

Abstract

The biomarker 5-chlorocytosine (5ClC) appears in the DNA of inflamed tissues. Replication of a site-specific 5ClC in a viral DNA genome results in C → T mutations, which is consistent with 5ClC acting as a thymine mimic in vivo. Direct damage of nucleic acids by immune-cell-derived hypochlorous acid is one mechanism by which 5ClC could appear in the genome. A second, nonmutually exclusive mechanism involves damage of cytosine nucleosides or nucleotides in the DNA precursor pool, with subsequent utilization of the 5ClC deoxynucleotide triphosphate as a precursor for DNA synthesis. The present work characterized the mutagenic properties of 5ClC in the nucleotide pool by exposing cells to the nucleoside 5-chloro-2′-deoxycytidine (5CldC). In both Escherichia coli and mouse embryonic fibroblasts (MEFs), 5CldC in the growth media was potently mutagenic, indicating that 5CldC enters cells and likely is erroneously incorporated into the genome from the nucleotide pool. High-resolution sequencing of DNA from MEFs derived from the gptΔ C57BL/6J mouse allowed qualitative and quantitative characterization of 5CldC-induced mutations; CG → TA transitions in 5′-GC(Y)-3′ contexts (Y = a pyrimidine) were dominant, while TA → CG transitions appeared at a much lower frequency. The high-resolution mutational spectrum of 5CldC revealed a notable similarity to the Catalogue of Somatic Mutations in Cancer mutational signatures SBS84 and SBS42, which appear in human lymphoid tumors and in occupationally induced cholangiocarcinomas, respectively. SBS84 is associated with the expression of activation-induced cytidine deaminase (AID), a cytosine deaminase associated with inflammation, as well as immunoglobulin gene diversification during antibody maturation. The similarity between the spectra of AID activation and 5CldC could be coincidental; however, the administration of 5CldC did induce some AID expression in MEFs, which have no inherent expression of its gene. In summary, this work shows that 5CldC induces a distinct pattern of mutations in cells. Moreover, that pattern resembles human mutational signatures induced by inflammatory processes, such as those triggered in certain malignancies.
Introduction
Inflammation is an important and essential consequence of immune system activation. One of the ways the inflammatory response combats invading pathogens is through the generation of reactive chemicals such as reactive oxygen, nitrogen, and halogen species.1 While acute inflammation is usually beneficial in the immediate defense against invading pathogens, prolonged exposures to inflammatory byproducts can lead to collateral damage in the surrounding otherwise healthy tissue, causing cell death or genetic and epigenetic modifications.1−4 In fact, it is well-established that chronic inflammation is a risk factor associated with the development of many human cancers.5−10 One of the chemicals closely associated with chronic inflammation is hypochlorous acid (HOCl), which is produced by myeloperoxidase in activated neutrophil granulocytes (Figure 1A).11−14 HOCl and HOCl-derived reactive chlorine species such as N-chloramines are potent chlorinators that, upon reaction with cellular macromolecules, give rise to damage products such as chlorohydrins in lipids, chlorinated tyrosines in proteins, and chlorinated nucleobases.15−18 5-Chlorocytosine (5ClC) is the most abundant chlorinated nucleobase found in inflamed tissues.17,19−21 Elevated levels of 5ClC are found in both murine models and human patients with inflammatory bowel diseases.22,23 The differences between the levels of 5ClC and other biomarkers of inflammation such as 7,8-dihydro-8-oxoguanine (8OG)22,23 suggest that 5ClC is inefficiently repaired, and therefore, its accumulation in the genomes of chronically inflamed tissues could play a role in the initiation or progression of malignancy, but it is still unclear as to how and to what extent it triggers and promotes these processes.
Figure 1.

Model depicting the mechanism by which a unique mutational spectrum is generated owing to the incorporation of 5CldC into the genome via the nucleotide pool. (A) Formation of HOCl through inflammation-induced neutrophil activation. (B) One of the ways HOCl can indirectly generate 5ClC in DNA is through reacting with dCTP in the nucleotide pool. Subsequently, 5CldCTP gets incorporated into the genome by DNA polymerases during replication. 5ClC in DNA can form either a mutagenic pairing with A or a nonmutagenic pairing with G. 5CldC provided in the media of either (C) E. coli or (D) MEFs in cell culture causes mutations in genomic DNA that were measured using two mutagenic end points. Namely, (E) the traditional gpt assay, which measures mutation frequency based on the 6-TG-resistant mutant phenotype, and (F) duplex sequencing, which reveals the types and context dependencies of mutations.
Previous studies have shown that 5ClC can induce both epigenetic and genetic changes. Formation of 5ClC in CpG islands can alter DNA methylation patterns through its ability to mimic the cognate epigenetic marker 5-methylcytosine (m5C), and thus, it misdirects the activity of one or more cytosine DNA methyl transferases,24 resulting in changes in gene expression. Moreover, in our previous work, we have shown that 5ClC, when present in a DNA template both in vitro and in replicating cells, is intrinsically mutagenic, inducing C → T transitions at frequencies of 3–9% depending on the polymerase employed.25 This finding is significant considering that cells in inflamed tissues facing an increased burden of chlorination damage, like 5ClC, also experience elevated levels of replication,26 which might explain the observed higher rate of mutagenesis and subsequent increase in the risk of progression toward cancer.
In addition to reacting directly with DNA, HOCl and N-chloramines can also chlorinate the nucleotide pool.15 Therefore, 5ClC could also appear in the genome due to the formation of 5-chloro-2′-deoxycytidine triphosphate (5CldCTP) and its subsequent use by replicative or repair polymerases (Figure 1B). Additionally, recent work on chlorinated nucleotides shows that, in addition to being predictive biomarkers of inflammation, they might also play a role in disease progression by increasing the expression of proinflammatory cytokines and chemokines.27
In the present work, we characterized the mutagenic consequences of 5ClC in the nucleotide pool. Specifically, we showed that supplementation of growth media with 5-chloro-2′-deoxycytidine (5CldC), a bioavailable deoxynucleoside, led to significantly increased mutant fractions in both Escherichia coli and gptΔ mouse embryonic fibroblasts (MEFs) (Figure 1C–E). High-resolution sequencing of genomic DNA collected from MEFs treated with 5CldC yielded a novel mutational pattern dominated by CG → TA transitions, alongside a minority of TA → CG transitions. Intriguingly, the pattern of CG → TA mutations featured a scarcity of mutations in CpG sites (the sites of m5C formation), with mutations occurring mainly in 5′-GC(Y)-3′ contexts (Y = a pyrimidine). The observation of context-dependent hot and cold spots for mutagenesis indicated a distinctive mechanism for 5CldC-induced mutations. When compared with the Catalogue of Somatic Mutations in Cancer (COSMIC) mutational signatures collection, the high-resolution mutational spectrum (HRMS) of 5CldC was found to be similar to mutational signatures SBS84 and SBS42, which appear in human lymphoid tumors28 and in occupationally induced cholangiocarcinomas,29 respectively. SBS84 is associated with the upregulation of activation-induced cytidine deaminase (AID),28 which functions to diversify immunoglobulin genes by inducing C → T mutations during antibody maturation. Interestingly, AID is also known to be highly expressed in inflammation-associated cancers.30−34 Further investigation revealed that 5CldC exposure actually induced the expression of AID in MEFs. Nevertheless, the timing and the magnitude of AID induction, together with other features of the mutational spectrum (i.e., mutational strand bias), suggested that, at least in MEFs under the present experimental conditions, 5CldC mutagenesis was largely AID-independent. In sum, the present work describes the mutational consequences of 5CldC administered to MEFs in growth media and showcases the MEF cell system coupled with duplex sequencing as a powerful tool for studying mutational processes and their associated patterns in living cells.
Experimental Procedures
Chemicals
5CldC was purchased from Alchem Laboratories (Alachua, FL). The identity and purity of 5CldC were confirmed by NMR (see Figures S1 and S2 in Supporting Information). Deoxycytidine (cat. no. D3897), rifampicin (cat. no. R7382), chloramphenicol, and 6-thioguanine (6-TG) were from Sigma-Aldrich (St. Louis, MO).
Bacterial Cell Culture
E. coli strain HK81 (AB1157 nalA) stocks were maintained at −80 °C in Luria–Bertani (LB) broth media containing 15% glycerol. Periodically, the stock was streaked on LB-agar plates and grown overnight at 37 °C to select single-cell colonies.
