

Abstract
Biomedical imaging techniques such as high content screening (HCS) are valuable for drug discovery, but high costs limit their use to pharmaceutical companies. To address this issue, The JUMP-CP consortium released a massive open image dataset of chemical and genetic perturbations, providing a valuable resource for deep learning research. In this work, we aim to utilize the JUMP-CP dataset to develop a universal representation model for HCS data, mainly data generated using U2OS cells and CellPainting protocol, using supervised and self-supervised learning approaches. We propose an evaluation protocol that assesses their performance on mode of action and property prediction tasks using a popular phenotypic screening dataset. Results show that the self-supervised approach that uses data from multiple consortium partners provides representation that is more robust to batch effects whilst simultaneously achieving performance on par with standard approaches. Together with other conclusions, it provides recommendations on the training strategy of a representation model for HCS images.
Keywords: High Content Screening, Self-supervised learning, Image representation, Deep Learning, Activity prediction
Graphical Abstract
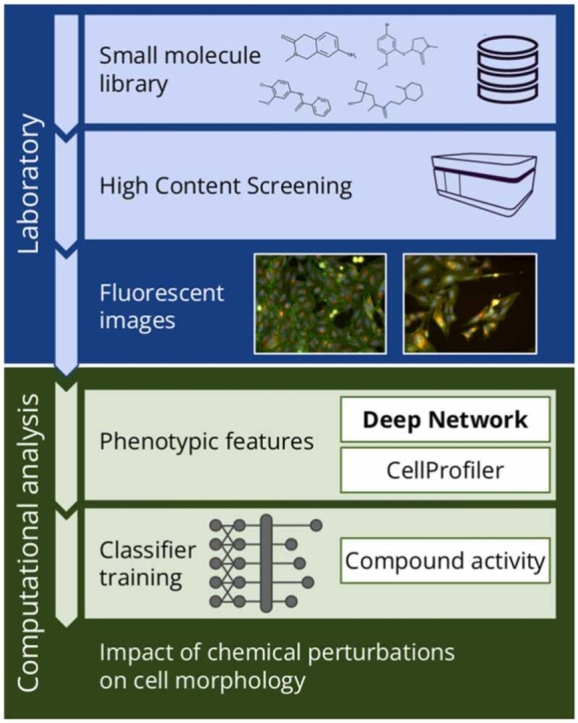
1. Introduction
Biomedical imaging techniques, including high content screening (HCS), have been recognized as valuable tools in drug discovery and biomedical research. Nevertheless, the high cost associated with these techniques has traditionally limited their implementation to pharmaceutical corporations, resulting in the creation of proprietary datasets with restricted or no access for the public or even academic researchers. Some progress has been made towards data sharing, exemplified by the release of public datasets such as those from the Broad Bioimage Benchmark Collection (BBBC) [1] and Recursion Pharmaceuticals RxRx datasets [2]. However, public datasets exhibit substantial variability stemming primarily from differences in the employed cell lines and the utilized experimental protocols. Therefore, it was practically impossible to create a universal model that generates an image representation for further classification or a model that can be fine-tuned to target tasks, like it was done for natural images with ImageNet [3].
This challenge might be solved by creating a dataset with diverse screening sources, such as the one generated by the Joint Undertaking in Morphological Profiling-Cell Painting (JUMP-CP) consortium [4]. It released a massive image dataset generated using chemical and genetic cell perturbations by 12 partners. Data was acquired using the same cell line, osteosarcoma cells (U2OS), commonly used in the drug discovery process [4], and the same protocol, cell painting [5], but with a variety of hardware. This dataset holds the potential to significantly advance research in high content screening, providing a valuable resource for developing more robust and generalizable deep learning models.
Our goal is to identify the most effective approach to using the JUMP-CP dataset to develop a representation model that is as expressive as possible. We focus on specific types of cells (U2OS) and protocol (cell painting) due to the nature of the dataset. To achieve this goal, we develop an evaluation protocol and investigate multiple supervised and self-supervised learning approaches. In the former, we analyze two auxiliary tasks that make use of labels that can be derived from images (e.g. CellProfiler [6] feature prediction) or from molecular structures (e.g. ECFP [7] prediction which simulates compound matching pretraining [8]). The usage of such labels allows training on the entire dataset. On the other hand, we exploit self-supervised learning that does not use annotations: SimCLR [9], DINO [10], and CLOOME [11].
We consider two baselines. The first one is a standard approach for HCS data, CellProfiler software [6], which generates hand-crafted features to represent a cell morphology. As the second, we investigate the expressiveness of features generated by an ImageNet-pretrained model, which was successful previously [12].
Our contributions can be summarized as follows:
-
●
We introduce the evaluation protocol that assesses the expressiveness of a representation model for CellPainting images of U2OS cells.
-
●
We extensively compare multiple approaches to develop a representation model, including auxiliary training and self-supervision.
-
●
We provide recommendations on using deep learning to boost the drug discovery process.
2. Related works
In recent years, there has been a significant surge in the popularity of utilizing deep learning methods for the analysis of images in High Content Screening (HCS). These methods encompass a diverse range of techniques, including Convolutional Neural Networks (CNN), transformers, Generative Adversarial Networks (GAN), and Variational AutoEncoders (VAE).
One of the first applications of CNNs for HCS images was a model trained to differentiate between control samples and three distinct predefined clusters [13]. Later works addressed the mode of action (MoA) prediction [14], [15] and leveraged the ImageNet pre-trained model by processing each fluorescent channel separately [14]. Following works fine-tuned a CNN on down-sampled images [15], employed multiple CNN architectures in a supervised manner [16]. Eventually, self-supervised methods started to emerge in the field [17].
