

Abstract
Objective
Over 100 DNA variants have been associated with osteoarthritis (OA), including rs1046934, located within a linkage disequilibrium block encompassing part of COLGALT2 and TSEN15. The present study was undertaken to determine the target gene(s) and the mechanism of action of the OA locus using human fetal cartilage, cartilage from OA and femoral neck fracture arthroplasty patients, and a chondrocyte cell model.
Methods
Genotyping and methylation array data of DNA from human OA cartilage samples (n = 87) were used to determine whether the rs1046934 genotype is associated with differential DNA methylation at proximal CpGs. Results were replicated in DNA from human arthroplasty (n = 132) and fetal (n = 77) cartilage samples using pyrosequencing. Allelic expression imbalance (AEI) measured the effects of genotype on COLGALT2 and TSEN15 expression. Reporter gene assays and epigenetic editing determined the functional role of regions harboring differentially methylated CpGs. In silico analyses complemented these experiments.
Results
Three differentially methylated CpGs residing within regulatory regions were detected in the human OA cartilage array data, and 2 of these were replicated in human arthroplasty and fetal cartilage. AEI was detected for COLGALT2 and TSEN15, with associations between expression and methylation for COLGALT2. Reporter gene assays confirmed that the CpGs are in chondrocyte enhancers, with epigenetic editing results directly linking methylation with COLGALT2 expression.
Conclusion
COLGALT2 is a target of this OA locus. We previously characterized another OA locus, marked by rs11583641, that independently targets COLGALT2. The genotype of rs1046934, like rs11583641, mediates its effect by modulating expression of COLGALT2 via methylation changes to CpGs located in enhancers. Although the single‐nucleotide polymorphisms, CpGs, and enhancers are distinct between the 2 independent OA risk loci, their effect on COLGALT2 is the same. COLGALT2 is the target of independent OA risk loci sharing a common mechanism of action.
INTRODUCTION
Genome‐wide association studies (GWAS) have identified over 100 DNA variants that are associated with osteoarthritis (OA) risk (1, 2, 3, 4). Biologic comprehension of GWAS signals requires elucidation of the molecular effects of the risk‐conferring alleles on their target genes (5, 6, 7, 8). Since the individual contribution of most variants to disease risk is small, assessing these effects is challenging (5, 6, 7, 8). Furthermore, determining the causal variants underpinning an association signal is not straightforward, as variants commonly occur within linkage disequilibrium (LD) blocks (5, 6, 7, 8). Despite these difficulties, the application of statistical fine mapping combined with laboratory‐based studies is generating functional insight into the molecular basis of OA genetic risk (3, 4, 9, 10, 11, 12, 13, 14, 15).
As with other polygenic diseases, most OA‐associated variants reside within the noncoding genome and contribute to disease by altering expression of genes within the same topologically associated domain (TAD), thereby acting as expression quantitative trait loci (eQTLs) (1). We have reported that DNA methylation (DNAm) at CpG dinucleotides is also often associated with genotype at OA‐associated variants, forming methylation QTLs (mQTLs), and that this epigenetic effect may act as an intermediate between the risk allele and the change in gene expression (16, 17, 18, 19, 20, 21).
One recent example was our investigation of the OA association signal marked by single‐nucleotide polymorphism (SNP) rs11583641 (22). This variant resides within the 3′‐untranslated region of COLGALT2, which encodes a galactosyltransferase that posttranslationally modifies collagen (22). We discovered that the OA risk allele of rs11583641 is associated with lower methylation levels of CpGs within an intronic enhancer of COLGALT2 and that this reduced methylation increases enhancer activity and COLGALT2 expression (22). Increased glycosylation of collagen reduces intermolecule crosslinking, leading to collagen fibrils with reduced diameters and lower tensile strength (23). We concluded that increased COLGALT2 expression, and therefore increased galactosyltransferase activity, could be detrimental to cartilage health via effects on collagen biosynthesis (22). We subsequently reported that for some OA risk loci, including rs11583641, genotype associations with gene expression and CpG methylation observed in human arthroplasty cartilage are also observed in human fetal cartilage (24). This implies that OA genetic risk may be programmed during development.
A second association signal was recently reported that maps close to COLGALT2 (4). Three SNPs were highlighted: rs12047271 and rs1327123, residing between COLGALT2 and TSEN15, and rs1046934, residing within TSEN15. The TSEN15 protein is a subunit of a transfer RNA (tRNA)–splicing endonuclease. The splicing of introns from pre‐tRNAs is performed by a heterotetrameric endonuclease composed of TSEN15, TSEN34, TSEN2, and TSEN54 (25, 26). TSEN15 and TSEN34 are the structural subunits of the endonuclease, whereas TSEN2 and TSEN54 form the catalytic domains (26). TSEN15 adopts a compact α‐α‐β‐β‐β‐β‐α‐β‐β fold, preceded by a disordered N‐terminal region, which has not been structurally resolved (25, 26).
The 3 SNPs rs12047271, rs1327123, and rs1046934 are part of an LD block containing 21 SNPs (r2 ≥ 0.8 in European ancestry cohorts) spanning 30 kb. Furthermore, they are in near perfect linkage equilibrium with rs11583641 (r2 = 0, D′ ≤0.08). This second COLGALT2 signal, which we henceforth refer to as the rs1046934 locus, is therefore genetically independent of the first COLGALT2 signal. Here, we set out to investigate the gene targets of this new OA locus using a range of techniques.
PATIENTS AND METHODS
Protein modeling
TSEN15 crystal structures were downloaded from the Protein Data Bank (Supplementary Table 1, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) and visualized in complex with TSEN34 (6Z9U) and as a monomeric structure (2GW6) using the PyMOL Molecular Graphics System (Schrödinger). The PyMOL Mutagenesis Wizard was used to perform in silico mutagenesis to model the missense variant Gln59‐His introduced by rs1046934. We used the tools gnomAD (27), PolyPhen, and Mutation Taster (Supplementary Table 1) to predict the effects of this variant and of Gly19‐Asp, introduced by rs2274432, on TSEN15 function.
Cartilage samples and ethics approval
Cartilage samples were obtained from 132 patients undergoing arthroplasty at the Newcastle upon Tyne NHS Foundation Trust hospitals for primary hip OA (n = 43), primary knee OA (n = 63), or femoral neck fracture (n = 26) (Supplementary Table 2, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). Ethics approval was granted by the NHS Health Research Authority, with donors providing written consent (19/LO/0389). Nucleic acids were extracted as previously described (20, 21, 22). Seventy‐seven matched fetal DNA and RNA samples (Supplementary Table 3, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) were provided by the Human Developmental Biology Resource (project 200363) (24). Nucleic acids were extracted as previously described (24).
Genotyping
Allelic quantification pyrosequencing assays were designed using PyroMark Assay Design (Qiagen) with oligonucleotide primers ordered from Integrated DNA Technologies (IDT). DNA encompassing the SNP of interest underwent polymerase chain reaction (PCR) amplification using the PyroMark PCR kit (Qiagen), with the genotype determined using the PyroMark Q24 Advanced System (Qiagen). Supplementary Table 4, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427, lists the oligonucleotide sequences.
