

Abstract
The E1 and E2 proteins are the only virus-encoded factors required for human papillomavirus (HPV) DNA replication. The E1 protein is a DNA helicase responsible for initiation of DNA replication at the viral origin. Its recruitment to the origin is facilitated by binding to E2, for which specific recognition elements are located at the origin. The remaining replication functions for the virus, provided by the host cell’s replication machinery, may be mediated by further interactions with E1 and E2. Histone H1 was identified as an HPV type 11 (HPV-11) E1-binding protein by far-Western blotting and by microsequence analyses of a 34-kDa protein purified by E1 affinity chromatography. E1 also bound in vitro to H1 isolated under native conditions in association with intact nucleosomes. In addition, E1 and H1 were coimmunoprecipitated by an E1 antiserum from a nuclear extract prepared from cells expressing recombinant E1. Bound H1 was displaced from HPV-11 DNA by the addition of E1, suggesting that E1 can promote replication initiation and elongation by alteration of viral chromatin structure and disruption of nucleosomes at the replication fork. Furthermore, a region of the HPV-11 genome containing the origin of replication was identified which had weaker affinity for H1 than that of the remaining genome. This result suggests that the presence of a DNA structure at or near the HPV origin facilitates initiation of DNA replication by exclusion of H1. These results are similar to those of studies of simian virus 40 DNA replication, in which a large T antigen-H1 interaction and an H1-resistant region at the origin of DNA replication have also been demonstrated.
Human papillomavirus type 11 (HPV-11) infects mucosal epithelia to induce benign anogenital and laryngeal warts. Vegetative DNA replication, late gene expression, and virus particle maturation are restricted to the upper layer of the epithelium, which is composed of differentiated cells (8, 20, 44, 61, 62). No culture system for growth of HPV-11 in tissue culture is available, and investigations of HPV-11 DNA replication have been limited to transient or cell-free methods. With either of these types of replication assays, the E1 and E2 proteins are the only virus-encoded factors which are required for replication of plasmids harboring the viral origin of replication (12, 17, 38). The E2 protein is a DNA-binding transcriptional transactivator for which specific recognition elements are located at the origin (27, 43). The E1 protein is a DNA helicase which initiates viral DNA synthesis from the origin (54, 71). Although E1 has a modest affinity for origin DNA, its recruitment to the origin is facilitated by binding to E2 (45, 70). The remaining required DNA replication proteins are provided by the host cell.
Support of viral DNA replication by cellular replication factors is commonly facilitated by interaction with viral replication proteins. For example, DNA polymerase α-primase is recruited to the simian virus 40 (SV40) and papillomavirus origins of replication by binding to the large T antigen and E1 protein, respectively (50, 57). To identify novel cellular proteins that might be involved in papillomavirus DNA replication, we looked for proteins that interact with the HPV-11 E1 protein. In these studies, histone H1 was identified as an E1-binding protein found in HeLa cell nuclei. Data presented here suggest that E1 facilitates papillomavirus replication by displacing H1 from DNA during the initiation and/or elongation phase of viral DNA replication. Furthermore, a region containing the HPV-11 origin of replication that excludes binding by H1, perhaps to facilitate initiation of replication, was identified.
MATERIALS AND METHODS
Cell lines, viruses, and antibodies.
Unless otherwise indicated, HeLa cells were used for all experiments and as the source for nuclear matrices and native nucleosome complexes. Human 143B cells were used for the selection of thymidine kinase (TK)-negative recombinants during the construction of the recombinant E1 vaccinia virus (vEE1). Both cell lines were maintained as monolayers in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum. For preparation of nuclear matrix extracts (NMEs), HeLa S3 cells were grown in suspension cultures in spinner flasks.
The WR strain of vaccinia virus was used to generate the vEE1 recombinant vaccinia virus. The recombinant vaccinia virus encoding the bacteriophage T7 RNA polymerase, vTF7-3 (24), was used to direct expression of E1 from vEE1. The recombinant vaccinia virus encoding the adenovirus fiber protein, 2F (31), was used for expression of the fiber protein in HeLa cells.
For immunoprecipitation and Western blot detection of the E1 protein, the rabbit polyclonal antiserum RL-070 (11), which recognizes the amino terminus of E1, was used; a 1:5,000 dilution was employed for Western blotting. For immunoprecipitation and Western blot detection of histone H1, the purified mouse monoclonal antibody AE-4 (Biogenesis Inc., Sandown, N.H.) was used. AE4 was used at a 1:1,000 dilution in Western blot analyses using the secondary avidin-biotin detection scheme for signal amplification. For immunoprecipitation and Western blot detection of the adenovirus fiber protein, the purified mouse monoclonal antibody 4D2 (32) was used; a 1:5,000 dilution was used for Western blotting. For detection of the chloramphenicol acetyltransferase (CAT) and E1N proteins in far-Western blot analyses, the rabbit polyclonal antiserum His-probe H-15 (Santa Cruz Biotechnology, Santa Cruz, Calif.), which recognizes the polyhistidine tags on those proteins, was used at a dilution of 1:5,000. All secondary goat anti-rabbit or goat anti-mouse immunoglobulin G antibodies conjugated to either alkaline phosphatase, biotin, or horseradish peroxidase (Pierce, Rockford, Ill.) were used at a dilution of 1:50,000. 35S-streptavidin (Amersham Life Sciences, Arlington Heights, Ill.) was used at a dilution of 1:1,000. Enhanced chemiluminescence (ECL; Amersham Life Sciences) was used for Western blot detection where indicated.
Construction of recombinant E1-expressing vaccinia virus and E1 expression.
The encephalomyocarditis virus (EMCV) untranslated region (UTR) was fused by overlapping PCR (22) to the 5′ end of the E1 open reading frame (ORF). The sequences of the oligonucleotide PCR primers used were as follows: 2235, 5′ CCGGATCCTAACGTTACTGGCCGAAGCCG 3′; 2953, 5′ CTCATTTTCTGTACCTGAATCGTCCGCCATATTATCATCGTGTTTTTCAAAGG 3′; 2951, 5′ CCTTGAAAAACACGATGATAATATGGCGGACGATTCAGGTACAGAAAATGAG 3′; and 3474, 5′ CCAGCGGATCCCTGCATGCTCTCGGGTGCTGTC 3′.
EMCV DNA and the E1 cDNA (from L. T. Chow) were used as templates in separate PCRs with oligonucleotide primer pairs 2235-2953 and 2951-3474, respectively. After purification to remove the unused primers, approximately 10-ng quantities of each of the PCR products from these reactions were combined and used together as PCR templates with oligonucleotides 2235 and 3474. This reaction fused the EMCV UTR to the N terminus of the E1 ORF, terminating at the SphI site (nucleotide 1399). The termini of the PCR product were digested with BamHI and ligated into pTF7.5 (24) linearized with BamHI to construct pTFEE1ΔSphI. The remaining 3′ portion of the E1 ORF was excised from the E1 DNA plasmid with SphI and ligated into pTFEE1ΔSphI linearized with SphI to construct pTFEE1. To recombine pTFEE1 into vaccinia virus, HeLa cells were infected with vaccinia virus strain WR at a multiplicity of infection of 0.05 and subsequently transfected with pTFEE1. At 48 h postinfection (p.i.), the infected cells were lysed by Dounce homogenization in hypotonic buffer. Virus in the lysate was plaque purified on human 143B cells and simultaneously selected for the TK-negative phenotype by maintenance in DMEM containing 250 mM bromodeoxyuridine. Virus was isolated from individual TK-negative plaques and screened for E1 expression by determining reactivity to the E1 RL-070 antiserum in Western blot analysis. Plaque purification of a positive isolate was repeated as described above, and a stock of this virus (vEE1) was prepared.
