

Abstract
Microglia and bone marrow‐derived monocytes are key elements of central nervous system (CNS) inflammation, both capable of enhancing and dampening immune‐mediated pathology. However, the study‐specific focus on individual cell types, disease models or experimental approaches has limited our ability to infer common and disease‐specific responses. This meta‐analysis integrates bulk and single‐cell transcriptomic datasets of microglia and monocytes from disease models of autoimmunity, neurodegeneration, sterile injury, and infection to build a comprehensive resource connecting myeloid responses across CNS disease. We demonstrate that the bulk microglial and monocyte program is highly contingent on the disease environment, challenging the notion of a universal microglial disease signature. Integration of six single‐cell RNA‐sequencing datasets revealed that these disease‐specific signatures are likely driven by differing proportions of unique myeloid subpopulations that were individually expanded in different disease settings. These subsets were functionally‐defined as neurodegeneration‐associated, inflammatory, interferon‐responsive, phagocytic, antigen‐presenting, and lipopolysaccharide‐responsive cellular states, revealing a core set of myeloid responses at the single‐cell level that are conserved across CNS pathology. Showcasing the predictive and practical value of this resource, we performed differential expression analysis on microglia and monocytes across disease and identified Cd81 as a new neuroinflammatory‐stable gene that accurately identified microglia and distinguished them from monocyte‐derived cells across all experimental models at both the bulk and single‐cell level. Together, this resource dissects the influence of disease environment on shared immune response programmes to build a unified perspective of myeloid behavior across CNS pathology.
Keywords: central nervous system pathology, high parameter data integration, microglia, monocyte‐derived cells, single‐cell RNA‐sequencing
Main Points
Microglia and monocytes adopt disease‐specific signatures in CNS disease. These are composed of differing proportions of unique subtypes.
Cd81 is an inflammatory‐stable, microglia‐enriched gene that accurately distinguishes them from monocytes.
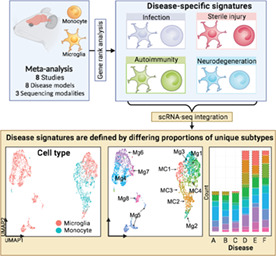
1. INTRODUCTION
Inflammation is a hallmark of many neuropathologies, irrespective of disease etiology (Spiteri & Wishart et al., 2022). Microglial activation and the infiltration of bone marrow (BM)‐derived monocytes into the central nervous system (CNS) are key elements of this inflammatory response, with both cell types capable of promoting tissue healing and repair, as well as disease pathogenesis and cell damage. However, despite the pervasive representation of these two cell types in CNS disease (Marioni et al., 2018; McQuade & Blurton‐Jones, 2019; Zhang et al., 2014), the interplay between resident microglia and recruited monocyte‐derived cells (MC), and the precise contribution to pathology of each cell remain poorly resolved.
Microglia and MCs are ontogenetically distinct cell types. Microglia are CNS‐resident tissue macrophages arising from uncommitted KIT+ erythromyeloid precursors (Kierdorf et al., 2013) that seed the CNS from the yolk sac during embryogenesis (Ginhoux et al., 2010). These CNS‐resident cells self‐renew in situ, independently of BM‐derived hematopoietic stem cells (HSC) (Ajami et al., 2007). By contrast, monocytes are derived from the fetal liver during embryogenesis and are continuously renewed throughout postnatal life from HSCs in the adult BM (Geissmann et al., 2010).
Despite microglia and MCs arising from precursors with distinct developmental trajectories, these cells often adopt similar phenotypes and morphologies during neuroinflammation, confounding definitive discrimination between them (Spiteri & Wishart et al., 2022; Spiteri et al., 2022; Spiteri et al., 2021; Getts et al., 2008). Recent advances in single‐cell RNA‐sequencing (scRNA‐seq) technologies have shed light on some uniquely‐expressed microglial genes, including Fc receptor‐like S (Fcrls), purinergic receptor P2Y G‐protein‐coupled 12 (P2ry12) (Butovsky et al., 2014), Spalt‐like transcription factor 1 (Sall1) (Buttgereit et al., 2016), sialic acid‐binding immunoglobulin‐type lectin H (Siglech) and transmembrane protein 119 (Tmem119) (Bennett et al., 2016). This has substantially aided the identification and resolution of these myeloid populations. During inflammation, however, many of these homeostatic markers are downregulated in microglia (Friedman et al., 2018; Jordão et al., 2019; Krasemann et al., 2017; Masuda et al., 2019; Vankriekelsvenne et al., 2022) or upregulated in MCs (H.‐R. Chen et al., 2020; Spiteri et al., 2021; Werner et al., 2020), making their transcriptomic and phenotypic profiles highly overlapping, highlighting the need for a more comprehensive insight into the biological processes defining distinct myeloid cells during homeostasis and across pathologies.
Along these lines, in the last few years transcriptomic sequencing uncovered a possible universal “disease‐associated microglia” (DAM) (Keren‐Shaul et al., 2017) or “microglial neurodegenerative” (MGnD) (Krasemann et al., 2017) signature that is argued to be conserved across neurological disease (Beuker et al., 2022; Cao et al., 2021; Y. Chen & Colonna, 2021; Deczkowska et al., 2018; Hunter et al., 2021; Plemel et al., 2020; Ramesha et al., 2021; Rexach et al., 2020; Safaiyan et al., 2021; Tay et al., 2018). These universal DAM/MGnD signatures characteristically downregulated homeostatic genes, including P2ry12, Tmem119 and Cx3cr1, and upregulated inflammatory program genes, including Trem2, Apoe, Axl, Lpl, Itgax and Clec7a (Keren‐Shaul et al., 2017; Krasemann et al., 2017).
In contrast, more recent findings suggest microglia develop heterogenous signatures highly specific to a disease state that may individually drive immune‐mediated pathology or promote tissue repair, rather than adopting a universal disease signature (Friedman et al., 2018; Olah et al., 2020; Sanin et al., 2022; Sobue et al., 2021; Sousa et al., 2018; Yang et al., 2021). However, many of these studies have focused on individual cell types in specific diseases, using disparate experimental approaches and sequencing platforms, which considerably limits our ability to convincingly compare the identified myeloid signatures and their associated functions more widely across CNS pathologies. Connecting these disease‐associated signatures across different studies is an essential unmet need of current research on neuroinflammation and would allow us to uncover intrinsic myeloid response programs to CNS perturbation, which could realistically inform the development of novel therapeutic and diagnostic tools.
To address this issue, we present a detailed meta‐analysis integrating the transcriptomes of resident and CNS‐infiltrating myeloid cells from demyelinating, ischemic, neurodegenerative, traumatic injury, and infectious conditions. Using high‐parameter data integration and visualization techniques, including clustering algorithms and dimensionality reduction techniques, we compared these unique profiles to understand the relationship between disease models and cell types. Specifically, we demonstrate that microglia and monocyte transcriptomes are highly divergent across pathologies, emphasizing the importance of individual disease environments in shaping myeloid immune responses. We suggest that these specialized disease‐specific signatures shown in bulk populations are driven by the differential proportional expansion of unique subpopulations, identified by scRNA‐seq integration, in different disease contexts. Further, differential expression analysis between microglia and monocytes across pathologies enabled the identification of the tetraspanin gene, Cd81, as a microglia‐enriched transcript that reliably discriminates between microglia and infiltrating monocytes across all examined CNS disease models, thereby showcasing the predictive and practical value of this resource.
2. METHODS
2.1. Resource availability
2.1.1. Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Professor Nicholas King (nicholas.king@sydney.edu.au).
2.1.2. Materials availability
This study did not generate new reagents or datasets.
2.2. Experimental model and subject details
2.2.1. Mice used for flow cytometry studies
Female 9–10 week‐old C57BL/6J mice were obtained from the Animal Resource Centre (Western Australia, Australia) and kept in individually ventilated cages under specific pathogen‐free conditions with access to food and water ad libitum in accordance with National Health and Medical Research Council's ethical guidelines and the University of Sydney Animal Ethics Committee under the animal ethics approval number 2019/1696. Mice were randomly assigned to an experimental condition, that is, non‐infected and infected, with at least three mice per group.
2.3. Method details
2.3.1. WNV infection
Mice were anesthetized with isoflurane before they were intranasally infected with 1.2 × 105 plaque‐forming units of West Nile virus (WNV) (Sarafend) delivered in 10 μl of sterile PBS, as previously described (Getts et al., 2008). The original virus stock was acquired from The John Curtin School of Medical Research (ACT, Australia).
2.3.2. Tissue preparation and staining for flow cytometry
Mice were anesthetized by intraperitoneal injection of avertin and perfused transcardially with ice cold sterile PBS before tissue collection at day 7 post‐infection. Spleen, BM, and brains were collected in ice cold PBS. For BM cells, mouse femurs were flushed using a 30‐gauge needle in PBS. Spleens were homogenized into single‐cell suspensions using 70 μm nylon mesh sieves and plastic syringe plungers. RBC lysis buffer (Invitrogen) was used to lyse erythrocytes in single cell suspensions of BM cells and splenocytes. Brains were processed into single‐cell suspensions in PBS and DNase I (0.05 mg/ml) and collagenase (5 mg/ml) (Sigma‐Aldrich, MO, USA) using the gentleMACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) at 37°C for 30 min. Brain cells were subsequently isolated from a 30%/80% Percoll gradient, as previously described (Spiteri et al., 2021; Spiteri et al., 2022). After tissue processing, live cells were counted with trypan blue (0.4%) on a hemocytometer. Single‐cell suspensions were incubated with purified anti‐CD16/32 and Zombie UV Fixable Viability Kit (Biolegend) for 30 min at 4°C in PBS and subsequently stained with a cocktail of fluorescently‐labeled antibodies (listed Table 1) in FACS buffer for 30 min at 4°C. Cells were washed twice and resuspended in fixation buffer (Biolegend). Anti‐CD206 (C068C2, Biolegend) was used to stain brain cells intracellularly after surface staining, fixation and incubation with Cytofix/Cytoperm (BD Biosciences).
TABLE 1.
Key resources table
| Reagent or resource | Source | Identifier |
|---|---|---|
| Antibodies | ||
| Brilliant Violet 510™ anti‐mouse 1‐A/I‐E | Biolegend | Cat#107635; Clone: M5/114.15.2 |
| Brilliant Violet 570™ anti‐mouse CD4 | Biolegend | Cat#100542; Clone: RM4‐5 |
| Brilliant Violet 605™ anti‐mouse Ly‐6C | Biolegend | Cat#128036; Clone: HK1.4 |
| Brilliant Violet 650™ anti‐mouse Ly‐6G | Biolegend | Cat#127641; Clone: 1A8 |
| Brilliant Violet 711™ anti‐mouse F4/80 | Biolegend | Cat#123147; Clone: BM8 |
| Brilliant Violet 785™ anti‐mouse CD11c | Biolegend | Cat#117336; Clone: N418 |
| PerCP/Cyanine5.5 anti‐mouse Siglec H | Biolegend | Cat#129614; Clone: 551 |
| PE anti‐mouse CD115 | Biolegend | Cat#135506; Clone: AFS98 |
| BD Horizon™ BUV395 Anti‐mouse CD11b | BD Biosciences | Cat#563553; Clone: M1/70 |
| PE anti‐P2RY12 | Biolegend | Cat#848004; Clone: S16007D |
| PE anti‐mouse/rat CD81 | Biolegend | Cat#104906; Clone: Eat‐2 |
| PE/Dazzle™ 594 anti‐mouse CD3ε | Biolegend | Cat#100348; Clone: 145‐2C11 |
| PE/Cyanine5 anti‐mouse NK‐1.1 | Biolegend | Cat#108716; Clone: PK136 |
| PE/Cyanine7 anti‐mouse CD64 (FcγRI) | Biolegend | Cat#139314; Clone: X54‐5/7.1 |
| PE/Cyanine7 anti‐mouse CD45 | Biolegend | Cat#103114; Clone: 30‐F11 |
| APC anti‐P2RY12 | Biolegend | Cat#848006; Clone: S16007D |
| APC anti‐mouse/rat CD81 | Biolegend | Cat#104910; Clone: Eat‐2 |
| BD Horizon™ BUV737 Anti‐mouse CD45R/B220 | BD Biosciences | Cat#612838; Clone: RA3‐6B2 |
| BD Horizon™ BUV737 Anti‐mouse CD11c | BD Biosciences | Cat#612796; Clone: HL3 |
| Alexa Fluor® 700 anti‐mouse CD45 Antibody | Biolegend | Cat#103128; Clone: 30‐F11 |
| APC/Cyanine 7 anti‐mouse CD86 | Biolegend | Cat#105030; Clone: GL‐1 |
| APC/Cyanine 7 anti‐mouse CD48 | Biolegend | Cat#103432; Clone: HM48‐1 |
| BD Horizon™ BUV805 Anti‐mouse CD8a | BD Biosciences | Cat#612898; Clone: 53–6.7 |
| Brilliant Violet 421™ anti‐mouse CX3CR1 | Biolegend | Cat#149023; Clone: SA011F11 |
| Brilliant Violet 421™ anti‐mouse CD117 (c‐Kit) | Biolegend | Cat#B105828; Clone: 2B8 |
| Brilliant Violet 785™ anti‐mouse CD206 (MMR) | Biolegend | Cat#141729; Clone: C068C2 |
| TruStain FcX™ anti‐mouse CD16/32 | Biolegend | Cat#101320; Clone: 93 |
| Bacterial and virus strains | ||
| WNV (Sarafend) | The John Curtin School of Medical Research (ACT, Australia) | |
| Chemicals, peptides, and recombinant proteins | ||
| Fixation Buffer | Biolegend | Cat#420801 |
| BD Cytofix/Cytoperm™ Fixation and Permeabilization Solution | BD Biosciences | Cat#554722 |
| Zombie UV™ Fixable Viability Kit | Biolegend | Cat#423108 |
| eBioscience™ 10× RBC Lysis Buffer | Invitrogen | Cat#00‐4300‐54 |
| Deposited data | ||
| Keren‐Shaul et al., 2017 | Gene Expression Omnibus | GSE98969 |
| Krasemann et al., 2017 | Gene Expression Omnibus | GSE101686 |
| Krasemann et al., 2017 | Gene Expression Omnibus | GSE101688 |
| Locatelli et al., 2018 | Gene Expression Omnibus | GSE107792 |
| Mendiola et al., 2020 | Gene Expression Omnibus | GSE146113 |
| Werner et al., 2020 | Gene Expression Omnibus | GSE120701 |
| Lewis et al., 2014 | Gene Expression Omnibus | GSE59725 |
| DePaula‐Silva et al., 2019 | Gene Expression Omnibus | GSE127233 |
| Hammond et al., 2019 | Gene Expression Omnibus | GSE121654 |
| Somebang et al., 2021 | Gene Expression Omnibus | GSE175430 |
| Milich et al., 2021 | Gene Expression Omnibus | GSE162610 |
| Experimental models: Organisms/strains | ||
| Mouse: C57BL/6J | Animal Resource Centre (ARC) (Western Australia, Australia) | |
| Software and algorithms | ||
| FlowJo v10.8 | BD Biosciences | https://www.flowjo.com |
| DESeq2 | Love et al., 2014; Galaxy Version 2.11.40.6 + galaxy1 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/deseq2/deseq2/2.11.40.6+galaxy1 |
| featureCounts | Liao et al., 2014; Galaxy Version 2.0.1 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/featurecounts/featurecounts/2.0.1 |
| annotatemyIDs | Galaxy Version 3.12.0 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/annotatemyids/annotatemyids/3.12.0 |
| CutAdapt | Martin, 2011; Galaxy Version 3.4 + galaxy0 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/lparsons/cutadapt/cutadapt/3.4+galaxy0 |
| HISAT2 | Kim et al., 2015; Galaxy Version 2.1.0 + galaxy7 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/iuc/hisat2/hisat2/2.1.0+galaxy7 |
| FastQC | Andrews, 2010; Galaxy Version 0.72 + galaxy1 | https://usegalaxy.org/root?tool_id=toolshed.g2.bx.psu.edu/repos/devteam/fastqc/fastqc/0.72+galaxy1 |
| Seurat v4 | Hao et al., 2021 | https://satijalab.org/seurat/ |
| Spectre | Ashhurst et al., 2021 | https://immunedynamics.io/spectre/ |
| LIGER v0.5.0 | Welch et al., 2019 | https://CRAN.R‐project.org/package=rliger |
| topGO v2.44.0 | Alexa & Rahnenfuhrer, 2022 | https://bioconductor.org/packages/topGO/ |
| pheatmap v1.0.12 | Kolde, 2012 | https://cran.r‐project.org/web/packages/pheatmap/index.html |
| Python v3.8.5 | Vanrossum & DeBoer, 1991 | https://www.python.org |
| TPMCalculator | Vera Alvarez et al., 2019 | https://github.com/ncbi/TPMCalculator |
| UpSetR v1.4.0 | Conway et al., 2017 | https://cran.r‐project.org/web/packages/UpSetR/index.html |
| ViSEAGO v1.6.0 | Brionne et al., 2019 | https://bioconductor.org/packages/ViSEAGO |
| RankProd 2.0 | Del Carratore et al., 2017 | https://www.bioconductor.org/packages/devel/bioc/html/RankProd.html |
| ggplot2 v3.3.5 | Wickham, 2006; Wickham & Wickham, 2007 | https://ggplot2.tidyverse.org/ |
| Galaxy | Afgan et al., 2016; Jalili et al., 2020 | https://usegalaxy.org.au |
| SuperExact test | Wang et al., 2015 | https://cran.r‐project.org/web/packages/SuperExactTest/index.html |
2.3.3. Acquisition and processing of flow cytometry data
Fluorescently‐labeled antibodies were measured using the 5‐laser Aurora, Spectral cytometer (Cytek Biosciences). Acquired data was analyzed in FlowJo (BD Biosciences, v10.8). Quality control gating including time, single cells, non‐debris/cells and Live/Dead staining was applied to exclude debris, doublets and dead cells prior to analysis. Populations shown in Figure 8 were gated as previously published (Spiteri et al., 2021). Histograms, dot plots (showing surface markers) and tSNE plots (using default settings) shown in Figure 8 were created in FlowJo (BD Biosciences, v10.8). The clustered heatmap shown in Figure 8 was made in R using the pheatmap package (v1.0.12) (Kolde, 2012).
