

Summary
The extracellular space of plant tissues contains hundreds of hydrolases that might harm colonising microbes. Successful pathogens may suppress these hydrolases to enable disease. Here, we report the dynamics of extracellular hydrolases in Nicotiana benthamiana upon infection with Pseudomonas syringae.
Using activity‐based proteomics with a cocktail of biotinylated probes, we simultaneously monitored 171 active hydrolases, including 109 serine hydrolases (SHs), 49 glycosidases (GHs) and 13 cysteine proteases (CPs).
The activity of 82 of these hydrolases (mostly SHs) increases during infection, while the activity of 60 hydrolases (mostly GHs and CPs) is suppressed during infection. Active β‐galactosidase‐1 (BGAL1) is amongst the suppressed hydrolases, consistent with production of the BGAL1 inhibitor by P. syringae. One of the other suppressed hydrolases, the pathogenesis‐related NbPR3, decreases bacterial growth when transiently overexpressed. This is dependent on its active site, revealing a role for NbPR3 activity in antibacterial immunity. Despite being annotated as a chitinase, NbPR3 does not possess chitinase activity and contains an E112Q active site substitution that is essential for antibacterial activity and is present only in Nicotiana species.
This study introduces a powerful approach to reveal novel components of extracellular immunity, exemplified by the discovery of the suppression of neo‐functionalised Nicotiana‐specific antibacterial NbPR3.
Keywords: activity‐based proteomics, apoplast, chitinase, glycosidase, hydrolases, Nicotiana benthamiana, PR3, Pseudomonas syringae
Introduction
Plant pathogens encounter a highly hydrolytic environment when they colonise the extracellular space (apoplast) in plant tissues. Plants secrete hundreds of hydrolytic enzymes including proteases, glucosidases and lipases, many of which accumulate to high levels during defence. These pathogenesis‐related (PR) proteins include chitinases, glucanases and proteases (van Loon et al., 2006; Doehlemann & Hemetsberger, 2013; Wang et al., 2020). In response, most pathogens secrete hydrolase inhibitors when colonising the apoplast to manipulate the host and cause disease. For instance, the oomycete plant pathogen Phytophthora infestans secretes Epi1 and Epi10, which are Kazal‐like protease inhibitors that suppress defence‐related subtilisin‐like protease P69B (Tian et al., 2004, 2005; Tian & Kamoun, 2005). Phytophthora infestans also secretes cystatin‐like protease inhibitors (EpiCs) that target papain‐like cysteine proteases (PLCPs) in the tomato apoplast (Tian et al., 2007). Tomato PLCPs are also suppressed by Cip1 and Avr2, secreted by the bacterium Pseudomonas syringae and the fungus Cladosporium fulvum, respectively (Rooney et al., 2005; Shabab et al., 2008; van Esse et al., 2008; Shindo et al., 2016). Hydrolase inhibition is not limited to proteases. Soybean pathogen P. sojae produces glucanase inhibitor GIP1 (Glucanase Inhibitory Protein 1; Rose et al., 2002) and the fungal corn pathogen Ustilago maydis produces Protein Essential during Penetration 1 (PEP1) to inhibit secreted maize peroxidase POX12, thereby preventing the oxidative burst associated with defence (Hemetsberger et al., 2012). Taken together, hydrolase inhibition in the apoplast by pathogen‐derived molecules is a common infection strategy used to manipulate the host.
We hypothesised that suppressed hydrolases play an important role in immunity. To test this, we used activity‐based protein profiling (ABPP), which allows monitoring of protein activities without previous knowledge of substrates or enzyme purification (Cravatt et al., 2008; Morimoto & van der Hoorn, 2016; Benns et al., 2021). ABPP involves the incubation of a proteome with a chemical probe, which contains a warhead that covalently reacts with the active site; a linker; and a tag to facilitate detection (Morimoto & van der Hoorn, 2016). In combination with analysis by mass spectrometry (MS), ABPP‐MS has been applied to study dynamic changes in the activities of serine hydrolases (SHs) in tomato upon infection with C. fulvum and Ralstonia solanacearum, as well as in Arabidopsis thaliana (Arabidopsis) upon infection with Botrytis cinerea (Kaschani et al., 2009; Sueldo et al., 2014; Planas‐Marquès et al., 2018). Here, we greatly increased the power of ABPP‐MS with a cocktail of biotinylated probes and taking advantage of a unique model pathosystem.
The interaction between Nicotiana benthamiana and the bacterial pathogen Pseudomonas syringae pv tomato DC3000 (PtoDC3000) provides an ideal system to study hydrolase suppression in the apoplast. The extraction of apoplastic fluid from N. benthamiana is a relatively simple procedure, ensuring high yield and purity. Nicotiana benthamiana is susceptible to the model plant pathogen PtoDC3000 when it lacks the type‐III effector hopQ1‐1 (ΔhQ), which would otherwise trigger immunity via the Roq1 immune receptor of N. benthamiana (Schultink et al., 2007; Wei et al., 2007). Using the N. benthamiana‐PtoDC3000 model pathosystem and a fluorescent probe targeting glycosidases, we previously discovered the suppression of BGAL1, an apoplast‐localised β‐galactosidase (Buscaill et al., 2019). BGAL1 participates in flagellin de‐glycosylation and thereby plays a key role in the release of the flagellin elicitor that is universally recognised in plants (Buscaill et al., 2019). The activity of BGAL1 is suppressed by a small molecule inhibitor produced by PtoDC3000 to promote disease (Buscaill et al., 2019). This discovery illustrates that suppressed hydrolases play important roles in plant immunity. Here, we explored hydrolase dynamics in apoplastic fluid of infected plants using ABPP‐MS with a cocktail of biotinylated probes to uncover additional suppressed hydrolases during infection.
Materials and Methods
Bacterial infection and apoplastic fluid isolation – Pseudomonas syringae
Van Hall pathovar tomato DC3000 lacking hopQ1‐1 (PtoDC3000(ΔhQ) was grown in liquid LB medium (rifampicin 25 μg ml−1) overnight 28°C, 220 rpm). The next morning, the bacterial density was measured, and the culture was brought to OD600 = 0.001 with distilled, sterile (MilliQ) water. Cultures were infiltrated in fully expanded leaves of 4–5‐wk‐old N. benthamiana Domin using a needleless syringe. Up to two leaves were infected per plant, and MilliQ water was used for the mock treatment. Apoplastic fluid was extracted from infected leaves, 2 d after bacterial infection, as described previously (Joosten, 2012; Hong & Van der Hoorn, 2014). Briefly, leaves were harvested and submerged in MilliQ water and ice, with the abaxial side facing down. A vacuum was applied with a pump and subsequently released to allow water intake. Leaves were then rolled into a 50‐ml syringe without plunger, placed into a 50 ml tube and centrifuged at 1500 g for 25 min at 4°C, with slow acceleration and de‐acceleration of the rotor. Apoplastic fluid was recovered from the bottom of the 50 ml tube and processed immediately.
Large‐scale labelling and affinity purification
The experiment involved four technical replicates for each biological treatment (mock, infected and no‐probe control), with each technical replicate corresponding to the apoplastic fluid isolated from 15 plants, two leaves per plant. The experiment was repeated in a subsequent year.
Four–five‐week‐old N. benthamiana plants were infiltrated with PtoDC3000(ΔhQ) or water (mock), and apoplastic fluid was extracted at 2 days postinoculation (dpi) as described previously. Three milliliters of the freshly obtained apoplastic fluid was labelled with the ‘probe cocktail’ (FP‐biotin, JJB111, DCG04 and DK‐D04) in a reaction mixture containing 50 mM NaAc pH 5, 5 mM DTT and 4 μM of each probe. Labelling was performed at room temperature for 4 h with constant rotation. For the no‐probe control, an equal volume of DMSO was added to a mixture of 1.5 ml of both AFs. To stop the labelling reaction, proteins were precipitated with chloroform : methanol (Wessel & Flügge, 1984) as follows: one volume of ice‐cold chloroform, three volumes of ice‐cold water and four volumes of ice‐cold methanol were added. The samples were vortexed and subsequently centrifuged (3000 g , 30 min, 4°C). The precipitated proteins were resuspended in 2 ml of 1.2% SDS in 1× PBS (Life Technologies) by pipetting, and the solution was further diluted to 0.2% SDS by adding 1× PBS buffer. Proteins were then denatured by heating at 95°C for 5 min. To precipitate labelled proteins, 130 μl of avidin beads (A9207; Sigma) was added to each labelling reaction and incubated for 1 h at room temperature while rotating, after which the beads were spun down for 10 min at 400 g . The supernatant was removed, and the beads were washed five times with 10 ml of 1% SDS and then twice with 10 ml of MS‐grade water. The beads were then transferred to a protein LoBind tube (Z666505‐100EA; Eppendorf, Hamburg, Germany).
Synthesis of DK‐D04
All chemicals and solvents were from Sigma‐Aldrich, ABCR (Karlsruhe, Germany) and Fluorochem (Hadfield, UK). Fmoc‐Asp‐AOMK was synthesised according to a literature procedure (Dolle et al., 1994). The synthesis of DK‐D04 (I939, Biotin‐PD‐AOMK, Supporting Information Fig. S1A) was carried out on solid support, according to a general SPPS procedure, following Fmoc‐strategy. Couplings have been conducted in a syringe reactor using the corresponding Fmoc‐building blocks (3 eq.), HOBt (3 eq.), DIC (3 eq.) and a reaction time of 2 h at room temperature, while the resin suspension was agitated on an orbital shaker. Fmoc‐deprotection was achieved by the addition of a 5% (v/v) solution of diethylamine in DMF and agitation of the resulting suspension for 15 min. The general washing procedure involved alternated washing of the resin with DMF (3×), MeOH (3×) and DCM (3×). 2‐Chlorotrityl resin (1 eq.) was placed in a flame‐dried flask under an argon atmosphere. Fmoc‐Asp‐AOMK (4 eq.) dissolved in anhydrous DCM and DIPEA (5 eq.) was added, and the suspension was shaken overnight at room temperature. Methanol was added, and stirring was continued for additional 30 min. The resin was transferred into a syringe reactor and washed according to the general procedure. Resin loading was determined by Fmoc‐loading test (0.8 mmol g−1). For this synthesis, 250 mg (0.2 mmol) of Fmoc‐Asp‐AOMK loaded 2‐Chlorotrityl resin was utilised. A Fmoc‐deprotection and general washing was carried out and a solution of Fmoc‐6‐Ahx‐OH (212 mg, 0.6 mmol), HOBt (81 mg, 0.6 mmol), DIC (76 mg, 0.6 mmol, 93 μl) in DMF (10 ml) was utilised for the next coupling step. Fmoc‐deprotection and general washing was carried out, and coupling was continued using a solution of Biotin (147 mg, 0.6 mmol), HOBt (81 mg, 0.6 mmol), DIC (76 mg, 0.6 mmol, 93 μl) in DMF (10 ml). The crude product was cleaved off the resin utilising TFA (3 ml) and agitation at room temperature for 1 h. The obtained crude product was purified by reversed‐phase HPLC (H2O : ACN, 0.1% TFA; gradient: from 3% to 80%). The desired product was isolated a colourless powder. Yield: 0.73 mg (0.9 μmol). LC–MS (ESI): m/z = calcd for C41H61N6O10S+ [M + H]+ 829.42, found 829.29. HRMS (ESI): m/z = calcd for C41H61N6O10S+ [M + H]+ 829.41644, found 829.41612.
