

Abstract
More than 50% of human tumors display hyperactivation of the serine/threonine kinase AKT. Despite evidence of clinical efficacy, the therapeutic window of the current generation of AKT inhibitors could be improved. Here, we report the development of a second-generation AKT degrader, INY-05–040, which outperformed catalytic AKT inhibition with respect to cellular suppression of AKT-dependent phenotypes in breast cancer cell lines. A growth inhibition screen with 288 cancer cell lines confirmed that INY-05–040 had a substantially higher potency than our first-generation AKT degrader (INY-03–041), with both compounds outperforming catalytic AKT inhibition by GDC-0068. Using multiomic profiling and causal network integration in breast cancer cells, we demonstrated that the enhanced efficacy of INY-05–040 was associated with sustained suppression of AKT signaling, which was followed by induction of the stress mitogen activated protein kinase (MAPK) c-Jun N-terminal kinase (JNK). Further integration of growth inhibition assays with publicly available transcriptomic, proteomic, and reverse phase protein array (RPPA) measurements established low basal JNK signaling as a biomarker for breast cancer sensitivity to AKT degradation. Together, our study presents a framework for mapping the network-wide signaling effects of therapeutically relevant compounds and identifies INY-05–040 as a potent pharmacological suppressor of AKT signaling.
Introduction
The phosphoinositide 3-kinase (PI3K)/AKT network has a fundamental role in the integration of extracellular growth stimuli to regulate cell metabolism, migration, proliferation, and survival1. Aberrant activation of this network is widespread in human cancers, particularly those of the female reproductive system2. Numerous therapies targeting PI3K/AKT pathway components have been developed and evaluated for their potential as cancer therapeutics, and some have been clinically approved, including the PI3Kα-specific inhibitor alpelisib (PIQRAY®) for ER+/HER2− breast cancer3. Because of its central role in mediating PI3K signaling and frequent hyperactivation across cancer types, the serine/threonine protein kinase AKT has become an attractive therapeutic target4–6. Several drugs targeting AKT have been developed and evaluated in clinical trials, including ATP-competitive, allosteric, and covalent pan-AKT inhibitors7–11. These inhibitors have yet to be approved for the treatment of cancer. Despite promising outcomes in some phase II and ongoing phase III clinical studies12, there is room to improve the therapeutic window of available AKT-targeting compounds. Moreover, conventional AKT inhibitors are largely cytostatic, not cytotoxic, thus failing to eradicate cancer cells as monotherapies. Consequently, there is an unmet need to identify more potent AKT-targeting drugs, in addition to uncovering cellular mechanisms that contribute to the efficacy of AKT inhibition.
Targeted protein degradation using small molecule degraders, also called PROTACs (proteolysis targeting chimeras), has emerged as a therapeutic modality and as a tool for the chemical depletion of proteins of interest13–16. In many cases, PROTACs display increased selectivity over the inhibitors from which they are designed, which presents advantages in limiting off-target toxicities17. Targeted protein degradation can also be used as a tool to understand network rewiring dynamics following near-complete and relatively acute depletion of the protein of interest. Potent and selective AKT-targeting PROTACs have been developed, with improved selectivity and potency over parental AKT inhibitors18–21.
Here, we report the development of a second-generation AKT degrader, INY-05–040, which selectively and rapidly (<5 h) degrades all three AKT isoforms and inhibits downstream signaling and cell proliferation in 288 cancer cell lines. Using a multiomics approach, combined with computational network modeling and experimental validation, we uncovered several degrader-selective cellular phenotypes in breast cancer cells, including potent activation of the stress mitogen activated protein kinase (MAPK) c-Jun N-terminal kinase 1 (JNK1). Additional breast cancer cell line analyses revealed that a signature of baseline JNK1 activation predicts lower sensitivity to AKT degradation, suggesting a potential biomarker for therapeutic stratification.
Results
INY-05–040 is an improved AKT degrader
We previously reported the development of an AKT-targeting degrader INY-03–041, a heterobifunctional degrader consisting of the catalytic AKT inhibitor GDC-0068 chemically linked to the Cereblon (CRBN) recruiter lenalidomide7. Despite the potency and selectivity of INY-03–041, this degrader exhibited relatively slow (12 h) cellular degradation kinetics for all three AKT isoforms20. We therefore developed an improved AKT degrader, INY-05–040, consisting of GDC-0068 chemically conjugated to a Von Hippel-Lindau (VHL) ligand with a ten-hydrocarbon linker (Fig. 1A). To generate the matched negative control compound INY-05–040-Neg (Fig. 1A), we incorporated a diastereoisomer of the VHL ligand that substantially loses activity towards VHL22. The biochemical selectivity of INY-050–040 was comparable to GDC-0068 across a panel of 468 kinases (Fig. S1A). A proteomic screen of the MOLT4 T lymphoblast cell line, which expresses all three AKT isoforms, confirmed pan-AKT downregulation following 4-h treatment with 250 nM INY-05–040 (Fig. S1B).
Figure 1. Design and characterization of INY-05–040.
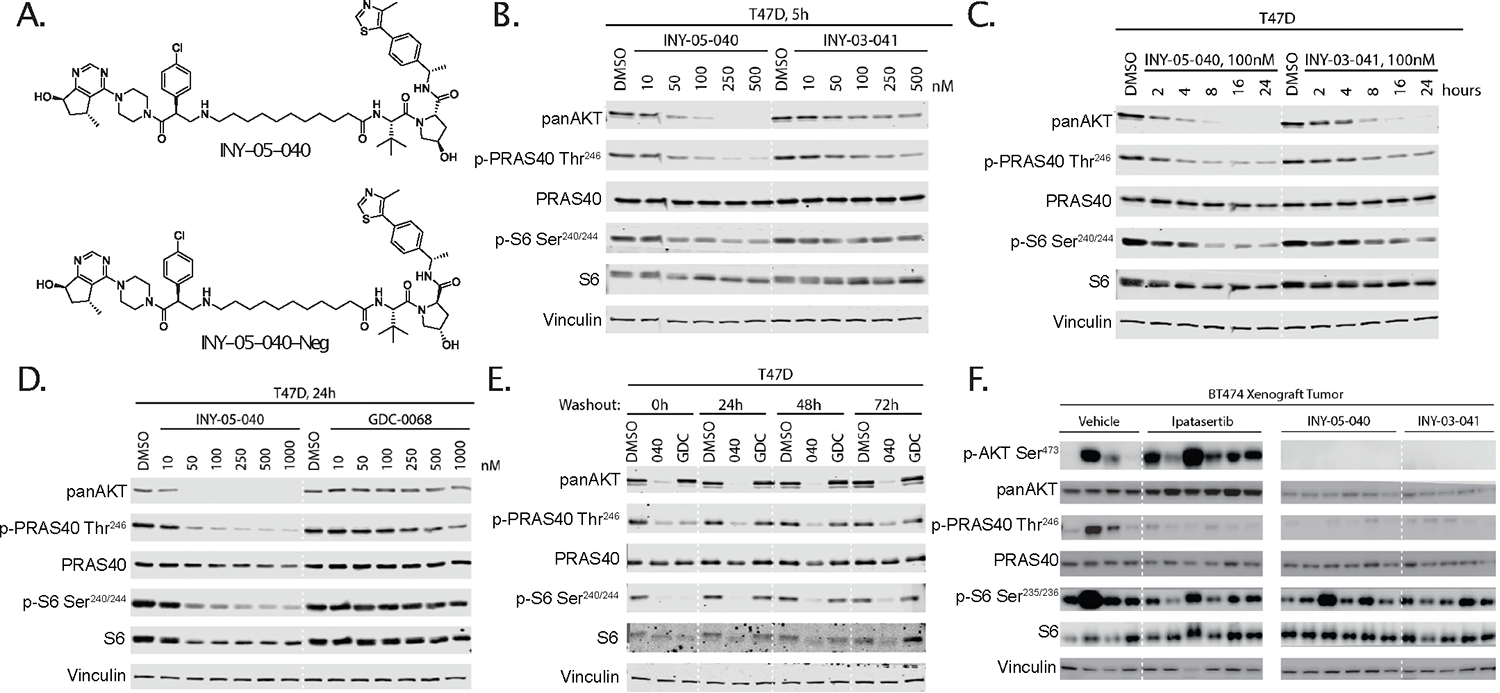
(A) Chemical structures of INY-05–040 and the negative control compound INY-05–040-Neg. (B) Immunoblots for pan-AKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-S6 (Ser240/244), total S6, and vinculin in T47D cells treated for 5 h with INY-05–040 or INY-03–041 at the indicated concentrations. Data are from a single experiment. (C) Immunoblots for pan-AKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-S6 (Ser240/244), total S6, and vinculin in T47D cells treated with INY-05–040 (100 nM) or INY-03–041 (100 nM) for the indicated times. Data are from a single experiment. (D) Immunoblots for panAKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-S6 (Ser240/244), total S6, and vinculin in T47D cells treated with INY-05–040 or GDC-0068 at the indicated concentrations for 24 hours. Data are from a single experiment. (E) Immunoblots for pan-AKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-S6 (Ser240/244), total S6, and vinculin in T47D cells treated with INY-05–040 (100 nM) or GDC-0068 (100 nM) for 5 h followed by washout for the indicated times. Data are from a single experiment. (F) Immunoblots for pan-AKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-S6 (Ser235/236), total S6, and vinculin in BT-474 mouse xenograft tumors treated with vehicle (10% DMSO, 25% kleptose), GDC-0068 (12.5 mg/kg), INY-05–040 (25 mg/kg), or INY-03–041 (25 mg/kg) for 3 days, with a terminal treatment 4 h prior to protein harvest. N = 4–6 mice per group as shown. Panels are from the same membrane but have been cropped for clarity, with a solid white line denoting the location of the crop. Additional supporting data related to this figure are included in Fig. S1.
All subsequent evaluation of INY-05–040 was performed as part of a screen of human breast cancer cell lines, due to the high prevalence of PI3K/AKT pathway activation. Exposure of the estrogen receptor-positive (ER+) and PIK3CAH1047R-mutant T47D cell line to increasing doses of INY-050–040 for 5 h (Fig.1B) or over a time course using a dose of 100 nM (Fig.1C) revealed an improved dose- and time-dependent reduction in total AKT levels compared to the first-generation degrader, INY-03–041. This was mirrored by suppression of downstream PRAS40 (Thr246) and S6 (Ser240/Ser244) phosphorylation (Fig.1B, 1C). INY-05–040 also outperformed GDC-0068 in T47D cells treated for 24 h, with >500 nM of GDC-0068 required to achieve comparable signaling suppression to that achieved with 50–100 nM INY-05–040 (Fig.1D). Because GDC-0068 is also a component of the negative control compound, INY-05–040-Neg, the latter suppressed signaling at higher concentrations (Fig. S1C), as expected. Unlike non-covalent, catalytic inhibition of AKT with GDC-0068, INY-05–040 treatment of T47D cells resulted in sustained AKT reduction and suppression of downstream signaling for at least 72 h following compound washout (Fig. 1E). Consistent with proteasome-dependent degradation, pharmacological abrogation of proteasomal function or neddylation prevented AKT degradation by INY-05–040 (Fig. S1F). We replicated these experiments in a screen of the PTEN-deficient triple-negative breast cancer (TNBC) MDA-MB-468 cell line (Fig. S1D, S1E, S1F, S1G), suggesting that the favorable cellular properties of INY-05–040 are generalizable across breast cancer cell lineages. Cells exposed to INY-05–040 also exhibited reduced levels of total ribosomal S6 protein, which was observed within the first 24 h of treatment as well as after compound wash-out (Fig. 1C, 1D, 1E, S1E). Consistent with rapid and long-term downregulation of AKT signaling, our second-generation AKT degrader INY-05–040 caused suppression of cell growth across four different breast cancer cell lines, at doses that were below those required for an equivalent response with catalytic inhibitors and lower or similar with respect to allosteric AKT inhibitors (Fig. S1H, S1I).
Furthermore, we tested the pharmacokinetic and pharmacodynamic properties of AKT degraders in vivo (Tables S1–S4). After 4 days of treatment in a BT-474C breast cancer xenograft model, both first- (INY-03–041) and second-generation (INY-05–040) degraders caused potent reduction in pan-AKT levels, which was accompanied by decreased phosphorylation of PRAS40 (Thr246) and pS6 (Ser240/244) (Fig. 1F). Likely due to incomplete AKT degradation in vivo, the observed suppression of downstream signaling was similar to that observed with GDC-0068. Together, these results show that INY-05–040 is a potent AKT degrader and inhibitor of downstream signaling output, outperforming both our first-generation AKT degrader and GDC-0068.
Multi-omic profiling reveals AKT degrader-selective responses
To identify mechanisms unique to AKT degradation relative to catalytic inhibition, we performed mRNA sequencing (RNAseq) analysis of T47D breast cancer cells that were treated for 5 or 10 h and grown in nutrient- and growth factor-replete cell culture media. To limit the confounding effect of differential potency, we determined the doses of INY-05–040 (100 nM) and GDC-0068 (500 nM) that resulted in comparable suppression of downstream signaling at these time points (Fig. S2).
