

Abstract
The hypoxia‐inducible factor (HIF) prolyl‐hydroxylases (human PHD1‐3) catalyze prolyl hydroxylation in oxygen‐dependent degradation (ODD) domains of HIFα isoforms, modifications that signal for HIFα proteasomal degradation in an oxygen‐dependent manner. PHD inhibitors are used for treatment of anemia in kidney disease. Increased erythropoietin (EPO) in patients with familial/idiopathic erythrocytosis and pulmonary hypertension is associated with mutations in EGLN1 (PHD2) and EPAS1 (HIF2α); a drug inhibiting HIF2α activity is used for clear cell renal cell carcinoma (ccRCC) treatment. We report crystal structures of PHD2 complexed with the C‐terminal HIF2α‐ODD in the presence of its 2‐oxoglutarate cosubstrate or N‐oxalylglycine inhibitor. Combined with the reported PHD2.HIFα‐ODD structures and biochemical studies, the results inform on the different PHD.HIFα‐ODD binding modes and the potential effects of clinically observed mutations in HIFα and PHD2 genes. They may help enable new therapeutic avenues, including PHD isoform‐selective inhibitors and sequestration of HIF2α by the PHDs for ccRCC treatment.
Keywords: Belzutifan, clear cell renal cell carcinoma, erythropoiesis, hypoxia‐inducible factor isoform 2‐alpha (HIF2α or EPAS1), prolyl hydroxylase domain (PHD or EGLN), Trichoplax adhaerens and Pseudomonas putida prolyl hydroxylase domain (TaPHD and PPHD), α‐ketoglutarate/2‐oxoglutarate oxygenase
1. INTRODUCTION
In humans and other animals, the hypoxia‐inducible factor (HIF) transcription factors play key roles in responses to limiting O2 availability by promoting context‐dependent expression of genes working to alleviate the effects of hypoxia. HIF is an α,β‐heterodimeric protein; the levels of HIFβ, also known as the aryl hydrocarbon receptor nuclear translocator protein, are not regulated directly by O2 concentrations. 1 By contrast, as a consequence of catalysis by the HIF prolyl hydroxylase domain enzymes (human PHD1‐3), HIFα levels are strongly regulated by O2 availability. 2 , 3 PHD1‐3 catalyze trans‐4‐prolyl hydroxylation of the N‐ and C‐terminal oxygen‐dependent degradation (NODD and CODD, respectively) domains in HIF1‐3α isoforms (note, HIF3α only contains a CODD) (Figure 1A). Such prolyl hydroxylation promotes binding of HIF1‐3α to the von Hippel–Lindau protein (pVHL) ubiquitin ligase complex, so signaling for proteasomal mediated hydrolysis of HIFα isoforms (Figure 1A). 4 , 5 , 6 , 7 A second HIFα hydroxylase, factor‐inhibiting HIF (FIH) catalyzes the C3 hydroxylation of an asparagine‐residue in the C‐terminal transcriptional activation domains of HIF1α and HIF2α (but not HIF3α), a modification that reduces the interaction of HIF with histone acetyltransferases (CREB binding protein and p300). 8 The FIH catalyzed modification inhibits HIF‐mediated transcription in an incompletely understood context‐dependent manner (Figure S1). 8 , 9 , 10 , 11
FIGURE 1.
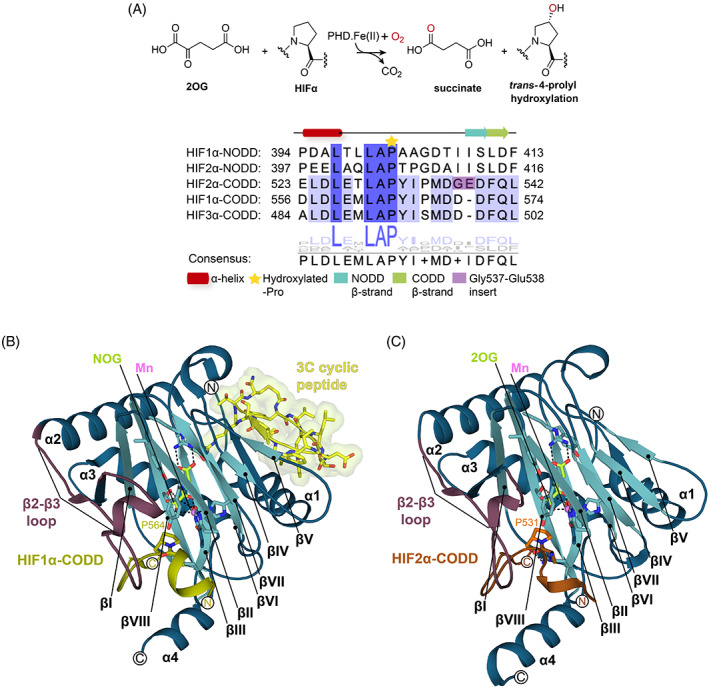
Overview of HIFα prolyl hydroxylase catalysis and view of the conserved double‐stranded β‐helix fold of the 2OG oxygenases. (A) PHD1‐3 catalyze 2OG‐dependent trans‐4‐prolyl hydroxylation of HIFα isoforms. Sequence alignment of the five human HIFα N‐/C‐terminal oxygen‐degradation domains. The secondary structure (α‐helix: red and β‐strand: orange‐NODD/green‐CODD) assignments are as observed in crystal structures of HIF1α394–413‐NODD and HIF1α556–574‐CODD in complex with the PHD2 catalytic domain (PDB: 5L9V and 5L9B). The hydroxylated proline is marked with a yellow star. (B, C) The distorted double‐stranded β‐helix core fold (teal βI‐βVIIΙ refer to the eight DSBH strands), the β2–β3 finger loop (red cartoon), and the N‐/C‐terminal extensions of the DSBH (N: α1–α3 and C: α4 helices‐blue) are labeled. (B) The PHD2181‐407.Mn(II).NOG.HIF1α‐CODD.3C (6YW3) complex is depicted as a cartoon showing the N‐terminal binding site of the co‐crystallized 3C cyclic peptide (yellow sticks with Connolly surface). (C) A view of the DSBH of the PHD2181–407.Mn(II).2OG.HIF2α‐CODD (7Q5X) complex, the main focus of this work. Key active site residues (teal), target proline (orange), and NOG (lemon) are represented as sticks. The HIF1α‐CODD (olive) and HIF2α‐CODD (orange) substrates are displayed as cartoons. Waters (red) and Mn (violet) are shown as spheres. CODD, C‐terminal oxygen‐dependent degradation; HIF, hypoxia‐inducible factor; NODD, N‐terminal oxygen‐dependent degradation.
In hypoxia, PHD1‐3 activity decreases and HIF1‐3α levels rise (Figure S1). 2 , 3 HIF1‐3α translocate to the nucleus and dimerize with HIFβ to form transcriptionally active α,β‐HIF heterodimers (Figure S1). 2 , 3 , 12 These bind to hypoxia response elements (HREs) associated with HIF target genes and consequently upregulate transcription of HIF controlled genes including those encoding for erythropoietin (EPO) and vascular endothelial growth factor (VEGF) (Figure S1). 1 , 2 , 3 , 13
The PHDs and FIH are both Fe(II) and 2‐oxoglutarate (2OG)‐dependent oxygenases that couple hydroxylation with the conversion of 2OG to succinate and CO2 (Figures 1A and S1). 3 , 8 , 11 , 14 , 15 Other human 2OG oxygenases have roles in the regulation of gene expression (e.g., the JmjC histone demethylases), and in other important cellular processes, including metabolism and collagen biosynthesis. 14 , 16 , 17 , 18 The biochemical properties of the PHDs are apparently suited to their roles as hypoxia/“O2‐sensors”; thus, they have unusually high K m values for O2 and react relatively slowly with O2, compared to FIH and most other 2OG oxygenases. 19 , 20 , 21 , 22 PHD2 also forms a relatively stable complex with Fe and 2OG, even after exposure to O2. Collectively, these observations suggest that the biochemical properties of the PHDs may be focused to sense O2 availability. 19 , 20 , 21 , 22 , 23 The PHDs are more sensitive than FIH to limiting O2 levels 24 and HIFα‐NODD hydroxylation is reported to be more sensitive than CODD hydroxylation to O2 levels. 24 , 25 The PHDs also show different selectivity toward the various HIFα‐ODDs, with PHD3 being reported to be particularly selective for the HIF1α‐ and HIF2α‐CODD domains (Figures 1A and S1). 12 , 22 , 26
Crystal structures of PHD2.HIFα‐ODD complexes and kinetic studies have revealed the importance of a conformationally mobile loop (the β2–β3 loop) that links β2 and β3 of the catalytic domain of PHD2 and which is involved in HIFα‐ODD binding and selectivity; in the PHD2.substrate complexes, the β2–β3 loop folds to isolate the HIFα‐ODD substrate proline at the active site. 11 , 27 , 28 , 29 , 30 Overall, these observations support the proposal that β2–β3 loop dynamics are important both in catalysis and determining PHD/HIFα‐ODD substrate selectivity, though the precise molecular details are undefined (Figure 1B,C). 27 , 29 , 30
VHL gene mutation is common in clear cell renal cell carcinoma (ccRCC) patients causing upregulation of HIFα isoforms, so increasing the expression of the HIF2α target VEGF, in a manner apparently promoting tumorigenesis and cancer progression. 31 , 32 Belzutifan (MK‐6482 or PT‐2977) inhibits HIF2α‐mediated expression and is used for ccRCC treatment (Figure S1). 33 Mutations in EPAS1 (encoding for HIF2α), EGLN1 (encoding for PHD2), are also linked to disease, including familial/idiopathic erythrocytosis and ccRCC (Figure S2A). 34 Thus, structural information of how the PHDs bind HIFα‐ODDs, and in particular HIF2α, may inform on the clinically observed pathologies of the mutant EPAS1‐related diseases.
Although structures of HIF1α‐CODD and NODD in complex with the catalytic domain of PHD2 are available, analogous structures with HIF2α‐CODD have not been reported. 11 , 27 , 28 The PHD.HIF2α‐CODD complexes are of particular interest because of the disease relevance of HIF2α and because of differences with HIF1α/2α‐NODD and HIF1α/3α‐CODD. In particular, HIF2α contains an ‘additional’ (Gly) residue on the C‐terminal side of the hydroxylated proline, compared to HIF1α/3α‐CODD and HIF1‐2α‐NODD (Figure 1A). In the respective position in HIF1/2α‐NODD, the polar Gly537/Glu538 unit in HIF2α is substituted by two non‐polar Ile‐residues (Figure 1A).
Here, we report high‐resolution crystal structures of the truncated catalytic domain of PHD2 (residues 181–407) in complex with HIF2α‐CODD (residues 523–542), a manganese ion, and 2OG or its close isostere N‐oxalylglycine (NOG) (Figures 1C and 2). The structures inform on differences in HIFα‐ODD binding that may influence the different selectivity of the PHDs.
FIGURE 2.
