

Abstract
Objective
The objective of this study was to aggregate data for the first genomewide association study meta‐analysis of cluster headache, to identify genetic risk variants, and gain biological insights.
Methods
A total of 4,777 cases (3,348 men and 1,429 women) with clinically diagnosed cluster headache were recruited from 10 European and 1 East Asian cohorts. We first performed an inverse‐variance genomewide association meta‐analysis of 4,043 cases and 21,729 controls of European ancestry. In a secondary trans‐ancestry meta‐analysis, we included 734 cases and 9,846 controls of East Asian ancestry. Candidate causal genes were prioritized by 5 complementary methods: expression quantitative trait loci, transcriptome‐wide association, fine‐mapping of causal gene sets, genetically driven DNA methylation, and effects on protein structure. Gene set and tissue enrichment analyses, genetic correlation, genetic risk score analysis, and Mendelian randomization were part of the downstream analyses.
Results
The estimated single nucleotide polymorphism (SNP)‐based heritability of cluster headache was 14.5%. We identified 9 independent signals in 7 genomewide significant loci in the primary meta‐analysis, and one additional locus in the trans‐ethnic meta‐analysis. Five of the loci were previously known. The 20 genes prioritized as potentially causal for cluster headache showed enrichment to artery and brain tissue. Cluster headache was genetically correlated with cigarette smoking, risk‐taking behavior, attention deficit hyperactivity disorder (ADHD), depression, and musculoskeletal pain. Mendelian randomization analysis indicated a causal effect of cigarette smoking intensity on cluster headache. Three of the identified loci were shared with migraine.
Interpretation
This first genomewide association study meta‐analysis gives clues to the biological basis of cluster headache and indicates that smoking is a causal risk factor. ANN NEUROL 2023;94:713–726
Cluster headache (CH) is a primary headache disorder that affects 0.1% of the population and is 4 times more common in men than in women. 1 It is characterized by episodes of excruciating unilateral pain centered around the eye or the temple. 2 The large majority of patients are either current or previous smokers and there is a higher prevalence of illicit drug use, depression, and sleep disorders among patients with CH than in the general population. 1 , 3
Much is unknown about the pathophysiology of CH, but hypothalamic, trigeminovascular, and autonomic nervous system dysfunction are likely involved. 1 , 4 Previous twin‐ and family‐based studies have suggested the involvement of genetic factors, 5 and 2 recent genomewide association studies (GWAS) in individuals of European ancestry 6 , 7 demonstrated robust genetic associations for CH, independently identifying 4 genetic risk loci on chromosome 1 (near the gene DUSP10), chromosome 2 (within MERTK and near SATB2), and chromosome 6 (within FHL5), with odds ratios (ORs) ranging from 1.30 to 1.61. A third GWAS in Han Chinese individuals replicated 2 of these loci (MERTK and SATB2) and reported an additional locus in the gene CAPN2. 8
To identify additional genetic factors and increase power for functional interpretation of the genetic signals, we established the International Consortium for Cluster Headache Genetics (CCG) and analyzed data from 10 European and 1 East Asian CH cohorts; those used in the 4 previous GWASs of CH 6 , 7 , 8 , 9 and 5 additional cohorts, increasing the sample size for analysis 3.2‐fold compared to the largest previous CH GWAS. 7
Methods
Cohorts and Phenotyping
For reference, acronyms are listed in Table S1. Data were obtained from 10 European and 1 East Asian cohorts (Table 1), with a combined sample size of 4,777 patients with CH (3,348 men and 1,429 women) and 31,575 controls, of which 4,043 patients (85%) were of European and 734 (15%) of East Asian ancestry. Cases were recruited between 2005 and 2022 through specialized headache clinics and diagnosed according to standardized International Classification of Headache Disorder (ICHD) criteria. 2 , 10 Details on the recruitment and phenotyping in each cohort are provided in Table S2. All studies were approved by local research ethics committees, and written informed consent was obtained from each study participant.
TABLE 1.
Cluster Headache GWAS Studies Included in the Meta‐Analysis
| Study | Cases (n) | Controls (n) | Cases: Men (%) | Cases: Current or previous smokers (%) |
|---|---|---|---|---|
| European ancestry | ||||
| Dutch Cluster Headache Cohorta | 943 | 1,424 | 68.2% | 81.1% |
| UK Cluster Headache Cohortb | 852 | 5,614 | 64.1% | NA |
| Swedish Cluster Headache Cohort 1b | 591 | 1,134 | 67.0% | 71.0% |
| German Cluster Headache Cohort | 477 | 938 | 72.5% | NA |
| Danish Cluster Headache Cohort | 492 | 9,658 | 65.9% | 0.76% |
| Swedish Cluster Headache Cohort 2 | 255 | 241 | 61.6% | 68.2% |
| Trondheim Cluster Headache Cohorta | 144 | 1,800 | 73.6% | NA |
| Greek Cluster Headache Cohort | 99 | 91 | 82.8% | NA |
| Barcelona Cluster Headache Cohort | 97 | 482 | 82.5% | NA |
| Italian Cluster Headache Cohortc | 93 | 347 | 82.6% | 90.3% |
| Total European ancestry samples | 4,043 | 21,729 | 68.2% | 77.3% |
| East Asian ancestry | ||||
| Taiwan Cluster Headache Cohortd | 734 | 9,846 | 80.5% | 58.9% |
| Total | 4,777 | 31,575 | 70.0% | 74.3% |
GWAS and Meta‐Analysis
A standardized quality control (QC) and analysis protocol was applied to each individual GWAS, while allowing for adaptations to comply with local data sharing regulations and analysis pipelines. Details are given in Table S3. Samples in each cohort were genotyped on genomewide arrays, and QC was performed on each dataset prior to imputation. Only variants with an imputation quality of ≥ 0.3 11 and a minor allele count of ≥ 12 were kept for further analysis. For X chromosome analyses, male patients were coded as diploid. Prior to the meta‐analysis, the per‐study allele labels and allele frequencies were compared with those of the imputation reference panels using EasyQC, 11 and removed or reconciled mismatches. The analysis of the Taiwanese cohort was performed separately. 8
We first conducted an inverse variance weighted fixed‐effects meta‐analysis of European ancestry cohorts using METAL, 12 without genomic control. A total of 14,860,930 variants were present in at least one cohort and included in the meta‐analysis, and 5,199,189 (35%) variants were present in all 10 cohorts. To identify additional loci, we next conducted a secondary trans‐ancestry GWAS meta‐analysis that also included the East Asian ancestry cohort, using MR‐MEGA with default settings, 13 which accounts for allelic heterogeneity between ancestries. Of 15,425,163 variants analyzed, 3,792,160 were present in the East Asian cohort. Of these, 3,225,258 (85%) were also present in at least one European cohort. Genomewide significance was set to p < 5 × 10−8.
Due to heterogeneity in allele frequencies and differences in linkage disequilibrium (LD) structure between European and East Asian populations, which complicates LD modeling, we focused subsequent fine‐mapping and functional analyses on data from the European ancestry GWAS.