Consistent with a shared target, the transcriptomes of GDC-0068- and INY-05–040-treated cells clustered closely together, separate from DMSO- and INY-05–040-Neg-treated controls, according to an unsupervised principal component analysis (PCA) (Fig. 2A). In agreement with the slower onset of AKT degradation, 5-h treatment with INY-05–040 resulted in differential abundance of only 194 transcripts (100 decreased, 94 increased; absolute fold-change ≥ 1.3), compared to 511 transcripts (249 decreased, 262 increased) with GDC-0068 during the same period (Fig. 2B). By contrast, after 10 h, INY-05–040 caused differential abundance of 1394 transcripts (626 decreased, 768 increased; absolute fold-change ≥ 1.3), whereas the extent of GDC-0068-induced transcriptional changes remained stable at 543 transcripts (243 decreased; 300 increased) (Fig. 2B). Across all differentially expressed transcripts after 10-h treatment, more than 700 were unique to INY-05–040, compared to less than 100 unique changes for GDC-0068 (Fig. S3A, S3B). No differential abundance was observed in response to treatment with the control compound INY-05–040-Neg (Fig. 2B).
Figure 2. Multiomic profiling of INY-05–040 and GDC-0068 in T47D breast cancer cells.
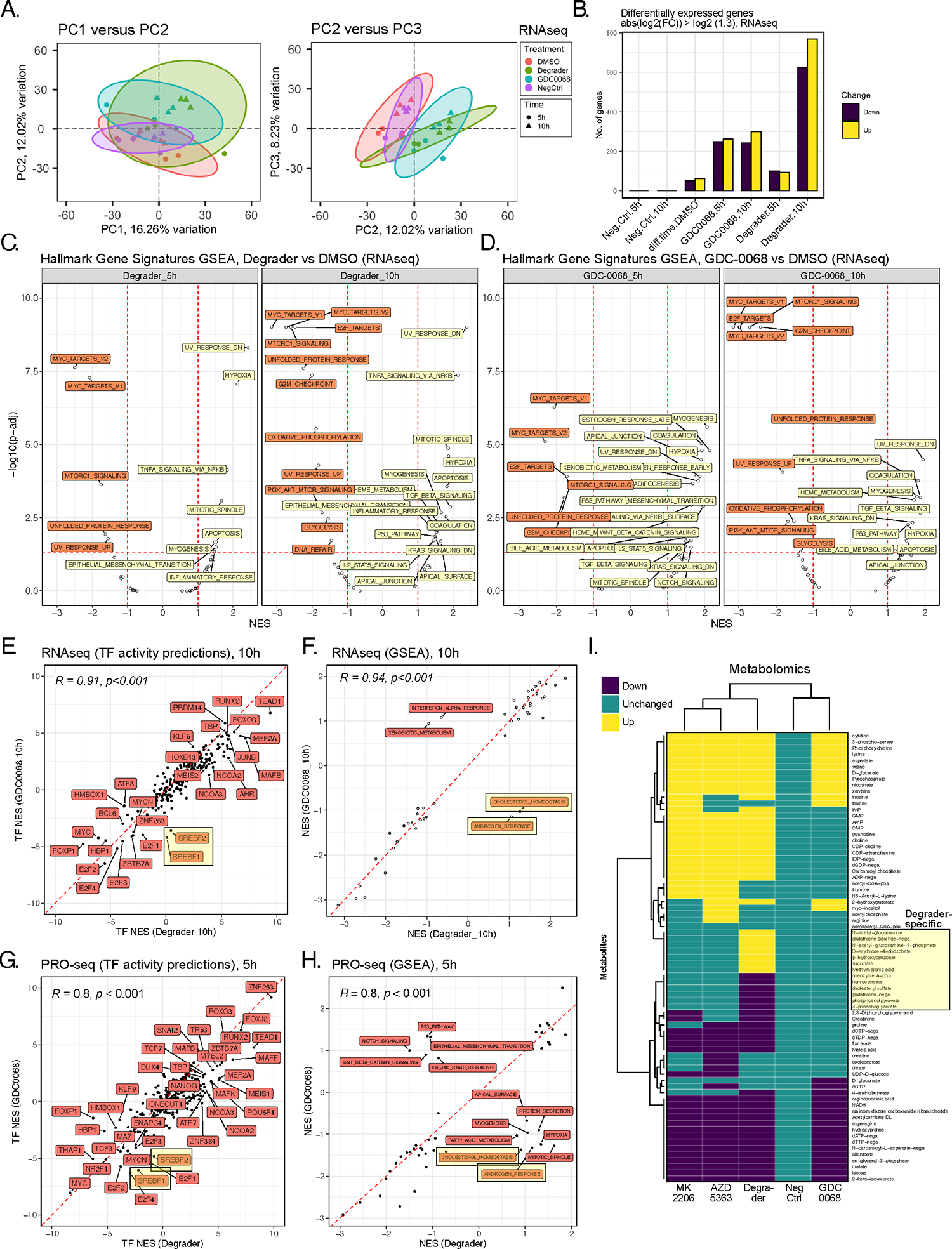
(A) Principal component analysis (PCA) projection of the transcriptomic dataset, comprising n=3 biological replicates per treatment (DMSO; degrader: 100 nM INY-05–040; 500 nM GDC-0068; NegCtrl: 100 nM INY-05–040-Neg) and time point (5 h and 10 h). Ellipses are drawn around each group at 95 % confidence level. The first three independent axes (PCs) of highest variation are shown. (B) Number of differentially up- and downregulated transcripts (absolute fold-change ≥ 1.3) following differential gene expression analysis (FDR ≤ 0.05) across the indicated comparisons. Comparisons are relative to the corresponding DMSO-treated control; for example, Neg.Ctrl.10h refers to the effect of 10 h treatment with INY-05–040-Neg vs 10 h treatment with DMSO. The exception is “diff.time.DMSO” which evaluates differential expression as a function of time in culture (treatment with DMSO for 10 h versus treatment with DMSO for 5 h). (C and D) Gene set enrichment analysis (GSEA) on the mSigDb HALLMARK collection, based on the ranked t values from all genes for the indicated treatments relative to the corresponding DMSO-treated controls. Gene sets are labelled if the absolute normalized enrichment score (NES) exceeds 1 and the adjusted p-value falls below 0.05 (FDR). (E) Spearman’s correlation analysis of transcription factor (TF) activity predictions from RNAseq data in cells treated for 10 h with either degrader or GDC-0068. TF footprint analyses were performed with DoRothEA. SREBF1 (protein name: SREBP1) and SREBF2 (protein name: SREBP2) activity predictions are highlighted due to their divergence between degrader and GDC-0068-treated cells, with lower activity predictions observed only in GDC-0068-treated cells. (F) Spearman’s correlation analysis of GSEA-derived NES for individual HALLMARK gene sets, based on RNAseq data from cells treated for 10 h with either degrader or GDC-0068. “CHOLESTEROL HOMEOSTASIS” and “ANDROGEN RESPONSE” hallmark gene sets are highlighted as having positive and negative NES in Degrader- and GDC-0068-treated cells, respectively. (G) Spearman’s correlation analysis of transcription factor (TF) activity predictions from PRO-seq data in T47D cells treated for 5 hours with either degrader and GDC-0068 relative to DMSO-treated control. TF activity predictions were calculated from t values from all genes following differential gene expression analysis (FDR ≤ 0.05; n = 2 biological replicates per treatment). (H) Spearman’s correlation analysis of GSEA-derived NES for individual HALLMARK gene sets, based on PROseq data from (G). (I) Hierarchical clustering (Euclidean distance) of differential metabolite abundance (FDR ≤ 0.05) following 24-h treatments of T47D with either AZD 5383 (capivasertib; catalytic pan-AKT inhibitor; 2 μM), degrader (INY-05–040; 100 nM), GDC-0068 (catalytic AKT inhibitor; 500–750 nM), MK2205 (allosteric pan-AKT inhibitor; 1 μM) or NegCtrl (INY-05–040-Neg; 100 nM). Differential abundance analysis was performed relative to DMSO-treated controls (n = 9 replicates per treatment, from 3 biological replicates with 3 technical replicates each). More than 85% of the observed differences in metabolite abundance for a given treatment corresponded to at least a 20% change relative to DMSO-treated cells. Metabolite levels that were changed only upon treatment with Degrader are highlighted. Additional supporting data related to this figure are included in Figs. S2, S3, S4.
We next conducted gene set enrichment analysis (GSEA) using the HALLMARK gene signature collection provided by the Broad Institute Molecular Signature Database (MSigDB)23. At 10 h, both INY-05–040 and GDC-0068 triggered a transcriptional footprint consistent with suppression of the cell cycle, glycolysis, oxidative phosphorylation, mTORC1 and the unfolded protein response (UPR) (Fig. 2C, 2D). Although 5-h treatment with GDC-0068 resulted in a larger number of distinct gene signatures with positive enrichment scores, most of these no longer reached statistical significance after 10 h (Fig. 2D), suggesting emerging adaptation to catalytic AKT inhibition. After 10-h treatment, positively enriched gene signatures were largely shared between degrader and catalytic inhibitor, but the underlying gene expression shifts were often more robust following AKT degradation, as evidenced by higher statistical significance despite equivalent sample size (Fig. 2C, 2D). Examples include gene signatures related to apoptosis, inflammatory signaling (including TNFα and NFκB) and the mitotic spindle (Fig. 2C, 2D).
We next used DoRothEA, a transcriptional footprint-based method featuring a curated gene regulatory network24, to predict differences in transcription factor regulation between INY-05–040 and GDC-0068 at 10 h. Overall, transcription factor activity predictions were highly concordant between the two compounds, with two notable exceptions. The lipid and sterol metabolism-regulating transcription factors, SREBP1 and SREBP2, were predicted as strongly inhibited upon catalytic AKT inhibition but not AKT degradation (Fig. 2E). A correlation analysis across the previously generated HALLMARK gene signature enrichments revealed a similar discordance with respect to cholesterol homeostasis and androgen response signatures (Fig. 2F). Of note, these two signatures shared four transcripts related to lipid and cholesterol synthesis: SCD, IDI1, HMGCR, and HMGCS1. Both HMGCR and HMGCS1 belong to the list of SREBP1 and SREBP2 targets whose mRNA levels were increased upon 10-h treatment with INY-05–040 but not GDC-0068 (Fig. S3C).
These findings were further supported by results from precision nuclear run-on sequencing (PRO-seq) analysis performed on T47D cells exposed to INY-05–040 or GDC-0068 for 5 h. PRO-seq analysis allows mapping of RNA polymerase active sites with base-pair resolution25, and changes in the expression of a transcript reflect immediate differences in active transcription, unlike RNAseq analysis, which captures steady-state mRNA levels. Similar to the bulk transcriptomes, PRO-seq datasets from degrader- and GDC-0068-treated samples clustered together and away from DMSO-treated controls by PCA (Fig. S3D). A substantially higher number of genes were differentially transcribed in response to AKT degradation (Fig. S3E, S3F), with further functional enrichment analyses supporting transcriptional regulation of SREBP1/2 and cholesterol homeostasis as defining differences between AKT degradation versus catalytic inhibition (Fig. 2G, 2H). Such activation of SREBP1 and SREBP2, despite potent AKT/mTORC1 inhibition, would be consistent with a phenotype of cholesterol depletion26.
Given evidence for altered metabolic homeostasis, we next assessed the metabolic profile of T47D cells treated with INY-05–040 and GDC-0068. For comparison, we also included an allosteric (MK-2206) and a second catalytic (AZD 5363) inhibitor. Treatments were performed for 24 h to allow capture of robust and persistent changes while minimizing the signaling rebound seen with GDC-0068 upon continuous treatment (Fig. S4). LC-MS-based metabolomics analysis showed that AKT degradation caused the largest number of differentially abundant metabolites (Fig. 2I). Many metabolite changes were shared across AKT-targeting compounds, especially MK-2206 and AZD 5363. Several nucleosides and their phosphorylated derivatives had increased in abundance, including inosine, guanosine, IMP, GMP, AMP and CMP. Metabolite changes unique to treatment with INY-05–040 included intermediates of the hexosamine biosynthesis pathway, the pentose phosphate pathway, glycolysis, the tricarboxylic acid cycle, glutathione and cholesterol metabolism (Fig. 2I). Only AKT degradation caused increased levels of methylmalonic acid (MMA), which is a potent inhibitor of the rate-limiting cholesterol biosynthesis enzyme HMGCR27. MMA accumulates if vitamin B12 levels are too low relative to the catabolism of branched chain amino acids and odd chain fatty acids28. Together, this multiomic approach supports a widespread perturbation of cellular homeostasis in breast cancer cells treated with INY-05–040, with distinct responses to AKT degradation pertaining to cholesterol homeostasis.
COSMOS analysis identifies altered stress MAPK signaling downstream of AKT degradation
We next reasoned that an integrated, transomic integration of the previous datasets may enable us to generate testable mechanistic hypotheses regarding previously unknown signaling changes downstream of AKT degradation. We applied the COSMOS (causal oriented search of multi-omic space) network analysis approach 29 to integrate transcriptomic and metabolomic datasets following treatment with the AKT degrader INY-05–040 or GDC-0068 for 10 h and 24 h, respectively (Fig. 3A). Briefly, COSMOS relies on an extensive prior knowledge network (PKN) of signaling pathways, transcriptional regulation and metabolic reactions, in combination with an integer linear programming (ILP) optimization strategy to identify the smallest coherent subnetwork causally connecting as many deregulated transcription factors and metabolites in the input data as possible29,30. Input data to COSMOS consisted of the background transcriptome of T47D cells, in addition to treatment-specific DoRothEA-derived transcription factor activity predictions and differentially abundant metabolites. The resulting networks enable identification of top degree signaling nodes or “hubs”, which are essential for holding a network together due to their high number of connections31. Replicate COSMOS runs identified MAPK1 (also known as ERK2) and/or MAPK3 (also known as ERK1) as top degree nodes in both INY-05–040 and GDC-0068 networks (Fig. 3B, 3C; S5A, S5B), consistent with the known compensatory RAS/MAPK signaling activation that follows potent PI3K/AKT pathway inhibition32,33. Focusing on unique differences, we noted that the stress MAPKs, MAPK8 (also known as JNK1) and MAPK14 (also known as p38α), were among the top degree nodes in the INY-05–040-specific networks (Fig. 3B). MAPK14 was identified as a top degree node in 10 out of 11 COSMOS runs with INY-05–040 input data but was never a top degree node in any of the eight COSMOS runs performed with GDC-0068 input data (Fig. 3B, 3C). In two out of eight GDC-0068-specific networks, MAPK14 was not part of the final network; in the remaining six, it had a maximum of two connections per network, suggesting a minor role for this kinase in the cellular response to GDC-0068.