For E1 expression, a vaccinia virus expressing the T7 RNA polymerase (vTF7-3) (24) and vEE1 were coadsorbed to HeLa cells in a 100-mm-diameter tissue culture dish (multiplicity of infection, 10 each) in 1 ml of serum-free DMEM for 1 h. Infected cells were maintained in complete medium until harvested. E1 expression was detected as early as 7 h p.i. All vaccinia virus stocks used in these studies were prepared as described by Moss and Earl (47).
Chicken erythrocyte histone H1.
Purified chicken erythrocyte histone H1 was a gift from Heisaburo Shindo and was purified as previously described (55).
Isolation of nucleosomes.
Native nucleosomes were prepared as described by von Holt et al. (69). Briefly, uninfected HeLa cell nuclei were digested with micrococcal nuclease in MN digestion buffer (50 mM Tris [pH 7.4], 25 mM KCl, 4 mM MgCl2, 1 mM CaCl2, 0.2 mM phenylmethylsulfonyl fluoride) for 30 min at 37°C and sedimented at 6,000 × g for 5 min at room temperature. The pellet was extracted by Dounce homogenization in chromatin extraction buffer (10 mM Tris [pH 7.4], 0.25 mM EDTA, 0.2 mM phenylmethylsulfonyl fluoride) and dialyzed overnight at 4°C against the same buffer. Residual proteins were removed by centrifugation at 10,000 × g for 20 min at 4°C. Soluble histones were recovered in the supernatant.
Preparation of nuclear matrices and NMEs.
Nuclei were isolated from uninfected or recombinant vaccinia virus-infected HeLa cells as described by Challberg and Kelly (9). To prepare nuclear matrices, nuclei were washed with digestion buffer (20 mM Tris-HCl [pH 7.4], 0.05 mM spermine, 0.125 mM spermidine, 20 mM KCl, 70 mM NaCl, 10 mM MgCl2) at 4°C and digested with DNase I at a concentration of 0.1 mg/ml for 1 h at the same temperature. Digested nuclei were sedimented by centrifugation at 13,000 × g for 5 min and extracted with digestion buffer containing 10 mM lithium-3,5-diiodosalicylate (LIS) at 4°C for 15 min. Nuclear matrices were sedimented as described above and washed three times with digestion buffer at 4°C to remove the LIS. Nuclear matrix protein concentrations were determined by the method of Bradford (7), in accordance with the manufacturer’s instructions (Bio-Rad Laboratories, Hercules, Calif.).
NME was prepared from uninfected HeLa cell nuclear matrix by a procedure similar to that described by Angeletti and Engler (4). Nuclear matrices were extracted with 4 ml of NME extraction buffer (digestion buffer supplemented with 8 M urea and 1% Triton X-100) per 10 mg of nuclear matrix protein for 15 min at 4°C. Residual proteins were removed by centrifugation at 13,000 × g and 4°C for 5 min. The supernatant was dialyzed successively against 250 volumes each of digestion buffer containing 4, 2, 1, and 0.1 M urea for 2 h each at 4°C; this was followed by a final dialysis against digestion buffer at 4°C overnight. Glycerol was added to the NME (final concentration, 10%), and it was stored at −100°C.
Far-Western blot analysis.
Nuclear matrix proteins were denatured by boiling in Laemmli sample buffer (39), and residual proteins were removed by centrifugation at 13,000 × g for 15 s at room temperature. Proteins were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on a 15% polyacrylamide gel and transblotted to nitrocellulose. Transblotted proteins were denatured, renatured, and blocked as described by Lee et al. (41). As probes, CAT and E1N were allowed to bind to blotted proteins in TBS-Tx (50 mM Tris [pH 7.5], 250 mM NaCl, 1% Triton X-100) containing 5% bovine serum albumin and 5% glycerol for 2 h at room temperature. The blots were washed five times with TBS-Tx for 10 min each at room temperature and then reacted with the His-probe antiserum in TBS-Tx containing 5% bovine serum albumin for 1 h at room temperature. The blots were washed as before and bound to secondary, horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G for 1 h at room temperature. The blots were washed, and bound proteins were detected by enhanced chemiluminescence (Amersham Life Sciences).
Escherichia coli-expressed E1N and CAT.
The pRSETE1 plasmid consisted of the full-length E1 ORF, fused at its N terminus to polyhistidine, in the pRSET vector (Invitrogen, Carlsbad, Calif.). pRSETE1N expressed the N-terminal portion of the E1 protein truncated at amino acid 185 (E1N). For its construction, the AccI restriction fragment between nucleotides 1376 and 1560 of the E1-coding region was excised. The cohesive termini were blunt ended with the Klenow fragment of E. coli DNA polymerase I, and the plasmid was recircularized by ligation. The pTrcHisCAT plasmid (Invitrogen) encoded the CAT protein fused to polyhistidine. To express E1N and CAT, E. coli BL21DE3plysS was transformed with each plasmid and grown to mid-log phase. Cultures were induced for protein expression with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at 37°C for 2 h. Bacteria were harvested by centrifugation at 5,000 × g for 5 min at 4°C, lysed with a French press in FP lysis buffer (50 mM NaPO4 [pH 8.0], 300 mM NaCl, 10% glycerol, 10 mM 2-mercaptoethanol), and cleared of residual protein by centrifugation at 13,000 × g for 30 min at 4°C. The lysates were applied to nickel-agarose affinity columns (Ni++-NTA; Qiagen, Santa Clarita, Calif.), which were washed with Ni2+ wash buffer (50 mM NaPO4 [pH 6.0], 300 mM NaCl, 10% glycerol, 10 mM 2-mercaptoethanol, 0.1% Triton X-100, 0.1% Nonidet P-40) and eluted with H1 binding buffer (10 mM Tris [pH 7.5], 100 mM NaCl) containing 200 mM imidazole. The imidazole was removed by dialysis at 4°C against H1 binding buffer. The final concentrations of E1N and CAT were 0.6 and 0.2 mg/ml, respectively.
E1 affinity chromatography.
The CAT and E1N affinity columns were constructed by binding purified CAT and E1N to Ni2+-agarose; CAT and E1N were used in excess to ensure saturation of the Ni2+-agarose. Unbound protein was removed with Ni2+ wash buffer. To confirm that equal amounts of CAT and E1N were bound, equal volumes of each resin were analyzed by SDS-PAGE and Coomassie blue staining. NME or nucleosomes were bound to the affinity resins in digestion buffer by the batch method overnight at 4°C. Samples were loaded onto columns plugged with siliconized glass wool, and unbound proteins were allowed to flow through the column. Columns were washed at 4°C with 50 volumes of digestion buffer and then with lysis buffer (50 mM Tris [pH 8], 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate) until eluting proteins were undetectable by measurement of optical density at 280 nm. For affinity chromatography of NME, columns were eluted at 4°C with lysis buffer containing 0.05% SDS. For affinity chromatography of nucleosomes, columns were washed successively at 4°C with lysis buffers containing 2 M NaCl and 8 M urea and then eluted at 4°C with lysis buffer at pH 6.0.
Microsequence analysis.