Allelic expression imbalance (AEI)
Proxy transcript SNPs (Supplementary Table 5, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) were used to investigate rs114661926 AEI for COLGALT2 (r2 = 0.79 with rs1046934) and rs2274432 AEI for TSEN15 (r2 = 1 with rs1046934). Patients' compound heterozygote at rs1046934 and the respective transcript SNP were investigated. Complementary DNA (cDNA) was reverse transcribed from 500 ng RNA using SuperScript IV (Invitrogen). The relative ratio of the risk to nonrisk allele at the SNPs was quantified by pyrosequencing in DNA and cDNA, as previously described (17, 20, 22). Oligonucleotides were obtained from IDT (Supplementary Table 4, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). Triplicate measures were performed and excluded if the difference was >5%. Allelic expression ratio in cDNA was normalized to allelic expression ratio in DNA for each patient.
Discovery of mQTLs
Genotype and methylation data previously generated from the cartilage DNA of 87 hip or knee arthroplasty OA patients (28) were used. We tested CpGs 200 kb upstream and 200 kb downstream of rs1046934, encompassing the TAD for the association signal.
Replication of mQTLs
CpGs with nominal P < 0.05 in the mQTL discovery were replicated in an independent cohort of cartilage arthroplasty samples and in fetal cartilage samples. DNAs were genotyped at rs1046934 by pyrosequencing. For methylation quantification, 500 ng of DNA was bisulfite converted using EZ DNA methylation kits (Zymo Research). The CpG regions were PCR amplified in bisulfite–converted DNA with methylation levels quantified using the PyroMark Q24 Platform (Qiagen). Duplicate measures were performed and excluded if the difference was >5%. Oligonucleotide sequences, which were generated by PyroMark Assay Design (Qiagen), were obtained from IDT (Supplementary Table 4, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427).
In silico analysis
The public databases that we used are listed in Supplementary Table 1 (available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). The Roadmap project and the 3D and University of California Santa Cruz Genome Browser databases were searched to identify regulatory functions of the regions encompassing associated SNPs and mQTL CpGs, focusing on human musculoskeletal cells: primary mesenchymal stem cells (MSCs), MSC‐derived chondrocytes (E049) and adipocytes, adipose‐derived MSCs, and primary osteoblasts. Pairwise LD between SNPs in European ancestry cohorts was determined using LDlink. Transcription factor (TF)–binding profiles and the predicted impact of SNP alleles on TF binding was assessed using the JASPAR and SNP2TFBS databases, respectively.
To assess whether SNPs or CpGs were in open or closed chromatin, we investigated ATAC‐sequencing data generated from the cartilage chondrocytes of 5 knee OA patients and 5 hip OA patients and from 6 fetal knee and 6 fetal hip samples (24) (GEO accession no. GSE214394). Expression of TFs was assessed using RNA‐sequencing data generated from the hip cartilage of 10 OA patients and 6 femoral neck fracture arthroplasty patients (29; GEO accession no. GSE111358).
Reporter gene assay
Regions surrounding cg15204595 (290 bp) and cg21606956 (260 bp) were cloned into the Lucia CpG‐free promoter vector (InvivoGen). The putative enhancers were amplified from DNA samples using oligonucleotides containing the required restriction enzyme sequences for cloning (Supplementary Table 6, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). The PCR products were cloned into the vector as previously described (21, 22). Plasmids were methylated or mock‐methylated in vitro using M.SssI (New England BioLabs). Cells from the human chondrocyte cell line Tc28a2 (30) were seeded at 5,000 cells/well in a 96‐well plate and transfected with 100 ng pCpG‐free promoter constructs and 10 ng pGL3‐promoter control vector (Promega) using Lipofectamine 2000 (Invitrogen). Cells were lysed after 24 hours, and luminescence was read using the Dual‐Luciferase Reporter Assay System (Promega) and analyzed as previously described (21).
Epigenetic modulation
Guide RNA 1 (gRNA1) and gRNA2, targeting cg15204595 and cg21606956, respectively, were designed using the CRISPR/Cas9 guide RNA design tool (IDT). The gRNA sequences (Supplementary Table 6, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) were synthesized as single‐stranded cDNA oligonucleotides (IDT) with overhangs to facilitate cloning. For methylation, oligonucleotides were annealed and ligated into pdCas9‐DNMT3a‐EGFP plasmid (31) (Addgene, plasmid no. 71666) and the catalytically inactivated control plasmid pdCas9‐DNMT3a‐EGFP (ANV) (31) (Addgene, plasmid no. 71685) as previously described (21, 22). For demethylation, the pdCas9‐DNMT3a‐EGFP plasmids containing the gRNAs were digested with PvuI and XbaI (New England Biolabs), and scaffold regions were subcloned into pSpdCas9‐huTET1CD‐T2A‐mCherry plasmid (Addgene, plasmid no. 129027) and the catalytically inactivated control plasmid pSpdCas9‐hudTET1CD‐T2A‐mCherry (Addgene, plasmid no. 129028), as previously described (31). Each construct (5 μg) was nucleofected into 1 × 106 Tc28a2 cells using the 4D Nucleofector kit (Lonza), with transfection confirmed after 24 hours by green fluorescent protein (for DNMT3a plasmids) or mCherry (for TET1 plasmids) visualization (Zeiss AxioVision).
Cells were harvested 72 hours after transfection. Nucleic acids were extracted using a DNA/RNA Purification Kit (Norgen Biotek). DNAm levels at cg15204595 and cg21606956 were measured using pyrosequencing. RNA (500 ng) was reverse transcribed using SuperScript IV Reverse Transcriptase (Invitrogen), and gene expression was measured by reverse transcription–quantitative PCR using Quant Studio 3 (Applied Biosystems). The expression of COLGALT2 and TSEN15, normalized to that of housekeeping genes 18S, GAPDH, and HPRT1, was calculated using the 2−ΔCt method (32). TaqMan assays were purchased from IDT (Supplementary Table 7, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427).
Statistical analysis
Wilcoxon's matched pairs signed rank test was used to calculate P values in AEI analysis. For graphical representations of DNAm data, methylation status was plotted in the form of β‐values, ranging from 0 (no methylation) to 1 (100% methylation). For statistical analysis of methylation data, β‐values were converted to M‐values (33). In mQTL analysis, linear regression was used to assess the relationship between CpG methylation and genotype (0, 1, or 2 copies of the minor allele) at rs1046934. For mQTL discovery, these calculations were performed using the Matrix eQTL package (34) in R, with age, sex, and joint site (hip or knee) used as covariates. Associations between AEI and DNAm and between expression of TFs and COLGALT2 were determined using linear regression. Mann‐Whitney U test was used to calculate P values when comparing methylation levels irrespective of genotype. For Lucia reporter gene assays, P values were calculated by paired and unpaired t‐tests. Paired t‐tests were used to calculate P values for changes in gene expression following epigenetic modulation. Unless stated otherwise, statistical tests were performed in GraphPad Prism.
RESULTS
Missense variants not predicted to affect the TSEN15 protein
The rs1046934 locus encompasses transcript SNPs that introduce amino acid (missense) substitutions into TSEN15: rs1046934 itself (A>C, pGln59–His) and rs2274432 (G>A, pGly19‐Asp). These SNPs are in perfect LD (r2 = 1). Gln59 falls within the α2‐helix of TSEN15 (Figures 1A and 1B). In silico mutagenesis of the residue predicts an outward‐facing position of the histidine side chain, away from the coiled‐coil interactions between the α1‐helix and α2‐helix (Figure 1C). This indicates that the variant is unlikely to affect the structure or stability of TSEN15. We could not undertake in silico mutagenesis of Gly19 since the Gly19–Asp variant resides within the structurally unresolved N‐terminal region of TSEN15. However, gnomAD, PolyPhen, and Mutation Taster all predicted this variant, as well as Gln59‐His, as benign. We conclude that the risk of OA residing at the rs1046934 locus is not driven by changes to TSEN15 protein function.
Figure 1.