FIGURE 8.
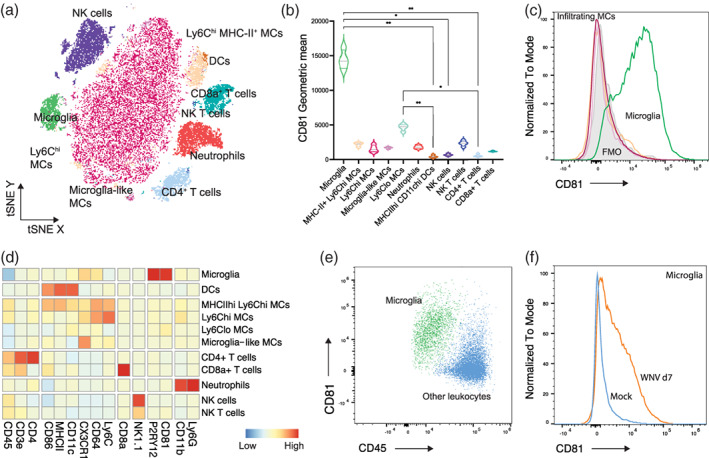
CD81 is a microglial‐enriched protein expressed in neuroinflammation. (a) tSNE plot showing West Nile virus‐infected brains with resident and infiltrating CD45+ leukocyte populations overlaid. (b) Violin plots showing the expression of CD81 on cell populations identified on the tSNE plot. (c) Histogram showing the expression of CD81 by microglia relative to infiltrating macrophage populations and fluorescence minus one (FMO) for CD81 in infected brains. See also Figures S3 and S4 showing CD81 expression by leukocyte populations in the bone marrow and spleen, respectively. (d) Heatmap showing the expression of markers on cell populations shown in (a). (e) FACs plot showing the expression of CD81 and CD45 on microglia and other CD45+ leukocytes in infected brains. (f) Histogram showing the expression of CD81 on microglia from mock and infected brains at day 7 post infection
2.4. Quantification and statistical analysis
2.4.1. Identification and selection of eligible gene expression datasets for meta‐analysis
Gene expression studies of acutely isolated microglia and MCs from adult mouse brains or spinal cords in various disease conditions were considered for our analysis. We systematically mined PubMed database for microarray, bulk RNA‐seq, and scRNA‐seq expression profiling. In addition, publicly availably transcriptomic data sets were searched in the NCBI Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). The following search terms were used: “neuropath*” [Title/Abstract], “neuroinflammation” [Title/Abstract], “neuroinflammatory diseases” [Title/Abstract], “cns pathology” [Title/Abstract], “spinal cord injury” [Title/Abstract], “stroke” [Title/Abstract], “viral encephalitis” [Title/Abstract], “multiple sclerosis” [Title/Abstract], “encephalitis” [Title/Abstract], “Alzheimer's disease” [Title/Abstract], “Parkinson's disease” [Title/Abstract], “amyotrophic lateral sclerosis” [Title/Abstract], “motor neuron disease” [Title/Abstract], “neurodegeneration” [Title/Abstract], “neurodegenerative disease” [Title/Abstract], “macrophage” [Title/Abstract], “myeloid” [Title/Abstract], “monocyte‐derived” [Title/Abstract], “monocyte” [Title/Abstract], “microglia” [Title/Abstract], “single cell rna seq” [Title/Abstract], “single cell rna sequencing” [Title/Abstract], “single cell rna seq” [Title/Abstract], “single cell rna sequencing” [Title/Abstract], “scRNA‐seq” [Title/Abstract], “single cell transcriptom*” [Title/Abstract], “single cell transcriptom*” [Title/Abstract], “RNA seq” [Title/Abstract], “RNA sequencing” [Title/Abstract], “Bulk transcriptome*” [Title/Abstract], “Gene Expression Profiling” [Title/Abstract], “Transcriptome” [Title/Abstract], “gene expression” [Title/Abstract]. References of identified articles were additionally searched. Studies were eligible for inclusion if: (1) raw data was accessible on a public data repository, (2) data was acquired by nanostring, bulk or single‐cell RNA‐sequencing technologies; and (3) data included either microglia in disease and control conditions or both microglia and MCs in disease conditions. Exclusion criteria for all eligible studies included (1) additional comorbidities (e.g., diabetes, obesity, etc.); (2) adoptively transferred microglia or monocytes; (3) gene knock‐out or mutant models but not excluding transgenic mice e.g., APP/PS1, 5xFAD, SOD1G93A; (4) transcriptomes from pharmacologically‐treated mice, unless data from non‐treated disease and homeostatic controls were available; (5) single‐cell RNA‐sequencing data included a minimum of 600 single cells passing quality‐control metrics (see 2.4.4 Single‐cell RNA‐sequencing analysis, Pre‐processing and normalization). Lists of excluded studies and reasons for exclusion, along with all study metadata characteristics for included studies (including time points at which mice were sacrificed, sorting methods and other possible confounding variables) are provided in Table S1. Excluded data included 19 samples from Keren‐Shaul et al., 2017 (see Table S1 for sample details and reasons for exclusion of each sample). Ten transcriptomic datasets from eight separate studies were integrated to identify conserved and disease‐specific transcriptional signatures and for differential expression analysis (see Table 1 and Table S1). The following information was recorded for each study: GEO expression number, cell type, disease model, mouse strain, mouse age (weeks), mouse sex, sequencing method, sequencing platform, layout (single vs. paired), scRNA‐seq protocol, library size, cell sorting strategy, anatomical region, and tissue processing method (Table S1). We additionally determined the FAST QC results for bulk RNA‐seq data sets and the median percent mitochondrial gene expression, median number of unique genes per cell, and total number of molecules per cell for all included scRNA‐seq data (Table S1).
2.4.2. Bulk RNA dataset pre‐processing and normalization
FASTQ files were downloaded from the Gene Expression Omnibus (Barrett et al., 2013) and loaded into the online Galaxy platform (Afgan et al., 2016; Jalili et al., 2020). Cutadapt (v 1.16.6) (Martin, 2011) was used to trim known adaptor and primer sequences from raw reads and filter low quality (minimum quality cutoff = 20) and short reads (minimum length = 20 bp after trimming). All reads passed quality control checks using FastQC (v0.72 + galaxy1) (Andrews, 2010). Trimmed and filtered reads were then aligned to the built‐in mm10 reference genome using HISAT2 (v2.1.0 + galaxy7) (Kim et al., 2015). FeatureCounts was used to generate counts from HISAT2 output files using the built‐in mm10 gene annotation file (v2.0.1) (Liao et al., 2014) and known genes were annotated with AnnotateMyIDs (v3.12.0). Transcripts per million mapped reads (TPM) values were calculated using TPMCalculator (Vera Alvarez et al., 2019) in Python (Vanrossum & DeBoer, 1991). The same pre‐processing, alignment and normalization pipeline was used for each bulk RNA‐sequencing study.
2.4.3. Nanostring data pre‐processing and normalization
Nanostring data was acquired from Krasemann et al. (2017) already pre‐processed and normalized using nSolver™ software. Data was not re‐analyzed from raw files since it was uniformly processed by a single study.
2.4.4. Single‐cell RNA sequencing analysis
Pre‐processing and normalization
Matrices of UMI counts were downloaded from the Gene Expression Omnibus (Barrett et al., 2013). The R toolkit Seurat v4.0 (Hao et al., 2021) was used for quality control processing, graph‐based clustering, visualizations and differential gene expression analyses of scRNA‐seq datasets and performed in R (v.4.0.3). Each scRNA‐seq dataset was processed separately. Samples were filtered to remove cells that were either empty droplets or possible doublets/multiples or had a higher percentage of reads mapping to the mitochondrial genome. Cells in each dataset were included if the number of unique genes detected per cell were between 250 and 25,000 and the percentage of mitochondrial reads were less than 5%. As mitochondrial genes were not present in Keren‐Shaul et al. (2017), this dataset was filtered only by the number of unique genes per cell. Low quality cells were prefiltered from Milich et al., (2021). The number of cells in each dataset before and after filtering is shown in Table 2. Normalization and variance stabilization of UMI counts were performed using the sctransform() function in Seurat (Hafemeister & Satija, 2019). Where possible, mitochondrial gene expression and total number of reads per cell were regressed out to control for confounding sources of variation. After normalization and variance stabilization, linear dimensionality reduction was performed using PCA on the top 2000 highly variable genes.
TABLE 2.
Number of single cells passing quality control metrics in each single‐cell RNA‐sequencing dataset
| First author | GSE # | Disease model | # Cells before QC | # Cells after QC | # QC myeloid cells |
|---|---|---|---|---|---|
| Mendiola et al. | GSE146113 | EAE | 9079 | 6859 | 3297 |
| Hammond et al. | GSE121654 | LPC‐induced demyelination | 5510 | 5408 | 5408 |
| Keren‐Shaul et al. | GSE98969 | 5xFAD transgenic mice | 10,146 | 9636 | 3958 |
| Keren‐Shaul et al. | GSE98969 | SOD1G93A transgenic mice | 2820 (day 80 only) | 2693 | 1091 |
| Milich et al. | GSE162610 | Spinal cord injury | 66,428 | 54,049 | 35,518 |
| Somebang et al. | GSE175430 | Traumatic brain injury | 99,769 | 90,998 | 17,097 |
Abbreviations: 5xFAD, 5 familial AD mutations; EAE, experimental autoimmune encephalomyelitis; LPC, lysophosphatidylcholine; QC, quality control.
Clustering and cell type annotation
To visualize the pre‐processed and normalized scRNA‐seq data in two‐dimensional space, Uniform Manifold Approximation and Projection (UMAP) was performed using the first 29 principal components. We used FindNeighbours() and FindClusters() functions in Seurat with default parameters to perform graph‐based clustering on a shared nearest‐neighbor graph. The optimal clustering resolution was determined by selecting the lowest resolution separating distinct clusters by a decision tree. Cell type annotation of clusters was performed using unbiased gene marker analysis. We performed differential expression analysis using the FindAllMarkers() function in Seurat with default parameters, which implements a Wilcoxon rank‐sum test comparing gene expression of cells within a given cluster versus all other cells. For a given cluster, genes with adjusted p < .05 and log2FC >0.25 were considered as markers for that cluster. Cell type annotation for myeloid cells were made based on expression of microglial‐ and MC‐specific genes. For downstream integration with LIGER and RankProduct analysis, datasets were subsetted so that only myeloid populations (“MC” or “microglial” clusters) were present in the datasets.
Integration of myeloid cells using LIGER
Prior to integration, data sets from each disease model were downsampled to the limiting cell number (469 microglia from Mendiola et al.) to ensure equal representation of each disease model in the integrated data set. Absolute cell numbers for each disease model prior to downsampling were 685 MCs and 1960 microglia in lysophosphatidylcholine (LPC)‐induced demyelination; 3234 MCs and 1468 microglia in experimental autoimmune encephalomyelitis (EAE); 182 MCs and 3620 microglia in 5xFAD; 72 MCs and 1136 microglia in SOD1G93A; 14,243 MCs and 17,739 microglia in spinal cord injury (SCI); and 612 MCs and 8434 microglia in traumatic brain injury (TBI). Microglia from each of the six disease models were downsampled to 469 cells (2814 microglia total) and 469 MCs were selected from EAE, TBI, SCI (1407 MC total), since MCs were not flow cytometrically sorted or sequenced by the studies investigating 5xFAD, SOD1G93A, or LPC‐induced demyelination disease models (Table S1).
The R package rliger v0.5.0 (Welch et al., 2019) was used to align and integrate the pre‐processed and subsetted myeloid cells from scRNA‐seq datasets. Only samples from disease conditions were included in this analysis. Data was integrated following a previously described protocol by the Welch Lab (Liu et al., 2020). In brief, normalization, variable gene selection, and scaling of individual genes were performed with default parameters on the combined dataset. The function suggestK (num.cors = 6, lamda = 5) was used to determine the optimal number of factors (k), and suggestLamda was used to determine the optimal regularization parameter, λ (Figure S1). Joint matrix factorization was performed on the normalized and scaled dataset using integrative non‐negative matrix factorization with optimal parameters k = 12 and λ = 5 to define dataset‐specific and shared metagenes (i.e., factors), which correspond to genes that define particular cell types and subsets. The resulting factors were then used to jointly cluster cells and perform quantile normalization by dataset, factor and cluster using the LIGER function quantile_norm(). Clustering of cells were visualized graphically by UMAP implemented in LIGER with default parameters. Cell type annotation for the four MC clusters and eight microglial clusters were made based on expression of known marker genes (Figure S1). The LIGER‐integrated dataset was converted to a Seurat object for downstream visualization and differential expression analysis. All plots of integrated scRNA‐seq data were constructed using Seurat (v4.0.1) or rliger (v0.5.0).
Differential expression testing and GO analysis
Identification of differentially expressed genes (DEGs) and gene ontology (GO) enrichment analysis was performed on myeloid clusters identified in the LIGER‐integrated dataset. To identify marker genes for each cluster, the FindAllMarkers() function implemented in the Seurat v4.0.1 package was implemented with default parameters. DEGs for myeloid clusters were calculated using the Seurat function FindAllMarkers() using and defined as positively enriched genes with an adjusted p < .01 and log2FC >0.25 (Wilcox sum rank test), which represents genes enriched in the cluster versus all other clusters.
