

Abstract
Background
Many observational studies support light-to-moderate alcohol intake as potentially protective against premature death. We used a genetic approach to evaluate the linear and nonlinear relationships between alcohol consumption and mortality from different underlying causes.
Methods
We used data from 278 093 white-British UK Biobank participants, aged 37–73 years at recruitment and with data on alcohol intake, genetic variants, and mortality. Habitual alcohol consumption was instrumented by 94 variants. Linear Mendelian randomization (MR) analyses were conducted using five complementary approaches, and nonlinear MR analyses by the doubly-ranked method.
Results
There were 20 834 deaths during the follow-up (median 12.6 years). In conventional analysis, the association between alcohol consumption and mortality outcomes was ‘J-shaped’. In contrast, MR analyses supported a positive linear association with premature mortality, with no evidence for curvature (Pnonlinearity ≥ 0.21 for all outcomes). The odds ratio [OR] for each standard unit increase in alcohol intake was 1.27 (95% confidence interval [CI] 1.16–1.39) for all-cause mortality, 1.30 (95% CI 1.10–1.53) for cardiovascular disease, 1.20 (95% CI 1.08–1.33) for cancer, and 2.06 (95% CI 1.36–3.12) for digestive disease mortality. These results were consistent across pleiotropy-robust methods. There was no clear evidence for an association between alcohol consumption and mortality from respiratory diseases or COVID-19 (1.32, 95% CI 0.96–1.83 and 1.46, 95% CI 0.99–2.16, respectively; Pnonlinearity ≥ 0.21).
Conclusion
Higher levels of genetically predicted alcohol consumption had a strong linear association with an increased risk of premature mortality with no evidence for any protective benefit at modest intake levels.
Keywords: Alcohol consumption, Mendelian randomization, nonlinear analysis, doubly-ranked method, mortality
Key Messages .
Genetically predicted alcohol intake was associated with an increased risk of premature death, including all leading underlying causes.
Genetic analysis did not provide evidence for a nonlinear association, failing to support observational studies suggesting benefits by modest alcohol intakes.
While the greatest mortality risks are seen with high intakes, lowering guidelines for safe alcohol consumption would benefit public health, as even moderate intake poses a risk.
Introduction
The relationship between alcohol consumption and mortality is complex.1 Excessive alcohol intake is associated with a higher risk of premature death.2 However, previous observational studies suggest a ‘J-shaped’ relationship between alcohol consumption and mortality, where light-to-moderate consumption has the lowest mortality, and both high levels and abstaining from alcohol are associated with an increased risk of death and morbidity.3–6 This has led to suggestions that light-to-moderate alcohol consumption might offer protection against premature death and to an ongoing debate about safe levels of alcohol intake.
Natural experiments using alcohol control policies, such as tax increases and availability restrictions, support a protective effect of lower alcohol intake on all-cause mortality.7 However, determining the causal link between moderate habitual alcohol consumption and mortality risk is challenging and while such studies have been attempted,8 we are not aware of any published randomized controlled trials (RCTs). Mendelian randomization (MR) is a statistical technique that uses naturally occurring genetic variation, typically single nucleotide polymorphisms (SNPs), to infer a causal relationship between an exposure (in this case, alcohol consumption) and an outcome (in this case, mortality risk)9 (Supplementary Figure S1, available as Supplementary data at IJE online). This approach assumes that genetic variants are not influenced by confounders and that they are randomly allocated at conception, thereby avoiding some of the limitations with traditional observational studies, such as confounding and reverse causality,10 and providing more robust evidence of causal links. There are several MR studies investigating the effect of alcohol intake on various health outcomes including cardiovascular diseases (CVD),11,12 stroke,13 and Alzheimer’s disease.14 These studies have typically relied on linear MR approaches, and, as such, could not have ascertained if a modest alcohol intake indeed had a beneficial protective effect. An interesting study is that conducted by Millwood et al.,15 which approximated average alcohol intakes based on known differences in consumption patterns between regions, with area-stratified MR analyses and cross-center comparisons suggesting similar increases in cardiovascular risk regardless of estimated alcohol intake. There is also a recent study looking at the association between alcohol intake and CVD outcomes which used a nonlinear MR approach in addition to conducting standard linear analyses. This study suggested a quadratic pattern to be the best fit for the associations between alcohol consumption and hypertension, coronary heart disease, and all-cause mortality.16 However, their analyses did not address associations of alcohol across cause-specific mortalities, and the inference on the shape of the association remains uncertain as subsequent methodological work suggests that there have been likely violations of model assumptions specific to the statistical approach used.17
In this study, we examine the genetic evidence for a causal relationship between alcohol consumption and mortality risk, including all-cause mortality and deaths caused by CVD, cancer, respiratory disease, digestive disease, and COVID-19. Our study used data from up to 278 093 UK Biobank participants with information on alcohol consumption, genetic variants, and mortality. The analyses were conducted using multiple complementary approaches to enhance the robustness of the findings.