Synthesis of TK009
Commercially available E64 Azide (Toronto Research Chemicals, Toronto, Ontario, Canada, 1 mg, 2.7 μmol, 1 eq.) was dissolved in acetonitrile (100 μl). An aliquot of an aqueous 100 mM CuI solution (16.2 μl, 1.6 μmol, 0.6 eq.) and DIPEA (1.9 μl, 10.8 μmol, 4 eq.) were added. In a second flask, commercially available BDP‐630/650‐alkyne (5 mg; Lumiprobe, Hannover, Germany) was dissolved in acetonitrile (100 μl) and an aliquot of this mixture (52.7 μl, 5.4 μmol, 2 eq.) was added to the first reaction solution. The resulting solution was stirred at room temperature for 16 h after which reaction control by LC–MS indicated completion of the reaction. The desired product was isolated from the solution by injection into a preparative HPLC equipped with a RP‐C18 column and run at a flow of 20 ml and with the following gradient program (all solvents contained 0.1% (v/v) TFA): 90% H2O/10% ACN to 30% H2O/70% ACN in 3 min, to 25% H2O/75% ACN in 20 min. Product‐containing fractions were pooled and lyophilised to yield 1.5 mg (64%) of TK009 (I912, Cy5‐E64, Fig. S1B). LC–MS (ESI): t R = 10.09 min, m/z calculated for C42H48BF2N8O7S [M + H]+: 857.34, found 857.20.
On‐bead trypsin digestion
The beads were resuspended in 250 μl of 8 M urea dissolved in 50 mM Tris–HCl pH 8. To reduce disulphide bridges, 12.5 μl of 200 mM DTT was added and beads were incubated at 65°C for 15 min while shaking. The beads were then cooled to 35°C. For the alkylation step, 12.5 μl of 400 mM IAA (iodoacetamide) was added and incubated at 35°C for 30 min while shaking and in the dark. Trypsin (Gold Mass spectrometry Grade; Promega, Madison, WI, USA) was reconstituted according to the manufacturers' instructions and added to the beads. Trypsin digestion was performed overnight at 37°C with shaking. After digestion, the beads were shortly spun down at low speed and the supernatant (containing the peptides) was transferred to a new protein LoBind tube. Beads were washed with 50 μl of MS‐grade water and spun down, and the supernatant was combined with the first supernatant. trifluoroacetic acid (TFA) was added to the peptides to a final concentration of 0.5–1% (v/v). Before mass spectrometry analysis, peptides were purified using Sep‐Pak C18 columns (WAT020515; Thermo Fisher, Waltham, MA, USA) following the instructions provided by the manufacturer.
Mass spectrometry analysis
Experiments were performed on an Orbitrap Elite instrument (Thermo Fisher Scientific, Waltham, MA, USA), coupled to an EASY‐nLC 1000 liquid chromatography (LC) system (Thermo) operated in the one‐column mode. The analytical column was a fused silica capillary (75 μm × 32 or 36 cm) with an integrated PicoFrit emitter (New Objective, Littleton, MA, USA) packed in‐house with Reprosil‐Pur 120 C18‐AQ 1.9 μm resin (Dr Maisch, Ammerbuch, Germany). The analytical column was encased by a column oven (Sonation GmbH, Biberach, Germany) and attached to a nanospray flex ion source (Thermo). The column oven temperature was adjusted to 45°C during data acquisition and at 30°C in all other modes. The LC was equipped with two mobile phases: solvent A (0.1% (v/v) formic acid, FA, in water) and solvent B (0.1% FA in acetonitrile). All solvents were of UPLC grade (Sigma). Peptides were directly loaded onto the analytical column with a flow rate c. 0.5–0.8 μl min−1, which did not exceed 980 bar. Peptides were subsequently separated on the analytical column by running a 140 min gradient of solvent A and solvent B (start with 7% (v/v) B; gradient 7–35% B for 120 min; gradient 35% to 100% B for 10 min and 100% B for 10 min) at a flow rate of 300 nl min−1. The mass spectrometer was set in the positive ion mode and operated using Xcalibur software (v.2.2 SP1.48). Precursor ion scanning was performed in the Orbitrap analyser (FTMS; Fourier Transform Mass Spectrometry) in the scan range of m/z 300–1500 or 1800 and at a resolution of 60 000 with the internal lock mass option turned on (lock mass was 445.120025 m/z, polysiloxane; Olsen et al., 2005). Product ion spectra were recorded in a data‐dependent fashion in the ion trap (ITMS) in a variable scan range and at a rapid scan rate. The ionsation potential was set to 1.8 kV. Peptides were analysed using a repeating cycle consisting of a full precursor ion scan (1.0 or 3.0 × 106 ions or 30 or 50 ms) followed by 15 product ion scans (1.0 × 104 ions or 50 ms) where peptides are isolated based on their intensity in the full survey scan (threshold of 500 counts) for tandem mass spectrum (MS2) generation that permits peptide sequencing and identification. The collision‐induced dissociation (CID) energy was set to 35% for the generation of MS2 spectra. During MS2 data acquisition, dynamic ion exclusion was set to 120 s with a maximum list of excluded ions consisting of 500 members and a repeat count of one. Ion injection time prediction, preview mode for the FTMS, monoisotopic precursor selection and charge state screening were enabled. Only charge states higher than 1 were considered for fragmentation.
Peptide and protein identification using MaxQuant
RAW spectra were submitted to an Andromeda (Cox et al., 2011) search using MaxQuant (v.1.6.10.43) using the default settings label‐free quantification (LFQ) and match between runs being activated (Cox et al., 2014). MS/MS spectra data were searched against the Uniprot Pseudomonas syringae pv tomato (strain ATCC BAA‐871/DC3000; UP000002515_223283.fasta; 5426 entries, downloaded 5/25/2020) reference Proteome and the Nicotiana benthamiana database (12864_2019_6058_MOESM10_ESM.fasta; 74 802 entries, downloaded 2/21/2020; Kourelis et al., 2019). To estimate the level of contamination, all searches included a contaminants database (as implemented in MaxQuant, 245 sequences) that contains known MS contaminants. Andromeda searches allowed for oxidation of methionine residues (16 Da) and acetylation of the protein N terminus (42 Da) as dynamic modifications and the static modification of cysteine (57 Da, alkylation with iodoacetamide). The digestion mode was set to ‘specific’, enzyme specificity was set to ‘Trypsin/P’ with two missed cleavages allowed, and the instrument type in Andromeda searches was set to Orbitrap and the precursor mass tolerance to ±20 ppm (first search) and ±4.5 ppm (main search). The MS/MS match tolerance was set to ±0.5 Da and the peptide spectrum match FDR and the protein FDR to 0.01 (based on target‐decoy approach and decoy mode ‘revert’). Minimum peptide length was seven amino acids.
The minimum score for unmodified peptides was set to 0. For protein quantification, modified peptides (minimum score 40) and unique and razor peptides were allowed. Further analysis and annotation of identified peptides was done in Perseus v.1.5.5.3 (Tyanova et al., 2016). Only protein groups with at least two identified unique peptides over all runs were considered for further analysis. For quantification, we combined related biological replicates to categorical groups and investigated only those proteins that were found in a minimum of one categorical group at least in three out of four biological replicas. Comparison of protein group quantities (relative quantification) between different MS runs is based solely on the LFQ's as calculated by MaxQuant (MaxLFQ algorithm). Briefly, label‐free protein quantification was switched on, and unique and razor peptides were considered for quantification with a minimum ratio count of 2. Retention times were recalibrated based on the built‐in nonlinear time rescaling algorithm. MS/MS identifications were transferred between LC–MS/MS runs with the ‘Match between runs’ option in which the maximal match time window was set to 0.7 min and the alignment time window set to 20 min. The quantification is based on the ‘value at maximum’ of the extracted ion current. At least two quantitation events were required for a quantifiable protein.
MS data analysis
Data were analysed with Perseus (Tyanova et al., 2016), and only proteins corresponding to Nicotiana benthamiana were included in the analysis. Proteins were only considered for analysis if they were identified in three of the four replicates for at least one treatment (i.e. mock or infected). Proteins enriched compared with the no‐probe control (NPC) were considered as labelled and further analysed. To identify differentially active hydrolases upon infection, we performed a t‐test with Benjamini–Hochberg correction for multiple testing (α = 0.05) comparing mock and infected samples. Final hydrolase list was manually curated using Pfam (El‐Gebal et al., 2019) to confirm correct annotation as probe target.
Cloning of hydrolases
Overexpression constructs for six candidate hydrolases were built by Golden Gate Assembly (Engler et al., 2014). The full‐length genes were amplified from cDNA using primers summarised in Table S1. Binary vectors were generated in a Golden Gate reaction with 35S promoter (pICH51288) and 35S terminator (pICH41414) into binary backbone pJK001c (Paulus et al., 2020) using Bsa1 restriction sites, resulting in binary clones summarised in Table S2. Binary vectors were transformed into Agrobacterium tumefaciens GV3101 (pMP90) by freeze‐thawing and selection for kanamycin and gentamicin resistance. The E92A and Q112E mutants of NbPR3 were generated by site‐directed mutagenesis using the primers listed in Table S1, using plasmid pAG001 as a template. The PCR product was digested with DpnI to remove template plasmid, and the remaining mutated product was transformed into E. coli. Sequence‐confirmed positive clones were selected and transformed into A. tumefaciens.