Proteins were resolved by SDS-PAGE and transblotted to a polyvinylidene difluoride membrane (Bio-Rad). The membrane was stained for protein with Ponceau S (Sigma Chemical Company, St. Louis, Mo.) in accordance with the procedure of Harlowe and Lane (28), and the 34-kDa band was excised from the membrane. The remaining protein analysis was performed by Harvard Microchem (Cambridge, Mass.). Protein in the excised membrane was digested with trypsin, and proteolytic peptide fragments were resolved by reverse-phase high-performance liquid chromatography. The amino acid sequences of four peptides were obtained by Edman degradation (40). A BLAST search (2) of the nonredundant protein database at the National Center for Biotechnology Information was used to identify sequences homologous to those obtained by amino acid sequencing.
Immunoprecipitations.
Two dishes each of HeLa cells were infected with 2F for expression of the adenovirus fiber protein or coinfected with vTF-7.3 and vEE1 for expression of the HPV-11 E1 protein. At 16 h p.i., the cells were lysed in 1 ml of lysis buffer, and nuclei were then sedimented at 400 × g for 5 min at 4°C. Nuclei were extracted with digestion buffer containing 700 mM NaCl, and residual proteins were removed by centrifugation as described above. The supernatant was treated with DNase I for 15 min at room temperature. Nucleosome extract aliquots of equal volume were reacted in solution with either the RL-070 E1 antiserum, the anti-H1 antibody AE4, or the antifiber antibody 4D2 for 2 h at room temperature. Immune complexes were precipitated by binding to formalin-fixed, heat-killed Staphylococcus aureus cells (Immunoprecipitin; GIBCO-BRL Life Technologies, Gaithersburg, Md.) for 30 min at room temperature and centrifuged at 400 × g for 3 min. Immunoprecipitates were washed four times with 0.5 ml of lysis buffer for 10 min each and finally centrifuged through a 3-ml cushion of 1 M sucrose-containing lysis buffer at 3,300 × g for 5 min at room temperature. Immunoprecipitates were analyzed by Western blot analysis. During the Western blot analysis, the blots were additionally blocked with unconjugated secondary goat anti-mouse or goat anti-rabbit antibodies to reduce the signals generated by immunoglobulins on the blots.
DNase I protection assays.
The histone H1-DNA binding and DNase I protection assays were adapted from the methods of Izaurralde et al. (33). The HPV-11 genome, cloned at its BamHI site into the pGEM-1 vector (Promega, Madison, Wis.), was digested with BamHI and NdeI, and the restriction fragments were end labeled by filling in the cohesive termini, using the Klenow fragment of DNA polymerase I and [α-32P]dATP. Unincorporated nucleotides were removed by gel filtration, using a Microspin S-400 HR column (Pharmacia Biotech, Piscataway, N.J.). Chicken erythrocyte histone H1 (200 ng) was bound to 200 ng of the labeled restriction fragments in 20 ml of H1 binding buffer for 30 min at room temperature. MgCl2 was added to the reaction mixtures (final concentration, 10 mM), and DNA was digested with 0.2 ng of DNase I for 5 min at room temperature. SDS and EDTA (final concentrations, 0.5% and 5 mM, respectively) were then added prior to digestion with 20 mg of proteinase K at 55°C for 2 h. Following extraction with phenol-chloroform-isoamyl alcohol (25:24:1) and ethanol precipitation, protected restriction fragments were analyzed by agarose gel electrophoresis followed by detection and analysis of the distribution of radioactivity, visualized on a Storm PhosphorImager (Molecular Dynamics, Sunnyvale, Calif.). For the E1 displacement experiment (see Fig. 6), E1N or CAT was added to the reaction mixtures following H1-DNA binding, and the mixtures were incubated for 15 min at room temperature. For the salmon sperm DNA competition experiment (see Fig. 7), salmon sperm DNA was added at the beginning of the H1-DNA reactions.
FIG. 6.

Displacement of H1 from HPV-11 DNA by E1. Two hundred nanograms of 32P-labeled HPV-11 DNA restriction fragments were bound to 200 ng of H1; this was followed by incubation with (+) or without (−) the indicated amounts (in micrograms) of E1N or CAT and, finally, digestion with DNase I. H1-bound restriction fragments were protected from digestion by DNase I; displacement of H1 by E1N is indicated by removal of labeled DNAs by DNase I digestion. The image was generated by PhosphorImager analysis (Molecular Dynamics). (B) Linearized map of HPV-11 DNA restriction fragments A to E. Arrows denote viral protein ORFs. The noncoding region between the E6 and L1 ORFs is the URR and contains the HPV-11 origin of DNA replication.
FIG. 7.

Differential affinities of HPV-11 restriction fragments for binding to histone H1. 32P-labeled HPV-11 DNA restriction fragments were bound to 200 ng of H1 in the presence (+) or absence (−) of the indicated amounts of salmon sperm DNA (ssDNA) followed by digestion with DNase I. H1-bound restriction fragments were protected from digestion by DNase I; competition for H1 binding by salmon sperm DNA resulted in removal of labeled DNAs by DNase I digestion. Differential affinity for H1 among the restriction fragments is indicated by the differing amount of competition at 60 μg of salmon sperm DNA. HPV DNA restriction fragments are labeled according to the map in Fig. 6B. The image was generated by PhosphorImager analysis (Molecular Dynamics).
RESULTS
Expression of the HPV-11 E1 protein from a recombinant vaccinia virus.
For expression of the HPV-11 E1 protein in mammalian cells, the T7 RNA polymerase-vaccinia virus expression system developed by Moss et al. was used (21, 24). An E1-encoding recombinant vaccinia virus (vEE1) that expressed E1 under the control of the T7 RNA polymerase promoter was constructed (Fig. 1A). The E1 ORF was fused at its 5′ end to the EMCV UTR to confer cap-independent protein translation on uncapped T7 RNA polymerase-generated mRNAs. Expression of E1 was achieved by coinfection of HeLa cells with vEE1 and vTF7-3, the recombinant vaccinia virus encoding T7 RNA polymerase (24). Western blotting of the infected-cell lysates with antiserum specific for E1 detected two proteins, with estimated molecular masses of 88 and 68 kDa (Fig. 1B). These bands represent E1-encoded proteins, since they were absent from cells infected with vTF7-3 alone. Except for their masses, the two E1 forms had no identifiable distinguishing features. Both proteins reacted with antisera raised against N- or C-terminal portions of HPV-11 E1 (data not shown). The 88-kDa species represents the full-length, 649-amino-acid protein encoded by the E1 ORF, while the 68-kDa form may result from E1 proteolysis or from internal protein translation initiation. Santucci et al. (52) also reported the expression of multiple forms of E1 from vaccinia virus; the major forms had estimated molecular masses of 72 and 88 kDa. In vitro translation of mRNA derived in vitro from plasmid pTFEE1 also yielded two major E1 products with the same molecular weights, indicating that the two E1 forms are not an artifact of expression by vaccinia virus (data not shown).
FIG. 1.

Expression of recombinant E1 from vEE1. (A) mRNA expression was under the control of the bacteriophage T7 RNA polymerase promoter (PT7) and terminator (T7ter). Cap-independent translation was directed by the EMCV UTR, which was fused to the 5′ end of the E1 ORF. The resulting operon was recombined into the TK gene (TKL and TKR) of vaccinia virus. The arrow indicates the direction of transcription, and the closed circle represents the T7 terminator of transcription. (B) Equal volumes of whole-cell lysates from HeLa cells infected with vTF-7.3 and vEE1 (+) or with vTF-7.3 alone (−) were resolved by SDS-PAGE on a 10% polyacrylamide gel and assayed for recombinant E1 protein by Western blot analysis with the E1 antiserum RL-070, biotinylated secondary antibody, and 35S-streptavidin. The image was generated by PhosphorImager analysis (Molecular Dynamics). The positions of molecular mass markers (in kilodaltons) are shown on the right.