Methods
The UK Biobank is a longitudinal study that initially included data from more than 500 000 participants from the general population of the United Kingdom. Participants were recruited between March 2006 and July 2010, aged 37 to 73 years.18 In this study, we restricted the study population to individuals of European ancestry who identified as white British, who were unrelated,19 and who had consistent information about self-reported and genetic sex and complete data on alcohol consumption, mortality, and covariates, leaving 278 093 participants for our analyses (Supplementary Figure S2, available as Supplementary data at IJE online).
We obtained mortality data from the National Health Service (NHS) Digital and the NHS Central Register (https://www.ukbiobank.ac.uk/) until November 12, 2021. Our study outcomes included both all-cause mortality and cause-specific mortalities from CVD, cancer, digestive, respiratory, and COVID-19 diseases. We defined causes of death using the tenth revision of the International Classification of Diseases (ICD)20 (Supplementary Table S1, available as Supplementary data at IJE online). As a sensitivity analysis to reduce potential bias from competing causes, we also restricted our analyses of all-cause mortality to deaths prior to the COVID-19 pandemic, including deaths until January 1, 2020.
Alcohol consumption was reported at baseline assessment using a touchscreen questionnaire. Participants reported their intake of different types of alcoholic drinks, and we calculated the amount of alcohol consumed as grams per day, by adding up the average consumption of various types of beverages. Intakes were reported as units of alcohol assuming the UK standard (1 unit = 8 grams of pure alcohol21 (Supplementary Methods, available as Supplementary data at IJE online). Information on covariates was collected through self-reported touchscreen questionnaires, except for the Townsend deprivation index (TDI) which was derived based on participants’ postcodes (Supplementary Methods, available as Supplementary data at IJE online).
Genetic variants were identified based on a recent genome-wide association (GWAS) meta-analysis on individuals of European ancestry.22 From the 99 SNPs associated with alcohol consumption, we used the 94 SNPs that had a directionally consistent association with alcohol intake in the UK Biobank (Supplementary Table S2, available as Supplementary data at IJE online). We extracted the SNPs from the UK Biobank and calculated weighted genetic risk scores (GRS) taking the weights from the corresponding SNP-alcohol consumption association estimates in the original discovery sample (Supplementary Methods, available as Supplementary data at IJE online).22
Statistical analysis
We used logistic regression to investigate the association of reported alcohol consumption with mortality. We used fractional polynomial models to determine the appropriate functional form comparing model fit between the best-fitting fractional polynomial model and the linear model, using the likelihood ratio test.23 All models were adjusted for age, sex, education level, assessment center, TDI, body mass index (BMI), smoking, physical activity, and self-perceived health and long-term illness.
We conducted linear and nonlinear MR analyses to examine the causal relationship between alcohol consumption and mortality, with the latter used to assess evidence for curvature (Supplementary Figure S3, available as Supplementary data at IJE online). In both approaches, the causal association was interrogated using the genetic variants associated with alcohol intake, adjusting for age, sex, assessment center, SNP array, birth location, and top 40 genetic principal components. In the linear MR analysis, the ratio-of-coefficients method was used to compute causal estimates,24 which requires GRS-exposure and GRS-outcomes association estimates as inputs. We used linear and logistic regression to obtain GRS-alcohol and GRS-mortality association estimates from the UK Biobank, respectively. In the nonlinear MR analysis, we first stratified the UK Biobank sample into 25 strata using the doubly-ranked stratification method,25 where strata are formed by firstly ranking participants into pre-strata based on their level of GRS and then by ranking participants within each pre-stratum according to their level of alcohol intake. Within each stratum, we computed stratum-specific GRS-alcohol and GRS-outcomes effect estimates and then applied the ratio-of-coefficients method (as described in the linear MR analysis) to calculate the localized average causal effect (LACE) estimate. To assemble the alcohol–mortality curve, we then carried out a meta-regression of LACE estimates against the stratum-specific mean alcohol consumption using a fractional polynomial function. The best-fitting fractional polynomial model was determined by the likelihood ratio test. The fractional polynomial test was reported for nonlinearity, which compares the best-fitting fractional polynomial of degree 1 against the linear model.25 Furthermore, for the linear MR analysis, where estimation via summary level data is also possible, as sensitivity analysis we repeated the analysis using summary-data-based method to test the robustness of the results to horizontal pleiotropy. We included five summary-data-based methods, including inverse variance weighted (IVW), MR-Egger, weighted median, weighted mode, and MR-PRESSO. Different methods made varying assumptions about horizontal pleiotropy, and consistent results across different methods suggest that the causal inference was more credible.26 Further details and information on additional sensitivity analyses are provided in Supplementary Methods (available as Supplementary data at IJE online).