Single nucleotide polymorphism (SNP)‐based heritability was calculated using LD‐Score Regression (LDSC) 14 after excluding variants that (1) were not present in the HapMap 3 reference panel, (2) explained > 1% of phenotype variation, or variants in LD (r 2 > 0.1) with these, and (3) were in the major histocompatibility complex region. Heritability estimates were converted to the liability scale assuming a population prevalence of CH of 0.1%. 1
Fine‐mapping for significant loci was performed using PICS2 15 with 1,000 Genomes EUR LD reference. Next, a stepwise conditional analysis was performed using FINEMAP. 16 , 17 Only biallelic, non‐indel variants were included, and a p value < 5 × 10−8 was used to define SNPs that were conditionally independent from the lead variant.
Candidate Gene Mapping
To prioritize candidate genes for a causal association to CH, 5 methods were applied: (1) expression quantitative trait locus (eQTL) analysis, (2) transcriptome‐wide association (FUSION), (3) fine‐mapping of causal gene sets (FOCUS), (4) association to genetically driven DNAm (MetaMeth), and (5) genes affected by protein‐altering variants in high LD with the lead CH variants.
eQTL Analysis
Association between variants and gene expression (cis‐eQTL) was estimated based on RNA sequencing and genotype data from 59,327 individuals (Table S4). 18 For each CH variant, it was tested whether the variant itself, or variants in high LD (r 2 ≥ 0.8), associated with one or more top cis‐eQTLs, defined as the variant with the lowest p value within a distance of 1 Mb from the gene for each gene and tissue. The significance threshold was determined at p < 1 × 10−9. Details on data sources and methods are described previously. 18
Transcriptome‐Wide Association Study Analysis
To identify genes whose expression is significantly associated with CH, the CH meta‐analysis results were integrated with gene expression data from single tissues (Table S5) using Transcriptome‐Wide Association Study Analysis (TWAS)‐FUSION. 19 TWAS expression weights were computed using 5 linear models (Table S5), followed by cross‐validation to determine the best performing model for a given gene. The imputed gene expression was then used to test for association with CH, taking into account the LD structure and Bonferroni correcting for the number of genes tested for the given tissue. A joint/conditional analysis was performed to test for the significance of GWAS signals after removing TWAS‐significant signals (expression weight from TWAS). Each variant association from the CH meta‐analysis was conditioned on the joint model and a p value for conditional analysis results was obtained by permutation testing.
Fine‐Mapping of Causal Gene Sets
Fine‐Mapping of Causal Gene Sets (FOCUS) 20 took as input the CH meta‐analysis results, the previously calculated TWAS expression prediction weights, and LD information for all SNPs in the risk regions, and estimated the probability for any given set of genes to explain the respective TWAS signal. FOCUS was run for chromosomes 1, 2, 6, 7, and 17, in which TWAS‐Fusion showed suggestive association of genes with tissues.
Genetically Driven DNA Methylation Scan (MetaMeth)
Association between CH and genetically driven DNA methylation (DNAm) was assessed using the MetaMeth function in EstiMeth (version 1.1). 21 EstiMeth includes 86,710 models reflecting a robust genetically driven signal at methylation of 5′‐C‐phosphate‐G‐3′ (CpG) sites in whole blood. 21 The approach was applied to the CH meta‐analysis results, and significance was set at p value < 0.05 after false discovery rate (FDR) correction. Each CpG was paired with its annotated gene(s) and represented in a Miami plot using the R‐project (https://www.R-project.org/) ggplot package. 22
Protein‐Altering Variants (VEP‐Ensembl)
At deCODE Genetics (Iceland), for each of the lead CH variants, it was determined if it was in high LD (r 2 > 0.80, based on the Icelandic genotype data) with protein‐altering (coding or splice) variants with moderate or high impact, as annotated using release 100 of the Ensembl Variant Effect Predictor (VEP‐Ensembl) tool. 23
Gene Set and Tissue Enrichment Analyses
Genes prioritized by at least one of the five methods were used as input to the GENE2FUNC tool implemented in FUMA 24 to examine enrichment in differentially expressed gene (DEG) sets for 54 tissues from GTEX version 8, 25 and in biological pathways and functional categories from MsigDB, WikiPathways, and the NHGRI GWAS catalog. 24 The p values < 9.26 × 10−4 (0.05/54 tests) were considered statistically significant. 24 We also applied 2 approaches based on variant‐level summary statistics: (1) DEPICT version 1.194 analysis 26 applied to independent variants with a nominal association to CH (p < 1 × 10−6), and (2) LD‐Score Regression applied to specifically expressed genes (LDSC‐SEG) version 1.0.1. 27 applied to the full set of summary statistics from the meta‐analysis. Both methods were run with default settings. FDR < 0.05 was considered statistically significant.
Drug Target Identification
For genes prioritized by at least 1 of the 5 methods, we examined their druggability status using the dataset from Finan et al 28 (Table S6). For detailed structured information about drugs and drug targets, we integrated information from the DrugBank online database (https://www.drugbank.com) 29 (version 5.1.9, released 2022‐01‐04).
Genetic Risk Score Analysis
Genetic risk scores (GRSs) were based on summary statistics from the meta‐analysis of all European ancestry cohorts except the given cohort to create independent test samples. In 3 cohorts (Dutch, Swedish cohort 1, and Danish) GRSs were calculated with LDpred2, 30 which uses genomewide data from the discovery dataset without applying a p value threshold. In the German cohort, GRS were calculated using PRSice2 31 (Table S7). Sample‐specific GRSs were normalized using the target sample mean and standard deviation. Using linear regression, adjusting for sex and the first 4 to 6 principal components, we examined the association of GRS in each cohort to case–control status, and among cases to episodic versus chronic CH, male versus female patients, age at onset, currently smoking yes versus no, and ever versus never smoked was examined for each cohort. The p values < 0.0024 (0.05/21 tests) were considered statistically significant.
Genetic Correlation
In a hypothesis‐free fashion, LDSC (version 1.0.1.) 14 was used to calculate pairwise genetic correlations between CH and 1,150 phenotypes from published GWASs (Table S8) based on GWAS summary statistics. Applying a stringent Bonferroni correction (0.05/1,150), the significance threshold was set at (p < 4.35 × 10−5). To evaluate differences in the correlation profiles for CH and migraine, the genetic correlation was calculated between migraine (48,975 migraine cases and 540,381 controls from Hautakangas et al, 17 not including 23andMe) and each of the traits that were significantly correlated with CH, whereas Bonferroni correcting for the number of tests (0.05/84, p < 5.95 × 10−5).
Colocalization Analysis
To test whether CH loci that were in close proximity to previously reported migraine loci share causal variants for both CH and migraine, the Bayesian colocalization procedure implemented in the R package “coloc” (version 5.1.0) was used with default settings 32 and the migraine dataset described above. Colocalization was tested for the region between the 2 nearest recombination hotspots (https://bitbucket.org/nygcresearch/ldetect-data/src/master/EUR/).