E1 binds to histone H1 in vitro.
A far-Western blot analysis was used to identify proteins in HeLa cell nuclei that bind to the E1 protein (Fig. 2). A truncated form of the E1 protein (E1N) was used as a probe in this analysis. E1N contains the amino-terminal 185 amino acids of E1 and was expressed in and purified from E. coli as a polyhistidine-tagged fusion protein. As a negative control in far-Western blot analysis, the CAT protein was used as a probe; it was also expressed and purified from E. coli as a polyhistidine-tagged fusion protein. An antibody (His-probe) that recognizes the polyhistidine tags present on both the E1N and CAT proteins was used to detect E1-reactive proteins on the far-Western blot. We used protein from the nuclear matrix fraction to probe for E1-binding proteins, since the nuclear matrix is known to be the site of eukaryotic and viral DNA replication (6, 15, 16, 35, 48) as well as E1 localization (63). The nuclear matrix is operationally defined as the subnuclear structure remaining after removal of chromatin from isolated nuclei by DNase I digestion and subsequent extraction with either 2 M NaCl or the nonionic detergent LIS. The nuclear matrix is a complex mixture of protein and nucleic acids and consists of the nuclear lamina, nuclear envelope, nucleoli, and internal core filaments as well as many other uncharacterized components (68). SV40 DNA replication, which relies on the host cell’s replication machinery to the same extent as that of papillomaviruses, also occurs in association with this nuclear substructure (29, 53, 60). In fact, the nuclear matrix has been implicated as the site of occurrence of most nuclear processes, including transcription and RNA processing (5, 34, 59, 66). The E1N protein bound to a doublet band migrating at 34 and 35 kDa. This interaction was specific for the E1N protein, since no reactivity of the CAT proteins with these bands was observed. The nonspecific reactive band at approximately 55 kDa on both the experimental and the control blots is a nuclear component that reacts with the His-probe antibody.
FIG. 2.
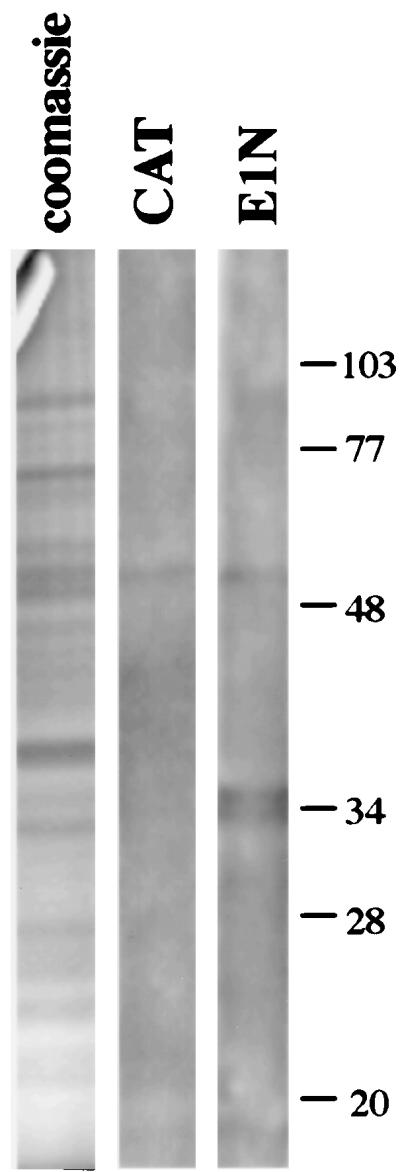
Far-Western blot analysis of HeLa cell nuclear proteins with the E1N protein. A 0.5-mg quantity of nuclear protein was resolved by SDS-PAGE on a 12.5% polyacrylamide slab gel, transblotted to nitrocellulose, and renatured. Strips cut from the blot were probed with the CAT or E1N protein. E1N-reactive proteins were detected by reaction with His-probe antiserum and enhanced chemiluminescence. The left strip is total nuclear matrix protein, stained with Coomassie blue, derived from the same gel as the far-Western blots. The positions of molecular mass markers (in kilodaltons) are shown on the right.
The E1-binding proteins were affinity purified from NME for their identification by microsequence analysis. NME was prepared by extraction of nuclear matrices with 8 M urea followed by removal of the urea by dialysis. An E1 affinity column was constructed by binding the E1N protein to nickel-agarose. A similar column, constructed with the CAT protein, was used as a control for nonspecific binding of NME proteins. The columns were loaded with NME, washed extensively to remove nonspecifically bound proteins, and eluted with 0.5% SDS. The silver-stained SDS-PAGE gel of the column fractions is shown in Fig. 3.
FIG. 3.

Purification of E1-bound proteins by E1 affinity chromatography. Protein from a dialyzed HeLa cell NME was loaded onto an E1N (+) or a CAT (−) affinity column. Bound proteins were washed with Ni2+ wash buffer and were eluted with lysis buffer containing 0.5% SDS. Equal volumes from the flowthrough, wash, and eluate fractions were resolved by SDS-PAGE and detected by silver staining. Crude NME was loaded in the far left lane. The positions of molecular mass markers (in kilodaltons) are shown on the left. The arrow indicates the position of the protein doublet.
The same protein doublet evident in Fig. 2 was bound by and eluted from the E1 column. Its association with the column was E1 dependent, since it was not bound by the CAT column. The 34-kDa band from the eluate was processed for microsequencing analysis by Harvard Microchem as described in Materials and Methods. The amino acid sequences of four tryptic peptides (SGVSLAALK, ALAAAGYDVEK, ELTDSGYGYSEVEAATQVEK, and GTLVQTK) were obtained. As shown by a BLAST search (2) of the nonredundant protein database at the National Center for Biotechnology Information, these sequences were identical to ones present in human histone H1. H1 is known to be a nucleus-localized protein for which six isoforms have been identified; they migrate as two distinct bands near 34 and 35 kDa in one-dimensional SDS-PAGE (13, 14). These data verify that H1 isolated from HeLa cell nuclei binds directly to E1 in vitro.
The relative abundance of histone H1 in the crude NME is likely exaggerated in Fig. 3 due to our silver staining method, since it was not evident in such relative abundance when stained with Coomassie blue (data not shown). Also, H1 was not present in such relative abundance in preparations of total nuclear matrix proteins, as shown in Fig. 2. As a result, we do not feel that the observed interaction between E1 and H1 is a nonspecific one attributable to the relative abundance of H1 in the extracts used in these experiments. Because H1 is a highly basic and lysine-rich protein, we assessed the contribution of lysines and ionic charges to the E1-H1 interaction by eluting the column with 0.5 M lysine and 2 M NaCl, respectively. H1 failed to elute from the column under either condition, indicating that the E1-H1 interaction is not mediated by nonspecific association between lysines or ionic charges (data not shown). Furthermore, as will be described below (see also Fig. 4), the E1-H1 interaction was also shown to be resistant to 8 M urea. These data suggest that the observed interaction between E1 and H1 is specific.
FIG. 4.