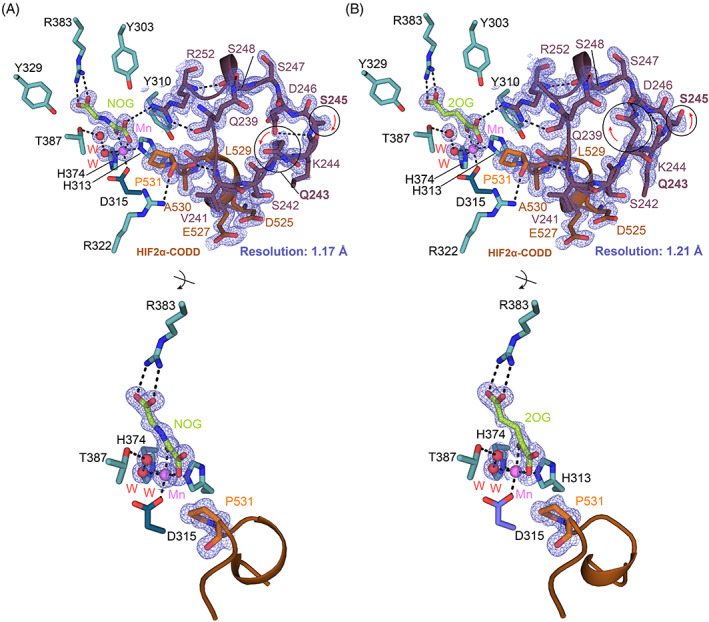
Structural basis for HIF2α‐CODD binding to PHD2. (A, B) Views from structures of PHD2181–407.Mn.2OG/NOG.HIF2α523–542‐CODD displayed as cartoons (PHD2181–407‐blue; HIF2α‐orange) (PDB: 7Q5V‐A and 7Q5X‐B). PHD2 residues (blue/teal), β2–β3 loop (red), 2OG/NOG (lime), and HIF2α (orange) residues are shown as sticks. The electron density map (contoured at 1.0 σ) is depicted as a mesh (blue). Key polar interactions are represented by black dashes. Waters (red) and Mn (violet) are displayed as spheres. Differences in the β2–β3 loop conformation in the two structures are highlighted by a black circle and a red arrow. Note the C4‐endo conformation of the substrate proline ring in both structures. CODD, C‐terminal oxygen‐dependent degradation; HIF, hypoxia‐inducible factor.
2. RESULTS
2.1. Crystallization and structural determination of the PHD2 181–407.HIF2α 523–542 complex
Recently, application of the random non‐standard Peptide Integrated Discovery (RaPID) platform has enabled the identification of a cyclic peptide (3C) that binds tightly to PHD2 in a non‐substrate competing manner. 28 , 35 3C binds to the N‐terminal region of the PHD2181‐426 catalytic domain (residues 185–214) and promotes crystallization of the PHD2181–426.HIF1α‐CODD complex (P21212 space group; PDB: 6YW3). 28 We added 3C (Figures 1B and S3B) with the aim of promoting crystallization of the PHD2181‐407.Mn.NOG.HIF2α‐CODD complex (Table S1). A 1 mm x 100 μm x 80 μm plate morphology crystal (P21212 space group) was obtained within 1 week that diffracted to 1.11 Å resolution at a synchrotron source (refined to 1.17 Å resolution) (PDB: 7Q5V) (Figure 2A and Table 1). These conditions also gave crystals of the analogous PHD2181‐407.Mn.2OG.HIF2α‐CODD complex (P21212 space group) that diffracted to 1.19 Å resolution (refined to 1.21 Å resolution) (PDB: 7Q5X) (Figure 2B and Table 1). Although addition of the 3C promoted crystallization of the PHD2181‐407.HIF2α‐CODD complexes, clear electron density for 3C was not observed in the crystal structures (Figure S3B), that is there was insufficient electron density to model in 3C as reported in the PHD2181‐426.HIF1α‐CODD complex structure (PDB: 6YW3).
TABLE 1.
Data collection and crystallographic processing statistics of the PHD2181–407.HIF2α complex structures.
| PHD2181–407.Mn(II).NOG.HIF2α (PDB:7Q5V) | PHD2181–407.Mn(II).2OG.HIF2α (PDB:7Q5X) | |
|---|---|---|
| Beamline | Diamond Light Source‐I24 | Diamond Light Source‐I24 |
| Detector | Dectris Pilatus3 6M | Dectris Pilatus3 6M |
| Data processing | Xia2 DIALS | Xia2 DIALS |
| Wavelength (Å) | 0.96861 | 0.97962 |
| Resolution range (Å) | 43.64–1.17 (1.21–1.17) | 43.48–1.21 (1.25–1.21) |
| Space group | P21212 | P21212 |
| Unit cell (Å) | 130.91 38.32 42.88 | 130.45 38.17 42.75 |
| Total reflections | 1 764 477 (121 954) | 808 554 (71 010) |
| Unique reflections | 73 707 (7148) | 66 188 (6526) |
| Multiplicity | 23.9 (15.4) | 12.2 (10.4) |
| Completeness (%) | 99.75 (98.07) | 99.94 (99.85) |
| Mean I/sigma(I) | 11.13 (1.20) | 10.05 (1.15) |
| Wilson B‐factor (Å2) | 13.75 | 14.13 |
| R‐merge a | 0.1467 (3.871) | 0.1317 (4.204) |
| R‐meas | 0.1499 (3.99) | 0.1376 (4.413) |
| R‐pim | 0.03046 (0.9422) | 0.03926 (1.322) |
| CC1/2 | 0.999 (0.39) | 0.999 (0.432) |
| CC* | 1 (0.749) | 1 (0.777) |
| Reflections used in refinement | 73654 (7115) | 66197 (6516) |
| Reflections used for R‐free | 3747 (345) | 3329 (334) |
| R‐work b | 0.1570 (0.3095) | 0.1570 (0.3118) |
| R‐free b | 0.1792 (0.3188) | 0.1759 (0.3075) |
| CC (work) | 0.975 (0.722) | 0.973 (0.775) |
| CC (free) | 0.961 (0.705) | 0.961 (0.805) |
| Number of non‐hydrogen atoms | 2416 | 2269 |
| Macromolecules | 2105 | 2036 |
| 2OG/NOG | 13 | 14 |
| Formic acid | 12 | 44 |
| Cl ion | 1 | 3 |
| Mg ion | 3 | 3 |
| PEG | 51 | 17 |
| Glycerol | 13 | 13 |
| Mn ion | 1 | 1 |
| Solvent | 277 | 159 |
| Protein residues | 243 | 240 |
| RMS (bonds, Å) | 0.008 | 0.011 |
| RMS (angles, °) | 1.02 | 1.22 |
| Ramachandran favored (%) | 97.91 | 97.88 |
| Ramachandran allowed (%) | 2.09 | 2.12 |
| Ramachandran outliers (%) | 0.00 | 0.00 |
| Rotamer outliers (%) | 1.37 | 1.43 |
| Clashscore | 5.94 | 4.63 |
| Average B‐factor (Å2) | 23.66 | 23.02 |
| Macromolecules | 18.5 | 20.92 |
| 2OG/NOG | 15.65 | 18.676 |
| Formic acid | 56.16 | 39.33 |
| Cl ion | 80.79 | 58.14 |
| Mg ion | 45.82 | 46.61 |
| PEG | 52.52 | 49.65 |
| Glycerol | 74.27 | 58.14 |
| Mn ion | 9.30 | 9.85 |
| Solvent | 32.16 | 33.86 |
| Number of TLS groups | 9 | 10 |
Note: Single crystal diffraction data were collected from samples at 100K with conventional, rotation‐based methods. Statistics for the highest‐resolution shell are in parentheses. R factor is equal to ∑hkl||Fobs(hkl)| − |Fcalc(hkl)||/ ∑hkl|Fobs(hkl)| and was calculated for the working set of reflections (R work).
R‐merge (or Rsym) is equal to ∑|I‐<I>|/∑I. R‐merge represents the data quality of merged reflection data. I is equal to the intensity of individual measurements and <I> is equal to the average of multiple measurements.
R free is the R factor for 5% of the reflections which were excluded during refinement.
Comparison of the overall PHD2181‐426/407.Mn.NOG/2OG.HIF2α‐CODD structures reveals conservation of the distorted double stranded β‐helix (DSBH) and associated HIFα‐ODD substrate binding elements (Figures 1B,C and 3). 11 , 27 , 28 , 36 , 37 The NOG and 2OG structures are very similar to each other (backbone RMSD: 0.078 Å) and, to a somewhat lesser extent in terms of details, with other PHD2.HIFα‐ODD structures (Figure 3). 11 , 27 , 28 In particular, variations in the conformations of α1, the β2–β3 loop, and the C‐terminal α4 regions are observed. Note that the constructs used vary in the length of their C‐terminus and in our case reversible binding of 3C may promote formation of, or stabilize, specific conformations that promote crystallization (Figure 3). 28
FIGURE 3.
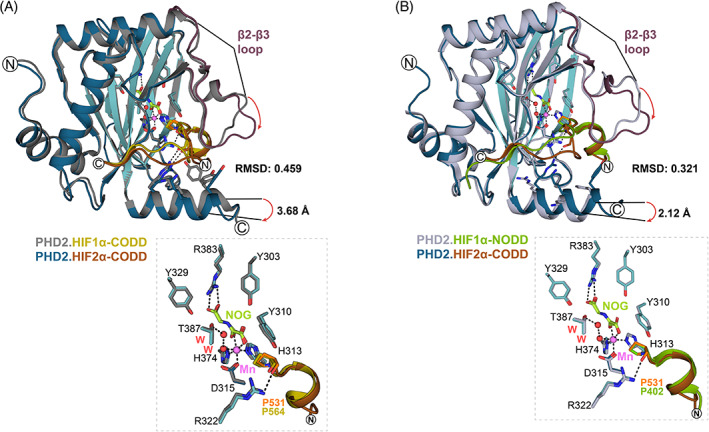
Comparison of crystal structure views of PHD2.NOG.HIF1α‐NODD/CODD complexes. (A, B) Views from structures of PHD2 (light blue‐5L9V, gray‐3HQR, and blue/cyan‐7Q5V) complexed with HIF1α394–413‐NODD (green‐5L9V), HIF1α556–564‐CODD (olive‐3HQR), and HIF2α523–542‐CODD (orange‐7Q5V). Polar interactions are represented by dashes (black). Waters (red) and Mn (violet) are displayed as spheres. RMSD values for aligned PHD2.HIF1α‐NODD/CODD complexes were calculated using PyMOL™. Variations in the β2–β3 loop and α4‐helix conformations are highlighted with red arrows. (A) Comparison of PHD2181–426.NOG.HIF1α‐CODD (3HQR) and PHD2181–407.NOG.HIF2α‐CODD (7Q5V) structures with a view of the active site residues and target prolines. (B) Comparison of PHD2181–426.NOG.HIF1α‐NODD (5L9V) and PHD2181–407.NOG.HIF2α‐CODD (7Q5V) structures with view of the active site residues and target prolines. CODD, C‐terminal oxygen‐dependent degradation; HIF, hypoxia‐inducible factor; NODD, N‐terminal oxygen‐dependent degradation.