However, a significant challenge arises from the scarcity of publicly available labeled data for robustly training image representations, especially for MoA prediction. To tackle this challenge, researchers have ventured into alternative methodologies including use of generative models such as generative adversarial networks [18] and variational autoencoders [19]. These techniques, beyond their application in MoA prediction, have also been harnessed for analyzing compound polypharmacology [20]. Other methods of tackling the lack of labels use auxiliary tasks, e.g. researchers trained the GAPNet model to predict compounds used to treat cells [8]. This concept was further extended in the subsequent work by introducing a causal framework that mitigates the influence of batch effects by treating them as confounding factors [21].
Alternatively, self-supervised methods can be exploited to develop a representation model for HCS images. E.g. paired cell inpainting was introduced where a model predicts fluorescent patterns in a cell given another cell as an example [22]. Another technique uses a weakly supervised approach for single-cell classification [23]. Additionally, applications of DIstillation with NO labels (DINO) [10] emerged [24], [25] together with its extension to weak supervision with WS-DINO [26].
The majority of research uses only one dataset, which results in non-transferable models. That is why we aim to leverage the scale of the JUMP-CP dataset to derive a representation model that can be further used on other datasets.
3. Materials and methods
3.1. Data
High content screening (HCS) is an approach to phenotypic screening which allows to capture fluorescent microscopic images of cells induced with either chemical (e.g. small molecules or oligonucleotides) or genetic perturbations (e.g. CRISPR [27]). In this work, we will focus on data perturbed with small molecules, also called chemical compounds. Data is collected from multiwell plates, and each well contains cells, solvent, and a compound of interest, see Fig. 1. Some wells are treated as a negative control because they do not contain any compound (only a solvent). Therefore, data from those wells can be used as a reference as they contain undisturbed cells. In order to observe changes in the cell, fluorescent dyes are added. Dyes can be specific to a research problem, e.g. a dye that binds to individual protein [28], [29], or can be somewhat universal, i.e. cell painting protocol [5]. This protocol uses 6 dyes that cover 8 cell components and are captured in 5 channels. Both datasets used in this work were created using cell painting.
Fig. 1.

Organization of high content screening data: a batch of plates is screened in one experiment. Each plate contains multiple wells (here 384) screened by an HCS imager containing a microscope. As a result, we obtain a set of 6–9 images per well, where each image is built from 5 fluorescent channels.
JUMP-CP dataset We used the dataset cpg0016-jump [4], available from the Cell Painting Gallery on the Registry of Open Data on AWS (https://registry.opendata.aws/cellpainting-gallery/). JUMP-CP dataset was created by JUMP-Cell Painting consortium, which includes 10 pharmaceutical and 2 non-profit partners. Each partner provided a set of genetic or chemical perturbations, which were then distributed across partners. Data was published at two different time points, and in this work, we use images released in the first one. In total, images for experiments for 116,000 compounds were obtained. Each compound was tested in 1–2 replicates by 3–5 different partners. The cell line used by the consortium is U2OS (osteosarcoma) and all partners used the same staining protocol (cell painting). However, data was acquired using different hardware, creating a massive dataset with high diversity. All images with their respective CellProfiler features are publicly available. Table 1.
Table 1.
Mode of action prediction results.Values in bold are statistically better across all methods according to the Wilcoxon signed-rank test with p = 0.05.
| Approach | Method | Avg ROC AUC | #tasks ≥ 0.9 | #tasks ≥ 0.8 | #tasks ≥ 0.7 |
|---|---|---|---|---|---|
| Baseline | CellProfiler | 0.6222 ± 0.0059 | 0.0 ± 0.0 | 1.2 ± 0.7 | 5.8 ± 0.7 |
| ResNet (ImageNet) | 0.6206 ± 0.0034 | 0.4 ± 0.2 | 1.4 ± 0.6 | 6.2 ± 0.5 | |
| Supervised | ResNet (JUMP tasks) | 0.5952 ± 0.0062 | 0.0 ± 0.0 | 0.6 ± 0.4 | 4.4 ± 0.4 |
| ResNet (ECFP) | 0.5302 ± 0.0033 | 0.0 ± 0.0 | 0.0 ± 0.0 | 0.2 ± 0.2 | |
| ResNet (CP) | 0.6243 ± 0.0071 | 0.4 ± 0.2 | 1.4 ± 0.7 | 6.2 ± 0.7 | |
| Self-supervised | SimCLR multiple sources | 0.6261 ± 0.0060 | 0.4 ± 0.2 | 1.2 ± 0.7 | 6.0 ± 0.6 |
| SimCLR single source | 0.6163 ± 0.0041 | 0.4 ± 0.2 | 1.0 ± 0.6 | 5.8 ± 0.4 |
Bray et al. dataset Bray et al. dataset [30] was published in 2017 as one of the first datasets created using cell painting protocol. It contains around 30,000 small-molecule perturbations, which are quite diverse and well-annotated compared to other datasets. There is a small intersection of 1591 compounds between Bray et al. and JUMP-CP data. Therefore we do not include those compounds when using JUMP-CP data for training. CellProfiler features derived from the Bray et al. dataset are publicly available in two versions. Here, we are using a newer version that was created using the JUMP-CP procedure and is consistent across both datasets.