Rifampicin Forward Mutation Assay
One colony of freshly streaked E. coli was collected with a sterile applicator and transferred to 1 mL of LB media. The titer of the resulting suspension was estimated by an optical density measurement at 600 nm (OD600). A volume of this stock corresponding to ∼500 cells was then used to inoculate 10 mL of LB (vehicle control) or 10 mL of LB containing 1 mM nucleosides [deoxycytidine (dC) or 5CldC]. The cultures were incubated at 37 °C on drum agitators for 7 h. Suitable dilutions of these cultures were then plated on LB-agar plates (to estimate total cell count) and on LB-agar plates containing 50 μg/mL rifampicin [to estimate number of rifampicin-resistant (RifR) colonies]. Each treatment was performed in a minimum of 3 biological replicates; each plating was done in 2 technical replicates. The mutant fraction was calculated as the ratio of number of RifR colonies over the total number of cells (per unit volume of culture).
Mammalian Cell Culture
SV40-immortalized MEFs were previously isolated from the gptΔC57BL/6J mouse as described in Thongararm et al.35 Immortalized MEFs were maintained in growth media [high glucose DMEM containing 200 mM GlutaMax (Gibco), 10% FBS (VWR Life Sciences), 1 mM sodium pyruvate (Gibco), 100 IU of penicillin, and 100 μg/mL streptomycin (Gibco)] in a humidified incubator at 37 °C with an atmosphere containing 5% CO2. Cells were shown to be mycoplasma-free by PCR.
ATP Luminescence Cell Viability Assay
Cells were seeded in 96-well plates at a density of 2.5 × 103 cells/well and incubated for 24 h. After 24 h, growth media were changed to fresh growth media (the untreated control), varied concentrations of dC in growth media, or varied concentrations of 5CldC in growth media. Cells were incubated for another 24, 48, or 72 h before luminescence signals were measured according to the protocol for Promega CellTiter-Glo 2.0. In short, 65 μL of CellTiter-Glo 2.0 Reagent was added to each well; then, the plate was shaken for 2 min before incubation at room temperature for 10 min. After incubation, the luminescence signals were measured using a SpectraMax M2e Microplate reader.
6-TG-Selected gpt Mutation Assay in MEFs
Cells were seeded in 100 mm tissue culture plates at a density of 5 × 105 cells/plate and incubated for 24 h. After 24 h, growth media were changed to fresh growth media, 0.3 mM dC in growth media, or 0.3 mM 5CldC in growth media. Cells were incubated for another 72 h, after which cells were harvested and washed twice with PBS (Gibco).
The mutational properties of 5CldC in mammalian cells were measured using a variant of the gpt assay developed by Nohmi et al.36 This is the traditional technique used to detect small mutations that occur in the 459 bp gpt transgene in the gptΔ mouse. MEFs that were isolated from the gptΔ mouse contain approximately 80 copies of the gpt transgene in the genome.35 For the traditionally described gpt assay, genomic DNA from 2 × 106 cells per group was prepared using the RecoverEase DNA Isolation Kit (Agilent Technologies). The λ-EG10 phages were packaged in vitro from genomic DNA using the Transpack packaging extract (Agilent Technologies) and then transfected into E. coli YG6020 expressing Cre-recombinase, generating a 6.4 kb plasmid carrying the gpt and chloramphenicol acetyltransferase genes. These bacteria were cultured on selective media containing 25 μg/mL chloramphenicol and 25 μl/mL 6-TG or 25 μg/mL chloramphenicol alone. 6-TG resistance was confirmed by the regrowth of colonies on plates containing chloramphenicol and 6-TG. The mutant frequency of each group was calculated as the ratio of total 6-TG-resistant colonies to the average of chloramphenicol-resistant colonies.
Duplex Consensus Sequencing
The gpt assay described above is often used for fast and accurate measurement of mutations in animal tissues and cell culture, but the mutation calling is limited only to mutations that can be phenotypically selected in the gpt gene.35,36 The advent of duplex consensus sequencing (DCS), however, allowed the identification of mutations in an unbiased manner (including unselected mutations) and in a much larger genomic target (6.4 kb), as we previously demonstrated.37,38 More recently, hybrid-capture technology expanded the genomic target to 48 kb, allowing for the evaluation of mutations across the entire mouse genome and not just at the transgenic locus (TwinStrand Biosciences, Mutagenesis Kit). This study compared mutational spectra in both 6.4 and 48 kb targets, and the results showed excellent concordance.
In the present work, genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen) according to the protocol from the manufacturer. Approximately 1 μg of genomic DNA was used to prepare the DCS libraries using a Mouse Mutagenesis Kit (TwinStrand Biosciences, Inc.) according to the manufacturer’s protocol. The libraries were sequenced on an Illumina NovaSeq 6000 DNA sequencer on an S4 flow cell using a 150 bp paired-end protocol (MIT BioMicro Center).
Data Analysis and Plotting
The data generated from the TwinStrand assay were processed using the manufacturer’s pipeline (TwinStrand DuplexSeq Mutagenesis App, v3.15.0) running on DNANexus. The resulting mutation-position (.mut) files were further analyzed using in-house custom Python scripts (available on GitHub at https://github.com/essigmannlab/5CldC_spectrum). For each sample, a list of unique mutations (a mutation at a given genomic location is counted only once) was generated from which trinucleotide mutational spectra were constructed and normalized. Normalization is performed by dividing the mutational frequency at each trinucleotide sequence context by the frequency of that context in the target genomic region. The cosine similarity metric was used to compare mutational spectra generated here with themselves and with the single-base substitution (SBS) mutational signatures available from the COSMIC repository (COSMIC SBS v3.2). To generate the probability LOGO (pLOGO) plots, 15mer sequence contexts (7 bases upstream and downstream of each mutated site) were extracted for each C → T mutation and analyzed using the Schwartz probability logo generator (https://plogo.uconn.edu/).
Quantification of Activation-Induced Cytidine Deaminase
Cells were seeded in 100 mm plates and incubated for 24 h. After 24 h, growth media were changed to 0.3 mM 5CldC (or the dC control compound) and allowed to incubate for 0–72 h, after which cells were harvested at a density of 5 × 106 cells/plate and washed twice with PBS (Gibco). Thymus tissue was obtained from 16 week old RaDR-gptΔ mice.
qRT-PCR analysis was used to quantify the levels of AID mRNA in the 5CldC-treated immortalized gptΔ MEF cells. The following primers were used for this analysis: Aicda: forward: 5′-TGCTACGTGGTGAAGAGGAG-3′, reverse: 5′-TCCCAGTCTGAGATGTAGCG-3′; Actb: forward: 5′-GGCTGTATTCCCCTCCATCG-3′, reverse: 5′-CCAGTTGGTAACAATGCCATGT-3′. Messenger RNA was extracted using the RNeasy Mini Kit (Qiagen, 74134) and the RNase-Free DNase Set (Qiagen, 79254) and converted to cDNA using the QuantiTect Reverse Transcription Kit (Qiagen, 205311). qPCR was performed with the QuantiTect SYBR Green PCR Kit (Qiagen, 204143) by preincubating at 95 °C for 15 min, followed by 45 cycles at 95 °C for 15 s, 65 °C for 30 s, and 72 °C for 30 s.
Results
5CldC-Induced Mutations in E. coli in a Forward Mutation Assay
The ability of 5CldC to induce mutations in replicating E. coli was measured using the rifampicin forward mutation assay. Rifampicin (Rif) is a bactericidal antibiotic that works by binding to rpoB (RNA polymerase beta), inhibiting transcription, and leading to cell death. However, even a single point mutation in the binding pocket of rpoB can lead to an amino acid change that prevents rifampicin binding without significant disruption to rpoB function. Cells harboring such point mutations in rpoB become resistant to rifampicin and can be identified on plates containing Rif in the media. Many such point mutation sites exist in the rpoB gene, which makes the Rif forward mutation assay very sensitive. When E. coli cells were replicated in the presence of 5CldC (supplied in LB media at 1 mM concentration), the resulting number of RifR colonies was two orders of magnitude higher, as compared to background (cells grown in LB media alone) or control treatment (cells grown in the presence of 1 mM deoxycytidine) (Figure 2A). Notably, the presence of 5CldC in the media had only a mild inhibitory effect on cell growth (Figure S3 in the Supporting Information).
Figure 2.