Analyses were conducted using STATA version 17.0 (StataCorp LP, College Station, Texas, USA) and R (version 4.2.0).27,28
Results
Up to 278 093 individuals were included in the analysis. The mean alcohol consumption was 18.20 grams per day (standard deviation [SD] 18.81). Male participants and those who smoked, engaged in intense physical activity, had completed NVQ/CSE/A-levels or a degree/professional education, or had the highest TDI, also had slightly higher average alcohol consumption compared to others. Participants who reported ‘fair’ self-rated health and those who did not have chronic illnesses consumed more alcohol compared to others (Table 1). The mortality rate varied across the socio-demographic, health, and lifestyle characteristics (Supplementary Table S3, available as Supplementary data at IJE online). Alcohol GRS was not associated with confounders except for smoking and TDI (Supplementary Table S4, available as Supplementary data at IJE online).
Table 1.
Alcohol consumption by baseline characteristics in UK Biobank
| Characteristic | Number of participants (%) | Alcohol intake in g/day, mean (SD) |
|---|---|---|
| Total | 278 093 (100) | 18.20 (18.81) |
| Age (years) | ||
| <65 | 222 626 (80.05) | 18.57 (19.05) |
| ≥65 | 55 467 (19.95) | 16.69 (17.74) |
| P-valuea | 8.17E-189 | |
| Sex | ||
| Male | 135 319 (48.66) | 24.51 (22.05) |
| Female | 142 774 (51.34) | 12.21 (12.45) |
| P-valuea | <1.0E-300 | |
| BMI (kg/m2) | ||
| <18.5 | 1341 (0.48) | 13.49 (18.74) |
| 18.5–25 | 91 655 (32.96) | 16.11 (16.51) |
| 25–30 | 120 519 (43.34) | 19.50 (19.10) |
| ≥30 | 63 722 (22.91) | 18.88 (20.95) |
| Missing | 856 (0.31) | 14.74 (19.91) |
| P-valuea | 9.45E-60 | |
| Smoking | ||
| Non-smokers | 148 824 (53.52) | 14.65 (15.36) |
| Ex-smokers | 100 892 (36.28) | 21.27 (19.66) |
| Smokersb | 7390 (2.66) | 25.27 (21.34) |
| Cigars/pipes | 1670 (0.60) | 32.12 (27.54) |
| ≤1 to 15 cigarettes/day | 10 966 (3.94) | 22.85 (23.97) |
| >15 cigarettes/day | 7423 (2.67) | 30.62 (33.23) |
| Missing | 928 (0.33) | 16.94 (17.07) |
| P-value a | <1.0E-300 | |
| Physical activity | ||
| Light | 81 647 (29.36) | 17.21 (18.68) |
| Moderate | 136 225 (48.99) | 18.26 (17.98) |
| High | 54 336 (19.54) | 19.59 (20.35) |
| Missing | 5885 (2.12) | 17.44 (23.27) |
| P-valuea | 4.61E-53 | |
| Education | ||
| Degree/professional | 131 960 (47.45) | 18.11 (17.38) |
| NVQ/CSE/A-levels | 98 291 (35.34) | 18.74 (19.73) |
| None of the above | 45 694 (16.43) | 17.42 (20.69) |
| Missing | 2148 (0.77) | 14.99 (17.18) |
| P-valuea | 3.61E-44 | |
| Townsend index | ||
| Quartile 1 (least deprived) | 70 647 (25.40) | 17.75 (16.88) |
| Quartile 2 | 70 429 (25.33) | 17.70 (17.44) |
| Quartile 3 | 69 692 (25.06) | 18.13 (18.71) |
| Quartile 4 (most deprived) | 66 993 (24.09) | 19.27 (21.92) |
| Missing | 332 (0.12) | 17.96 (21.41) |
| P-valuea | 2.95E-41 | |
| Self-rated health | ||
| Excellent | 47 534 (17.09) | 17.68 (16.01) |
| Good | 163 846 (58.92) | 18.06 (17.83) |
| Fair | 55 063 (19.80) | 19.07 (21.76) |
| Poor | 10 770 (3.87) | 18.04 (26.40) |
| Missing | 880 (0.32) | 18.85 (23.34) |
| P-valuea | 6.79E-17 | |
| Long-term illness | ||
| No | 186 033 (66.90) | 18.50 (18.04) |
| Yes | 86 100 (30.96) | 17.60 (20.31) |
| Missing | 5960 (2.14) | 17.25 (19.68) |
| P-valuea | 2.21E-88 |
BMI, body mass index; CSE, Certificate of Secondary Education; NVQ, National Vocational Qualification; SD, standard deviation.
P values are from the linear regression test with missing category excluded. We included age, sex, and assessment center, for adjustment.
Smokers without information on types of tobacco that they smoke.
Figure 1 illustrates a ‘J’-shaped association between the reported alcohol consumption and all-cause mortality. Those who did not drink or drank in small amounts had a higher risk of premature death compared to moderate drinkers, and heavy drinkers had an even greater risk. The curved pattern was consistent across CVD, cancer, digestive, and respiratory mortality. A similar pattern was observed when limiting the analysis to deaths occurring before the onset of the COVID-19 pandemic (up until January 1, 2020) (Supplementary Figure S4 and Supplementary Table S5, available as Supplementary data at IJE online). There was no statistical association between alcohol intake and COVID-19 mortality (P-value = 0.66 and Pnonlinearity = 0.77). Sensitivity analyses removing the BMI adjustment provided similar results (Supplementary Table S5, available as Supplementary data at IJE online). We also conducted sensitivity analyses stratifying by age- and self-reported health which provided support for some influences by selection bias and reverse causality in the conventional observational analyses (Supplementary Table S6, available as Supplementary data at IJE online).
