used to help enable robust characterization of potential future inhibitors for low clearance reaction phenotyping.
Palbociclib was the first orally available CDK4/6 inhibitor to receive approval as a therapy to treat advanced or metastatic hormone receptor positive (HR+) and HER2 negative (HER2‐) breast cancers and is often administered in combination with other drugs. The metabolic clearance for palbociclib is reported to be primarily mediated by CYP3A and SULT2A1. 32 Earlier this year, Arvinas reported a delay in the clinical development of ARV‐471 due to an unexpected increase in palbociclib exposure (50% area under the curve) leading to grade 3/4 neutropenia during the phase Ib study (https://endpts.com/arvinas‐reports‐delay‐for‐pfizer‐partnered‐protein‐degradation‐program/). Based on the reported metabolic clearance pathways of palbociclib, it could be theorized that ARV‐471 is an inhibitor of either CYP3A, SULT2A1, or both.
Given that palbociclib is a low clearance drug, a long‐term co‐culture hepatocyte model, Hμrel, was used to explore the mechanisms behind this clinical DDI. The Hμrel system is a co‐culture model that is routinely applied within AstraZeneca to derive clearance parameters for low clearance compounds and retains functional metabolic enzymes and transporters over several weeks. Co‐treatment of palbociclib with ARV‐471 resulted in concentration‐dependent decreases in palbociclib Clint, with an IC50 of 5 μM. In the same study, there was no direct or time‐dependent inhibition of CYP3A (measured by 1'hydroxylation of midazolam). Inhibition of SULT2A1 in Hμrel or using recombinant SULT2A1 (measured by sulfation of DHEA) was observed, but at concentrations far exceeding clinical relevance (IC50 > 35 μM). Additionally, ARV‐471 resulted in time‐dependent increases in 1'hydroxylation of midazolam, suggesting that it is a CYP3A inducer, and increased formation of palbociclib‐sulfate was also observed with ARV‐471.
Metabolite profiling of samples generated from two separate low clearance hepatocyte models demonstrated that glucuronidation is an important pathway for metabolic clearance of palbociclib and that ARV‐471 extensively inhibits both the oxidative N‐dealkylation and glucuronidation of palbociclib. Other pathways were shown to increase due to a combination of reduced secondary conjugation and metabolic switching. Considering the change in Clint the magnitude of the observed clinical interaction could be recapitulated in these hepatocyte models, suggesting their value as tools for evaluating the DDI potential of co‐medicants. Neither the reaction phenotyping data nor the results from the human absorption, distribution, metabolism, and excretion (ADME) study of palbociclib have been published. Data from the NDA demonstrate that the contribution of CYP mediated metabolism versus sulfotransferase mediated metabolism was determined by using the hepatocyte relay method in the presence of 1 mM ABT. As the data here demonstrate that 1 mM ABT extensively inhibits multiple UGTs, it can be postulated that the potential importance of glucuronidation was missed due to the experimental conditions used (NDA 207103). Indeed, the most abundant circulating metabolite following a single oral dose of [14C]‐palbociclib to healthy subjects was reported to be a glucuronide metabolite (14.8% of circulating radioactivity), which was not observed in excreta (NDA 207103). This pathway was considered to be minor when taking into account the recovered radioactivity of unchanged palbociclib during the human ADME study, however, the metabolite profiling suggests that several secondary metabolites are glucuronide conjugated. It is possible that primary glucuronide metabolites could undergo additional metabolism and be recovered in feces without glucuronide due to gut microbiota‐mediated hydrolysis. Preliminary reaction phenotyping studies using a novel approach indicate that glucuronidation could account for ~25%–30% of palbociclib clearance in hepatocytes (manuscript under preparation). Additional work is warranted to confirm the palbociclib f m,gluc to better context the DDI risk of NCE. Further characterization of the N‐dealkylation pathway is also relevant as ARV‐471 was shown to inhibit the production of this metabolite as well. However, inhibition of glucuronidation was determined to be an important mechanism behind the observed interaction with ARV‐471. Given that increased exposure of palbociclib can lead to high incidence of neutropenia along with the widespread use of palbociclib in combination therapies, it is prudent to consider the potential for UGT inhibition when partnering with other oncology drugs. This example highlights the consequence of using ABT to conduct reaction phenotyping studies of low clearance molecules and identifies the opportunity for improved methods in this space.
AUTHOR CONTRIBUTIONS
K.C.A. and D.R. wrote the manuscript. D.R. designed the research. K.C.A., M.S., Y.T., A.W.‐J., and D.R. performed the research. K.C.A., M.S., Y.T., A.W.‐J., Z.Z., N.H., and D.R. analyzed the data.
FUNDING INFORMATION
No funding was received for this work.