Figure 3. COSMOS-based integration of transcriptomic and metabolomic datasets to identify treatment-specific networks.
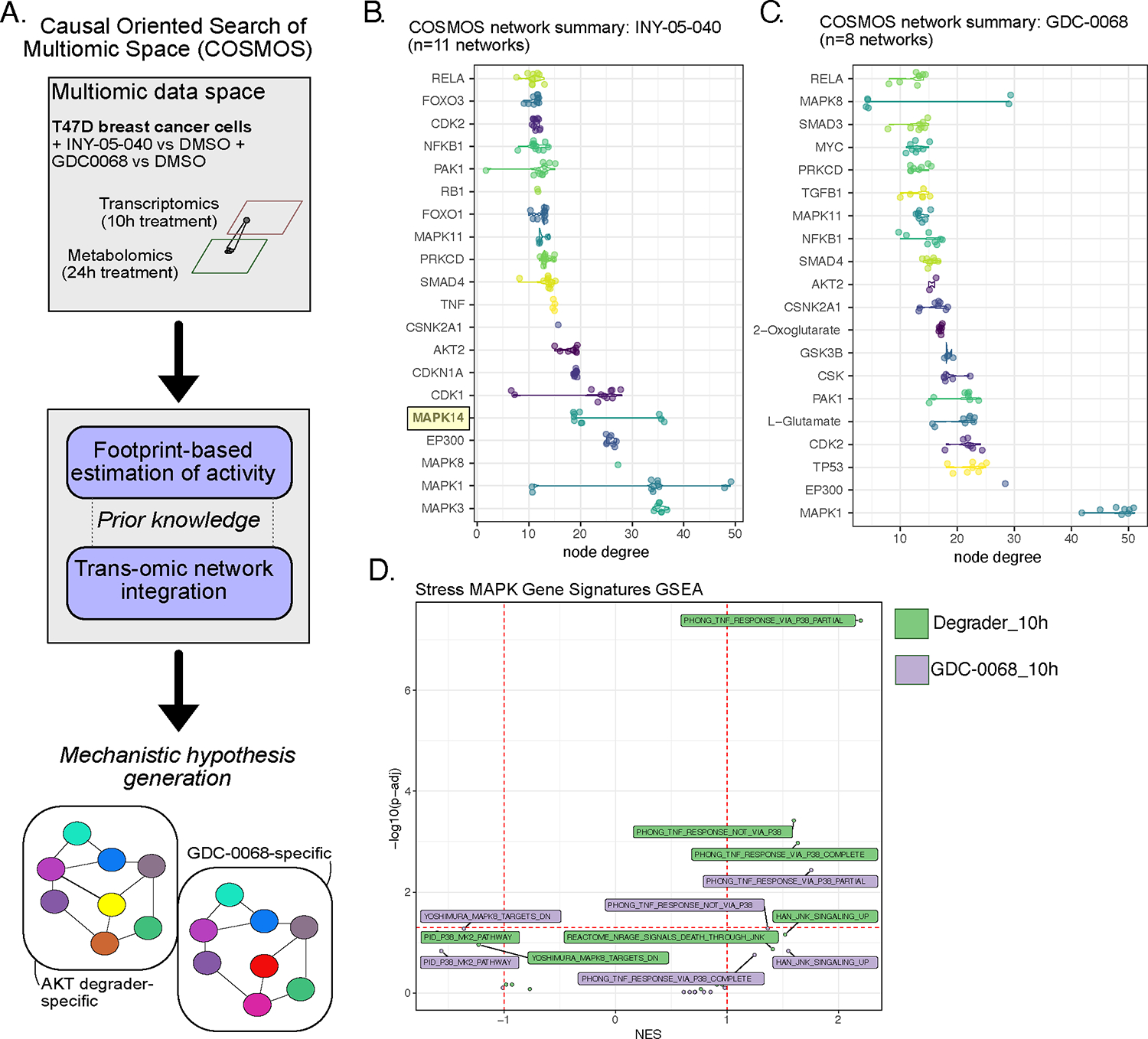
(A) Schematic illustrating the principle of COSMOS and the datasets used for multiomic integration and predictions of treatment-specific signaling networks. (B and C) Top degree nodes from degrader- and GDC-0068-specific networks plotted in increasing order. MAPK14 (protein: p38𝛂) is highlighted as a degrader network-specific top degree node. The raw COSMOS networks are included in Fig. S5 (n = 11 independent runs using degrader data; n = 8 independent runs for GDC-0068 data). (D) Complementary GSEA analyses using stress MAPK-related gene sets (mSigDb C2 collection), based on the ranked t values from all genes for the indicated treatments relative to the corresponding DMSO treatment. Gene sets are labelled if the absolute normalized enrichment score (NES) exceeds 1.
To corroborate these findings, we next retrieved all MSigDb curated gene sets (C2 collection) featuring transcriptional changes downstream of JNK/p38 perturbation and performed GSEA using the RNAseq dataset. Three gene signatures related to TNFα signaling were positively and significantly enriched in INY-05–040-treated T47D cells after 10 h, with two of the signatures representing transcriptional changes that are either completely or partially dependent on p38 (Fig. 3D). These signatures originated from a study examining the response of lung cancer cells to TNFα in the presence or absence of the pan-p38 inhibitor LY47975434. Only one of the two p38-dependent signatures were significantly enriched for with a positive score in GDC-0068-treated cells; however, neither the significance nor the magnitude of enrichment were as strong as that observed in INY-05–040-treated cells (Fig. 3D). This is also consistent with a much weaker enrichment of the hallmark gene signature “TNFA_signaling_via_NFκB” in response to 10-h treatment with GDC-0068 compared to INY-05–40 (Fig. 2C, 2D). Together, these integrated analyses point towards potent AKT degradation-induced activation of stress MAPK signaling and inflammatory gene signatures.
Activation of stress MAPK signaling in response to AKT degradation
To validate the COSMOS predictions, we screened the kinetics of p38 and JNK signaling over a time course in a panel of breast cancer cell lines (Fig. 4A–C, S6A–C). Cells exhibited distinct p38 and JNK signaling kinetics and magnitude in response to INY-05–040 compared to GDC-0068. Consistently, AKT degradation resulted in more robust induction of p38α phosphorylation (Fig. 4A–C, Fig. S6A–C), supporting the COSMOS-based prediction of differential activity at the level of p38 (also known as MAPK14) when comparing INY-05–040- and GDC0068-specific networks (Fig. 3B, 3C).
Figure 4. Validation of COSMOS-generated prediction of MAPK stress kinase signaling.
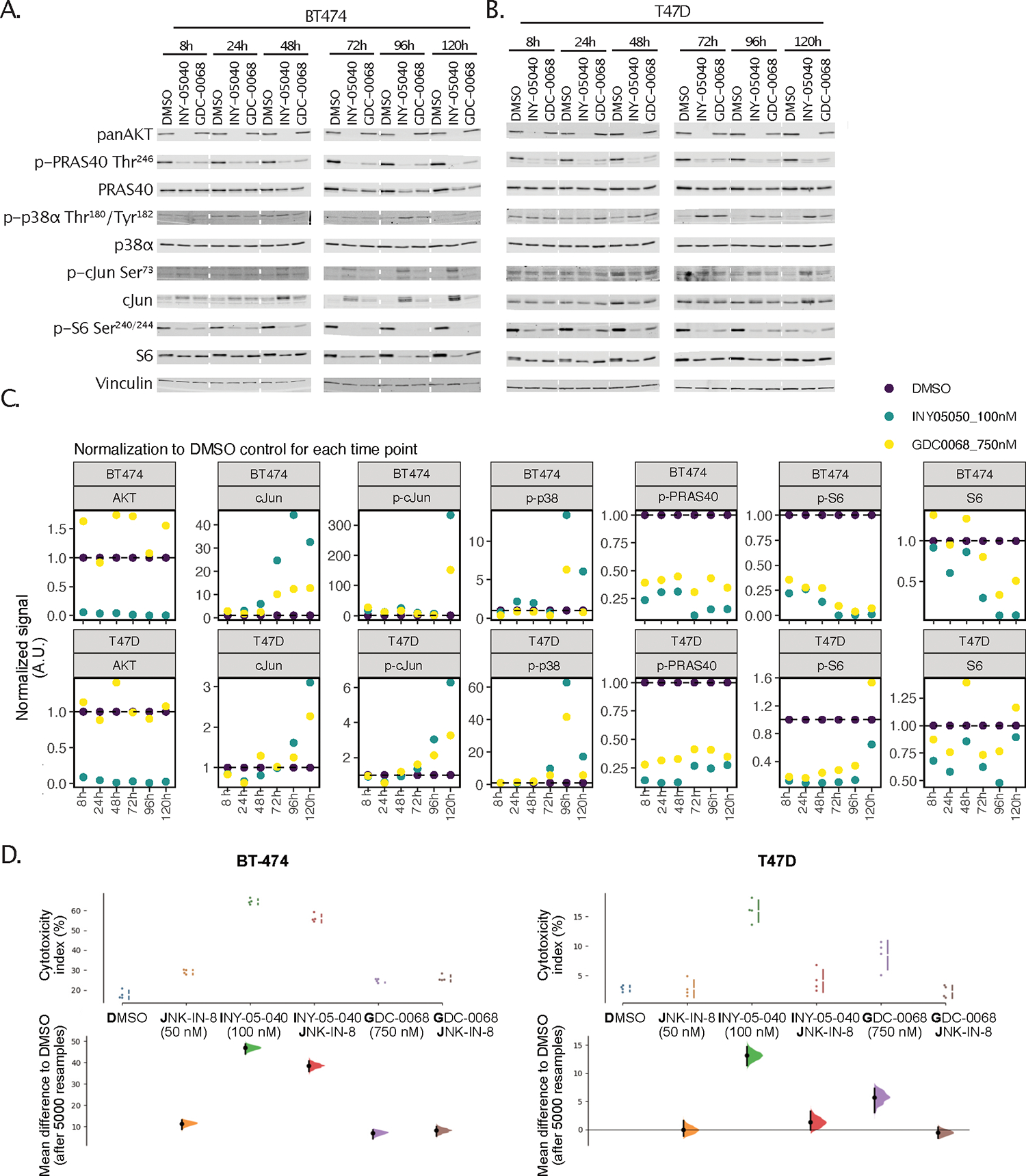
(A and B) Immunoblots for panAKT, phospho-PRAS40 (Thr246), total PRAS40, phospho-p38α (Thr180/Tyr182), total p38α, phospho-c-Jun (Ser73), total c-Jun, phospho-S6 (Ser240/244), total S6, and vinculin after treatment of BT-474 (A) or T47D (B) cells for the indicated times with DMSO, 100 nM INY-05–040, or 750 nM GDC-0068. Data are from a single experiment. (C) Quantification of total AKT (normalized to vinculin), c-Jun (normalized to vinculin), phospho-c-Jun (pJun) Ser73 (normalized to vinculin), phospho-p38 (pP38) Thr180/Tyr182 (normalized to total p38), phospho-PRAS40 (pPRAS40) Thr246 (normalized to total PRAS40), phospho-S6 (pS6) Ser240/244 (normalized to vinculin) and total S6 (normalized to vinculin), including normalization to the respective DMSO control signal for each time point and cell line. Note that phospho-c-Jun and phospho-S6 were normalized to vinculin given changes in the levels of the respective total proteins. Stippled white lines are added to aid interpretation of samples loaded on the same membrane; white blocks separate samples run on different membranes. Supporting data for additional cell lines (MCF7 and MD-MB-468) are included in Fig. S6. (C) Cytotoxicity index assayed using CellTox Green, in BT-474 or T47D cells treated for 24 h with either DMSO or 50 nM JNK-IN-8, followed by 120-h co-treatment with either DMSO, INY-05–040 (100 nM) or GDC-0068 (750 nM). The cytotoxicity index represents cytotoxicity values corrected for background fluorescence and normalized to total signal following chemical permeabilization (used as proxy measure for total cell number). The data are displayed as Cumming plots following bootstrap-coupled estimation of effect size for each condition relative to DMSO. The upper plots display the raw data alongside standard deviations indicated with gapped lines. The plots beneath display the estimated effect sizes, sampling distribution and bootstrap 95% (percentile) confidence intervals. For accurate interpretation, please note differences in y-axis scaling. The data are from a single experiment performed with four technical replicates per condition; data from a separate experimental replicate, including a JNK-IN-8 dose curve, are in Fig. S7.