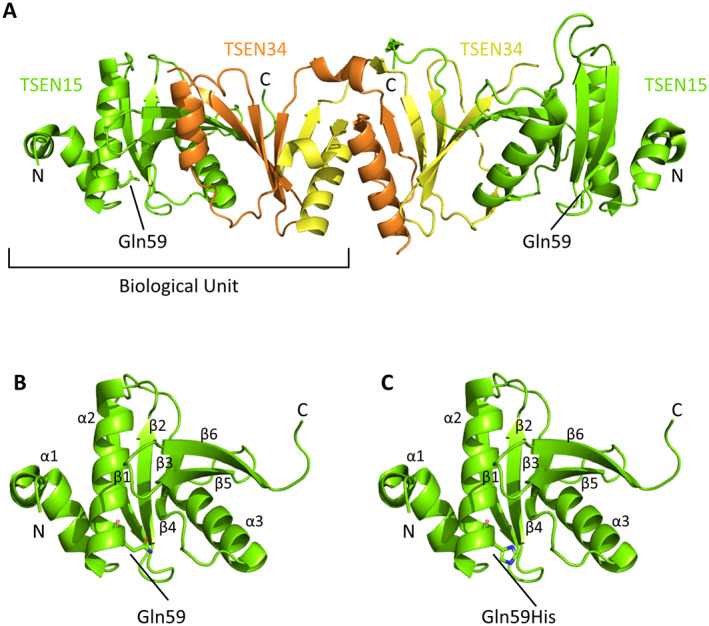
TSEN15 protein structure. A, Crystal structure of the TSEN15 (green)–TSEN34 (yellow and orange) heterodimer (Protein Data Bank, identification no. 6Z9U). The position of the Gln59 residue is highlighted, and the amino acid side chain is displayed. B, Monomeric crystal structure of TSEN15 (Protein Data Bank, identification no. 2GW6), showing numbering of the α‐α‐β‐β‐β‐β‐α‐β‐β fold (α1–3 and β1–6). Gln59 is labeled, and the side chain is displayed. C, TSEN15 structure, shown as in B, following in silico mutagenesis to predict the conformation of Gln59‐His (labeled). In B and C, red shows oxygen atoms and blue shows nitrogen atoms. The PyMOL Molecular Graphics System was used to view structures and to perform mutagenesis.
Association between the genotype at rs1046934 and expression of COLGALT2 and TSEN15 in human arthroplasty cartilage
The rs1046934 OA association signal and the observation that the genotype at the SNP is associated with expression of COLGALT2 and TSEN15 were reported in a range of tissues in the Genotype‐Tissue Expression (GTEx) portal, forming eQTLs (4). None of the tissues comprising the GTEx portal originate from articulating joints. Therefore, we undertook an AEI analysis in cartilage samples from OA patients to assess whether the rs1046934 genotype was associated with expression of either COLGALT2 or TSEN15 in this disease relevant tissue.
Both genes demonstrated AEI (Figure 2), with OA risk allele C of COLGALT2 transcript SNP rs114661926 showing an average 1.21‐fold increase in COLGALT2 expression (P = 0.003) and OA risk allele G of TSEN15 transcript SNP rs2274432 showing an average 1.09‐fold increase in TSEN15 expression (P = 0.02).
Figure 2.
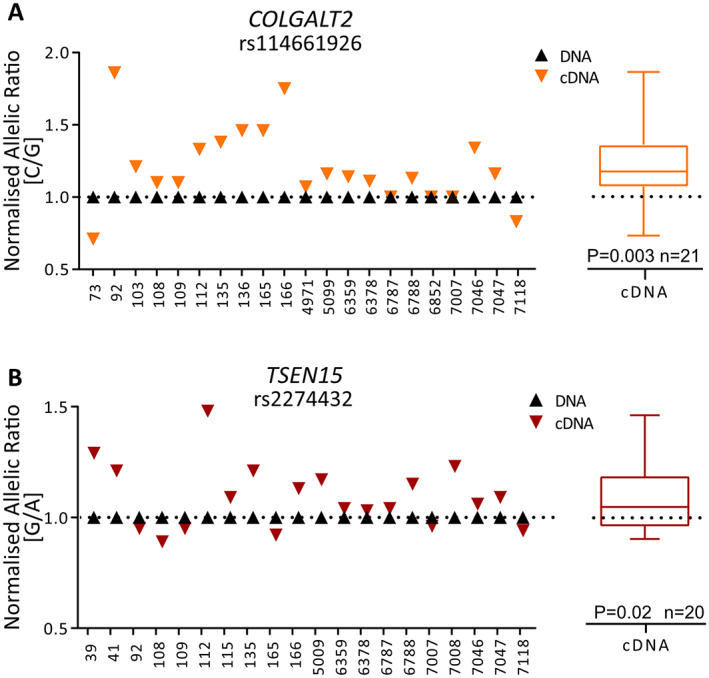
Allelic expression imbalance analysis of COLGALT2 and TSEN15 in human arthroplasty cartilage samples. A, Allelic ratios for COLGALT2 transcript single‐nucleotide polymorphism (SNP) rs114661926 (C/G; C = osteoarthritis [OA] risk allele). B, Allelic ratios for TSEN15 transcript SNP rs2274432 (G/A; G = OA risk allele). Numbers on x‐axes indicate patient sample identification numbers. Symbols in graphs represent the mean of 3 technical replicates. Mean complementary DNA (cDNA) values measured across all samples are shown as box plots. Each box represents the interquartile range. Lines inside the box represent the median. Whiskers represent the minimum and maximum values. Dashed lines represent the allele ratios in DNA. P values were calculated using Wilcoxon's matched pairs signed rank test. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42427/abstract.
Identification of rs1046934 mQTLs operating within putative enhancers in human arthroplasty cartilage
We next analyzed an arthroplasty cartilage epigenome‐wide DNAm data set derived from the cartilage of 87 OA patients (28) to assess whether the rs1046934 genotype was associated with proximal DNAm levels. We analyzed 58 CpGs in a 400‐kb interval surrounding rs1046934 (Supplementary Table 8, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) and identified 3 CpGs with methylation status that was nominally (P < 0.05) associated with genotype, forming mQTLs: cg15204595 (P = 0.005), cg01436608 (P = 0.04), and cg21606956 (P = 0.002). At all 3 CpGs, the OA risk allele A of rs1046934 was associated with reduced methylation (Figure 3A).
Figure 3.