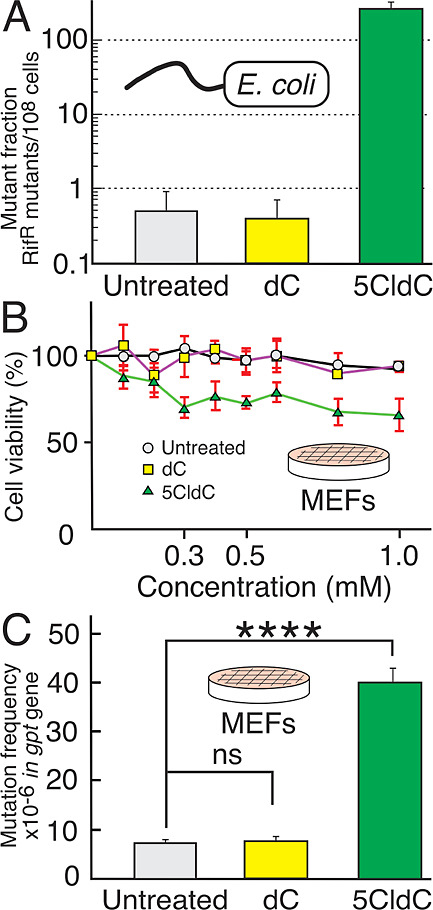
Cytotoxicity and mutagenicity of 5CldC. (A) 5CldC induced an increase in mutation frequency in E. coli as compared to untreated cells and cells treated with dC. (B) Effect of 5CldC at various concentrations on MEF growth and survival, measured at the 72 h time point by using the ATP assay described in the text. When compared to untreated and dC-treated controls, treatment with 5CldC induced a dose-dependent decrease in cell growth and survival up to a dose of 0.3 mM; the dose-dependent effect diminished at higher concentrations. (C) Mutation frequencies in MEFs as measured by the gpt assay described in the text. Untreated cells and dC controls showed a similarly low level of mutations at around 7–8 mutants/106gpt genes. By contrast, 5CldC-treated MEFs induced a large increase (∼5-fold) in mutation frequency as compared to the untreated cells and to the dC controls at the same concentration and time point (0.3 mM at 72 h).
5CldC-Induced Growth Inhibition and Point Mutations in a MEF Cell Line
Growth inhibition of 5CldC toward the MEFs was evaluated by an ATP cell viability assay. The results of this experiment provided the data needed to determine a proper treatment concentration and duration for the subsequent high-resolution mutational assay using DCS. Cells were treated with 5CldC in concentrations ranging from 0 to 1 mM. Cell viability was measured every 24 h (see Figure S4 in the Supporting Information). After 72 h at a 5CldC concentration of 0.3 mM, approximately 60–70% of the cells were viable when compared to the untreated control (Figure 2B). Based on historical experience with the assay, this treatment would likely produce enough independent mutants for enumeration and later sequencing. The treated cells at 0.3 mM were then harvested for mutational analysis. Notably, the dose-dependent effect of 5CldC toward cell growth and viability plateaus at doses greater than 0.3 mM, which indicates a mild toxic effect on the cells.
The ability of 5CldC to induce point mutations was evaluated initially by the gpt assay. The MEF cell line used in this study was isolated from the gptΔ C57BL/6J transgenic mouse, and it is therefore isogenic to the mouse model regularly used for in vivo evaluation of mutagenic compounds.38−40 These MEFs were also previously shown to be a useful tool for evaluating the mutagenicity of the toxicants N-methyl-N-nitrosourea, streptozotocin, and temozolomide.35,40 Untreated control, dC treated, and 5CldC-treated MEFs were collected at the 72 h time point and used for DNA isolation. The results from the gpt assay showed that 5CldC, when added to the medium at the concentration of 0.3 mM for 72 h, was highly mutagenic, generating more than a 5-fold increase in mutation frequency when compared with the untreated control. By contrast, there was no significant effect observed from the dC-treated MEFs at the same concentration, as compared to the untreated control (Figure 2C).
5CldC Induces a Distinctive Mutational Pattern in MEFs
DNA samples were isolated from the untreated control MEFs, 0.3 mM 5CldC, and dC-treated MEFs at 72 h. DCS data show that the mutational spectrum generated by 5CldC was dominated by CG → TA, with a lower frequency of TA → CG mutations (Table 1A). There were no significant differences between the mutation types observed in the control and dC-treated cells, although the level of CG → AT mutations was slightly higher than the levels of other types of mutations in these controls.
Table 1. (A) Relative Amounts of Point Mutation Types Observed in the Genomes of Untreated Control, dC-Treated, and 5CldC-Treated MEFs; (B) Relative Amounts of C → T Substitution Mutations Observed in Each DNA Strand in the Genome of MEFs Treated with 5CldC.
| (A) types of substitution mutations induced by 5CldC | |||
|---|---|---|---|
| substitution type | control (%) | dC (%) | 5CldC (%) |
| Transitions | |||
| CG → TA | 39.1 | 26.8 | 86.2 |
| TA → CG | 9.3 | 16.8 | 7.9 |
| Transversion | |||
| CG → AT | 30.0 | 28.3 | 4.1 |
| CG → GC | 8.0 | 10.2 | 0.5 |
| TA → AT | 5.4 | 9.0 | 0.9 |
| TA → GC | 8.2 | 8.8 | 0.5 |
| total number of substitutions | 100 | 100 | 100 |
| (B) strand bias in 5CldC-treated MEFs | |||
|---|---|---|---|
| coding strand | noncoding strand | ratio | |
| number of C → T substitutions | 60.9% | 39.1% | 1.56 |
While the dominant mutation from 5CldC was always CG → TA, we did observe a significant strand bias in the frequency of the mutations. Specifically, the coding strand had 1.5 times more C → T mutations than the noncoding strand (Table 1B). This result might reflect the preferential repair of 5CldC in the DNA strand transcribed by the RNA polymerase (i.e., the noncoding strand).
Data from DCS also allow for further understanding of the types of mutations generated by 5CldC through the construction of an HRMS (Figure 3A,B), where the sequenced mutation data are plotted in three-base contexts 5′-NXN-3′ [the mutated base (X) is presented along with its 3′ and 5′ neighboring bases (N)]. This manner of data presentation can help to associate computationally a unique mutational spectrum generated by a mutagen with a specific mutational process. The DCS data, when plotted using the three-base context convention, showed that treatment with 0.3 mM 5CldC for 72 h induced a unique mutational spectrum in MEFs that was not seen in the untreated control (see Figure S5 in Supporting Information) nor in the dC-treated cells at the same concentration and time (Figure 3C). The unique mutational pattern was dominated by CG → TA transitions that occurred predominantly in 5′-CY-3′ contexts (where Y = a pyrimidine and the underscored C indicates the position of the mutation) (Figure 3A,B).
Figure 3.

HRMS obtained from DCS of DNA from MEFs treated with 5CldC and control compounds. The HRMS is plotted in three-base contexts, 5′-NXN-3′, where the mutated base (X) is presented along with its neighboring bases (N). There are 6 point mutation types and 16 three-base contexts for each mutation type, so there are (6 × 16) = 96 possible three-base context mutation types in total. (A,B) present independent experiments using two different strategies of duplex sequencing. In both, the HRMS was obtained from treating the MEFs with 5CldC at 0.3 mM. The results show a unique mutational pattern for 5CldC mutagenesis, dominated by CG → TA transitions, followed at a lower frequency by TA → CG mutations. (C) HRMS of dC-treated MEFs. (D,E) show human cancer mutational signatures SBS84 (D) and SBS42 (E). (F) is a heat map depicting quantitative comparisons expressed as pairwise cosine similarities among the HRMS of 5CldC-treated cells and human COSMIC mutational signatures.
Next, to get a more comprehensive understanding of the influence of neighboring bases toward 5CldC-induced mutagenesis, the 15-base sequence contexts with C → T mutations fixed at the zero (central) position were extracted from the DCS data. The extracted data were visualized using the pLOGO plot41 (Figure 4), where G and T were found to be overrepresented on the 5′ and 3′ flanks, respectively, of the observed C → T mutations in the 5CldC-treated samples (Figure 4A,B). By contrast, the most abundantly overrepresented base observed in the untreated control was G at the +1 position (Figure 4C), which we hypothesize to be due to the spontaneous deamination of 5-methylcytosine at CpG sites. Unexpectedly, the dC-treated samples did not show any overrepresented base (Figure 4D) compared to the untreated control.
Figure 4.