Mendelian Randomization Analysis
To test for a causal effect of smoking on CH, we performed a summary statistics‐based 2‐sample inverse‐variance weighted (IVW) Mendelian randomization analysis, 33 using as instrumental variables 40 independent variants significantly (p < 5 × 10−8) associated with “Cigarettes smoked per day” in a previous GWAS, 34 as an indication for smoking intensity (Table S9). Because the IVW method assumes the absence of horizontal pleiotropy, several sensitivity analyses were used to exclude pleiotropy. Cochran's Q tests were used to detect heterogeneity. 35 In addition, the Mendelian randomization‐Egger intercept was used to detect directional pleiotropy. 35 , 36 Both models were fit using robust regression and assuming a t‐distribution of the fitted parameters. Analyses were performed using the Mendelian Randomization package (version 0.5.1) in R (version 3.6.3). To verify the causality between smoking and CH, we applied a latent causal variable (LCV) model to estimate the genetic causality proportion (GCP). 37 Here, a latent variable mediates the genetic correlation, avoiding false positives due to genetic correlations when determining causality. A GCP of 0 is interpreted as no, and GCP of 1 as complete genetic causality.
Results
European Ancestry GWAS Meta‐Analysis
Seven independent genomewide significant CH associated (p < 5 × 10−8) risk loci (Table 2, Figs 1 and 2) were identified. Associations were consistent across the 10 cohorts (heterogeneity p > 0.10; see Tables 2 and S10). Named by their nearest protein‐coding gene, 4 of the risk loci were previously reported 6 , 7 (DUSP10, MERTK, FTCDNL1, and FHL5), whereas 3 are novel (WNT2, PLCE1, and LRP1). A stepwise conditional analysis using FINEMAP 16 revealed that 2 of the identified loci (MERTK and WNT2) contained additional independent signals, increasing the number of independent association signals to 9 (Table S11). Fine‐mapping with PICS2 15 suggested that the lead signal in the LRP1 locus (rs11172113) is most likely the causal variant (posterior probability 65.8%). Five other variants in 3 other loci had PICS2 posterior probability > 10% for being causal (Table S12).
TABLE 2.
Summary of the Genomic Loci Associated with Cluster Headache in Primary Meta‐Analysis of Ten European‐Ancestry Cohorts
| Locus name | Lead variant (Chr:Pos) | EA/NEA (EAF) | OR (95% CI) | p (Het p) | Variant type [Prioritized genes] |
|---|---|---|---|---|---|
| DUSP10 | rs17011182 (1:222164327) | G/A (0.793) | 1.38 (1.29–1.48) | 7.76 × 10−21 (0.58) | Regulatory region [DUSP10] |
| MERTK | rs13399108 (2:112747123) | G/A (0.627) | 1.41 (1.33–1.50) | 1.74 × 10−30 (0.16) | Intron [ MERTK , TMEM87B , FBLN7 , SLC20A1] |
| FTCDNL1 | rs6714578 (2:200485487) | A/G (0.655) | 1.53 (1.43–1.63) | 2.83 × 10−37 (0.65) | Intergenic [ SATB2 ] |
| FHL5 | rs9486725 (6:97061159) | T/C (0.346) | 1.29 (1.21–1.36) | 2.50 × 10−17 (0.29) | Intron [ UFL1 , FHL5, KLHL32, and NDUFAF4] |
| WNT2 | rs2402176 (7:116908448) | C/G (0.291) | 1.20 (1.12–1.27) | 2.61 × 10−8 (0.51) | Intergenic [ CFTR , CAPZA2, ST7] |
| PLCE1 | rs57866767 (10:96023077) | T/C (0.588) | 1.18 (1.12–1.25) | 4.45 × 10−9 (0.51) | Intron [PLCE1] |
| LRP1 | rs11172113 (12:57527283) | T/C (0.600) | 1.18 (1.12–1.25) | 5.15 × 10−9 (0.52) | Intron [ LRP1 ] |
Note: Locus name = the closest protein‐coding gene within a 250‐Kb window. Chr = chromosome; CI = confidence interval; EA = effect allele, which here is set to correspond with the risk allele; EAF = effect allele frequency; Het p = p value from Cochran's Q‐test for heterogeneity; NEA = non‐effect allele; OR = odds ratio; Pos = position (hg19).
Prioritized genes = genes prioritized by at least 1 of 5 complementary methods: (1) expression quantitative trait (eQTL) analysis, (2) transcriptome‐wide association analysis using FUSION, (3) fine‐mapping of causal gene sets (FOCUS), (4) association to genetically driven DNAm (MetaMeth), and (5) protein‐altering variants in high LD (r 2 > 0.8) with lead variant. Genes identified by ≥ 2 of the methods are marked in bold.
FIGURE 1.
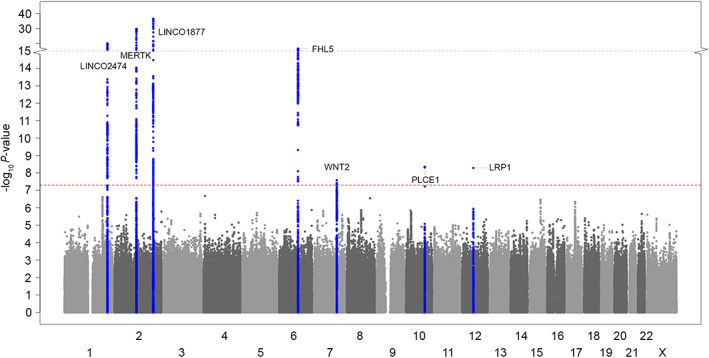
Manhattan plot showing genomewide significant loci associated with cluster headache in meta‐analysis of 10 European cohorts (4,043 cases and 21,729 controls). The horizontal axis shows the chromosomal position and the vertical axis shows the significance (−log10 p value) of tested markers. Each dot represents a genetic variant. The threshold for genomewide significance (p < 5 × 10−8) is indicated by a red dotted line, and genomewide significance loci are shown in blue. [Color figure can be viewed at www.annalsofneurology.org]
FIGURE 2.
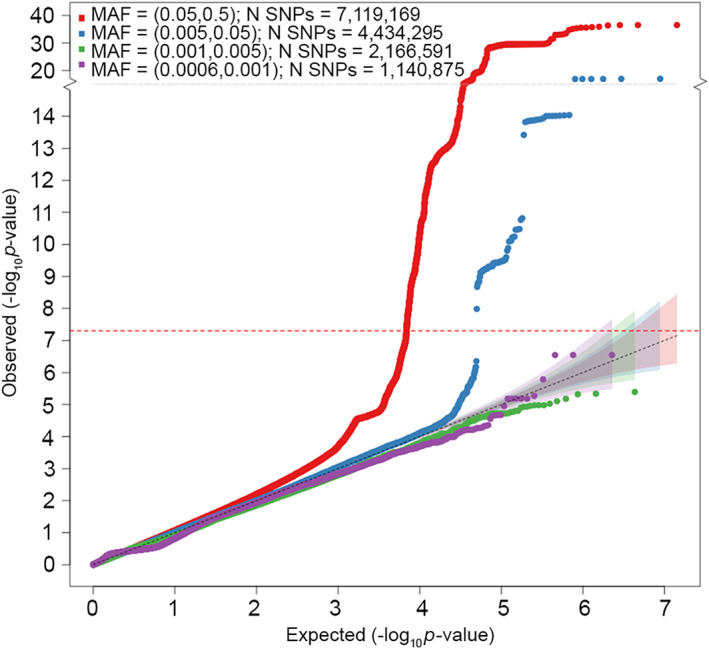
Quantile‐quantile (Q‐Q) plot for association with cluster headache in meta‐analysis of 10 European cohorts (4,043 cases and 21,729 controls). The horizontal axis shows −log10 p values expected under the null distribution. The vertical axis shows observed −log10 p values. Genomic inflation factor (λ) = 1.086. Red = common variants (MAF ≥ 5%), blue = low frequency variants (MAF = 0.5–5%), green = rare variants (MAF = 0.1–0.5%), purple = very rare variants (MAF < 0.1%). MAF, minor allele frequency; SNPs, single nucleotide polymorphisms. [Color figure can be viewed at www.annalsofneurology.org]
The genomic inflation factor (λ) f was 1.086, whereas the LD score regression intercept was 1.004 (SE = 0.007), with a ratio of 0.033 (SE = 0.062), indicating that 96.7% of the observed signal is caused by true polygenic heritability rather than confounding factors, such as population stratification. The estimated SNP‐based heritability (h 2) of CH was 14.5% (SE = 1.74%) on the liability scale.