The previously reported PHD2 active site chemistry is also conserved in the PHD2181‐407.Mn.NOG/2OG.HIF2α‐CODD structures (Figure 3), 37 with a single manganese ion (substituting for iron) being coordinated by the side chains of His313, Asp315, and His374, as well as a well‐defined water molecule/hydroxide ion. 11 , 27 , 28 The use of Mn(II) in PHD2 crystallization/inhibition is of interest given links between disease associated with Mn metabolism and erythropoiesis. 38 2OG and NOG bind the manganese ion in a bidentate manner via their C1 carboxylate and C2 carboxylate oxygens. The 2OG and NOG C5 carboxylates are positioned to interact with the guanidino group of Arg383 (Figures 2 and 3). The 2OG C1 carboxylate coordinates the manganese ion in the position adjacent to Pro531HIF2α, i.e., 2OG coordination is in an off‐line mode, suggesting that at some stage a metal‐centered rearrangement may be required to present the reactive ferryl adjacent to the oxidized Pro531HIF2α C‐H bond. The pyrrolidine ring of Pro531HIF2α is clearly observed in the C4‐endo conformation, as observed in previous PHD2‐substrate complex structures. 11 , 27 , 39 However, the C4 of Pro564HIF1α‐CODD is ~0.5 Å closer to the metal than the Pro531HIF2α‐CODD, though whether this has any kinetic relevance is unclear (Figure 3A). 11
Collectively, these observations reveal a conserved mode of binding for HIFα‐ODD substrate proline‐residues at the active site, including with respect to the substrate proline‐ring conformation and off‐line 2OG binding. The overall binding mode is also conserved in PHD type prolyl hydroxylases in Trichoplax adhaerens (TaPHD), 40 including bacteria (Pseudomonas putida PHD (PPHD) and Bacillus anthracis prolyl‐4‐hydroxylase (BaP4H)), which catalyze prolyl hydroxylation of elongation factor‐thermally unstable (EF‐Tu) (Figure S4). 41 , 42 This conservation is important because these features are proposed to be involved in the HIF/PHD/VHL “O2‐sensing” mechanism. Thus, off‐line 2OG binding may help enable the slow reaction of the PHDs with O2, a property proposed to be important in their “O2‐sensing” role. 43 C4 proline hydroxylation is proposed to enable a stereoelectronic preference for the C4‐exo over the C4‐endo proline ring conformation, with the former being observed in PHD.HIFα‐ODD complexes and the latter in VHL.hydroxylated‐HIFα‐ODD complexes. 11 , 39 , 44
Comparison of reported PHD2181‐426.NOG.HIF1α‐ODD complex structures (PDB: 3HQR and 5L9V) with the new PHD2181–407.NOG.HIF2α‐CODD (PDB: 7Q5V) structure reveals a shift in the position of the C‐terminal α4‐helix relative to the core DSBH fold, which is manifests in differences in the positions of PHD2 Thr405 and Lys402 as observed in overlaid structures. Analysis of the Thr405 Cα positions reveals a shift of ~3.7 Å in the new PHD2181‐407.HIF2α‐CODD complex structure (PDB: 7Q5V) compared to the PHD2181‐426.HIF1α‐CODD complex structure (PDB: 3HQR). Comparison of the PHD2181‐407.HIF2α‐CODD structure (PDB: 7Q5V) with the PHD2181‐426.HIF1α‐NODD structure (PDB: 5L9V) a reveals a shift of ~2.1 Å for the Lys402 Cα (Figure 3). These differences in α4 might, in part, reflect variations in the HIFα‐ODD substrate binding modes at the C‐terminal region of PHD2 and their impact on catalysis. 27 , 29 , 30 However, it cannot be ruled out if these are caused by variations in crystal lattice packing, possibly relating to 3C binding. 28
The β2–β3 loops of the PHDs are important in catalysis and in determining HIFα‐ODD substrate selectivity. 27 , 30 , 37 In the absence of HIFα‐ODD substrates, the β2–β3 loop is likely conformationally mobile/disordered and principally adopts conformations that are not near the active site, including those observed by crystallography. 11 , 27 , 28 , 29 , 45 In all reported PHD2.HIFα‐ODD structures, the β2–β3 loop folds to enclose the substrate proline residue in the active site, as is observed in our PHD2181‐407.HIF2α‐CODD structures (Figure 2).
Although the overall PHD2181‐407.HIF2α‐CODD structures with NOG and 2OG are very similar (RMSD: 0.078 Å), there are some differences in the conformations of the β2–β3 loop involving PHD2 residues Gln243‐Asp246 (Figure 2). In the 2OG.HIF2α‐CODD complex, the side chain amide NΗ2 group of PHD2 Gln243β2–β3 is positioned to form a hydrogen bond (2.74 Å) with the main chain carbonyl O atom of PHD2 Asp246 (Figure 2B). This hydrogen bond is, however, not observed in the NOG.HIF2α‐CODD complex, where Gln243β2–β3 is oriented away from the loop and adopts a more solvent‐exposed position (Figure 2A).
Although further work is required, given the 2OG and isostreric NOG structures have the same space group and similar crystal packing, the differences in the β2–β3 loop between them suggests that small differences at the active site region may influence the conformation of relatively distant structural elements within PHD2181‐407. This observation is interesting in part because recent studies on the mechanism of isopenicillin N synthase, which is structurally and mechanistically related to the 2OG‐dependent oxygenases, imply that conformational changes distant from the active site are involved in catalysis. 46 It is also of interest because it supports the previous proposal that inhibition by 2OG mimetics involves effects on structural dynamics in addition to simple blockade of 2OG binding in the active site. 30 , 47 , 48 Modeling studies on 2OG oxygenases, including demethylases, also imply the relevance of conformational changes both at and relatively distant from the active site during catalysis. 49 , 50 However, defining the precise effects of Fe‐binding inhibitors on the overall structural dynamics (and in some cases including complexed substrate) is technically challenging, requiring room temperature solution as well as low‐temperature biophysical crystallographic studies. 48 Hence, in addition to studies with isolated PHDs, empirical optimization of inhibitors in a cellular context is desirable.
We compared the β2–β3 loop conformations in PHD2181–407.2OG/NOG.HIF2α‐CODD with those of the other PHD2.HIFα‐ODD complexes. An intra‐loop Gln243 hydrogen bond with the main chain carbonyl O of Asp246 is observed in the PHD2181‐426.NOG.HIF1α‐NODD (PDB: 5L9V), PHD2181‐426.2OG.HIF1α‐CODD (PDB: 5L9B), and our PHD2181‐407.2OG.HIF2α‐CODD complexes. 27 In the case of the PHD2181‐426.NOG.HIF1α‐CODD (PDB: 3HQR) 11 and PHD2181‐407.NOG.HIF1α‐CODD.3C (PDB: 6YW3) 28 complexes, the side chain of Gln243 is not involved in hydrogen bonding; instead, Gln243 is in a more solvent‐exposed conformation, as observed in our PHD2.NOG.HIF2α‐CODD structure (similarly, the side chain of Ser245 adopts a different conformation in the 2OG complexed structure (PDB: 7Q5X)) when compared to the NOG complex (PDB: 7Q5V) (Figure 2). Although these crystallographic observations likely reflect snapshots of β2–β3 loop conformations in solution, they further highlight the importance of the mobility of the β2–β3 loop in catalysis.
Binding of the residues to the N‐terminal side of the substrate proline residue in the structures of PHD2 in complex with HIF1α556–574.CODD and HIF1α394–413.NODD involves interactions with βII, βII/III loop, β2–β3 loop, βIII, βVI–VII, and βVIII (βI–βVIII refer to the eight β‐strands of the DSBH). 27 , 36 , 37 Binding of the residues to the C‐terminal side of the substrate proline residues of these peptides involves interactions with βVIII, βIII, helix α3, and the α3‐βI loop. Notably, the structures show that the CODD substrates are observed to make more polar and hydrophobic interactions with the C‐terminal α4 compared to HIF1α‐NODD (Lys400PHD2‐Asp571HIF1α‐CODD/Asp538HIF2α‐CODD; Tyr403PHD2‐Asp536HIF2α‐CODD; Tyr403PHD2‐Met568HIF1α‐CODD/Met535HIF2α‐CODD), though the mobile C‐terminal region is likely involved in catalysis in all cases. 11 , 27 Notably, residues Val241, Ser242, Lys244, and Ile251 of the β2–β3 loop interact with Glu560/Met561HIF1α‐CODD and Thr398/Leu399HIF1α‐NODD, via interactions with the “XX” residues of the conserved LXXLAP motif present in all HIFα‐ODDs, highlighting the role of the β2–β3 loop in productive positioning of the different HIFα‐ODD substrates (Figure 4A,B ). 11 , 27
FIGURE 4.
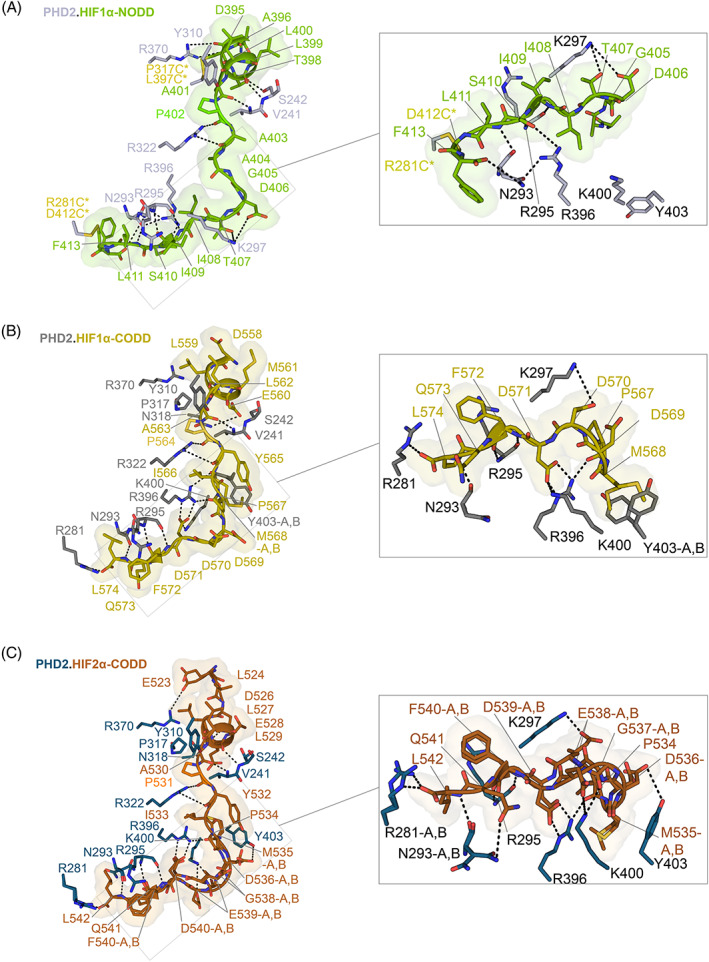
Comparison of PHD.HIFα‐ODD binding interactions. (A–C) Views showing the conformations of HIF1‐2α (HIF1α‐NODD‐green, HIF1α‐CODD‐olive, and HIF2α‐CODD‐orange) as observed by crystallography in complex with truncated PHD2 displayed as sticks with solvent‐excluded surface representation (Connolly) (PDB: 5L9V‐gray, 7Q5V‐blue, and 3HQR‐dark gray). HIF1‐2α‐ODDs are displayed as cartoons and sticks. Hydrogen bonding and electrostatic interactions are represented by black dashes. (A) Disulfide cross‐linked residues in (A), produced to enable stable complex formation, are shown as yellow sticks. CODD, C‐terminal oxygen‐dependent degradation; HIF, hypoxia‐inducible factor; NODD, N‐terminal oxygen‐dependent degradation.