Agroinfiltation
Agrobacterium cultures were grown in LB media (kanamycin 50 μg ml−1 and gentamicin 10 μg ml−1) overnight at 28°C. The next day, cultures were spun at 4000 g for 10 min at room temperature and resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES, 150 μM acetosyringone) to a final OD600 0.5. Cultures carrying NbPR3 were co‐infiltrated with cultures carrying silencing suppressor P19 (Van der Hoorn et al., 2003), and P19 combined with empty vector (EV) was used as negative control. N. benthamiana plants were infiltrated at 4–5 wk old, and apoplastic fluid was extracted at 4 dpi as indicated previously (Joosten, 2012; Hong & Van der Hoorn, 2014).
Protein analysis
Apoplastic fluid was isolated from N. benthamiana plants transiently overexpressing hydrolases at 4 dpi. The presence of active serine hydrolases and cysteine proteases was measured by ABPP using the FP‐TAMRA probe (Thermo Fisher Scientific) and TK009 (see above), respectively. To label hydrolases, apoplastic fluid samples were incubated in a 50 μl reaction with 0.2 μM FP‐TAMRA (for serine hydrolases), or a 250 μl reaction with 2 μM I912 (for cysteine proteases), in the presence of 5 mM DTT and 50 mM sodium acetate pH 5 at room temperature for 1 h (serine hydrolases) or 4 h (for cysteine proteases). After labelling, the reaction was stopped by precipitation in 4× ice‐cold acetone followed by resuspension in 4× gel loading buffer and heating at 90°C for 5 min. Samples were separated by SDS‐PAGE and visualised using a Typhoon scanner at Cy3 filter (serine hydrolases) or Cy5 filter (cysteine proteases). Coomassie staining was used to check protein loading. NbPR3 was detected by western blot using anti‐PR3 antibody designed for tobacco PR3 isoforms (1 : 2500 in PBS‐T; Agrisera, Vännäs, Sweden) and an‐rabbit‐HRP secondary antibody (GE Healthcare, Chicago, IL, USA).
Agromonas infection assay
Agrobacterium cultures were grown in LB media (kanamycin 50 μg ml−1 and gentamicin 10 μg ml−1) overnight at 28°C. The next day, cultures were spun at 4000 g for 10 min at room temperature and resuspended in infiltration buffer (10 mM MgCl2, 10 mM MES, 150 μM acetosyringone) to a final OD600 0.5. All constructs were co‐infiltrated with the silencing suppressor P19 (Van der Hoorn et al., 2003), and P19 with empty vector was used as mock control. Nicotiana benthamiana plants were infiltrated when 3–4 wk old, and 2–3 fully expanded leaves were infiltrated per plant. Six plants were used per overexpression construct. Plants were covered overnight with a transparent lid to increase humidity. Two days later, agroinfiltrated leaves were infiltrated with 106 CFU ml−1 PtoDC3000(ΔhQ) as indicated previously. Three days later, leaf discs were punched with a cork borer from each infected leaf and surface‐sterilised with 15% hydrogen peroxide for 2 min. Leaf discs were then washed twice in MilliQ and dried. Leaf discs were placed into a 1.5 ml safe‐lock Eppendorf tube with three 3 mm diameter metal beads and 1 ml of MilliQ. Tubes were placed in tissue lyser for 5 min at 30 Hertz s−1. 200 μl of the lysed tissue was transferred to the first row (A) of a 96‐well plate, and then serial 10‐fold dilutions were made until the last row (20 μl tissue +180 μl MilliQ water). Twenty microliters of undiluted tissue and serial dilutions was plated on LB‐agar plates containing Pseudomonas CFC Agar Supplement (10 μg ml−1 cetrimide, 10 μg ml−1 fucidin and 50 μg ml−1 cephaloridine, SR0103; Thermo Fisher Scientific). Plates were allowed to dry and incubated at 28°C for 2 d, and then colonies were counted.
Endochitinase activity
Apoplastic fluid was used as an enzyme source to test endochitinase activity as described before (Hollis et al., 1997; Libantová et al., 2009). Briefly, 20 μl of apoplastic fluid was combined with 30 μl of substrate solution (4‐methylumbelliferyl‐β‐d‐N,N′,N′′‐triacetylchitotrioside; Sigma) to a final substrate concentration of 18 μM. Reactions were performed in a black 96‐well plate and incubated for 1 h at 37°C in the plate reader. Measurements were taken every minute for 1 h using 355 nm excitation and 450 nm emission. The positive control contained commercial chitinase from Trichoderma viride at a final concentration of 0.04 mg ml−1 (C8241; Sigma).
Lysozyme assay
Apoplastic fluid isolated from leaves transiently expressing NbPR3 was used to test lysozyme activity following the instructions of the lysozyme manufacturer. Briefly, 10 μl apoplastic fluid was mixed with 250 μl Micrococcus lysodeikticus cells resuspended in potassium phosphate buffer at 0.15 mg ml−1. Absorbance at 450 nm was recorded in Tecan plate reader at 15‐s intervals for 5 min at 25°C. The maximum linear rate of A450 decrease was calculated and converted into enzymatic units per μg protein. The positive control contained 0.1 μg commercial lysozyme from chicken egg white (L6876; Sigma).
Results
The apoplast is rich in hydrolases
To investigate the accumulation of hydrolases in the apoplast of infected and noninfected plants, N. benthamiana was infiltrated with water (mock) and PtoDC3000(ΔhQ). We extracted apoplastic fluids (AFs) at 2‐d postinfection (2 dpi), then digested the proteomes with trypsin and analysed them by LC–MS/MS. We robustly identified 217 proteins carrying a predicted signal peptide (SP, by SignalP, Almagro Armenteros et al., 2019). Classification of these 217 proteins using Pfam (El‐Gebal et al., 2019) revealed that 48% of these apoplastic proteins are hydrolases (105 proteins, Fig. 1a; Table S3). The other 112 apoplastic proteins are diverse and include eight inhibitors and 13 peroxidases.
Fig. 1.
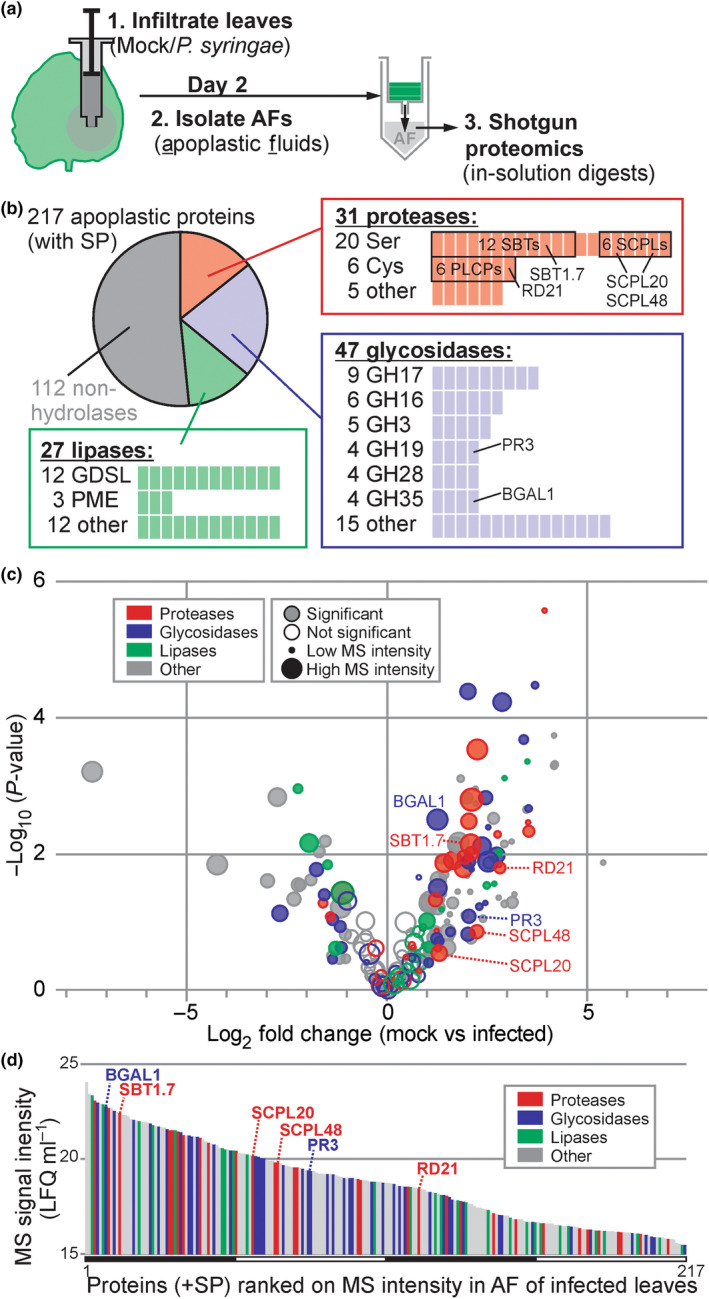
Many plant hydrolases accumulate in the apoplast following infection. (a) Experimental design. Nicotiana benthamiana leaves were inoculated with PtoDC3000(ΔhQ) (infected) or water (Mock) and apoplastic fluid (AF) was collected at 2‐d postinoculation (2 dpi). Proteins were digested with trypsin and analysed by mass spectrometry. (b) Many detected secreted proteins are hydrolases. Detected proteins that have a signal peptide predicted by SignalP were annotated with Pfam and classified into proteases (red), glycosidases (blue) and lipases (green) and subdivided into protein families. (c) More hydrolytic enzymes accumulate in apoplast upon infection. The P‐values of the t‐test were plotted against the fold change. The circle diameter reflects the sum of the mass spectrometry (MS) intensities, and filled circles identify proteins with a significant differential accumulation. (d) Hydrolytic enzymes are relatively abundant in the apoplast. All 217 signal peptide (SP)‐containing proteins detected by MS were ranked on average protein intensities detected in AF from mock and infected samples. The different hydrolase classes highlighted in colours, and six hydrolases are highlighted. (a–d) AF isolated from mock‐ and PtoDC3000(ΔhQ)‐infected plants were isolated from four independent experiments and analysed by mass spectrometry. label‐free quantitation (LFQ) intensities were corrected for protein concentrations measured in the respective AF samples to calculate LFQ ml−1. Plant proteins having a predicted SP and detected in all eight samples were retained and used for graphs (b–d). See Supporting Information Table S3 for all detected proteins from this ACE_0276 experiment.