CONFLICT OF INTEREST STATEMENT
The authors declared no competing interests for this work.
Supporting information
Appendix S1
ACKNOWLEDGMENTS
The authors thank Drs. Dermot McGinnity and Abhishek Srivastava for helpful discussions on the data, Dr. Jess Griffith for critical review of the manuscript, and Cameron Parsons for contributions to the UGT inhibition experiments.
Aluri KC, Slavsky M, Tan Y, et al. Aminobenzotriazole inhibits and induces several key drug metabolizing enzymes complicating its utility as a pan CYP inhibitor for reaction phenotyping. Clin Transl Sci. 2024;17:e13746. doi: 10.1111/cts.13746
Krishna C. Aluri, Marina Slavsky, and Diane Ramsden contributed equally to this study.
REFERENCES
- 1. Zhao M, Ma J, Li M, et al. Cytochrome P450 enzymes and drug metabolism in humans. Int J Mol Sci. 2021;22:12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Masimirembwa CM, Thompson R, Andersson TB. In vitro high throughput screening of compounds for favorable metabolic properties in drug discovery. Comb Chem High Throughput Screen. 2001;4:245‐263. [DOI] [PubMed] [Google Scholar]
- 3. Center for drug evaluation and research . In vitro drug interaction studies – cytochrome P450 enzyme‐ and transporter mediated drug interactions guidance for industry. 2020.
- 4. ICH M12 on Drug Interaction Studies . International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. 2022.
- 5. Al‐Jahdari WS, Yamamoto K, Hiraoka H, Nakamura K, Goto F, Horiuchi R. Prediction of total propofol clearance based on enzyme activities in microsomes from human kidney and liver. Eur J Clin Pharmacol. 2006;62:527‐533. [DOI] [PubMed] [Google Scholar]
- 6. Kilford PJ, Stringer R, Sohal B, Houston JB, Galetin A. Prediction of drug clearance by glucuronidation from in vitro data: use of combined cytochrome P450 and UDP‐glucuronosyltransferase cofactors in alamethicin‐activated human liver microsomes. Drug Metab Dispos. 2009;37:82‐89. [DOI] [PubMed] [Google Scholar]
- 7. Klammers F, Goetschi A, Ekiciler A, et al. Estimation of fraction metabolized by cytochrome P450 enzymes using long‐term cocultured human hepatocytes. Drug Metab Dispos. 2022;50:566‐575. [DOI] [PubMed] [Google Scholar]
- 8. Petersson C, Zhou X, Berghausen J, et al. Current approaches for predicting human PK for small molecule development candidates: findings from the IQ human PK prediction working group survey. AAPS J. 2022;24:1‐16. [DOI] [PubMed] [Google Scholar]
- 9. Waters NJ. Quantitative approaches to human clearance projection in drug research and development. Handbook of drug metabolism. CRC Press; 2019:253‐278. [Google Scholar]
- 10. Hultman I, Vedin C, Abrahamsson A, Winiwarter S, Darnell M. Use of HΜREL human coculture system for prediction of intrinsic clearance and metabolite formation for slowly metabolized compounds. Mol Pharm. 2016;13:2796‐2807. [DOI] [PubMed] [Google Scholar]
- 11. Chan TS, Yu H, Moore A, Khetani SR, Tweedie D. Meeting the challenge of predicting hepatic clearance of compounds slowly metabolized by cytochrome P450 using a novel hepatocyte model, hepatopac. Drug Metab Dispos. 2013;41:2024‐2032. [DOI] [PubMed] [Google Scholar]
- 12. Chan TS, Scaringella YS, Raymond K, Taub ME. Evaluation of erythromycin as a tool to assess CYP3A contribution of low clearance compounds in a long‐term hepatocyte culture. Drug Metab Dispos. 2020;48:690‐697. [DOI] [PubMed] [Google Scholar]
- 13. Smith S, Lyman M, Ma B, Tweedie D, Menzel K. Reaction phenotyping of low‐turnover compounds in long‐term hepatocyte cultures through persistent selective inhibition of cytochromes P450. Drug Metab Dispos. 2021;49:995‐1002. [DOI] [PubMed] [Google Scholar]
- 14. Harper TW, Brassil PJ. Reaction phenotyping: current industry efforts to identify enzymes responsible for metabolizing drug candidates. AAPS J. 2008;10:200‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang X, Atkinson K, Di L. Novel cytochrome P450 reaction phenotyping for low‐clearance compounds using the hepatocyte relay method. Drug Metab Dispos. 2016;44:460‐465. [DOI] [PubMed] [Google Scholar]