Figure 1.
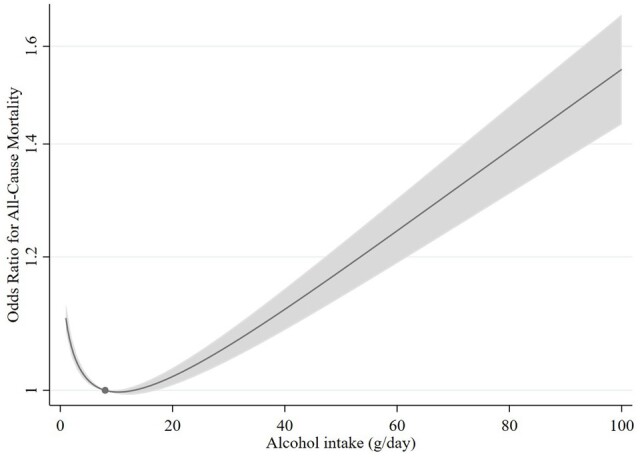
Nonlinear relationship between reported average daily alcohol consumption and all-cause mortality. The dot in the figure represents the reference point (8 grams per day), and the shaded region is the 95% confidence interval. The analyses are adjusted for sex, age, assessment center, birth location, educational status, Townsend deprivation index, body mass index, physical activity, and smoking
Figure 2 shows the findings from the linear MR analysis on genetically predicted alcohol consumption and mortality. The results support a positive relationship between alcohol consumption and all-cause mortality (OR by 8 grams higher daily alcohol intake 1.27, 95% confidence interval [CI] 1.16–1.39), CVD mortality (1.30, 1.10–1.53), cancer mortality (1.20, 1.08–1.33), and digestive mortality (2.06, 1.36–3.12). The associations between genetically predicted alcohol consumption and respiratory (1.32, 0.96–1.83) and COVID-19 disease mortality (1.46, 0.99–2.16) were imprecise. Findings from analyses using the GRS-based and summary-data-based approaches were generally consistent for all outcomes (Figure 2). Adjustment for smoking and TDI did not affect the results (Supplementary Table S7, available as Supplementary data at IJE online).
Figure 2.

Linear Mendelian randomization analyses of alcohol consumption with all-cause and cause-specific mortality. The point estimates are represented by squares and the 95% confidence intervals by horizontal bars. Adjustments were made for age, sex, assessment center, birth location, single nucleotide polymorphism array, and the top 40 genetic principal components. IVW—inverse-variance weighted Mendelian randomization; MR-PRESSO—Mendelian Randomization Pleiotropy RESidual Sum and Outlier; W-Median—weighted median Mendelian randomization; W-Mode—weighted mode Mendelian randomization; CVD—cardiovascular disease. g/day—grams per day
Nonlinear MR analyses did not provide any evidence to support the J-shaped association between alcohol intake and mortality suggested in the conventional observational analysis. In contrast, the doubly-ranked method confirmed the linear association, revealing that higher genetically predicted alcohol consumption was associated with increased mortality from all causes, including respiratory and COVID-19 diseases (Figure 3). Notably, the association between alcohol intake and all-cause mortality remained similar when restricted to deaths before the COVID-19 pandemic (Supplementary Figure S5, available as Supplementary data at IJE online).
Figure 3.

Genetically predicted alcohol intake and mortality by the mean level of alcohol intake from nonlinear Mendelian randomization analyses. (A) All-cause mortality; (B) CVD mortality; (C) cancer mortality; (D) respiratory mortality; (E) digestive mortality (F) COVID-19 mortality in the UK Biobank. For all models, Pnon-linearity ≥ 0.21 suggesting the linear model had the best fit. The odds ratio with the 95% confidence intervals for genetically predicted alcohol intake falls in the shaded area, and the dot represents the reference point (8 grams per day). Associations were adjusted for age, sex, assessment center, birth location, single nucleotide polymorphism array, and the top 40 genetic principal components. CVD—cardiovascular disease
Genetic effects on alcohol intake were stronger in strata with higher mean alcohol intake (Supplementary Figure S6, available as Supplementary data at IJE online), supporting the use of a doubly-ranked MR approach for the nonlinear analyses. Further details for instrument validation, stratum-specific estimates, and sensitivity analyses are provided in the Supplementary text pages 2–6, Supplementary Figures S7–S11, and Supplementary Table S8 (available as Supplementary data at IJE online).