3.2. Evaluation protocol
To evaluate trained representations, we simulate the real-life scenario presented in Fig. 2. An entity, e.g., a company or a research group, screens its proprietary data and wants to use a pre-trained model to generate representations of its images that can be later used for prediction tasks. The Bray et al. dataset represents data generated by such an entity, and the JUMP-CP dataset corresponds to the available public data. To achieve this simulation, we first train a representation model on the JUMP-CP dataset, and then we evaluate that representation using the second dataset and two sets of prediction tasks. Details of prediction tasks can be found in Supp. (Mode of Action) and Supp. Table 6 (properties). Training protocols and experimental setup of representation models are described in Section 5, and the evaluation protocol is described below.
Fig. 2.

Evaluation protocol: a representation model is trained on the main dataset, JUMP-CP, which contains images derived from compounds shared across partners, resulting in multiple replicates of the same compound. In the next step, a representation model is used to generate representations of images.
For each image, we generate a representation vector using one from the considered representation learning methods. Firstly, an image is normalized using the mean and standard deviation of the negative control. Models were modified to accept input with five channels, except for the ViT-based model, which accepts the images as five patches of RGB values. Features are generated for five crops of the image (using torchvision FiveCrop transform [31]) and then averaged to ensure that crops cover parts of the image containing cells. We do not perform further normalization of features generated in this process because it was performed at the extraction step by the image normalization. In the next step, a shallow neural network is trained to predict selected tasks. The neural network has five linear layers with ReLU activation and dropout with p = 0.2. These parameters were chosen in preliminary experiments: we’ve trained four classifiers with increasing size to predict properties based on features from baseline methods.
The results are presented in the Supp. Table 2. We observe that the best performance is given by a neural network with five layers, and a further increase in the network size does not result in an increase in the average ROC AUC. The dimension of the input of a classifier is dependent on the type of features used for classification and the dimension of the output layer is 30 for mode of action tasks and 121 for property prediction tasks.
Table 2.
Property prediction results.Values in bold are statistically better across all methods according to the Wilcoxon signed-rank test with p = 0.05.
| Approach | Method | Avg ROC AUC | #tasks ≥ 0.9 | #tasks ≥ 0.8 | #tasks ≥ 0.7 |
|---|---|---|---|---|---|
| Baseline | CellProfiler | 0.5826 ± 0.0065 | 1.8 ± 0.7 | 5.4 ± 1.0 | 18.4 ± 0.9 |
| ResNet (ImageNet) | 0.5825 ± 0.0055 | 1.6 ± 0.6 | 4.6 ± 1.4 | 16.8 ± 1.0 | |
| Supervised | ResNet (JUMP tasks) | 0.5519 ± 0.0068 | 0.4 ± 0.4 | 2.0 ± 1.0 | 7.0 ± 0.9 |
| ResNet (ECFP) | 0.5236 ± 0.0028 | 0.0 ± 0.0 | 0.8 ± 0.4 | 3.4 ± 0.8 | |
| ResNet (CP) | 0.5825 ± 0.0055 | 1.2 ± 0.6 | 4.8 ± 1.2 | 17.6 ± 0.8 | |
| Self-supervised | SimCLR multiple sources | 0.5741 ± 0.0060 | 1.2 ± 0.4 | 4.4 ± 1.3 | 15.6 ± 1.0 |
| SimCLR single source | 0.5704 ± 0.0046 | 0.4 ± 0.4 | 2.6 ± 1.2 | 12.4 ± 0.7 |
To measure the effectiveness of representation models, we build classifiers for two sets of tasks commonly used in the field [32], [33]: mode of action (MoA) prediction and property prediction. Labels for those tasks were pooled from the ChEMBL database and are described as 1: active/present, − 1: inactive/absent, 0: unknown. We use tasks that have at least 25 active and 25 inactive labels in the dataset. However, due to the train/test split, some tasks are missing from the test set. All data points are used in training, and the loss function is masked by binary cross entropy calculated only for known labels. The training was performed in 5-fold cross-validation, and folds were created using a hierarchical scaffold split to ensure that structurally similar compounds were not in the test and the training dataset. Details regarding the number of compounds, plates, and wells can be found in Supp. Additionally, we removed tasks that have low ROC AUC across all methods (below 0.5) from the evaluation as it is expected that not all chemical properties can be seen in the cell painting assay or on a specific cell line (more detailed analysis in subsection 5.1). Metrics used for evaluation are in line with [16] and include:
-
●
ROC AUC averaged over all tasks (Avg ROC AUC),
-
●
number of tasks with ROC AUC over 0.9 (#tasks ≥ 0.9),
-
●
number of tasks with ROC AUC over 0.8 (#tasks ≥ 0.8),
-
●
number of tasks with ROC AUC over 0.7 (#tasks ≥ 0.7).
We use the additional thresholded metrics because usage of average ROC AUC alone may not properly capture the effectiveness of a model. For example, a small change in the average ROC AUC can translate to better performance of multiple tasks. As benchmark methods, we use one deep learning approach (ResNet-18 [34] pretrained on ImageNet data), and one deterministic method (CellProfiler [6]). CellProfiler is an open-source software widely used in the HCS community that generates hand-crafted features that describe the morphology of the cell, e.g. texture, size, shape, intensity values, etc. CellProfiler features are calculated at a single cell level, aggregated per well, and then filtered, as in [35], resulting in a 4501-dimensional vector describing a single well from a plate. As the last step, we perform normalization of the CellProfiler feature to negative control following the same method as for image normalization.
from the Bray et al. dataset [30], which is a single source dataset and simulates a real-life case. To evaluate generated representations, a classifier is trained to predict chemical property tasks which are described by a sparse activity matrix (predictions). The performance of the classifier, the average ROC AUC over all tasks, corresponds to the performance of the representation model.