In the screen of BT-474 and T47D breast cancer cells, INY-05–040 induced sustained phosphorylation of the JNK target cJun at Ser73, as well as increased total c-Jun protein levels, which is a marker for JNK activation35 (Fig. 4A–C). Particularly at later time points (>96 h), BT-474 cells responded with a near-binary difference in stress MAPK activation in response to AKT degradation compared to catalytic inhibition (Fig. 4B, 4C). We therefore tested whether induction of stress MAPK signaling was associated with AKT degrader-induced cytotoxicity. BT-474 and T47D cells were pre-treated with a low-dose (50 nM) of the covalent JNK1/2/3 inhibitor JNK-IN-8 for 24 h, then with either GDC-0068 or INY-05–040 for another 120 h. The two cell lines were chosen for screening as models for a potent (BT-474) versus modest (T47D) cytotoxic response to AKT degradation and a substantially lower magnitude of GDC-0068-induced cell death (fig. S7A). Consistently, the INY-05–040-induced cytotoxic response in T47D cells was neutralized by JNK inhibition (Fig. 4D, Fig. S7A). In BT-474 cells, however, combined AKT degradation and JNK inhibition only led to a small, partial rescue of cytotoxicity (Fig. 4D, Fig. S7A); the increased levels of cleaved PARP, a marker of apoptosis, in BT-474 cells treated with AKT degrader were also not reduced by co-treatment with JNK-IN-8 (Fig. S7B). We therefore conclude that although sustained activation of stress MAPK correlates with INY-05–040-induced toxicity, complete ablation of this mechanism is not sufficient to rescue cell viability, suggesting the existence of other contributing factors.
Global cell line screening identifies stress MAPK-associated resistance biomarkers
Given the improved cellular potency of INY-05–040, including robust downstream transcriptional and metabolic changes, we next undertook global cancer cell line profiling to determine whether INY-05–040 causes more potent growth suppression relative to GDC-0068 and the first-generation AKT degrader INY-03–041. Across 288 cancer cell lines, spanning a total of 18 different cancer lineages, INY-05–040 exhibited superior growth-inhibitory activity (Fig. S8A). This was based on calculation of the drug concentration required to reduce overall growth by 50% (GI50adj, Fig. 5A), which includes adjustment for cell number at the start of the assay36. Although GI50adj calculation was possible for all cell lines treated with the second-generation degrader and for 282 cell lines treated with the first-generation degrader, it was not possible for 161 cell lines treated with GDC-0068 due to lack of sufficient growth suppression (Fig. S8A). Consequently, the median GI50adj value for GDC-0068 in our screen was higher than 10 μM, compared to 1.1 μM for INY-05–040 and 3.1 μM for INY-03–041.
Figure 5. Integration of cell line screen data with publicly available omics datasets to identify sensitivity biomarkers for INY-05–040.
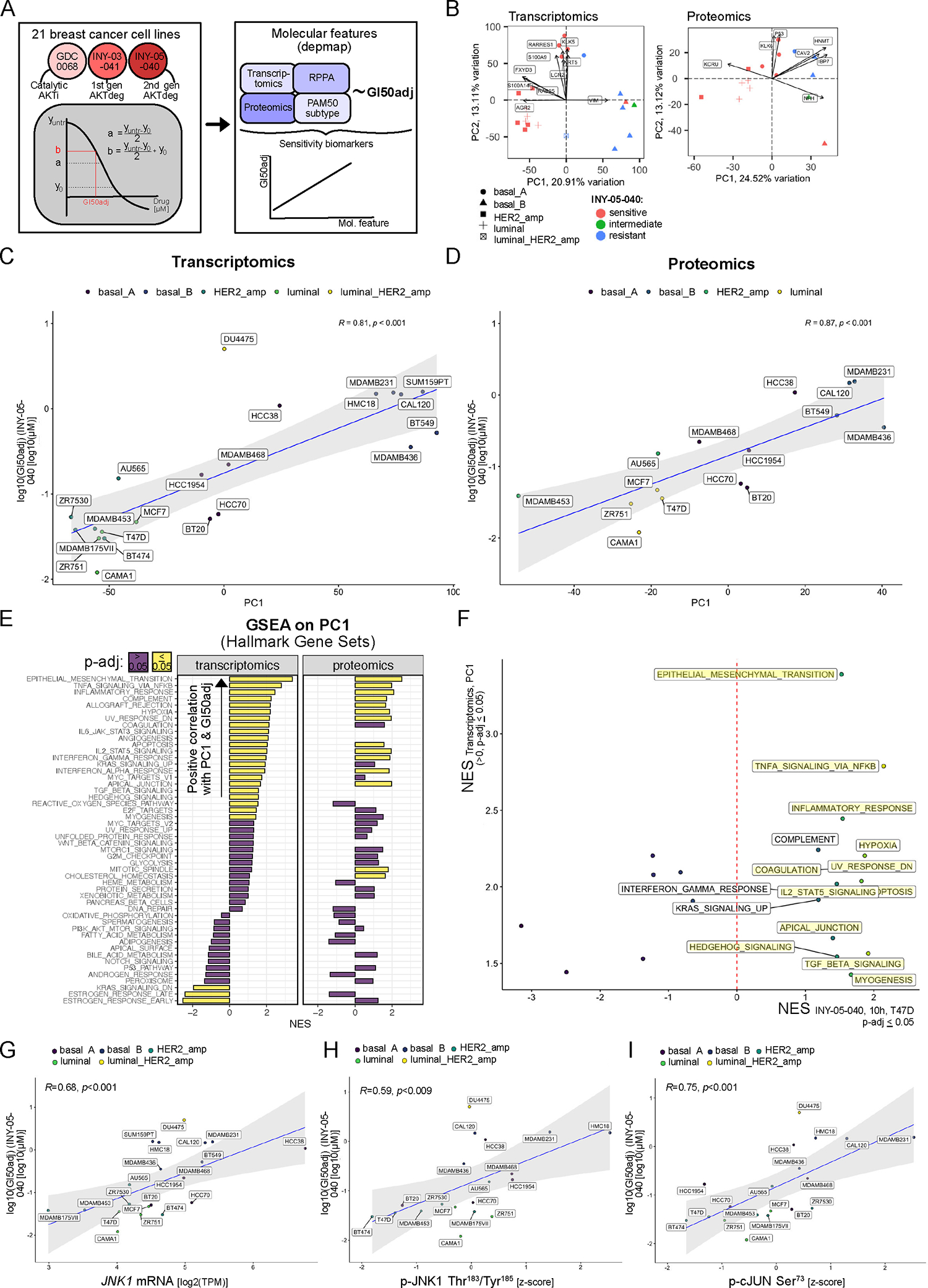
(A) Analytical workflow for cell line screen processing and subsequent integration of the growth response metric (GI50adj) with publicly available cell line omics data from the DepMap project. A total of 288 cancer cell lines were profiled with GDC-0068, INY-03–41 and INY-05–040, with the full set of responses included in Fig. S8A. Subsequent integrative analyses focused on breast cancer cell lines only. Note that the applied growth response metric (GI50adj) takes into account cell line growth which is a known confounder in drug sensitivity measurements67. The final output corresponds to the concentration of drug that results in 50 % cell growth inhibition. (B) PCA on breast cancer-specific transcriptomics and proteomics data, with coloring according to sensitivity to INY-05–040 (sensitive: GI50adj < 0.5 μM; intermediate: 0.5 μM ≤ GI50adj ≤ 1 μM; resistant: GI50adj > 1 μM; see also Fig. S8B). The PAM50 subtype of each cell line is specified by shape. Transcripts and proteins contributing the most to the observed data structure alongside PC1 and PC2 are labelled. (C and D) Spearman’s correlation analysis of PC1 values for each cell line and the corresponding GI50adj value for INY-05–040. A linear regression line with 95% confidence intervals (shaded area) is included in each analysis, demonstrating that cell line-specific PC1 scores can be used as proxy measures for INY-05–040 sensitivity (meaning the higher the PC1 score, the more resistant the cell line). (E) GSEA (mSigDb HALLMARK gene sets) using transcript and protein loading values alongside the respective PC1, a proxy measure for sensitivity to INY-05–040; FDR ≤ 0.05. NES: normalized enrichment score. (F) A plot of all gene sets that were significantly and positively enriched across PC1 loadings from the DepMap transcriptomic data, and the corresponding NES from the T47D dataset following 10 h treatment with INY-05–040 (see also Fig. 2). Highlighted gene signatures were also statistically significant (FDR ≤ 0.05) in the T47D dataset. (G to I) Spearman’s correlation analysis of JNK1 mRNA expression (G), pJNK1 (T183/Y187) (H) and p-cJun (S73) with the cell line-specific GI50adj value for INY-05–040. A linear regression line with 95% confidence intervals (shaded area) is included in each analysis. Reverse phase protein phosphorylation (RPPA) data were obtained from the DepMap project and subset for the signals of interest. Additional supporting data related to this figure are in Figs. S8 and S9.
To identify functional biomarkers predictive of sensitivity to INY-05–040 in the 21 breast cancer cell lines profiled, we took advantage of the measured GI50adj values and the corresponding baseline transcriptomic, proteomic and reverse phase protein array (RPPA) data publicly available through the Cancer Dependency Map project (Fig. 5A)37,38. We classified breast cancer cells into sensitive, intermediate, and resistant if the measured GI50adj was less than 0.5 μM, between 0.5 to 1 μM, and higher than 1 μM (Fig. S8B), respectively. Subsequent unsupervised PCA using either transcriptomic or proteomic datasets revealed a separation of INY-05–040-sensitive from -resistant breast cancer cells, which was not simply driven by ER expression as assessed by PAM50 status (Fig. 5B). Except for one mixed-subtype HER2-amplified-luminal breast cancer cell line (DU4475), all examined HER2-amplified and luminal breast cancer cells were sensitive to INY-05–040. This sensitivity was also observed for 4 out of 5 breast cancer cells belonging to the basal A subtype. By contrast, only 1 out of 6 basal B breast cancer cell lines were sensitive to INY-05–040, with 4 out of 6 exhibiting overt resistance (Fig. 5B).
Using the PC1 loadings from the transcriptomic and proteomic data, we then correlated these to the measured GI50adj values. This revealed strong and statistically significant correlations for either comparison, with higher PC1 loadings associated with higher GI50adj values and thus resistance to INY-05–040 (Fig. 5C, 5D). To identify the underlying molecular features, we performed GSEA on the two PC1 loadings (transcriptomic and proteomic data). Gene sets that were positively enriched for alongside either PC1 were highly concordant and characterized by strong enrichment for epithelial mesenchymal transition and inflammatory signaling (Fig. 5E). Most of these positive enrichments overlapped with those observed upon acute 10-h treatment of T47D breast cancer cells with INY-05–040 (Fig. 5F). Based on our mechanistic data on acute JNK activation and sensitivity to INY-05–040, we reasoned that the correlation between inflammatory gene signatures and INY-05–040 resistance in the breast cancer cell panel may reflect an already high baseline JNK activation and thus stress MAPK signaling. Accordingly, we found that both JNK1 mRNA levels (Fig. 5G), JNK1 phosphorylation (Thr183/187) (Fig. 5H) and cJun phosphorylation (Ser73) (Fig. 5I) exhibited a positive and statistically significant correlation with INY-05–040 GI50adj values.
The BT-474 breast cancer cell line, which exhibits a strong cytotoxic response to INY-05–040 (Fig. 4C, S7A), had the lowest GI50adj value and the lowest values for markers of baseline JNK1 activation, followed by T47D cells (Fig. 5H, 5I). The AKT degrader-induced cell death response in T47D cells was modest compared to that in BT-474 cells and nearly on par with that observed following treatment with GDC-0068 (Fig. 4C and S7A). Thus, the relative cytotoxic response to INY-05–040 correlated with the GI50adj-based sensitivity rankings predicted by baseline protein-level measurements of JNK1 activity markers. To test this relationship, we next applied INY-05–040 and GDC-0068 to two distinct breast cancer cell lines, HCC-1395 (TNBC, with loss of PTEN) and HCC-1143 (TNBC, with amplification of AKT1), that were not part of the initial breast cancer cell line screen. Based on available RPPA data, the two cell lines ranked higher than T47D for baseline JNK1 activation (Fig. S9A), and our model would therefore predict a low cytotoxic response to AKT degradation/inhibition, with a lower magnitude relative to T47D cells (Fig. 4C, S7A). Consistent with this prediction, both HCC-1395 and HCC-1143 exhibited minimal cytotoxicity to either treatment (Fig. S9B,S9C), and failed to induce further stress MAPK signaling relative to a higher baseline (Fig. S9D). Together, these data demonstrate superior potency of INY-05–040-induced AKT degradation over AKT catalytic inhibition across cancer cell lines, with evidence for a cytotoxic response in the context of low baseline yet potent and sustained induction of stress MAPK signaling in breast cancer cells.
Discussion
Targeted protein degradation has emerged as both a therapeutic approach and a powerful experimental tool to evaluate the effects of acute protein depletion on cellular networks. Here we have reported the development of a potent and highly selective second-generation pan-AKT degrader, INY-05–040, which we used as a tool to uncover AKT biology. Using a multiomic approach in breast cancer cell models, we found that AKT degradation led to distinct transcriptomic and metabolomic changes, suppression of downstream AKT signaling, concomitant with activation of stress MAPK signaling. Furthermore, in a set of breast cancer cell lines, low baseline levels of JNK activation were associated with increased sensitivity to AKT degradation.
The ongoing search for targeted agents to treat patients with PI3K pathway hyperactivation has focused on the identification of more selective compounds and effective combinations to limit toxicity and improvements in patient selection39. The PI3Kα-selective inhibitor alpelisib (PIQRAY®) is approved for the treatment of advanced hormone receptor-positive, HER2-negative breast cancer, in combination with the ER antagonist fulvestrant40. Alpelisib (VIJOICE®) is also approved for the treatment of developmental overgrowth disorders collectively known as PIK3CA-related overgrowth spectrum (PROS)41,42. Despite this progress, more treatment options are urgently needed for both cancers and diseases of PI3K pathway activation to address issues of resistance and/or poor tolerability. Independent lines of evidence, including the current study, indicate that targeted protein degradation of PI3K pathway components may represent a distinct therapeutic strategy, with the added benefit of sustained inhibition of downstream signaling18,43,44. This property may partly be explained by the inability of various negative feedback mechanisms within the PI3K/AKT pathway to overcome inhibition when a critical downstream transducer is absent. Prolonged cellular stress can also suppress AKT/mTORC1 activity, alongside a more complete shutdown of protein translation, which may contribute to a self-sustained feedforward loop of continued suppression of AKT signaling despite removal of the AKT degrader. This hypothesis is supported by washout screen experiments in which pathway reactivation was not observed or remained low for at least 72 h after degrader removal, in contrast to the corresponding findings with catalytic AKT inhibition with GDC-0068 (Fig. 1E, Fig. S1G).