Visual representation of the C → T mutations in a 15-base sequence context. The C → T mutations are shown fixed at the zero position along with the letter code of the adjacent bases scaled in size in accord with their statistical significance. The horizontal red lines correspond to the p = 0.05 (log-odds value of ±3.05) statistically significant threshold according to a Bonferroni correction. (A) presents 5CldC-induced mutations obtained from mouse genomic DNA [n(fg) = 1,533, n(bg) = 10,776]. (B) presents 5CldC-induced mutations obtained from the gpt transgene [n(fg) = 601, n(bg) = 11,757]. (C) presents untreated control mutations obtained from mouse genomic DNA [n(fg) = 49, n(bg) = 10,776]. (D) presents dC control mutations obtained from mouse genomic DNA [n(fg) = 71, n(bg) = 10,776]. For the above, n(fg) denotes number of (foreground) 15mer sequence contexts where the central C mutates to a T; n(bg) denotes number of (background) 15mer sequence contexts within the sequenced space that contain a central C.
Cosine Similarity Comparison between the 5CldC-Induced Mutational Pattern and Mutational Signatures Obtained from Human Cancers
To address the biomedical relevance of the unique 5CldC mutational pattern, we compared the acquired pattern to the current collection of human-derived COSMIC mutational signatures. The COSMIC patterns were generated by nonnegative matrix factorization of tens of thousands of sequenced cancer genomes.42 Typically, one uses the metric of cosine similarity to compare complex mutational spectra, such as that of 5CldC, to the collection of mutational signatures from cancer genome databases. The mutational spectrum obtained from the treatment of the MEFs with 5CldC showed a significant cosine similarity to COSMIC signatures SBS84 and SBS42 (Figure 3 D–E), with a higher association (cosine similarity = 0.85) observed between our 5CldC spectra and signature SBS84 (Figure 3F). Additionally, when one considers only the C → T mutation portion of the spectrum, the cosine similarity score between the obtained mutational pattern and SBS84 increased to 0.90 (Figure 3F). A weaker but still significant (cosine similarity = 0.78) association was observed between the mutational pattern induced by 5CldC and that represented by SBS42 (Figure 3E). Signature SBS42 has been associated with the development of early onset cholangiocarcinoma in workers exposed to haloalkanes.29 The existence of a mechanistic link between 5CldC and cholangiocarcinoma is unknown at this time.
Additional investigative focus was put upon the association between the obtained mutational spectrum and signature SBS84, which is a mutational signature associated with the activity of AID, the activation-induced cytidine deaminase gene editing enzyme. AID expression has been linked to various inflammation-related cancers.30−34 In the present study, the induction of Aicda (the gene encoding the AID enzyme) over time was measured by RT-qPCR to address the possibility that AID may have been induced or perhaps could have contributed to the observed 5CldC-generated mutational spectrum. As a control, the mRNA expression of Aicda was detected in constitutively expressed positive control mouse thymus cells and in our MEFs treated with 0.3 mM 5CldC for 48 and 72 h; no expression was detected in the untreated control and in the 5CldC-treated MEFs at 0 and 24 h (Figure 5), as shown in melting curve plots (see Figure S6 in Supporting Information). There was, however, a 1.5-fold increase in the expression of Aicda between 48 and 72 h after treatment with 5CldC (Figure 5). The level of Aicda expression in the mouse thymus control was much higher as compared to the expression seen in the 5CldC-treated cells (Figure 5), a result that was expected, because this gene is known to be active in some T-cell populations resident in the thymus.43 While the observed enhanced expression of the gene responsible for AID was modest in our experiment, the results do leave open the possibility that the transcription of Aicda can be induced by 5CldC in the MEFs, a cellular system that would not ordinarily be expected to express this gene.
Figure 5.

Aicda expression in 5CldC-treated MEFs. MEFs were treated with 0.3 mM 5CldC, and subsequently, mRNA was isolated at various time points. Aicda expression was not detected (or below the detection limit) in the untreated control, the 0.3 mM dC-treated control, and the 24 h time points (see Figure S6 in Supporting Information). Expression of the Aicda gene was detected, however, at the 48 and 72 h time points in the 5CldC samples, showing an approximately 1.5-fold increase between the two time points. Although reliably detected, these levels of AID expression are significantly lower than what was observed in the positive control mouse thymus cells, which had expression of ∼50-fold higher than that in the 5CldC-treated MEFs at 48 h.
Discussion
Chronic inflammation is a known risk factor associated with the development of various cancers.5−10 Despite this established association, there is still significant uncertainty regarding the precise mechanisms by which prolonged inflammation contributes to the initiation or progression of malignancy. Many biological processes are hypothesized to be involved, such as macrophage-regulated angiogenesis,44 lymphatic metastases,45 and induction of cellular proliferation through chemokine-associated pathways.46,47 In this study, we focused on genetic instability triggered by 5ClC, a known biomarker of chronic inflammation and a known mutagen.22,48 Typically, 5ClC is produced when neutrophils generate HOCl during inflammatory episodes.15 While HOCl (or HOCl-derived reactive chloramines) can directly damage DNA forming 5ClC (Figure 1), an alternative pathway by which 5ClC could appear in the genome would be if HOCl were to react with deoxycytidine triphosphate (dCTP), or precursors to dCTP, in the nucleotide pool, leading to insertion of the chlorinated cytosine into the genome by a polymerase-mediated event. Similarly, the well-established mutagenic base, 8OG, is also mutagenic both when it forms in DNA49 and when it enters DNA via the nucleotide pool,50 illustrating the general importance of the nucleotide pool as a major source of mutations. Because many nucleic acid bases or nucleosides and several chemically similar agents used in anticancer or antiviral therapies enter cells by salvage pathways, in our experiments, we supplied the deoxynucleoside 5CldC in the growth medium. Previous work has shown that this route successfully leads to the incorporation of 5CldC into DNA.27
The results show that the provision of 5CldC in the growth medium produced a distinctive pattern of mutations in MEFs derived from the gptΔ C57BL/6J transgenic mouse. Mutagenicity analysis of 5CldC determined by the gpt assay revealed it to be a potent mutagen (Figure 2). DNA from treated cells was submitted to a detailed mutational analysis by DCS, a tool that has emerged in recent years as affording a better than 1000-fold increase in accuracy over conventional next-generation sequencing methods. The DCS data show that 5CldC caused mostly CG → TA and, to a lesser extent, TA → CG mutations (Figure 3 and Table 1). Figure 6 presents a model to help explain the origins of these mutations. The types of mutations observed were qualitatively in accord with predictions from replication studies on single 5CldC residues inserted at known positions in defined-sequence oligonucleotides in vitro and in viral genomes in vivo.25 It was found in this earlier work that 5CldC in DNA templates mispairs with adenine at a rate of around 5% depending on the DNA synthesis system used.25 Thus, 5CldC can pair with either guanine (the canonical base-pairing partner of cytosine) or adenine, which would generate a mutation. In the present work, we surmise that when 5CldCTP enters DNA from the pool, in principle, it can be incorporated either as a cytosine mimic opposite guanine or as a thymine mimic opposite adenine, which, taken together, would produce C → T and T → C mutations, respectively. In our experiments, we observed that C → T mutations highly dominated the mutational distribution (Table 1). It is thus likely that 5CldCTPs in the pool were incorporated mostly as cytosines during the first round of replication, and then, some of the inserted 5ClC lesions mispaired with adenine during subsequent rounds of replication, generating ultimately C → T mutations in the genome, as shown in Figure 6. This notion is reinforced by the observation that DNA polymerases do not display a significant preference when incorporating 5CldCTP as compared to dCTP opposite G.24 In contrast, 5CldCTP is much less favorably incorporated, as compared with dTTP, opposite adenine, as indicated by the much lower percentage of T → C mutations induced (Table 1).
Figure 6.

Proposed mechanism of 5CldC-induced mutagenesis. 5CldC enters the cell presumably by salvage uptake pathways and is phosphorylated to 5CldCTP. During replication (symbolized by R inside a circle), 5CldCTP is incorporated into DNA primarily opposite a G in the template strand forming a G:5ClC base pair. During a second round of replication, 5ClC in the nascent (blue) strand can pair either with a G or, less frequently, with an A, yielding either a G:5ClC or an A:5ClC base pair. The box inset to the lower left is based on the crystal structure previously determined to explain this mutagenic event.25 The duplex that contains the A:5ClC base pair then engages in a third round of DNA replication. A pairs with T during this replication cycle, thereby causing the mutation; an A:T base pair has replaced the G:C at the position where the replicative event originally inserted the 5ClC base. For convenience, the starting G:C and final A:T base pairs are included in yellow boxes. Also note that the A/G:5ClC base pairs in the right-most box will continue to generate C → T mutations unless the 5ClC base is repaired. Black circles under the duplexes denote the parental DNA strands, and the blue circles track the fate of the nascent, 5ClC-containing strand. The green lollipop depicts the chlorine group on 5ClC.