4. Research questions
-
•
The goal of this work is to evaluate whether it is possible to create a deep learning representation model that can be applied in future drug discovery research, e.g. to predict properties of compounds in a newly-performed screening of chemical perturbations. We state five research questions addressed in the following subsections:
-
•
What training strategy should be employed to acquire the most expressive representation model in a supervised learning framework?
-
•
Is self-supervision a viable approach for constructing a meaningful representation model for high-content screening data?
-
•
Does using multiple data sources in training improve the performance of a representation model?
-
•
Among the available methods, which one proves to be the most effective in mitigating batch effects induced by an experiment design?
-
•
Which methods are complementary to CellProfiler and can be integrated along those features to enhance the information conveyed within the representation?
5. Results and discussion
5.1. What training strategy should be employed to acquire the most expressive representation model in a supervised learning framework?
Experimental setup We test three different training strategies for the supervised approach: chemical activity prediction, molecular feature prediction, and hand-crafted morphological features prediction. The representation model used in the supervised approach is a convolutional neural network, ResNet-18 [34].
The first model (JUMP tasks) is trained to predict chemical activity, a task similar to the one used in the evaluation but with a different set of tasks pooled from the publicly available database ChEMBL [36] using the preprocessing step described in Section 3.2. There are 184 tasks, which results in almost 300,000 data points (around 8% of JUMP-CP data). In this task, we use the masked binary cross entropy loss.
The second training strategy is the prediction of molecular structure feature vector, i.e. Extended-Connectivity Fingerprints (ECFP) [7]. ECFPs are high-dimensional vectors characterizing the molecule through circular topological fingerprints. They were generated using the RDKit package with radius = 3 and nBits = 1024. As ECFP is a binary vector, we use the regular binary cross entropy loss.
The last strategy is CellProfiler feature prediction with L1 loss. We attempted to include a fourth task and train a model to predict a compound. The direct application of the training strategy presented in [8] is virtually impossible due to the size of the output of such a model. Given over 116,000 compounds, the training was unstable, and the solution collapsed.
All models were trained for up to 10,000 iterations with batch size 512 and early stopping with a window of 1000 iterations as well as a learning rate scheduler (ReduceLROnPlateau with default parameters [37]). Unless mentioned, we use data from 6 partners of JUMP-CP dataset (partners: 2, 3, 5, 6, 8, 10 as described in [4]). Images were randomly cropped to 224 × 224 pixels and normalized using negative control from the same plate as it is a standard practice [8], [38]. For each image, a random control image was selected, and its mean and standard deviation per channel were used to normalize the training image using torchvision Normalize transform [31]). This intervention increases the diversity of the training dataset while simultaneously removing some negative influence of batch effects.
We compare results of average ROC AUC using the pairwise Wilcoxon signed-rank test [39]. We first find the best average ROC AUC values and then use the test to analyze if these results are significantly better than the remaining ones. This procedure follows previous works in the field [40].
Results Results of Mode of action and property prediction are presented in Table 1 and Table 2, respectively. Out of all supervised approaches, the ResNet (CP) model achieves the highest score for both tasks. The results of both baselines are comparable to the ResNet (CP) model, showing that those representations are already a good description of the data. The ResNet (ECFP) representation is limited to describe the chemical structure, which may explain lower results as the Bray et al. compound set is distinct from the JUMP-CP compound set. The performance of representation given by the ResNet (JUMP tasks) model is inferior to other representations. This can be caused by the lower amount of training data (around 8% of JUMP-CP dataset) as well as by the difference between the tasks in the representation training step and the evaluation step.
In our evaluation, we excluded low-performing tasks, such as predicting activity towards adenosine, serotonin, cannabinoid, and dopamine receptors. These receptors are not expressed in bone tissue, which serves as the source of the U2OS cell line, causing a model’s inability to predict those tasks. This limitation also extends to other tissue-specific proteins, including: thyroid hormone, glucagon receptors, acetylcholinesterase, and monoamine oxidase.
5.2 Is self-supervision a viable approach for constructing a meaningful representation model for High-Content Screening data?
Experimental setup We analyzed three self-supervised learning approaches: SimCLR [9], DINO [10], and CLOOME [11]. SimCLR is an example of a contrastive learning approach which assumes that different augmentations of one image have similar representations. DINO trains a vision transformer [41] by employing the self-distillation technique, and CLOOME utilizes both image and structure to train a backbone model in the contrastive matter. As SimCLR uses ResNet-18 as a backbone, it is comparable to supervised methods.
To create positive pairs for SimCLR, we use a compound as a label [42]. This is different from regular self-supervision, which does not use any labels at all. However, in the case of data such as HCS images, the compound information is inherently present, and the data cannot exist without it. Similarly, standardized protocol for CT scans of lungs can be exploited to extract an auxiliary task, e.g. based on an anatomical location [43]. Due to this reason, we use different images of the same compound as positive pairs where images come from: from different sources (p = 0.7), the same source but different wells (p = 0.2), and the same well (p = 0.1). Values in parentheses present the probability of creating a given pair and were chosen to ensure the majority of pairs were created using different sources. In training, we use random crops of size 224 × 224 pixels, to accommodate CNN input size, which creates vast amounts of possible training images, so we do not use additional augmentation on those images as they can introduce morphological changes to the image of a cell. For example, augmentations of cell size are unsafe because they can be interpreted by the model as an influence of chemical perturbation. Nevertheless, in other aspects, we follow the training procedure of supervised models, as described in subsection 5.1.