Trans‐Ancestry GWAS Meta‐Analysis
One additional genomewide significant CH locus, in CAPN2, was identified when adding the East Asian cohort in an ancestry‐adjusted GWAS meta‐analysis (Table 3, Table S13, Fig 3). This locus, previously reported and internally replicated within the East Asian cohort, 8 was exclusively driven by the same cohort in our analysis (see Table S13). However, a nearby locus reached nominal significance in the European ancestry meta‐analysis, with lead variant rs68046706 (OR = 1.76, 95% confidence interval [CI] = 1.10–1.26, p = 3.86 × 10−6) 86 kb away from rs10916600. The WNT2 locus identified in the European ancestry meta‐analysis, for which the lead variant was not present in the East Asian cohort, fell below significance (p = 5.91 × 10−7). At the PLCE1 locus, the new lead variant was a missense variant (rs2274224) in PLCE1. Cohort‐wise associations for all the identified loci are given in Tables S10 and S13.
TABLE 3.
Summary of the Genomic Loci Associated with Cluster Headache in Trans‐Ancestry GWAS
| Locus name | Lead variant (Chr:Pos) | EA/NEA (EAF) | χ 2 (df) | p association | p ancestry | Variant type |
|---|---|---|---|---|---|---|
| DUSP10 | rs12129860 (1:222153461) | G/T (0.196) | 90.86 (4) | 8.64 × 10−19 | 9.59 × 10−3 | Regulatory region |
| CAPN2 | rs10916600 (1:223897012) | T/C (0.214) | 66.43 (4) | 1.28 × 10−13 | 1.80 × 10−11 | Intron |
| MERTK | rs10188642 (2:112741099) | A/G (0.521) | 182.48 (4) | 2.19 × 10−38 | 0.40 | Intron |
| FTCDNL1 | rs4673382 (2:200492346) | G/A (0.653) | 164.96 (4) | 1.26 × 10−34 | 0.52 | Intergenic |
| FHL5 | rs11153085 (6:97066355) | T/A (0.326) | 88.53 (4) | 2.70 × 10−18 | 0.30 | Regulatory region |
| PLCE1 | rs2274224 (10:96039597) | G/C (0.551) | 58.68 (4) | 5.48 × 10−12 | 0.28 | Missense variant |
| LRP1 | rs11172113 (12:57527283) | T/C (0.635) | 46.08 (4) | 2.37 × 10−9 | 0.40 | Intron |
Note: Locus name = the closest protein‐coding gene within a 250‐Kb window.
Abbreviation: χ 2 = chi square test statistic from MR‐MEGA; Ancestry = heterogeneity due to different ancestry; Chr = chromosome; df = number degrees of freedom; EA = effect allele, which here is set to correspond with the risk allele; EAF = effect allele frequency; NEA = non‐effect allele; Pos = position (hg19).
FIGURE 3.
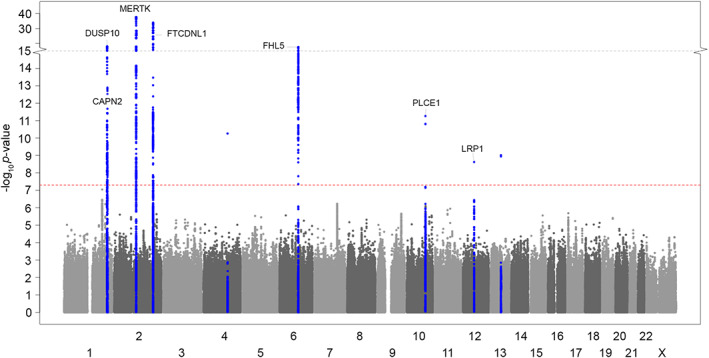
Manhattan plot showing genomewide significant loci associated with cluster headache in trans‐ancestry meta‐analysis (4,777 cases and 31,575 controls). The horizontal axis shows the chromosomal position and the vertical axis shows the significance (−log10 p value) of tested markers. Each dot represents a genetic variant. The threshold for genomewide significance (p < 5 × 10−8) is indicated by a red dotted line, and genomewide significance loci are shown in blue. Three genomewide significant variants (rs9307511 on chr4 and rs338106 and rs747974 on chr 13) were considered spurious associations as they lacked a supporting LD structure, were driven by the East Asian cohort alone, and were previously interpreted as being spurious associations in this cohort. 8 LD = linkage disequilibrium. [Color figure can be viewed at www.annalsofneurology.org]
All the 5 previously reported GWAS‐significant loci were re‐identified in our study, whereas none of the associations reported from candidate gene studies were replicated (Table S14).
Candidate Gene Mapping and Functional Characterization
The subsequent downstream analyses were based on the European ancestry meta‐analysis. To prioritize candidate genes for a causal association with CH, we applied 5 methods. (1) The eQTL analysis found that at the MERTK locus, three variants in high LD (r 2 > 0.92) with the lead variant rs13399108 modulate the expression of TMEM87B (in fibroblasts and aortic artery) and SLC20A1 (in whole blood). At the FHL5 locus, 2 variants (r 2 > 0.84 with the lead variant rs9486725) associate with the expression of UFL1 (in whole blood, white blood cells, and tibial artery). At the LRP1 locus, the T allele of lead variant rs11172113 associates with an increased LRP1 mRNA expression in aortic artery, adipose tissue, and tibial artery (Table S15). (2) The transcriptome‐wide association study (TWAS‐FUSION) identified 8 candidate genes at 5 loci with a significant TWAS p value ≤ 1.0 × 10−6 (Table S5). (3) Fine‐mapping by FOCUS identified 8 candidate genes based on posterior inclusion probability (PIP) >0.5 (Table S16). Four genes (MERTK, TMEM87B, SATB2, and CFTR) were prioritized by both TWAS‐FUSION and FOCUS with high confidence (PIP > 0.99 in the same tissue in both analyses). (4) Using MetaMeth, 13 CpG sites at 9 genes were predicted to be hypo‐ or hypermethylated in CH (Table S17, Fig 4). (5) At 2 loci, the lead variant was in high LD with protein‐altering missense variants. That is, at the FHL5 locus, the intronic lead variant rs9486725 is in strong LD (r 2 ≥ 0.98) with p.Arg204Gly (rs2273621) and p.Ser243Arg (rs9373985) in FHL5; and at the PLCE1 locus the intronic lead variant rs57866767 is in strong LD (r 2 = 1) with p.Arg1267Pro (rs2274224) in PLCE1 (Table S18).