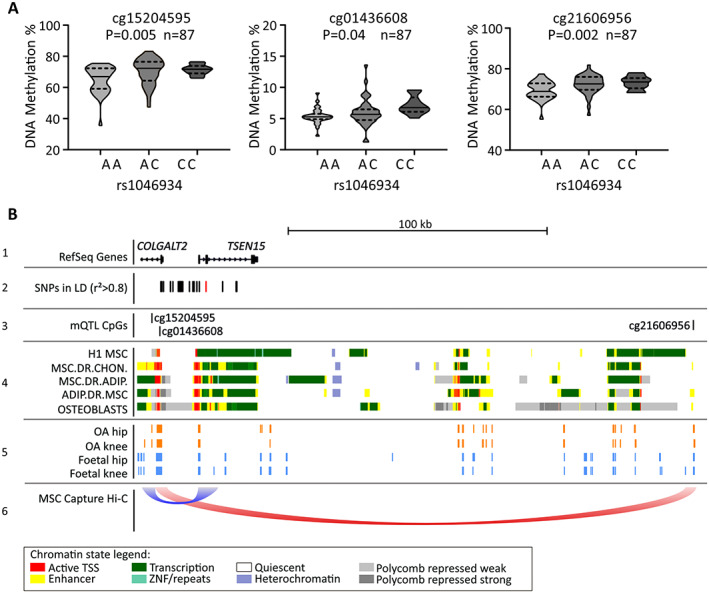
Methylation quantitative trait locus (mQTL) discovery and in silico analysis. A, DNA methylation (DNAm) values at cg15204595, cg01436608, and cg21606956 stratified by rs1046934 genotype, using hip or knee arthroplasty cartilage samples from osteoarthritis (OA) patients (n = 87), are shown as violin plots. Solid lines and dashed lines inside the plots represent median and interquartile range, respectively. P values were calculated by linear regression. B, Schematic overview of the rs1046934 locus. Panel 1, Relative genomic position of the 5' end of COLGALT2 and all of TSEN15, visualized using the University of California Santa Cruz Genome Browser (hg19). Panel 2, Red line indicates position of rs1046934, and black lines indicate single‐nucleotide polymorphisms (SNPs) in high linkage disequilibrium (LD) (pairwise r2 > 0.8). SNPs comprise a 30‐kb block. Panel 3, Black lines indicate positions of cg15204595, cg01436608, and cg21606956. Panel 4, Chromatin state data, as determined from the Roadmap project, for primary human mesenchymal stem cells (H1 MSC), MSC‐derived chondrocytes (MSC.DR.CHON) and adipocytes (MSC.DR.ADIP), adipose‐derived MSCs (ADIP.DR.MSC), and human osteoblasts. Colors correspond to different chromatin states, as indicated in legend at bottom. Panel 5, Open regions marked by orange blocks indicate ATAC‐sequencing peaks generated from OA hip and knee chondrocytes; open regions marked by blue blocks indicate ATC‐sequencing peaks generated from fetal hip and knee chondrocytes. Panel 6, Loops indicate capture Hi‐C chromatin interactions from the 3D Genome Browser in human MSCs, with the flat ends of loops spanning the width of the interacting regions. TSS = transcription start site. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42427/abstract.
We observed that rs1046934 and the 20 SNPs in high pairwise LD (r2 > 0.8) form a 30‐kb block that encompasses the 5′‐untranslated region and promoter of COLGALT2, the promoter and part of the gene body of TSEN15, and the intergenic region between the 2 genes (Figure 3B, panels 1 and 2). Two of the mQTL CpGs, cg15204595 and cg01436608, are 2.35‐kb apart and located within intron 1 of COLGALT2 (Figure 3B, panels 1 and 3). Both are close to the LD block, with cg01436608 being 595 bp from rs74767794, the most upstream variant in the block. Both cg15204595 and cg01436608 reside within a region that is marked as an enhancer and a transcriptionally active site in a range of relevant human cell types (primary MSCs, MSC‐derived chondrocytes and adipocytes, adipose‐derived MSCs, and primary osteoblasts) (Figure 3B, panel 4) and marked as an open chromatin region in OA and fetal chondrocytes (Figure 3B, panel 5). Conversely, cg21606956 is distal to the LD block and over 200 kb from cg15204595 and cg01436608 (Figure 3B, panels 1–3), falling within an intergenic enhancer (Figure 3B, panels 3 and 4) that is marked as an open chromatin region in OA and fetal chondrocytes (Figure 3B, panel 5). MSC capture Hi‐C data showed physical interactions between a broad region encompassing rs1046934 and the enhancer containing cg15204595 (Figure 3B, panel 6). Additional interactions were observed between the COLGALT2 promoter and the enhancer containing cg21606956 (Figure 3B, panel 6).
Replication of mQTLs using cartilage DNA from an independent human arthroplasty cohort
Because none of the CpGs from the analysis of the 87 patients would have significant P values following multiple correction testing, we set out to replicate the 3 mQTLs in an independent cohort of arthroplasty cartilage DNAs. We were able to design pyrosequencing assays for cg15204595 and cg21606956 but not for cg01436608, due to a long run of thymine bases following bisulfite conversion and subsequent PCR amplification. The cg15204595 and cg21606956 mQTLs replicated (P < 0.0001 for each) and confirmed the association between the OA risk allele A of rs1046934 and reduced methylation (Figure 4A).
Figure 4.
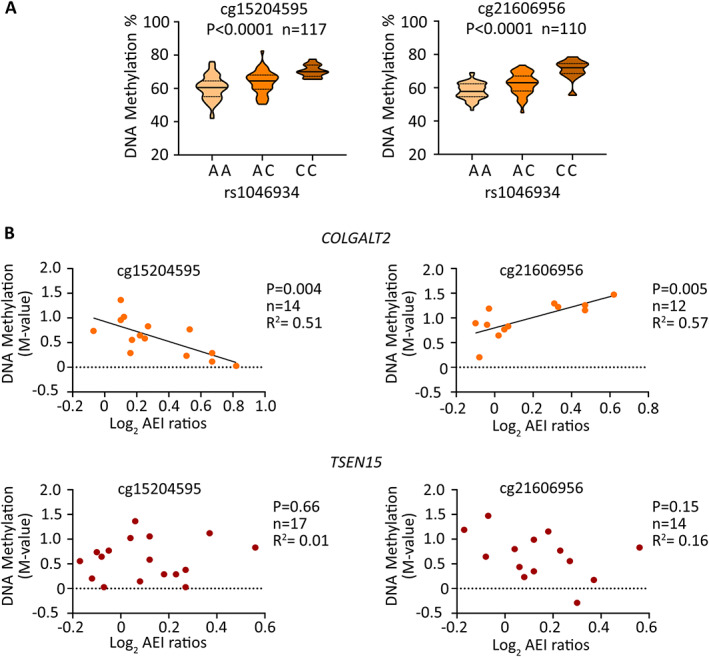
Replication of mQTLs and discovery of methylation–expression QTLs (meQTLs) in arthroplasty cartilage. A, DNAm values at cg15204595 and cg21606956 stratified by genotype at rs1046934, using DNA derived from cartilage from hip and knee OA patients and femoral neck fracture patients, are shown as violin plots. Solid lines and dashed lines inside the plots represent the median and interquartile range, respectively. P values were calculated by linear regression. B, Graphs show allelic expression imbalance (AEI) allelic ratios (log2) for COLGALT2 (rs114661926) and TSEN15 (rs2274432) plotted against matched DNAm levels (M‐values) at cg15204595 and cg21606956. Symbols represent data from 1 individual. P values were calculated by linear regression. See Figure 3 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42427/abstract.
The cartilage DNAs used for replication were derived from OA (hip and knee) patients and femoral neck fracture patients. After stratification by disease state (OA or femoral neck fracture; Supplementary Figure 1A, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427), mQTLs were detectable in both patient groups, indicating that differential methylation is not a consequence of OA disease state. When the data were stratified by disease state irrespective of rs1046934 genotype (Supplementary Figure 1B), methylation at cg15204595 was significantly higher in the femoral neck fracture group than in the OA group (P = 0.003). Patients with femoral neck fracture were on average older at surgery (77.35 years of age) than patients with OA at surgery (65.32 years of age for knee OA and 66.51 years of age for hip OA) (Supplementary Table 2, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). We found no significant association between age and DNAm status (P > 0.05; Supplementary Figure 2, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427).
Association between CpG methylation and COLGALT2 expression
We subsequently assessed whether there were associations between DNAm and gene expression in samples with matched data (Figure 4B). For COLGALT2, significant associations were observed at cg15204595 (r2 = 0.51, P = 0.004) and cg21606956 (r2 = 0.57, P = 0.005), marking methylation–expression QTLs (meQTLs). No significant associations were shown between either of the CpGs and TSEN15.