To train DINO, we employ a similar approach for pair construction. DINO uses global and local views of the image for training, which we expand by using replicates of one compound in the same training batch, and we use the same sampling strategy as for SimCLR. We then mix global and local views between the pair to take advantage of the compound information. Another method included in the benchmark is CLOOME, a multi-modal contrastive learning method. Analogously to SimCLR, this model is trained using pairs of samples, being the objective that representations of the positive pairs are close to each other in the embedding space. However, in this case, the pair is not composed of two microscopy images. Instead, one of the objects of the pair is a microscopy image of treated cells, and the other one is the molecular fingerprint of the corresponding compound. The images were randomly cropped to a resolution of 520 × 520, and the batch size used was 1024.
Results Results of SimCLR models are presented in Table 1 and Table 2. As DINO and CLOOME are not directly comparable to SimCLR models due to their architecture, we provide the results of those models separately in Table 3 and Table 4. Self-supervised models achieve performance close to the supervised approaches and CellProfiler. In the case of the mode of action prediction, DINO obtains the highest score across all metrics, which can be caused by its more advanced architecture, vision transformer. However, all self-supervised methods perform on par according to the Wilcoxon signed-rank test. CLOOME’s performance is lower than DINO’s, which can suggest that introducing information about structure to the representation makes its training more challenging. This is consistent with the results of the ResNet (ECFP) model, which also achieved lower results and was trained with structure information. Table 5.
Table 3.
Mode of action prediction results for other models. Values in bold are statistically better according to the Wilcoxon signed-rank test with p = 0.05.
| Method | Avg ROC AUC | #tasks ≥ 0.9 | #tasks ≥ 0.8 | #tasks ≥ 0.7 |
|---|---|---|---|---|
| DINO | 0.6264 ± 0.0031 | 0.4 ± 0.2 | 1.2 ± 0.6 | 7.2 ± 0.9 |
| DINO +CP | 0.6370 ± 0.0031 | 0.4 ± 0.3 | 1.8 ± 0.6 | 8.8 ± 0.6 |
| CLOOME | 0.6084 ± 0.0044 | 0.2 ± 0.2 | 0.4 ± 0.2 | 3.2 ± 0.4 |
| CLOOME + CP | 0.6287 ± 0.0085 | 0.4 ± 0.3 | 1.2 ± 0.6 | 6.0 ± 0.8 |
Table 4.
Property prediction results for other models. Values in bold are statistically better according to the Wilcoxon signed-rank test with p = 0.05.
| Method | Avg ROC AUC | #tasks ≥ 0.9 | #tasks ≥ 0.8 | #tasks ≥ 0.7 |
|---|---|---|---|---|
| DINO | 0.5802 ± 0.0052 | 1.4 ± 0.6 | 4.8 ± 1.4 | 17.4 ± 1.4 |
| DINO + CP | 0.5918 ± 0.0057 | 3.6 ± 1.1 | 8.2 ± 1.4 | 24.4 ± 1.2 |
| CLOOME | 0.5306 ± 0.0035 | 0.2 ± 0.2 | 0.4 ± 0.4 | 5.2 ± 1.1 |
| CLOOME + CP | 0.5868 ± 0.0069 | 1.8 ± 0.9 | 6.4 ± 1.6 | 19.8 ± 0.8 |
Table 5.
Batch effects metrics.
| Method | Wassertein distance ↓ | Batch ASW ↑ |
|---|---|---|
| ResNet (ImageNet) | 1.909 | 0.8513 |
| ResNet (JUMP tasks) | 1.356 | 0.7263 |
| ResNet (ECFP) | 2.584 | 0.7037 |
| ResNet (CP) | 9.030 | 0.7749 |
| SimCLR multiple sources | 1.205 | 0.7904 |
| SimCLR single source | 3.515 | 0.7694 |
| DINO | 3.967 | 0.7794 |
| CLOOME | 1.308 | 0.8550 |
5.2. Does using multiple data sources in training improve the performance of a representation model?
Experimental setup We run two versions of the SimCLR model: one using data from multiple partners (see: 5.2) and the second one using data from a randomly selected partner (partner 2).
Results As seen in Table 1 and Table 2, using more data sources improves the average ROC AUC and increases the number of well-modeled tasks. This is expected due to the bigger amount of the data and higher diversity of this data. However, the increase is not statistically significant, showing that a good representation can be obtained even from a single source of data. Nevertheless, there are other aspects of HCS data that need to be taken into account, i.e. batch effects, which are investigated in the next subsection.
5.4 Among the available methods, which one proves to be the most effective in mitigating batch effects induced by an experiment design?
Experimental setup We used two metrics to analyze batch effects. Firstly, we use a distribution of distance between replicates of 100 randomly selected compounds. We compare two distributions: distance of replicates on the same plate, usually the same well, and on different plates. To measure the difference between those distributions, we used Wasserstein distance [44]. The second method is the modified Average Silhouette Width of batch (batch ASW) calculated using the scib package [45] on 30 random plates constituting 50665 data points. We compared them across all methods, except CellProfiler, due to different aggregation of the data on the well level.