Functional GO enrichment analysis from DEGs of each population (by cluster or by bulk cell population for each disease model) was performed using the VISEAGO (v1.4.0) and topGO (v2.42.0) packages in R. GO biological process term enrichment was performed using the VISEAGO create_topGOdata() relative to the background gene expression, which was defined as the full list of genes expressed in the LIGER‐integrated data set. Enrichment tests were performed with Fisher's exact test using both the “classic” and “elim” algorithms. Enriched GO terms were defined as terms with a minimum of 10 genes mapping to a term and an adjusted p‐value greater than .01. For visualization of GO terms, GO terms for each population were combined into a single matrix using the ViSEAGO function build_GO_SS() (Brionne et al., 2019) and annotated using the Bioconductor org.Mm.eg.db database package for the mouse species (Carlson, 2019). UpSet visualization was performed on the significantly enriched GO term matrices using the UpSetR package (v1.4.0) (Conway et al., 2017), and the significance of intersections was calculated using the SuperExactTest (v1.0.7) R package, which reports one‐tailed p values (Wang et al., 2015).
2.4.5. Using a rank product statistic to generate gene ranks
To compare datasets from different origins and sequencing platforms, we applied a rank product (RP) statistic to genes common to all studies (319 genes). To increase our gene list (7804 genes) we excluded the dataset with the limiting number of genes (Krasemann et al., 2017) and reapplied the same RP analysis. A RP is a non‐parametric statistical test that ranks gene by their fold‐change values, adjusted p‐value and percentage of false discovery (Del Carratore et al., 2017; Hong et al., 2006). The RP output consists of two tables: genes ranked in order of upregulation and genes ranked in order of downregulation. Genes more likely to be up‐ or down‐regulated in each table have a lower RP value and thus are more highly ranked. A RP was applied to normalized expression values using a two‐class setting (i.e., control vs disease) and a single‐ (samples originating from one study) function. Only studies with a minimum of two replicates per condition could be included in this analysis (this necessitated the exclusion of DePaula‐Silva et al., 2019) (Table S1).
2.4.6. 2D visualization of gene ranks
Table 1 from the single‐origin RP analysis on the common gene list A (319 genes) were used for hierarchical clustering and heatmap visualization, bubble plots and UMAPs. Clustered heatmaps were generated in R Studio using the pheatmap package (v1.0.12) (Kolde, 2012). Rows and/or columns were clustered on for hierarchical clustering using the complete parameter for clustering_method() and Euclidean parameter for clustering_distance(). Bubble plots were made using the ggplots2 package (v3.3.3) in R studio (Wickham, 2006; Wickham & Wickham, 2007). The R package, Spectre (Ashhurst et al., 2021), was used to perform dimensionality reduction by UMAP on data clustered by k‐means clustering with FlowSOM (xdim = 5; ydim = 5; meta.k = auto) on the common gene list.
2.4.7. GO analysis and UpSet visualization of gene ranks
Functional GO enrichment analysis was performed on differentially expressed or upregulated genes (log2FC >1, p value <.05). The enriched biological process results were obtained using a Fisher's exact test with elim algorithm developed in the topGO package. Significantly enriched GO terms were defined as terms with a minimum of ten significantly enriched genes mapping to a term and an enrichment p < .05 relative to the gene background (all expressed genes in the population). For GO analysis in Figure 3, GO terms for each population were combined into a single matrix using the ViSEAGO function build_GO_SS() (Brionne et al., 2019) and annotated using the Bioconductor org.Mm.eg.db database package for the mouse species (Carlson, 2019). UpSet visualization was performed on the significantly enriched GO term matrices using the UpSetR package (Conway et al., 2017).
FIGURE 3.
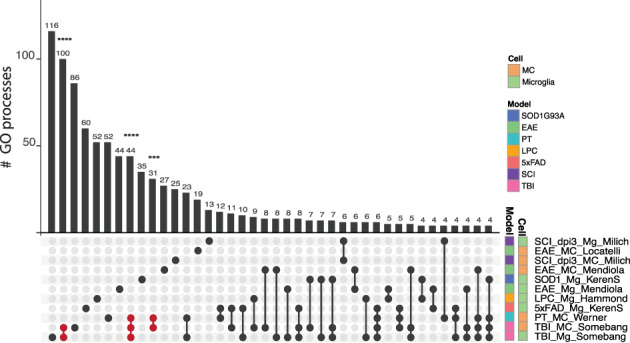
Cell type‐specific programs are dependent on disease‐specific perturbation. UpSet plot showing shared and unique gene ontology (GO) biological processes associated with genes significantly upregulated (p > .05, log2 > 1) by microglial and MC populations out of the 7804 genes of the Common Gene set B. Populations are colored by cell type and disease‐model membership and arranged in order of increasing to decreasing number of biological processes associated with each population. The number of shared or unique GO biological processes (p < .05) are shown by the bar graph and the corresponding population(s) are indicated by the dot plot panel below. Lines connecting populations indicate shared GO processes. Individual populations are indicated by a singular dark circle with no intersecting lines. Significant intersections are shown in red and were determined using the SuperExact test. See Table S3 for a list of the GO terms and significance of each intersection shown
2.4.8. Identification of differentially enriched genes
To define cell type‐enriched markers, only studies that processed RNA from MCs and microglia in the same disease state were included in our analysis. These included (1) Locatelli et al. (2018), (2) Mendiola et al. (2020), (3) Werner et al. (2020), (4) Milich et al. (2021), (5) Somebang et al. (2021) and (6) DePaula‐Silva et al. (2019). For all bulk RNA‐seq datasets, differential expression analysis comparing MCs to microglia was performed in DESeq2 on the online Galaxy platform from HISAT2 outputs (v2.11.40.6 + galaxy1) (Love et al., 2014). For the scRNA‐seq dataset, fold‐change values from RP outputs were used, since scRNA‐seq data was incompatible with DESeq2 in the online Galaxy platform. DEGs common to all studies were identified and filtered for genes with a log2FC > 1 and p < .05. Membrane‐expressed DEGs were identified by annotating gene names in Uniprot (UniProt, 2021). Total enriched genes were filtered for (1) the term cell membrane in the Uniprot category subcellular location and (2) the terms integral component of plasma membrane or cell surface in the Uniprot category Gene ontology. The expression of identified DEGs by microglia and MCs was confirmed at the single‐cell level in the LIGER‐integrated scRNA‐seq dataset.
3. RESULTS
3.1. Integrating studies across CNS disease with gene rank analysis
To determine how microglial and MC responses converge or diverge across different pathological settings, we performed an integrative meta‐analysis on eight published studies from independent laboratories describing different disease models and/or different experimental approaches. In our analysis, we included transcriptomic datasets from murine models of acute inflammation and chronic neurodegeneration, including photothrombosis (PT)‐induced focal ischemia (Werner et al., 2020), spinal cord injury (SCI) (Milich et al., 2021) and traumatic brain injury (TBI) (Somebang et al., 2021) models of sterile injury, experimental autoimmune encephalomyelitis (EAE) (Krasemann et al., 2017; Locatelli et al., 2018; Mendiola et al., 2020) and lysophosphatidylcholine (LPC)‐induced demyelination (Hammond et al., 2019) models of multiple sclerosis (MS), 5xFAD (Keren‐Shaul et al., 2017) and APP/PS1 (Krasemann et al., 2017) transgenic models of Alzheimer's disease (AD), and the SOD1G93A model of amyotrophic lateral sclerosis (ALS) (Keren‐Shaul et al., 2017; Krasemann et al., 2017) (Figure 1a,b and Table S1). This data was obtained from three different sequencing platforms, including nanostring, scRNA‐seq or bulk RNA‐sequencing technologies (Figure 1b).
FIGURE 1.
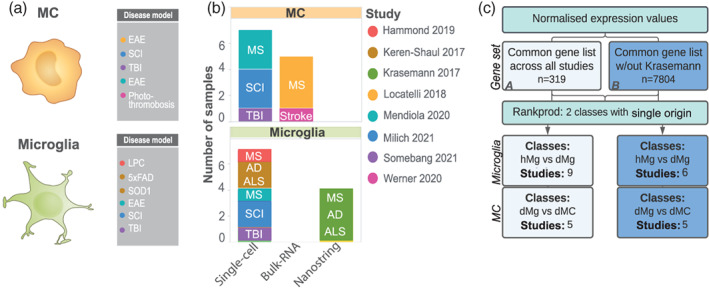
Integrating studies across CNS disease with gene rank analysis. (a–b) Summary of datasets used in the single origin rank product (RP) analysis including cell type, disease and disease model, number of samples, study origin and sequencing modality used. (c) Flow chart of analysis pipeline. Single origin RP analysis was performed on normalized expression values on a common gene list from eight datasets (n = 319 genes, Common Gene set A) or seven datasets (n = 7804 genes, Common Gene set B), including or excluding Krasemann et al., 2017, respectively. Microglia fold‐change calculations were made between microglia from disease (dMg) and homeostatic/control conditions (hMg). MC fold‐change calculations were made between MCs from disease conditions (dMC) and microglia from disease conditions (dMg)
Direct comparison of gene expression or fold‐change data acquired from different sequencing technologies, laboratories and disease models is highly challenging, as global normalization often fails to overcome lab‐specific batch effects. To surmount this, we integrated data sets from multiple origins using a rank product approach on genes common to all studies (Figure 1c). This method employs a simple non‐parametric test to generate a single‐origin rank product (RP) statistic, which ranks genes in order of upregulation and downregulation for each study using fold‐change, adjusted p‐value and percentage of false predictions (Del Carratore et al., 2017; Hong et al., 2006). Thus, a comparison of gene ranks or RP values across different studies enables a reliable determination of which genes are likely to be regulated across different CNS disease models.
We performed a single‐origin RP analysis on transcriptomic data sequenced from both microglia and MCs (Figure 1a–c). Within each study, fold‐change calculations were made between microglia from disease (dMg) and homeostatic/control conditions (hMg) (Figure 1c). However, the absence of peripherally‐derived MCs in homeostatic brains meant this control was unavailable for MCs in these studies. Therefore, fold‐change calculations were instead made between MCs from disease conditions (dMC) and microglia from disease conditions (dMg) (Figure 1c). While disease‐related microglia are a suboptimal control for disease‐related MCs, such comparisons are used to assess MC gene expression in the brain in most studies, as more suitable controls (i.e., blood or BM‐derived monocytes) are rarely included in published work. However, this decision illustrates the intrinsic challenges underlying a comprehensive meta‐analysis of published data. Thus, while this approach aligns with previously published work (DePaula‐Silva et al., 2019; Schlachetzki et al., 2018; Yamasaki et al., 2014), accurately understanding MC biology will require consensus MC controls to be included by future investigators.
Integrating data sets by genes common to all eight studies narrowed our gene list to 319 genes (Common Gene set A). This limited number of genetic candidates was dictated by the small dataset published by Krasemann et al., 2017. Thus, to broaden the scope of our analysis, we performed a parallel analysis where we excluded the smallest data set (Krasemann et al., 2017, n = 411 genes), thereby increasing our gene list 24‐fold (n = 7804 genes) (Common Gene set B), but reducing the number of studies to 7 (Figure 1c).
3.2. Cell type‐specific programs are dependent on disease‐specific perturbation for microglia and MCs
To compare the disease signature of microglia and monocyte‐derived cells to the nominal universal microglial signature, as defined in Krasemann et al., 2017 we first used the Common Gene set A (n = 319 genes). Strikingly, we observed few similarities between disease‐specific signatures. This is shown by the minimal overlap in the top 10 highest and lowest ranked genes and the trajectory of gene ranking, as visualized by the plateau phases and slopes of the bubble plots (Figure 2a). Notably, no commonly upregulated genes were identified across diseases between either microglia or MC populations. This suggests that microglia and MCs adopt separate, highly specific responses to unique disease settings, with minimal overlap across pathologies that is distinct from the universal microglial signature identified in Krasemann et al., 2017.
FIGURE 2.
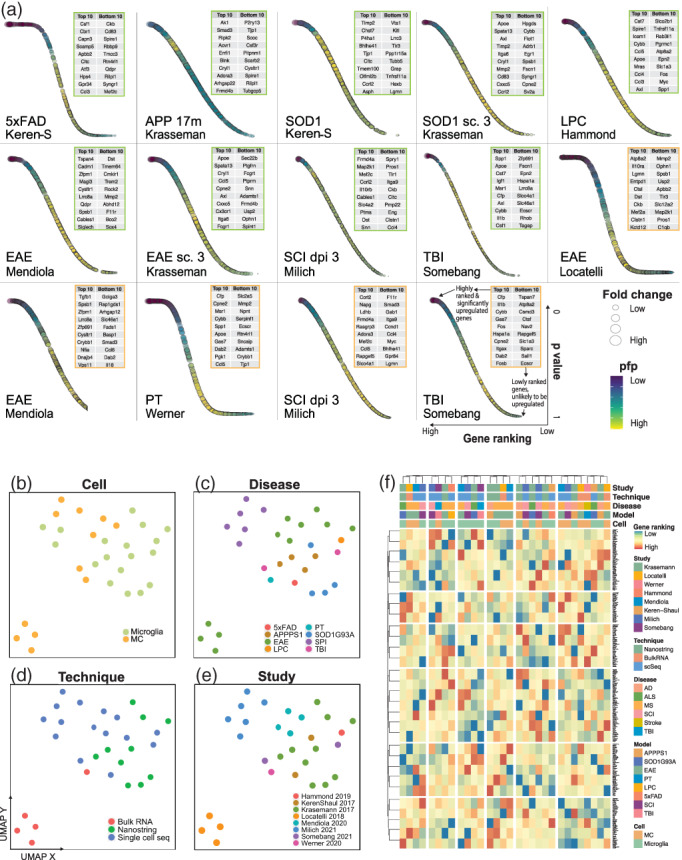
Clustering analysis connects the transcriptomic responses adopted by microglia and MCs in disease. (a) Bubble plots showing gene ranking using Common Gene set A by order of upregulation (x‐axis) with associated p‐values (y‐axis), fold‐change (bubble size) and percentage of false predictions (pfp) values (bubble color) for selected MC (the cell type indicated by orange boxes) and microglia (indicated by green boxes) populations. The top‐ and bottom‐ranked genes are listed in these boxes, placed at the top right of each plot, for MC (orange boxes) and microglia (green boxes). (b–e) UMAP plots of 18 microglia and 12 MCs samples pseudocolored by cell type (b), disease (c), sequencing technology (d) or study (e). UMAPs were run on gene rank values for the 319 genes of Common Gene set A. See also Table S2 for a list of populations used with their study and disease origins. (f) Heatmap showing gene rank values for the 319 genes of Common Gene set A in order of upregulation for MC and microglial populations. Clustering was performed on both rows (genes) and columns (populations). See also Table S2 for a list of populations used with their study and disease origins in order of the clustering arrangement shown in the heatmap
Next, to confirm these disease‐specific responses we coupled our gene rank analysis with an unbiased approach using unsupervised dimensionality reduction, k‐means clustering and hierarchical clustering (Figure 2b–f). Importantly, clustering analysis of individual myeloid populations showed intermixing of different studies and sequencing platforms, suggesting little or no bias specific to sequencing technology or laboratory in this analysis (Figure 2d–f and Table S2). Notably, MC and microglial populations did not cluster by cell type or by disease model (Figure 2b,c,f), supporting the notion that myeloid populations adopt unique profiles.
We confirmed disease‐specific signatures in Common Gene set B (n = 7804 genes) (Table S3), demonstrating that these differences are not specific to the smaller dataset. We then assessed functional similarities between these unique transcriptional profiles by gene ontology (GO) enrichment analysis on the upregulated genes (log2 fold‐change >1, p‐value <.05) from the larger gene set (Common Gene set B, n = 7804 genes) and evaluated overlap in GO terms between diseases using an UpSet plot (Figure 3). We observed no significant overlapping biological functions between any two microglia populations in any diseases. Significant overlap in these functions, however existed between microglia and MCs in TBI and between MCs in both TBI and stroke (Figure 3, and Table S3). This emphasizes that the transcriptional profiles of microglia and MCs are contingent upon the disease environment, but may converge in a disease‐dependent manner. Overall, however, the limited set of significant intersecting lines between populations indicate more uniquely‐enriched than shared functions across diseases (Figure 3).