FIGURE 4.
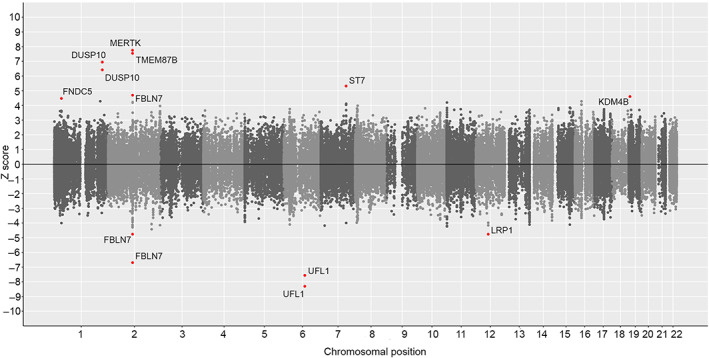
Miami plot of genetically driven DNA methylation genes in cluster headache. Computational prediction of genetically driven CpG methylation associated with cluster headache, using MetaMeth. Genes annotated to significant CpGs are shown (FDR‐corrected p value < 0.05). Horizontal axis shows the chromosomal position and the vertical axis shows significance (−log10 p value). The top panel shows predicted hypermethylation, whereas the bottom panel shows predicted hypomethylation. CpG = 5′‐C‐phosphate‐G‐3′; FDR = false discovery rate. [Color figure can be viewed at www.annalsofneurology.org]
Twenty genes were prioritized by at least 1 of the 5 methods. A summary of the gene prioritization results is given in Table S19.
When considering the 20 prioritized genes, FUMA 24 found a significant enrichment for genes differentially expressed in artery (tibial artery) and brain (substantia nigra; Fig 5 and Table S20), and a significant overlap with genes reported in the GWAS catalog for 10 traits, most significantly for headache and migraine (Table S21). The summary statistics‐based enrichment analyses DEPICT and LDSC‐SEG did not yield significant enrichment for gene sets or tissues after correcting for multiple testing (Tables S22–S26). Of the 20 prioritized genes (Table S19), 10 are highlighted as druggable in the druggable genome database. 28 Of these, 5 encode targets of 33 existing drugs registered in DrugBank 29 (Table S6), including 3 genes that were implicated in CH by at least 2 gene prioritization methods (ie, MERTK, CFTR and LRP1). Calpain 2, encoded by CAPN2 in the trans‐ancestry locus, was not registered in DrugBank.
FIGURE 5.
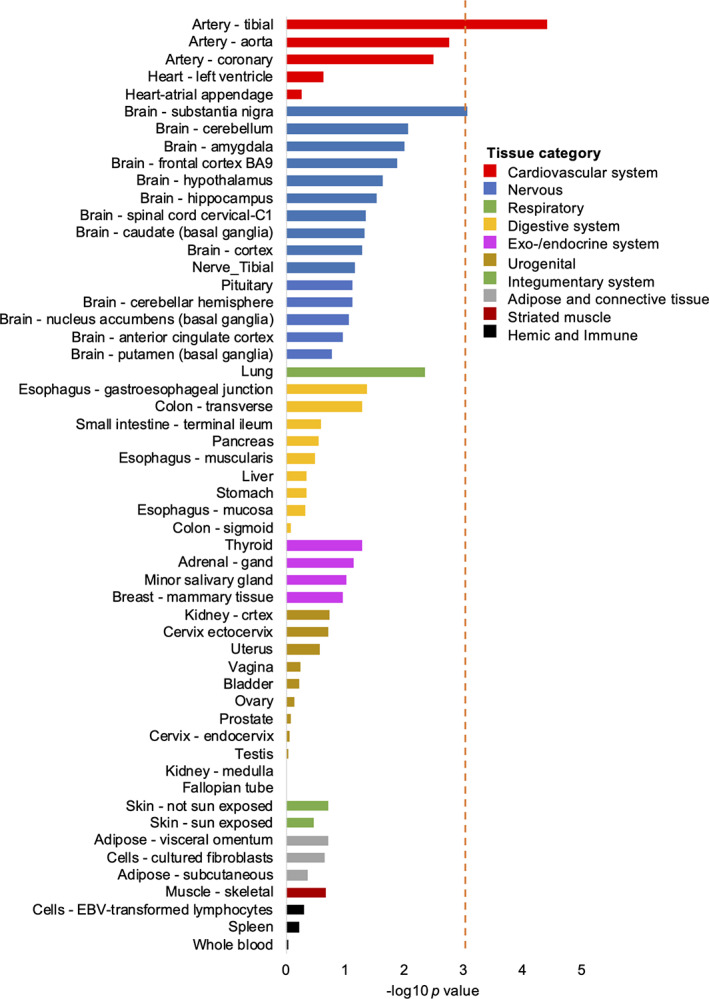
Tissue enrichment for the putative causal genes. Enrichment of the 20 genes with supportive evidence for implication in cluster headache in DEG sets for 54 tissues from GTEX version 8. The analysis was performed using FUMA and based on pre‐calculated DEG sets defined by a two‐sided t test per tissue versus all other tissues. The red line shows the significance threshold after adjustment for multiple testing by Bonferroni correction (p = 0.05/54 tests = 9.26 × 10−4). DEG = differentially expressed gene; EBV = Epstein–Barr virus. [Color figure can be viewed at www.annalsofneurology.org]
Genetic Risk Score Analysis
GRS for CH were associated with case–control status in leave‐one‐out analyses in each of the 4 tested independent cohorts. Among cases with CH, no association was seen between GRS and episodic versus chronic CH, age‐at‐onset, sex, current smoking, or ever smoking (Table S7).
Genetic Correlation
After correcting for multiple testing, CH was genetically correlated with 84 traits (Table S8). The strongest correlation was with “cigarettes per day” 34 (rg = 0.36, p = 6.32 × 10−18). Notably, 10 (12%) of the correlated traits were related to smoking behavior. CH was also positively correlated with measures of risk‐taking behavior, attention deficit hyperactivity disorder (ADHD), mood disorders, musculoskeletal pain, migraine, and with unfavorable lifestyle factors, including low physical activity, low nutritional diet, and lower educational attainment (Table S8). When examining the correlation of the same 84 traits to migraine, the genetic correlations to pain, depression, and ADHD were similar to those seen for CH, whereas no correlation was observed between migraine and smoking traits or measures of risk‐taking behavior.
Three of the CH loci are near previously identified risk loci for migraine (ie, FHL5, PLCE1, and LRP1 17 ; Table S27). Colocalization analysis indicated that CH and migraine are caused by the same causal variant at each of the 3 loci (posterior probability 98.6% for FHL5 locus, 99.6% for PLCE1 locus, and 100% for LRP1 locus). Effect sizes were, however, consistently higher for CH (ORs = 1.29, 1.18, 1.18) than for migraine (1.09, 1.06, 1.11) with non‐overlapping CIs for the ORs (Table S28). Among 122 loci associated with migraine in the most recent GWAS, 17 no other migraine variant was associated with CH after Bonferroni correction (Table S29). The effect sizes (beta) for association to migraine and CH were not significantly correlated (Pearson r = 0.16, p = 0.074) for the remaining 119 variants, after excluding the 3 overlapping loci.