As an alternative hypothesis for the origin of the infrequent T → C mutations (the green bars in Figure 3A,B), it has been reported that 5CldC inside cells can be enzymatically deaminated to its uracil derivative, which could contribute to the TA → CG mutations upon replication.51,52 The extent of the conversion of 5CldC to 5CldU in the nucleotide pool relative to the conversion of 5CldC to 5CldCTP remains an important consideration that warrants further investigation. The dominance of the CG → TA mutations that we observe argues against 5CldU incorporation as a major mutagenic contributor. A few explanations are plausible. It is possible that the MEF cells that we are working with have relatively low levels of pool deaminases. Greer and co-workers have noted, in fact, that the levels of the deaminases that convert 5CldC to 5CldU vary considerably between different tissues and cell types.51,53,54 Another possibility is that even if a substantial portion of 5CldC is converted to 5CldU, enough 5CldCTP remains to result in the mutational pattern observed. Moreover, the incorporation of 5CldUTP from the pool may be mutagenically less consequential because of the DNA repair. The 5CldU:G mismatch is actually a great substrate for base excision repair initiated by glycosylases such as MUG and TDG.52,55
Interestingly, the dominant CG → TA mutations in the 5CldC spectrum also show a strong strand bias; we found 50% more C → T mutations (as opposed to G → A mutations) in the coding (nontranscribed) strand than in the noncoding (transcribed) strand (Table 1). Asymmetry in the distribution of mutations between the coding and noncoding DNA strands is usually a sign of the involvement of certain types of transcription-coupled DNA repair. DNA repair enzymes preferentially repair the noncoding (or transcribed) strand of the duplex. Therefore, if 5CldC were inserted equally in each of the strands of a transcriptionally active duplex, one would expect to find fewer mutations coming from the transcribed strand, which is what was observed. However, to date, there have been no reports of any repair factor for 5ClC in DNA. In support of the view that the adducts might be poorly repaired in vivo, it is noteworthy that a high level of 5ClC is observed in chronically inflamed tissues, relative to other biomarkers of inflammation such as 8OG and other oxidized DNA lesions.22,48 While mutagenesis is a serious concern for any naturally produced DNA lesion, 5CldC has additional concerning properties, including the possibility that it participates in reprogramming the epigenome. Because the 5-chloro group of 5CldC is at the same position as the methyl group of m5C, investigators have looked at the ability of 5CldC to affect the patterns of natural gene methylation, mediated by m5C DNA methyltransferases.24 Administration of the triphosphate nucleotide of 5CldC to cells results in an increase in the content of m5C at certain CpG sites, accompanied by transcriptional silencing. The overall level of m5C in the genome, however, is not noticeably affected.24
It is becoming increasingly clear that carcinogenesis involves a series of chemical and biological processes, each with a distinctive mutational spectrum that evolves increasing complexity over time.56 The individual mutational spectra, when combined, can theoretically reconstitute the composite spectrum for any sequenced tumor. It is therefore of central importance to uncover the mutational fingerprints of each biological and chemical step in malignant transformation. The unique HRMS of 5CldC presented as the distribution of point mutations across sequence contexts throughout the genome (Figure 3A,B) likely reflects a specific mutational process. Point mutations typically are displayed in three-base contexts, with the mutated base in the middle.42,57 Presentation of mutations in their three-base contexts stems from our understanding that point mutations are not distributed uniformly throughout the genome because each step of the mutational process (formation of the DNA lesion, lesion repair avoidance, and errors upon attempted replication past the lesion) is influenced by the DNA bases proximal to the mutated base.57−61 As shown in Figure 3A,B, 5CldC induced a unique mutational spectrum that is dominated by C → T mutations specifically in the context 5′-GCY-3′ (where C = the presumed site of mutation and Y = a pyrimidine); this pattern was not observed in either the untreated control or the dC-treated sample. Consequently, this unique mutational pattern reflected a mutational process that was triggered by exposure to 5CldC.
When compared with the COSMIC mutational signatures that are computationally derived from human tumors, the 5CldC-induced HRMS shows the closest similarity to SBS84 (Figure 3D). SBS84 was derived from chronic lymphocytic leukemia cases, and it has features that associate it with the expression of canonical AID.28,62,63 The AID-associated mutational signature has also been suggested to be involved in the etiology of certain B-cell lymphomas.62,63 AID is an enzyme primarily associated with the development of adaptive immunity and is encoded by the Aicda gene. When functioning normally, AID deaminates cytosines into uracils causing mutations in the variable (V) and switch (S) regions in the immunoglobulin loci of B cells.64 It is hypothesized to be the master regulator of secondary antibody diversification. AID activity generates a mutational signature that is characterized by C → T/G mutations at RCY motifs28 (R = purine, Y = pyrimidine). Due to its highly mutagenic nature, it is not surprising that AID is tightly regulated at multiple levels. Interestingly, AID expression has also been linked to various types of chronic inflammation-related cancers, such as colorectal cancer in patients with inflammatory bowel diseases,30−32 hepatocellular carcinoma in patients with an HCV infection,33 and gastric cancer in Helicobacter pylori-infected patients.34
In addition to finding an AID-like mutational pattern in the genome of our 5CldC-treated MEFs, we found that Aicda expression at the RNA level was slightly upregulated in the MEFs (Figure 5). Though not as high as the expression level of Aicda in immune cells (e.g., the thymus tissue used as a control), the observed overexpression might help to explain the distinctive 5CldC-induced mutational spectra observed here. As mentioned previously, we observe a strong strand bias in C → T mutations induced by 5CldC that might be explained by transcription-coupled repair. The same strand bias is seen for the SBS84 signature associated with AID activation, but the magnitude of strand bias is much lower.28 Therefore, it is possible that 5CldC-dependent mutagenesis and AID-dependent mutagenesis might be mechanistically linked with AID being potentially activated by 5CldC exposure.
The observation of an AID-like mutational spectrum and the slight induction of the Aicda gene beg several questions such as how does 5CldC exposure induce the expression of AID, and to what extent does 5CldC contribute to the development of inflammation-related cancer? Several studies have been carried out to investigate the role of chlorinated nucleosides in the induction of inflammation. It has been observed that exposure to chlorinated nucleosides, especially 5CldC and 8-chlorodeoxyguanosine, induces the expression of proinflammatory cytokines, including IL-1B expression and secretion in exposed macrophages.27 Additionally, the proinflammatory cytokines, IL-1B and TNF-α, have been shown to induce the expression of AID in human hepatocytes through NF-kB activation.65 Taken together, these findings raise the possibility that chlorinated nucleosides, specifically 5CldC, could contribute to carcinogenesis through mechanisms unrelated to their intrinsic mutagenic property. With the strong link between chronic inflammation and cancer, however, further studies on the biological properties of these chlorinated nucleosides might significantly improve our ability to understand the big picture underlying inflammation-related carcinogenesis while also providing novel targets for preventing or treating these types of diseases.
Glossary
Abbreviations
- HOCl
hypochlorous acid
- 5ClC
5-chlorocytosine
- 8OG
7,8-dihydro-8-oxoguanine
- m5C
5-methylcytosine
- 5CldCTP
5-chloro-2′-deoxycytidine triphosphate
- 5CldC
5-chloro-2′-deoxycytidine
- E. coli
Escherichia coli
- MEFs
mouse embryonic fibroblasts
- HRMS
high-resolution mutational spectrum
- AID
activation-induced cytidine deaminase
- 6-TG
6-thioguanine
- LB
Luria–Bertani
- OD600
optical density measurement at 600 nm
- dC
deoxycytidine
- RifR
rifampicin-resistant
- DCS
duplex consensus sequencing
- SBS
single-base substitution
- pLOGO
probability LOGO
- Rif
rifampicin
- rpoB
RNA polymerase beta
- dCTP
deoxycytidine triphosphate
Data Availability Statement
The sequencing data presented in this work are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) repository under the BioProject PRJNA1035149 (http://www.ncbi.nlm.nih.gov/bioproject/1035149). The Python scripts used for data analysis and visualization are available on GitHub (https://github.com/essigmannlab/5CldC_spectrum/).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.chemrestox.3c00358.