Association of histone H1, isolated under native conditions, with the E1 affinity column. Protein from a nucleosome extract (lanes 3 and 4) or nucleosome extract buffer alone (lanes 5 and 6) was loaded onto an E1N (lanes 4 and 6) or a CAT (lanes 3 and 5) affinity column, washed with Ni2+ wash buffer supplemented with 8 M urea, and eluted with pH 6.0 lysis buffer. Equal volumes of protein from the columns’ eluates were resolved by SDS-PAGE on a 15% polyacrylamide gel. Resolved proteins were detected by Coomassie brilliant blue staining. Proteins from crude NME and the flowthrough from the NME-loaded E1 column were loaded in lanes 1 and 2, respectively. Protein molecular mass standards (in kilodaltons) were loaded in the far-left lane.
In both the far-Western blot analysis and affinity purification, the analyzed proteins were denatured at some point prior to E1 binding. Although protein renaturation was attempted in both cases, the native state of H1 was not affirmed. In a protocol employing no protein denaturants, H1 was isolated in its native state in association with intact nucleosomes from micrococcal-nuclease-treated nuclei, as described by von Holt et al. (69). The nucleosome preparation contained H1 as well as core histones, and its DNA composition was characteristic of mononucleosomal DNA (data not shown). The nucleosome extract was applied to an E1 column, which was washed and eluted with 8 M urea followed by a low-pH (6.0) buffer. A duplicate E1 column was loaded with nucleosome buffer alone and processed in the same manner; this control was designed to detect eluting proteins which might originate from the affinity column itself instead of the extract. Interestingly, H1 failed to elute from the column with 8 M urea (data not shown). Conditions tested that eluted H1 were 0.5% SDS and low pH (pH 6.0). Figure 4 shows the Coomassie-stained SDS-PAGE gel of the low-pH eluates. The identification of the proteins in the E1 column eluate as H1 was confirmed as follows. (i) They were recognized by an H1 antiserum in a Western blot analysis (data not shown). (ii) They were absent in the eluate from the column loaded with buffer alone, indicating that they originated from the nucleosome preparation. The binding of H1 to the affinity column was E1 dependent, since H1 did not bind to CAT protein immobilized on a similar nickel column. The association of H1 with E1 was not mediated by core histones, since they were not present in the eluate. The 28-kDa protein seen in the CAT column eluate is a fraction of the CAT protein which dissociated from the nickel-agarose at low pH. This behavior was not observed with the E1N column, since no E1 protein was detected in its eluate by Western blot analysis (data not shown). These data demonstrate that the E1 protein bound in vitro to H1 which was isolated under native conditions in association with nucleosomes. Moreover, taken with the resistance to high ionic strength, the pH dependence of the interaction may reflect a dependence on protein conformation.
E1 was associated with histone H1 in vivo.
To demonstrate the E1-H1 complex in vivo, we coimmunoprecipitated E1 and H1 from a nuclear extract made from HeLa cells expressing recombinant E1. Nuclear extracts were prepared from HeLa cell cultures expressing the HPV-11 E1 protein or the adenovirus fiber protein, each from recombinant vaccinia viruses, and immunoprecipitated with the E1 antiserum RL-070, the anti-H1 antibody AE4, or the antifiber antibody 4D2. The Western blots of the immunoprecipitates are shown in Fig. 5. The E1 antiserum coimmunoprecipitated H1 with E1 from the lysate derived from E1-expressing HeLa cells. H1’s association with E1 immune complexes did not result from nonspecific interaction with the E1 antiserum, since H1 was not immunoprecipitated from lysates derived from cells expressing the fiber protein instead. In addition, the anti-H1 antibody coimmunoprecipitated E1 with H1 from cells expressing E1. Immunoprecipitation of E1 or H1 by their respective immune reagents was specific, as shown by their absence from immune complexes formed with heterologous antibodies. To dismiss the possibility that the observed E1-H1 complexes are an artifact of overexpression from vaccinia virus and result from nonspecific interactions, we assayed for coimmunoprecipitation of a different nucleus-localized protein expressed from vaccinia virus. The adenovirus fiber protein was expressed in HeLa cells from vaccinia virus. Immune complexes derived from a nuclear extract of the fiber-expressing cells were formed by using an antifiber or anti-H1 antibody, and the presence of the fiber protein or H1 was determined by Western blot analysis (Fig. 5C). Coimmunoprecipitation of the fiber protein and H1 was not observed with either antibody, indicating that the observed E1-H1 complexes were not an artifact of E1 overexpression from vaccinia virus. These data support the notion of an intranuclear complex formed specifically between the E1 protein and histone H1 in vivo.
FIG. 5.

Coimmunoprecipitation of histone H1 and HPV-11 E1. Nuclear extracts derived from HeLa cells expressing the HPV-11 E1 (vEE1) or adenovirus fiber (2F) protein, each from vaccinia virus, were incubated with (+) or without (−) E1 antiserum (α-E1), anti-H1 antibody (α-H1), and antifiber antibody (α-fiber). Immunoprecipitates were assayed for H1 (A), E1 (B), or fiber (C) by Western blot analysis and enhanced chemiluminescence. The asterisks denote the heavy and light chains of immunoglobulins used in the immunoprecipitations. The band in panel B denoted by the uppermost asterisk is derived from the E1 antiserum and is likely incompletely denatured immunoglobulin. Positions of molecular mass markers (in kilodaltons) are shown on the left.
E1 displaces H1 from DNA.
H1 is a linker histone that binds to regions of DNA between adjacent nucleosomes, condensing chromatin into a more highly ordered structure. Alteration of H1 binding to chromatin alters the more highly ordered structure of chromatin, which can affect chromatin-dependent processes such as gene expression and DNA replication (64). We tested the hypothesis that binding by E1 alters the association of H1 with DNA and influences chromatin structure. To assay for H1 association with DNA, we used the H1-DNA binding and DNase I protection assays describe by Izaurralde et al. (33). H1 binds nonspecifically but tightly to DNA, protecting it from digestion by DNase I. Because this assay only assesses H1’s association with naked DNA, and papillomavirus DNA exists as chromatin in vivo, the physiological relevance of this assay is questionable. However, H1’s association with chromatin occurs via direct interaction with DNA; the core histones comprising the octamer do not mediate or stabilize the association. We therefore feel that this assay suffices to assess DNA binding by H1 and that results obtained with this assay may be extended to chromatin in vivo. 32P-labeled HPV-11 DNA restriction fragments were bound to purified chicken erythrocyte H1 and subsequently incubated with increasing amounts E1N. The samples were then digested with DNase I and analyzed by agarose gel electrophoresis and autoradiography. The autoradiograph of the agarose gel is shown in Fig. 6. E1N diminished the association of H1 with the DNA fragments in a dose-dependent manner. Protection from DNase I digestion was eliminated with 800 ng of E1, whereas up to 2 μg of the CAT protein had a minimal effect on the association of H1 with the DNA fragments. These data indicate that interaction between E1 and histone H1 results in displacement of H1 from DNA.
Regions within the HPV-11 genome have differential affinities for H1.
Certain DNAs have been shown to have differential affinities for H1 (33). Regions within the SV40 genome, at the origin of replication and early promoter, with resistance to H1 binding were previously identified (30). We looked for such regions within the HPV-11 genome by using the DNase I protection assay described in the legend to Fig. 6. H1 was incubated with the labeled HPV-11 restriction fragments in the presence of increasing amounts of salmon sperm DNA as a nonspecific DNA binding competitor. Samples were digested with DNase I and analyzed by agarose gel electrophoresis and autoradiography. The results are shown in Fig. 7. Salmon sperm DNA competed with H1 for association with each of the restriction fragments. However, regions with differential affinities for H1 were identified at lower concentrations of competing salmon sperm DNA. Fragments 3 and 5 were weaker-binding fragments. These fragments span nucleotides 7658 to 1101, a sequence which contains the 5′ portion of the protein coding region and the 3′ portion of the upstream regulatory region (URR), including the origin of replication (Fig. 7). They are not contiguous and are separated by a 117-bp restriction fragment.