3.3. Integration of scRNA‐seq data connects myeloid signatures across disease
We next wanted to evaluate whether these disease‐specific signatures were reflected at the single cell level. We therefore used LIGER (Liu et al., 2020; Welch et al., 2019) to integrate myeloid populations from six scRNA‐seq datasets investigating LPC‐induced demyelination (Hammond et al., 2019), EAE (Mendiola et al., 2020), 5xFAD, SOD1G93A (Keren‐Shaul et al., 2017), TBI (Somebang et al., 2021) and SCI (Milich et al., 2021) (Figure 4a). This approach enabled us to (1) assess gene expression data independent of gene ranking, (2) assess MCs independently of their comparison to disease‐related microglia, and (3) examine microglia and MC subpopulations that may be unique to, or conserved across, multiple disease states, which may be missed at the bulk population level. Prior to data integration, all scRNA‐seq studies underwent the same pre‐processing, normalization, and clustering workflow. Microglia and MCs were defined by their expression of microglia‐specific genes, P2ry12, Tmem119, Sparc, Hexb, Fcrls, Siglech and monocyte‐specific/enriched genes, Ly6c2, Plac8, Vim, Cd44, respectively, and the lack of expression of border‐ and CNS‐associated macrophage‐specific genes (e.g., Cd163, Cd206) (Figure S1) and other cell lineage markers. Identified cell populations did not cluster by anatomical region, suggesting these populations are not spatially‐dependent (Figure S1). Pre‐defined myeloid populations from the above disease conditions were then integrated into a single dataset and downsampled to the limiting cell number. Joint matrix factorization, quantile normalization, dimensionality reduction and joint clustering were then performed on 2814 microglia and 1407 MCs (Figure 4a and Figure S1) in our integrated dataset.
FIGURE 4.
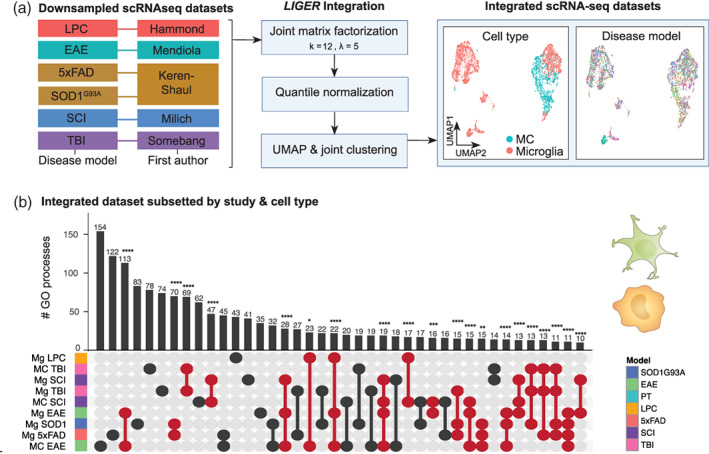
Single‐cell RNA‐sequencing integration workflow with LIGER. (a) Myeloid cells from six disease models from five separate studies were integrated using LIGER. Data sets were downsampled prior to integration to ensure equal representation across disease models (469 microglia from each six disease models, 469 MCs from EAE, TBI, and SCI). Optimal k and lambda values were determined prior to clustering (Figure S1). Datasets were then integrated with LIGER and subjected to joint matrix factorization (k = 12, lambda = 5), quantile normalization, dimensionality reduction and joint clustering (data set alignment = 0.878). UMAPs of the LIGER‐integrated dataset show representation of both cell types intermixed across the six disease models. (b) UpSet plot showing overlap in the number of enriched GO terms for microglia and/or MCs from six disease models from the LIGER‐integrated dataset. GO term enrichment was performed on the differentially expressed genes on pseudo‐bulk cell populations from each disease versus every other disease. Samples are colored by disease‐model membership and arranged in order of increasing to decreasing number of biological processes associated with each population. Populations that share GO terms are indicated by connecting lines in the dot plot. Individual populations are indicated by a singular dark circle with no intersecting lines. Significant intersections are shown in red and were determined using the SuperExact test.
To compare our integrated scRNA‐seq dataset to our gene rank analysis on bulk populations, we first created pseudo‐bulk microglia/MC populations by grouping single cells into cell type, study and disease model. Differential gene expression and GO term enrichment analysis was then perfomed on these pseudo‐bulk populations (Figure 4b). This revealed a similar pattern to our gene rank approach, demonstrating overall that the majority of bulk myeloid populations had more unique than shared biological functions across pathology. When more shared than unique processes existed, these were between microglia and MCs in the same disease setting, e.g., microglia and MCs in EAE, TBI and SCI (Figure 4b). Supporting our RP analysis, this suggests that microglia and MC responses are principally disease‐ and disease model‐specific, with the disease environment bearing an important role in shaping similar profiles of these cells within the same disease.
3.4. Differing proportions of unique myeloid subtypes drive disease‐specific signatures in bulk populations
To understand what drives the identified disease‐specific signatures, we performed clustering analysis on individual single‐cells in the LIGER‐integrated scRNA‐seq. This enabled us to identify myeloid subpopulations that are shared across or unique to each of the six disease models (Figure 5a).
FIGURE 5.
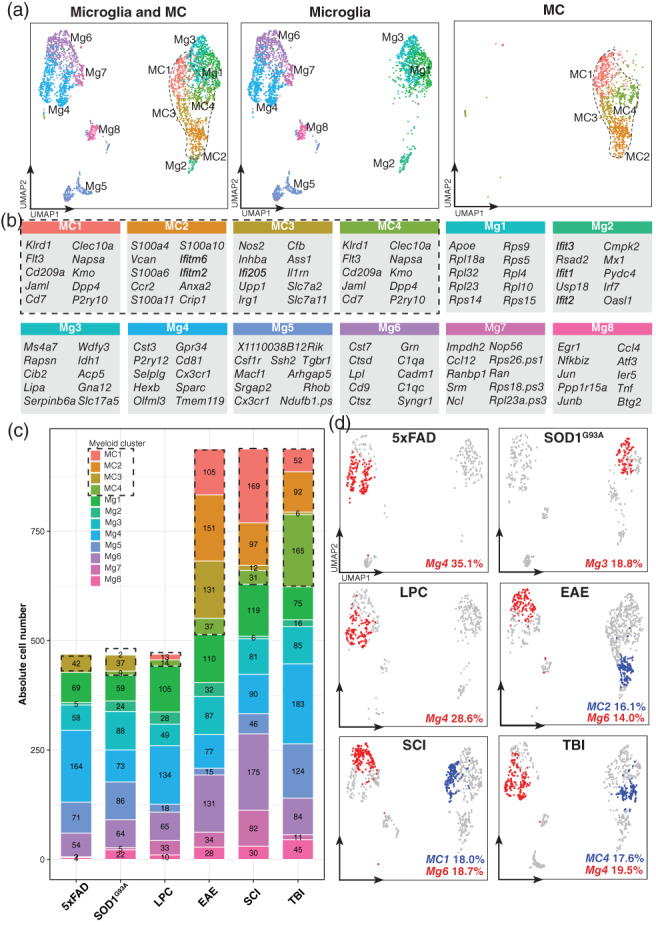
Integrative analysis of single‐cell RNA‐sequencing datasets identifies conserved and disease‐specific MC and microglial clusters across six disease models. (a) UMAPs of myeloid cells from the downsampled LIGER‐integrated dataset overlaid by either microglia (2814 cells) and/or MC clusters (1407 cells). See also Figure S1 for the expression of cell type‐specific genes used to annotate cell types. (b) Top 10 differentially expressed genes per cluster. Differentially expressed genes were defined as genes enriched in a cluster versus all other clusters (log2 fold‐change >0.25, p value <.01) by Wilcoxon rank sum test. (c) Absolute cell numbers of microglial and MC clusters across the six disease models. Datasets were downsampled prior to integration to ensure equal representation across disease models (469 microglia from each six disease models, 469 MCs from EAE, TBI, and SCI). Absolute cell numbers for each disease model prior to downsampling were 685 MCs and 1960 microglia in LPC‐induced demyelination; 3234 MCs and 1468 microglia in EAE; 182 MCs and 3620 microglia in 5xFAD; and 72 MCs and 1136 microglia in SOD1G93A, 14,243 MCs and 17,739 microglia in SCI; and 612 MCs and 8434 microglia in TBI. (d) Highest proportional representation of MC (blue) and microglia (red) clusters for each disease model overlaid onto UMAPs of myeloid cells from each disease model. Numbers represent the relative percentage of each of the myeloid clusters of the total myeloid pool in each disease model. Disease models 5xFAD, SOD1G93A, and LPC‐induced demyelination contain only microglia (469 cells per disease model), whereas EAE, TBI and SCI contain both microglia and MCs (938 cells per disease model)
This revealed a core set of microglia and MC clusters, determined by their differential expression of subset‐defining markers (Figure 5b) and enriched GO pathways (Figure 6 and Table S4). Among the 8 microglial clusters, we identified interferon‐related (Mg2), inflammatory (Mg3, Mg8), phagocytic (Mg4) and DAM‐related (Mg6) microglia, each of which displayed unique genetic and functional programs (Figure 6a,c). Although we identified the DAM‐related cluster (Mg6) (Figure S2), argued to be a universal microglia disease state, this was only one of 8 microglia subpopulations that was shared across diseases, suggesting that there are other signatures that are conserved. For MCs, we identified four clusters that included inflammatory (MC3), antigen‐presenting (MC4), and lipopolysaccharide (LPS)‐responsive (MC2) states. Importantly, these clusters were consistently detected in each disease model (Figure 5c), demonstrating that microglia and MCs can acquire a variety of functionally different and unique states throughout the course of a disease. However, the proportions of these clusters differed in each disease (Figure 5c,d), suggesting functionally distinct populations are individually expanded in response to different disease settings.
FIGURE 6.
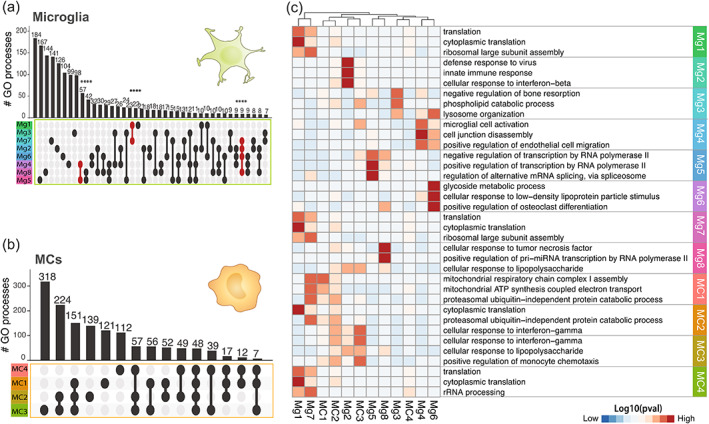
Differing proportions of unique myeloid subtypes drive disease‐specific signatures in bulk populations. (a and b) UpSet plot showing overlap in the number of enriched GO terms for each of the (a) eight microglia (n = 2814 single cells) or (b) four MC (n = 1407 single cells) clusters identified in the LIGER‐integrated scRNA‐seq dataset from six disease models. GO term enrichment was performed on the differentially expressed genes for each cluster (log2FC >0.25, p value <.01) and enriched GO terms were defined as terms with more than 10 genes mapping to a term and a p value less than .01 (Fisher's exact test, elim algorithm). Populations that share GO terms are indicated by connecting lines in the dot plot. Individual populations are indicated by a singular dark circle with no intersecting lines. Significant intersections are shown in red and were determined using the SuperExact test. See also Table S5 for a list of the significance of each intersection shown. Samples are colored by cell cluster and arranged in order of increasing to decreasing number of biological processes associated with each population. (c) Heatmap showing the top three GO terms for each microglia and MC cluster. See also Table S4 for a list of all GO processes. Clustering was applied to columns only
We observed that the expansion of individual myeloid subtypes was highly specialized to microenvironmental cues in each disease model. For instance, Mg4 (phagocytic Mg) was the most prevalent microglial subset in 5xFAD (35.1% of all myeloid cells), which was shared with LPC‐induced demyelination (28.6%) and TBI (19.5%) (Figure 5c,d), potentially representing a cell state important for debris clearance in these disease models. In contrast, Mg3 (inflammatory Mg) was the most common microglia population (18.8%) (Figure 5c,d) in SOD1G93A, pointing to important differences between this model and other neurodegenerative diseases, such as the 5xFAD model of AD that may induce a more phagocytic cellular state. Mg2 clustered separately from the other microglia (Figure 5a) and was enriched for interferon‐related genes (Ifit3, Ifit2, Ifit1, Irf7) (Figure 5b) and genes related to the negative regulation of viral genome replication, response to bacterium, cellular response to LPS, cellular response interferon‐γ, and defense response to protozoan (Table S4), potentially suggesting that this population may be uniquely expanded in response to infectious stimuli (e.g., viruses, bacteria, and protozoa), but persists at low levels in neurodegeneration, sterile injury, and demyelinating disorders. Despite filtering out low quality cells (i.e., cells with high mitochondrial gene expression and low relative RNA content), microglial populations, Mg1 and Mg7 show a high differential expression of ribosomal genes (Figure 5b). These populations could represent cells important for transcriptional regulation of terminal cell differentiation.
In contrast to microglia, the four MC clusters demonstrated overlap in their transcriptional and functional states, despite unique MC clusters dominating each disease model (Figure 5c,d). In TBI, for instance, MC4 was the most prevalent MC population (17.6% of all myeloid cells) and was enriched for genes related to antigen processing and presentation of exogenous peptide antigen, a functional state partially shared with MC3, represented at much lower numbers in in TBI (Figure 5c,d and Table S4). MC2, on the other hand, was the most prominent MC subset in EAE (16.1% of all myeloid cells) and was enriched for genes related to cellular response to interferon‐β and LPS (Figure 5c,d and Table S4). MC1 was the most prevalent population in SCI (18.0% of all myeloid cells) and was enriched for genes related to cell redox homeostasis and aerobic respiration (Figure 5c,d and Table S4). Notably, UpSet analysis on enriched GO terms for each cluster demonstrated 48 shared functional pathways between the four MC clusters, which included interferon‐responsive, oxidative stress, innate immune responses, cytokine production, and inflammatory signaling cascade pathways (Figure 6b,c, Tables S4 and S5). We observed small variations in this core MC state between clusters, often with pathway overlap between two or more clusters (Table S5), possibly indicating a continuous population rather than a series of distinct MC subtypes.
Together, this work suggests that the microenvironment drives the expansion of unique myeloid subtypes that drive disease‐specific signatures in bulk populations.
3.5. Cross‐disease comparison reveals CD81 as a microglia‐enriched marker in neuroinflammation
Considering that some nominal microglia‐specific markers are downregulated during pathology (Krasemann et al., 2017; Spiteri et al., 2021; Vankriekelsvenne et al., 2022), we sought to find a novel and stably‐expressed microglia marker to reliably identify these cells in disease at both RNA (Werner et al., 2020) and protein level. We performed differential expression analysis on individual bulk and scRNA‐seq studies to evaluate the genes highly enriched in microglia versus MCs across CNS disease (Figure 7a). For this analysis only studies that examined both microglia and MCs in the CNS simultaneously were used. This included the additional infectious disease model, using Theiler's encephalomyelitis virus.
FIGURE 7.