Identity and purity evaluation by NMR of the 5CldC nucleoside, growth inhibition properties of 5CldC in E. coli and MEF cells, background mutational spectrum of MEF cells, and validation of the qPCR probes used for Aicda expression (PDF)
Author Contributions
CRediT: Marisa Chancharoen conceptualization, formal analysis, investigation, methodology, validation, visualization, writing-original draft; Zhiyu Yang formal analysis, investigation, methodology, resources, validation; Esha D. Dalvie conceptualization, data curation, formal analysis, methodology, resources, validation, visualization; Nina Gubina data curation, formal analysis, investigation, methodology, resources, validation; Mathuros Ruchirawat funding acquisition, resources, supervision; Robert G. Croy data curation, formal analysis, investigation, methodology, software, supervision, writing-review & editing; Bogdan I. Fedeles conceptualization, data curation, formal analysis, investigation, methodology, resources, software, supervision, validation, visualization, writing-review & editing; John M. Essigmann conceptualization, data curation, funding acquisition, project administration, resources, supervision, visualization, writing-original draft, writing-review & editing.
The authors acknowledge financial support from grants from the U.S. National Institutes of Health: P30-ES002109, R01-CA080024, T32-ES007020; and by way of a Chulabhorn Graduate Institute Scholarship (CGS): CGS(2020)/02 to the first author.
The authors declare no competing financial interest.
Supplementary Material
References
- Mangerich A.; Dedon P. C.; Fox J. G.; Tannenbaum S. R.; Wogan G. N. Chemistry meets biology in colitis-associated carcinogenesis. Free Radical Res. 2013, 47 (11), 958–986. 10.3109/10715762.2013.832239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov S. I.; Greten F. R.; Karin M. Immunity, inflammation, and cancer. Cell 2010, 140 (6), 883–899. 10.1016/j.cell.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonkar P.; Dedon P. C. Reactive species and DNA damage in chronic inflammation: reconciling chemical mechanisms and biological fates. Int. J. Cancer 2011, 128 (9), 1999–2009. 10.1002/ijc.25815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter S.; Gupta S. C.; Chaturvedi M. M.; Aggarwal B. B. Oxidative stress, inflammation, and cancer: how are they linked?. Free Radical Biol. Med. 2010, 49 (11), 1603–1616. 10.1016/j.freeradbiomed.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coussens L. M.; Werb Z. Inflammation and cancer. Nature 2002, 420 (6917), 860–867. 10.1038/nature01322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balkwill F.; Charles K. A.; Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell 2005, 7 (3), 211–217. 10.1016/j.ccr.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Perwez Hussain S.; Harris C. C. Inflammation and cancer: an ancient link with novel potentials. Int. J. Cancer 2007, 121 (11), 2373–2380. 10.1002/ijc.23173. [DOI] [PubMed] [Google Scholar]
- Kundu J. K.; Surh Y.-J. Inflammation: gearing the journey to cancer. Mutat. Res., Rev. Mutat. Res. 2008, 659 (1–2), 15–30. 10.1016/j.mrrev.2008.03.002. [DOI] [PubMed] [Google Scholar]
- Elinav E.; Nowarski R.; Thaiss C. A.; Hu B.; Jin C.; Flavell R. A. Inflammation-induced cancer: crosstalk between tumours, immune cells and microorganisms. Nat. Rev. Cancer 2013, 13 (11), 759–771. 10.1038/nrc3611. [DOI] [PubMed] [Google Scholar]
- Brenner D. R.; Scherer D.; Muir K.; Schildkraut J.; Boffetta P.; Spitz M. R.; Le Marchand L.; Chan A. T.; Goode E. L.; Ulrich C. M.; et al. A Review of the Application of Inflammatory Biomarkers in Epidemiologic Cancer Research. Cancer Epidemiol., Biomarkers Prev. 2014, 23 (9), 1729–1751. 10.1158/1055-9965.epi-14-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantovani A.; Cassatella M. A.; Costantini C.; Jaillon S. Neutrophils in the activation and regulation of innate and adaptive immunity. Nat. Rev. Immunol. 2011, 11 (8), 519–531. 10.1038/nri3024. [DOI] [PubMed] [Google Scholar]
- Van der Veen B. S.; de Winther M. P.; Heeringa P. Myeloperoxidase: molecular mechanisms of action and their relevance to human health and disease. Antioxid. Redox Signaling 2009, 11 (11), 2899–2937. 10.1089/ars.2009.2538. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Hampton M. B.; Livesey J. H.; Kettle A. J. Modeling the reactions of superoxide and myeloperoxidase in the neutrophil phagosome: implications for microbial killing. J. Biol. Chem. 2006, 281 (52), 39860–39869. 10.1074/jbc.M605898200. [DOI] [PubMed] [Google Scholar]
- Henderson J. P.; Byun J.; Heinecke J. W. Molecular chlorine generated by the myeloperoxidase-hydrogen peroxide-chloride system of phagocytes produces 5-chlorocytosine in bacterial RNA. J. Biol. Chem. 1999, 274 (47), 33440–33448. 10.1074/jbc.274.47.33440. [DOI] [PubMed] [Google Scholar]
- Stanley N. R.; Pattison D. I.; Hawkins C. L. Ability of hypochlorous acid and N-chloramines to chlorinate DNA and its constituents. Chem. Res. Toxicol. 2010, 23 (7), 1293. 10.1021/tx100188b. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. Biological reactivity and biomarkers of the neutrophil oxidant, hypochlorous acid. Toxicology 2002, 181–182, 223–227. 10.1016/S0300-483X(02)00286-X. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C.; Kettle A. J. Biomarkers of myeloperoxidase-derived hypochlorous acid. Free Radical Biol. Med. 2000, 29 (5), 403–409. 10.1016/S0891-5849(00)00204-5. [DOI] [PubMed] [Google Scholar]
- Andrés C. M. C.; Pérez de la Lastra J. M.; Juan C. A.; Plou F. J.; Pérez-Lebeña E. Hypochlorous Acid Chemistry in Mammalian Cells-Influence on Infection and Role in Various Pathologies. Int. J. Mol. Sci. 2022, 23 (18), 10735. 10.3390/ijms231810735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai Y.; Morinaga H.; Kondo H.; Miyoshi N.; Nakamura Y.; Uchida K.; Osawa T. Endogenous formation of novel halogenated 2′-deoxycytidine: hypohalous acid-mediated DNA modification at the site of inflammation. J. Biol. Chem. 2004, 279 (49), 51241–51249. 10.1074/jbc.M408210200. [DOI] [PubMed] [Google Scholar]
- Whiteman M.; Jenner A.; Halliwell B. Hypochlorous acid-induced base modifications in isolated calf thymus DNA. Chem. Res. Toxicol. 1997, 10 (11), 1240–1246. 10.1021/tx970086i. [DOI] [PubMed] [Google Scholar]
- Kang J. I. Jr.; Sowers L. C. Examination of hypochlorous acid-induced damage to cytosine residues in a CpG dinucleotide in DNA. Chem. Res. Toxicol. 2008, 21 (6), 1211–1218. 10.1021/tx800037h. [DOI] [PubMed] [Google Scholar]