ResultsFig. 3 presents distributions of distances between replicates, and Table 5 presents Wasserstein distance (lower is better) and batch ASW metrics (higher is better). All methods exhibit batch effects and distribution separation. This effect is most prominent in ResNet (CP), ResNet (ECFP), and SimCLR single source. Mixed metrics are obtained by ResNet (JUMP tasks) and DINO. The representations most robust to batch effects are SimCLR multiple sources, ResNet (ImageNet), and CLOOME. The low result of ResNet (CP) can be caused by the CNN being trained to predict features already influenced by batch effects. In contrast, SimCLR trained on multiple sources is better than SimCLR single source, which shows the positive effect of contrastive training with positive pairs defined between different sources. ResNet (ImageNet) was trained on natural images, so it did not learn any information regarding a compound or batch influence on data, which, combined with its high average ROC AUC, proves a good representation. Conversely, the high result of the CLOOME method reaffirms that contrastive learning is an appropriate approach to this problem. Additionally, this could suggest a positive influence of the structural information. On the other hand, ResNet (ECFP) achieves the lowest Batch ASW, which contradicts the aforementioned thesis.
Fig. 3.

Distribution of distances on the same plate and between plates for 100 random compounds and Wasserstein distance (wd). Plot for ResNet (CP) has a different scale than plots for other methods. We can observe that distributions given by SimCLR trained on multiple sources are the closest, meaning that there is some mitigation of batch effects, while ResNet (CP) obtains representation the least robust to batch effects.
5.5 Which methods are complementary to CellProfiler and can be integrated along those features to enhance the information conveyed within the representation?
Experimental setup To analyze how complementary CellProfiler features are to the deep learning methods, we concatenated both types of features and tested them using our evaluation protocol.
ResultsFig. 4 presents the results of those models, compared to single feature models and CellProfiler. Detailed values are presented in Supp. Table 3 and Supp. Table 4 for ResNet-18 based methods and in Table 3 and Table 4 for other methods. The biggest difference is observed when using ResNet (ECFP) and CLOOME features. These models were trained with information about the chemical structure of the compound rather than its function. ResNet (ECFP) is the worst-performing model on its own. This suggests that the best results can be obtained by joining structural and image information which is consistent with previous works [46], [47]. DINO with CP features performs better than CellProfiler alone across both mode of action and property prediction tasks. For the majority of models, adding the CP information is not significantly beneficial or detrimental. This suggests that those methods already capture cell morphology similarly to CellProfiler. This claim is additionally supported by the increase in number of well-predicted tasks (#tasks ≥ 0.9, #tasks ≥ 0.8, #tasks ≥ 0.7) across all methods. However, there is an advantage of using only a pre-trained Deep Learning model over CellProfiler, i.e. the ability to accelerate the feature extraction by the use of GPU capabilities.
Fig. 4.

Comparison between obtained representation (dot) and its concatenation with CellProfiler (diamond). The addition of CP features provides a slight increase in performance, however not.
significantly, except for the model trained to predict ECFP and the DINO model that seem to be complementary to CellProfiler.
6. Conclusions
In this study, we propose the evaluation protocol that assesses representations of high content screening data on mode of action and property prediction tasks. We use this protocol to analyze various supervised and self-supervised representation models trained on the JUMP-CP dataset, delivering a set of conclusions.
Firstly, the results demonstrate that the Deep Learning (DL) and CellProfiler (CP) features are interchangeable in the MoA and property prediction tasks. However, generating CellProfiler features requires much more time [46]. Therefore, DL features are preferable for time-sensitive applications. Secondly, combining CP and DL features can statistically increase the performance of the chemical property prediction model. Thirdly, training self-supervised models on multiple sources has a positive impact on mitigating batch effects.
There's a need for future work in this field, including high-content screening for other cell lines and staining protocols.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Jacek Tabor reports financial support was provided by National Science Centre Poland. Dawid Rymarczyk reports financial support was provided by National Science Centre Poland. Lukasz Struski reports financial support was provided by National Science Centre Poland. Bartosz Zielinski reports financial support was provided by National Science Centre Poland. If there are other authors, they declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This research was partially funded by the National Science Centre, Poland, grants no. 2021/41/B/ST6/01370 (work by Jacek Tabor), 2022/45/N/ST6/04147 (work by Dawid Rymarczyk), 2020/39/D/ST6/01332 (work by Łukasz Struski), and 2022/47/B/ST6/03397 (work by Bartosz Zieliński). Moreover, Dawid Rymarczyk received an incentive scholarship from the funds of the program Excellence Initiative - Research University at the Jagiellonian University in Kraków.
Author Agreement Statement
We the undersigned declare that this manuscript is original, has not been published before and is not currently being considered for publication elsewhere.
We confirm that the manuscript has been read and approved by all named authors and that there are no other persons who satisfied the criteria for authorship but are not listed. We further confirm that the order of authors listed in the manuscript has been approved by all of us.
We understand that the Corresponding Author is the sole contact for the Editorial process. lie/she is responsible for communicating with the other authors about progress, submissions of revisions and final approval of proofs.
Footnotes
Supplementary data associated with this article can be found in the online version at doi:10.1016/j.csbj.2024.02.022.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Ljosa V., Sokolnicki K.L., Carpenter A.E. Annotated high-throughput microscopy image sets for validation. Nat. Methods. 2012;9(7) doi: 10.1038/nmeth.2083. 637–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sypetkowski M., Rezanejad M., Saberian S., Kraus O., Urbanik J., Taylor J., et al. Rxrx1: a dataset for evaluating experimental batch correction methods : Proc IEEE/CVF Conf Comput Vis Pattern Recognit. 2023:4284–4293. [Google Scholar]
- 3.Deng J., Dong W., Socher R., Li L.J., Li K., Fei-Fei L. Imagenet: A large-scale hierarchical image database. In: 2009 IEEE conference on computer vision and pattern recognition. Ieee; 2009. p. 248–255.