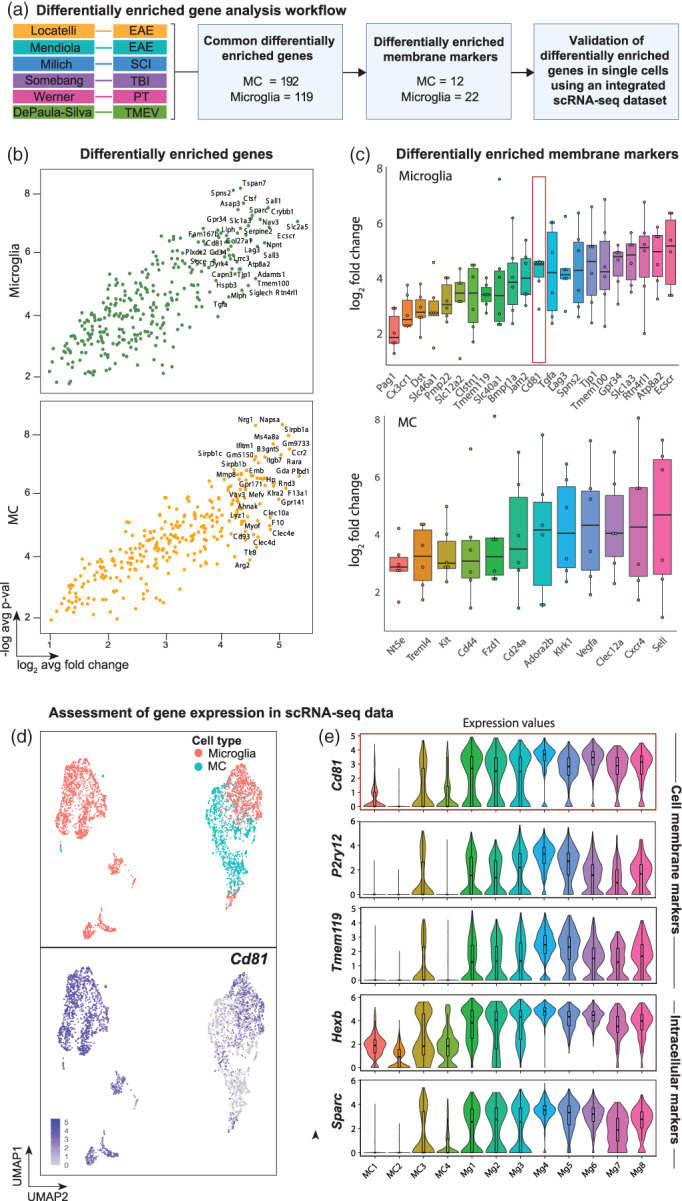
Cross‐disease analysis reveals Cd81 as a microglial‐enriched gene in neuroinflammation. (a) Flow chart analysis pipeline used to identify differentially enriched microglial and MC genes in the inflamed brain. Six studies that simultaneously analyzed microglia and MCs in the CNS were used to identify 192 and 119 differentially enriched (p > .05, log2 > 1) MC and microglia genes, respectively. This gene list was filtered in Uniprot to identify membrane expressed genes, which were subsequently validated with expression data using an integrated scRNA dataset. (b) Volcano plots showing all genes differentially enriched by microglia and MCs in EAE, SCI, TBI, PT and TMEV. The top 35 differentially enriched genes are annotated. See also Table S6 for a list of these genes. (c) Box plots showing differentially enriched surface/plasma membrane‐expressed genes for microglia and MCs. (d) UMAPs showing single‐cell gene expression overlays for Cd81 in MC and microglial clusters identified in the integrated scRNA‐seq dataset. (e) Violin plots showing the expression of Cd81 relative to nominal microglia‐specific genes in the 12 myeloid clusters
This analysis identified 192 and 119 genes differentially enriched in MCs and microglia, respectively, that were conserved across five models of CNS pathology, including EAE, SCI, TBI, PT and Theiler's encephalomyelitis virus infection (Figure 7a and Table S6). Filtering these genes by their cellular localization on the plasma membrane revealed 12 and 22 MC and microglia differentially enriched membrane markers, respectively (Figure 7a–c), including previously identified MC markers Cxcr4 and Cd44 that have been used to distinguish these cells from microglia in stroke (Werner et al., 2020) and EAE (Lewis et al., 2014) (Figure 7c). Of interest, Cd81 was stably expressed by microglia across all models of CNS inflammation, with a 25‐fold higher median expression relative to MCs (Figure 7c). This marker is a species‐conserved microglial‐enriched tetraspanin gene (Geirsdottir et al., 2020) expressed on the cell surface, reagents for which are commercially available for flow cytometric and RNA detection, making this a potentially suitable marker for detecting these cells multimodally across species.
We confirmed the differential expression of Cd81 at the single‐cell level in our LIGER‐integrated scRNA‐seq dataset and observed that Cd81 remained highly expressed by all microglial clusters (i.e., Mg1 to Mg8), but remained expressed only at low levels by MCs during neuroinflammatory conditions (Figure 7d,e). Importantly, previously identified membrane markers for yolk‐sac‐derived microglia (e.g., P2ry12, Tmem119) showed variable expression in all microglia clusters relative to Cd81 expression (Figure 7e). Although genes such as Sparc (Jordão et al., 2019) and Hexb (Masuda et al., 2020) were also enriched in all microglia populations (Figure 7e), the intracellular localization of their encoded protein products do not allow antibody binding in live cells without lethal cell permeabilization, thus precluding downstream live cell experimental approaches (e.g., after flow cytometric sorting) in the absence of specific genetic manipulation (Masuda et al., 2020). As CD81 is a surface expressed protein, this marker can be used for live cell sorting, providing an advantage over the previously identified microglial‐enriched genes, Sparc and Hexb.
To validate Cd81 as a microglia‐enriched marker for flow cytometry during pathology, we investigated the expression of CD81 protein on microglia and CNS‐infiltrating MCs in a mouse model of flaviviral encephalitis (Figure 8). Since many microglia‐specific markers are either downregulated by microglia or upregulated by MCs during inflammation, we used a highly inflammatory model of CNS disease with a significant monocytic infiltrate to validate the differential protein expression of this marker on these cells. West Nile virus (WNV) encephalitis causes a severe inflammatory response characterized by a ~10‐fold increase in the number of leukocytes in the brain, with approximately 50% of this infiltrate comprising Ly6Chi inflammatory macrophages (Getts et al., 2007; Getts et al., 2008; Getts et al., 2012; Getts et al., 2014; Spiteri et al., 2021; Spiteri & Wishart et al., 2022; Terry et al., 2012; Terry et al., 2015). Examination of CD45+ cells in the virus‐infected brain on day 7 post‐infection confirmed that CD81 was expressed at high levels only on microglia (Figure 8b–e) and not on Ly6Chi MCs or microglia‐like MCs (i.e., myeloid cells of non‐microglial origin present in the infected brain with a CD45+, CX3CR1+ and CD11b+ profile similar to microglia, but absent in the homeostatic brain) (Spiteri et al., 2021). Interestingly, CD81 was not detectable on microglia during homeostasis (Figure 8f), despite the homeostatic expression of RNA for this molecule (Geirsdottir et al., 2020), suggesting that the induction of protein expression occurs during inflammation. Furthermore, while CD81 is microglia‐enriched out of all CNS resident and infiltrating leukocytes, this protein is expressed by a number of cells in the bone marrow and spleen, suggesting that CD81 is downregulated by infiltrating cells upon entry into the CNS (Figure 8b, Figures S3 and S4). Taken together, while other disease models need to be investigated, this data suggests that under highly inflammatory conditions, the differential expression of CD81 in combination with previously reported CD45, Ly6C, CX3CR1 or P2RY12 (Spiteri et al., 2021) in the CNS, enables a novel and simple method for discriminating microglia from other leukocytes by flow cytometry, as well as for live cell sorting and imaging.
4. DISCUSSION
Despite decades of research and recent technical advances, the differential and shared contributions of microglia and MCs to disease and disease resolution are still poorly understood. While their immense functional and phenotypic diversity make this problem intrinsically complex, this is further exacerbated by the wide range of technologies and analytical approaches used to describe these cells in different fields of research. To address this problem, we have systematically integrated the transcriptional and functional profiles of resident and CNS‐infiltrating myeloid cells obtained from demyelinating, neurodegenerative, ischemic, traumatic and infectious conditions across three sequencing modalities. Concatenating the gene expression profiles across these models identified a quorum of genes and their associated functions that were principally disease‐specific.
In contrast to the proposed universal microglia signature, we observed that individual disease environments drive microglia to adopt highly specialized signatures with minimal overlap across CNS conditions. Analysis of bulk microglia populations demonstrate limited overlap in gene expression profiles and biological functions between diseases. More detailed examination of scRNA‐seq populations suggest that these disease‐specific bulk populations may be a result of the distinct composition of differing proportions of unique subpopulations in each disease setting, including interferon‐related, inflammatory, phagocytic, and DAM‐related microglia identified in this report. Indeed, concatenating microglia populations from each scRNA‐seq study demonstrated more unique than shared biological functions across pathologies, confirming that although each disease state shared these subpopulations, the differing proportions drove unique signatures when pooled together. While these disease‐specific microglia populations showed limited overlap across pathologies, they demonstrated convergence with MC populations in the same disease, suggesting that various activation states are regulated by the disease setting. The notion of diverse myeloid activation signatures has been previously proposed by other studies including a meta‐analysis integrating various animal disease models (Friedman et al., 2018) and in human AD, where they identified homeostatic, interferon‐, LPS‐, neurodegeneration‐ or proliferation‐related modules (Olah et al., 2020), closely resembling the activation states reported here. It remains an important task to understand the signaling pathways driving the distinct functional specialization of microglia. This could help restore beneficial functions such as phagocytic functions in the handling of myelin debris associated with senescence (Safaiyan et al., 2016) or amyloid plaques that arise during AD (Ennerfelt et al., 2022; Grubman et al., 2021), or prevent maladaptive responses such as the production of reactive oxygen species or reactive nitrogen species that may underlie tissue damage in MS (Mendiola et al., 2020) or viral encephalitis (Getts et al., 2012), respectively.
Intriguingly, microglia and MCs adopt more distinct than shared expression profiles and functions across pathologies, despite being exposed to similar inflammatory settings. This may relate to their unique ontogenies and regulatory signaling patterns (Bennett et al., 2018; Butovsky et al., 2014; Buttgereit et al., 2016). In contrast to microglia, MCs showed more similarities across models of acute neuroinflammation, potentially relating to the limited time spent in the CNS, compared to microglia, that would normally be required for adaptation and specialization to environmental cues. Previous single‐cell studies show multiple transcriptionally diverse MCs within a single disease (Giladi et al., 2020; Jordão et al., 2019; Mendiola et al., 2020). The accumulation of CNS‐infiltrating monocytes in anatomically distinct CNS compartments may contribute to this previously reported transcriptomic heterogeneity (Ivan et al., 2021; Jordão et al., 2019; Locatelli et al., 2018). Alternatively, these distinct transcriptional programs may represent a trajectory of maturation within the CNS, where MCs adapt more subtly in response to changes in the local microenvironment with disease progression (Locatelli et al., 2018) from an initially conserved, functional program that generally enables rapid, if generic, control of an acute insult (Sanin et al., 2022).
Single‐cell transcriptomics has uncovered nominally microglia‐specific markers. However, several of these markers are downregulated during inflammation (Jordão et al., 2019; Keren‐Shaul et al., 2017; Krasemann et al., 2017) or also expressed by peripherally‐derived cells (Chen et al., 2020; Spiteri et al., 2021), making them unreliable for the unambiguous identification of microglia in the inflamed CNS. Highlighting the practical value of our cross‐disease analysis, we here identified Cd81 as a novel and conserved RNA marker distinguishing microglia from MCs during neuroinflammation. Indeed, interrogation of other publicly available human and mouse datasets showed a stable expression of Cd81 across disease states (Li et al., 2018; Srinivasan et al., 2020; Zhang et al., 2014). Notably, Cd81 is a microglial marker conserved at least across the evolutionary span of sheep, mice and humans (Geirsdottir et al., 2020). It is not upregulated by monocytes that engraft the brain (Cronk et al., 2018), suggesting that Cd81, in combination with other microglia‐enriched genes, is a robust marker for distinguishing microglia from MCs under a range of disease conditions.
Importantly, of all the CD45+ cells in the inflamed brain, CD81 protein was highly expressed only on microglia. While other disease models require further investigation, this work validates the use of CD81 along with other differentially expressed microglia proteins for cytometric identification of these cells during severe inflammatory conditions, such as WNV encephalitis. While other cells also express this tetraspanin (Maecker et al., 1997), its function appears to be cell type‐specific (Dijkstra et al., 2000; Mordica et al., 2009). The upregulation and expression of CD81 by microglia in the WNV‐infected brain (Figure 8), human AD (Mathys et al., 2019) and in spinal cord injury in the rat (Dijkstra et al., 2000), may relate to their activation/transformation (Dijkstra et al., 2000), enhanced proliferative capacity (Dijkstra et al., 2001; Geisert et al., 2002; Ma et al., 2010) and mobility (Maecker et al., 1997) and/or the release of extracellular vesicles (Clayton et al., 2021; Muraoka et al., 2021; Paolicelli et al., 2019) during disease.
While our analysis is informative, it has several limitations. Firstly, we were unable to dissect the contribution of gender or mouse strain per se to the identified gene profiles. Secondly, given our stringent criteria for data incorporation (see methods and Table S1), several studies were not included, restricting our evaluation of other cell types and diseases. For example, the unexplored contribution of border‐associated macrophages to disease (Ivan et al., 2021) and how their gene expression profiles may relate to those of microglia and MCs remains unclear. Thirdly, we were unable to investigate microglia or MCs in certain diseases either due to the lack of the relevant homeostatic controls or due to a statistically insufficient number of biological replicates. Fourthly, further characterization of these myeloid clusters is still required, including functional analyses, to determine the biological importance of these subsets. It is also critical to determine whether specific myeloid clusters identified here are spatially localized and/or timepoint dependent. Lastly, to translate these findings into a clinically relevant setting, parallel analysis of human data would be required, however the potential discordance of animal models and human disease will require further integrative techniques that can accurately align biological differences between humans and mice. While additional characterization is required, we believe this work provides a cornerstone for building a consensus to enable more accurate classification of myeloid populations in pathological settings.
Additionally, the technical and methodological discrepancies between incorporated datasets have limited more direct comparisons. This argues for a unified approach in processing, sorting, and analysis of these cell types to enable integration across broader, often segregated disease research fields. For instance, the use of standard tissue processing procedures, standard gating strategies to identify and sort populations for subsequent transcriptomic analysis, and in the absence of appropriate controls, the use of homeostatic, peripheral blood monocytes as standardized controls for CNS‐infiltrating populations in the inflamed brain, would resolve many of these issues. A consensus defining a set of standards for the field to unify experimental and analytical procedures would go a considerable way to begin this process, as previously attempted for macrophage activation nomenclature, transcriptomic profiling of brain barriers and flow cytometry standards (Cossarizza et al., 2019; Francisco et al., 2020; Murray et al., 2014).
Together, our meta‐analytical approach integrates data across fields of research and technology to enable comparison of myeloid behavior in different experimental models of neuroinflammation. As a resource, this work highlights the importance of cross‐disease comparison to better characterize the conserved and divergent microglial and MC responses in CNS disease and contributes to the creation of a unified perspective of the behavior of these cells across CNS conditions. We demonstrate that microglia and monocyte transcriptomes are disease‐dependent, with differing proportions of unique myeloid subpopulations driving these bulk population signatures. These include 8 microglia clusters and 4 monocyte clusters that were functionally‐defined and conserved across disease settings, identifying a core set of myeloid responses. This paves the way for identifying myeloid “archetypes” across CNS pathologies, which may enable us more precisely to dissect the influence of disease environments on functional programs. This is also crucial to inform the ongoing development of targeted therapeutic approaches.
AUTHOR CONTRIBUTIONS
Conceptualization: Claire L. Wishart, Alanna G. Spiteri, Giuseppe Locatelli, and Nicholas J. C. King. Data curation: Claire L. Wishart. Investigation: Alanna G. Spiteri. Formal analysis, software and visualization: Claire L. Wishart and Alanna G. Spiteri. Funding acquisition and supervision: Nicholas J. C. King. Writing – original draft: Claire L. Wishart, Alanna G. Spiteri, Giuseppe Locatelli, and Nicholas J. C. King. Writing – review and editing: Claire L. Wishart, Alanna G. Spiteri, Giuseppe Locatelli, and Nicholas J. C. King.