- Mangerich A.; Knutson C. G.; Parry N. M.; Muthupalani S.; Ye W.; Prestwich E.; Cui L.; McFaline J. L.; Mobley M.; Ge Z.; Taghizadeh K.; Wishnok J. S.; Wogan G. N.; Fox J. G.; Tannenbaum S. R.; Dedon P. C. Infection-induced colitis in mice causes dynamic and tissue-specific changes in stress response and DNA damage leading to colon cancer. Proc. Natl. Acad. Sci. U.S.A. 2012, 109 (27), E1820 10.1073/pnas.1207829109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson C. G.; Mangerich A.; Zeng Y.; Raczynski A. R.; Liberman R. G.; Kang P.; Ye W.; Prestwich E. G.; Lu K.; Wishnok J. S.; et al. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (26), E2332 10.1073/pnas.1222669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lao V. V.; Herring J. L.; Kim C. H.; Darwanto A.; Soto U.; Sowers L. C. Incorporation of 5-chlorocytosine into mammalian DNA results in heritable gene silencing and altered cytosine methylation patterns. Carcinogenesis 2009, 30 (5), 886. 10.1093/carcin/bgp060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedeles B. I.; Freudenthal B. D.; Yau E.; Singh V.; Chang S. C.; Li D.; Delaney J. C.; Wilson S. H.; Essigmann J. M. Intrinsic mutagenic properties of 5-chlorocytosine: A mechanistic connection between chronic inflammation and cancer. Proc. Natl. Acad. Sci. U.S.A. 2015, 112 (33), E4571–80 10.1073/pnas.1507709112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiraly O.; Gong G.; Olipitz W.; Muthupalani S.; Engelward B. P. Inflammation-Induced Cell Proliferation Potentiates DNA Damage-Induced Mutations In Vivo. PLoS Genet. 2015, 11 (2), e1004901 10.1371/journal.pgen.1004901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macer-Wright J. L.; Stanley N. R.; Portman N.; Tan J. T.; Bursill C.; Rayner B. S.; Hawkins C. L. A Role for Chlorinated Nucleosides in the Perturbation of Macrophage Function and Promotion of Inflammation. Chem. Res. Toxicol. 2019, 32 (6), 1223–1234. 10.1021/acs.chemrestox.9b00044. [DOI] [PubMed] [Google Scholar]
- Kasar S.; Kim J.; Improgo R.; Tiao G.; Polak P.; Haradhvala N.; Lawrence M. S.; Kiezun A.; Fernandes S. M.; Bahl S.; Sougnez C.; Gabriel S.; Lander E. S.; Kim H. T.; Getz G.; Brown J. R. Whole-genome sequencing reveals activation-induced cytidine deaminase signatures during indolent chronic lymphocytic leukaemia evolution. Nat. Commun. 2015, 6 (1), 8866. 10.1038/ncomms9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimaki S.; Totsuka Y.; Suzuki Y.; Nakai C.; Goto M.; Kojima M.; Arakawa H.; Takemura S.; Tanaka S.; Marubashi S.; Kinoshita M.; Matsuda T.; Shibata T.; Nakagama H.; Ochiai A.; Kubo S.; Nakamori S.; Esumi H.; Tsuchihara K. Hypermutation and unique mutational signatures of occupational cholangiocarcinoma in printing workers exposed to haloalkanes. Carcinogenesis 2016, 37 (8), 817–826. 10.1093/carcin/bgw066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo Y.; Marusawa H.; Kou T.; Nakase H.; Fujii S.; Fujimori T.; Kinoshita K.; Honjo T.; Chiba T. Activation-induced cytidine deaminase links between inflammation and the development of colitis-associated colorectal cancers. Gastroenterology 2008, 135 (3), 889–898.e3. 10.1053/j.gastro.2008.06.091. [DOI] [PubMed] [Google Scholar]
- Takai A.; Marusawa H.; Minaki Y.; Watanabe T.; Nakase H.; Kinoshita K.; Tsujimoto G.; Chiba T. Targeting activation-induced cytidine deaminase prevents colon cancer development despite persistent colonic inflammation. Oncogene 2012, 31 (13), 1733. 10.1038/onc.2011.352. [DOI] [PubMed] [Google Scholar]
- Endo Y.; Marusawa H.; Chiba T. Involvement of activation-induced cytidine deaminase in the development of colitis-associated colorectal cancers. J. Gastroenterol. 2011, 46 (S1), 6–10. 10.1007/s00535-010-0326-1. [DOI] [PubMed] [Google Scholar]
- Kou T.; Marusawa H.; Kinoshita K.; Endo Y.; Okazaki I.-m.; Ueda Y.; Kodama Y.; Haga H.; Ikai I.; Chiba T. Expression of activation-induced cytidine deaminase in human hepatocytes during hepatocarcinogenesis. Int. J. Cancer 2007, 120 (3), 469–476. 10.1002/ijc.22292. [DOI] [PubMed] [Google Scholar]
- Kim C. J.; Song J. H.; Cho Y. G.; Cao Z.; Kim S. Y.; Nam S. W.; Lee J. Y.; Park W. S. Activation-induced cytidine deaminase expression in gastric cancer. Tumour Biol. 2007, 28 (6), 333. 10.1159/000124239. [DOI] [PubMed] [Google Scholar]
- Thongararm P.; Fedeles B. I.; Khumsubdee S.; Armijo A. L.; Kim L.; Thiantanawat A.; Promvijit J.; Navasumrit P.; Ruchirawat M.; Croy R. G.; Essigmann J. M. Modulation of N-Methyl-N-nitrosourea Mutagenesis in Mouse Embryo Fibroblasts Derived from the gpt Delta Mouse by an Inhibitor of the O6-Methylguanine Methyltransferase, MGMT. Chem. Res. Toxicol. 2020, 33 (2), 625–633. 10.1021/acs.chemrestox.9b00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nohmi T.; Katoh M.; Suzuki H.; Matsui M.; Yamada M.; Watanabe M.; Suzuki M.; Horiya N.; Ueda O.; Shibuya T.; Ikeda H.; Sofuni T. Other transgenic mutation assays: A new transgenic mouse mutagenesis test system using Spi- and 6-thioguanine selections. Environ. Mol. Mutagen. 1996, 28 (4), 465–470. 10.1002/(SICI)1098-2280(1996)28:4<465::AID-EM24>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Valentine C. C.; Young R. R.; Fielden M. R.; Kulkarni R.; Williams L. N.; Li T.; Minocherhomji S.; Salk J. J. Direct quantification of in vivo mutagenesis and carcinogenesis using duplex sequencing. Proc. Natl. Acad. Sci. U.S.A. 2020, 117 (52), 33414–33425. 10.1073/pnas.2013724117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawanthayatham S.; Valentine C. C. 3rd; Fedeles B. I.; Fox E. J.; Loeb L. A.; Levine S. S.; Slocum S. L.; Wogan G. N.; Croy R. G.; Essigmann J. M. Mutational spectra of aflatoxin B(1) in vivo establish biomarkers of exposure for human hepatocellular carcinoma. Proc. Natl. Acad. Sci. U.S.A. 2017, 114 (15), E3101 10.1073/pnas.1700759114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawanthayatham S.; Thiantanawat A.; Egner P. A.; Groopman J. D.; Wogan G. N.; Croy R. G.; Essigmann J. M. Prenatal exposure of mice to the human liver carcinogen aflatoxin B1 reveals a critical window of susceptibility to genetic change. Int. J. Cancer 2015, 136 (6), 1254. 10.1002/ijc.29102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armijo A. L.; Thongararm P.; Fedeles B. I.; Yau J.; Kay J. E.; Corrigan J. J.; Chancharoen M.; Chawanthayatham S.; Samson L. D.; Carrasco S. E.; Engelward B. P.; Fox J. G.; Croy R. G.; Essigmann J. M. Molecular origins of mutational spectra produced by the environmental carcinogen N-nitrosodimethylamine and S(N)1 chemotherapeutic agents. NAR Cancer 2023, 5 (2), zcad015. 10.1093/narcan/zcad015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea J. P.; Chou M. F.; Quader S. A.; Ryan J. K.; Church G. M.; Schwartz D. pLogo: a probabilistic approach to visualizing sequence motifs. Nat. Methods 2013, 10 (12), 1211–1212. 10.1038/nmeth.2646. [DOI] [PubMed] [Google Scholar]