DISCUSSION
Histone H1 binds to nucleosome-organized chromatin at regions of DNA between adjacent nucleosomes, condensing chromatin into a more highly ordered structure (64). The SV40 and bovine papillomavirus type 1 (BPV-1) genomes are known to be complexed with nucleosomes and to be organized into conventional chromatin structures (23, 36, 49). However, replication of both of these viral DNAs in vitro is repressed by the presence of nucleosomes on the template (10, 19, 37, 56). Each of these viruses must have a means of overcoming this repression (18). Nucleosomal repression of DNA replication is postulated to occur during both initiation and chain elongation. During initiation, nucleosomes interfere with interactions between replication factors and the origin (10, 56). They must be displaced for site-specific DNA interactions to occur. They also act as obstacles for the DNA polymerase during chain elongation by impeding progression of the replication fork. They must be disrupted in front of the replication fork and reassembled behind it, onto daughter strands, once the fork has passed (1, 26, 46, 67). Work by Ramsperger and Stahl (51) suggested that these tasks are accomplished by the large T antigen during SV40 DNA replication. T antigen was shown to bind nucleosomal SV40 origin DNA and displace core octamers in the process. It also unwound nucleosomal DNA in a helicase assay. Furthermore, T antigen bound directly to histones H1 and H3, suggesting that nucleosome disruption by T antigen is facilitated through these interactions. Li and Botchan (42) demonstrated that nucleosomal repression of in vitro BPV-1 DNA replication was relieved by site-specific binding of the E2 protein to viral DNA. This result indicates that E2, not E1, is responsible for nucleosomal disruption during BPV-1 DNA replication and that E2 plays an additional role in replication initiation besides recruitment of E1 to the origin. Perhaps our failure to observe an interaction between E1 and the core histones is a further indication that E1 plays no role in nucleosome disruption.
What is the function of the interaction of E1 with H1? Li and Botchan did not assess the effect of H1-organized nucleosomes on replication, since their templates contained no H1. The presence of H1 on the template presents an additional obstacle during DNA replication. Accessibility of the origin to replication factors should be further constrained, and nucleosome disruption during chain elongation should be impaired. During initiation of HPV DNA synthesis, the chromatin structure at the viral origin may be critical and require loosening for recognition by the E1-E2 complex to occur. This could be facilitated by displacement of H1 from DNA by E1. Most studies have implicated the E1-E2 complex as the form of E1 likely to function during initiation of HPV DNA synthesis (12, 17, 58, 65, 70), and both E1 and E2 were required to initiate in vitro BPV-1 DNA synthesis on nucleosome-organized templates (42). Consequently, E1 may displace H1 from DNA while complexed with E2. Because we did not assess the ability of E1-E2 complexes to displace H1, the physiological relevance of the experiment of Fig. 6 to replication initiation is limited. Recent work by Gasser et al. (25) suggested a model for nucleosome disruption during the elongation phase of DNA synthesis in which H1 dissociates from the first one or two nucleosomes ahead of the replication fork. E1 may serve a role during chain elongation in addition to its DNA helicase activity by displacing H1 ahead of the replication fork. E2’s only identified role in replication is origin recognition, occurring during initiation. No role for E2 during chain elongation has yet been suggested. In fact, E2 may dissociate from E1 and the replication complex following initiation and be absent from the replication fork altogether during chain elongation. As a result, H1 displacement during chain elongation may be induced by E1 in the absence of E2, and the H1 displacement seen in Fig. 6 may be more representative of events occurring during chain elongation than of those taking place during initiation; however, it does not rule out the possibility that E1 displaces H1 while complexed with E2. T antigen may provide similar functions for SV40 DNA replication through its interaction with H1, although an ability to affect the interaction of H1 with DNA has not been demonstrated. The data presented in this paper suggest that both E1 and T antigen share this additional function apart from their roles as DNA helicases.
Late in infection, SV40 DNA is depleted of nucleosomes at the origin (3). Although this observation may be explained by occupation of the origin by T antigen, work by Hendrickson and Cole (30) suggested that the DNA structure around the origin may also contribute to this nucleosome depletion. Restriction fragments containing the SV40 origin were resistant to binding of histones H1 and H4 relative to other regions of the genome. Nucleotides flanking the AT element within the origin were underprotected from DNase I digestion by H1 and hypersensitive to hydroxyl radical cleavage (30). In this paper, we showed that a 511-bp restriction fragment containing the HPV-11 origin of replication was also resistant to H1 binding relative to the majority of other regions of the genome. This property is not limited to sequences around the origin, since a second fragment containing a region 3′ to the URR behaved similarly. Although these two fragments are not contiguous and are separated by 117 bp, the structure responsible for this resistance may extend into both regions contained in the fragments. These data correlate with those presented by Hendrickson and Cole (30) for SV40 DNA, which showed that a similar resistance was conferred on an origin-containing fragment of 366 bp. They suggested that regions around papovavirus origins of DNA replication have a common DNA structure that excludes histone H1 from binding.
ACKNOWLEDGMENTS
We thank Louise T. Chow for the gifts of numerous plasmids and antisera against HPV-11 E1. Plasmid pTF7.5 and recombinant vaccinia viruses (wild type and vTF7-3) were obtained from Bernard Moss. We thank Willie Thomas for assisting us with the large-scale culturing of HeLa cells.
This work was supported by NIH grant AI20408 (to J.A.E.). C.S.S. was supported in part by NIH training grant T32 CA09467 (to E. Hunter). Support for the synthesis of oligonucleotides used in construction of clones was provided though NCI grant CA13148 to the Comprehensive Cancer Center.