The 105 detected hydrolases include 31 proteases, 47 glycosidases and 27 lipases (Fig. 1b). Detected apoplastic proteases belonged to the mechanistic class of Ser proteases (SPs, 20 proteins), Cys proteases (CPs, six proteins) and other proteases (five proteins; Fig. 1b). The 20 Ser proteases include 12 subtilisin‐like proteases (SBTs, S8 family) and six Ser carboxypeptidase‐like proteases (SCPLs, S10). The six Cys proteases are all papain‐like Cys proteases (PLCPs, C01). The 47 glycosidases belong to 18 different glycosyl hydrolase (GH) families, which include nine GH17 glycosidases, six GH16 glycosidases, five GH3 glycosidases and four glycosidases each from families GH19, GH28 and GH35. Finally, the 27 lipases include 12 GDSL‐lipases, three pectinacetylesterases (PAEs) and 12 other lipases. This proteome composition is similar to that of a previously reported proteome (Buscaill et al., 2019, ACE_0058).
Protein concentrations in the apoplast can increase 10‐fold upon infection (Table S4), so we corrected our proteomics dataset for this by calculating the MS intensity per ml of apoplastic fluid to facilitate the comparative analysis of protein concentrations. When plotted in a volcano plot, the protein concentrations of abundant hydrolases significantly increase upon infection (Fig. 1c), consistent with the well‐known accumulation of PR proteins, such as chitinases and glucanases (van Loon et al., 2006). Consequently, apoplastic hydrolases are a major component of the apoplastic proteome of infected plants. When ranked by MS intensity, which is an approximation for protein abundance (Cox et al., 2014), 30 hydrolases are amongst the top quartile of most abundant proteins in the apoplast of infected plants (Fig. 1d). In conclusion, a large proportion of the extracellular proteins encountered by pathogens during infection are hydrolytic enzymes.
Large‐scale activity profiling of the apoplast upon bacterial infection
To investigate changes in hydrolase activities in the apoplast caused by bacteria, we investigated changes in active hydrolases upon bacterial infection with ABPP‐MS. To display the active proteome, we labelled the apoplastic proteome with a cocktail of biotinylated activity‐based probes, including FP‐biotin (Liu et al., 1999), JJB111 (Chandrasekar et al., 2014) and DCG04 (Greenbaum et al., 2000) to display the active serine hydrolases (SHs), glycosyl hydrolases (GHs) and cysteine proteases (CPs), respectively. We also included the custom‐made Biotin‐PD‐AOMK (DK‐D04, Fig. S1A) to detect vacuolar processing enzymes (VPEs), which have been detected in apoplastic fluids (Sueldo et al., 2014). Biotinylated proteins were purified and analysed by MS in n = 4 replicates (Fig. 2a).
Fig. 2.
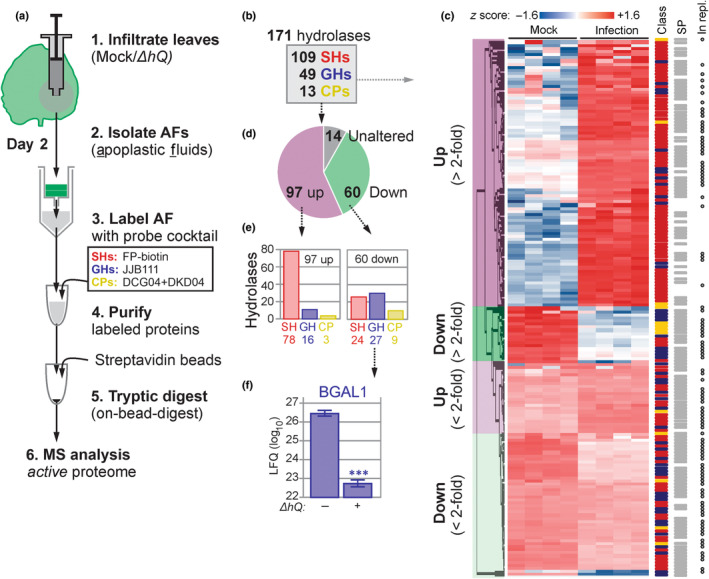
Activity‐based proteomics displays hydrolase activity dynamics upon infection. (a) Experimental procedure. Nicotiana benthamiana plants were infiltrated with PtoDC3000(ΔhQ) or water (Mock) and apoplastic fluid (AF) was collected at 2‐d postinfection (2 dpi). AFs were labelled with a cocktail activity‐based probes targeting active serine hydrolases (SHs, red), glycosyl hydrolases (GHs, blue) and cysteine proteases (CPs, yellow). Labelled proteins were purified and identified by mass spectrometry (MS) for n = 4 replicates. (b) Classification of 171 robustly detected labelled hydrolases. (c) Heatmap of the 171 detected active hydrolases detected by activity‐based proteomics (experiment ACE136), grouped by category (left) and annotated for being SH/GH/CP; having a SignalP‐predicted signal peptide (SP); and for being detected in an independent replicate MS experiment (ACE236) (right). (d) Summary of the detected differential active hydrolases showing statistically significant up or downregulation upon bacterial infection. (e) Summary of the numbers of active hydrolases belonging to each class that are up or downregulated. (f) β‐galactosidase‐1 (BGAL1) labelling is significantly downregulated upon infection. The label‐free quantitation intensities for this positive control were extracted from this dataset (ACE_0236). Error bars represent standard error from n = 4 biological replicates. ***, P < 0.001 (Students' t‐test).
The annotation of ABPP‐MS spectra to the proteome of N. benthamiana (Kourelis et al., 2019) enabled the detection of 171 predicted probe targets that were all enriched (α = < 0.05) compared with the no‐probe control (Table S5). These probe targets included 109 SHs, 49 GHs and 13 CPs (Fig. 2b). Overall, 157 target proteins were differentially labelled between mock and PtoDC3000(ΔhQ) treatments (α = 0.05), indicating that 92% of the detected active proteome changes significantly during bacterial infection (Fig. 2c,d). Of the 97 activities that increased upon infection, 78 were SHs, 16 were GHs, and three CPs (Fig. 2e). Furthermore, we detected 60 reduced hydrolytic activities, including 24 SHs, 27 GHs and nine CPs (Fig. 2e). Overall, bacterial infection induces mostly active SHs and reduces mostly active GHs and CPs. Importantly, the suppressed hydrolases include BGAL1 (3.67‐fold downregulated, P‐value = 1.65E‐07), as described previously (Fig. 2f, Buscaill et al., 2019). These results indicate that the active apoplastic proteome undergoes large changes during bacterial infection, including 57% of the hydrolases having increased activity and 35% having reduced activity. Of the 97 increased active hydrolases, 74 had a fold change of at least 2 (FC ≥ 2), consisting of 63 SHs, nine GHs and two CPs. Likewise, 25 of the 60 reduced active hydrolases also had FC ≥ 2, consisting of six SH, 14 GHs and five CPs.
Although the general trend is that active SHs are induced and active GH and CPs are reduced, we observed many differences within each hydrolase subfamily (Fig. 2e). We identified 28 active GDSL‐like lipases, 23 (most) of which showed increased activity in infected tissues. We also identified 21 subtilisin‐like serine proteases (S8, SBTs), of which nine showed decreased activity and nine increased activity upon bacterial infection. Furthermore, we identified 18 serine carboxypeptidase‐like proteases (S10, SCPLs) of which 12 had increased activity during infection. We also detected 10 carboxylesterases (CXEs) and six pectin acetylesterases (PAEs), all of which were more active upon infection. Of the 17 detected active GH3s, seven were less active in infected tissue. We also detected six active GH79s and six GH35s, including BGAL1 (NbD029635; Buscaill et al., 2019). Eight of the 13 detected papain‐like proteases (C01) had a reduced activity upon infection. Both detected VPEs are less active in infected tissue. Altogether, these data demonstrate that the active apoplastic proteome changes drastically upon bacterial infection.
One of the tested suppressed hydrolases inhibits bacterial growth
An independent experiment confirmed differential activities for 90 of the detected hydrolases (Figs 3, S2; Table S6). We chose five hydrolases with a predicted SP (Almagro Armenteros et al., 2019) that showed robustly suppressed activities upon bacterial infection (Fig. 3). We selected three SHs (NbSBT1.7, NbSCPL20 and NbSCPL48), one GH (NbPR3) and one PLCP (NbRD21). Because depletion of suppressed hydrolases is less likely to cause disease phenotypes, we tested whether increased expression could overcome the suppression and uncover the roles of these hydrolases in immunity. We therefore took advantage of the recently developed ‘agromonas’ assay (Buscaill et al., 2021), which is based on infections of agroinfiltrated tissues with PtoDC3000(ΔhQ). We cloned and transiently expressed the five selected hydrolases through agroinfiltration and infected the agroinfiltrated leaves 2 d later with PtoDC3000(ΔhQ). Bacterial growth was determined 3 d later by plating out dilution series of extracts of infected leaves on selective media. Hydrolase overexpression was confirmed for all the tested hydrolases (Fig. S3A), but only transient expression of NbPR3 also suppressed bacterial growth (Figs 4a, S3B), suggesting a role for NbPR3 in antibacterial immunity. This ‘agromonas’ infection assay was repeated 10 times, and increased immunity to PtoDC3000(ΔhQ) upon expression of NbPR3 was found in each of these experiments (Fig. S4; P = 8.74 × 10−9 over all 10 experiments). Transient expression of NbPR3 also increased resistance to pv tabaci 6605 (Pta6605), but not against pv syringae B728a (PsyB728a; Fig. 4a), indicating that NbPR3‐based immunity might be strain specific.
Fig. 3.