Mendelian Randomization Analysis
Using the random‐effect IVW method, we observed a strong association between the instrumental variables for smoking intensity and CH (β = 1.11, SE = 0.43, p = 6.3 × 10−6). The direction and magnitude were similar in the Mendelian randomization‐Egger analysis (β = 1.04, SE = 0.55, p = 4.6 × 10−4). The Cochran's Q test statistic was significant (p = 0.03), indicative of some heterogeneity, but the Mendelian randomization‐Egger intercept showed no evidence for bias caused by directional pleiotropy (p = 0.79). Mendelian randomization may, however, yield false positive results in the presence of genetic correlation between the 2 traits examined. 37 To test for this, we performed a latent causal variable model, finding that smoking intensity had a nearly full (> 0.6) genetic causality with CH (pLCV = 8.57 × 10−10, GCP = 0.74 ± 0.18). Combined, the results strongly support a causal effect of smoking intensity on CH. Full results are presented in Tables S30 to S32.
Discussion
In a GWAS meta‐analysis for CH in European ancestry cohorts, we identified 9 independent associations in 7 risk loci and confirm the strong associations at 4 loci (ORs = 1.29–1.53) reported in recent smaller GWAS. 6 , 7 , 8 One additional locus, previously reported and internally replicated in the East Asian cohort, 8 was identified in a subsequent trans‐ancestry GWAS meta‐analysis that included this cohort.
We estimate that common genetic variants explain 14.5% of CH's phenotypic variance. Twenty genes were prioritized as candidates for being involved in CH. These showed enrichment for arterial tissue, in addition to brain, fueling the idea that CH may have a vascular involvement. 1 Still, because no significant tissues were identified by summary statistics‐based enrichment analyses (using DEPICT and LDSC‐SEG), more evidence is needed to draw definite conclusions. Several of the 20 prioritized genes encode targets for existing drugs, and may represent candidates for repurposing studies. The clinical utility of GRS remains to be explored. We found no association between GRS and specific clinical phenotypes, suggesting that the signal is not driven by any of the subgroups.
Differences in CH clinical presentation between Asian and European populations, such as reduced restlessness and circadian rhythmicity, may indicate distinct genetic predispositions. 38 The CAPN2 locus was selectively driven by the East Asian cohort, and may exemplify how the contribution of individual risk loci varies between populations. Future well‐powered trans‐ancestral studies should further explore ancestry‐related risk loci, and whether these are related to differences in clinical presentation.
In our hypothesis‐free genetic correlation analysis, CH was correlated with several traits, including smoking, risk‐taking behavior, ADHD, mood disorders, musculoskeletal pain, and migraine. The strongest genetic correlation was with smoking, which is consistent with the observation that as many as 70 to 90% of patients with CH smoke, 1 , 3 , 39 seen also in our cohorts (Table S2). The high proportion of smokers among patients with CH may theoretically be explained by smoking causing CH or vice versa, or because they have shared causal factors. Whether smoking is causing CH is heavily debated. On the one hand, smoking initiation typically predates the onset of CH3 and, among those with CH who have never smoked, the majority were exposed to parental smoking in childhood. 40 Furthermore, it seems that smoking is associated with more severe manifestations of CH1 and some data suggest that the prevalence of CH has followed trends in smoking prevalence. 39 On the other hand, arguments against a causal effect of smoking include the typically long latency between smoking onset and CH debut (> 15 years). 3 In addition, in retrospective studies, patients with CH who stopped smoking several years earlier did not experience an improvement in their CH. 1 , 39
To investigate the potential causality of smoking on CH, we performed a Mendelian randomization and LCV analysis. 41 The analyses indicated a causal effect of smoking intensity on CH, with high statistical confidence. Of note, the high observed proportion of smokers among cases with CH is expected if smoking is a causal risk factor. Because cases were recruited independently of smoking status, and the proportion of smokers is similar to previous reports, we find it unlikely that recruitment bias explains the results.
Although our study cannot give definite answers regarding mechanisms linking smoking to CH, we note that several of the prioritized genes are influenced by smoking. Cigarette smoking leads to overexpression of MERTK 42 and reduced expression and function of CFTR in airway tissues. 43 Notably, our TWAS also revealed an increased expression for MERTK and reduced expression for CFTR in CH. It has been shown that smoking can induce epigenetic changes that persist even 30 years after smoking cessation, 44 therefore, the observation that patients who stop smoking do not experience an improvement of their CH might be explained by stable epigenetic modifications. In a large study, DNA methylation at 2,568 CpG sites related to 1,450 genes were found to be associated with former smoking at FDR < 0.05. 44 Four of our prioritized genes are among these (ie, FBLN7, SLC20A1, KDM4B, and ST7), that is 4 of 20 versus 1,450 of 23,300 genes (post hoc 1‐tailed binomial p = 0.033). More detailed molecular studies in relevant tissues are needed to identify mechanisms linking smoking to CH.
The suggestion that smoking is a causal risk factor for CH has potential clinical implications. Smoking is a modifiable risk factor, and it gives a further impetus to promoting smoking cessation in this group of patients. The long‐term effect of smoking cessation on CH should be carefully revisited by well‐designed prospective studies.
Notably, CH was to some extent genetically correlated with measures of risk‐taking behavior apart from smoking. Although our results support a causal effect of smoking on the development of CH, it is possible that patients with CH are also more likely to start smoking because of a tendency toward risk‐taking, as has been suggested. 39 , 45 The genetic correlations to smoking and risk‐taking behavior were not seen for migraine.
Whereas primary headache disorders are among the top causes of disability worldwide, 46 it is unknown to what extent they represent biologically distinct disorders or rather variations in clinical presentation with a shared biological basis. 47 Migraine is the only other primary headache disorder that has been explored in well‐powered GWAS. 17 We found that 3 of the 8 risk loci for CH are shared with migraine, and colocalization analyses give a high probability that the same causal variants in these loci give rise to both disorders. Notably, the remaining 5 CH loci show no association to migraine (p values > 0.10). Likewise, apart from the 3 overlapping loci, none of the other 119 known migraine loci 17 show association with CH. Our results, therefore, suggest that CH and migraine have a partly shared and partly distinct genetic basis, likely reflecting partly shared and partly distinct biological mechanisms. This corresponds well with the clinical impression of the 2 disorders as being distinct entities, but with certain shared clinical characteristics, including unilateral headache cranial autonomic symptoms, and response to some of the same medications. 47 , 48 Future studies with deep phenotyping should explore if the shared genetic risk factors are directly related to shared clinical features, such as prominent autonomic symptoms in some patients with migraine. 49
We note that for all 3 shared loci, the effect sizes were higher for CH (ORs = 1.18–1.29) than for migraine (ORs = 1.06–1.11) with nonoverlapping CIs. Even for the most consistently identified migraine risk locus, LRP1 (p value 1.38 × 10−90 in the latest migraine GWAS), 17 the effect size was higher for CH (1.18 vs 1.11). This holds true also when comparing to GWAS of clinic‐based migraine cohorts (OR = 1.11). 50 The larger effect sizes suggest that the 3 shared loci are stronger drivers of disease susceptibility in CH than in migraine, and also makes it unlikely that the observed associations are a result of misclassification of patients with migraine as having CH.