In our PHD2181‐407.HIF2α523–542‐CODD structures, interactions of the HIFα‐ODD residues with residues both on the N‐terminal and C‐terminal sides of the target proline peptide with PHD2 are conserved, including the interaction with α4 (Arg396PHD2‐Asp539HIF2α‐CODD) (Figure 4C). The β2–β3 loop residues (Val241, Ser242, Lys244, and Ile251) interact with Glu527/Thr528HIF2α‐CODD (“XX” residues of the LXXLAP motif of HIF2α‐CODD) in a similar fashion to the previously reported PHD2 structures with HIF1α‐NODD/‐CODD. 27
These combined observations further support a role for the β2–β3 loop in positioning the HIFα‐ODD substrates at the PHD active site, notably via interactions with the conserved LXXLAP motif in HIFα‐ODDs. Despite most of the interactions appearing to be conserved in the different HIFα‐ODDs, a striking conformational feature is observed at the C‐terminal site of HIF2α523–542‐CODD in both the 2OG and NOG PHD2181‐407.HIF2α‐CODD complex structures (PDB: 7Q5V and 7Q5X). Glu538HIF2α‐CODD is observed to adopt two conformations in both structures, one of which, conformation‐A, is less solvent exposed and one of which, conformation‐B, is more solvent exposed. In conformation‐B, Glu538HIF2α‐CODD projects towards a symmetry‐related chain forming a hydrogen bond with Glu538HIF2α‐CODD in a symmetry‐related molecule (Figure S3A). It is possible that the conformational flexibility of Gly537HIF2α/Glu538HIF2α unit relates to the presence of the additional Gly537 in HIF2α‐CODD on the C‐terminal side of the hydroxylated proline, compared to HIF1α/3α‐CODD and HIF1/2α‐NODD (Figure 1A). When compared with HIF1/2α‐NODD, hydrophobic Ile‐residues are in the same position as the polar Gly/Glu unit of HIF2α‐CODD; these may alter the dynamics of the enzyme‐substrate interaction (Figure 1A). 27
The above‐described differences may, at least to some extent, influence PHD2 HIFα‐isoform selectivity. To investigate the preference of PHD2 towards HIFα‐CODD substrates, we carried out assays comparing the PHD2 catalyzed hydroxylation of HIF1‐3α CODD peptides, both individually and as a mixture (Figure 5). The results with PHD2 and individual peptides showed no clear preference for the HIF1‐3α‐CODD. However, when conducting the reaction with a 1:1:1 mixture of HIF1‐3α‐CODD peptides, PHD2 showed a clear preference for HIF1α‐ over the HIF3α‐ and HIF2α‐CODDs (Figure 5A). This result supports the proposal that the presence of the additional Gly537/Glu538 unit in HIF2α‐CODD on the C‐terminal side of the hydroxylated proline, compared to HIF1α/3α‐CODD causes PHD2 to preferentially catalyze hydroxylation of HIF1α peptide over HIF2‐3α‐CODDs peptides. 51 The Gly537/Glu538 unit in HIF2α‐CODD may also reflect differences in crystallization conditions required for the various PHD2.HIFα‐ODD complexes (Table S1).
FIGURE 5.
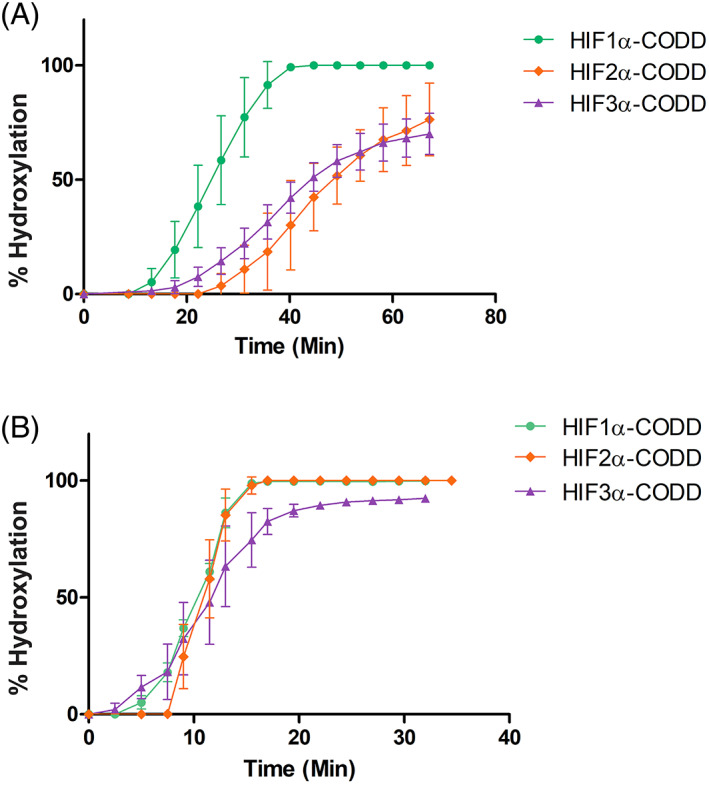
PHD2.HIFα‐CODD hydroxylation assays. (A, B) Studies on the HIFα‐CODD selectivity of PHD2181–426. The following CODD peptides (with C‐terminal amides) were used: HIF1α556‐574‐CODD (DLDLEMLAPYIPMDDDFQL), HIF2α523–542‐CODD (ELDLETLAPYIPMDGEDFQL) and HIF3α484–505‐CODD (ALDLEMLAPYISMDDDFQLN). (A) 200 μM sodium‐l‐ascorbate, 20 μM 2OG, 20 μM (NH4)2Fe(II)SO4, and HIF1‐3α‐CODD peptides (each at 10 μM) were mixed in a ratio of 1:1 with 300 nM PHD2181–426. Hydroxylation (%) was measured in real time (min) by SPE‐MS‐based enzymatic assays. (B) Single peptide control reaction conditions: 200 μM sodium‐l‐ascorbate, 20 μM 2OG, 20 μM (NH4)2Fe(II)(SO4), and individual HIFα‐CODD peptides (10 μM) with 300 nM of PHD2181–426. CODD, C‐terminal oxygen‐dependent degradation; HIF, hypoxia‐inducible factor; NODD, N‐terminal oxygen‐dependent degradation.
2.2. Structural comparison of PHD1‐3.HIF1‐3α between crystallographic and AlphaFold predicted structures
Although structures of PHD2 in complex with HIF1‐2α‐CODD and HIF1α‐NODD are available, 11 , 27 , 52 analogous structures of PHD1 and PHD3 complexed with HIFα‐ODDs are not reported. To gain insight into how structural differences between PHDs may influence isoform selectivity toward HIFα‐substrates, we built AlphaFold models 53 of PHD1 (UniProt: Q96KS0) and PHD3 (UniProt: Q9H6Z9) and compared these with the PHD2 crystal structures (Figures S5 and S6). The structural alignments imply variations in the conformations of residues in the DSBH βII/βIII loop and β2–β3 loop regions (Figures 1B,C, S5, and S6). The predicted PHD1 DSBH βII/βIII loop region differs from that of PHD2 at two residues (Lys291PHD2/Val297PHD1 and Asn318PHD2/His302PHD1) and at four residues in the β2–β3 loop region (Ser247PHD2/Pro231PHD1, Ser248PHD2/Pro232PHD1, Asp250PHD2/Ser234PHD1, Asp246/Ile230PHD1). The predicted PHD3 β2–β3 loop differs at eight residues compared with PHD2 (Gln243PHD2/Ala66PHD3, Leu244PHD2/Arg65PHD3, Ser242PHD2/Pro64PHD3, Arg281PHD2/Leu103PHD3, Asn293PHD2/Lys115PHD3, Lys291PHD2/Tyr113PHD3, Gly294PHD2/Glu116PHD3, Tyr403PHD2/Phe225PHD3) (Figure S5B).
Comparison of the binding modes of HIFα‐ODDs C‐terminal to the proline substrate PHD1‐3 implies that most of the key protein:substrate interactions are conserved in the analogous PHD1 and PHD2 complexes (Figure S5). By contrast, although care should be taken not to over interpret the preliminary models, more apparent differences are observed between PHD3 and PHD2 (and by implication PHD1). In the PHD2181‐426/407.HIF1‐2α‐CODD structures (PDB: 3HQR and 7Q5V), Arg281PHD2 interacts with residues C‐terminal to the target prolines of the HIF1‐2α‐CODD substrates, that is Leu574HIF1α and Leu542HIF2α, respectively. These interactions may not occur (or occur differently/less efficiently) in PHD3 where Arg281 is replaced by Leu103 (Figure S5D,E). Additionally, in the PHD2181–426.HIF1α‐NODD complex structure (PDB: 5L9V), Arg396PHD2 is positioned to form a polar interaction with Ser410HIF1α‐NODD (Figure 5B). In the PHD3 model, Ser410HIF1α‐NODD is positioned to interact with Glu116PHD3. Further, in the PHD3 model, the Tyr113PHD3 and Lys115PHD3 side chains clash with Phe413HIF1α‐NODD (Figure S5C). The relative lack of predicted interactions between PHD3 and HIF1α‐NODD might, in part, rationalize the preference of PHD3 for HIF1‐2α‐CODD over NODD. 26
Comparison of the HIFα‐ODD binding modes of PHD1‐3 with respect to the N‐terminal sides of the target substrate prolines (Figure S6) also implies differences in the PHD.HIFα‐ODD interactions between PHD1/PHD3 models and the PHD2 crystal structures. In PHD2181‐426/407.HIF1‐2α‐CODD structures (PDB: 3HQR and 7Q5V), Asn318PHD2 interacts with Glu560HIF1α‐CODD and Asp525/Glu527HIF2α‐CODD; the Asn318PHD2 side chain is in different conformations in the HIF1α‐ and HIF2α‐CODD structures. In the predicted PHD1 model, Asn318PHD2 (DSBH βII/βIII loop) is replaced by His302PHD1 (Figure S6), which may interact differently with the HIF1‐2α‐CODDs compared to Asn318PHD2. In the PHD1 model with HIF1α‐NODD, Thr398HIF1α‐NODD likely forms a polar interaction with His302PHD1; however, in the covalently cross‐linked PHD2181‐426.HIF‐1α‐NODD complex residues Thr398HIF1α‐NODD and Asn318PHD2 are 5.46 Å apart, which suggests a weak interaction at this position (PDB: 5L9V). Similarly, in all reported PHD2.HIFα‐ODD complexes, Ser242PHD2 interacts with Leu397HIF1α‐NODD, Glu560HIF1α‐CODD, and Asp390HIF2α‐CODD. In PHD3, residue Ser242PHD2 (β2–β3 loop) is substituted by Pro64PHD3, the latter of which cannot make the same polar interactions (Figure S6). Pro64PHD3 may also alter the dynamics of the β2–β3 loop during catalysis compared with PHD1‐2.
The predicted weaker interactions of PHD3.NODD residues both on the N‐ and C‐terminal sides of the proline substrate residue may explain the low level of PHD3.HIF1‐2α‐NODD turnover observed from these substrates. 26 , 27 Due to the preliminary nature of the models and given the multitude of interactions in the PHD.HIFα‐ODD complexes, to what extent these structural conformational changes/induced fit processes directly influence catalysis and PHD isoform selectivity remains unclear.
3. DISCUSSION
Interactions between the PHDs and the HIFα‐ODDs play a central role in the hypoxic response in humans and other animals. PHD‐like prolyl‐hydroxylases are also present in certain non‐animal eukaryotes and prokaryotes, though to date these identified substrates are not HIF (like) transcription factors. In early metazoan PHD/HIF‐containing organisms, there is typically only one PHD and one HIFα, as exemplified by studies on T. adhaerens (Figure S4). 40 , 54 However, in humans and other complex HIF containing animals, there are commonly more than one PHD isoform and more than one HIFα isoform, though (typically) likely only one von Hippel–Lindau protein and one FIH. 54 There are some subsequent bioinformatic studies that have revealed multiple PHDs and HIFα‐ODDs present in complex animals, at least in part, a reflection of the need for context‐dependent regulation of the hypoxic response. 54 , 55 This increased complexity may introduce vulnerabilities with respect to mutations enabling specific disease states including cancer, for example, by using modulation of one HIFα isoform in tumor progression whilst maintaining an ability to execute a robust hypoxic response, with another HIFα isoform. 31 , 56 , 57 In this regard, the link between HIF2α upregulation in ccRCC (most commonly associated with VHL gene mutation) and diseases related to erythrocytosis is of interest.
Reduction of HIF2α mediated expression is the mode of action of Belzutifan which is used to treat ccRCC. 31 , 33 However, there is a need for new treatments for ccRCC and other diseases associated with VHL/HIFα/PHD gene mutations. Such treatments could, in principle, involve modulation of PHD.HIFα‐ODD interactions, for example, by sequestering a HIF2α‐CODD in complex with PHD, possibly in a manner that signals for a non‐VHL mediated protein degradation process, by using a small‐molecule and/or metal ion that promotes the PHD.HIF2α‐CODD interaction. The structures presented here may help in the design of such small molecules.