- 4.Chandrasekaran S.N., et al. JUMP Cell Painting dataset: morphological impact of 136,000 chemical and genetic perturbations. bioRxiv; 2023. doi:10.1101/2023.03.23.534.
- 5.Bray M.A., Singh S., Han H., Davis C.T., Borgeson B., Hartland C., et al. Cell Painting, a high-content image-based assay for morphological profiling using multiplexed fluorescent dyes. Nat Protoc. 2016;11(9):1757–1774. doi: 10.1038/nprot.2016.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stirling D.R., Swain-Bowden M.J., Lucas A.M., Carpenter A.E., Cimini B.A., Goodman A. CellProfiler 4: improvements in speed, utility and usability. BMC Bioinforma. 2021;22(1):11. doi: 10.1186/s12859-021-04344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rogers D., Hahn M. Extended-connectivity fingerprints. J. Chem Inf Model. 2010;50(5):742–754. doi: 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
- 8.Caicedo J.C., McQuin C., Goodman A., Singh S., Carpenter A.E. Weakly supervised learning of single-cell feature embeddings : Proc IEEE Conf Comput Vis Pattern Recognit. 2018:9309–9318. doi: 10.1109/CVPR.2018.00970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen T., Kornblith S., Norouzi M., Hinton G. A simple framework for contrastive learning of visual representations. In: International conference on machine learning. PMLR. 2020:1597–1607. [Google Scholar]
- 10.Caron M., Touvron H., Misra I., J´egou H., Mairal J., Bojanowski P., et al. Emerging properties in self-supervised vision transformers : Proc IEEE/CVF Int. Conf Comput Vis. 2021:9650–9660. [Google Scholar]
- 11.Sánchez-Fernández A., Rumetshofer E., Hochreiter S., Klambauer G. CLOOME: contrastive learning unlocks bioimaging databases for queries with chemical structures. Nat. Commun. 2023;14(1):7339. doi: 10.1038/s41467-023-42328-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pawlowski N., Caicedo J.C., Singh S., Carpenter A.E., Storkey A. Automating Morphological Profiling with Generic Deep Convolutional Networks. bioRxiv; 2016. doi:10.1101/085118.
- 13.Dürr O., Sick B. Single-cell phenotype classification using deep convolutional neural networks. J. Biomol Screen. 2016;21(9):998–1003. doi: 10.1177/1087057116631284. [DOI] [PubMed] [Google Scholar]
- 14.Ando D.M., McLean C.Y., Berndl M. Improving phenotypic measurements in high-content imaging screens. BioRxiv. 2017 [Google Scholar]
- 15.Kensert A., Harrison P., Spjuth O. Transfer Learning with Deep Convolutional Neural Networks for Classifying Cellular Morphological Changes. SLAS DISCOVERY: Advancing Life Sciences RD. 2019;24:247255521881875. doi:10.1177/2472555218818756. [DOI] [PMC free article] [PubMed]
- 16.Hofmarcher M., Rumetshofer E., Clevert D.A., Hochreiter S., Klambauer G. Accurate prediction of biological assays with high-throughput microscopy images and convolutional networks. J Chem Inf Model. 2019;59(3):1163–1171. doi: 10.1021/acs.jcim.8b00670. [DOI] [PubMed] [Google Scholar]
- 17.Masud U., Cohen E., Bendidi I., Bollot G., Genovesio A. In: European Conference on Computer Vision. Springer,; 2022. Comparison of semi-supervised learning methods for high content screening quality control; pp. 395–405. [Google Scholar]
- 18.Goldsborough P., Pawlowski N., Caicedo J.C., Singh S., Carpenter A.E. CytoGAN: generative modeling of cell images. BioRxiv. 2017 [Google Scholar]
- 19.Lafarge M.W., Caicedo J.C., Carpenter A.E., Pluim J.P., Singh S., Veta M. Capturing single-cell phenotypic variation via unsupervised representation learning : Int Conf Med Imaging Deep Learn PMLR. 2019:315–325. [PMC free article] [PubMed] [Google Scholar]
- 20.Chow Y.L., Singh S., Carpenter A.E., Way G.P. Predicting drug polypharmacology from cell morphology readouts using variational autoencoder latent space arithmetic. PLoS Comput Biol. 2022;18(2) doi: 10.1371/journal.pcbi.1009888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moshkov N., Bornholdt M., Benoit S., Smith M., McQuin C., Goodman A., et al. Learning representations for image-based profiling of perturbations. Biorxiv. 2022 doi: 10.1038/s41467-024-45999-1. 2022–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lu A.X., Kraus O.Z., Cooper S., Moses A.M. Learning unsupervised feature representations for single cell microscopy images with paired cell inpainting. PLoS Comput Biol. 2019;15(9) doi: 10.1371/journal.pcbi.1007348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Borowa A., Kruczek S., Tabor J., Zieliński B. In: International Conference on Computational Science. Springer,; 2022. Weakly-supervised cell classification for effective high content screening; pp. 318–330. [Google Scholar]
- 24.Doron M., Moutakanni T., Chen Z.S., Moshkov N., Caron M., Touvron H., et al. Unbiased single-cell morphology with self-supervised vision transformers. bioRxiv. 2023; p. 2023–06.