A major strength of our study is the substantially larger sample size compared to previous studies, which allows for downstream functional analyses, and clinical diagnoses made according to ICHD criteria. 2 , 10 This was made possible through the establishment of the CCG, which has brought together 16 headache research groups from 13 countries (www.clusterheadachegenetics.org). A limitation of the current study is that it included only a single non‐European cohort, from East Asia, limiting the possibility for conducting ancestry‐specific meta‐analyses and downstream analyses, for non‐European ancestries. This highlights the need for future, well‐powered trans‐ancestry genetic studies in CH.
In conclusion, in this GWAS meta‐analysis, we identify 9 independent associations in 7 risk loci for CH in European ancestry samples and one additional locus in East Asian samples. The prioritized genes show enrichment in arterial and brain tissues. CH shares certain risk loci with migraine, and is most strongly genetically correlated with smoking. Of clinical interest, Mendelian randomization analysis indicates a causal effect of cigarette smoking on the development of CH.
Author Contributions
B.S.W., A.V.H., C.R., M.A.C., M.C.D., G.M.T., P.P.R., A.C.B., M.M., A.M.J.M.vdM., T.F.H., A.R., and J.Z. contributed to the conception and design of the study. B.S.W., A.V.H., C.R., M.A.C., M.C.D., E.F., K.P.T., E.B., S.B., C.F., A.S.P., L.S.V., S.H.M., E.O., G.B., P.H., Y.F.W., I.C., T.K., V.J.G., I.dB., F.J., K.H., N.L., L.F.T., C.L.H., M.P., H.Ha., M.R., S.G., P.S., J.Y., A.H., F.K., S.R.O., O.B.P., E.S.K., A.E.M., M.S.A., S.L., M.M.C., J.A., O.Q., C.G., A.C., A.E.V., S.H.H., A.H.S., M.E.G., L.A.W., D.D., D.M., D.F.G., F.R.R., K.W.vD., R.F., M.W., M.S., H.G., K.Sl., O.A.S., L.P., M.Z., J.A.R.Q., E.D., A.S., S.R.H., C.S., T.E.T., H.S., L.S., R.C.T., J.V., R.N., K.P., K.St., C.S.J.F., E.W., E.T., R.H.J., S.C., H.Ho., G.M.T., C.K., E.M., M.V., P.P.R., A.C.B., M.M., A.M.J.M.vdM., T.F.H., A.R., and J.Z. contributed to the acquisition and analysis of data. B.S.W., A.V.H., C.R., M.A.C., M.C.D., E.F., K.P.T., and S.H.M. contributed to drafting the text or preparing the figures. Members of “HUNT All‐In Headache” are available in Table S34. Members of “The International Headache Genetics Consortium” are available in Table S35. Members of “DBDS Genomic Consortium” are available in Table S36.
Potential Conflicts of Interest
Nothing to report.
Supporting information
Data S1. Supporting information.
Acknowledgments
The authors want to thank both all participating patients and their general practitioners for their good collaboration. Additional acknowledgements are found in Table S33. This work was funded by the South‐Eastern Norway Regional Health Authority (#2020034); the Swedish National Infrastructure for Computing (SNIC) at Uppmax, Uppsala University partially funded by the Swedish Research Council through grant agreement no. 2018‐05973; the Wellcome Trust under award 076113, 085475, and 090355; the Instituto de Salud Carlos III (PI18/01788, PI19/01224, and PI20/00041); the European Regional Development Fund (ERDF); the Biomedical Network Research Center on Mental Health (CIBERSAM, Madrid, Spain); the Agència de Gestió d'Ajuts Universitaris i de Recerca‐AGAUR, Generalitat de Catalunya (2017SGR1461); the Netherlands Organization for Scientific Research, ie, the Center of Medical System Biology established by the Netherlands Genomics Initiative/Netherlands Organization for Scientific Research (to A.M.J.M.v.d.M.); the EU‐funded FP7 “EUROHEADPAIN” (grant no. 6026337 to A.M.J.M.v.d.M.); The NEO study, which comprised of the Dutch controls, was supported by the participating Departments, the Division and the Board of Directors of the Leiden University Medical Centre, and by the Leiden University, Research Profile Area “Vascular and Regenerative Medicine”; A.V.E. Harder was sponsored by the Leiden University Fund/Fonds Mr. J.J. van Enter “Pro Universitate”, www.luf.nl (grant W212163‐2‐64). Part of the genotyping of the German sample was funded by the JPND EADB grant to AR (German Federal Ministry of Education and Research [BMBF] grant: 01ED1619A); the BMBF (grants KND: 01GI0102, 01GI0420, 01GI0422, 01GI0423, 01GI0429, 01GI0431, 01GI0433, and 01GI0434; grants KNDD: 01GI0710, 01GI0711, 01GI0712, 01GI0713, 01GI0714, 01GI0715, and 01GI0716; grants Health Service Research Initiative: 01GY1322A, 01GY1322B, 01GY1322C, 01GY1322D, 01GY1322E, 01GY1322F, and 01GY1322G); PainFACT (H2020‐2020‐848099) to T.E.T; Italian Ministry of Health (RF2009‐1549619); The Research Funding Pool at Rigshospitalet to M.A.C.; The Swedish Brain Foundation and the Mellby Gård Foundation (FO2020‐0006 and FO2022‐0001); Karolinska Institutet Research Funds (2018‐01738, 2020‐01411, and 2022‐01781); The Swedish Research Council (2017‐01096); the Region Stockholm (ALF project 20200095); the Ministry of Science and Technology, Taiwan [MOST‐108‐2314‐B‐010‐022‐MY3 and 110‐2326‐B‐A49A‐501‐MY3]. The funders had no role in the design and conduct of the study; collection, management, analysis and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Bendik S. Winsvold, Aster V. E. Harder, Caroline Ran, Mona A. Chalmer, Maria Carolina Dalmasso, Egil Ferkingstad these authors contributed equally to this work and should be considered co‐first authors.
Andrea C. Belin, Manjit Matharu, Arn M.J.M. van den Maagdenberg, Thomas F. Hansen, Alfredo Ramirez, John‐Anker Zwart these authors contributed equally to this work and should be considered co‐last authors.