Since many mutations to the catalytic domain of PHD2 and HIF2α‐ODDs have been identified, 58 , 59 , 60 , 61 , 62 , 63 , 64 understanding how these impact on PHD2.HIF2α‐ODD interactions is of interest in terms of understanding the molecular basis of associated diseases (Figure S2). At least some of the observed EPAS1/HIF2α mutations will likely impact on PHD catalysis as they involve residues that interact with the PHD2 active site as shown by our PHD2181‐407.NOG/2OG.HIF2α‐CODD structures (PDB: 7Q5V and 7Q5X) and inferred by models of PHD1‐3.HIFα‐ODD complexes (Figure S2). Strikingly, some of the clinically observed mutations (M535V, M535T, G537W, and G537R) affect the Gly537/Glu538 unit, thus likely altering PHD.HIFα‐ODD binding potentially in a manner affecting catalytic efficiency and/or HIFα‐ODD selectivity in a disease‐relevant manner. Differences in PHD1‐3.HIF1‐3α‐ODD related interactions are also important in the normal hypoxic response and knowledge of them may help enable treatments including modulation of specific sets of HIF target genes.
The combined crystallographic and NMR studies, further highlight the importance of the conformationally mobile β2–β3 loop and the C‐terminal PHD region in HIFα‐ODD hydroxylation and selectivity (Figures 2 and 4). 11 , 27 , 29 , 30 , 36 , 45 However, the available evidence also supports the dynamic nature of PHD.HIFα‐ODD interactions, at least in certain stages of the catalytic cycle. This means that structure‐based attempts to modulate PHD.HIFα‐ODD interactions, for example, to alter PHD isoform selectivity, should be coupled with empirical approaches in cells (note PHD.HIFα interactions likely involve other components and regions beyond the immediate PHD catalytic domain and HIFα‐ODD reactions).
The presence of an additional residue (Gly537/Glu538 unit) in HIF2α‐CODD compared to other HIFα‐ODDs, likely results in increased flexibility of HIF2α‐CODD, possibly weakening its binding to PHD2 (Figure S3A). This may, at least partially, explain the preference of PHD2 for HIF1α > HIF3α > HIF2α‐CODDs as observed in our biochemical studies (Figure 5). However, it is important to note that multiple interactions occur between the PHDs and the HIFα‐ODDs and given the dynamic nature of at least some of these interactions, it is difficult to predict the effects of individual residue changes with confidence.
By contrast, the dynamic and multivalent interaction of the PHDs with the overall HIFα‐ODDs, the chemistry in the immediate active site vicinity appears to be highly conserved in the PHDs, an observation which even extends, at least substantially, to PHD‐like enzymes with non‐HIFα substrates (Figure S4C). 41 , 42 The conservation includes with respect to the nature of Fe(II) and 2OG binding, including the positioning of the 2OG C1 carboxylate adjacent to the methylene of the proline‐residue that undergoes hydroxylation, an arrangement that is likely partially responsible for the unusually slow reaction of the PHDs with O2, though other factors also likely impact this aspect of the mechanism. 43 Another chemically relevant conservation is the conformation of the unhydroxylated substrate proline ring at the active site, which to date has always been observed in the C4‐endo formation, at least in the PHD2 substrate complexes. PHD catalyzed trans‐4‐hydroxylation results in a bias of the proline ring to the C4‐exo protein conformation, due to operation of a stereoelectronic effect, as observed in pVHL‐hydroxylated‐HIFα‐ODD complex structures. 39
Comparison of AlphaFold models of PHD1 and PHD3 with PHD2.HIF1‐2α‐CODD and PHD2.HIF1α‐NODD structures predict differences in the β2–β3 loop and C‐terminal regions apparently linked to differences in the HIFα‐ODDs binding modes, with overall fewer interactions between the HIFα‐ODDs and PHD1/3 models compared to PHD2 (Figures S5 and S6). Testing the consequences of these differences for PHD catalysis with wild‐type and clinically relevant mutated HIFα‐ODDs is the subject of ongoing work.
4. EXPERIMENTAL SECTION
4.1. Materials
Reagents, chemicals, and solvents were from Sigma‐Aldrich (Merck), Apollo Scientific, or Thermo Fisher Scientific, except where stated. The HIF1α556–574‐CODD (DLDLEMLAPYIPMDDDFQL), HIF2α523–542‐CODD (ELDLETLAPYIPMDGEDFQL), and HIF3α484–505‐CODD (ALDLEMLAPYISMDDDFQLN) and 3C cyclic (d‐YVWLTDTWVLSRTC) 28 , 35 peptides were from GL Biochem (prepared with a C‐terminal amide). Water used for cell culture was purified using a Millipore Elix® 10 system (Merck Life Sciences) and autoclave sterilized (Crystal 300‐RP25, Rodwell Engineering Group). Water used for buffers and molecular experiments was filtered purified by a 0.22‐μm Milli‐Q filtration system (Milli‐Q, Merck Life Sciences). Kanamycin (final concentration 62 mM) was prepared in water sterilized by a benchtop autoclave (LTE TouchClave II, LTE Scientific; program: 121°C and 1 Bar for 15 min) and filtered for impurities with a 0.22‐μm syringe filter (Sarstedt).
4.2. Expression and protein purification
The PHD2181–407‐pET‐28a(+) plasmid was expressed in Escherichia coli BL21(DE3) cells (New England Biolab Inc.). 27 Expression was induced with 0.5 mM isopropyl β‐d‐1‐thiogalactopyranoside (IPTG) (OD600 nm 0.6–1.2) at 28°C for 3–4 h. 27 Cells were harvested and stored at −80°C until purification. Cells were freeze‐thawed at 4°C in the lysis buffer (20 mM Tris–HCl pH 7.5 RT, 0.5 M NaCl, 5 mM imidazole, and 5% (vol/vol) glycerol). 27 DNaseI and ethylenediaminetetraacetic acid (EDTA)‐free protease inhibitor tablet (Roche) were added. Sonication (10 min total elapsed time, 3 s on/off pulse) was used for lysis (Cole‐Parmer®‐500‐Watt ultrasonic homogenizer, Cole‐Parmer). Cell lysates were then centrifuged (20,000 rpm, 4°C, JA‐25.50 rotor‐Avanti‐JHC centrifuge, Beckman Coulter); the supernatant was loaded onto a 5‐mL HisTrap™ column (GE Life Sciences) for Ni(II) affinity chromatography. A 5‐mL HisTrap™ column was charged with 5 column volumes (CVs) of 100 mM Ni(II)SO4, then washed with 5 CV of lysis buffer, followed by 5 CV of elution buffer (20 mM Tris–HCl pH 7.5 room temperature [RT], 0.5 M NaCl, 0.5 M imidazole, and 5% [vol/vol] glycerol), finally with 5 CV of lysis buffer. 30 The loaded column was washed with 30 CV of wash buffer (20 mM Tris–HCl pH 7.5 RT, 0.5 M NaCl, 30 mM imidazole, and 5% [vol/vol] glycerol). Tagged PHD2181–407 was eluted using a step gradient (5 CV each step) of increasing elution buffer (16% [vol/vol], 34% [vol/vol], and 100% [vol/vol]). 30 The purity of the protein fractions was analyzed by SDS‐PAGE (>90% pure material was used). His6‐PHD2181–407 fractions were concentrated to 5–6 mL volume with a concentrator (10 kDa cutoff, Amicon) at 4000 rpm and 4°C. To cleave His6‐tag 0.25 units of restriction grade thrombin (Novagen, Merck) and 1× thrombin cleavage buffer (10× stock of 200 mM Tris–HCl pH 8.4, 1.5 M NaCl, and 25 mM CaCl2, Novagen, Merck) were added to tagged PHD2181–407. The cleaved PHD2181–407 was loaded onto a Superdex® 75 gel filtration column (GE Life Sciences) pre‐equilibrated with 1 CV of 50 mM Tris–HCl pH 7.5 RT, 100 mM NaCl, and 1% (vol/vol) glycerol. Proteins were eluted with an isocratic gradient and fractions were collected and analyzed for purity by SDS‐PAGE (estimated >90%). 30 PHD2181–407 was buffer exchanged with a PD‐10 desalting column (GE Life Sciences) into the final storage/crystallization buffer (50 mM Tris–HCl pH 7.5 RT, and 1% [vol/vol] glycerol).
4.3. PHD2.HIF2α‐CODD complex preparation
Highly purified PHD2181–407 via a two‐column purification strategy (affinity and SEC chromatography) was used to obtain the PHD2181–407.Mn(II).NOG/2OG.HIF2α523–542‐CODD complex crystals. Stocks of Mn(II) (100 mM), NOG pH 7–8 (80 mM), and 2OG disodium salt (100 mM) were prepared in deionized water (filter sterilized with 0.22 μm Milli‐Q filtration system, Merck Life Sciences). Cofactors/inhibitors were diluted to final concentrations of 1.2 mM‐Mn(II) and 2 mM‐NOG/2OG in the protein solution. The protein.metal.ligand mixture was pipetted directly onto lyophilized 3C cyclic peptide (weighed out at a final concentration of 2 mM into a 70‐μL final mixture volume) and left to equilibrate on a Cole‐Parmer® tube rotator (Cole‐Parmer) at 4°C for 2.5 h. PHD2181–407 was centrifuged at 12000 rpm (9600g) for 10 min at 4°C. The protein (1 mM), dissolved cofactors (1.2 mM‐Mn(II), 2 mM‐NOG/2OG), and 3C (2 mM) mixture were added to lyophilized HIF2α523–542‐CODD peptide (weighed out for a final concentration of 2–4 mM into a 70‐μL final mixture volume). The protein‐substrate mixtures were left overnight to equilibrate on the tube rotator at 4°C. The next day, the incubated sample was centrifuged at 14,000 rpm (18,800 g) and the supernatant was harvested for crystallization. The protein‐substrate sample volume was adjusted with crystallization buffer (50 mM Tris–HCl pH 7.5 RT, 1% [vol/vol] glycerol) to a final volume of 70 μL before preparation of the crystallization plates.
4.4. Crystallization of the PHD2181 –407.Mn(II).NOG/2OG.HIF2α523–542‐CODD complexes
The PHD2181–407.Mn(II).NOG or 2OG.HIF2α523–542‐CODD.3C mixtures were screened against 0.25–0.39 M magnesium formate disodium salt and 18%–22% (wt/vol) poly‐ethylene glycol (PEG) 3350 pH 7.0 (precipitant solutions were filtered; 0.22‐μm filter, Sarstedt). 28 The protein‐substrate mixture was prepared with 1 mM PHD2181–407, 1 mM Mn(II)Cl2, 2 mM NOG/or 2OG, 2–4 mM HIF2α523–542‐CODD peptide, and 2 mM 3C peptide dispensed into crystallization plates (300 nL drops at 2:1 and 1:2 ratios and 200 nL drop at 1:1 ratio in Intelli‐plates, Art Robbins) with a Phoenix robot (Art Robbins) at 4°C and stored at 298K. A 1 mm × 100 μm × 80 μm (PDB: 7Q5V) and a 240 μm × 50 μm × 30 μm (PDB: 7Q5X) plate‐like crystals appeared after 1‐week of equilibration in 0.31 M magnesium formate and 16.6% (wt/vol) PEG 3350 (200 nL, 1:1 protein‐to‐well ratio, 298K). Crystals were exposed to the cryo‐protectant (reservoir solution supplemented with 20% (vol/vol) glycerol), manually looped, and cryo‐cooled into liquid‐N2. Crystals were stored under liquid‐N2 until data collection at the Diamond Light Source.