Discussion
The proposed benefits of low-to-moderate alcohol intakes6,29 have been a source of long-standing debate and controversy. We investigated the association between alcohol intake and mortality using a genetic approach, confirming the risks associated with heavy consumption but no evidence to support any benefits for moderate intakes. Indeed, we observed linear increases in the risk of premature mortality across different contributing causes, suggesting that a reduction in the amount of alcohol consumed is likely to be beneficial regardless of the level of current intake.
Conventional observational analyses often show a ‘J-shaped’ relationship between alcohol consumption and mortality,5,11 which was also observed in our analyses. Previous studies have focused on cause-specific mortality risk,11 for example, finding evidence for a lower risk of CVD and cancer mortality in light or moderate drinkers compared to non-drinkers. However, benefits have not been consistently observed,30 and many have argued that the impact of light-to-moderate alcohol consumption on premature death is likely to be driven by biases such as reverse causality, confounding, and selection bias. Supporting this notion, a meta-analysis of 87 studies found that low-volume drinking may have protective effects, but only when selection biases and study quality are not taken into account.31 Here we demonstrate that the alcohol–mortality relationship differs between younger and older individuals and is strongly affected by adjustment to potential confounders and indicators of poor health, supporting the role of selection bias and reverse causality. We used an MR approach which can, at least to some extent, address these biases. All but one of the previous MR studies have assumed linear effects, using an analytical approach that would not have been able to detect protective effects from modest alcohol intake, should they exist. In the earlier nonlinear MR study, the authors observed a convex association,16 but the analytical method used assumed a constant association between the genetic instrument and alcohol intake, which we now show is strongly violated in this context (Supplementary Figure S6, available as Supplementary data at IJE online). In our analyses using the doubly-ranked approach,25 we can relax this assumption, and while we may not be able to fully discount possible nonlinearity, results obtained across all our analyses are more compatible with a linear effect. This was also suggested by an earlier study that approximated alcohol intakes based on regional location and showed that the alcohol-related cardiovascular risks are similar regardless of the average level of intake.15
Multiple mechanisms may contribute to the harmful effects of alcohol on cells and tissues, and the mediating pathways may differ between different causes of death. One mechanism with a possible broad impact involves the impact of alcohol metabolism on the body's systemic oxidative and inflammatory state, which generates toxic intermediates and metabolic stress.32–34 Additionally, alcohol exerts a direct effect on cellular components, which alters their biological function.32 Consequently, alcohol intake may induce chronic inflammation and metabolic changes, which can increase the risk of several types of deaths. Particularly relevant for cardiovascular health, drinking alcohol can raise blood pressure,35 and if consumed excessively, these effects may be aggravated by inflammation and harm to the heart muscles.36 Contrary to popular belief that moderate alcohol consumption may enhance heart health by increasing high-density lipoprotein (HDL) cholesterol and adiponectin levels while lowering fibrinogen levels,37 studies have shown that alcohol intake can have the opposite effect. It can raise low-density lipoprotein (LDL) cholesterol levels and decrease HDL cholesterol levels in the bloodstream.38 Furthermore, multiple MR studies and RCTs have found no evidence linking elevated levels of HDL cholesterol or fibrinogen to a reduced risk of CVD.39 Acetaldehyde, the toxic by-product of alcohol metabolism, can damage DNA and other cellular components, contributing to cancer mortality. Moreover, ethanol consumption can suppress the immune system, alter hormonal and chemical levels, and facilitate the development of cancer in various organs.40,41 Alcohol can also impair liver function, increase inflammation in the digestive tract, interfere with nutrient absorption, lead to deficiencies, and cause digestive problems such as acid reflux and heartburn, or irritate and damage the digestive tract lining.42 Lastly, alcohol can disrupt the functioning of airway cilia and alveolar macrophages, induce mast cells, impair the immune system, and cause inflammation in the respiratory tract.43,44
Strengths of our study include the large sample and the genetic analysis approach which allowed us to explore the association between moderate alcohol consumption and mortality risk largely avoiding influences from reverse causality and confounding which commonly bias conventional epidemiological analyses of observational studies. Our approach also allows for the exploration of causal associations at any level of intake without subjecting participants to potential harm, which is unlikely to apply to any RCT conducted in this context. To ensure the consistency of our estimates, we utilized multiple linear MR methods. To the best of our understanding, this is the first study of its kind to employ the doubly-ranked method for nonlinear MR analysis to examine multiple cause-specific mortality types. This new method allows us to overcome the assumption that the association between the genetic instrument (here, alcohol GRS) remains constant across differing levels of alcohol consumption, which may be violated and lead to a bias in previous nonlinear MR studies.17,25 However, our study also has some limitations. As information on alcohol consumption is typically obtained through self-report, calculated amounts may not be entirely accurate. This may introduce bias into the stratification of individuals into different strata and on weights used to calculate the genetically predicted intakes. Although we utilized several MR methods to address issues of pleiotropy and weak instrument bias, we cannot fully discount-related effects. Indeed, the alcohol GRS was associated with TDI and smoking, but it is uncertain whether this reflects pleiotropy or downstream effects of higher alcohol intakes. Adjustment for TDI and smoking did not materially affect results. Furthermore, while there is some evidence supporting the relevance of our findings to other ethnic groups,11 we restricted the sample to white-British individuals, which reduces the generalizability of our results to individuals from other ancestries. Like all MR studies, our use of genetic instruments to approximate average effects over the life course may not fully capture the true biological association between alcohol consumption and mortality risk, which could vary in shape and strength at different life stages and be more complex than reflected in our study. MR cannot eliminate bias from competing risks, hence, causal estimates for cause-specific mortalities may be biased toward the null, especially for conditions that typically affect people of older age. The UK Biobank, despite its large sample size, has only a 5% response rate and may not be representative of the general public in the UK. While selection bias is likely to affect findings from linear MR studies less compared to other designs,45 it is difficult to determine the extent to which selection may have affected our MR analysis. In particular, differential selection bias can induce genetic associations within strata of the population, even for the doubly-ranked method.46 We have adjusted for age and sex, as these are the strongest predictors of selection, which should mitigate the influence of differential selection based on these variables.47 Nevertheless, some residual bias due to different selections cannot be ruled out.