REFERENCES
- 1.Adams C C, Workman J L. Nucleosome displacement in transcription. Cell. 1993;72:305–308. doi: 10.1016/0092-8674(93)90109-4. [DOI] [PubMed] [Google Scholar]
- 2.Altschul S F, Gish W, Miller W, Myers E W, Lipman D J. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 3.Ambrose C, Blasquez V, Bina M. A block in initiation of simian virus 40 assembly results in the accumulation of minichromosomes containing an exposed regulatory region. Proc Natl Acad Sci USA. 1986;83:3287–3291. doi: 10.1073/pnas.83.10.3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Angeletti P C, Engler J A. Tyrosine kinase-dependent release of an adenovirus preterminal protein complex from the nuclear matrix. J Virol. 1996;70:3060–3067. doi: 10.1128/jvi.70.5.3060-3067.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berezney R. The nuclear matrix: a heuristic model for investigating genomic organization and function in the cell nucleus. J Cell Biochem. 1991;47:109–123. doi: 10.1002/jcb.240470204. [DOI] [PubMed] [Google Scholar]
- 6.Berezney R. Organization and function of the nuclear matrix. Boca Raton, Fla: CRC Press; 1984. [Google Scholar]
- 7.Bradford M M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 8.Broker T R, Chow L T, Chin M T, Rhodes C, Wolinsky S M, Whitbeck A, Stoler M H. A molecular portrait of human papillomavirus carcinogenesis. Cancer Cells. 1989;7:197–208. [Google Scholar]
- 9.Challberg M D, Kelly T J. Adenovirus DNA replication in vitro. Proc Natl Acad Sci USA. 1979;76:655–659. doi: 10.1073/pnas.76.2.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng L, Kelly T J. Transcriptional activator nuclear factor I stimulates the replication of SV40 minichromosomes in vivo and in vitro. Cell. 1989;59:541–551. doi: 10.1016/0092-8674(89)90037-8. [DOI] [PubMed] [Google Scholar]
- 11.Chiang C-M, Broker T R, Chow L T. An E1M∧E2C fusion protein encoded by human papillomavirus type 11 is a sequence-specific transcription repressor. J Virol. 1991;65:3317–3329. doi: 10.1128/jvi.65.6.3317-3329.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang C-M, Ustav M, Stenlund A, Ho T F, Broker T R, Chow L T. Viral E1 and E2 proteins support replication of homologous and heterologous papillomaviral origins. Proc Natl Acad Sci USA. 1992;89:5799–5803. doi: 10.1073/pnas.89.13.5799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cole R D. Microheterogeneity in H1 histones and its consequences. Int J Pept Protein Res. 1987;30:433–444. doi: 10.1111/j.1399-3011.1987.tb03352.x. [DOI] [PubMed] [Google Scholar]
- 14.Cole R D. A minireview of microheterogeneity in H1 histone and its possible significance. Anal Biochem. 1984;136:24–30. doi: 10.1016/0003-2697(84)90303-8. [DOI] [PubMed] [Google Scholar]
- 15.Cook P R. The nucleoskeleton and the topology of replication. Cell. 1991;66:627–635. doi: 10.1016/0092-8674(91)90109-c. [DOI] [PubMed] [Google Scholar]
- 16.DeBruyn Kops A, Knipe D M. Formation of DNA replication structures in herpes virus-infected cells requires a viral DNA binding protein. Cell. 1988;55:857–868. doi: 10.1016/0092-8674(88)90141-9. [DOI] [PubMed] [Google Scholar]
- 17.Del Vecchio A M, Romanczuk H, Howley P M, Baker C C. Transient replication of human papillomavirus DNAs. J Virol. 1992;66:5949–5958. doi: 10.1128/jvi.66.10.5949-5958.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DePamphilis M L. Eukaryotic DNA replication: anatomy of an origin. Annu Rev Biochem. 1993;62:29–63. doi: 10.1146/annurev.bi.62.070193.000333. [DOI] [PubMed] [Google Scholar]
- 19.DePamphilis M L, Bradley M K. Replication of SV40 and polyomavirus chromosomes. In: Salzman N P, editor. The polyomaviruses. Vol. 1. New York, N.Y: Plenum Publishing Corporation; 1986. pp. 99–246. [Google Scholar]
- 20.Dollard S C, Wilson J L, Demeter L M, Bonnez W, Reichman R C, Broker T R, Chow L T. Production of human papillomavirus and modulation of the infectious program in epithelial raft cultures. Genes Dev. 1992;6:1131–1142. doi: 10.1101/gad.6.7.1131. [DOI] [PubMed] [Google Scholar]
- 21.Elroy-Stein O, Fuerst T R, Moss B. Cap-independent translation of mRNA conferred by encephalomyocarditis virus 5′ sequence improves the performance of the vaccinia virus/bacteriophage T7 hybrid expression system. Proc Natl Acad Sci USA. 1989;86:6126–6130. doi: 10.1073/pnas.86.16.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erlich H A. PCR technology: principles and applications for DNA amplification. W. H. New York, N.Y: Freeman and Company; 1992. [Google Scholar]
- 23.Favre M, Breitburd F, Croissant O, Orth G. Chromatin-like structures obtained after alkaline disruption of bovine and human papillomaviruses. J Virol. 1977;21:1205–1209. doi: 10.1128/jvi.21.3.1205-1209.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuerst T R, Niles E G, Studier F W, Moss B. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc Natl Acad Sci USA. 1986;83:8122–8126. doi: 10.1073/pnas.83.21.8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gasser R, Koller T, Sogo J M. The stability of nucleosomes at the replication fork. J Mol Biol. 1996;258:224–239. doi: 10.1006/jmbi.1996.0245. [DOI] [PubMed] [Google Scholar]
- 26.Gruss C, Sogo J M. Chromatin replication. Bioessays. 1992;14:1–8. doi: 10.1002/bies.950140102. [DOI] [PubMed] [Google Scholar]
- 27.Ham J, Dostatni N, Gauthier J M, Yaniv M. The papillomavirus E2 protein: a factor with many talents. Trends Biochem Sci. 1991;16:440–444. doi: 10.1016/0968-0004(91)90172-r. [DOI] [PubMed] [Google Scholar]
- 28.Harlowe E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor, N.Y: Cold Spring Harbor Laboratory; 1988. [Google Scholar]
- 29.Harper F, Florentin Y, Puvion E. Large T antigen-rich viral DNA replication loci in SV40-infected monkey kidney cells. Exp Cell Res. 1985;161:434–444. doi: 10.1016/0014-4827(85)90099-0. [DOI] [PubMed] [Google Scholar]
- 30.Hendrickson F M, Cole R D. Selectivity in the interaction of various DNA sequences with H1 histone. Biochemistry. 1994;33:2997–3006. doi: 10.1021/bi00176a032. [DOI] [PubMed] [Google Scholar]
- 31.Hong J S, Engler J A. The amino terminus of the adenovirus fiber protein encodes the nuclear localization signal. Virology. 1991;185:758–767. doi: 10.1016/0042-6822(91)90547-o. [DOI] [PubMed] [Google Scholar]
- 32.Hong J S, Engler J A. Domains required for assembly of adenovirus type 2 fiber trimers. J Virol. 1996;70:7071–7078. doi: 10.1128/jvi.70.10.7071-7078.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Izaurralde E, Kas E, Laemmli U K. Highly preferential nucleation of histone H1 assembly on scaffold-associated regions. J Mol Biol. 1989;210:573–585. doi: 10.1016/0022-2836(89)90133-2. [DOI] [PubMed] [Google Scholar]
- 34.Jackson D A. Structure-function relationships in eukaryotic nuclei. Bioessays. 1991;13:1–10. doi: 10.1002/bies.950130102. [DOI] [PubMed] [Google Scholar]
- 35.Jackson D A, Cook P R. Replication occurs at a nucleoskeleton. EMBO J. 1986;5:1403–1410. doi: 10.1002/j.1460-2075.1986.tb04374.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Keller W, Muller U, Eicken I, Wendel I, Zentgraf H. Biochemical and ultrastructural analysis of SV40 chromatin. Cold Spring Harbor Symp Quant Biol. 1978;42:227–244. doi: 10.1101/sqb.1978.042.01.025. [DOI] [PubMed] [Google Scholar]
- 37.Kelly T J. SV40 DNA replication. J Biol Chem. 1988;263:17889–17892. [PubMed] [Google Scholar]