4.5. Solid phase extraction‐MS based enzymatic activity assays
Activity assays were conducted using a RapidFire® RF360 sampling robot (Agilent Technologies). Samples were loaded onto a C4 SPE cartridge (Agilent Technologies) and peptides were eluted with 85% (vol/vol) acetonitrile and 15% (vol/vol) water mixture added with 0.1% (v/v) formic acid. Real‐time activity assays were performed in reaction buffer containing 50 mM Tris–HCl pH 7.8 and 50 mM NaCl. Stock solutions of each component were made freshly. 100 mM stock solution of sodium‐l‐ascorbate and 50 mM stock solution of 2OG were made in water (LC–MS Grade, LiChrosolv®). 10 mM peptides stock solution were made in dimethyl sulfoxide (DMSO). To limit oxidation of Fe(II) to Fe(III), a 100 mM stock solution of (NH4)2Fe(II)(SO4) was made in HCl (20 mM), then diluted to 10 mM with water (LC–MS Grade, LiChrosolv®). 1 mL final volume solutions containing 200 μM sodium‐l‐ascorbate, 20 μM 2OG, 20 μM (NH4)2Fe(II)(SO4) and 10 μM peptide (HIF1α556–574‐CODD, HIF2α523–542‐CODD, or HIF3α484–505‐CODD) were prepared as control reactions. 1 mL solutions containing 200 μM sodium‐l‐ascorbate, 20 μM 2OG, 20 μM (NH4)2Fe(II)(SO4), and 10 μM of a 1:1:1 mixture of peptides (HIF1α556–574‐CODD, HIF2α523–542‐CODD, and HIF3α484–505‐CODD) were prepared for the competition reactions. About 500 μL of the substrate mixture was transferred into 96‐well polypropylene plates (Agilent Technologies). After a first injection onto the C4 SPE cartridge (Agilent Technologies), data acquisition was paused, then 500 μL of 300 nM PHD2181–426 in reaction buffer was added into the well to initiate the reaction. The control reactions were monitored for 32 min (one injection every 2.5 min). The competition reactions were monitored for 67 min (one injection every 5 min). The positive ion mode was used to monitor peptide charge states. RapidFire Integrator software (Agilent Technologies) was used to integrate the area of the peaks extracted from the chromatogram. Excel was used to calculate percent (%) hydroxylation of the CODD peptide substrates using the formula: % hydroxylated substrate = 100 × hydroxylated/(hydroxylated + non‐hydroxylated peptide). Oxidation of the methionine residues in the CODD sequences was 4%–6% in the no enzyme control. Every data set was normalized to a no enzyme buffer control.
4.6. X‐ray data analysis and software
X‐ray diffraction data were collected at Diamond Light Source synchrotron at I24 MX beamline and autoprocessed with Xia2 (DIALS, Diamond Light Source Ltd.). 65 PHENIX.Xtriage was used to assess the data quality of the reflections. 66 Phaser‐Molecular replacement (PHASER‐MR) was used to phase the processed diffraction data for the structures (PDB: 7Q5V and 7Q5X). 67 A previously determined structure of PHD2 (PDB: 3HQR) was used as a search model for MR‐phasing. 11 COOT (version 0.9.5, CCP4) was used to semi‐manual model build based on the overlaid 2mFo‐DFc and difference mFo‐DFc electron density maps from the phased structures (PDB: 7Q5V and 7Q5X). 68 , 69 The geometry of the model was adjusted based on calculated electron density maps and was improved in COOT with subsequent refinement cycles using PHENIX.Refine. 66 , 70 Three cycles were typically run for each refinement round before manual fitting. PHENIX.Refine was used to modify and improve the model in iterative cycles. 70 Model improvement was assessed by the decrease in, and convergence of R work/R Free values between cycles of refinement. MolProbity was used to assess the geometric quality of the refined model and to guide re‐building in COOT. 68 , 71 Resolution was defined depending on the completeness of the resolution bin (>95% in all resolution bins). PDB extract online tool (version 3.24, Research Collaboratory for Structural Bioinformatics PDB) was used to prepare coordinate and structure factors files in macromolecular CIF format (mmCIF) to be uploaded to Onedep for PDB deposition. 72 PyMOL™ (Schrodinger) was used for graphical representation and structure alignment.
4.7. Quantification and statistical analysis
GraphPad prism (version 6.0) was used to plot hydroxylation over time. Multiple sequence alignments of HIF2α523–542‐CODD (EPAS1) sequences employed ClustalOmega (EMBL‐EBI) using the default settings. JalView (version 2.10.5) was used to generate figures.
AUTHOR CONTRIBUTIONS
William D. Figg, Jr.: Conceptualization; methodology; software; data curation; investigation; validation; formal analysis; visualization; writing – original draft; writing – review and editing. Giorgia Fiorini: Methodology; software; data curation; investigation; validation; formal analysis; visualization; writing – original draft; writing – review and editing. Rasheduzzaman Chowdhury: Conceptualization; methodology; investigation; validation; writing – review and editing. Yu Nakashima: Software; investigation; validation; writing – review and editing. Anthony Tumber: Methodology; software; investigation; validation. Michael A. McDonough: Conceptualization; methodology; software; investigation; validation; formal analysis. Christopher J. Schofield: Conceptualization; methodology; investigation; validation; formal analysis; supervision; funding acquisition; visualization; project administration; resources; writing – original draft; writing – review and editing.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting information
FIGURE S1. Overview of the role of protein hydroxylations in the HIF‐mediated hypoxic response pathway. In the presence of sufficient O2, PHD1‐3, and FIH efficiently hydroxylate HIFα isoforms. PHD catalysis promotes degradation of HIFα via the ubiquitin‐proteasomal pathway in which the von Hippel–Lindau protein/elongin B/C complex plays a key role. HIF‐mediated transcription is inhibited by FIH catalysis which hinders binding of HIF to the CBP/p300 acetyltransferases. In moderate hypoxia, the PHDs are less active than FIH. In hypoxia, HIF hydroxylase activity is reduced so enabling increased levels of HIFα and formation of the transcriptionally active HIFα,β‐heterodimer. 1 , 2 , 3
FIGURE S2. Structural locations of selected clinically observed HIF2α‐CODD variants and sequence conservation of the EPAS1 (HIF2α) C‐terminal oxygen dependent degradation domains in a set of eukaryotic organisms. (A) Predicted locations of selected predicted clinically observed HIF2α523‐542‐CODD variants (sticks‐yellow) on the basis of the PHD2181–407.Mn(II).NOG.HIF2α523–542‐CODD complex structure (sticks‐blue) (PDB: 7Q5V). 4 , 5 , 6 , 7 , 8 , 9 , 10 PHD2 (blue) and HIF2α‐CODD (orange) are depicted as cartoons. Key polar interactions are represented by black dashes. Waters (red) and Mn (violet) are displayed as spheres. (B) Alignment of the HIF2α (EPAS1) CODD with HIF2α sequences from selected eukaryotic organisms. The percentage identities compared with the shown human HIF2α sequence are given.
FIGURE S3. View of the binding modes of the glycine‐glutamate unit in PHD2181–407.Mn.NOG.HIF2α523–542‐CODD and of the 3C cyclic peptide binding site. (A) Lattice packing view of the PHD2181–407.Mn.NOG.HIF2α523–542‐CODD (orange) complex crystal structure (PDB: 7Q5V). Electron density is shown as a blue mesh (contoured at 1.0 σ). Interactions of HIF2α Glu538 (conformation B) (orange) with the same residue (HIF2α Glu538, conformation B) in a symmetry‐related molecule (tan) are shown. (B) Comparison of the PHD2.Mn.NOG.HIF1α.3C (gray‐6YW3) and the PHD2181–407.Mn.NOG.HIF2α523–542‐CODD (blue/teal‐7Q5V) complex structures comparing residues that interact with the 3C cyclic peptide in the former case (density for the 3C cyclic peptide was not observed in the PHD2181–407.HIF2α‐CODD complex structure). Polar interactions are represented by black dashes. Waters (red) and Mn (violet) are displayed as spheres.
FIGURE S4. Comparison of substrate binding modes in the Trichoplax adhaerens PHD.TaODD, PHD2181–407.NOG.HIF2α‐CODD, and Pseudomonas putida PHD.NOG.EF‐Tu complex crystal structures. (A) Comparison structures of TaPHD21–257.Mn(II).NOG.TaODD477–497 (PDB: 6F0W) and PHD2181–407.Mn(II).NOG.HIF2α523–542‐CODD (PDB: 7Q5V). Enzymes (TaPHD‐green and PHD2‐blue) and substrates (TaODD‐pink and HIF2α‐orange) are shown as cartoons. The active sites, β2–β3 loops (TaPHD‐green cyan and PHD2‐red), and ligands (NOG‐yellow) are displayed with key residues as sticks. Waters (red) and Mn (violet) are displayed as spheres. (B) Comparison of the TaODD477–497 (pink‐sticks) and HIF2α (orange‐sticks) substrate binding showing solvent‐excluded surface representation (Connolly). (A, B) Hydrogen bonding and electrostatic/polar interactions are in black dashes. (C) Two views of the off‐line NOG binding mode in the PHD2181–407.HIF2α‐CODD, TaPHD.TaODD, and PPHD (purple).EFTu (yellow‐4IW3) complex structures. Note the conservation of the C4 endo‐conformation of all the substrate proline residues.
FIGURE S5. Comparison of binding modes of HIFα‐ODDs to PHD1‐3 involving residues to the C‐terminal side of the substrate proline residue. (A) Residues to the N‐terminal and C‐terminal sides of the substrate proline residue in HIFα‐ODD peptides are displayed on PHD1/3 model overlays. (A, B) Comparison of the conformation of the β2–β3 loop in the PHD2181–407.HIF2α523‐542 with AlphaFold (AF) models of PHD1 (cyan sticks/cartoon; UniProt: Q96KS0) and PHD3 (purple sticks/cartoon; UniProt: Q62630). (C–E) Views from crystal structures of PHD2181–426.HIF1α394–413‐NODD (PDB: 5L9V), PHD2181–426.HIF1α556–574‐CODD (PDB: 3HQR), and PHD2181–407.HIF2α523–542‐CODD (PDB: 7Q5V) displayed as cartoons and sticks (HIF1α‐NODD‐green, HIF1α‐CODD‐olive, and HIF2α‐CODD‐orange). The PHD1 (cyan; UniProt: Q96KS0) and PHD3 (purple; UniProt: Q62630) stick views are derived from AF models and are aligned with crystals for PHD2.HIFα complexes. Polar interactions in the experimentally determined PHD2 structures are represented by black dashes to enable comparison with the predicted residue conformations in the PHD1 and PHD3 models.
FIGURE S6. Comparison of the binding modes of HIFα‐ODDs to PHD1‐3 involving residues to the N‐terminal side of the substrate proline residue. (A–C) Views from crystal structures of cross‐linked‐PHD2181–426.HIF1α394–413‐NODD (PDB: 5L9V), PHD2181–426.HIF1α556–574‐CODD (PDB: 3HQR), and PHD2181–407.HIF2α523–542‐CODD (PDB: 7Q5V) displayed as cartoons (HIFα‐peptides) and stick views (HIF1α‐NODD‐green, HIF1α‐CODD‐olive, and HIF2α‐CODD‐orange). The PHD1 (cyan; UniProt: Q96KS0) and PHD3 (purple; UniProt: Q62630) stick views are derived from AlphaFold (AF) models aligned with crystal structures for PHD2.HIFα complexes. Polar interactions in the experimentally determined PHD2 structures are represented by black dashes to enable comparison with the predicted residue conformations in the PHD1 and PHD3 models.
TABLE S1. Crystallization conditions for PHD2.HIFα‐substrate complexes.
ACKNOWLEDGMENTS
The authors thank our funding bodies the Wellcome Trust (091857/7/10/7), the Biotechnology and Biological Sciences Research Council (BB/L000121/1, BB/J001694/2, and BB/R013829/1) the Cancer Research UK (C8717/A18245) and a Newton Abraham studentship (to GF) for for support of this work. The authors thank Diamond Light Source for the allocated beamtime (proposal‐visit: MX‐18069‐63 and MX‐18069‐86), and the staff of the I24 beamline for assistance with crystal screening and data collection.