Using a network biology framework, COSMOS, we demonstrated how systematic integration of a prior knowledge with context-specific transcriptomic and metabolomic data can be used to identify and subsequently test mechanistic hypotheses on AKT degradation-selective signaling outcomes. This approach identified the stress MAPKs, p38α (which is encoded by MAPK14) and JNK1 (which is encoded by MAPK8), as differentially activated in breast cancer cells treated with the AKT degrader INY-05–040. The observed quantitative differences would have been challenging to resolve with conventional approaches, emphasizing the power of computational integration of multiomics data and temporal analyses.
The involvement of stress MAPK and inflammatory signaling in the cellular response to AKT degradation was further supported by integration of growth inhibition measurements with publicly available omics data. The observation that the same transcriptional and signaling signatures induced upon degrader treatment of T47D cells were already elevated at baseline in breast cancer cell lines with lower sensitivity to INY-05–040 suggests that low baseline stress MAPK and inflammatory signaling activity may be a prerequisite for potent cell growth suppression following AKT degradation. Our data furthermore suggest that a low baseline yet strong and sustained stress MAPK activation upon AKT degradation predicts cytotoxicity in response to AKT degradation.
At present, the precise mechanistic link between AKT degradation and stress MAPK activation remains undescribed. We speculate that ribosomal stress may contribute to the induction of stress MAPKs because AKT and mTORC1 promote ribosome biogenesis through transcriptional and translational mechanisms. Conversely, disruption of any given step in ribosome biogenesis causes ribosomal stress45. Accordingly, AKT degradation but not catalytic inhibition leads to a potent and sustained reduction in total ribosomal S6 protein, which would be consistent with the low stability of ribosomal proteins in the absence of functional ribosome formation45,46. Aberrant cholesterol metabolism may also contribute to the cellular stress observed upon AKT degradation. Low cholesterol is linked to increased NFκB activation and cell death in fibroblasts through a p38 MAPK-dependent mechanism47,48; accordingly, activating transcriptomic signatures for both inflammatory and stress MAPK pathways were strongly enriched for in AKT degrader-treated cells. Additional studies are required to understand this putative crosstalk.
Several other AKT degraders have been developed to date, including the VHL-recruiting AZD5363-based AKT degrader MS2118. Like INY-05–040, MS21 also outperformed its parental AKT kinase inhibitor in cancer cell growth and signaling assays18. Additional side-by-side comparisons of MS21 and INY-05–040 are needed to determine whether both compounds share similar cellular mechanisms of action downstream of AKT degradation, given the subtle but important differences in their biochemical profiles. The six cell lines identified as more sensitive to AKT degradation with MS21 compared to inhibition with AZD5363 all had lower than average levels of phosphorylated JNK as measured by RPPA18, consistent with our results. In summary, we demonstrate improved suppression of cancer cell growth with a potent second-generation AKT degrader and illustrate how protein degraders, in combination with integrated systems-level analyses, can be used to uncover new biology of a widely-studied signaling kinase.
MATERIALS AND METHODS
A complete list of all reagents used in this work is included in the Supplementary Materials (Tables S5–S9).
Biochemical Selectivity Assay
Biochemical selectivity across 468 kinases was measured through the scanMAX kinase assay panel provided through Eurofins Discovery.
Cell Culture
BT-474, T47D, MCF-7, MDA-MB-468, HCC1143, and HCC1395 cells were obtained from ATCC and cultured in RPMI media supplemented with 10% heat inactivated fetal bovine serum without antibiotics at 37 °C in the presence of 5 % CO2. Cells were maintained in Corning TC-treated 15 cm culture dishes (Corning Cat. # 08–772-24) in 20 mL medium. Medium was replenished every 3 days, until cells reached 70–90% confluence. To passage, cells were washed once with 10 mL PBS and incubated for 5–10 min at 37 °C with 0.25% Trypsin 0.1% EDTA and passaged up to 5 times in the same dish. Cells were maintained in culture for up to one month. Cells were routinely tested for mycoplasma using a Mycoplasma Detection Kit (Lonza Cat. # LT07–218).
Cell Death Experiments
T47D and BT-474 cells were plated at 4,000 or 6,000 cells/well in 80 μl RPMI + 10% FBS medium in black-walled clear-bottom 96-well plates (Fisher Cat # 12–566-70). Medium was exchanged the following day with 90 μl medium plus 10 μl of drug containing medium for a 24-h pre-treatment. Two days after plating, medium was exchanged with 80 μl complete medium plus 20 μl of drug containing medium. After an additional three days, 100 μl of drug-containing medium was replenished without removing existing medium to prevent nutrient depletion until assay endpoint.
HCC1143 and HCC1395 cells were plated at 2,000 or 6,000 cells//well in 80 μl RPMI + 10% FBS medium in black-walled clear-bottom 96-well plates (Fisher Cat # 12–566-70). The following day, medium was exchanged with 90 μl complete medium plus 10 μl of drug containing medium. After an additional three days, 100 μl of drug-containing medium was replenished without removing existing medium, to prevent nutrient depletion until assay endpoint, for a total of 120 hours compound treatment.
Cellular Signaling
Depending on the length of the experiment, cells were plated at 150,000–250,000 cells/mL (MDA-MB-468), 200,000–300,000 cells/mL (BT-474, T47D) in RPMI medium with 10% serum at 2 mL per well in 6-well treated tissue culture plates (Greiner, Cat. # TCG-657160) and incubated overnight. The next day, medium was exchanged, and cells were treated with the indicated compounds at the appropriate concentration and protein lysates were harvested at the times specified. Time courses were conducted in reverse by drugging cells for the longest time point first, which was followed by drugging cells for the shorter time points such that all samples were collected at the same time. At the time of harvest, cells were washed once with 2 mL of ice-cold PBS and either snap frozen on dry ice and stored at −80 °C or harvested immediately.
RNAseq analysis
Cells were plated at 300,000 cells/mL in RPMI medium with 10% serum at 2 mL per well in 6-well treated tissue culture plates (Greiner, Cat. # TCG-657160) to achieve 75% density the following day and incubated overnight. The following day, media stocks containing indicated compounds were prepared and used to treat all conditions at respective time points, stored at 4°C between treatments. After 5 h and 10 h of treatment, wells were washed once with 2 mL ice cold PBS and aspirated completely, snap frozen on dry ice, and stored at −80 °C until all replicates were collected. Three independent biological replicates were plated on sequential days. In parallel, samples were also collected for protein harvest for confirmation of consistent drug effect on cellular signaling.
PRO-seq analysis
T47D cells were seeded at 8 × 106 cells per 15 cm plates (Corning Cat. # 08–772-24) in 15 mL medium for PRO-seq samples or at 3 × 106 cells per 10 cm plates (Westnet Cat. # 353003) in 8 mL medium for protein samples to achieve 70% confluence. The following day, a stock of drug-containing medium was prepared and used to treat technical triplicates for each condition. Technical triplicate protein replicates treated the same way were collected in parallel. Protein plates were washed once with 8 mL of ice-cold PBS and snap frozen on dry ice, then stored at −80 °C until all replicates were collected.
Metabolomics analysis
Cells were plated at 2 × 106 cells/plate in RPMI medium with 10% in 3 mL per plate in 60 mm treated tissue culture plates (Corning, Cat. # 430166) and incubated overnight. The next day, stocks of medium were prepared containing the indicated compounds at the appropriate concentration, and medium was exchanged for drug-containing medium. Three independent biological replicates were performed for metabolomics experiments, each comprising technical triplicates for metabolite plates and technical duplicate of parallel protein samples used to assess suppression of signaling and normalize metabolite levels to total protein content. Due to an apparent loss of potency, the dose of GDC-0068 was increased to 750 nM in Trial 3, compared to 500 nM in Trials 1 and 2, to ensure consistent biochemical signaling suppression across all runs.
Proliferation Assays
T47D, MDA-MB-468, MCF-7 or BT-474 cells were plated in 384 well plates at 250 cells per well. After 24 hours, cells were treated with GDC-0068, AZD5363, MK-2206, ARQ-092, INY-03–041, INY-05–040, INY-05–040-Neg, or VH032 compounds at the indicated concentrations for 72 hours. The anti-proliferative effects of these compounds were assessed using the Cell Titer Glo assay kit (Promega Cat. # G7570) following the manufacturer’s protocol. EC50 values were determined using GraphPad Prism using nonlinear regression curve fitting.
CellTox Green Cell Death Assay
Cell viability was assayed with a CellTox Green cell death assay. Cells in 96-well plates (ThermoFisher Cat. # 165305) were treated with a 1:1000 dilution (in assay buffer) of CellTox Green dye for 30 min at room temperature, protected from light. Fluorescence intensity, corresponding to binding of CellTox Green dye to double-stranded DNA from dead cells, was measured on a SpectraMax iD3 Microplate Reader (485 nm excitation/520 nm emission) from the bottom, with an integration time of 400 ms and 9 multi-point readings per well. To estimate the total number of cells for subsequent normalization, all wells were subsequently permeabilized with 0.1% Triton X-100 (Fisher Scientific Cat. # BP151–100) and enough CellTox Green reagent to maintain “1X” final concentration. After incubating for 30 min at room temperature protected from light, the final fluorescence intensity was measured as above. Readings from each well were averaged and corrected by subtracting the average background signal from wells with medium and CellTox Green and no cells. The cytotoxicity index was calculated for treatments of interest by dividing background-corrected non-permeabilized readings by the corresponding permeabilized readings to assess the percentage of cell death. Each assay run was quality checked by inclusion of a standard curve of increasing cell number, which was followed by permeabilization and measurement of the CellTox Green signal. All raw data and annotated analysis scripts are available on the associated OSF project website (https://osf.io/fasqp/).
In parallel, cell health and CellTox Green uptake were also assessed by light microscopy, with image capture on a Keyence BZ-X800 (brightfield and 488 nm) and an ECHO Scope (brightfield only; 10X). These images were used as internal QC and are not incorporated in the final manuscript but have been deposited on the OSF project website (https://osf.io/fasqp/) as further supporting evidence.
Immunoblotting
Cells were washed once in 1x PBS then lysed in RIPA buffer (150 mM Tris-HCl, 150 mM NaCl, 0.5% (w/v) sodium deoxycholate, 1% (v/v) NP-40, pH 7.5) containing 0.1% (w/v) sodium dodecyl sulfate, 1 mM sodium pyrophosphate, 20 mM sodium fluoride, 50 nM calyculin, and 0.5% (v/v) protease inhibitor cocktail (Sigma-Aldrich Cat. # P8340–5ML) for 15 min. Cell extracts were precleared by centrifugation at 18,800 × g for 10 min at 4 °C. The Bio-Rad DC protein assay was used to assess protein concentration as per the manufacturer’s instructions, and sample concentration was normalized using 2x SDS sample buffer. Next, 20 μg of protein lysates and PageRuler Plus (Fisher Cat. # PI26619) prestained protein ladder were resolved on 10% acrylamide gels by SDS-polyacrylamide gel electrophoresis and electrophoretically transferred to nitrocellulose membrane (BioRad Cat. # 1620112) at 100 volts for 90 min. Membranes were blocked in 5% (w/v) nonfat dry milk (Fisher Cat# NC9022655/190915ASC) or 5% (w/v) bovine serum albumin (Boston Bioproducts Cat. # P-753) in Tris-buffered saline (TBS) for 1 h, then incubated with specific primary antibodies diluted 1:1000 in 5% (w/v) bovine serum albumin in TBS-T (TBS with 0.05% Tween-20) at 4 °C overnight, shaking. The next day, membranes were washed 3 times for 5 min each with TBS-T then incubated for 1 h at room temperature with fluorophore-conjugated secondary antibodies (LI-COR Biosciences) in 5% (w/v) nonfat dry milk, protected from light. The membrane was washed again 3 times for 5 min each with TBS-T, followed by a final 5-min wash in TBS, then imaged with a LI-COR Odyssey CLx Imaging System (LI-COR Biosciences).
For blots in Figure S7, medium containing dead or floating cells was collected from each well and centrifuged for 5 min at 300 × rcf. Medium was aspirated, and the pellet lysed in RIPA buffer and combined with protein harvested from corresponding adherent cells as described above.
Quantification was performed in ImageStudioLite Software (Licor Biosciences) by drawing rectangles around bands to capture band signal intensities, which were calculated as total pixel intensity minus background pixel intensity. Relative phospho-protein signal was performed for each lane by dividing phospho-protein signal intensity by corresponding total protein signal intensity. Relative AKT signal was calculated by dividing AKT signal intensity by Vinculin signal intensity. Normalization to DMSO samples was performed by dividing relative signal intensity for each condition by the corresponding DMSO signal intensity values. Dotted white lines were used throughout the figures to aid the reader in separating different treatments and not as an indicator of lane splicing, unless indicated in the figure legend.