Identification of OA genetic risk mechanisms at the rs1046934 locus in human fetal development
As noted earlier, we recently reported that, for several OA SNPs, the associations between gene expression and CpG methylation observed in human arthroplasty cartilage also occur in fetal cartilage (24). Therefore, we determined whether the rs1046934 AEI, mQTL, and meQTL effects were also detectable in fetal cartilage.
AEI was detected for both genes (Figure 5A) in fetal cartilage, in the same direction as that observed in human arthroplasty cartilage (Figure 2), with OA risk allele C at rs114661926 showing an average 1.35‐fold increase in COLGALT2 expression (P < 0.0001) and OA risk allele G at rs2274432 showing an average 1.03‐fold increase in TSEN15 expression (P = 0.04).
Figure 5.
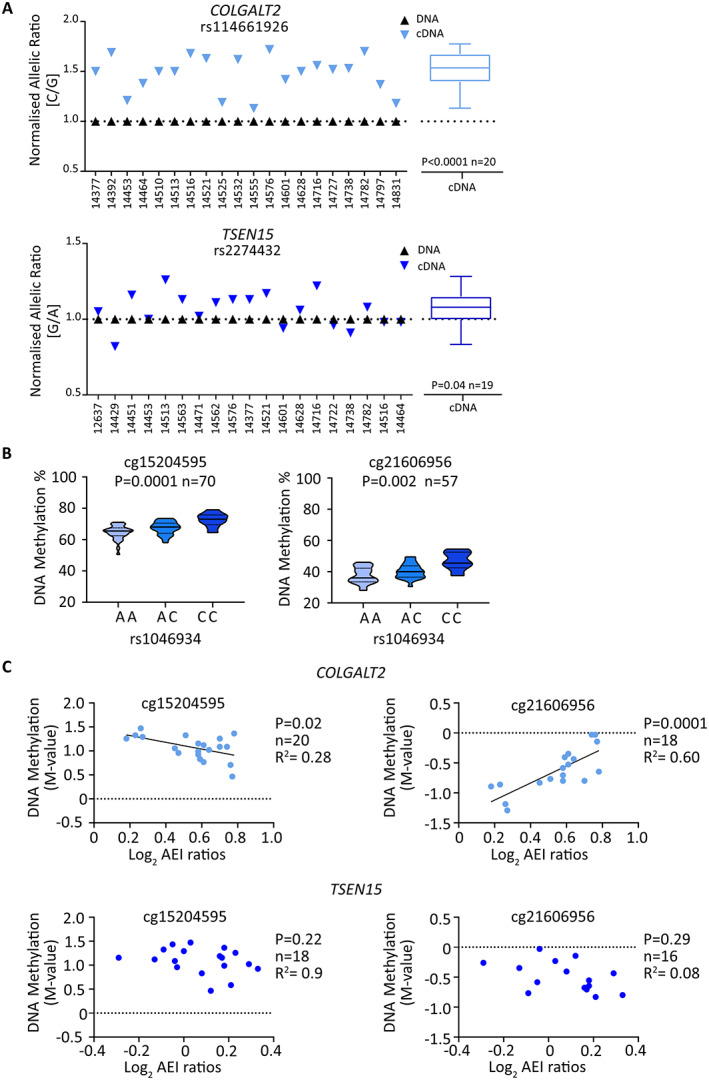
AEI, mQTL, and meQTL analyses in fetal cartilage. A, Allelic ratios for COLGALT2 transcript SNP rs114661926 (C/G; C = OA risk allele) and for TSEN15 transcript SNP rs2274432 (G/A; G = OA risk allele). Numbers on x‐axes indicate patient sample identification numbers. Symbols in graphs represent the mean of 3 technical replicates. Mean complementary DNA (cDNA) values measured across all samples are shown as box plots. Each box represents the interquartile range. Lines inside the box represent the median. Whiskers represent the minimum and maximum values. Dashed lines represent the allele ratios in DNA. P values were calculated using Wilcoxon's matched pairs signed rank test. B, DNAm values at cg15204595 and cg21606956 stratified by genotype at rs1046934, using DNA derived from fetal cartilage, are shown as violin plots. Solid lines and dashed lines inside the plots represent the median and interquartile range, respectively. P values were calculated by linear regression. C, Graphs show AEI allelic ratios (log2) for COLGALT2 (rs114661926) and TSEN15 (rs2274432) plotted against matched DNAm levels (M‐values) at cg15204595 and cg21606956. Symbols represent data from 1 individual. P values were calculated by linear regression. See Figure 3 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42427/abstract.
In fetal DNA, cg15204595 and cg21606956 displayed mQTL effects (Figure 5B) in the same direction as observed in human arthroplasty DNA (Figure 4A), with OA risk allele A of rs1046934 showing an association with reduced methylation. Mean DNAm levels at cg15204595 were higher in fetal cartilage (66.7%) than in arthroplasty cartilage (62.8%) (P = 0.0002), with the opposite observed at cg21606956, with mean values of 40.0% in fetal cartilage and 61.1% in arthroplasty cartilage (P < 0.0001) (Supplementary Figure 3, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). In fetal cartilage, meQTLs were observed for COLGALT2 but not for TSEN15 (Figure 5C), consistent with our observations in arthroplasty cartilage (Figure 4B).
In both arthroplasty and fetal cartilage, the slopes of the COLGALT2 meQTLs at cg15204595 and cg21606956 were in opposite directions. At cg15204595, high M‐values were associated with low AEI ratios, whereas, for cg21606956, high M‐values were associated with high AEI ratios (Figure 4B, Figure 5C). A proposed model describing this is presented in the Supplementary Text and in Supplementary Figure 4, available on the Arthritis & Rheumatology website at https://onlinelibrary.wiley.com/doi/10.1002/art.42427.
Presence of cg15204595 and cg21606956 in enhancers and effects of their demethylation on COLGALT2 expression
Using the chondrocyte cell line Tc28a2, we cloned the regions surrounding cg15204595 and cg21606956 into the CpG‐free Lucia reporter gene vector and tested for enhancer activity in methylated and unmethylated states. No other CpGs were captured within the cloned regions. For cg15204595, the unmethylated and methylated constructs showed increased Lucia readings compared with that shown in reporter gene assays with the empty control vectors, with an average increase in activity of 1.36‐fold (P < 0.01) and 1.35‐fold (P < 0.01) respectively (Figure 6A, left). The region encompassing cg21606956 also acted as an enhancer, with an average 1.41‐fold (P < 0.01) and 1.32‐fold (P < 0.001) increase in Lucia activity in the unmethylated and methylated constructs, respectively (Figure 6A, right). In vitro methylation status had no significant effect on the function of the enhancers.
Figure 6.