Figg WD Jr., Fiorini G, Chowdhury R, et al. Structural basis for binding of the renal carcinoma target hypoxia‐inducible factor 2α to prolyl hydroxylase domain 2. Proteins. 2023;91(11):1510‐1524. doi: 10.1002/prot.26541
DATA AVAILABILITY STATEMENT
Coordinates and structure factors for PHD2.HIF2α‐CODD complex structures were deposited in the RCSB Protein Data bank as: PHD2181–407.Mn.NOG.HIF2α523–542‐CODD, PDB: 7Q5V and PHD2181–407.Mn.2OG.HIF2α523‐542‐CODD, PDB: 7Q5X. AlphaFold models and HIFα sequences were accessed using UniProt: Q96KS0‐PHD1, Q9H6Z9‐PHD3, Q16665‐HIF1α, Q99814‐HIF2α, and Q9Y2N7‐HIF3α.
REFERENCES
- 1. Semenza GL. Hypoxia‐inducible factor 1: master regulator of O2 homeostasis. Curr Opin Genet Dev. 1998;8(5):588‐594. [DOI] [PubMed] [Google Scholar]
- 2. Kaelin WG, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393‐402. [DOI] [PubMed] [Google Scholar]
- 3. Schofield CJ, Ratcliffe PJ. Oxygen sensing by HIF hydroxylases. Nat Rev Mol Cell Biol. 2004;5(5):343‐354. [DOI] [PubMed] [Google Scholar]
- 4. Chowdhury R, Hardy A, Schofield CJ. The human oxygen sensing machinery and its manipulation. Chem Soc Rev. 2008;37(7):1308‐1319. [DOI] [PubMed] [Google Scholar]
- 5. Maxwell PH, Wlesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia‐inducible factors for oxygen‐dependent proteolysis. Nature. 1999;399(6733):271‐275. [DOI] [PubMed] [Google Scholar]
- 6. Stebbins CE, Kaelin WG, Pavletich NP. Structure of the VHL‐elonginC‐elonginB complex: implications for VHL tumor suppressor function. Science. 1999;284(5413):455‐461. [DOI] [PubMed] [Google Scholar]
- 7. Ivan M, Kondo K, Yang H, et al. HIFα targeted for VHL‐mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292(5516):464‐468. [DOI] [PubMed] [Google Scholar]
- 8. Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML. Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science. 2002;295(5556):858‐861. [DOI] [PubMed] [Google Scholar]
- 9. Chan MC, Ilott NE, Schödel J, et al. Tuning the transcriptional response to hypoxia by inhibiting hypoxia‐inducible factor (HIF) prolyl and asparaginyl hydroxylases. J Biol Chem. 2016;291(39):20661‐20673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elkins JM, Hewitson KS, McNeill LA, et al. Structure of factor‐inhibiting hypoxia‐inducible factor (HIF) reveals mechanism of oxidative modification of HIF‐1α. J Biol Chem. 2003;278(3):1802‐1806. [DOI] [PubMed] [Google Scholar]
- 11. Chowdhury R, McDonough MA, Mecinović J, et al. Structural basis for binding of hypoxia‐inducible factor to the oxygen‐sensing prolyl hydroxylases. Structure. 2009;17(7):981‐989. [DOI] [PubMed] [Google Scholar]
- 12. Epstein ACR, Gleadle JM, McNeill LA, et al. C. elegans EGL‐9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107(1):43‐54. [DOI] [PubMed] [Google Scholar]
- 13. Bruick RK, McKnight SL. A conserved family of prolyl‐4‐hydroxylases that modify HIF. Science. 2001;294(5545):1337‐1340. [DOI] [PubMed] [Google Scholar]
- 14. Islam MS, Leissing TM, Chowdhury R, Hopkinson RJ, Schofield CJ. 2‐Oxoglutarate‐dependent oxygenases. Annu Rev Biochem. 2018;87(1):585‐620. [DOI] [PubMed] [Google Scholar]
- 15. Hewitson KS, McNeill LA, Riordan MV, et al. Hypoxia‐inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem. 2002;277(29):26351‐26355. [DOI] [PubMed] [Google Scholar]
- 16. Tsukada Y, Fang J, Erdjument‐Bromage H, et al. Histone demethylation by a family of JmjC domain‐containing proteins. Nature. 2006;439(7078):811‐816. [DOI] [PubMed] [Google Scholar]
- 17. Klose RJ, Kallin EM, Zhang Y. JmjC‐domain‐containing proteins and histone demethylation. Nat Rev Genet. 2006;7(9):715‐727. [DOI] [PubMed] [Google Scholar]
- 18. Myllyharju DJ. Prolyl 4‐hydroxylases, key enzymes in the synthesis of collagens and regulation of the response to hypoxia, and their roles as treatment targets. Ann Med. 2008;40(6):402‐417. [DOI] [PubMed] [Google Scholar]
- 19. Flashman E, Hoffart LM, Hamed RB, Bollinger JM, Krebs C, Schofield CJ. Evidence for the slow reaction of hypoxia‐inducible factor prolyl hydroxylase 2 with oxygen. FEBS J. 2010;277(19):4089‐4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tarhonskaya H, Chowdhury R, Leung IKH, et al. Investigating the contribution of the active site environment to the slow reaction of hypoxia‐inducible factor prolyl hydroxylase domain 2 with oxygen. Biochem J. 2014;463(3):363‐372. [DOI] [PubMed] [Google Scholar]
- 21. Tarhonskaya H, Hardy AP, Howe EA, et al. Kinetic investigations of the role of factor inhibiting hypoxia‐inducible factor (FIH) as an oxygen sensor. J Biol Chem. 2015;290(32):19726‐19742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hirsilä M, Koivunen P, Günzler V, Kivirikko KI, Myllyharju J. Characterization of the human prolyl 4‐hydroxylases that modify the hypoxia‐inducible factor. J Biol Chem. 2003;278(33):30772‐30780. [DOI] [PubMed] [Google Scholar]
- 23. McNeill LA, Hewitson KS, Claridge TD, Seibel JF, Horsfall LE, Schofield CJ. Hypoxia‐inducible factor asparaginyl hydroxylase (FIH‐1) catalyses hydroxylation at the β‐carbon of asparagine‐803. Biochem J. 2002;367(3):571‐575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tian YM, Yeoh KK, Lee MK, et al. Differential sensitivity of hypoxia inducible factor hydroxylation sites to hypoxia and hydroxylase inhibitors. J Biol Chem. 2011;286(15):13041‐13051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan DA, Sutphin PD, Yen SE, Giaccia AJ. Coordinate regulation of the oxygen‐dependent degradation domains of hypoxia‐inducible factor 1α. Mol Cell Biol. 2005;25(15):6415‐6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Appelhoffl RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia‐inducible factor. J Biol Chem. 2004;279(37):38458‐38465. [DOI] [PubMed] [Google Scholar]
- 27. Chowdhury R, Leung IKH, Tian YM, et al. Structural basis for oxygen degradation domain selectivity of the HIF prolyl hydroxylases. Nat Commun. 2016;7(1):12673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chowdhury R, Abboud MI, McAllister TE, et al. Use of cyclic peptides to induce crystallization: case study with prolyl hydroxylase domain 2. Sci Rep. 2020;10(1):21964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Abboud MI, Chowdhury R, Leung IKH, et al. Studies on the substrate selectivity of the hypoxia‐inducible factor prolyl hydroxylase 2 catalytic domain. ChemBioChem. 2018;19(21):2262‐2267. [DOI] [PubMed] [Google Scholar]
- 30. Figg WD Jr, McDonough MA, Chowdhury R, et al. Structural basis of prolyl hydroxylase domain inhibition by Molidustat. ChemMedChem. 2021;16(13):2082‐2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choueiri TK, Kaelin WG. Targeting the HIF2–VEGF axis in renal cell carcinoma. Nat Med. 2020;26(10):1519‐1530. [DOI] [PubMed] [Google Scholar]
- 32. Lee YS, Vortmeyer AO, Lubensky IA, et al. Coexpression of erythropoietin and erythropoietin receptor in Von Hippel‐Lindau disease‐associated renal cysts and renal cell carcinoma. Clin Cancer Res. 2005;11(3):1059‐1064. [PubMed] [Google Scholar]
- 33. Xu R, Wang K, Rizzi JP, et al. 3‐[(1S,2S,3R)‐2,3‐Difluoro‐1‐hydroxy‐7‐methylsulfonylindan‐4‐yl]oxy‐5‐fluorobenzonitrile (PT2977), a hypoxia‐inducible factor 2α (HIF‐2α) inhibitor for the treatment of clear cell renal cell carcinoma. J Med Chem. 2019;62(15):6876‐6893. [DOI] [PubMed] [Google Scholar]
- 34. Ohh M, Taber CC, Ferens FG, Tarade D. Hypoxia‐inducible factor underlies von Hippel‐Lindau disease stigmata. eLife. 2022;11:e80774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McAllister TE, Yeh TL, Abboud MI, et al. Non‐competitive cyclic peptides for targeting enzyme‐substrate complexes. Chem Sci. 2018;9(20):4569‐4578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Clifton IJ, McDonough MA, Ehrismann D, Kershaw NJ, Granatino N, Schofield CJ. Structural studies on 2‐oxoglutarate oxygenases and related double‐stranded β‐helix fold proteins. J Inorg Biochem. 2006;100(4):644‐669. [DOI] [PubMed] [Google Scholar]
- 37. McDonough MA, Li V, Flashman E, et al. Cellular oxygen sensing: crystal structure of hypoxia‐inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci U S A. 2006;103(26):9814‐9819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Anagianni S, Tuschl K. Genetic disorders of manganese metabolism. Curr Neurol Neurosci Rep. 2019;19(6):33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Loenarz C, Mecinović J, Chowdhury R, McNeill LA, Flashman E, Schofield CJ. Evidence for a stereoelectronic effect in human oxygen sensing. Angew Chem Int Ed Engl. 2009;48(10):1784‐1787. [DOI] [PubMed] [Google Scholar]
- 40. Lippl K, Boleininger A, McDonough M, et al. Born to sense: biophysical analyses of the oxygen sensing prolyl hydroxylase from the simplest animal Trichoplax adhaerens . Hypoxia. 2018;6:57‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Scotti JS, Leung IKH, Ge W, et al. Human oxygen sensing may have origins in prokaryotic elongation factor Tu prolyl‐hydroxylation. Proc Natl Acad Sci U S A. 2014;111(37):13331‐13336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schnicker NJ, Dey M. Bacillus anthracis prolyl 4‐hydroxylase modifies collagen‐like substrates in asymmetric patterns. J Biol Chem. 2016;291(25):13360‐13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Domene C, Jorgensen C, Schofield CJ. Mechanism of molecular oxygen diffusion in a hypoxia‐sensing prolyl hydroxylase using multiscale simulation. J Am Chem Soc. 2020;142(5):2253‐2263. [DOI] [PubMed] [Google Scholar]
- 44. Hon WC, Wilson MI, Harlos K, et al. Structural basis for the recognition of hydroxyproline in HIF‐1α by pVHL. Nature. 2002;417(6892):975‐978. [DOI] [PubMed] [Google Scholar]
- 45. Holt‐Martyn JP, Chowdhury R, Tumber A, et al. Structure‐activity relationship and crystallographic studies on 4‐hydroxypyrimidine HIF prolyl hydroxylase domain inhibitors. ChemMedChem. 2020;15(3):270‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rabe P, Kamps JJAG, Sutherlin KD, et al. X‐ray free‐electron laser studies reveal correlated motion during isopenicillin N synthase catalysis. Sci Adv. 2021;7(34):eabh0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Islam MS, Markoulides M, Chowdhury R, Schofield CJ. Structural analysis of the 2‐oxoglutarate binding site of the circadian rhythm linked oxygenase JMJD5. Sci Rep. 2022;12(1):20680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yeh TL, Leissing TM, Abboud MI, et al. Molecular and cellular mechanisms of HIF prolyl hydroxylase inhibitors in clinical trials. Chem Sci. 2017;8(11):7651‐7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ramanan R, Chaturvedi SS, Lehnert N, Schofield CJ, Karabencheva‐Christova TG, Christov CZ. Catalysis by the JmjC histone demethylase KDM4A integrates substrate dynamics correlated motions and molecular orbital control. Chem Sci. 2020;11(36):9950‐9961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ramanan R, Waheed SO, Schofield CJ, Christov CZ. What is the catalytic mechanism of enzymatic histone N‐methyl arginine demethylation and can it be influenced by an external electric field? Chem Eur J. 2021;27(46):11750. [DOI] [PubMed] [Google Scholar]