MOLT4 Cell Culture and Sample Preparation for Proteomics Analysis
MOLT4 cells (T lymphoblast cell line established from a 19-year-old male patient with acute lymphoblastic leukemia in relapse) were grown in RPMI-1640 media including 2mM L-glutamine (Gibco) and supplemented with 10% fetal bovine serum (Gibco) in a 37°C incubator with 5% CO2. MOLT4 cells were treated with DMSO or 250 nM INY-05–040 for 4 h. Cells were harvested by centrifugation and lysis buffer (8 M rrea, 50 mM NaCl, 50 mM 4-(2hydroxyethyl)-1-piperazineethanesulfonic acid (EPPS) pH 8.5) with 1x cOmplete protease inhibitor (Roche) and 1x PhosphoStop (Roche) was added. Cells were subsequently homogenized by 20 passes through a 21-gauge (1.25 inch long) needle to achieve a cell lysate with a protein concentration between 0.5–4 mg/mL. The homogenized sample was clarified by centrifugation at 20,000 × g for 10 min at 4°C. A Bradford assay was used to determine the final protein concentration in the cell lysate. 200 μg protein for each sample were reduced, alkylated precipitated using methanol/chloroform and dried as previously described49. Precipitated protein was resuspended in 4 M urea, 50 mM HEPES pH 7.4 then diluted to 1 M urea with the addition of 200 mM EPPS pH 8 for digestion with LysC (1:50; enzyme:protein) for 12 h at room temperature. The LysC digestion was diluted to 0.5 M urea, 200 mM EPPS pH 8 and digested with trypsin (1:50; enzyme:protein) for 6 h at 37°C. Tandem mass tag (TMT) reagents (Thermo Fisher Scientific) were dissolved in anhydrous acetonitrile (ACN) according to the manufacturer’s instructions. Anhydrous ACN was added to each peptide sample to a final concentration of 30% v/v, and labeling was induced with the addition of TMT reagent to each sample at a ratio of 1:4 peptide:TMT label. The 11-plex labeling reactions were performed for 1.5 h at room temperature and the reaction quenched by the addition of 0.3% hydroxylamine for 15 minutes at room temperature. The sample channels were combined at a 1:1 ratio, desalted using C18 solid phase extraction cartridges (Waters) and analyzed by LC-MS for channel ratio comparison. Samples were combined using the adjusted volumes determined in the channel ratio analysis and dried down in a speed vacuum. The combined sample was resuspended in 1% formic acid and acidified to pH 2–3 before being subjected to desalting with C18 SPE (Sep-Pak, Waters). Samples were offline fractionated into 96 fractions by high pH reverse-phase HPLC (Agilent LC1260) through an Aeris peptide XB-C18 column (phenomenex) with mobile phase A containing 5% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O, and mobile phase B containing 90% acetonitrile and 10 mM NH4HCO3 in LC-MS grade H2O (both pH 8.0). The 96 resulting fractions were pooled in a non-contiguous manner into 24 fractions, desalted using solid phase extraction plates (SOLA, Thermo Fisher Scientific), and subjected to mass spectrometry analysis.
Data were collected using an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA, USA) coupled with a Proxeon EASY-nLC 1200 LC pump (Thermo Fisher Scientific). Peptides were separated on a 50 cm and 75 μm inner diameter Easyspray column (ES803a, Thermo Fisher Scientific). Peptides were separated using a 190 min gradient of 6 – 27% acetonitrile in 1.0% formic acid with a flow rate of 300 nL/min. Each analysis used an MS3-based TMT method as described previously50. The data were acquired using a mass range of m/z 340 – 1350, resolution 120,000, AGC target 5 × 105, maximum injection time 100 ms, dynamic exclusion of 120 seconds for the peptide measurements in the Orbitrap. Data dependent MS2 spectra were acquired in the ion trap with a normalized collision energy (NCE) set at 35%, AGC target set to 1.8 × 104 and a maximum injection time of 120 ms. MS3 scans were acquired in the Orbitrap with a HCD collision energy set to 55%, AGC target set to 2 × 105, maximum injection time of 150 ms, resolution at 50,000 and with a maximum synchronous precursor selection (SPS) precursors set to 10.
Proteome Discoverer 2.4 (Thermo Fisher) was used for .RAW file processing and controlling peptide and protein level false discovery rates, assembling proteins from peptides, and protein quantification from peptides. MS/MS spectra were searched against a Swissprot human database (February 2020) with both the forward and reverse sequences. Database search criteria are as follows: tryptic with two missed cleavages, a precursor mass tolerance of 10 ppm, fragment ion mass tolerance of 0.6 Da, static alkylation of cysteine (57.02146 Da), static TMT labeling of lysine residues and N-termini of peptides (229.16293 Da), variable phosphorylation of serine, threonine and tyrosine (79.966 Da), and variable oxidation of methionine (15.99491 Da). TMT reporter ion intensities were measured using a 0.003 Da window around the theoretical m/z for each reporter ion in the MS3 scan. Peptide spectral matches with poor quality MS3 spectra were excluded from quantitation (summed signal-to-noise across 11 channels < 100 and precursor isolation specificity < 0.5). Only proteins containing at least two unique peptides identified in the experiment were included in the final quantitation. Reporter ion intensities were normalized and scaled using in-house scripts in the R framework51.
The proteomics experiment was part of a standard screening workflow for new degrader molecules and therefore only has triplicate measurements for the control-treated samples and a single measurement for the degrader molecule. This precludes formal statistical analyses of fold-changes beyond confirming that the expected target molecules (AKT1, AKT2, AKT3 in this case) have been depleted.
RNA sequencing analysis
Snap-frozen cells were thawed on ice and RNA extracted with Takara’s Nucleospin RNA Plus kit (Takara Cat. # 740984.50) according to the manufacturer’s instructions. RNA integrity was assessed for quantity and purity by Nanodrop 1000. Samples were submitted to Novogene for integrity assessment (Agilent 2100 analysis), mRNA library preparation (unstranded), and paired-end (150 bp) sequencing on a NovaSeq S4 flow cell.
Raw read processing was performed with the Nextflow (version 20.07.1) nf-core RNAseq pipeline (version 1.4.2)52, with Spliced Transcripts Alignment to a Reference (STAR)53 for read alignment to the human genome (Homo_sapiens.GRCh38.96.gtf) and featureCounts54 for counting of mapped reads (multimapped reads were discarded). All subsequent data processing was performed in R, with differential gene expression analysis following the limma-voom method55. Filtering of low gene expression counts was performed with the TCGAbiolinks package with quantile value 0.75 (chosen empirically based on the observed count distribution). Next, read count normalization was performed with the trimmed mean of M (TMM) method56. PCA was done using the PCAtools package. The mean-variance relationship was modelled with voom(), which was followed by linear modelling and computation of moderated t-statistics using the lmFit() and eBayes() functions in the limma package55. Experimental replicate was included as a batch effect term in the model. The associated p-values for assessment of differential gene expression were adjusted for multiple comparisons with the Benjamini-Hochberg method at a false-discovery rate (FDR) = 0.0557. Adjustments were performed separately for each contrast of interest. Subsequent gene annotations were performed with BioMart within R58, using the associated ENSEMBL Gene IDs as key values. Intersection plots and heatmaps were generated using the ComplexHeatmap package59. Clustering was performed using the Ward.D2 method. Columns were clustered according to Euclidean distance, and rows (genes) were clustered according to Spearman’s correlation (meaning patterns of change as opposed to maximum values).
The msigdbr package was used to retrieve the indicated gene signatures. GSEA was performed with the fgsea package60, using the list of all genes ranked according to their t statistic for a comparison of interest. The choice to use the t statistic ensures that the gene ranking considers signal magnitude (fold-change) as well as uncertainty of estimation. Normalized enrichment values and associated p-values were calculated with the fgseaMultilevel() function, using default settings. The normalized enrichment score computed by the algorithm corresponds to the enrichment score normalized to mean enrichment of random samples, using the same gene set size.
The voom-normalized counts were used to predict transcription factor activities with DoRothEA24, choosing regulons within confidence groups “A”, “B” and “C” (low-confidence regulons in groups “D” and “E” were therefore not considered). As per the developer’s recommendations, the “minsize” argument in the options was set to “5”, and “eset.filter” was set to “FALSE”. Exact details can be retrieved from the deposited code. Annotated scripts for all analysis steps post-read processing are provided on the OSF project webpage (https://osf.io/3f2m5/).
Precision nuclear run-on sequencing (PRO-seq) analysis
To harvest cell pellets for PRO-seq analysis, cells were washed once with 8 mL room temperature 1X PBS and trypsinized for 5 min. Trypsin was quenched with ice cold DMEM + 10% FBS and cells were collected in a 50 mL conical tube and placed onto ice immediately. Cells were spun at 300 × g for 4 min at 4°C, supernatant was removed, and cells were resuspended in 250 μL Buffer W (10 mM Tris-Cl, pH 8.0; 10 mM KCl; 250 mM Sucrose; 5 mM MgCl2; 1 mM EGTA; 0.5 mM DTT; 10 % (v/v) Glycerol; Protease inhibitor tablet (EDTA-free), 0.02% SUPERase-IN RNAse inhibitor) to obtain a single-cell suspension by pipetting. 10 mL of Buffer P (10 mM Tris-Cl, pH 8.0; 10 mM KCl; 250 mM Sucrose; 5 mM MgCl2; 1 mM EGTA; 0.1 % (v/v) Igepal CA-630; 0.5 mM DTT; 0.05 % (v/v) Tween-20; 10 % (v/v) Glycerol; Protease inhibitor tablet (EDTA-free), 0.02% SUPERase-IN RNAse inhibitor) was added and cells were incubated on ice for 5 min and spun at 400 × g for 4 min at 4 °C. Supernatant was removed and Buffer W was added and pipetted gently 2–3 times to resuspend cells. An additional 9 mL of Buffer W was added to each tube, and cells were spun at 400 × g for 4 min at 4 °C. An additional wash with Buffer W was completed as above and supernatant was decanted so cell pellets were not disturbed. Pellets were resuspended in Buffer F (50 mM Tris-Cl, pH 8.0; 40 % (v/v) glycerol; 5 mM MgCl2; 1.1 mM EDTA; 0.5 mM DTT, and SUPERase-IN RNAse inhibitor) and transferred to a 1.5 mL tube. The 50 mL tube was rinsed again with 250 μl of Buffer F and added to the corresponding 1.5 mL tube for a final volume of 500 μl per sample. 10μl was reserved for counting after dilution 1:10 and 1:20 in PBS, both with and without trypan blue to calculate the fraction of permeabilized cells. Cells were diluted to 1 × 106 permeabilized cells per 100 μl and a total of 5 × 106 cells were aliquoted in 500 μl of Buffer F and snap frozen in liquid nitrogen and stored at −80°C until further processing. RNAse-free water was used to make all reagents and solutions, and solutions were filter sterilized with 0.2 μM filters into RNAse-free plastic bottles. Two independent biological replicates were collected, alongside the corresponding protein samples to confirm drug action at the signaling level.
Aliquots of frozen (−80 °C) permeabilized cells were thawed on ice and pipetted gently to fully resuspend. Aliquots were removed and permeabilized cells were counted using a Logos Biosystems Luna II instrument. For each sample, 1 million permeabilized cells were used for nuclear run-on, with 50,000 permeabilized Drosophila S2 cells added to each sample for normalization. Nuclear run-on assays and library preparation were performed essentially as previously described 61 with the following modifications: 2X nuclear run-on buffer consisted of (10 mM Tris (pH 8), 10 mM MgCl2, 1 mM DTT, 300 mM KCl, 40uM/ea biotin-11-NTPs (Perkin Elmer), 0.8 U/μL SuperaseIN (Thermo), 1% sarkosyl). Run-on reactions were performed at 37 °C. Adenylated 3’ adapter was prepared using the 5’ DNA adenylation kit (NEB) and ligated using T4 RNA ligase 2, truncated KQ (NEB, per manufacturer’s instructions with 15% PEG-8000 final) and incubated at 16 °C overnight. 180 μL of betaine blocking buffer (1.42 g of betaine brought to 10 mL with binding buffer supplemented with 0.6 μM blocking oligo (TCCGACGATCCCACGTTCCCGTGG/3InvdT/)) was mixed with the ligation reactions and incubated 5 min at 65°C and 2 min on ice prior to addition of streptavidin beads. After T4 polynucleotide kinase (NEB) treatment, beads were washed once each with high salt, low salt, and blocking oligo wash (0.25X T4 RNA ligase buffer (NEB), 0.3 μM blocking oligo) solutions and resuspended in 5’ adapter mix (10 pmol 5’ adapter, 30 pmol blocking oligo, water). The 5’ adapter ligation was as previously described 61 but contained 15% PEG-8000. Eluted cDNA was amplified for 5 cycles (NEBNext Ultra II Q5 master mix (NEB) with Illumina TruSeq PCR primers RP-1 and RPI-X) following the manufacturer’s suggested cycling protocol for library construction. The product (preCR) was serially diluted and used for test amplification to determine the optimal PCR conditions for the final libraries. The pooled libraries were paired-end sequenced using the Illumina NovaSeq platform.
All custom scripts described herein are available on the AdelmanLab Github (https://github.com/AdelmanLab/NIH_scripts). Using a custom script (trim_and_filter_PE.pl), FASTQ read pairs were trimmed to 41bp per mate, and read pairs with a minimum average base quality score of 20 retained. Read pairs were further trimmed using cutadapt 1.14 to remove adapter sequences and low-quality 3’ bases (--match-read-wildcards -m 20 -q 10). R1 reads, corresponding to RNA 3’ ends, were then aligned to the spiked in Drosophila genome index (dm3) using Bowtie 1.2.2 (-v 2 -p 6 –best –un), with those reads not mapping to the spike genome serving as input to the primary genome alignment step (using Bowtie 1.2.2 options -v 2 –best). Reads mapping to the hg38 reference genome were then sorted with samtools 1.3.1 (-n) and subsequently converted to bedGraph format using a custom script (bowtie2stdBedGraph.pl) that counts each read once at the exact 3’ end of the nascent RNA. Because R1 in PRO-seq reveals the position of the RNA 3’ end, the “+” and “-“ strands were swapped to generate bedGraphs representing 3’ end positions at single nucleotide resolution.