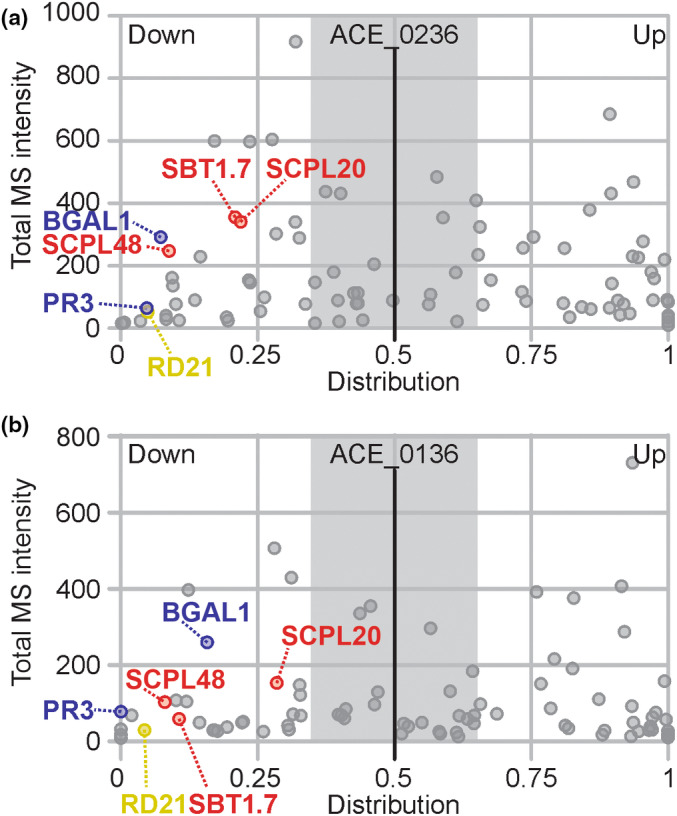
Several secreted hydrolases are consistently suppressed during infection. Distribution graphs of the active proteome from two independent experiments. Data derived from Fig. 2 (experiment ACE_0236) (a) were compared with a replicate experiment (ACE_0136, Supporting Information Fig. S3) (b) and used to select consistently suppressed hydrolases. Total mass spectrometry (MS)/MS spectra for mock and infected samples were combined and plotted against the distribution of each protein in mock and infected samples. The distribution was calculated as label‐free quantitation (LFQ)infected/(LFQmock + LFQinfected). Proteins are represented as dots, and only proteins showing the same behaviour in both biological replicates are shown. Six hydrolases (NbSBT1.7a, NbSCPL20, NbSCPL48, NbPR3, NbRD21 and NbBGAL1) are indicated and coloured red, blue and yellow according to hydrolase class (SH, GH and CP, respectively).
Fig. 4.
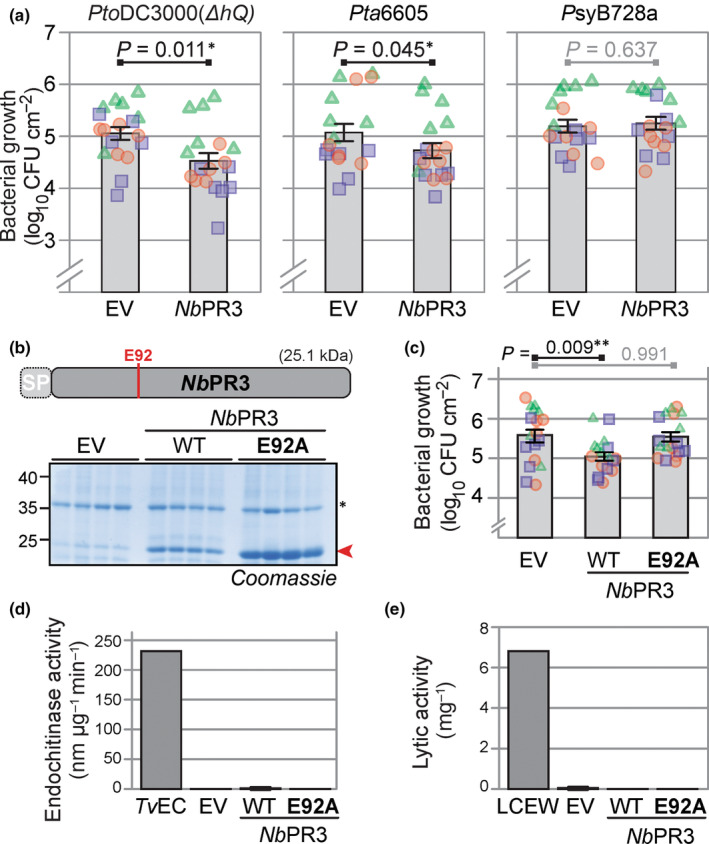
NbPR3 requires its active site to reduce bacterial growth but lacks chitinase and lysozyme activity. (a) Transient NbPR3 expression decreases susceptibility to Pseudomonas syringae pathovars tomato DC3000(ΔhQ) and tabaci 6605, but not syringae B728a. NbPR3 and the empty vector (EV) control were transiently expressed in Nicotiana benthamiana by agroinfiltration. Two days later, the same leaves were infiltrated with 106 CFU ml−1 bacteria and bacterial population densities in log10CFU cm−2 were determined after 3 d. Bars show the mean value of 18 biological replicates performed over three separate experiments, and error intervals represent the SE. P‐values were calculated by two‐way ANOVA followed by post hoc comparison using the Dunnett test to examine the effect of NbPR3 overexpression on bacterial growth. (b) The NbPR3(E92) mutant accumulates in the apoplast of agroinfiltrated leaves. NbPR3 and its E92A mutant and the empty vector (EV) control were agroinfiltrated and apoplastic fluid (AF) was isolated at Day 4 from four different replicates, separated on protein gel and analysed by Coomassie staining. The red arrowhead indicates NbPR3 protein. *, Endogenous PR2 protein, induced by agroinfiltration. (c) Transient expression of NbPR3 but not its catalytic mutant suppresses bacterial growth of PtoDC3000(ΔhQ). NbPR3, its catalytic site E92A mutant, and the empty vector (EV) control were transiently expressed by agroinfiltration. Two days later, the same leaf was infiltrated with 106 CFU ml−1 PtoDC3000(ΔhQ) and bacterial population densities in log10CFU cm−2 were determined after 3 d. Bars show the mean value of 18 biological replicates performed over three separate experiments, and error intervals represent the SE. P‐values were calculated by two‐way ANOVA followed by post hoc comparison using the Dunnett test to examine the effect of NbPR3 overexpression on bacterial growth. **, P < 0.01. (d) NbPR3 lacks endochitinase activity. AF from plants transiently expressing NbPR3 or its E92A mutant were incubated with 4‐MU‐GlcNac3, and the rate of hydrolysis was measured at 355ex/450em and calculated per microgram NbPR3 protein estimated by Coomassie staining. The endochitinase of Trichoderma viride (TvEC) was included as a positive control. (e) NbPR3 lacks lysozyme activity. AF from plants transiently expressing NbPR3 or its E92A mutant were incubated with Micrococcus lysodeikticus cells. The change in A450 was measured and converted to units per μg NbPR3 protein estimated by Coomassie staining. The lysozyme of chicken egg white (LCEW) was included as a positive control.
To determine whether antibacterial immunity is dependent on NbPR3 activity, we generated an E92A substitution mutant of NbPR3, replacing the active site Glu by an Ala residue. This NbPR3E92A protein was successfully expressed upon agroinfiltration (Fig. 4b), yet was unable to suppress bacterial growth (Fig. 4c), demonstrating that the intact active site is essential for antibacterial immunity.
NbPR3 belongs to the GH19 hydrolase family and is annotated as an acidic endochitinase. We therefore tested the ability of NbPR3 to degrade a N‐acetylglucosamine polymer in an in vitro fluorogenic reaction using apoplastic fluid from agroinfiltrated leaves expressing either NbPR3 or its E92A mutant as negative control. A commercial chitinase from the fungus T. viride could degrade this substrate, but NbPR3 could not (Fig. 4d), indicating that NbPR3 does not have endochitinase activity.
The cell wall of bacteria contains peptidoglycan, of which the glycan polymer typically consists of alternating residues of β‐(1,4) linked N‐acetylglucosamine and N‐acetylmuramic acid units. To test whether NbPR3 can hydrolyse peptidoglycan, we monitored the change in the absorbance upon lysis of Micrococcus lysodeikticus, which is an established assay for peptidoglycan hydrolysis (Lee & Yang, 2002). However, in contrast to lysozyme from chicken egg white, AF containing NbPR3 could not lyse the bacteria (Fig. 4e), indicating that NbPR3 does not have lysozyme activity.
Nicotiana‐specific Q112 is required for antibacterial activity
We next compared the protein sequence of NbPR3 with chitinase A (CHN‐A) from Nicotiana tabacum, the closest related sequence with documented endochitinase activity (Suarez et al., 2001). Eight residues have been identified in CHN‐A as being important for endochitinase activity (Garcia‐Casado et al., 1998; Tang et al., 2004; Chaudet et al., 2014; Han et al., 2016), and only two of these eight residues are different in NbPR3 (Figs 5a, S5). Notably, whereas the predicted catalytic general acid Glu residue is present in both sequences (NbPR3E92 and CHN‐AE145), the predicted catalytic general base is absent from NbPR3 (NbPR3Q112 vs CHN‐AE167). Furthermore, Ser residue S198 in CHN‐A is a Thr residue in NbPR3 (T128), though this substitution is common in the plant kingdom. These sequence polymorphisms indicate that NbPR3 may have a catalytic activity that is different from CHN‐A, but is still likely to bind carbohydrates because the other residues relevant for chitinase activity are conserved. Indeed, structural modelling indicates that NbPR3 may have a similar fold as chitinase‐I (Kezuka et al., 2010), but the region carrying Q112 is different from chitinases (Fig. 5b).
Fig. 5.