- 16. De Montellano PRO. 1‐AMINOBENZOTRIAZOLE: a mechanism‐based cytochrome P450 inhibitor and probe of cytochrome P450 biology. Med Chem. 2018;8: 038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Di L. Reaction phenotyping to assess victim drug‐drug interaction risks. Expert Opin Drug Discov. 2017;12:1105‐1115. [DOI] [PubMed] [Google Scholar]
- 18. Kozminski KD, Zientek MA. Differentiation of cytochrome P450‐mediated from non‐CYP‐mediated metabolism: aldehyde oxidase and xanthine oxidoreductase. Cytochrome P450. Springer; 2021:277‐289. [Google Scholar]
- 19. Kimoto E, Obach RS, Varma MV. Identification and quantitation of enzyme and transporter contributions to hepatic clearance for the assessment of potential drug–drug interactions. Drug Metab Pharmacokinet. 2020;35:18‐29. [DOI] [PubMed] [Google Scholar]
- 20. Argikar UA, Potter PM, Hutzler JM, Marathe PH. Challenges and opportunities with non‐CYP enzymes aldehyde oxidase, carboxylesterase, and UDP‐glucuronosyltransferase: focus on reaction phenotyping and prediction of human clearance. AAPS J. 2016;18:1391‐1405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang K, Hye Koh K, Jeong H. Induction of CYP2B6 and CYP3A4 expression by 1‐AMINOBENZOTRIAZOLE (ABT) in human hepatocytes. Drug Metab Lett. 2010;4:129‐133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mugford CA, Mortillo M, Mico BA, Tarloff JB. 1‐Aminobenzotriazole‐induced destruction of hepatic and renal cytochromes P450 in male SPRAGUE‐DAWLEY rats. Toxicol Sci. 1992;19:43‐49. [DOI] [PubMed] [Google Scholar]
- 23. Miners JO, Rowland A, Novak JJ, Lapham k, Goosen TC. Evidence‐based strategies for the characterisation of human drug and chemical glucuronidation in vitro and UDP‐glucuronosyltransferase reaction phenotyping. Pharmacol Therapeut. 2021;218:107689. [DOI] [PubMed] [Google Scholar]
- 24. Sun Q, Harper TW, Dierks EA, et al. 1‐Aminobenzotriazole, a known cytochrome P450 inhibitor, is a substrate and inhibitor of N‐acetyltransferase. Drug Metab Dispos. 2011;39:1674‐1679. [DOI] [PubMed] [Google Scholar]
- 25. Dhillon S. Palbociclib: first global approval. Drugs. 2015;75:543‐551. [DOI] [PubMed] [Google Scholar]
- 26. Hamilton EP, Schott AF, Nanda R, et al. Arv‐471, an estrogen receptor (ER) PROTAC degrader, combined with PALBOCICLIB in advanced ER+/human epidermal growth factor receptor 2–negative (HER2‐) breast cancer: phase 1B cohort (part C) of a phase 1/2 study. J Clin Oncol. 2022;40:16. [Google Scholar]
- 27. Busse D, Leandersson S, Amberntsson S, Darnell M, Hilgendorf C. Industrial approach to determine the relative contribution of seven major UGT isoforms to hepatic glucuronidation. J Pharm Sci. 2020;109:2309‐2320. [DOI] [PubMed] [Google Scholar]
- 28. Kilford PJ, Gertz M, Houston JB, Galetin A. Hepatocellular binding of drugs: correction for unbound fraction in hepatocyte incubations using microsomal binding or drug lipophilicity data. Drug Metab Dispos. 2008;36:1194‐1197. [DOI] [PubMed] [Google Scholar]
- 29. Uchaipichat V, Mackenzie PI, Guo X‐H, et al. Human UDP‐glucuronosyltransferases: isoform selectivity and kinetics of 4‐methylumbelliferone and 1‐naphthol glucuronidation, effects of organic solvents, and inhibition by diclofenac and probenecid. Drug Metab Dispos. 2004;32:413‐423. [DOI] [PubMed] [Google Scholar]
- 30. Giri P, Gupta L, Rathod A, et al. ZY12201, a potent TGR5 agonist: identification of a novel pan CYP450 inhibitor tool compound for in‐vitro assessment. Drug Metab Letters. 2022;15:116‐132. [DOI] [PubMed] [Google Scholar]
- 31. El‐Kattan AF, Poe J, Buchholz L, Thomas HV, Brodfuehrer J, Clark A. The use of 1‐aminobenzotriazole in differentiating the role of cyp mediated first pass metabolism and absorption in limiting drug oral bioavailability: a Case study. Drug Metab Lett. 2008;2:120‐124. [DOI] [PubMed] [Google Scholar]
- 32. Yu Y, Loi CM, Hoffman J, Wang D. Physiologically based pharmacokinetic modeling of palbociclib. J Clin Pharmacol. 2017;57:173‐184. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1