Annotated transcription start sites were obtained from human (GRCh38.99) GTFs from Ensembl. After removing transcripts with {immunoglobulin, Mt_tRNA, Mt_rRNA} biotypes, PRO-seq signal in each sample was calculated in the window from the annotated TSS to +150 nt downstream, using a custom script (make_heatmap.pl). Given good agreement between replicates and similar return of spike-in reads, bedGraphs were merged within conditions and depth-normalized to generate bigwig files binned at 10 bp.
The corresponding paired-end RNA-seq reads were mapped to the hg38 reference genome with HISAT2 v2.2.1 (--known-splicesite-infile). To select gene-level features for differential expression analysis and for pairing with PRO-seq data, we assigned a single, dominant TSS and transcription end site (TES) to each active gene. This was accomplished using a custom script, get_gene_annotations.sh (available at https://github.com/AdelmanLab/GeneAnnotationScripts), which uses RNAseq read abundance and PRO-seq R2 reads (RNA 5’ ends) to identify dominant TSSs, and RNAseq profiles to define most commonly used TESs. RNAseq and PRO-seq data from all conditions were used for this analysis, to capture gene activity in these samples. Reads were summed within the TSS to TES window for each active gene using the make_heatmap script (https://github.com/AdelmanLab/NIH_scripts), which counts each read once at the exact 3’ end location of the nascent RNA.
All subsequent processing of the PRO-seq count data were as described above for the RNAseq count data. Filtering of low counts was performed with the TCGAbiolinks package with quantile value 0.1.
Metabolomics analysis
For metabolite extraction, media was aspirated and cells were washed once with ice-cold PBS on wet ice. Ice-cold 80% (v/v) mass spectrometry-grade methanol was added, the plate was transferred to dry ice and scraped, and the resulting solution was collected. Protein samples were collected in duplicate for normalization to protein content and signaling validation as described above. Insoluble material was pelleted by centrifugation at 20,000 × g for 5 min, and the resulting supernatant was evaporated under nitrogen gas. Samples were resuspended in 20 mL HPLC-grade water for LC/MS analysis.
For polar metabolite profiling, 5 μL from each sample were injected and analyzed using a 5500 QTRAP hybrid triple quadrupole mass spectrometer (AB/SCIEX) coupled to a Prominence UFLC HPLC system (Shimadzu) with HILIC chromatography (Waters Amide XBridge), by selected reaction monitoring (SRM) with polarity switching. A total of 295 endogenous water-soluble metabolites were targeted for steady-state analyses. Electrospray source voltage was +4950 V in positive ion mode and −4500 V in negative ion mode. The dwell time was 3 ms per SRM transition 32. Peak areas from the total ion current for each metabolite were integrated using MultiQuant v2.1.1 software (AB/SCIEX).
Prior to differential abundance analysis, the raw metabolomics data were preprocessed as follows. Untrusted metabolites were removed from the datasets, including SBP, shikimate, shikimate-3-phosphate, spermidine, spermine, succinyl-CoA-methylmalonyl-CoA-nega, trehalose-6-phosphate, trehalose-sucrose, malonyl-CoA-nega, N-acetyl spermidine, N-acetyl spermine, acetylputrescine, NAD+_nega, NADH-nega, NADP+_nega, NADPH-nega, O8P-O1P, OBP, propionyl-CoA-neg, putrescine, acetoacetyl-CoA_neg, acetyl-CoA_neg, cellobiose, coenzyme A_nega, glutathione, glutathione disulfide-posi. Next, metabolites with low peak intensities (<10,000) across at least 50% of the samples were removed. Finally, all metabolites with 0 intensity in more than 3 samples were also removed, and any metabolites with 0 intensity in < 3 samples were removed in the final differential abundance analysis steps.
Metabolomics data normalized to matched protein samples from three independent experiments, each including three separate cell cultures per treatment, were combined into one dataset. Metabolites with missing (“NA”) or negative values in at least one trial were removed, resulting in 169 metabolites included in the final analyses. These were processed for differential abundance testing using the limma-voom method, with quantile normalization due to significant heteroscedascity. Subsequent linear modelling and computation of moderated t-statistics was performed with lmFit() and eBayes() as for the RNAseq data, including experimental replicate as blocking factor due to a noticeable batch effect. Heatmap generation and clustering of differentially abundant metabolites was performed as described for the RNAseq data.
Causal Oriented Search of Multi-Omic Space (COSMOS)
The RNAseq input data for COSMOS consisted of transcription factor t values from DoRothEA and the limma-voom-based t statistic for all genes, irrespective of significance, for a given contrast of interest (GDC-0068 compared to DMSO; INY-05–040 compared to DMSO). The latter served as additional constraints on the solver. Metabolite data for COSMOS consisted of the limma-voom-based t statistic for metabolites with unadjusted p-value ≤ 0.05, resulting in 58 metabolites for GDC-0068 and 77 metabolites for INY-05–040. The decision to use unadjusted p-values for filtering was made a priori due to well-known high correlation across groups of metabolites, thus making the resulting corrections for multiple comparisons overly restrictive. Metabolite names had to be mapped to their corresponding PubChem ID, which was facilitated by the R packages KEGGREST and webchem62.
Exact code for generation of both RNAseq and metabolite values in the correct format for COSMOS, as well as extensive details on all required installations and subsequent code for running COSMOS on a high-performance computer cluster, are provided on the accompanying OSF project page (https://osf.io/tdvur/). Briefly, the algorithm relies on CARNIVAL’s Integer Linear Programming (ILP) optimization, which was rerun multiple times for each dataset to determine the most consistent network predictions. Settings for each run, including the resulting network gap values, are provided in an accompanying table on the OSF project page. Differences included explicit indication of AKT1/2 inhibition (AKT3 was not expressed in T47D cells) as well as shuffling of individual t values for the background transcriptome, thus artificially forcing the solver to initiate the optimization from different starting points.
A “forward” optimization run to connect deregulated transcription factors (“signaling” input) as starting points to metabolites was performed first, followed by a “backward” optimization run connecting metabolites to signaling components. These optimization runs were used as the basis for the actual forward and final runs defining the output of the algorithm. Time limits for solving were set empirically, ensuring that the gap values of the resulting networks were ≤ 5% (indicative of a good fit). This was achieved for all runs except for one backward run (gap = 9.68%) using GDC-0068 input data. For each network run, we have provided the COSMOS script and its output as separate text files, including all run-specific settings and final gap values (https://osf.io/tdvur/).
Subsequent network analysis and visualization was performed in R, using the rCy363 package to interface with Cytoscape64. For the final visualization, a filter was applied such that text was only displayed for nodes with betweenness values of ≥ 0.05, the size of the text is indicative of the degree, and the color of the node indicative of its COSMOS-derived activation value. Betweenness is a measure of the number of shortest paths going through a node, or how much a node acts as point of connection or information transmission31.
Cancer Cell Line Growth Inhibition Screen
The high throughput cell line screen was outsourced to Horizon by Astra Zeneca. A detailed description of the protocol, alongside cell-line specific culture conditions and GI50 curve fits, are included on the OSF project webpage (https://osf.io/us45v/). Briefly, the 288 cell lines were thawed and expanded until they reached their expected doubling times, at which point the screening was begun. Cells were seeded in 25 μL of growth media in black 384-well tissue culture and equilibrated at 37°C and 5% CO2 for 24 h before treatment. At the time of treatment, a set of assay plates were collected for initial (V_0) measurements of ATP (used as proxy for viability) using the luminescence-based CellTiter Glo 2.0 (Promega) assay and an Envision plate reader (Perkin Elmer). Compounds were transferred to the remaining treatment plates using an Echo acoustic liquid handling system; 25 nL of each compound was added at the appropriate concentration for all dose points. Plates were incubated with compound for 6 days, and ATP was measured with CellTiter Glo. Data points were collected through automated processes, subjected to quality control, and analyzed with Horizon’s proprietary software.
Horizon utilizes Growth Inhibition (GI) as a measure of cell growth. The GI percentages are calculated by applying the following test and equation:
where T is the signal measure for a test drug, V is the untreated/vehicle-treated control measure, and V_0 is the untreated/vehicle control measure at time zero (see above). This formula is derived from the growth inhibition calculation used in the National Cancer Institute’s NCI-60 high throughput screen.
Publicly available transcriptomic, proteomic and RPPA data and the relevant metadata for breast cancer cells of interest were retrieved from DepMap using the depmap R package (doi: 10.18129/B9.bioc.depmap), with access to the following data versions: 21Q1 for metadata and transcriptomic data; 20Q2 for proteomic data; 19Q3 for RPPA. PCA, GSEA, hierarchical clustering and heatmap generation as part of subsequent integration with experimental GI50adj data were performed as described for RNA sequencing analysis. RNAseq data were obtained as transcripts per million (TPM) and were subjected to quantile-based filtering (quantile threshold = 0.25) using the TCGAbiolinks package65 to remove genes with low expression. We used non-parametric Spearman’s correlation to measure the strength of association between variables of interest. GI50adj values were log-transformed (base10) for visualization. For RPPA data, all antibodies labeled with “Caution” were excluded from analysis and the remaining antibody measurements were converted to z-scores prior to visualization.
Pharmacokinetic and Pharmacodynamic Analyses in BT474C Xenografts
Plasma concentrations of GDC-0068, INY-05–040, and INY-03–041 were evaluated over a period of 24 hours. Blood samples were drawn 0.5, 2, 6, 12, and 24 hours following a single dose of GDC-0068 at 12.5 mg/kg, INY-05–040 25 mg/kg, or INY-03–041 25 mg/kg. BT474C pharmacodynamic animal studies were conducted according to AstraZeneca’s Global Bioethics Policy in accordance with the PREPARE and ARRIVE guidelines. Female nude mice were surgically implanted with a 0.36 mg/60d 17β-estradiol pellet (Innovative Research of America) into the left subcutaneous flank. The following day, BT474C cells were implanted at 5 × 106 cells per mouse (suspended in 50% DMEM:50% Matrigel) into the right subcutaneous flank. Mouse weights were monitored twice weekly up until dosing, after which mouse weights were monitored daily. Tumors were measured twice weekly by caliper, with tumor volumes calculated using the formula:
The experiments were performed on adult mice weighing more than 18 g at time of first procedure. Mice were randomized by tumor volume into either control or treatment groups when average tumor volume reached 0.5 cm3. Tumor volumes were excluded if outside of the desired range (range used was 0.191–0.851 cm3 with an average of 0.354 cm3).
GDC-00068 was dosed perorally twice a day for 4 days at 12.5 mg/kg (5 mL/kg)(0.5% HPMC, 0.1% Tween 80). INY-05–040 and INY-03–041 were dosed for 4 days as a once daily intraperitoneal injection at 25mg/kg (5mL/kg) (10% DMSO/20% Captisol, pH 5.0 with gluconic acid). On the final day of dosing, 4 h after dosing AM dose, mice were humanely killed, and tumor tissue was collected and immediately snap frozen in liquid nitrogen before storage at −80°C.
Protein was extracted from snap-frozen tumor fragments by adding 900 μL of extraction buffer (20 mM Tris at pH7.5, 137 mM NaCl, 10% Glycerol, 50 mM NaF, 1 mM Na3VO4, 1% SDS, 1% NP40 substitute) with complete protease inhibitor cocktail (Roche Cat. #11836145001; 1 tablet per 50 mL). Samples were homogenized twice for 30 seconds at 6.5m/s in a fast-prep machine with an incubation at 4°C for 5 min between runs. Lysates were sonicated in a chilled Diagenode Bioruptor for two cycles (setting: HIGH) of 30 s ON/30s OFF. Lysates were cleared twice by centrifugation and protein concentrations were estimated with the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific Cat. # 23227). Approximately 40 μg of protein was run on a NuPAGE 4–12% Bis-Tris gel (Thermo Fisher Scientific) using standard methods. Following protein separation, proteins were transferred onto nitrocellulose membranes using dry transfer with iBlot2 (Thermo Fisher Scientific #IB21001). Primary antibodies were diluted in Tris-buffered saline (TBS)/0.05% Tween (TBS/T) supplemented with 5% Marvel and incubated overnight at 4 °C. The membranes were washed three times for 15 min each in 20 mL of TBS/T. A secondary horseradish peroxidase (HRP)-linked antibody was diluted 1:2000 in TBS/T supplemented with 5 % Marvel and incubated for 1 h at room temperature. The membranes were washed three times for 15 min each in 20 mL of TBS/T, and signals were detected using chemiluminesent SuperSignal West Dura Extended Duration Substrate (Thermo Fisher Scientific).
Statistics and Reproducibility
Based on the Statistical Health Check and Principles of Good Statistical Practice, Cohen’s D was used to estimate the required number of animals for in vivo experiments, taking into account the variability of the assay and the expected effect size. In this case, n of 6 mice was determined as the appropriate sample size for the endpoint.
Sample size for other experiments was not pre-determined. Statistical analyses on multidimensional datasets are detailed in the relevant sections. To avoid the pitfalls of dichotomous significance testing on conventional, low-throughput biological datasets, we used estimation statistics (Data Analysis using Bootstrap-Coupled ESTimation)66 for data in Figure 4 and Figure S9. The default settings were used (5000 resamples, BCa boostrap)66. A key advantage of this approach is the ability to focus on effect sizes and relative confidence intervals derived from bootstrapping; however, we note that the small sample size of the typical cell biological experiment is a general limitation also when it comes to reliable bootstrapping. Nevertheless, the observation of similar trends across independent experiments (for example, compare Figure S7A to Figure 4) gives us confidence in our conclusions.