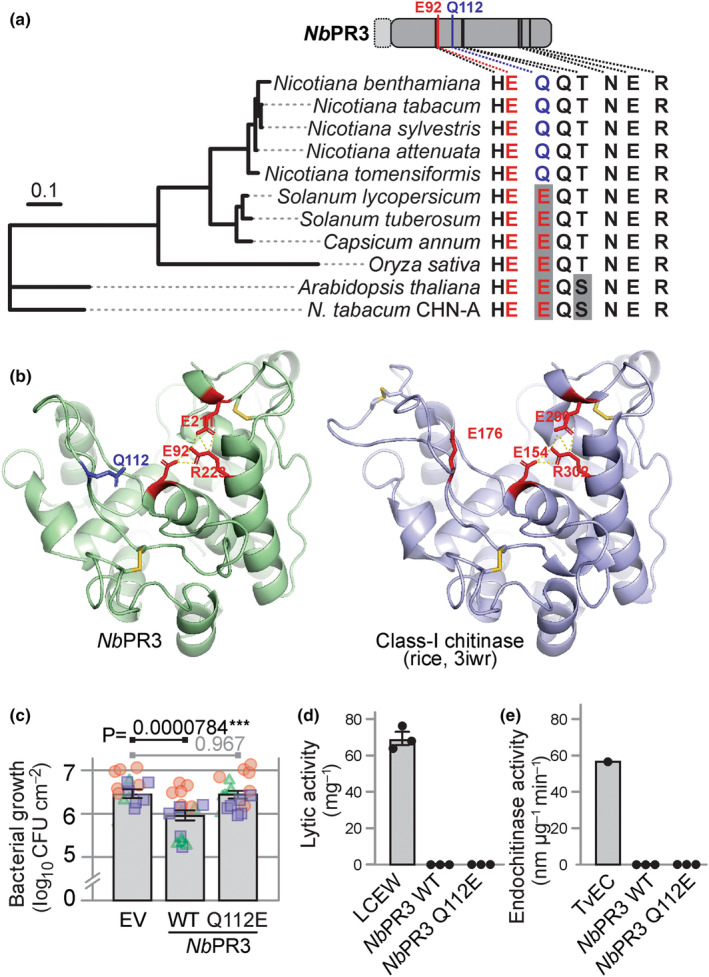
Nicotiana‐specific active site Q112 in NbPR3 is essential for antibacterial activity. (a) Conservation of chitinase‐relevant residues in NbPR3 homologs. The second catalytic glutamate of chitinases (red) is replaced by glutamine (blue) in all NbPR3 orthologs of Nicotiana. NbPR3 orthologs were identified by Blast searches, aligned by ClustalO (Supporting Information Fig. S6), and used to construct a maximum likelihood phylogenetic tree. Chitinase A (CHN‐A) from Nicotiana tabacum was included as the closest related enzyme for which chitinase activity has been demonstrated. The residues relevant for chitinase activity are summarised on the right, showing that the Gln is conserved in Nicotiana and is specific for this genus. (b) Model of NbPR3 structure, compared with rice class‐I chitinase. A model of NbPR3 was generated with SWISS Model, using protein data bank 3iwr (Kezuka et al., 2010) as a template. Catalytically important residues (red), and the noncanonical Q112 (blue), and three cysteine bridges (yellow) are indicated. (c) The Nicotiana‐specific Q112 is essential for antibacterial activity. NbPR3, its Q112E substitution mutant, and the empty vector (EV) control were transiently expressed in Nicotiana benthamiana by agroinfiltration, and 2 d later, the same leaf was infiltrated with 106 CFU ml−1 PtoDC3000(ΔhQ). Bacterial population sizes 3 d later are shown in log10CFU cm−2. Bars show the mean value of 18 biological replicates performed over three separate experiments, and error intervals represent the SE. P‐values were calculated by two‐way ANOVA followed by post hoc comparison using the Dunnett test to examine the effect of NbPR3 expression on bacterial growth. ***, P < 0.001. (d) The Q112E substitution in NbPR3 does not increase lytic activity. Apoplastic fluid (AF) from plants transiently expressing NbPR3 or its Q112E mutant was incubated with Micrococcus lysodeikticus cells. The change in A450 was measured and converted to units μg−1 NbPR3 protein estimated by Coomassie staining. The lysozyme of chicken egg white (LCEW) was included as a positive control. (e) The Q112E substitution in NbPR3 does not increase endochitinase. AF from plants transiently expressing NbPR3 or its Q112E mutant was incubated with 4‐MU‐GlcNac3, and the rate of hydrolysis was measured at 355ex/450em and calculated per μg NbPR3 protein estimated by Coomassie staining. The endochitinase of Trichoderma viride (TvEC) was included as a positive control.
Interestingly, Q112 is present in all putative NbPR3 orthologs of the Nicotiana genus but is absent in paralogs and in closely related Solanum species or Arabidopsis and rice (Oryza sativa; Figs 5a, S6), suggesting that this active site substitution occurred in the Nicotiana clade. To test the relevance of Q112 for antibacterial activity of NbPR3, we generated the Q112E mutant and tested its ability to suppress the growth of PtoDC3000(ΔhQ) in the agromonas assay. Importantly, while NbPR3 suppresses bacterial growth, the Q112E substitution abolishes this activity (Fig. 5c), demonstrating that the Nicotiana‐specific Q112 is relevant for antibacterial activity. The Q112E mutant of NbPR3 does, however, not gain lytic or endochitinase activity (Fig. 5d), indicating that also other residues contribute to these activities.
NbPR3 promotes immunity without inducing PR protein accumulation
Previous work on PR‐Q, the tobacco ortholog of NbPR3, has revealed that transgenic tobacco overexpressing PR‐Q also constitutively accumulate PR proteins (Tang et al., 2017). To investigate whether NbPR3 expression also induces PR protein accumulation, we monitored the accumulation of PR2, an abundant secreted PR protein in N. benthamiana, upon agroinfiltration with empty vector (EV) and NbPR3 and its E92A and Q122E mutant derivatives. The NahG transgenic N. benthamiana (Wulff et al., 2004) was included to determine whether PR protein accumulation was dependent on salicylic acid (SA), which cannot accumulate in NahG transgenic plants. Notably, NbPR3 does not induce PR2 levels and the E92A and Q122E mutants of NbPR3 have similar levels of PR2 when compared to EV and NbPR3 (Fig. 6a). In fact, expression of NbPR3 and its mutants even reduces PR2 levels (Fig. 6b), possibly caused by competition on translation and secretion. PR2 levels upon agroinfiltration are much reduced in NahG transgenic plants when compared to WT plants and again not increased upon NbPR3 expression (Fig. 6). NbPR3 signals are two–threefold higher in the apoplast of plants transiently expressing NbPR3 and derived mutants (Fig. 6b). Reduced expression of NbPR3 is detected upon agroinfiltration of NahG plants, presumably because there is no SA‐regulated expression of endogenous NbPR3, and because the 35S promoter used to express NbPR3 contains a SA‐responsive as‐1 element (Redman et al., 2002). In conclusion, unlike PR‐Q overexpression in tobacco, NbPR3 overexpression by agroinfiltration does not induce PR protein accumulation.
Fig. 6.
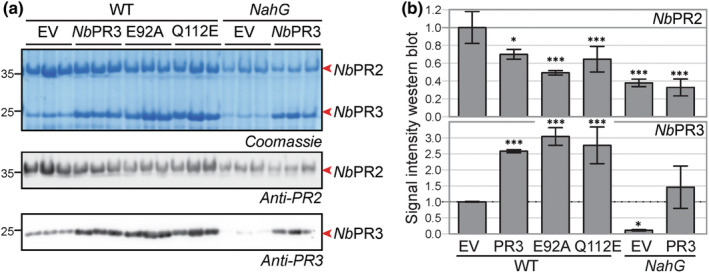
NbPR3 does not promote PR2 protein accumulation. (a) NbPR3 and its mutant derivatives were transiently expressed in WT and NahG transgenic Nicotiana benthamiana in n = 3 independent plants. Apoplastic fluids were isolated 4 d later and analysed by Coomassie staining and western blotting using anti‐PR2 and anti‐PR3 antibodies. (b) Western blot signals were quantified and normalised for the empty vector (EV) control in WT plants. Error bars represent SD of the n = 3 replicates. *, P < 0.5; ***, P < 0.001. P‐values are from a one‐way ANOVA with Dunnett correction for multiple comparisons.
To investigate whether NbPR3 has a direct effect on bacterial growth, we incubated PtoDC3000 bacteria in AF isolated from plants expressing EV, NbPR3 and its E92A mutant and monitored bacterial growth over time. Lysozyme mixed with AF of plants agroinfiltrated with EV was included as control. Despite being abundant in AF, NbPR3 did not impact bacterial growth when compared to the EV and E92A controls, whereas lysozyme significantly reduces bacterial growth (Fig. S7), indicating that NbPR3‐derived immunity is not direct.
Discussion
Most SP‐containing proteins secreted into the apoplast are hydrolytic enzymes. Here, we explored the apoplast of a model plant–pathogen interaction with activity‐based probes and were able to monitor 171 active hydrolases, of which 82 showed increased activity, and 60 had reduced activity upon infection. We have summarised the suppressed hydrolases in Table S7. Disease assays on leaves transiently overexpressing five selected suppressed hydrolases revealed that NbPR3 has antibacterial immunity that requires its active site residue. Despite its annotation as chitinase, NbPR3 has no chitinase or lysozyme activity, presumably because of a E112Q substitution in the active site. This substitution is conserved in Nicotiana and is essential for antibacterial activity.
As pathogenic bacteria have adapted to a susceptible host, we anticipate that they will actively suppress secreted hydrolases that may directly or indirectly harm them. Therefore, consistently suppressed hydrolases might be components of the plant immune system that are targeted by pathogen‐derived inhibitors. We identified 60 suppressed hydrolases that may play an active role in immunity. Massive changes in protein activities can be caused by altered pH or ion strength, but the influence of these factors was prevented by buffering the AF. Many of the suppressed hydrolases are Cys proteases, which might be suppressed by increased levels of reactive oxygen species (ROS). However, not all Cys proteases are suppressed, indicating that the suppression is caused by selective inhibition rather than ROS. Indeed, we previously showed that PtoDC3000(ΔhQ) produces Cip1, which is selectively targeting immune‐related Cys proteases of tomato (Shindo et al., 2016). Besides Cys proteases, several glycosidases are also suppressed. This includes BGAL1, which is inhibited by a small molecule produced by PtoDC3000(ΔhQ) (Buscaill et al., 2019). However, given the large proportion of changes, we cannot rule out a general apoplastic regulator (i.e. metabolites, specific ions and post‐translational modifications) that might influence global protein activities.
Secreted hydrolases that are suppressed during infection can be important novel components of the extracellular immune system of plants. To test this hypothesis, we overexpressed the hydrolases to overcome the threshold of suppression during infection. Indeed, we found that transient overexpression of NbPR3 results in increased immunity to PtoDC3000(ΔhQ), confirming that supressed hydrolases may act in immunity. Likewise, we found that transient overexpression of BGAL1 increases bacterial resistance (Buscaill et al., 2021). However, not all suppressed hydrolases reduced bacterial growth upon overexpression in our agromonas assay. This might be because their overexpression does not overcome the suppression mechanism or because they act in immunity without reducing bacterial growth in the agromonas assay.