- 51. Tarade D, Lee JE, Ohh M. Evolution of metazoan oxygen‐sensing involved a conserved divergence of VHL affinity for HIF1α and HIF2α. Nat Commun. 2019;10(1):3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Chowdhury R, Abboud MI, Wiley J, Tumber A, Markolovic S, Schofield CJ. Conservation of the unusual dimeric JmjC fold of JMJD7 from Drosophila melanogaster to humans. Sci Rep. 2022;12(1):6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jumper J, Evans R, Pritzel A, et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596(7873):583‐589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Loenarz C, Coleman ML, Boleininger A, et al. The hypoxia‐inducible transcription factor pathway regulates oxygen sensing in the simplest animal, Trichoplax adhaerens . EMBO Rep. 2011;12(1):63‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rytkönen KT, Williams TA, Renshaw GM, Primmer CR, Nikinmaa M. Molecular evolution of the metazoan PHD–HIF oxygen‐sensing system. Mol Biol Evol. 2011;28(6):1913‐1926. [DOI] [PubMed] [Google Scholar]
- 56. Mohlin S, von Stedingk K, Pietras A, Påhlman S. No reason to reconsider HIF‐2 as an oncogene in neuroblastoma and other cancer forms. Proc Natl Acad Sci U S A. 2017;114(51):E10856‐E10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu D, Potluri N, Lu J, Kim Y, Rastinejad F. Structural integration in hypoxia‐inducible factors. Nature. 2015;524(7565):303‐308. [DOI] [PubMed] [Google Scholar]
- 58. Percy MJ, Furlow PW, Beer PA, Lappin TRJ, Mcmullin MF, Lee FS. A novel erythrocytosis‐associated PHD2 mutation suggests the location of a HIF binding groove. Blood. 2007;110(6):2193‐2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Furlow PW, Percy MJ, Sutherland S, et al. Erythrocytosis‐associated HIF‐2α mutations demonstrate a critical role for residues C‐terminal to the hydroxylacceptor proline. J Biol Chem. 2009;284(14):9050‐9058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Percy MJ, Furlow PW, Lucas GS, et al. A gain‐of‐function mutation in the HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(2):162‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Perrotta S, della Regione F. The HIF2A gene in familial erythrocytosis. N Engl J Med. 2008;358(18):1965‐1967. [PubMed] [Google Scholar]
- 62. Gale DP, Harten SK, Reid CDL, Tuddenham EGD, Maxwell PH. Autosomal dominant erythrocytosis and pulmonary arterial hypertension associated with an activating HIF2α mutation. Blood. 2008;112(3):919‐921. [DOI] [PubMed] [Google Scholar]
- 63. Martini M, Teofili L, Cenci T, et al. A novel heterozygous HIF2AM535I mutation reinforces the role of oxygen sensing pathway disturbances in the pathogenesis of familial erythrocytosis. Haematologica. 2008;93(7):1068‐1071. [DOI] [PubMed] [Google Scholar]
- 64. van Wijk R, Sutherland S, Van Wesel ACW, et al. Erythrocytosis associated with a novel missense mutation in the HIF2A gene. Haematologica. 2010;95(5):829‐832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Winter G, Waterman DG, Parkhurst JM, et al. DIALS: implementation and evaluation of a new integration package. Acta Crystallogr D Struct Biol. 2018;74(2):85‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Adams PD, Afonine PV, Bunkóczi G, et al. PHENIX: a comprehensive Python‐based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66(2):213‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. McCoy AJ, Grosse‐Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst. 2007;40(4):658‐674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Emsley P, Cowtan K. Coot: model‐building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 12 Pt 1):2126‐2132. [DOI] [PubMed] [Google Scholar]
- 69. Winn MD, Ballard CC, Cowtan KD, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67(4):235‐242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Afonine PV, Grosse‐Kunstleve RW, Echols N, et al. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68(4):352‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Chen VB, Arendall WB, Headd JJ, et al. MolProbity: all‐atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66(1):12‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Young JY, Westbrook JD, Feng Z, et al. OneDep: unified wwPDB system for deposition, biocuration, and validation of macromolecular structures in the PDB archive. Structure. 2017;25(3):536‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1. Overview of the role of protein hydroxylations in the HIF‐mediated hypoxic response pathway. In the presence of sufficient O2, PHD1‐3, and FIH efficiently hydroxylate HIFα isoforms. PHD catalysis promotes degradation of HIFα via the ubiquitin‐proteasomal pathway in which the von Hippel–Lindau protein/elongin B/C complex plays a key role. HIF‐mediated transcription is inhibited by FIH catalysis which hinders binding of HIF to the CBP/p300 acetyltransferases. In moderate hypoxia, the PHDs are less active than FIH. In hypoxia, HIF hydroxylase activity is reduced so enabling increased levels of HIFα and formation of the transcriptionally active HIFα,β‐heterodimer. 1 , 2 , 3
FIGURE S2. Structural locations of selected clinically observed HIF2α‐CODD variants and sequence conservation of the EPAS1 (HIF2α) C‐terminal oxygen dependent degradation domains in a set of eukaryotic organisms. (A) Predicted locations of selected predicted clinically observed HIF2α523‐542‐CODD variants (sticks‐yellow) on the basis of the PHD2181–407.Mn(II).NOG.HIF2α523–542‐CODD complex structure (sticks‐blue) (PDB: 7Q5V). 4 , 5 , 6 , 7 , 8 , 9 , 10 PHD2 (blue) and HIF2α‐CODD (orange) are depicted as cartoons. Key polar interactions are represented by black dashes. Waters (red) and Mn (violet) are displayed as spheres. (B) Alignment of the HIF2α (EPAS1) CODD with HIF2α sequences from selected eukaryotic organisms. The percentage identities compared with the shown human HIF2α sequence are given.
FIGURE S3. View of the binding modes of the glycine‐glutamate unit in PHD2181–407.Mn.NOG.HIF2α523–542‐CODD and of the 3C cyclic peptide binding site. (A) Lattice packing view of the PHD2181–407.Mn.NOG.HIF2α523–542‐CODD (orange) complex crystal structure (PDB: 7Q5V). Electron density is shown as a blue mesh (contoured at 1.0 σ). Interactions of HIF2α Glu538 (conformation B) (orange) with the same residue (HIF2α Glu538, conformation B) in a symmetry‐related molecule (tan) are shown. (B) Comparison of the PHD2.Mn.NOG.HIF1α.3C (gray‐6YW3) and the PHD2181–407.Mn.NOG.HIF2α523–542‐CODD (blue/teal‐7Q5V) complex structures comparing residues that interact with the 3C cyclic peptide in the former case (density for the 3C cyclic peptide was not observed in the PHD2181–407.HIF2α‐CODD complex structure). Polar interactions are represented by black dashes. Waters (red) and Mn (violet) are displayed as spheres.
FIGURE S4. Comparison of substrate binding modes in the Trichoplax adhaerens PHD.TaODD, PHD2181–407.NOG.HIF2α‐CODD, and Pseudomonas putida PHD.NOG.EF‐Tu complex crystal structures. (A) Comparison structures of TaPHD21–257.Mn(II).NOG.TaODD477–497 (PDB: 6F0W) and PHD2181–407.Mn(II).NOG.HIF2α523–542‐CODD (PDB: 7Q5V). Enzymes (TaPHD‐green and PHD2‐blue) and substrates (TaODD‐pink and HIF2α‐orange) are shown as cartoons. The active sites, β2–β3 loops (TaPHD‐green cyan and PHD2‐red), and ligands (NOG‐yellow) are displayed with key residues as sticks. Waters (red) and Mn (violet) are displayed as spheres. (B) Comparison of the TaODD477–497 (pink‐sticks) and HIF2α (orange‐sticks) substrate binding showing solvent‐excluded surface representation (Connolly). (A, B) Hydrogen bonding and electrostatic/polar interactions are in black dashes. (C) Two views of the off‐line NOG binding mode in the PHD2181–407.HIF2α‐CODD, TaPHD.TaODD, and PPHD (purple).EFTu (yellow‐4IW3) complex structures. Note the conservation of the C4 endo‐conformation of all the substrate proline residues.
FIGURE S5. Comparison of binding modes of HIFα‐ODDs to PHD1‐3 involving residues to the C‐terminal side of the substrate proline residue. (A) Residues to the N‐terminal and C‐terminal sides of the substrate proline residue in HIFα‐ODD peptides are displayed on PHD1/3 model overlays. (A, B) Comparison of the conformation of the β2–β3 loop in the PHD2181–407.HIF2α523‐542 with AlphaFold (AF) models of PHD1 (cyan sticks/cartoon; UniProt: Q96KS0) and PHD3 (purple sticks/cartoon; UniProt: Q62630). (C–E) Views from crystal structures of PHD2181–426.HIF1α394–413‐NODD (PDB: 5L9V), PHD2181–426.HIF1α556–574‐CODD (PDB: 3HQR), and PHD2181–407.HIF2α523–542‐CODD (PDB: 7Q5V) displayed as cartoons and sticks (HIF1α‐NODD‐green, HIF1α‐CODD‐olive, and HIF2α‐CODD‐orange). The PHD1 (cyan; UniProt: Q96KS0) and PHD3 (purple; UniProt: Q62630) stick views are derived from AF models and are aligned with crystals for PHD2.HIFα complexes. Polar interactions in the experimentally determined PHD2 structures are represented by black dashes to enable comparison with the predicted residue conformations in the PHD1 and PHD3 models.
FIGURE S6. Comparison of the binding modes of HIFα‐ODDs to PHD1‐3 involving residues to the N‐terminal side of the substrate proline residue. (A–C) Views from crystal structures of cross‐linked‐PHD2181–426.HIF1α394–413‐NODD (PDB: 5L9V), PHD2181–426.HIF1α556–574‐CODD (PDB: 3HQR), and PHD2181–407.HIF2α523–542‐CODD (PDB: 7Q5V) displayed as cartoons (HIFα‐peptides) and stick views (HIF1α‐NODD‐green, HIF1α‐CODD‐olive, and HIF2α‐CODD‐orange). The PHD1 (cyan; UniProt: Q96KS0) and PHD3 (purple; UniProt: Q62630) stick views are derived from AlphaFold (AF) models aligned with crystal structures for PHD2.HIFα complexes. Polar interactions in the experimentally determined PHD2 structures are represented by black dashes to enable comparison with the predicted residue conformations in the PHD1 and PHD3 models.
TABLE S1. Crystallization conditions for PHD2.HIFα‐substrate complexes.
Data Availability Statement
Coordinates and structure factors for PHD2.HIF2α‐CODD complex structures were deposited in the RCSB Protein Data bank as: PHD2181–407.Mn.NOG.HIF2α523–542‐CODD, PDB: 7Q5V and PHD2181–407.Mn.2OG.HIF2α523‐542‐CODD, PDB: 7Q5X. AlphaFold models and HIFα sequences were accessed using UniProt: Q96KS0‐PHD1, Q9H6Z9‐PHD3, Q16665‐HIF1α, Q99814‐HIF2α, and Q9Y2N7‐HIF3α.