The exact number of technical and biological replicates are specified in the relevant figure legends. We use biological replicates to refer to independent experimental repeats or tumor samples from different mice. Technical replicates refer to individual samples exposed to the same treatment within the same experimental replicate. The biological responses deduced from the Western blots shown in Figures 1 and 4 were reproduced across independent biological contexts (Figures S1 and S6, respectively); for all other Western blots performed in T47D cells only, all independent experimental replicates are included in the Supplementary Materials as indicated. Raw Western blot images are available on the OSF project webpage: https://osf.io/maq7k/.
Supplementary Material
Acknowledgments:
We thank members of the Toker laboratory, Joan Brugge (Harvard Medical School), Steve Elledge (Harvard Medical School), Kevin Haigis (Dana Farber Cancer Institute), Taru Muranen (Harvard Medical School), Philip Cole (Harvard Medical School), and Benjamin Turk (Yale School of Medicine), for helpful advice and suggestions; John Asara and Min Yuan for technical support with metabolomics. We would also like to thank Karen Adelman, Seth Goldman, and the Nascent transcriptomics Core (Harvard Medical School) for assistance with PRO-Seq library construction and data analysis. This work was conducted with support from Harvard Catalyst | The Harvard Clinical and Translational Science Center (National Center for Research Resources and the National Center for Advancing Translational Sciences, National Institutes of Health Award UL1 TR002541 and financial contributions from Harvard University and its affiliated academic health care centers. The content is solely the responsibility of the authors and does not necessarily represent the official views of Harvard Catalyst, Harvard University and its affiliated academic health care centers, or the National Institutes of Health.
Funding:
Research support was derived in part from NIH (R35 CA253097, A. T.), and the Ludwig Center at Harvard (A.T.). E.C.E. was supported by a F31 predoctoral fellowship (5F31CA254000–02). R.R.M. was supported by a Sir Henry Wellcome Fellowship (220464/Z/20/Z). This work was supported in part by the NIH (R01 CA218278, E.S.F. and N.S.G.) N.S.G., I.Y. and E.S.F. were supported by NIH grant (5 R01 CA218278–03).
Footnotes
Competing interests: K.A.D is a consultant to Kronos Bio and Neomorph Inc. E.S.F. is a founder, member of the scientific advisory board (SAB), and equity holder of Civetta Therapeutics, Proximity Therapeutics, and Neomorph Inc (also board of directors), science advisory board (SAB) member and equity holder in Avilar Therapeutics and Photys Therapeutics equity holder in Lighthorse Therapeutics, and a consultant to Astellas, Sanofi, Novartis, Deerfield, EcoR1 capital, Odyssey and Ajax Therapeutics. The Fischer laboratory receives or has received research funding from Novartis, Deerfield, Ajax, Interline, and Astellas. N.S.G. is a founder, SAB member and equity holder in Syros, C4, Allorion, Lighthorse, Voronoi, Inception, Matchpoint, CobroVentures, GSK, Larkspur (board member) and Soltego (board member). The Gray lab receives or has received research funding from Novartis, Takeda, Astellas, Taiho, Jansen, Kinogen, Arbella, Deerfield, Springworks, Interline and Sanofi. S.R., J.I.M., R.E.Z., S.W., C.C., J.W.J., and S.T.B. are employees and shareholders of AstraZeneca.
Data and materials availability:
Transcriptomic data from the RNAseq and PRO-seq analyses have been deposited with GEO under series accession number GSE206389. Proteomics data have been deposited under PRIDE accession number PXD036614. Additional source data are available at the OSF project website (https://osf.io/3ay2w/) with the following subcomponents: RNAseq and metabolomics raw and processed data (https://osf.io/3f2m5/); PROseq raw and processed data (https://osf.io/3f2m5/); raw and processed data for CellToxGreen-based cytotoxicity assays (https://osf.io/fasqp/); raw and processed data from the cell line screen and detailed protocol information, cell line-specific culture conditions and individual GI50 curve fits (https://osf.io/us45v/); and raw Western blot images (https://osf.io/maq7k/). Custom code for implementing COSMOS is available on Zenodo (doi: 10.5281/zenodo.10510991). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Any outstanding information regarding the computational work will be provided by R.R.M. upon request. All custom reagents are available from A.T. and N.G. under a material transfer agreement with Harvard Medical School.
References and Notes
- 1.Manning, et al. AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sanchez-Vega, et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 173, 321–337.e10 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Castel, et al. The present and future of PI3K inhibitors for cancer therapy. Nature Cancer vol. 2 587–597 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner, et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proc. Natl. Acad. Sci. 112, 3421–3426 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies, et al. A novel AKT3 mutation in melanoma tumours and cell lines. Br. J. Cancer 99, 1265–1268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hyman, et al. AKT inhibition in solid tumors with AKT1 mutations. J. Clin. Oncol. 35, 2251–2259 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nitulescu, et al. Akt inhibitors in cancer treatment: The long journey from drug discovery to clinical use (Review). Int. J. Oncol. 48, 869–885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Uhlenbrock, et al. Structural and chemical insights into the covalent-allosteric inhibition of the protein kinase Akt †. (2019) doi: 10.1039/c8sc05212c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oliveira, et al. FAIRLANE, a double-blind placebo-controlled randomized phase II trial of neoadjuvant ipatasertib plus paclitaxel for early triple-negative breast cancer. Ann. Oncol. 30, 1289–1297 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Turner, et al. BEECH: a dose-finding run-in followed by a randomised phase II study assessing the efficacy of AKT inhibitor capivasertib (AZD5363) combined with paclitaxel in patients with estrogen receptor-positive advanced or metastatic breast cancer, and in a PIK3CA. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 30, 774–780 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huck, et al. Recent progress towards clinically relevant ATP-competitive Akt inhibitors. Bioorganic and Medicinal Chemistry Letters vol. 27 2838–2848 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Martorana, et al. AKT Inhibitors: New Weapons in the Fight Against Breast Cancer? Frontiers in Pharmacology vol. 12 546 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nalawansha, et al. PROTACs: An Emerging Therapeutic Modality in Precision Medicine. Cell Chem. Biol. 27, 998–1014 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burslem, et al. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 181, 102–114 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Konstantinidou, et al. PROTACs– a game-changing technology. Expert Opin. Drug Discov. 14, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cromm, et al. Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell Chem. Biol. 24, 1181–1190 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang, et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem. Biol. 25, 88–99.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu, et al. AKT degradation selectively inhibits the growth of PI3K/PTEN pathway mutant cancers with wild-type KRAS and BRAF by destabilizing Aurora kinase B. Cancer Discov candisc.0815.2020 (2021) doi: 10.1158/2159-8290.cd-20-0815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu, et al. Design, Synthesis, and Evaluation of Potent, Selective, and Bioavailable AKT Kinase Degraders. J. Med. Chem. 64, 18054–18081 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.You, et al. Discovery of an AKT Degrader with Prolonged Inhibition of Downstream Signaling. Cell Chem. Biol. 27, 66–73.e7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhu, et al. Structure-based rational design enables efficient discovery of a new selective and potent AKT PROTAC degrader. Eur. J. Med. Chem. 238, 114459 (2022). [DOI] [PubMed] [Google Scholar]
- 22.Bricelj, et al. E3 Ligase Ligands in Successful PROTACs: An Overview of Syntheses and Linker Attachment Points. Front. Chem. 9, 1–46 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liberzon, et al. The Molecular Signatures Database Hallmark Gene Set Collection. Cell Syst. 1, 417–425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garcia-Alonso, et al. Transcription factor activities enhance markers of drug sensitivity in cancer. Cancer Res. 78, 769–780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mahat, et al. Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat. Protoc. 11, 1455–1476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shimano, et al. SREBP-regulated lipid metabolism: convergent physiology — divergent pathophysiology. Nat. Rev. Endocrinol. 13, 710–730 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Goedeke, et al. MMAB promotes negative feedback control of cholesterol homeostasis. Nat. Commun. 12, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chandler, et al. Genetic and genomic systems to study methylmalonic acidemia. Mol. Genet. Metab. 86, 34–43 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dugourd, et al. Causal integration of multi-omics data with prior knowledge to generate mechanistic hypotheses. Mol. Syst. Biol. 17, 1–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu, et al. From expression footprints to causal pathways: contextualizing large signaling networks with CARNIVAL. Syst. Biol. Appl. 5, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Barabási, et al. Network medicine: a network-based approach to human disease. Nat. Rev. Genet. 12, 56–68 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carracedo, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Invest. 118, 3065–3074 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zmajkovicova, et al. MEK1 Is Required for PTEN Membrane Recruitment, AKT Regulation, and the Maintenance of Peripheral Tolerance. Mol. Cell 50, 43–55 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phong, et al. p38 Mitogen-Activated Protein Kinase Promotes Cell Survival in Response to DNA Damage but Is Not Required for the G 2 DNA Damage Checkpoint in Human Cancer Cells. Mol. Cell. Biol. 30, 3816–3826 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angel, et al. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell 55, 875–885 (1988). [DOI] [PubMed] [Google Scholar]
- 36.Brooks, et al. Applicability of drug response metrics for cancer studies using biomaterials. Philos. Trans. R. Soc. B Biol. Sci. 374, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tsherniak, et al. Defining a Cancer Dependency Map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nusinow, et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 180, 387–402.e16 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jansen, et al. Is there a future for AKT inhibitors in the treatment of cancer? Clin. Cancer Res. 22, 2599–2601 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.André, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 380, 1929–1940 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Madsen, et al. Cancer-Associated PIK3CA Mutations in Overgrowth Disorders. Trends in Molecular Medicine vol. 24 856–870 (Trends Mol Med, 2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Madsen, et al. PIK3CA-related overgrowth: silver bullets from the cancer arsenal? Trends Mol. Med. xx, 1–3 (2022). [DOI] [PubMed] [Google Scholar]
- 43.Song, et al. RTK-Dependent Inducible Degradation of Mutant PI3Ka Drives GDC-0077 (Inavolisib) Efficacy. Cancer Discov. 0077, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vanhaesebroeck, et al. Precision Targeting of Mutant PI3Kα in Cancer by Selective Degradation. Cancer Discov. 12, 20–22 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou, et al. Ribosomal proteins: Functions beyond the ribosome. J. Mol. Cell Biol. 7, 92–104 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Warner. In the absence of ribosomal RNA synthesis, the ribosomal proteins of HeLa Cells are synthesized normally and degraded rapidly. J. Mol. Biol. 115, 315–333 (1977). [DOI] [PubMed] [Google Scholar]
- 47.Calleros, et al. RhoA and p38 MAPK mediate apoptosis induced by cellular cholesterol depletion. Apoptosis 11, 1161–1173 (2006). [DOI] [PubMed] [Google Scholar]
- 48.Calleros, et al. Low cell cholesterol levels increase NFκB activity through a p38 MAPK-dependent mechanism. Cell. Signal. 18, 2292–2301 (2006). [DOI] [PubMed] [Google Scholar]
- 49.Donovan, et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane radial ray syndrome. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McAlister, et al. MultiNotch MS3 Enables Accurate, Sensitive, and Multiplexed Detection of Differential Expression across Cancer Cell Line Proteomes. Anal. Chem. 86, 7150–7158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing; (2019). [Google Scholar]
- 52.Ewels, et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020 383 38, 276–278 (2020). [DOI] [PubMed] [Google Scholar]
- 53.Dobin, et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liao, et al. FeatureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 55.Ritchie, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47–e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Robinson, et al. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, 1–9 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benjamini, et al. Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Methodological 57, 289–300 (1995). [Google Scholar]
- 58.Durinck, et al. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 21, 3439–3440 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Gu, et al. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849 (2016). [DOI] [PubMed] [Google Scholar]
- 60.Sergushichev. An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv 060012 (2016). [Google Scholar]
- 61.Reimer, et al. Co-transcriptional splicing regulates 3′ end cleavage during mammalian erythropoiesis. Mol. Cell 81, 998–1012.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Szöcs, et al. Webchem: An R package to retrieve chemical information from the web. J. Stat. Softw. 93, (2020). [Google Scholar]
- 63.Gustavsen, et al. RCy3: Network biology using Cytoscape from within R. F1000Research 8, 1774 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cline, et al. Integration of biological networks and gene expression data using cytoscape. Nat. Protoc. 2, 2366–2382 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mounir, et al. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEX. PLoS Comput. Biol. 15, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ho, et al. Moving beyond P values: data analysis with estimation graphics. Nat. Methods 16, 565–566 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Hafner, et al. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 13, 521–527 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Transcriptomic data from the RNAseq and PRO-seq analyses have been deposited with GEO under series accession number GSE206389. Proteomics data have been deposited under PRIDE accession number PXD036614. Additional source data are available at the OSF project website (https://osf.io/3ay2w/) with the following subcomponents: RNAseq and metabolomics raw and processed data (https://osf.io/3f2m5/); PROseq raw and processed data (https://osf.io/3f2m5/); raw and processed data for CellToxGreen-based cytotoxicity assays (https://osf.io/fasqp/); raw and processed data from the cell line screen and detailed protocol information, cell line-specific culture conditions and individual GI50 curve fits (https://osf.io/us45v/); and raw Western blot images (https://osf.io/maq7k/). Custom code for implementing COSMOS is available on Zenodo (doi: 10.5281/zenodo.10510991). All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Any outstanding information regarding the computational work will be provided by R.R.M. upon request. All custom reagents are available from A.T. and N.G. under a material transfer agreement with Harvard Medical School.