- 38.Kuo S R, Liu J S, Broker T R, Chow L T. Cell-free replication of the human papillomavirus DNA with homologous viral E1 and E2 proteins and human cell extracts. J Biol Chem. 1994;269:24058–24065. [PubMed] [Google Scholar]
- 39.Laemmli U K. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 40.Lane W S. Complete amino acid sequence of the FK506 and rapamycin binding protein, FKBP, isolated from calf thymus. J Protein Chem. 1991;10:151–160. doi: 10.1007/BF01024778. [DOI] [PubMed] [Google Scholar]
- 41.Lee W S, Kao C C, Bryant G O, Liu X, Berk A J. Adenovirus E1A activation domain binds the basic repeat in the TATA box transcription factor. Cell. 1991;67:365–376. doi: 10.1016/0092-8674(91)90188-5. [DOI] [PubMed] [Google Scholar]
- 42.Li R, Botchan M R. Acidic transcription factors alleviate nucleosome-mediated repression of DNA replication of bovine papillomavirus type 1. Proc Natl Acad Sci USA. 1994;91:7051–7055. doi: 10.1073/pnas.91.15.7051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McBride A A, Romanczuk H, Howley P M. The papillomavirus E2 regulatory proteins. J Biol Chem. 1991;266:18411–18414. [PubMed] [Google Scholar]
- 44.Meyers C, Frattini M G, Hudson J B, Laimins L A. Biosynthesis of human papillomavirus from a continuous cell line upon epithelial differentiation. Science. 1992;257:971–973. doi: 10.1126/science.1323879. [DOI] [PubMed] [Google Scholar]
- 45.Mohr I J, Clark R, Sun S, Androphy E J, Macpherson P, Botchan M R. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science. 1990;250:1694–1699. doi: 10.1126/science.2176744. [DOI] [PubMed] [Google Scholar]
- 46.Morse R H. Transcribed chromatin. Trends Biochem Sci. 1992;17:23–26. doi: 10.1016/0968-0004(92)90422-6. [DOI] [PubMed] [Google Scholar]
- 47.Moss B, Earl P E. –1997. Expression of proteins in mammalian cells using vaccinia viral vectors. In: Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K, editors. Current protocols in molecular biology. Vol. 2. New York, N.Y: John Wiley & Sons, Inc.; 1987. pp. 16.15.1–16.19.9. [Google Scholar]
- 48.Nakayasu H, Berezney R. Mapping replication sites in the eukaryotic cell nucleus. J Cell Biol. 1989;108:1–11. doi: 10.1083/jcb.108.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nedospasov S A, Bakayev V V, Georgiev G P. Chromosome of the mature virion of simian virus 40 contains H1 histone. Nucleic Acids Res. 1978;5:2847–2860. doi: 10.1093/nar/5.8.2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Park P, Copeland W, Yang L, Wang T, Botchan M R, Mohr I J. The cellular DNA polymerase alpha-primase is required for papillomavirus DNA replication and associates with the viral E1 helicase. Proc Natl Acad Sci USA. 1994;91:8700–8704. doi: 10.1073/pnas.91.18.8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramsperger U, Stahl H. Unwinding of chromatin by the SV40 large T antigen DNA helicase. EMBO J. 1995;14:3215–3225. doi: 10.1002/j.1460-2075.1995.tb07324.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Santucci S, Androphy E J, Bonne-Andréa C, Clertant P. Proteins encoded by the bovine papillomavirus E1 open reading frame: expression in heterologous systems and in virally transformed cells. J Virol. 1990;64:6027–6039. doi: 10.1128/jvi.64.12.6027-6039.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schirmbeck R, Deppert W. Structural topography of simian virus 40 DNA replication. J Virol. 1991;65:2578–2588. doi: 10.1128/jvi.65.5.2578-2588.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seo Y S, Muller F, Lusky M, Hurwitz J. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc Natl Acad Sci USA. 1993;90:702–706. doi: 10.1073/pnas.90.2.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Simon R H, Felsenfeld G. A new procedure for purifying histone pairs H2A + H2B and H3 + H4 from chromatin using hydroxylapatite. Nucleic Acids Res. 1979;6:689–696. doi: 10.1093/nar/6.2.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Simpson R T. Nucleosome positioning can affect the function of a cis-acting DNA element in vivo. Nature. 1990;343:387–389. doi: 10.1038/343387a0. [DOI] [PubMed] [Google Scholar]
- 57.Smale S T, Tjian R. T-antigen-DNA polymerase α complex implicated in simian virus 40 DNA replication. Mol Cell Biol. 1986;6:4077–4087. doi: 10.1128/mcb.6.11.4077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Spalholz B A, McBride A A, Sarafi T, Quintero J. Binding of bovine papillomavirus E1 to the origin is not sufficient for DNA replication. Virology. 1993;193:201–212. doi: 10.1006/viro.1993.1116. [DOI] [PubMed] [Google Scholar]
- 59.Spector D L. Macromolecular domains within the cell nucleus. Annu Rev Cell Biol. 1993;9:265–315. doi: 10.1146/annurev.cb.09.110193.001405. [DOI] [PubMed] [Google Scholar]
- 60.Staufenbiel M, Deppert W. Different structural systems of the nucleus are targets for SV40 large T antigen. Cell. 1983;33:173–181. doi: 10.1016/0092-8674(83)90346-x. [DOI] [PubMed] [Google Scholar]
- 61.Stoler M H, Whitbeck A, Wolinsky S M, Broker T R, Chow L T, Howett M K, Kreider J W. Infectious cycle of human papillomavirus type 11 in human foreskin xenografts in nude mice. J Virol. 1990;64:3310–3318. doi: 10.1128/jvi.64.7.3310-3318.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stoler M H, Wolinsky S M, Whitbeck A, Broker T R, Chow L T. Differentiation-linked human papillomavirus types 6 and 11 transcription in genital condylomata revealed by in situ hybridization with message-specific RNA probes. Virology. 1989;172:331–340. doi: 10.1016/0042-6822(89)90135-9. [DOI] [PubMed] [Google Scholar]
- 63.Swindle, C. S., and J. A. Engler. Unpublished results.
- 64.Thoma F, Koller T H, Klug A. Involvement of histone H1 in the organization of the nucleosome and of the salt-dependent superstructures of chromatin. J Cell Biol. 1979;83:403–427. doi: 10.1083/jcb.83.2.403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ustav M, Stenlund A. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J. 1991;10:449–457. doi: 10.1002/j.1460-2075.1991.tb07967.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van Driel R, Humbel B, de Jong L. The nucleus: a black box being opened. J Cell Biochem. 1991;47:311–316. doi: 10.1002/jcb.240470405. [DOI] [PubMed] [Google Scholar]
- 67.van Holde K E, Lohr D E, Robert C. What happens to nucleosomes during transcription? J Biol Chem. 1992;267:2837–2840. [PubMed] [Google Scholar]
- 68.Verheijen R, Venroij W V, Ramaekers F. The nuclear matrix: structure and composition. J Cell Sci. 1988;90:11–36. doi: 10.1242/jcs.90.1.11. [DOI] [PubMed] [Google Scholar]
- 69.von Holt C, Brandt W F, Greyling H J, Lindsey G G, Retief J D, Rodrigues J D, Schwager S, Sewell B T. Isolation and characterization of histones. Methods Enzymol. 1989;170:431–503. doi: 10.1016/0076-6879(89)70061-6. [DOI] [PubMed] [Google Scholar]
- 70.Yang L, Li R, Mohr I J, Clark R, Botchan M R. Activation of BPV-1 replication in vitro by the transcription factor E2. Nature. 1991;353:628–632. doi: 10.1038/353628a0. [DOI] [PubMed] [Google Scholar]
- 71.Yang L, Mohr I, Fouts E, Lim D A, Nohaile M, Botchan M B. The E1 protein of bovine papilloma virus 1 is an ATP-dependent DNA helicase. Proc Natl Acad Sci USA. 1993;90:5086–5090. doi: 10.1073/pnas.90.11.5086. [DOI] [PMC free article] [PubMed] [Google Scholar]