Genetic evidence strongly suggests that as alcohol consumption increases, there is a linear increase in the risk of premature death, including from specific causes such as CVD, cancer, and digestive illness, with no evidence for any protection by modest intakes. While the greatest mortality risks are associated with heavy drinking, public health initiatives should prioritize efforts to reduce alcohol intakes at all levels of consumption. A re-evaluation of current public policies regarding drinking guidelines may be warranted.
Ethics approval
This research was conducted under UK Biobank project number 20175. The UK Biobank was approved by the National Information Governance Board for Health and Social Care and the North West Multicentre Research Ethics Committee (11/NW/0382).
Supplementary Material
Contributor Information
Nigussie Assefa Kassaw, Australian Centre for Precision Health, University of South Australia, Adelaide, Australia; Clinical & Health Sciences, University of South Australia, Adelaide, Australia; School of Public Health, Addis Ababa University, Addis Ababa, Ethiopia; South Australian Health and Medical Research Institute, Adelaide, Australia.
Ang Zhou, Australian Centre for Precision Health, University of South Australia, Adelaide, Australia; Clinical & Health Sciences, University of South Australia, Adelaide, Australia; South Australian Health and Medical Research Institute, Adelaide, Australia; Medical Research Council Biostatistics Unit, University of Cambridge, Cambridge, UK.
Anwar Mulugeta, Australian Centre for Precision Health, University of South Australia, Adelaide, Australia; Clinical & Health Sciences, University of South Australia, Adelaide, Australia; South Australian Health and Medical Research Institute, Adelaide, Australia; Department of Pharmacology, College of Health Sciences, Addis Ababa University, Addis Ababa, Ethiopia.
Sang Hong Lee, Australian Centre for Precision Health, University of South Australia, Adelaide, Australia; South Australian Health and Medical Research Institute, Adelaide, Australia; Allied Health & Human Performance, University of South Australia, Adelaide, Australia.
Stephen Burgess, Medical Research Council Biostatistics Unit, University of Cambridge, Cambridge, UK; British Heart Foundation Cardiovascular Epidemiology Unit, University of Cambridge, Cambridge, UK.
Elina Hyppönen, Australian Centre for Precision Health, University of South Australia, Adelaide, Australia; Clinical & Health Sciences, University of South Australia, Adelaide, Australia; South Australian Health and Medical Research Institute, Adelaide, Australia.
Data availability
The data used in this research can be accessed by researchers who meet the required criteria and obtain necessary approvals from the UK Biobank access management committee at the University of Oxford. More information about accessing the research database can be found on the UK Biobank website at https://www.ukbiobank.ac.uk/.
Supplementary data
Supplementary data are available at IJE online.
Authors contributions
Conception and design: N.A.K. and E.H. Data access: E.H. Curated and managed the data: N.A.K., A.Z., and A.M. Analyses and interpretation of the findings: N.A.K., A.Z., A.M, H.L., S.B., and E.H. Wrote the first draft of the article: N.A.K. Critical revision of the manuscript and ensure important intellectual content: A.Z., A.M, H.L., S.B., and E.H. Approval of the final version of the manuscript: N.A.K., A.Z., A.M, H.L., S.B., and E.H. Study supervision: A.Z., A.M., H.L., and E.H. Obtaining funding: E.H. Guarantor: N.A.K. and E.H.
Funding
This work was supported by the Australian Government Research Training Program Scholarship to N.A.K.; National Health and Medical Research Council of Australia (Grant numbers GT1123603 and GT2025349); Medical Research Future Fund of Australia (Grant number MRF2007431) to E.H.; and Wellcome Trust (Grant number 225790/Z/22/Z); United Kingdom Research and Innovation Medical Research Council (Grant number MC_UU_00002/7); and National Institute for Health Research Cambridge Biomedical Research Centre (Grant number NIHR203312) to SB.