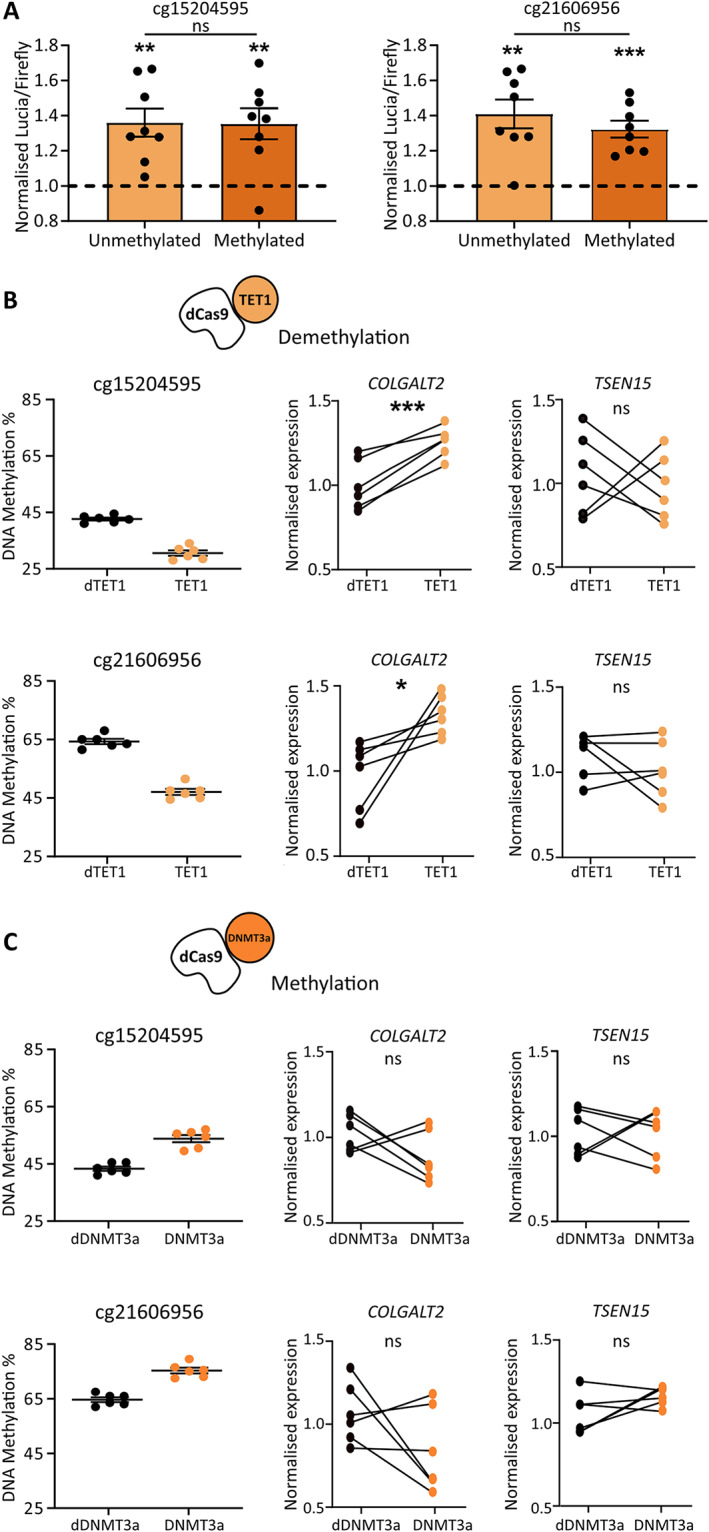
cg15204595 and cg21606956 reside in enhancers and increase COLGALT2 expression when demethylated. A, Lucia luminescence in Tc28a2 chondrocytes following transfection with constructs containing cg15204595 (left) or cg21606956 (right) in unmethylated or methylated states. Symbols represent biologic replicates (n = 8). Dashed lines represent cells with empty control vectors. B, Left, DNAm levels at cg15204595 (top) and cg21606956 (bottom) in Tc28a2 chondrocytes following transfection of guide RNAs (gRNAs) with dead Cas9 (dCas9) coupled with dead TET1 (dTET1) in controls (black symbols) or with active TET1 (orange symbols) (n = 6 biologic replicates/treatment). Right, Effect of demethylation on gene expression. Values were normalized to mean control values. C, Left, DNAm levels at cg15204595 (top) and cg21606956 (bottom) in Tc28a2 chondrocytes following transfection of gRNAs with dCas9 coupled with dead DNMT3a (dDNMT3a) in controls (black symbols) or with active DNMT3a (orange symbols) (n = 6 biologic replicates/treatment). Right, Effect of methylation on gene expression. Values were normalized to mean control values. A–C, Bars show mean ± SEM. P values were calculated using paired t‐test for empty control versus insert and unpaired t‐test for unmethylated versus methylated insert (A) or were calculated using paired t‐test (B and C). * = P < 0.05; ** = P < 0.01; *** = P < 0.001; ns = not significant (P > 0.05). See Figure 3 for other definitions. Color figure can be viewed in the online issue, which is available at http://onlinelibrary.wiley.com/doi/10.1002/art.42427/abstract.
Targeted demethylation and methylation of cg15204595 and cg21606956 in Tc28a2 cells was performed to investigate the effects of DNAm on COLGALT2 and TSEN15 expression using catalytically dead Cas9 (dCas9) protein coupled with catalytically active TET1 (to demethylate) or DNMT3a (to methylate). Control cells were transfected with the same gRNAs coupled with dCas9 and dead TET1 (dTET1) or dead DNMT3a (dDNMT3a). Mean reductions in methylation at cg15204595 of 12.8% and cg21606956 of 17.3% were achieved using TET1 (Figure 6B, left). This resulted in 1.3‐fold (P = 0.0009) and 1.2‐fold (P = 0.01) increases in COLGALT2 expression but no significant change in TSEN15 expression (Figure 6B, right). Mean increases in methylation at cg15204595 of 10.5% and cg21606956 of 10.7% were achieved using DNMT3a (Figure 6C, left). Increases in methylation did not significantly alter expression of either gene (Figure 6C, right).
Investigation of the OA risk SNPs affecting TF binding at the locus
Noncoding SNPs can mediate their effects on expression of a target gene through alteration of the consensus sequence of DNA binding proteins, including TFs (5, 6, 7, 8). This variance can lead to differential protein binding that directly or indirectly (i.e., with a DNAm intermediary) alters transcription (5, 6, 7, 8). One of the major hurdles of post‐GWAS functional studies is identifying the causal variants within the risk haplotype, marked by the association signal. These SNPs can exert their functional effects in concert through shared or distinct mechanisms (5, 6, 7, 8).
To investigate which of the 21 SNPs in the rs1046934 LD block may alter TF binding, we used SNP2TFBS (Supplementary Table 1, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). Six SNPs were identified that were predicted to impact the binding of 8 TFs, 4 of which are expressed in human arthroplasty cartilage (transcripts per million >10) (Supplementary Table 9, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). The expression levels of these genes (STAT1, STAT2, SP2, and ZNF263) were regressed against COLGALT2 expression (Supplementary Figure 5, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427). A significant association was identified between COLGALT2 and SP2 (P = 0.001). Interestingly, 1 of the 2 SNPs predicted to disrupt SP2 binding (rs74767794) is located within the COLGALT2 promoter. This potentially points toward an additional, direct genetic impact on COLGALT2 conferred by the risk haplotype.
Our targeted epigenetic modulation demonstrated that demethylation of cg15204595 and cg21606956 has direct effects on the function of their respective enhancer regions (Figure 6B). Methylation at CpGs has the potential to alter the binding efficiency of TFs to DNA, modulating enhancer activity (35, 36). We hypothesized that these CpGs also fall within protein binding motifs and can influence TF recruitment to the enhancers. To assess this, we searched the JASPAR database (Supplementary Table 1, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427) and identified multiple TFs predicted to bind at or near the CpGs (Supplementary Figures 6A and B, available at https://onlinelibrary.wiley.com/doi/10.1002/art.42427), many of which are expressed in human cartilage (Supplementary Figure 6C).