CONFLICT OF INTEREST
The authors declare no competing interests.
Supporting information
Figure S1. Clustering parameters used for defining myeloid subtypes of the LIGER‐integrated scRNA‐seq data set. (A) Parameter selection for the number of factors k versus the median Kullback–Leibler (KL) value across all cells for the LIGER‐integrated data set of six disease models. The tuning parameter, lambda, was kept at the recommended value of 5 (Liu et al., 2020). The optimal value of k is defined as the “elbow” of the plot, that is, the k value at which the median KL value becomes negligible (defined between k = 10–15, dashed lines). (B) UMAP showing the clusters for two optimal k values, k = 12 (alignment = 0.878) and k = 15 (alignment = 0.837). The optimal number of factors chosen was k = 12 based on improved alignment between datasets and based on prior biological knowledge of clustering (Liu et al., 2020). (C) UMAP of myeloid cells from the downsampled LIGER‐integrated dataset (k = 12) overlaid by either microglia (2814 cells) and/or MC clusters (1407 cells). (D) UMAP showing the anatomical location from which myeloid subclusters were isolated during CNS tissue processing and cell sorting. (E) Dot plot heatmap showing the average expression of microglia‐specific and MC‐enriched genes in identified clusters. Clusters have been ordered by unsupervised hierarchical clustering. (F) UMAPs showing single‐cell gene expression overlays for microglia‐specific genes, P2ry12, Tmem119, Hexb and Sparc, in MC and microglial clusters.
Figure S2. Expression of universal microglial signatures by single‐cell RNA‐seq myeloid clusters.
(A) UMAP of 4221 myeloid cells (downsampled) with clusters annotated and pseudocolored based on cell type cluster. (B) Dot plot heatmap showing the average expression of disease‐associated microglial (DAM) and microglial neurodegenerative (MGnD) genes in all myeloid clusters. Clusters have been ordered by unsupervised hierarchical clustering. Microglial clusters with high expression of DAM/MGnD genes are annotated as DAM‐like microglia (Mg6, red box). (C) DAM‐like microglia Mg6 (red) overlaid onto UMAPs of microglia clusters from each disease model. Numbers represent the relative frequency percentage of Mg6 out of the microglial pool within each disease model.
Figure S3. Histograms showing the expression of CD81 on CD45+ cell types in the non‐infected and WNV‐infected bone marrow at dpi 7.
Figure S4. Histograms showing the expression of CD81 on CD45+ cell types in the non‐infected and WNV‐infected spleen at dpi 7.
Table S1. Study metadata for included and excluded studies.
Table S2. Hierarchical clustering arrangement of microglial and MC samples from disparate studies, disease‐models and sequencing technologies.
Table S3. Top ranked genes and GO terms associated with single‐origin RP analysis for each microglia and MC population from six disease models.
Table S4. Top GO terms associated with scRNA‐seq myeloid clusters across six disease models.
Table S5. GO intersections for scRNA‐seq myeloid clusters.
Table S6. Differentially enriched microglial and MC genes and their associated GO enrichment terms conserved across seven disease models.
ACKNOWLEDGMENTS
N.J.C.K. was supported by a National Health and Medical Research Council Project Grant 1088242 and the Merridew Foundation. A.G.S. is supported by the Australian Government Research Training Stipend Scholarship and the University of Sydney Postgraduate Merit Award. C.L.W. is supported by a Merridew Foundation PhD Scholarship. G.L. is supported from the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska‐Curie grant agreement No. 813294 (ENTRAIN), from the Swiss Multiple Sclerosis Society, and from the Italian Multiple Sclerosis Society (FISM 2019/R‐Single/001). We would also like to acknowledge Sydney Cytometry Core Research facility, a joint initiative of Centenary Institute and the University of Sydney, the University of Sydney's Laboratory animal services, Dr. Tracy Chew from the Sydney Informatics Hub at the University of Sydney for her advice on data analysis and integration and Dr. Thomas Ashhurst, Briunca Spiteri and Laine Wishart for their assistance with code, excel and/or software. Open access publishing facilitated by The University of Sydney, as part of the Wiley ‐ The University of Sydney agreement via the Council of Australian University Librarians.
Wishart, C. L. , Spiteri, A. G. , Locatelli, G. , & King, N. J. C. (2023). Integrating transcriptomic datasets across neurological disease identifies unique myeloid subpopulations driving disease‐specific signatures. Glia, 71(4), 904–925. 10.1002/glia.24314
Claire L. Wishart and Alanna G. Spiteri contributed equally to this work.
Funding information Australian Government Research Training Stipend Scholarship; University of Sydney Postgraduate Merit Award; Associazione Italiana Sclerosi Multipla, Grant/Award Number: FISM 2019/R‐Single/001; European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska‐Curie grant agreement, Grant/Award Number: 813294; Merridew Foundation; National Health and Medical Research Council, Grant/Award Number: 1088242; Swiss Multiple Sclerosis Society
DATA AVAILABILITY STATEMENT
All RNA datasets were analyzed from existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table (Table 1). All code was adapted from existing code listed in Table 1. Flow cytometry data will be shared by the lead contact upon reasonable request. Any additional information required to re‐analyze the data reported in this paper is available from the lead contact upon request.
REFERENCES
- Afgan, E. , Baker, D. , van den Beek, M. , Blankenberg, D. , Bouvier, D. , Čech, M. , Chilton, J. , Clements, D. , Coraor, N. , Eberhard, C. , Grüning, B. , Guerler, A. , Hillman‐Jackson, J. , von Kuster, G. , Rasche, E. , Soranzo, N. , Turaga, N. , Taylor, J. , Nekrutenko, A. , & Goecks, J. (2016). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research, 44(W1), W3–W10. 10.1093/nar/gkw343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajami, B. , Bennett, J. L. , Krieger, C. , Tetzlaff, W. , & Rossi, F. M. (2007). Local self‐renewal can sustain CNS microglia maintenance and function throughout adult life. Nature Neuroscience, 10(12), 1538–1543. [DOI] [PubMed] [Google Scholar]
- Alexa, A., & Rahnenfuhrer, J. (2022). topGO: Enrichment analysis for gene ontology. R package version 2.50.0.
- Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. In Babraham bioinformatics. Babraham Institute. [Google Scholar]
- Ashhurst, T. M. , Marsh‐Wakefield, F. , Putri, G. H. , Spiteri, A. G. , Shinko, D. , Read, M. N. , Smith, A. L. , & King, N. J. C. (2021). Integration, exploration, and analysis of high‐dimensional single‐cell cytometry data using spectre. Cytometry. Part A, 101, 237–253. 10.1002/cyto.a.24350 [DOI] [PubMed] [Google Scholar]
- Barrett, T. , Wilhite, S. E. , Ledoux, P. , Evangelista, C. , Kim, I. F. , Tomashevsky, M. , Marshall, K. A. , Phillippy, K. H. , Sherman, P. M. , Holko, M. , Yefanov, A. , Lee, H. , Zhang, N. , Robertson, C. L. , Serova, N. , Davis, S. , & Soboleva, A. (2013). NCBI GEO: Archive for functional genomics data sets—Update. Nucleic Acids Research, 41(Database issue), D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, F. C. , Bennett, M. L. , Yaqoob, F. , Mulinyawe, S. B. , Grant, G. A. , Hayden Gephart, M. , Plowey, E. D. , & Barres, B. A. (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron, 98(6), 1170–1183.e1178. 10.1016/j.neuron.2018.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L. , Bennett, F. C. , Liddelow, S. A. , Ajami, B. , Zamanian, J. L. , Fernhoff, N. B. , Mulinyawe, S. B. , Bohlen, C. J. , Adil, A. , Tucker, A. , Weissman, I. L. , Chang, E. F. , Li, G. , Grant, G. A. , Hayden Gephart, M. G. , & Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113(12), E1738–E1746. 10.1073/pnas.1525528113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuker, C. , Schafflick, D. , Strecker, J.‐K. , Heming, M. , Li, X. , Wolbert, J. , Schmidt‐Pogoda, A. , Thomas, C. , Kuhlmann, T. , Aranda‐Pardos, I. , A‐Gonzalez, N. , Kumar, P. A. , Werner, Y. , Kilic, E. , Hermann, D. M. , Wiendl, H. , Stumm, R. , Meyer zu Hörste, G. , & Minnerup, J. (2022). Stroke induces disease‐specific myeloid cells in the brain parenchyma and pia. Nature Communications, 13(1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brionne, A. , Juanchich, A. , & Hennequet‐Antier, C. (2019). ViSEAGO: A bioconductor package for clustering biological functions using gene ontology and semantic similarity. BioData Mining, 12, 16. 10.1186/s13040-019-0204-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky, O. , Jedrychowski, M. P. , Moore, C. S. , Cialic, R. , Lanser, A. J. , Gabriely, G. , Koeglsperger, T. , Dake, B. , Wu, P. M. , Doykan, C. E. , Fanek, Z. , Liu, L. P. , Chen, Z. , Rothstein, J. D. , Ransohoff, R. M. , Gygi, S. P. , Antel, J. P. , & Weiner, H. L. (2014). Identification of a unique TGF‐β‐dependent molecular and functional signature in microglia. Nature Neuroscience, 17(1), 131–143. 10.1038/nn.3599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buttgereit, A. , Lelios, I. , Yu, X. , Vrohlings, M. , Krakoski, N. R. , Gautier, E. L. , Nishinakamura, R. , Becher, B. , & Greter, M. (2016). Sall1 is a transcriptional regulator defining microglia identity and function. Nature Immunology, 17(12), 1397–1406. 10.1038/ni.3585 [DOI] [PubMed] [Google Scholar]
- Cao, Z. , Harvey, S. S. , Chiang, T. , Foltz, A. G. , Lee, A. G. , Cheng, M. Y. , & Steinberg, G. K. (2021). Unique subtype of microglia in degenerative thalamus after cortical stroke. Stroke, 52(2), 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson, M. (2019). org.Mm.eg.db: Genome wide annotation for mouse.
- Chen, H.‐R. , Sun, Y.‐Y. , Chen, C.‐W. , Kuo, Y.‐M. , Kuan, I. S. , Tiger Li, Z.‐R. , Short‐Miller, J. C. , Smucker, M. R. , & Kuan, C.‐Y. (2020). Fate mapping via CCR2‐CreER mice reveals monocyte‐to‐microglia transition in development and neonatal stroke. Science Advances, 6(35), eabb2119. 10.1126/sciadv.abb2119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y. , & Colonna, M. (2021). Microglia in Alzheimer's disease at single‐cell level. Are there common patterns in humans and mice? Journal of Experimental Medicine, 218(9), e20202717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton, K. , Delpech, J. C. , Herron, S. , Iwahara, N. , Ericsson, M. , Saito, T. , Saido, T. C. , Ikezu, S. , & Ikezu, T. (2021). Plaque associated microglia hyper‐secrete extracellular vesicles and accelerate tau propagation in a humanized APP mouse model. Molecular Neurodegeneration, 16(1), 18. 10.1186/s13024-021-00440-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway, J. R. , Lex, A. , & Gehlenborg, N. (2017). UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics, 33, 2938–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossarizza, A. , Chang, H. D. , Radbruch, A. , Acs, A. , Adam, D. , Adam‐Klages, S. , Agace, W. W. , Aghaeepour, N. , Akdis, M. , Allez, M. , Almeida, L. N. , Alvisi, G. , Anderson, G. , Andrä, I. , Annunziato, F. , Anselmo, A. , Bacher, P. , Baldari, C. T. , Bari, S. , … Zychlinsky, A. (2019). Guidelines for the use of flow cytometry and cell sorting in immunological studies (second edition). European Journal of Immunology, 49(10), 1457–1973. 10.1002/eji.201970107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cronk, J. C. , Filiano, A. J. , Louveau, A. , Marin, I. , Marsh, R. , Ji, E. , Goldman, D. H. , Smirnov, I. , Geraci, N. , Acton, S. , Overall, C. C. , & Kipnis, J. (2018). Peripherally derived macrophages can engraft the brain independent of irradiation and maintain an identity distinct from microglia. The Journal of Experimental Medicine, 215(6), 1627–1647. 10.1084/jem.20180247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deczkowska, A. , Keren‐Shaul, H. , Weiner, A. , Colonna, M. , Schwartz, M. , & Amit, I. (2018). Disease‐associated microglia: A universal immune sensor of neurodegeneration. Cell, 173(5), 1073–1081. [DOI] [PubMed] [Google Scholar]
- Del Carratore, F. , Jankevics, A. , Eisinga, R. , Heskes, T. , Hong, F. , & Breitling, R. (2017). RankProd 2.0: A refactored bioconductor package for detecting differentially expressed features in molecular profiling datasets. Bioinformatics, 33(17), 2774–2775. 10.1093/bioinformatics/btx292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaula‐Silva, A. B. , Gorbea, C. , Doty, D. J. , Libbey, J. E. , Sanchez, J. M. S. , Hanak, T. J. , Cazalla, D. , & Fujinami, R. S. (2019). Differential transcriptional profiles identify microglial‐ and macrophage‐specific gene markers expressed during virus‐induced neuroinflammation. Journal of Neuroinflammation, 16(1), 152. 10.1186/s12974-019-1545-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkstra, S. , Geisert, E. J. , Gispen, W. H. , Bar, P. R. , & Joosten, E. A. (2000). Up‐regulation of CD81 (target of the antiproliferative antibody; TAPA) by reactive microglia and astrocytes after spinal cord injury in the rat. Journal of Comparative Neurology, 428(2), 266–277. [DOI] [PubMed] [Google Scholar]
- Dijkstra, S. , Geisert, E. E., Jr. , Dijkstra, C. D. , Bar, P. R. , & Joosten, E. A. (2001). CD81 and microglial activation in vitro: Proliferation, phagocytosis and nitric oxide production. Journal of Neuroimmunology, 114(1–2), 151–159. 10.1016/s0165-5728(01)00240-5 [DOI] [PubMed] [Google Scholar]
- Ennerfelt, H. , Frost, E. L. , Shapiro, D. A. , Holliday, C. , Zengeler, K. E. , Voithofer, G. , Bolte, A. C. , Lammert, C. R. , Kulas, J. A. , Ulland, T. K. , & Lukens, J. R. (2022). SYK coordinates neuroprotective microglial responses in neurodegenerative disease. Cell, 185(22), 4135–4152.e4122. 10.1016/j.cell.2022.09.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco, D. M. F. , Marchetti, L. , Rodríguez‐Lorenzo, S. , Frías‐Anaya, E. , Figueiredo, R. M. , BtRAIN Network , Heymanns, M. , Culot, M. , Santa‐Maria, A. R. , Deli, M. A. , Germano, R. F. V. , Vanhollebeke, B. , Kakogiannos, N. , Giannotta, M. , Dejana, E. , Dominguez‐Belloso, A. , Liebner, S. , Schuster, M. , Klok, H. A. , … Bruggmann, R. (2020). Advancing brain barriers RNA sequencing: Guidelines from experimental design to publication. Fluids and Barriers of the CNS, 17(1), 51. 10.1186/s12987-020-00207-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, B. A. , Srinivasan, K. , Ayalon, G. , Meilandt, W. J. , Lin, H. , Huntley, M. A. , Cao, Y. , Lee, S. H. , Haddick, P. C. G. , Ngu, H. , Modrusan, Z. , Larson, J. L. , Kaminker, J. S. , van der Brug, M. P. , & Hansen, D. V. (2018). Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer's disease not evident in mouse models. Cell Reports, 22(3), 832–847. 10.1016/j.celrep.2017.12.066 [DOI] [PubMed] [Google Scholar]