Conflict of interest
None declared.
References
- 1. Bryazka D, Reitsma MB, Griswold MG. et al. Population-level risks of alcohol consumption by amount, geography, age, sex, and year: a systematic analysis for the Global Burden of Disease Study 2020. Lancet 2022;400:185–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Esser MB, Leung G, Sherk A. et al. Estimated deaths attributable to excessive alcohol use among US adults aged 20 to 64 years, 2015 to 2019. JAMA Netw Open 2022;5:e2239485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bergmann MM, Rehm J, Klipstein-Grobusch K. et al. The association of pattern of lifetime alcohol use and cause of death in the European prospective investigation into cancer and nutrition (EPIC) study. Int J Epidemiol 2013;42:1772–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schröder H, Masabeu A, Marti MJ. et al. ; REGICOR Investigators. Myocardial infarction and alcohol consumption: a population-based case-control study. Nutr Metab Cardiovasc Dis 2007;17:609–15. [DOI] [PubMed] [Google Scholar]
- 5. Petrone AB, Gaziano JM, Djoussé L.. Alcohol consumption and risk of death in male physicians with heart failure. Am J Cardiol 2014;114:1065–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. van der Heide FCT, Eussen S, Houben A. et al. Alcohol consumption and microvascular dysfunction: a J-shaped association: The Maastricht Study. Cardiovasc Diabetol 2023;22:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaitkevičiūtė J, Gobiņa I, Janik-Koncewicz K. et al. Alcohol control policies reduce all-cause mortality in Baltic Countries and Poland between 2001 and 2020. Sci Rep 2023;13:6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. US NIH Advisory Committee to the Director (ACD). ACD Working Group for Review of the Moderate Alcohol and Cardiovascular Health Trial. 2018. https://acd.od.nih.gov/working-groups/machtrial.html (11 December 2023, date last accessed).
- 9. Davies NM, Holmes MV, Davey Smith G.. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 2018;362:k601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Davey Smith G, Ebrahim S.. ‘Mendelian randomization’: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol 2003;32:1–22. [DOI] [PubMed] [Google Scholar]
- 11. Hu C, Huang C, Li J. et al. Causal associations of alcohol consumption with cardiovascular diseases and all-cause mortality among Chinese males. Am J Clin Nutr 2022;116:771–79. [DOI] [PubMed] [Google Scholar]
- 12. Lankester J, Zanetti D, Ingelsson E, Assimes TL.. Alcohol use and cardiometabolic risk in the UK Biobank: a Mendelian randomization study. PLoS One 2021;16:e0255801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee SJ, Cho YJ, Kim JG. et al. ; CRCS-5 Investigators. Moderate alcohol intake reduces risk of ischemic stroke in Korea. Neurology 2015;85:1950–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Andrews SJ, Goate A, Anstey KJ.. Association between alcohol consumption and Alzheimer's disease: A Mendelian randomization study. Alzheimers Dement 2019;16:345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Millwood IY, Walters RG, Mei XW. et al. Conventional and genetic evidence on alcohol and vascular disease aetiology: a prospective study of 500 000 men and women in China. Lancet 2019;393:1831–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Biddinger KJ, Emdin CA, Haas ME. et al. Association of habitual alcohol intake with risk of cardiovascular disease. JAMA Netw Open 2022;5:e223849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Burgess S. Violation of the constant genetic effect assumption can result in biased estimates for non-linear Mendelian randomization. Hum Hered 2023;88:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sudlow C, Gallacher J, Allen N. et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med 2015;12:e1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bycroft C, Freeman C, Petkova D. et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018;562:203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Steindel SJ. International classification of diseases, 10th edition, clinical modification and procedure coding system: descriptive overview of the next generation HIPAA code sets. J Am Med Inform Assoc 2010;17:274–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Department of Health. Alcohol Guidelines Review–Report from the Guidelines Development Group to the UK Chief Medical Officers. Department of Health London. 2016. https://assets.publishing.service.gov.uk/media/5a7507d8ed915d3c7d529d6f/GDG_report-Jan2016.pdf (11 December 2023, date last accessed).