Data Availability
Summary statistics generated by the International Consortium for Cluster Headache Genetics are available for academic use from www.clusterheadachegenetics.org/access/
References
- 1. May A, Schwedt TJ, Magis D, et al. Cluster headache. Nat Rev Dis Primers 2018;4:18006. [DOI] [PubMed] [Google Scholar]
- 2. Headache Classification Committee of the International Headache Society (IHS) . The international classification of headache disorders, 3rd edition. Cephalalgia 2018;38:1–211. [DOI] [PubMed] [Google Scholar]
- 3. Lund N, Petersen A, Snoer A, et al. Cluster headache is associated with unhealthy lifestyle and lifestyle‐related comorbid diseases: results from the Danish cluster headache survey. Cephalalgia 2019;39:254–263. [DOI] [PubMed] [Google Scholar]
- 4. Wei DY, Goadsby PJ. Cluster headache pathophysiology–insights from current and emerging treatments. Nat Rev Neurol 2021;17:308–324. [DOI] [PubMed] [Google Scholar]
- 5. Sjaastad O, Shen JM, Stovner LJ, Elsås T. Cluster headache in identical twins. Headache 1993;33:214–217. [DOI] [PubMed] [Google Scholar]
- 6. Harder AVE, Winsvold BS, Noordam R, et al. Genetic susceptibility loci in genomewide association study of cluster headache. Ann Neurol 2021;90:203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. O'Connor E, Fourier C, Ran C, et al. Genome‐wide association study identifies risk loci for cluster headache. Ann Neurol 2021;90:193–202. [DOI] [PubMed] [Google Scholar]
- 8. Chen SP, Hsu CL, Wang YF, et al. Genome‐wide analyses identify novel risk loci for cluster headache in Han Chinese residing in Taiwan. J Headache Pain 2022;23:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bacchelli E, Cainazzo MM, Cameli C, et al. A genome‐wide analysis in cluster headache points to neprilysin and PACAP receptor gene variants. J Headache Pain 2016;17:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Headache Classification Subcommittee of the International Headache Society . The international classification of headache disorders: 2nd edition. Cephalalgia 2004;24:9–160. [DOI] [PubMed] [Google Scholar]
- 11. Winkler TW, Day FR, Croteau‐Chonka DC, et al. Quality control and conduct of genome‐wide association meta‐analyses. Nat Protoc 2014;9:1192–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta‐analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mägi R, Horikoshi M, Sofer T, et al. Trans‐ethnic meta‐regression of genome‐wide association studies accounting for ancestry increases power for discovery and improves fine‐mapping resolution. Hum Mol Genet 2017;26:3639–3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bulik‐Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor KE, Ansel KM, Marson A, et al. PICS2: next‐generation fine mapping via probabilistic identification of causal SNPs. Bioinformatics 2021;37:3004–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Benner C, Spencer CC, Havulinna AS, et al. FINEMAP: efficient variable selection using summary data from genome‐wide association studies. Bioinformatics 2016;32:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hautakangas H, Winsvold BS, Ruotsalainen SE, et al. Genome‐wide analysis of 102,084 migraine cases identifies 123 risk loci and subtype‐specific risk alleles. Nat Genet 2022;54:152–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ferkingstad E, Sulem P, Atlason BA, et al. Large‐scale integration of the plasma proteome with genetics and disease. Nat Genet 2021;53:1712–1721. [DOI] [PubMed] [Google Scholar]
- 19. Gusev A, Ko A, Shi H, et al. Integrative approaches for large‐scale transcriptome‐wide association studies. Nat Genet 2016;48:245–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Mancuso N, Freund MK, Johnson R, et al. Probabilistic fine‐mapping of transcriptome‐wide association studies. Nat Genet 2019;51:675–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Freytag V, Vukojevic V, Wagner‐Thelen H, et al. Genetic estimators of DNA methylation provide insights into the molecular basis of polygenic traits. Transl Psychiatry 2018;8:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer, 2016. [Google Scholar]
- 23. McLaren W, Gil L, Hunt SE, et al. The Ensembl variant effect predictor. Genome Biol 2016;17:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017;8:1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. GTEx Consortium . The genotype‐tissue expression (GTEx) project. Nat Genet 2013;45:580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pers TH, Karjalainen JM, Chan Y, et al. Biological interpretation of genome‐wide association studies using predicted gene functions. Nat Commun 2015;6:5890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Finucane HK, Reshef YA, Anttila V, et al. Heritability enrichment of specifically expressed genes identifies disease‐relevant tissues and cell types. Nat Genet 2018;50:621–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Finan C, Gaulton A, Kruger FA, et al. The druggable genome and support for target identification and validation in drug development. Sci Transl Med 2017;9:eaag1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wishart DS, Feunang YD, Guo AC, et al. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res 2018;46:D1074–D1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Privé F, Arbel J, Vilhjálmsson BJ. LDpred2: better, faster, stronger. Bioinformatics 2020;36:5424–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi SW, O'Reilly PF. PRSice‐2: polygenic risk score software for biobank‐scale data. Gigascience 2019;8:giz082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Giambartolomei C, Vukcevic D, Schadt EE, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014;10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hemani G, Zheng J, Elsworth B, et al. The MR‐base platform supports systematic causal inference across the human phenome. Elife 2018;7:e34408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu M, Jiang Y, Wedow R, et al. Association studies of up to 1.2 million individuals yield new insights into the genetic etiology of tobacco and alcohol use. Nat Genet 2019;51:237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bowden J, Del Greco MF, Minelli C, et al. A framework for the investigation of pleiotropy in two‐sample summary data Mendelian randomization. Stat Med 2017;36:1783–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bowden J, Davey Smith G, Burgess S. Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int J Epidemiol 2015;44:512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. O'Connor LJ, Price AL. Distinguishing genetic correlation from causation across 52 diseases and complex traits. Nat Genet 2018;50:1728–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Peng KP, Takizawa T, Lee MJ. Cluster headache in Asian populations: similarities, disparities, and a narrative review of the mechanisms of the chronic subtype. Cephalalgia 2020;40:1104–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ferrari A, Zappaterra M, Righi F, et al. Impact of continuing or quitting smoking on episodic cluster headache: a pilot survey. J Headache Pain 2013;14:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rozen TD. Cluster headache as the result of secondhand cigarette smoke exposure during childhood. Headache 2010;50:130–132. [DOI] [PubMed] [Google Scholar]
- 41. Sanderson E, Glymour MM, Holmes MV, et al. Mendelian randomization. Nat Rev Methods Primers 2022;2:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kazeros A, Harvey BG, Carolan BJ, et al. Overexpression of apoptotic cell removal receptor MERTK in alveolar macrophages of cigarette smokers. Am J Respir Cell Mol Biol 2008;39:747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bodas M, Min T, Vij N. Critical role of CFTR‐dependent lipid rafts in cigarette smoke‐induced lung epithelial injury. Am J Physiol Lung Cell Mol Physiol 2011;300:L811–L820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Joehanes R, Just AC, Marioni RE, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet 2016;9:436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lambru G, Castellini P, Manzoni GC, Torelli P. Mode of occurrence of traumatic head injuries in male patients with cluster headache or migraine: is there a connection with lifestyle. Cephalalgia 2010;30:1502–1508. [DOI] [PubMed] [Google Scholar]
- 46. GBD 2015 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the global burden of disease study 2015. Lancet 2016;388:1545–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vollesen AL, Benemei S, Cortese F, et al. Migraine and cluster headache–the common link. J Headache Pain 2018;19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chwolka M, Goadsby PJ, Gantenbein AR. Comorbidity or combination–more evidence for cluster‐migraine. Cephalalgia 2023;43. [DOI] [PubMed] [Google Scholar]
- 49. Lai TH, Fuh JL, Wang SJ. Cranial autonomic symptoms in migraine: characteristics and comparison with cluster headache. J Neurol Neurosurg Psychiatry 2009;80:1116–1119. [DOI] [PubMed] [Google Scholar]
- 50. Gormley P, Anttila V, Winsvold BS, et al. Meta‐analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet 2016;48:856–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supporting information.
Data Availability Statement
Summary statistics generated by the International Consortium for Cluster Headache Genetics are available for academic use from www.clusterheadachegenetics.org/access/