- 25.Kim V., Adaloglou N., Osterland M., Morelli F., Zapata P.A.M. Self-supervision advances morphological profiling by unlocking powerful image representations. bioRxiv. 2023; p. 2023–04.
- 26.Cross-Zamirski J.O., Williams G., Mouchet E., Schönlieb C.B., Turkki R., Wang Y. Self-supervised learning of phenotypic representations from cell images with weak labels. arXiv preprint arXiv:220907819. 2022;.
- 27.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable Dual-RNA–Guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hertzberg R.P., Pope A.J. High-throughput screening: new technology for the 21st century. Curr Opin Chem Biol. 2000;4(4):445–451. doi: 10.1016/s1367-5931(00)00110-1. [DOI] [PubMed] [Google Scholar]
- 29.Giuliano K.A., DeBiasio R.L., Dunlay R.T., Gough A., Volosky J.M., Zock J., et al. High-content screening: a new approach to easing key bottlenecks in the drug discovery process. SLAS Discov. 1997;2(4):249–259. [Google Scholar]
- 30.Bray M.A., Gustafsdottir S.M., Rohban M.H., Singh S., Ljosa V., Sokolnicki K.L., et al. A dataset of images and morphological profiles of 30 000 small-molecule treatments using the Cell Painting assay. Gigascience. 2017;6(12) doi: 10.1093/gigascience/giw014. giw014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marcel S., Rodriguez Y. Torchvision the machine-vision package of torch : Proc 18th ACM Int Conf Multimed. 2010:1485–1488. [Google Scholar]
- 32.Chandrasekaran S.N., Ceulemans H., Boyd J.D., Carpenter A.E. Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat. Rev Drug Discov. 2021;20(2):145–159. doi: 10.1038/s41573-020-00117-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rose F., Basu S., Rexhepaj E., Chauchereau A., Nery E., Genovesio A. Compound functional prediction using multiple unrelated morphological profiling assays. SLAS TECHNOLOGY: Transl Life Sci Innov. 2017;23 doi: 10.1177/2472630317740831. 247263031774083. doi:10.1177/2472630317740831. [DOI] [PubMed] [Google Scholar]
- 34.He K., Zhang X., Ren S., Sun J. Deep residual learning for image recognition. In: Proceedings of the IEEE conference on computer vision and pattern recognition; 2016. p. 770–778.
- 35.Caicedo J.C., Cooper S., Heigwer F., Warchal S., Qiu P., Molnar C., et al. Data-analysis strategies for image-based cell profiling. Nat Methods. 2017;14(9):849–863. doi: 10.1038/nmeth.4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mendez D., Gaulton A., Bento A.P., Chambers J., De Veij M., F´elix E., et al. ChEMBL: towards direct deposition of bioassay data. Nucleic Acids Res. 2019;47(D1):D930–D940. doi: 10.1093/nar/gky1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Paszke A., Gross S., Massa F., Lerer A., Bradbury J., Chanan G., et al. Pytorch: an imperative style, high-performance deep learning library. Adv Neural Inf Process Syst. 2019:32. [Google Scholar]
- 38.Mpindi J.P., Swapnil P., Dmitrii B., Jani S., Saeed K., Wennerberg K., et al. Impact of normalization methods on high-throughput screening data with high hit rates and drug testing with dose–response data. Bioinformatics. 2015;31(23):3815–3821. doi: 10.1093/bioinformatics/btv455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blair R.C., Higgins J.J. Comparison of the power of the paired samples t test to that of Wilcoxon’s signed-ranks test under various population shapes. Psychol Bull. 1985;97(1):119. [Google Scholar]
- 40.Koutsoukas A., Monaghan K.J., Li X., Huan J. Deep-learning: investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Chemin- 2017;9(1):1–13. doi: 10.1186/s13321-017-0226-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dosovitskiy A., Beyer L., Kolesnikov A., Weissenborn D., Zhai X., Unterthiner T., et al. An Image is Worth 16×16 Words: Transformers for Image Recognition at Scale. ICLR. 2021;.
- 42.Haslum J.F., Matsoukas C., Leuchowius K.J., Müllers E., Smith K. Metadata-guided Consistency Learning for High Content Images. Medical Imaging with Deep Learning (MIDL). 2022;.
- 43.Haghighi F., Hosseinzadeh Taher M.R., Zhou Z., Gotway M.B., Liang J. Learning semantics-enriched representation via self-discovery, self-classification, and self-restoration. In: Medical Image Computing and Computer Assisted Intervention–MICCAI 2020: 23rd International Conference, Lima, Peru, October 4–8, 2020, Proceedings, Part I 23. Springer; 2020. p. 137–147. [DOI] [PMC free article] [PubMed]
- 44.Vallender S. Calculation of the Wasserstein distance between probability distributions on the line. Theory Probab Its Appl. 1974;18(4):784–786. [Google Scholar]
- 45.Luecken M.D., Büttner M., Chaichoompu K., Danese A., Interlandi M., Müller M.F., et al. Benchmarking atlas-level data integration in single-cell genomics. Nat Methods. 2022;19(1):41–50. doi: 10.1038/s41592-021-01336-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koziarski M., Gaiński P., Rataj K., Borowa A., Wójtowicz K., Gwóźdź J., et al. Multimodal Approach to MoA Prediction Based on Cell Painting Imaging and Chemical Structure Data. ELRIG. 2022;
- 47.Tian G., Harrison P.J., Sreenivasan A.P., Carreras-Puigvert J., Spjuth O. Combining molecular and cell painting image data for mechanism of action prediction. Artif Intell Life Sci. 2023;3 100060. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material