- 22. Liu M, Jiang Y, Wedow R. et al. ; 23andMe Research Team; HUNT All-In Psychiatry. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019;51:237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Royston P, Altman DG.. Regression using fractional polynomials of continuous covariates—parsimonious parametric modeling. J R Stat Soc Ser C Appl Stat 1994;43:429–67. [Google Scholar]
- 24. Palmer TM, Sterne JA, Harbord RM. et al. Instrumental variable estimation of causal risk ratios and causal odds ratios in Mendelian randomization analyses. Am J Epidemiol 2011;173:1392–403. [DOI] [PubMed] [Google Scholar]
- 25. Tian H, Mason AM, Liu C, Burgess S.. Relaxing parametric assumptions for non-linear Mendelian randomization using a doubly-ranked stratification method. PLoS Genet 2023;19:e1010823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Slob EAW, Burgess S.. A comparison of robust Mendelian randomization methods using summary data. Genet Epidemiol 2020;44:313–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hemani G, Zheng J, Elsworth B. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mason AM, Burgess S.. Software Application Profile: SUMnlmr, an R package that facilitates flexible and reproducible non-linear Mendelian randomization analyses. Int J Epidemiol 2022;51:2014–19. [Google Scholar]
- 29. Facchini F, Chen YD, Reaven GM.. Light-to-moderate alcohol intake is associated with enhanced insulin sensitivity. Diabetes Care 1994;17:115–19. [DOI] [PubMed] [Google Scholar]
- 30. Wood AM, Kaptoge S, Butterworth AS. et al. ; Emerging Risk Factors Collaboration/EPIC-CVD/UK Biobank Alcohol Study Group. Risk thresholds for alcohol consumption: combined analysis of individual-participant data for 599 912 current drinkers in 83 prospective studies. Lancet 2018;391:1513–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stockwell T, Zhao J, Panwar S, Roemer A, Naimi T, Chikritzhs T.. Do “moderate” drinkers have reduced mortality risk? A systematic review and meta-analysis of alcohol consumption and all-cause mortality. J Stud Alcohol Drugs 2016;77:185–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Molina PE, Hoek JB, Nelson S. et al. Mechanisms of alcohol-induced tissue injury. Alcohol Clin Exp Res 2003;27:563–75. [DOI] [PubMed] [Google Scholar]
- 33. Wang HJ, Zakhari S, Jung MK.. Alcohol, inflammation, and gut-liver-brain interactions in tissue damage and disease development. World J Gastroenterol 2010;16:1304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wu D, Cederbaum AI.. Alcohol, oxidative stress, and free radical damage. Alcohol Res Health 2003;27:277–84. [PMC free article] [PubMed] [Google Scholar]
- 35. Chen L, Davey Smith G, Harbord RM, Lewis SJ.. Alcohol intake and blood pressure: a systematic review implementing a mendelian randomization approach. PLoS Med 2008;5:e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zakhari S. Alcohol and the cardiovascular system: molecular mechanisms for beneficial and harmful action. Alcohol Health Res World 1997;21:21–29. [PMC free article] [PubMed] [Google Scholar]
- 37. Nova E, San Mauro-Martín I, Díaz-Prieto LE, Marcos A.. Wine and beer within a moderate alcohol intake is associated with higher levels of HDL-c and adiponectin. Nutr Res 2019;63:42–50. [DOI] [PubMed] [Google Scholar]
- 38. Brien SE, Ronksley PE, Turner BJ, Mukamal KJ, Ghali WA.. Effect of alcohol consumption on biological markers associated with risk of coronary heart disease: systematic review and meta-analysis of interventional studies. BMJ 2011;342:d636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van de Luitgaarden IAT, van Oort S, Bouman EJ. et al. Alcohol consumption in relation to cardiovascular diseases and mortality: a systematic review of Mendelian randomization studies. Eur J Epidemiol 2022;37:655–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Seitz HK, Stickel F.. Molecular mechanisms of alcohol-mediated carcinogenesis. Nat Rev Cancer 2007;7:599–612. [DOI] [PubMed] [Google Scholar]
- 41. Boffetta P, Hashibe M.. Alcohol and cancer. Lancet Oncol 2006;7:149–56. [DOI] [PubMed] [Google Scholar]
- 42. Pohl K, Moodley P, Dhanda AD.. Alcohol’s impact on the gut and liver. Nutrients 2021;13:3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Simet SM, Sisson JH.. Alcohol's effects on lung health and immunity. Alcohol Res 2015;37:199–208. [PMC free article] [PubMed] [Google Scholar]
- 44. Matsuse H, Fukushima C, Shimoda T, Sadahiro A, Kohno S.. Effects of acetaldehyde on human airway constriction and inflammation. Novartis Found Symp 2007;285:97–106. [DOI] [PubMed] [Google Scholar]
- 45. Gkatzionis A, Burgess S.. Contextualizing selection bias in Mendelian randomization: how bad is it likely to be? Int J Epidemiol 2019;48:691–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hamilton FW, Hughes DA, Spiller W, Tilling K, Smith GD.. Non-linear Mendelian randomization: evaluation of biases using negative controls with a focus on BMI and Vitamin D. medRxiv; doi: 10.1101/2023.08.21.23293658, 23 August 2023, preprint: not peer reviewed. [DOI] [Google Scholar]
- 47. Burgess S, Sun Y-Q, Zhou A, Buck C, Mason AM, Mai X-M.. Body mass index and all-cause mortality in HUNT and UK Biobank studies: revised non-linear Mendelian randomization analyses. medRxiv; doi: 10.1101/2023.10.31.23297612; 31 October 2023, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data used in this research can be accessed by researchers who meet the required criteria and obtain necessary approvals from the UK Biobank access management committee at the University of Oxford. More information about accessing the research database can be found on the UK Biobank website at https://www.ukbiobank.ac.uk/.