- Geirsdottir, L. , David, E. , Keren‐Shaul, H. , Weiner, A. , Bohlen, S. C. , Neuber, J. , Balic, A. , Giladi, A. , Sheban, F. , Dutertre, C. A. , Pfeifle, C. , Peri, F. , Raffo‐Romero, A. , Vizioli, J. , Matiasek, K. , Scheiwe, C. , Meckel, S. , Mätz‐Rensing, K. , van der Meer, F. , … Prinz, M. (2020). Cross‐species single‐cell analysis reveals divergence of the primate microglia program. Cell, 181(3), 746. 10.1016/j.cell.2020.04.002 [DOI] [PubMed] [Google Scholar]
- Geisert, E. E., Jr. , Williams, R. W. , Geisert, G. R. , Fan, L. , Asbury, A. M. , Maecker, H. T. , Deng, J. , & Levy, S. (2002). Increased brain size and glial cell number in CD81‐null mice. Journal of Comparative Neurology, 453(1), 22–32. 10.1002/cne.10364 [DOI] [PubMed] [Google Scholar]
- Geissmann, F. , Manz, M. G. , Jung, S. , Sieweke, M. H. , Merad, M. , & Ley, K. (2010). Development of monocytes, macrophages, and dendritic cells. Science, 327(5966), 656–661. 10.1126/science.1178331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts, D. R. , Matsumoto, I. , Müller, M. , Getts, M. T. , Radford, J. , Shrestha, B. , Campbell, I. L. , & King, N. J. (2007). Role of IFN‐γ in an experimental murine model of West Nile virus‐induced seizures. Journal of Neurochemistry, 103(3), 1019–1030. [DOI] [PubMed] [Google Scholar]
- Getts, D. R. , Terry, R. L. , Getts, M. T. , Deffrasnes, C. , Müller, M. , van Vreden, C. , Ashhurst, T. M. , Chami, B. , McCarthy, D. , Wu, H. , Ma, J. , Martin, A. , Shae, L. D. , Witting, P. , Kansas, G. S. , Kühn, J. , Hafezi, W. , Campbell, I. L. , Reilly, D. , … King, N. J. C. (2014). Therapeutic inflammatory monocyte modulation using immune‐modifying microparticles. Science Translational Medicine, 6(219), 219ra217–219ra217, 219ra7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts, D. R. , Terry, R. L. , Getts, M. T. , Müller, M. , Rana, S. , Deffrasnes, C. , Ashhurst, T. M. , Radford, J. , Hofer, M. , Thomas, S. , Campbell, I. L. , & King, N. J. C. (2012). Targeted blockade in lethal West Nile virus encephalitis indicates a crucial role for very late antigen (VLA)‐4‐dependent recruitment of nitric oxide‐producing macrophages. Journal of Neuroinflammation, 9(1), 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Getts, D. R. , Terry, R. L. , Getts, M. T. , Müller, M. , Rana, S. , Shrestha, B. , Radford, J. , van Rooijen, N. , Campbell, I. L. , & King, N. J. C. (2008). Ly6c+ “inflammatory monocytes” are microglial precursors recruited in a pathogenic manner in West Nile virus encephalitis. The Journal of Experimental Medicine, 205(10), 2319–2337. 10.1084/jem.20080421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi, A. , Wagner, L. K. , Li, H. , Dörr, D. , Medaglia, C. , Paul, F. , Shemer, A. , Jung, S. , Yona, S. , Mack, M. , Leutz, A. , Amit, I. , & Mildner, A. (2020). Cxcl10+ monocytes define a pathogenic subset in the central nervous system during autoimmune neuroinflammation. Nature Immunology, 21(5), 525–534. [DOI] [PubMed] [Google Scholar]
- Ginhoux, F. , Greter, M. , Leboeuf, M. , Nandi, S. , See, P. , Gokhan, S. , Mehler, M. F. , Conway, S. J. , Ng, L. G. , Stanley, E. R. , Samokhvalov, I. M. , & Merad, M. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubman, A. , Choo, X. Y. , Chew, G. , Ouyang, J. F. , Sun, G. , Croft, N. P. , Rossello, F. J. , Simmons, R. , Buckberry, S. , Landin, D. V. , Pflueger, J. , Vandekolk, T. H. , Abay, Z. , Zhou, Y. , Liu, X. , Chen, J. , Larcombe, M. , Haynes, J. M. , McLean, C. , … Polo, J. M. (2021). Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nature Communications, 12(1), 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafemeister, C. , & Satija, R. (2019). Normalization and variance stabilization of single‐cell RNA‐seq data using regularized negative binomial regression. Genome Biology, 20(1), 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, T. R. , Dufort, C. , Dissing‐Olesen, L. , Giera, S. , Young, A. , Wysoker, A. , Walker, A. J. , Gergits, F. , Segel, M. , Nemesh, J. , Marsh, S. E. , Saunders, A. , Macosko, E. , Ginhoux, F. , Chen, J. , Franklin, R. J. M. , Piao, X. , McCarroll, S. A. , & Stevens, B. (2019). Single‐cell RNA sequencing of microglia throughout the mouse lifespan and in the injured brain reveals complex cell‐state changes. Immunity, 50(1), 253–271.e256. 10.1016/j.immuni.2018.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, Y. , Hao, S. , Andersen‐Nissen, E. , Mauck, W. M., III , Zheng, S. , Butler, A. , Lee, M. J. , Wilk, A. J. , Darby, C. , Zager, M. , Hoffman, P. , Stoeckius, M. , Papalexi, E. , Mimitou, E. P. , Jain, J. , Srivastava, A. , Stuart, T. , Fleming, L. M. , Yeung, B. , … Satija, R. (2021). Integrated analysis of multimodal single‐cell data. Cell, 184, 3573–3587.e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong, F. , Breitling, R. , McEntee, C. W. , Wittner, B. S. , Nemhauser, J. L. , & Chory, J. (2006). RankProd: A bioconductor package for detecting differentially expressed genes in meta‐analysis. Bioinformatics, 22(22), 2825–2827. 10.1093/bioinformatics/btl476 [DOI] [PubMed] [Google Scholar]
- Hunter, M. , Spiller, K. J. , Dominique, M. A. , Xu, H. , Hunter, F. W. , Fang, T. C. , Canter, R. G. , Roberts, C. J. , Ransohoff, R. M. , Trojanowski, J. Q. , & Lee, V. M. Y. (2021). Microglial transcriptome analysis in the rNLS8 mouse model of TDP‐43 proteinopathy reveals discrete expression profiles associated with neurodegenerative progression and recovery. Acta Neuropathologica Communications, 9(1), 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan, D. C. , Walthert, S. , & Locatelli, G. (2021). Central nervous system barriers impact distribution and expression of iNOS and arginase‐1 in infiltrating macrophages during neuroinflammation. Frontiers in Immunology, 1238. 10.3389/fimmu.2021.666961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jalili, V. , Afgan, E. , Gu, Q. , Clements, D. , Blankenberg, D. , Goecks, J. , Taylor, J. , & Nekrutenko, A. (2020). Corrigendum: The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Research, 48(14), 8205–8207. 10.1093/nar/gkaa554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordão, M. J. C. , Sankowski, R. , Brendecke, S. M. , Sagar, Locatelli, G. , Tai, Y. H. , Tay, T. L. , Schramm, E. , Armbruster, S. , Hagemeyer, N. , Groß, O. , Mai, D. , Çiçek, Ö. , Falk, T. , Kerschensteiner, M. , Grün, D. , & Prinz, M. (2019). Single‐cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science, 363(6425), eaat7554. 10.1126/science.aat7554 [DOI] [PubMed] [Google Scholar]
- Keren‐Shaul, H. , Spinrad, A. , Weiner, A. , Matcovitch‐Natan, O. , Dvir‐Szternfeld, R. , Ulland, T. K. , Amit, I. , Baruch, K. , Lara‐Astaiso, D. , Toth, B. , Itzkovitz, S. , Colonna, M. , Schwartz, M. , & Amit, I. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell, 169(7), 1276–1290 e1217. 10.1016/j.cell.2017.05.018 [DOI] [PubMed] [Google Scholar]
- Kierdorf, K. , Erny, D. , Goldmann, T. , Sander, V. , Schulz, C. , Perdiguero, E. G. , Wieghofer, P. , Heinrich, A. , Riemke, P. , Hölscher, C. , Müller, D. N. , Luckow, B. , Brocker, T. , Debowski, K. , Fritz, G. , Opdenakker, G. , Diefenbach, A. , Biber, K. , Heikenwalder, M. , … Prinz, M. (2013). Microglia emerge from erythromyeloid precursors via Pu.1‐ and Irf8‐dependent pathways. Nature Neuroscience, 16(3), 273–280. 10.1038/nn.3318 [DOI] [PubMed] [Google Scholar]
- Kim, D. , Langmead, B. , & Salzberg, S. L. (2015). HISAT: A fast spliced aligner with low memory requirements. Nature Methods, 12(4), 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolde, R. (2012). Pheatmap: Pretty heatmaps. R package version, 1(2).
- Krasemann, S. , Madore, C. , Cialic, R. , Baufeld, C. , Calcagno, N. , el Fatimy, R. , Beckers, L. , O'Loughlin, E. , Xu, Y. , Fanek, Z. , Greco, D. J. , Smith, S. T. , Tweet, G. , Humulock, Z. , Zrzavy, T. , Conde‐Sanroman, P. , Gacias, M. , Weng, Z. , Chen, H. , … Butovsky, O. (2017). The TREM2‐APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity, 47(3), 566–581 e569. 10.1016/j.immuni.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis, N. D. , Hill, J. D. , Juchem, K. W. , Stefanopoulos, D. E. , & Modis, L. K. (2014). RNA sequencing of microglia and monocyte‐derived macrophages from mice with experimental autoimmune encephalomyelitis illustrates a changing phenotype with disease course. Journal of Neuroimmunology, 277(1–2), 26–38. 10.1016/j.jneuroim.2014.09.014 [DOI] [PubMed] [Google Scholar]
- Li, M. , Santpere, G. , Imamura Kawasawa, Y. , Evgrafov, O. V. , Gulden, F. O. , Pochareddy, S. , Sunkin, S. M. , Li, Z. , Shin, Y. , Zhu, Y. , Sousa, A. M. M. , Werling, D. M. , Kitchen, R. R. , Kang, H. J. , Pletikos, M. , Choi, J. , Muchnik, S. , Xu, X. , Wang, D. , … Li, Z. (2018). Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science, 362(6420), aat7615. 10.1126/science.aat7615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao, Y. , Smyth, G. K. , & Shi, W. (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30(7), 923–930. 10.1093/bioinformatics/btt656 [DOI] [PubMed] [Google Scholar]
- Liu, J. , Gao, C. , Sodicoff, J. , Kozareva, V. , Macosko, E. Z. , & Welch, J. D. (2020). Jointly defining cell types from multiple single‐cell datasets using LIGER. Nature Protocols, 15(11), 3632–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locatelli, G. , Theodorou, D. , Kendirli, A. , Jordão, M. J. C. , Staszewski, O. , Phulphagar, K. , Cantuti‐Castelvetri, L. , Dagkalis, A. , Bessis, A. , Simons, M. , Meissner, F. , Prinz, M. , & Kerschensteiner, M. (2018). Mononuclear phagocytes locally specify and adapt their phenotype in a multiple sclerosis model. Nature Neuroscience, 21(9), 1196–1208. 10.1038/s41593-018-0212-3 [DOI] [PubMed] [Google Scholar]
- Love, M. I. , Huber, W. , & Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biology, 15(12), 550. 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, J. , Liu, R. , Peng, H. , Zhou, J. , & Li, H. (2010). CD81 inhibits the proliferation of astrocytes by inducing G(0)/G (1) arrest in vitro. Journal of Huazhong University of Science and Technology. Medical Sciences, 30(2), 201–205. 10.1007/s11596-010-0214-1 [DOI] [PubMed] [Google Scholar]
- Maecker, H. T. , Todd, S. C. , & Levy, S. (1997). The tetraspanin superfamily: Molecular facilitators. FASEB Journal, 11(6), 428–442. [PubMed] [Google Scholar]
- Marioni, R. E. , Harris, S. E. , Zhang, Q. , McRae, A. F. , Hagenaars, S. P. , Hill, W. D. , Davies, G. , Ritchie, C. W. , Gale, C. R. , Starr, J. M. , Goate, A. M. , Porteous, D. J. , Yang, J. , Evans, K. L. , Deary, I. J. , Wray, N. R. , & Visscher, P. M. (2018). GWAS on family history of Alzheimer's disease. Translational Psychiatry, 8(1), 99. 10.1038/s41398-018-0150-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, M. (2011). Cutadapt removes adapter sequences from high‐throughput sequencing reads. EMBnet. Journal, 17(1), 10–12. [Google Scholar]
- Masuda, T. , Amann, L. , Sankowski, R. , Staszewski, O. , Lenz, M. , d'Errico, P. , Snaidero, N. , Costa Jordão, M. J. , Böttcher, C. , Kierdorf, K. , Jung, S. , Priller, J. , Misgeld, T. , Vlachos, A. , Meyer‐Luehmann, M. , Knobeloch, K. P. , & Prinz, M. (2020). Novel Hexb‐based tools for studying microglia in the CNS. Nature Immunology, 21(7), 802–815. 10.1038/s41590-020-0707-4 [DOI] [PubMed] [Google Scholar]
- Masuda, T. , Sankowski, R. , Staszewski, O. , Böttcher, C. , Amann, L. , Sagar, Scheiwe, C. , Nessler, S. , Kunz, P. , van Loo, G. , Coenen, V. A. , Reinacher, P. C. , Michel, A. , Sure, U. , Gold, R. , Grün, D. , Priller, J. , Stadelmann, C. , & Prinz, M. (2019). Spatial and temporal heterogeneity of mouse and human microglia at single‐cell resolution. Nature, 566(7744), 388–392. 10.1038/s41586-019-0924-x [DOI] [PubMed] [Google Scholar]
- Mathys, H. , Davila‐Velderrain, J. , Peng, Z. , Gao, F. , Mohammadi, S. , Young, J. Z. , Menon, M. , He, L. , Abdurrob, F. , Jiang, X. , Martorell, A. J. , Ransohoff, R. M. , Hafler, B. P. , Bennett, D. A. , Kellis, M. , & Tsai, L. H. (2019). Single‐cell transcriptomic analysis of Alzheimer's disease. Nature, 570(7761), 332–337. 10.1038/s41586-019-1195-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuade, A. , & Blurton‐Jones, M. (2019). Microglia in Alzheimer's disease: Exploring how genetics and phenotype influence risk. Journal of Molecular Biology, 431(9), 1805–1817. 10.1016/j.jmb.2019.01.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendiola, A. S. , Ryu, J. K. , Bardehle, S. , Meyer‐Franke, A. , Ang, K. K. , Wilson, C. , Baeten, K. M. , Hanspers, K. , Merlini, M. , Thomas, S. , Petersen, M. A. , Williams, A. , Thomas, R. , Rafalski, V. A. , Meza‐Acevedo, R. , Tognatta, R. , Yan, Z. , Pfaff, S. J. , Machado, M. R. , … Akassoglou, K. (2020). Transcriptional profiling and therapeutic targeting of oxidative stress in neuroinflammation. Nature Immunology, 21(5), 513–524. 10.1038/s41590-020-0654-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milich, L. M. , Choi, J. S. , Ryan, C. , Cerqueira, S. R. , Benavides, S. , Yahn, S. L. , Tsoulfas, P. , & Lee, J. K. (2021). Single‐cell analysis of the cellular heterogeneity and interactions in the injured mouse spinal cord. The Journal of Experimental Medicine, 218(8), e20210040. 10.1084/jem.20210040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mordica, W. J. , Woods, K. M. , Clem, R. J. , Passarelli, A. L. , & Chapes, S. K. (2009). Macrophage cell lines use CD81 in cell growth regulation. In Vitro Cellular & Developmental Biology. Animal, 45(5–6), 213–225. 10.1007/s11626-008-9167-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraoka, S. , Jedrychowski, M. P. , Iwahara, N. , Abdullah, M. , Onos, K. D. , Keezer, K. J. , Hu, J. , Ikezu, S. , Howell, G. R. , Gygi, S. P. , & Ikezu, T. (2021). Enrichment of neurodegenerative microglia signature in brain‐derived extracellular vesicles isolated from Alzheimer's disease mouse models. Journal of Proteome Research, 20(3), 1733–1743. 10.1021/acs.jproteome.0c00934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray, P. J. , Allen, J. E. , Biswas, S. K. , Fisher, E. A. , Gilroy, D. W. , Goerdt, S. , Gordon, S. , Hamilton, J. A. , Ivashkiv, L. B. , Lawrence, T. , Locati, M. , Mantovani, A. , Martinez, F. O. , Mege, J. L. , Mosser, D. M. , Natoli, G. , Saeij, J. P. , Schultze, J. L. , Shirey, K. A. , … Wynn, T. A. (2014). Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity, 41(1), 14–20. 10.1016/j.immuni.2014.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olah, M. , Menon, V. , Habib, N. , Taga, M. F. , Ma, Y. , Yung, C. J. , Cimpean, M. , Khairallah, A. , Coronas‐Samano, G. , Sankowski, R. , Grün, D. , Kroshilina, A. A. , Dionne, D. , Sarkis, R. A. , Cosgrove, G. R. , Helgager, J. , Golden, J. A. , Pennell, P. B. , Prinz, M. , … de Jager, P. L. (2020). Single cell RNA sequencing of human microglia uncovers a subset associated with Alzheimer's disease. Nature Communications, 11(1), 6129. 10.1038/s41467-020-19737-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli, R. C. , Bergamini, G. , & Rajendran, L. (2019). Cell‐to‐cell communication by extracellular vesicles: Focus on microglia. Neuroscience, 405, 148–157. 10.1016/j.neuroscience.2018.04.003 [DOI] [PubMed] [Google Scholar]