DISCUSSION
We used a range of techniques to study a novel OA association locus marked by rs1046934. This signal maps close to COLGALT2, a gene that we had previously highlighted as a target of a completely independent OA risk locus, marked by rs11583641 (22). We discovered that the rs1046934 locus, like the rs11583641 locus, mediates its effect by modulating the expression of COLGALT2 via methylation changes to CpGs located in enhancers. The associated SNPs, the CpGs, and the enhancers are entirely distinct between the 2 loci, but their ultimate effect on COLGALT2 is the same.
Our analysis of human arthroplasty cartilage showed that the OA risk allele A of rs1046934 was associated with increased COLGALT2 expression and decreased methylation of CpGs cg15204595, cg01436608, and cg21606956, with the methylation effects observed at cg15204595 and cg21606956 replicated in an independent cohort. Importantly, we identified significant associations between methylation and COLGALT2 expression. Epigenetic modulation demonstrated this to be a direct causal link, with demethylation increasing expression. Furthermore, reporter gene assays confirmed that the genomic regions harboring cg15204595 and cg21606956 are enhancers in chondrocytes. In silico data revealed that the CpGs reside in or close to TF binding sites and in open chromatin regions in chondrocytes, further supporting their functional role. MSC capture Hi‐C data highlighted the physical interactions encompassing the associated SNPs, the cg15204595 and cg21606956 enhancers, and the COLGALT2 promoter. Therefore, we conclude that these enhancers interact with COLGALT2 to regulate its expression, with genotype at the association signal modulating the methylation status and consequently the function of the enhancers.
Although OA is a disease of older people, OA susceptibility has been reported to have developmental origins, with many OA SNPs associating with joint shape phenotypes (38, 39, 40, 41, 42, 43). This implies that a proportion of OA genetic risk is functionally active during skeletogenesis and early postnatal life and manifests with aging (44, 45, 46). We previously investigated this by assessing AEI and mQTLs at OA risk loci in fetal cartilage samples (24). For a proportion of the studied loci, the AEI and mQTLs observed in arthroplasty cartilage were also observed in fetal cartilage (24). This included the rs11583641 COLGALT2 locus (24), which prompted us to investigate fetal cartilage at the rs1046934 locus. The rs1046934 AEI, mQTL, and meQTL effects detected in human arthroplasty cartilage were also detected in fetal cartilage, implying that this locus is one in which the molecular effects on a target gene are activated during development. Our cohort of arthroplasty cartilage samples included cartilage from patients with femoral neck fracture. Although femoral neck fracture patients lack OA cartilage lesions in their hip joints, we detected mQTLs at cg15204595 and cg21606956 in their DNA. Combined, our fetal and femoral neck fracture data imply that the molecular effects of the rs1046934 signal on COLGALT2 are not dependent on age or OA disease status yet contribute to this highly polygenic disease across the life course.
In our dCas9 experiment, demethylation of cg15204595 and cg21606956 had significant effects on COLGALT2 expression. Demethylating cg15204595 and cg21606956 in vitro mimics the effect of the risk‐conferring allele A of rs1046934 in cartilage, which is associated with reduced methylation of the CpGs and with increased COLGALT2 expression. We propose that the enhancers harboring cg15204595 and cg21606956 are particularly sensitive to decreased methylation, accounting for the changes in COLGALT2 expression, which were only measured when DNAm levels were reduced and not increased.
Throughout our study, we investigated TSEN15 alongside COLGALT2, as both genes were highlighted in the discovery GWAS as potential targets of the association signal, primarily due to rs1046934 eQTLs shown at each gene in the GTEx portal (4). We observed AEI at TSEN15, albeit the fold differences in expression between risk and nonrisk alleles were not as large as those for COLGALT2. However, we did not observe meQTLs for TSEN15, and the epigenetic modulation of cg15204595 and cg21606956 did not significantly alter TSEN15 expression. Furthermore, our in silico analyses of the TSEN15 missense variants did not indicate that the changes affected protein structure or function. Despite these observations, we cannot definitively exclude TSEN15 as an additional target of the association signal.
Clinical exploitation of OA genetic discoveries will require an understanding of the molecular mechanism by which risk‐conferring alleles impact their target genes (1, 46, 47). In this study, we undertook a detailed analysis of the OA locus marked by rs1046934, highlighting its effect on the expression of COLGALT2 via 2 distal enhancers that are epigenetically regulated. For the first time to our knowledge, our data provide compelling evidence of a target gene being impacted in a nearly identical manner by 2 genetically independent OA association signals and disease‐relevant gene enhancers. This increases confidence in COLGALT2 and its encoded enzyme as targets of OA risk and therefore prioritizes them for future translational investigation.
AUTHOR CONTRIBUTIONS
All authors were involved in drafting the article or revising it critically for important intellectual content, and all authors approved the final version to be published. Prof. Loughlin had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Study conception and design
Wilkinson, Rice, Loughlin.
Acquisition of data
Kehayova, Rice.
Analysis and interpretation of data
Kehayova, Wilkinson, Rice, Loughlin.
Supporting information
Disclosure Form
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
Cartilage tissue was provided by the Newcastle Bone and Joint Biobank, supported by the NIHR Newcastle Biomedical Research Centre, awarded to the Newcastle‐upon‐Tyne NHS Foundation Trust and Newcastle University. We thank the surgeons and research nurses at the NHS Foundation Trust for providing us with access to these samples. The human embryonic and fetal material was provided by the Joint MRC/Wellcome Trust (grant MR/R006237/1) Human Developmental Biology Resource (http://hdbr.org).
Supported by Versus Arthritis grants 20771 and 22615, by Medical Research Council and Versus Arthritis Centre for Integrated Research into Musculoskeletal Ageing grants MR/P020941/1 and MR/R502182/1, and by the Ruth and Lionel Jacobson Charitable Trust.
Author disclosures are available at https://onlinelibrary.wiley.com/action/downloadSupplement?doi=10.1002%2Fart.42427&file=art42427‐sup‐0001‐Disclosureform.pdf.
Contributor Information
Sarah J. Rice, Email: sarah.rice@ncl.ac.uk.
John Loughlin, Email: john.loughlin@ncl.ac.uk.