The mechanism by which NbPR3 mediates antibacterial immunity, however, remains mysterious. Overexpression of PR‐Q in tobacco upregulates multiple defence‐related genes, an observation which was thought to originate from an unfolded protein response (UPR) caused by the presumed accumulation of unfolded proteins in the ER (Tang et al., 2017). We, however, found that the E92A and Q112E mutants of NbPR3 accumulate equally well but do not trigger immunity, ruling out the UPR being responsible for the observed resistance. That the catalytically important glutamate E92 is essential for the role of NbPR3 in our agromonas assays, implies that catalysis by NbPR3 is required for immunity. However, despite its annotation as an endochitinase, NbPR3 lacks endochitinase activity, at least in our assays. The absence of bacteriolytic activity indicates that NbPR3 does also not hydrolyse peptidoglycan, unlike Arabidopsis LYS1, a secreted GH18 lysozyme that releases immunogenic fragments from peptidoglycan (Liu et al., 2014).
The absence of chitinase and lysozyme activities of NbPR3 is probably at least in part caused by the absence of the second catalytic glutamate (E), which is important for chitinase activity (Tang et al., 2004) but is a glutamine (Q112) in NbPR3. Interestingly, Q112 is conserved in all Nicotiana NbPR3 orthologs, including tobacco PR‐Q (Payne et al., 1990), but is absent in other species. The closest PR3 family members from other angiosperm plants, including rice and Arabidopsis, all have a catalytic glutamate (E) at this position, which indicates that the E112Q substitution occurred in a Nicotiana ancestor within the nightshade family c. 50 million years ago and was maintained upon speciation. Importantly, we showed that Q112 is essential for antibacterial activity, which implies that an antifungal chitinase in the Nicotiana ancestor was neo‐functionalised to gain antibacterial activity. This neo‐functionalisation must have involved additional substitutions because we found that the Q112E mutant of NbPR3 is insufficient to restore chitinase activity. The fact that the catalytic E92 is still essential for antibacterial activity and that other residues relevant for chitin binding are still present in NbPR3 and its orthologs indicates that NbPR3 might act on a bacterial glycan.
We discovered that the level of active NbPR3 is consistently downregulated upon infection with virulent PtoDC3000(ΔhQ) (Fig. 3), even though the level of the NbPR3 protein increases upon infection (Fig. S8), consistent with being a PR protein. This observation indicates that apoplastic NbPR3 is inhibited during infection by a small molecule or protein secreted by PtoDC3000(ΔhQ). The active suppression of an antibacterial enzyme is similar to the suppression of BGAL1 by a small molecule inhibitor produced by PtoDC3000(ΔhQ) (Buscaill et al., 2019) and indicates that exploring the apoplastic battlefield with activity‐based proteomics to identify these suppressed hydrolases is an exciting new approach to discover novel components of extracellular immunity in plants.
Competing interests
None declared.
Author contributions
DJS, AG and RALvdH planned and designed the research; DJS, AG and PB performed experiments; FK and MK performed proteomic analysis; DK, TK, CJS and MK synthesised probes; DJS and RALvdH wrote the manuscript with help of all authors. DJS and AG contributed equally to this work.
Supporting information
Fig. S1 Structure of additional activity‐based probes.
Fig. S2 Replicate activity‐based proteomics experiment.
Fig. S3 Transient hydrolase expression and agromonas assays.
Fig. S4 Replicates of infection assays show increased resistance upon NbPR3 expression.
Fig. S5 Protein sequence alignment of NbPR3 and endochitinase CHN‐A.
Fig. S6 Protein alignment of putative NbPR3 orthologs from diverse plant species.
Fig. S7 NbPR3 does not impact bacterial growth in vitro.
Fig. S8 NbPR3 accumulates in the apoplast upon infection.
Table S1 Used oligonucleotides.
Table S2 Used plasmids.
Table S3 In‐solution digest (ACE_0276).
Table S4 Protein concentrations in apoplastic fluid.
Table S5 On‐bead digest (ACE_0236).
Table S6 On‐bead digest (ACE_0136).
Table S7 Summary of suppressed hydrolases.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Acknowledgements
We thank Ursula Pyzio for excellent plant care; Sarah Rodgers and Caroline O'Brian for technical assistance; and Friederike Grosse‐Holz, Jiorgos Kourelis and Mariana Schuster for constructive discussions. We thank Sylvestre Marillonnet and Nicola Patron for providing pICH41414 and pICH51288 via Addgene; Jonathan Jones for providing seeds of NahG transgenic N. benthamiana; and Hermen Overkleeft for providing JJB111 until 2019. This project was financially supported by the European Research Council grant (ERC‐AdG‐2020) 101019324 ‘ExtraImmune’; the BBSRC research grant ‘GH35’ (BB/R017913/1); and the Interdisciplinary Doctoral Training Program (DTC) of the BBSRC (DDT00060).
Data availability
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (www.ebi.ac.uk/pride/, Perez‐Riverol et al., 2022) under accessions PXD039897 (in‐solution digest ACE0276) and PXD034869 (on‐bead digests ACE0136 and ACE0236).
References
- Almagro Armenteros JJ, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, von Heijne G, Nielsen H. 2019. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology 37: 420–423. [DOI] [PubMed] [Google Scholar]
- Benns HJ, Wincott CJ, Tate EW, Child MA. 2021. Activity‐ and reactivity‐based proteomics: recent technological advances and applications in drug discovery. Current Opinion in Chemical Biology 60: 20–29. [DOI] [PubMed] [Google Scholar]
- Buscaill P, Chandrasekar B, Sanguankiattichai N, Kourelis J, Kaschani F, Thomas EL, Morimoto K, Kaiser M, Preston GM, Ichinose Y et al. 2019. Glycosidase and glycan polymorphism control hydrolytic release of immunogenic flagellin peptides. Science 364: 145. [DOI] [PubMed] [Google Scholar]
- Buscaill P, Sanguankiattichai N, Lee YJ, Kourelis J, Preston G, van der Hoorn RAL. 2021. Agromonas: a rapid disease assay for Pseudomonas syringae growth in agroinfiltrated leaves. The Plant Journal 105: 831–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekar B, Colby T, Emon AEK, Jiang J, Hong TN, Villamor JG, Harzen A, Overkleeft HS, Van der Hoorn RAL. 2014. Broad range glycosidase activity profiling. Molecular & Cellular Proteomics 13: 2787–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudet MM, Naumann TA, Price NP, Rose DR. 2014. Crystallographic structure of ChitA, a glycoside hydrolase family 19, plant class IV chitinase from Zea mays . Protein Science 23: 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Hein MY, Luber CA, Paron I, Nagaraj N, Mann M. 2014. Accurate proteome‐wide label‐free quantification by delayed normalization and maximal peptide ratio extraction, termed MaxLFQ. Molecular & Cellular Proteomics 13: 2513–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox J, Neuhauser N, Michalski A, Scheltema RA, Olsen JV, Mann M. 2011. Andromeda: a peptide search engine integrated into the MaxQuant environment. Journal of Proteome Research 10: 1794–1805. [DOI] [PubMed] [Google Scholar]
- Cravatt BF, Wright AT, Kozarich JW. 2008. Activity‐based protein profiling: from enzyme chemistry to proteomic chemistry. Annual Review in Biochemistry 77: 383–414. [DOI] [PubMed] [Google Scholar]
- Doehlemann G, Hemetsberger C. 2013. Apoplastic immunity and its suppression by filamentous plant pathogens. New Phytologist 198: 1001–1016. [DOI] [PubMed] [Google Scholar]
- Dolle RE, Hoyer D, Prasad CV, Schmidt SJ, Helaszek CT, Miller RE, Ator MA. 1994. P1 aspartate‐based peptide alpha‐((2,6‐dichlorobenzoyl)oxy)methyl ketones as potent time‐dependent inhibitors of interleukin‐1 beta‐converting enzyme. Journal of Medicinal Chemistry 37: 563–564. [DOI] [PubMed] [Google Scholar]
- El‐Gebal S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A et al. 2019. The Pfam protein families database in 2019. Nucleic Acids Research 47: D427–D423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler C, Youles M, Gruetzner R, Ehnert TM, Werner S, Jones JD, Patron NJ, Marillonnet S. 2014. A golden gate modular cloning toolbox for plants. ACS Synthetic Biology 3: 839–843. [DOI] [PubMed] [Google Scholar]
- van Esse HP, van't Klooster JW, Bolton MD, Yadeta KA, van Baarlen P, Boeren S, Vervoort J, de Wit PJGM, Thomma BPHJ. 2008. The Cladosporium fulvum virulence protein Avr2 inhibits host proteases required for basal defense. Plant Cell 20: 1948–1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia‐Casado G, Collada C, Allona I, Casado R, Pacios LF, Aragoncillo C, Gomez L. 1998. Site‐directed mutagenesis of active site residues in a class I endochitinase from chestnut seeds. Glycobiology 8: 1021–1028. [DOI] [PubMed] [Google Scholar]
- Greenbaum D, Medzihradszky KF, Burlingame A, Bogyo M. 2000. Epoxide electrophiles as activity‐dependent cysteine protease profiling and discovery tools. Chemistry & Biology 7: 569–581. [DOI] [PubMed] [Google Scholar]
- Han B, Zhou K, Li Z, Sun B, Ni Q, Meng X, Pan G, Li C, Long M, Li T et al. 2016. Characterization of the first fungal glycosyl hydrolase family 19 chitinase (NbchiA) from Nosema bombycis (Nb). Journal of Eukaryotic Microbiology 63: 37–45. [DOI] [PubMed] [Google Scholar]
- Hemetsberger C, Herrberger C, Zechmann B, Hillmer M, Doehlemann G. 2012. The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathogens 8: e1002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollis T, Honda Y, Fukamizo T, Marcotte E, Day PJ, Robertus JD. 1997. Kinetic analysis of barley chitinase. Archives of Biochemistry & Biophysics 344: 335–342. [DOI] [PubMed] [Google Scholar]
- Hong TN, Van der Hoorns RAL. 2014. DIGE‐ABPP by click chemistry: pairwise comparison of serine hydrolase activities from the apoplast of infected plants. Methods in Molecular Biology 1127: 183–194. [DOI] [PubMed] [Google Scholar]