- Plemel, J. R. , Stratton, J. A. , Michaels, N. J. , Rawji, K. S. , Zhang, E. , Sinha, S. , Baaklini, C. S. , Dong, Y. , Ho, M. , Thorburn, K. , Friedman, T. N. , Jawad, S. , Silva, C. , Caprariello, A. V. , Hoghooghi, V. , Yue, J. , Jaffer, A. , Lee, K. , Kerr, B. J. , … Yong, V. W. (2020). Microglia response following acute demyelination is heterogeneous and limits infiltrating macrophage dispersion. Science Advances, 6(3), eaay6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramesha, S. , Rayaprolu, S. , Bowen, C. A. , Giver, C. R. , Bitarafan, S. , Nguyen, H. M. , Gao, T. , Chen, M. J. , Nwabueze, N. , Dammer, E. B. , Engstrom, A. K. , Xiao, H. , Pennati, A. , Seyfried, N. T. , Katz, D. J. , Galipeau, J. , Wulff, H. , Waller, E. K. , Wood, L. B. , … Rangaraju, S. (2021). Unique molecular characteristics and microglial origin of Kv1. 3 channel–positive brain myeloid cells in Alzheimer's disease. Proceedings of the National Academy of Sciences, 118(11), e2013545118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexach, J. E. , Polioudakis, D. , Yin, A. , Swarup, V. , Chang, T. S. , Nguyen, T. , Sarkar, A. , Chen, L. , Huang, J. , Lin, L. C. , Seeley, W. , Trojanowski, J. Q. , Malhotra, D. , & Geschwind, D. H. (2020). Tau pathology drives dementia risk‐associated gene networks toward chronic inflammatory states and immunosuppression. Cell Reports, 33(7), 108398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safaiyan, S. , Besson‐Girard, S. , Kaya, T. , Cantuti‐Castelvetri, L. , Liu, L. , Ji, H. , Schifferer, M. , Gouna, G. , Usifo, F. , Kannaiyan, N. , Fitzner, D. , Xiang, X. , Rossner, M. J. , Brendel, M. , Gokce, O. , & Simons, M. (2021). White matter aging drives microglial diversity. Neuron, 109(7), 1100–1117.e1110. [DOI] [PubMed] [Google Scholar]
- Safaiyan, S. , Kannaiyan, N. , Snaidero, N. , Brioschi, S. , Biber, K. , Yona, S. , Edinger, A. L. , Jung, S. , Rossner, M. J. , & Simons, M. (2016). Age‐related myelin degradation burdens the clearance function of microglia during aging. Nature Neuroscience, 19(8), 995–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanin, D. E. , Ge, Y. , Marinkovic, E. , Kabat, A. M. , Castoldi, A. , Caputa, G. , Grzes, K. M. , Curtis, J. D. , Thompson, E. A. , Willenborg, S. , Dichtl, S. , Reinhardt, S. , Dahl, A. , Pearce, E. L. , Eming, S. A. , Gerbaulet, A. , Roers, A. , Murray, P. J. , & Pearce, E. J. (2022). A common framework of monocyte‐derived macrophage activation. Science Immunology, 7(70), eabl7482. 10.1126/sciimmunol.abl7482 [DOI] [PubMed] [Google Scholar]
- Schlachetzki, J. C. M. , Prots, I. , Tao, J. , Chun, H. B. , Saijo, K. , Gosselin, D. , Winner, B. , Glass, C. K. , & Winkler, J. (2018). A monocyte gene expression signature in the early clinical course of Parkinson's disease. Scientific Reports, 8(1), 10757. 10.1038/s41598-018-28986-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobue, A. , Komine, O. , Hara, Y. , Endo, F. , Mizoguchi, H. , Watanabe, S. , Murayama, S. , Saito, T. , Saido, T. C. , Sahara, N. , Higuchi, M. , Ogi, T. , & Yamanaka, K. (2021). Microglial gene signature reveals loss of homeostatic microglia associated with neurodegeneration of Alzheimer's disease. Acta Neuropathologica Communications, 9(1), 1. 10.1186/s40478-020-01099-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somebang, K. , Rudolph, J. , Imhof, I. , Li, L. , Niemi, E. C. , Shigenaga, J. , Tran, H. , Gill, T. M. , Lo, I. , Zabel, B. A. , Schmajuk, G. , Wipke, B. T. , Gyoneva, S. , Jandreski, L. , Craft, M. , Benedetto, G. , Plowey, E. D. , Charo, I. , Campbell, J. , … Hsieh, C. L. (2021). CCR2 deficiency alters activation of microglia subsets in traumatic brain injury. Cell Reports, 36(12), 109727. 10.1016/j.celrep.2021.109727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa, C. , Golebiewska, A. , Poovathingal, S. K. , Kaoma, T. , Pires‐Afonso, Y. , Martina, S. , Coowar, D. , Azuaje, F. , Skupin, A. , Balling, R. , Biber, K. , Niclou, S. P. , & Michelucci, A. (2018). Single‐cell transcriptomics reveals distinct inflammation‐induced microglia signatures. EMBO Reports, 19(11), e46171. 10.15252/embr.201846171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri, A. G. , Ni, D. , Ling, Z. L. , Macia, L. , Campbell, I. L. , Hofer, M. J. , & King, N. J. C. (2022). PLX5622 reduces disease severity in lethal CNS infection by off‐target inhibition of peripheral inflammatory monocyte production. Frontiers in Immunology, 13. 10.3389/fimmu.2022.851556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri, A. G. , Terry, R. L. , Wishart, C. L. , Ashhurst, T. M. , Campbell, I. L. , Hofer, M. J. , & King, N. J. C. (2021). High‐parameter cytometry unmasks microglial cell spatio‐temporal response kinetics in severe neuroinflammatory disease. Journal of Neuroinflammation, 18(1), 166. 10.1186/s12974-021-02214-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiteri, A. G. , Wishart, C. L. , Pamphlett, R. , Locatelli, G. , & King, N. J. C. (2022). Microglia and monocytes in inflammatory CNS disease: Integrating phenotype and function. Acta Neuropathologica, 143(2), 179–224. 10.1007/s00401-021-02384-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, K. , Friedman, B. A. , Etxeberria, A. , Huntley, M. A. , van der Brug, M. P. , Foreman, O. , Paw, J. S. , Modrusan, Z. , Beach, T. G. , Serrano, G. E. , & Hansen, D. V. (2020). Alzheimer's patient microglia exhibit enhanced aging and unique transcriptional activation. Cell Reports, 31(13), 107843. 10.1016/j.celrep.2020.107843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay, T. L. , Sagar, Dautzenberg, J. , Grun, D. , & Prinz, M. (2018). Unique microglia recovery population revealed by single‐cell RNAseq following neurodegeneration. Acta Neuropathologica Communications, 6(1), 87. 10.1186/s40478-018-0584-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, R. L. , Deffrasnes, C. , Getts, D. R. , Minten, C. , van Vreden, C. , Ashhurst, T. M. , Getts, M. T. , Xie, R. D. V. , Campbell, I. L. , & King, N. J. (2015). Defective inflammatory monocyte development in IRF8‐deficient mice abrogates migration to the West Nile virus‐infected brain. Journal of Innate Immunity, 7(1), 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry, R. L. , Getts, D. R. , Deffrasnes, C. , van Vreden, C. , Campbell, I. L. , & King, N. J. (2012). Inflammatory monocytes and the pathogenesis of viral encephalitis. Journal of Neuroinflammation, 9, 270. 10.1186/1742-2094-9-270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt, C. (2021). UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Research, 49(D1), D480–D489. 10.1093/nar/gkaa1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vankriekelsvenne, E. , Chrzanowski, U. , Manzhula, K. , Greiner, T. , Wree, A. , Hawlitschka, A. , Llovera, G. , Zhan, J. , Joost, S. , Schmitz, C. , Ponsaerts, P. , Amor, S. , Nutma, E. , Kipp, M. , & Kaddatz, H. (2022). Transmembrane protein 119 is neither a specific nor a reliable marker for microglia. Glia, 70(6), 1170–1190. 10.1002/glia.24164 [DOI] [PubMed] [Google Scholar]
- Vanrossum, G. , & DeBoer, J. (1991). Interactively testing remote servers using the Python programming language. Centrum voor Wiskunde en Informatica, 4, 283–304. [Google Scholar]
- Vera Alvarez, R. , Pongor, L. S. , Marino‐Ramirez, L. , & Landsman, D. (2019). TPMCalculator: One‐step software to quantify mRNA abundance of genomic features. Bioinformatics, 35(11), 1960–1962. 10.1093/bioinformatics/bty896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Zhao, Y. , & Zhang, B. (2015). Efficient test and visualization of multi‐set intersections. Scientific Reports, 5, 16923. 10.1038/srep16923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch, J. D. , Kozareva, V. , Ferreira, A. , Vanderburg, C. , Martin, C. , & Macosko, E. Z. (2019). Single‐cell multi‐omic integration compares and contrasts features of brain cell identity. Cell, 177(7), 1873–1887.e1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, Y. , Mass, E. , Ashok Kumar, P. , Ulas, T. , Händler, K. , Horne, A. , Klee, K. , Lupp, A. , Schütz, D. , Saaber, F. , Redecker, C. , Schultze, J. L. , Geissmann, F. , & Stumm, R. (2020). Cxcr4 distinguishes HSC‐derived monocytes from microglia and reveals monocyte immune responses to experimental stroke. Nature Neuroscience, 23(3), 351–362. 10.1038/s41593-020-0585-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham, H. (2006). An introduction to ggplot: An implementation of the grammar of graphics in R. Statistics. [Google Scholar]
- Wickham, H. , & Wickham, M. H. (2007). The ggplot package. Google Scholar. [Google Scholar]
- Yamasaki, R. , Lu, H. , Butovsky, O. , Ohno, N. , Rietsch, A. M. , Cialic, R. , Wu, P. M. , Doykan, C. E. , Lin, J. , Cotleur, A. C. , Kidd, G. , Zorlu, M. M. , Sun, N. , Hu, W. , Liu, L. P. , Lee, J. C. , Taylor, S. E. , Uehlein, L. , Dixon, D. , … Ransohoff, R. M. (2014). Differential roles of microglia and monocytes in the inflamed central nervous system. The Journal of Experimental Medicine, 211(8), 1533–1549. 10.1084/jem.20132477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, H. S. , Onos, K. D. , Choi, K. , Keezer, K. J. , Skelly, D. A. , Carter, G. W. , & Howell, G. R. (2021). Natural genetic variation determines microglia heterogeneity in wild‐derived mouse models of Alzheimer's disease. Cell Reports, 34(6), 108739. 10.1016/j.celrep.2021.108739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Chen, K. , Sloan, S. A. , Bennett, M. L. , Scholze, A. R. , O'Keeffe, S. , Phatnani, H. P. , Guarnieri, P. , Caneda, C. , Ruderisch, N. , Deng, S. , Liddelow, S. A. , Zhang, C. , Daneman, R. , Maniatis, T. , Barres, B. A. , & Wu, J. Q. (2014). An RNA‐sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. The Journal of Neuroscience, 34(36), 11929–11947. 10.1523/JNEUROSCI.1860-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Clustering parameters used for defining myeloid subtypes of the LIGER‐integrated scRNA‐seq data set. (A) Parameter selection for the number of factors k versus the median Kullback–Leibler (KL) value across all cells for the LIGER‐integrated data set of six disease models. The tuning parameter, lambda, was kept at the recommended value of 5 (Liu et al., 2020). The optimal value of k is defined as the “elbow” of the plot, that is, the k value at which the median KL value becomes negligible (defined between k = 10–15, dashed lines). (B) UMAP showing the clusters for two optimal k values, k = 12 (alignment = 0.878) and k = 15 (alignment = 0.837). The optimal number of factors chosen was k = 12 based on improved alignment between datasets and based on prior biological knowledge of clustering (Liu et al., 2020). (C) UMAP of myeloid cells from the downsampled LIGER‐integrated dataset (k = 12) overlaid by either microglia (2814 cells) and/or MC clusters (1407 cells). (D) UMAP showing the anatomical location from which myeloid subclusters were isolated during CNS tissue processing and cell sorting. (E) Dot plot heatmap showing the average expression of microglia‐specific and MC‐enriched genes in identified clusters. Clusters have been ordered by unsupervised hierarchical clustering. (F) UMAPs showing single‐cell gene expression overlays for microglia‐specific genes, P2ry12, Tmem119, Hexb and Sparc, in MC and microglial clusters.
Figure S2. Expression of universal microglial signatures by single‐cell RNA‐seq myeloid clusters.
(A) UMAP of 4221 myeloid cells (downsampled) with clusters annotated and pseudocolored based on cell type cluster. (B) Dot plot heatmap showing the average expression of disease‐associated microglial (DAM) and microglial neurodegenerative (MGnD) genes in all myeloid clusters. Clusters have been ordered by unsupervised hierarchical clustering. Microglial clusters with high expression of DAM/MGnD genes are annotated as DAM‐like microglia (Mg6, red box). (C) DAM‐like microglia Mg6 (red) overlaid onto UMAPs of microglia clusters from each disease model. Numbers represent the relative frequency percentage of Mg6 out of the microglial pool within each disease model.
Figure S3. Histograms showing the expression of CD81 on CD45+ cell types in the non‐infected and WNV‐infected bone marrow at dpi 7.
Figure S4. Histograms showing the expression of CD81 on CD45+ cell types in the non‐infected and WNV‐infected spleen at dpi 7.
Table S1. Study metadata for included and excluded studies.
Table S2. Hierarchical clustering arrangement of microglial and MC samples from disparate studies, disease‐models and sequencing technologies.
Table S3. Top ranked genes and GO terms associated with single‐origin RP analysis for each microglia and MC population from six disease models.
Table S4. Top GO terms associated with scRNA‐seq myeloid clusters across six disease models.
Table S5. GO intersections for scRNA‐seq myeloid clusters.
Table S6. Differentially enriched microglial and MC genes and their associated GO enrichment terms conserved across seven disease models.
Data Availability Statement
All RNA datasets were analyzed from existing, publicly available data. These accession numbers for the datasets are listed in the Key Resources Table (Table 1). All code was adapted from existing code listed in Table 1. Flow cytometry data will be shared by the lead contact upon reasonable request. Any additional information required to re‐analyze the data reported in this paper is available from the lead contact upon request.