REFERENCES
- 1. Aubourg G, Rice SJ, Bruce‐Wootton P, et al. Genetics of osteoarthritis. Osteoarthritis Cartilage 2022;30:636–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Styrkarsdottir U, Lund SH, Thorleifsson G, et al. Meta‐analysis of Icelandic and UK data sets identifies missense variants in SMO, IL11, COL11A1 and 13 more new loci associated with osteoarthritis. Nat Genet 2018;50:1681–7. [DOI] [PubMed] [Google Scholar]
- 3. Tachmazidou I, Hatzikotoulas K, Southam L, et al. Identification of new therapeutic targets for osteoarthritis through genome‐wide analyses of UK Biobank data. Nat Genet 2019;51:230–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boer CG, Hatzikotoulas K, Southam L, et al. Deciphering osteoarthritis genetics across 826,690 individuals from 9 populations. Cell 2021;184:4784–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gallagher MD, Chen‐Plotkin AS. The post‐GWAS era: from association to function. Am J Hum Genet 2018;102:717–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cano‐Gamez E, Trynka G. From GWAS to function: using functional genomics to identify the mechanisms underlying complex diseases. Front Genet 2020;11:424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lichou F, Trynka G. Functional studies of GWAS variants are gaining momentum. Nat Commun 2020;11:6283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lappalainen T, MacArthur DG. From variant to function in human disease genetics. Science 2021;373:1464–8. [DOI] [PubMed] [Google Scholar]
- 9. Steinberg J, Ritchie GR, Roumeliotis TI, et al. Integrative epigenomics, transcriptomics and proteomics of patient chondrocytes reveal genes and pathways involved in osteoarthritis. Sci Rep 2017;7:8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Liu Y, Chang JC, Hon CC, et al. Chromatin accessibility landscape of articular knee cartilage reveals aberrant enhancer regulation on osteoarthritis. Sci Rep 2018;8:15499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shepherd C, Zhu D, Skelton AJ, et al. Functional characterization of the osteoarthritis genetic risk residing at ALDH1A2 identifies rs12915901 as a key target variant. Arthritis Rheumatol 2018;70:1577–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Den Hollander W, Pulyakhina I, Boer C, et al. Annotating transcriptional effects of genetic variants in disease‐relevant tissue: transcriptome‐wide allelic imbalance in osteoarthritic cartilage. Arthritis Rheumatol 2019;71:561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shepherd C, Reese AE, Reynard LN, et al. Expression analysis of the osteoarthritis genetic susceptibility mapping to the matrix Gla protein gene MGP. Arthritis Res Ther 2019;21:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Klein JC, Keith A, Rice SJ, et al. Functional testing of thousands of osteoarthritis‐associated variants for regulatory activity. Nat Commun 2019;10:2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Steinberg J, Southam L, Roumeliotis TI, et al. A molecular quantitative trait locus map for osteoarthritis. Nat Commun 2021;12:1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rice S, Aubourg G, Sorial A, et al. Identification of a novel, methylation‐dependent, RUNX2 regulatory region associated with osteoarthritis risk. Hum Mol Genet 2018;27:3464–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rice SJ, Tselepi M, Sorial AK, et al. Prioritization of PLEC and GRINA as osteoarthritis risk genes through the identification and characterization of novel methylation quantitative trait loci. Arthritis Rheumatol 2019;71:1285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sorial AK, Hofer IM, Tselepi M, et al. Multi‐tissue epigenetic analysis of the osteoarthritis susceptibility locus mapping to the plectin gene PLEC. Osteoarthritis Cartilage 2020;28:1448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rice SJ, Beier F, Young DA, et al. Interplay between genetics and epigenetics in osteoarthritis. Nat Rev Rheumatol 2020;16:268–81. [DOI] [PubMed] [Google Scholar]
- 20. Parker E, Hofer IM, Rice SJ, et al. Multi‐tissue epigenetic and gene expression analysis combined with epigenome modulation identifies RWDD2B as a target of osteoarthritis susceptibility. Arthritis Rheumatol 2021;73:100–9. [DOI] [PubMed] [Google Scholar]
- 21. Rice SJ, Roberts JB, Tselepi M, et al. Genetic and epigenetic fine‐tuning of TGFB1 expression within the human osteoarthritic joint. Arthritis Rheumatol 2021;73:1866–77. [DOI] [PubMed] [Google Scholar]
- 22. Kehayova YS, Watson E, Wilkinson JM, et al. Genetic and epigenetic interplay within a COLGALT2 enhancer associated with osteoarthritis. Arthritis Rheumatol 2021;73:1856–65. [DOI] [PubMed] [Google Scholar]
- 23. Dominguez LJ, Barbagallo M, Moro L. Collagen overglycosylation: a biochemical feature that may contribute to bone quality. Biochem Biophys Res Commun 2005;330:1–4. [DOI] [PubMed] [Google Scholar]
- 24. Rice SJ, Brumwell A, Falk J, et al. Genetic risk of osteoarthritis operates during human skeletogenesis. Hum Mol Genet 2022. DOI: 10.1093/hmg/ddac251. E‐pub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song J, Markley JL. Three‐dimensional structure determined for a subunit of human tRNA splicing endonuclease (Sen15) reveals a novel dimeric fold. J Mol Biol 2007;366:155–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Sekulovski, S. Devant P, Panizza S, et al. Assembly defects of human tRNA splicing endonuclease contribute to impaired pre‐tRNA processing in pontocerebellar hypoplasia. Nature Commun 2021;12:5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Rice SJ, Cheung K, Reynard LN, et al. Discovery and analysis of methylation quantitative trait loci (mQTLs) mapping to novel osteoarthritis genetic risk signals. Osteoarthritis Cartilage 2019;27:1545–56. [DOI] [PubMed] [Google Scholar]
- 29. Ajekigbe B, Cheung K, Xu Y, et al. Identification of long non‐coding RNAs expressed in knee and hip osteoarthritic cartilage. Osteoarthritis Cartilage 2019;27:694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kokenyesi R, Tan L, Robbins JR, et al. Proteoglycan production by immortalized human chondrocyte cell lines cultured under conditions that promote expression of the differentiated phenotype. Arch Biochem Biophys 2000;383:79–90. [DOI] [PubMed] [Google Scholar]
- 31. Vojta A, Dobrinic P, Tadic V, et al. Repurposing the CRISPR‐Cas9 system for targeted DNA methylation. Nucleic Acids Res 2016;44:5615–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2−DDCT method. Methods 2001;25:402–8. [DOI] [PubMed] [Google Scholar]
- 33. Du P, Zhang X, Huang CC, et al. Comparison of Beta‐value and M‐value methods for quantifying methylation levels by microarray analysis. BMC Bioinformatics 2010;11:587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shabalin AA. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 2012;28:1353–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhu H, Wang G, Qian J. Transcription factors as readers and effectors of DNA methylation. Nat Rev Genet 2016;17:551–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yin Y, Morgunova E, Jolma A, et al. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 2017;356:eaaj2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Castro‐Mondragon JA, Riudavets‐Puig R, Rauluseviciute I, et al. JASPAR 2022: the 9th release of the open‐access database of transcription factor binding profiles. Nucleic Acids Res 2022;50:D165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Slagboom E, Meulenbelt I. Genetics of osteoarthritis: early developmental clues to an old disease. Nat Clin Pract Rheumatol 2008;4:563. [DOI] [PubMed] [Google Scholar]
- 39. Pitsillides AA, Beier F. Cartilage biology in osteoarthritis—lessons from developmental biology [review]. Nat Rev Rheumatol 2011;7:654–63. [DOI] [PubMed] [Google Scholar]
- 40. Sandell LJ. Etiology of osteoarthritis: genetics and synovial joint development. Nat Rev Rheumatol 2012;8:77–89. [DOI] [PubMed] [Google Scholar]
- 41. Aspden RM, Saunders FR. Osteoarthritis as an organ disease: from the cradle to the grave. Eur Cell Mater 2019;37:74–87. [DOI] [PubMed] [Google Scholar]
- 42. Tangredi BP, Lawler DF. Osteoarthritis from evolutionary and mechanistic perspectives. Anat Rec (Hoboken) 2020;303:2967–76. [DOI] [PubMed] [Google Scholar]
- 43. Wilkinson JM, Zeggini E. The genetic epidemiology of joint shape and the development of osteoarthritis. Calcif Tissue Int 2021;109:257–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Richard D, Liu Z, Cao J, et al. Evolutionary selection and constraint on human knee chondrocyte regulation impacts osteoarthritis risk. Cell 2020;181:362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Muthuirulan P, Zhao D, Young M, et al. Joint disease‐specificity at the regulatory base‐pair level. Nat Commun 2021;12:4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Loughlin J. Translating osteoarthritis genetics research: challenging times ahead. Trends Mol Med 2022;28:176–82. [DOI] [PubMed] [Google Scholar]
- 47. Young DA, Barter MJ, Soul J. Osteoarthritis year in review: genetics, genomics, epigenetics. Osteoarthritis Cartilage 2022;30:216–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Disclosure Form
Appendix S1: Supplementary Information