- Alexandrov L. B.; Nik-Zainal S.; Wedge D. C.; Aparicio S. A. J. R.; Behjati S.; Biankin A. V.; Bignell G. R.; Bolli N.; Borg A.; Børresen-Dale A. L.; Boyault S.; Burkhardt B.; Butler A. P.; Caldas C.; Davies H. R.; Desmedt C.; Eils R.; Eyfjörd J. E.; Foekens J. A.; Greaves M.; Hosoda F.; Hutter B.; Ilicic T.; Imbeaud S.; Imielinski M.; Jäger N.; Jones D. T. W.; Jones D.; Knappskog S.; Kool M.; Lakhani S. R.; López-Otín C.; Martin S.; Munshi N. C.; Nakamura H.; Northcott P. A.; Pajic M.; Papaemmanuil E.; Paradiso A.; Pearson J. V.; Puente X. S.; Raine K.; Ramakrishna M.; Richardson A. L.; Richter J.; Rosenstiel P.; Schlesner M.; Schumacher T. N.; Span P. N.; Teague J. W.; Totoki Y.; Tutt A. N. J.; Valdés-Mas R.; van Buuren M. M.; van’t Veer L.; Vincent-Salomon A.; Waddell N.; Yates L. R.; Zucman-Rossi J.; Andrew Futreal P.; McDermott U.; Lichter P.; Meyerson M.; Grimmond S. M.; Siebert R.; Campo E.; Shibata T.; Pfister S. M.; Campbell P. J.; Stratton M. R.; Shibata T.; Pfister S. M.; Campbell P. J.; Stratton M. R. Signatures of mutational processes in human cancer. Nature 2013, 500 (7463), 415–421. 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin H.; Suzuki K.; Nakata M.; Chikuma S.; Izumi N.; Thi Huong L.; Maruya M.; Fagarasan S.; Busslinger M.; Honjo T.; Nagaoka H. Activation-Induced Cytidine Deaminase Expression in CD4+ T Cells is Associated with a Unique IL-10-Producing Subset that Increases with Age. PLoS One 2011, 6 (12), e29141 10.1371/journal.pone.0029141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono M.; Torisu H.; Fukushi J.; Nishie A.; Kuwano M. Biological implications of macrophage infiltration in human tumor angiogenesis. Cancer Chemother. Pharmacol. 1999, 43, S69–S71. 10.1007/s002800051101. [DOI] [PubMed] [Google Scholar]
- Schoppmann S. F.; Birner P.; Stöckl J.; Kalt R.; Ullrich R.; Caucig C.; Kriehuber E.; Nagy K.; Alitalo K.; Kerjaschki D. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am. J. Pathol. 2002, 161 (3), 947. 10.1016/S0002-9440(10)64255-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S.; Singh A. P.; Sharma B.; Owen L. B.; Singh R. K. CXCL8 and its cognate receptors in melanoma progression and metastasis. Future Oncol. 2010, 6 (1), 111. 10.2217/fon.09.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgauer J.; Metzner B.; Schraufstätter I. Expression and growth-promoting function of the IL-8 receptor beta in human melanoma cells. J. Immunol. 1996, 156 (3), 1132. 10.4049/jimmunol.156.3.1132. [DOI] [PubMed] [Google Scholar]
- Knutson C. G.; Mangerich A.; Zeng Y.; Raczynski A. R.; Liberman R. G.; Kang P.; Ye W.; Prestwich E. G.; Lu K.; Wishnok J. S.; Korzenik J. R.; Wogan G. N.; Fox J. G.; Dedon P. C.; Tannenbaum S. R. Chemical and cytokine features of innate immunity characterize serum and tissue profiles in inflammatory bowel disease. Proc. Natl. Acad. Sci. U.S.A. 2013, 110 (26), E2332 10.1073/pnas.1222669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood M. L.; Dizdaroglu M.; Gajewski E.; Essigmann J. M. Mechanistic studies of ionizing radiation and oxidative mutagenesis: genetic effects of a single 8-hydroxyguanine (7-hydro-8-oxoguanine) residue inserted at a unique site in a viral genome. Biochemistry 1990, 29 (30), 7024–7032. 10.1021/bi00482a011. [DOI] [PubMed] [Google Scholar]
- Maki H.; Sekiguchi M. MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis. Nature 1992, 355 (6357), 273–275. 10.1038/355273a0. [DOI] [PubMed] [Google Scholar]
- Greer S.; Alvarez M.; Mas M.; Wozniak C.; Arnold D.; Knapinska A.; Norris C.; Burk R.; Aller A.; Dauphinée M. Five-chlorodeoxycytidine, a tumor-selective enzyme-driven radiosensitizer, effectively controls five advanced human tumors in nude mice. Int. J. Radiat. Oncol., Biol., Phys. 2001, 51 (3), 791–806. 10.1016/S0360-3016(01)01706-0. [DOI] [PubMed] [Google Scholar]
- Kim C. H.; Darwanto A.; Theruvathu J. A.; Herring J. L.; Sowers L. C. Polymerase incorporation and miscoding properties of 5-chlorouracil. Chem. Res. Toxicol. 2010, 23 (4), 740. 10.1021/tx900302j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer S.; Han T.; Dieguez C.; McLean N.; Saer R.; Reis I.; Levi J.; Marquez V. E. Enzyme-Driven Chemo-and Radiation-Therapy with 12 Pyrimidine Nucleoside Analogs Not Yet in the Clinic. Anticancer Agents Med. Chem. 2017, 17 (2), 250–264. 10.2174/1871520616666161013145853. [DOI] [PubMed] [Google Scholar]
- Santos O.; Perez L. M.; Briggle T. V.; Boothman D. A.; Greer S. B. Radiation, pool size and incorporation studies in mice with 5-chloro-2’-deoxycytidine. Int. J. Radiat. Oncol., Biol., Phys. 1990, 19 (2), 357. 10.1016/0360-3016(90)90544-T. [DOI] [PubMed] [Google Scholar]
- Morgan M. T.; Bennett M. T.; Drohat A. C. Excision of 5-Halogenated Uracils by Human Thymine DNA Glycosylase. J. Biol. Chem. 2007, 282 (38), 27578. 10.1074/jbc.M704253200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L. B.; Nik-Zainal S.; Wedge D. C.; Campbell P. J.; Stratton M. R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013, 3 (1), 246. 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney J. C.; Essigmann J. M. Effect of sequence context on O(6)-methylguanine repair and replication in vivo. Biochemistry 2001, 40 (49), 14968. 10.1021/bi015578f. [DOI] [PubMed] [Google Scholar]
- Singer B.; Chavez F.; Goodman M. F.; Essigmann J. M.; Dosanjh M. K. Effect of 3′ flanking neighbors on kinetics of pairing of dCTP or dTTP opposite O6-methylguanine in a defined primed oligonucleotide when Escherichia coli DNA polymerase I is used. Proc. Natl. Acad. Sci. U.S.A. 1989, 86 (21), 8271. 10.1073/pnas.86.21.8271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatahet Z.; Zhou M.; Reha-Krantz L. J.; Morrical S. W.; Wallace S. S. In search of a mutational hotspot. Proc. Natl. Acad. Sci. U.S.A. 1998, 95 (15), 8556–8561. 10.1073/pnas.95.15.8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibutani S.; Suzuki N.; Tan X.; Johnson F.; Grollman A. P. Influence of flanking sequence context on the mutagenicity of acetylaminofluorene-derived DNA adducts in mammalian cells. Biochemistry 2001, 40 (12), 3717. 10.1021/bi0027581. [DOI] [PubMed] [Google Scholar]
- Fedeles B. I.; Essigmann J. M.. Mutational Spectra Provide Insight into the Mechanisms Bridging DNA Damage to Genetic Disease; Royal Society of Chemistry: London, 2020; Vol. 2. [Google Scholar]
- Ye X.; Ren W.; Liu D.; Li X.; Li W.; Wang X.; Meng F.-L.; Yeap L.-S.; Hou Y.; Zhu S.; Casellas R.; Zhang H.; Wu K.; Pan-Hammarström Q. Genome-wide mutational signatures revealed distinct developmental paths for human B cell lymphomas. J. Exp. Med. 2020, 218 (2), e20200573 10.1084/jem.20200573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J. C.; Kamath-Loeb A. S.; Kohrn B. F.; Loeb K. R.; Preston B. D.; Loeb L. A. A high-resolution landscape of mutations in the BCL6 super-enhancer in normal human B cells. Proc. Natl. Acad. Sci. U.S.A. 2019, 116 (49), 24779–24785. 10.1073/pnas.1914163116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLennan I. C.; Gray D. Antigen-driven selection of virgin and memory B cells. Immunol. Rev. 1986, 91, 61–86. 10.1111/j.1600-065X.1986.tb01484.x. [DOI] [PubMed] [Google Scholar]
- Endo Y.; Marusawa H.; Kinoshita K.; Morisawa T.; Sakurai T.; Okazaki I. M.; Watashi K.; Shimotohno K.; Honjo T.; Chiba T. Expression of activation-induced cytidine deaminase in human hepatocytes via NF-κB signaling. Oncogene 2007, 26 (38), 5587–5595. 10.1038/sj.onc.1210344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data presented in this work are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) repository under the BioProject PRJNA1035149 (http://www.ncbi.nlm.nih.gov/bioproject/1035149). The Python scripts used for data analysis and visualization are available on GitHub (https://github.com/essigmannlab/5CldC_spectrum/).