- Joosten MHAJ. 2012. Isolation of apoplastic fluid from leaf tissue by the vacuum infiltration‐centrifugation technique. Plant Fungal Pathogens: Methods and Protocols 835: 603–610. [DOI] [PubMed] [Google Scholar]
- Kaschani F, Gu C, Niessen S, Hoover H, Cravatt BF, Van der Hoorn RAL. 2009. Diversity of serine hydrolase activities of non‐challenged and Botrytis‐infected Arabidopsis thaliana . Molecular & Cellular Proteomics. 8: 1082–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kezuka Y, Kojima M, Mizuno R, Suzuki K, Watanabe T, Nonaka T. 2010. Structure of full‐length class I chitinase from rice revealed by X‐ray crystallography and small‐angle X‐ray scattering. Proteins 78: 2295–2305. [DOI] [PubMed] [Google Scholar]
- Kourelis J, Kaschani F, Grosse‐Holz FM, Homma F, Kaiser M, Van der Hoorn RAL. 2019. A homology‐guided, genome‐based proteome for improved proteomics in the alloploid Nicotiana benthamiana . BMC Genomics 20: 722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YC, Yang D. 2002. Determination of lysozyme activities in a microplate format. Analytical Biochemistry 310: 223–224. [DOI] [PubMed] [Google Scholar]
- Libantová J, Kämäräinen T, Moravcíková J, Matusíková I, Salaj J. 2009. Detection of chitinolytic enzymes with different substrate specificity in tissues of intact sundew (Drosera rotundifolia L.): chitinases in sundew tissues. Molecular Biology Reporter 36: 851–856. [DOI] [PubMed] [Google Scholar]
- Liu X, Grabherr HM, Willmann R, Kolb D, Brunner F, Bertsche U, Kühner D, Franz‐Wachtel M, Amin B, Felix G et al. 2014. Host‐induced bacterial cell wall decomposition mediates pattern‐triggered immunity in Arabidopsis. eLife 3: e01990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Patricelli MP, Cravatt BF. 1999. Activity‐based protein profiling: the serine hydrolases. Proceedings of the National Academy of Sciences, USA 96: 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Loon LC, Rep M, Pieterse CM. 2006. Significance of inducible defense‐related proteins in infected plants. Annual Review in Phytopathology 44: 135–162. [DOI] [PubMed] [Google Scholar]
- Morimoto K, van der Hoorn RAL. 2016. The increasing impact of activity‐based protein profiling in plant science. Plant Cell Physiology 57: 446–461. [DOI] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. 2005. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C‐trap. Molecular & Cellular Proteomics 4: 2010–2021. [DOI] [PubMed] [Google Scholar]
- Paulus JK, Kourelis J, Ramasubramanian S, Homma F, Godson A, Hörger AC, Hong TN, Krahn D, Ossorio Carballo L, Wang S et al. 2020. Extracellular proteolytic cascade in tomato activates immune protease Rcr3. Proceedings of the National Academy of Sciences, USA 117: 17409–17417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne G, Ahl P, Moyer M, Harper A, Beck J, Meins F Jr, Ryals J. 1990. Isolation of complementary DNA clones encoding pathogenesis‐related proteins P and Q, two acidic chitinases from tobacco. Proceedings of the National Academy of Sciences USA 87: 98–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez‐Riverol Y, Bai J, Bandla C, García‐Seisdedos D, Hewapathirana S, Kamatchinathan S, Kundu DJ, Prakash A, Frericks‐Zipper A, Eisenacher M et al. 2022. The PRIDE database resources in 2022: a hub for mass spectrometry‐based proteomics evidences. Nucleic Acids Research 50: D543–D552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planas‐Marquès M, Bernardo‐Faura M, Paulus J, Kaschani F, Kaiser M, Valls M, van der Hoorn RAL, Coll NS. 2018. Protease activities triggered by Ralstonia solanacearum infection in susceptible and tolerant tomato lines. Molecular & Cellular Proteomics 17: 1112–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman J, Whitcraft J, Johnson C, Arias J. 2002. Abiotic and biotic stress differentially stimulate as‐1 element activity in Arabidopsis. Plant Cell Reports 21: 180–185. [Google Scholar]
- Rooney HC, Van't Klooster JW, van der Hoorn RAL, Joosten MHAJ, Jones JDG, de Wit PJGM. 2005. Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf‐2‐dependent disease resistance. Science 308: 1783–1786. [DOI] [PubMed] [Google Scholar]
- Rose JK, Ham KS, Darvill AG, Albersheim P. 2002. Molecular cloning and characterization of glucanase inhibitor proteins: coevolution of a counterdefense mechanism by plant pathogens. Plant Cell 14: 1329–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultink A, Qi T, Lee A, Steinbrenner AD, Staskawicz B. 2007. Roq1 mediates recognition of the Xanthomonas and Pseudomonas effector proteins XopQ and HopQ1. The Plant Journal 92: 787–795. [DOI] [PubMed] [Google Scholar]
- Shabab M, Shindo T, Gu C, Kaschani F, Pansuriya T, Chintha R, Harzen A, Colby T, Kamoun S, van der Hoorn RAL. 2008. Fungal effector protein AVR2 targets diversifying defense‐related Cys proteases of tomato. Plant Cell 20: 1169–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo T, Kaschani F, Yang F, Kovacs J, Tian F, Kourelis J, Hong TN, Colby T, Shabab M, Chawla R et al. 2016. Screen of non‐annotated small secreted proteins of Pseudomonas syringae reveals a virulence factor that inhibits tomato immune proteases. PLoS Pathogens 12: e1005874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez V, Staehelin C, Arango R, Holtorf H, Hofsteenge J, Meins F Jr. 2001. Substrate specificity and antifungal activity of recombinant tobacco class I chitinases. Plant Molecular Biology 45: 609–618. [DOI] [PubMed] [Google Scholar]
- Sueldo D, Ahmed A, Misas‐Villamil J, Colby T, Tameling W, Joosten MHAJ, van der Hoorn RAL. 2014. Dynamic hydrolase activities precede hypersensitive tissue collapse in tomato seedlings. New Phytologist 203: 913–925. [DOI] [PubMed] [Google Scholar]
- Tang CM, Chye ML, Ramalingam S, Ouyang SW, Zhao KJ, Ubhayasekera W, Mowbray SL. 2004. Functional analyses of the chitin‐binding domains and the catalytic domain of Brassica juncea chitinase BjCHI1. Plant Molecular Biology 56: 285–298. [DOI] [PubMed] [Google Scholar]
- Tang Y, Liu Q, Liu Y, Zhang L, Ding W. 2017. Overexpression of NtPR‐Q up‐regulates multiple defense‐related genes in Nicotiana tabacum and enhances plant resistance to Ralstonia solanacearum . Frontiers in Plant Science 8: 1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MY, Benedetti B, Kamoun S. 2005. A second kazal‐like protease inhibitor from Phytophthora infestans inhibits and interacts with the apoplastic pathogenesis‐related protease P69B of tomato. Plant Physiology 138: 1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MY, Huitema E, da Cunha L, Torto‐Alalibo T, Kamoun S. 2004. A Kazal‐like extracellular serine protease inhibitor from Phytophthora infestans targets the tomato pathogenesis‐related protease P69B. Journal of Biological Chemistry 279: 26370–26377. [DOI] [PubMed] [Google Scholar]
- Tian MY, Kamoun S. 2005. A two disulfide bridge Kazal domain from Phytophthora exhibits stable inhibitory activity against serine proteases of the subtilisin family. BMC Biochemistry 6: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian MY, Win J, Song J, van der Hoorn R, van der Knaap E, Kamoun S. 2007. A Phytophthora infestans cystatin‐like protein targets a novel tomato papain‐like apoplastic protease. Plant Physiology 143: 364–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein M, Geiger T, Mann CJ. 2016. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nature Methods 13: 731–740. [DOI] [PubMed] [Google Scholar]
- Van der Hoorn RAL, Rivas S, Wulff BB, Jones JDG, Joosten MHAJ. 2003. Rapid migration in gel filtration of the Cf‐4 and Cf‐9 resistance proteins is an intrinsic property of Cf proteins and not because of their association with high‐molecular‐weight proteins. The Plant Journal 35: 305–315. [DOI] [PubMed] [Google Scholar]
- Wang Y, Wang Y, Wang Y. 2020. Apoplastic proteases: powerful weapons against pathogen infection in plants. Plant Communications 12: 100085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei CF, Kvitko BH, Shimizu R, Crabill E, Alfano JR, Lin NC, Martin GB, Huang HC, Collmer A. 2007. A Pseudomonas syringae pv tomato DC3000 mutant lacking the type III effector HopQ1‐1 is able to cause disease in the model plant Nicotiana benthamiana . The Plant Journal 51: 32–46. [DOI] [PubMed] [Google Scholar]
- Wessel D, Flügge UI. 1984. A method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Analytical Biochemistry 138: 141–143. [DOI] [PubMed] [Google Scholar]
- Wulff BBH, Kruijt M, Collins PL, Thomas CM, Ludwig AA, De Wit PJGM, Jones JDG. 2004. Gene shuffling‐generated and natural variants of the tomato resistance gene Cf‐9 exhibit different auto‐necrosis‐inducing activities in Nicotiana species. The Plant Journal 40: 942–956. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Structure of additional activity‐based probes.
Fig. S2 Replicate activity‐based proteomics experiment.
Fig. S3 Transient hydrolase expression and agromonas assays.
Fig. S4 Replicates of infection assays show increased resistance upon NbPR3 expression.
Fig. S5 Protein sequence alignment of NbPR3 and endochitinase CHN‐A.
Fig. S6 Protein alignment of putative NbPR3 orthologs from diverse plant species.
Fig. S7 NbPR3 does not impact bacterial growth in vitro.
Fig. S8 NbPR3 accumulates in the apoplast upon infection.
Table S1 Used oligonucleotides.
Table S2 Used plasmids.
Table S3 In‐solution digest (ACE_0276).
Table S4 Protein concentrations in apoplastic fluid.
Table S5 On‐bead digest (ACE_0236).
Table S6 On‐bead digest (ACE_0136).
Table S7 Summary of suppressed hydrolases.
Please note: Wiley is not responsible for the content or functionality of any Supporting Information supplied by the authors. Any queries (other than missing material) should be directed to the New Phytologist Central Office.
Data Availability Statement
The proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository (www.ebi.ac.uk/pride/, Perez‐Riverol et al., 2022) under accessions PXD039897 (in‐solution digest ACE0276) and PXD034869 (on‐bead digests ACE0136 and ACE0236).