

Abstract
Since 1997, highly pathogenic avian influenza viruses, such as H5N1, have been recognized as a possible pandemic hazard to men and the poultry business. The rapid rate of mutation of H5N1 viruses makes the whole process of designing vaccines extremely challenging. Here, we used an in silico approach to design a multi-epitope vaccine against H5N1 influenza A virus using hemagglutinin (HA) and neuraminidase (NA) antigens. B-cell epitopes, Cytotoxic T lymphocyte (CTL) and Helper T lymphocyte (HTL) were predicted via IEDB, NetMHC-4 and NetMHCII-2.3 respectively. Two adjuvants consisting of Human β-defensin-3 (HβD-3) along with pan HLA DR-binding epitope (PADRE) have been chosen to induce more immune response. Linkers including KK, AAY, HEYGAEALERAG, GPGPGPG and double EAAAK were utilized to link epitopes and adjuvants. This construct encodes a protein having 350 amino acids and 38.46 kDa molecular weight. Antigenicity of ~ 1, the allergenicity of non-allergen, toxicity of negative and solubility of appropriate were confirmed through Vaxigen, AllerTOP, ToxDL and DeepSoluE, respectively. The 3D structure of H5N1 was refined and validated with a Z-Score of − 0.87 and an overall Ramachandran of 99.7%. Docking analysis showed H5N1 could interact with TLR7 (docking score of − 374.08 and by 4 hydrogen bonds) and TLR8 (docking score of − 414.39 and by 3 hydrogen bonds). Molecular dynamics simulations results showed RMSD and RMSF of 0.25 nm and 0.2 for H5N1-TLR7 as well as RMSD and RMSF of 0.45 nm and 0.4 for H5N1-TLR8 complexes, respectively. Molecular Mechanics Poisson-Boltzmann Surface Area (MM/PBSA) confirmed stability and continuity of interaction between H5N1-TLR7 with the total binding energy of − 29.97 kJ/mol and H5N1-TLR8 with the total binding energy of − 23.9 kJ/mol. Investigating immune response simulation predicted evidence of the ability to stimulate T and B cells of the immunity system that shows the merits of this H5N1 vaccine proposed candidate for clinical trials.
Supplementary Information
The online version contains supplementary material available at 10.1186/s12985-024-02337-7.
Keywords: Docking, Immunoinformatics, Influenza, Molecular dynamics, Vaccine
Introduction
A member of the Orthomyxoviridae family is influenza virus A [1]. This virus is responsible for causing acute respiratory disease and consistently changes while circulating among various animal hosts, such as wild birds, pigs, poultry, and horses as well as humans. The differentiation of these viruses is primarily according to the surface glycoproteins, specifically Hemagglutinin (HA) along with Neuraminidase (NA) [2]. There exist eighteen distinct variations of HA as well as eleven different NAs, each of which can be distinguished serologically. Notably, antibodies produced against a virus genotype don’t exhibit a reaction through other genotypes [3].
H5 influenza viruses have been typically found in avian populations and are naturally hosted by birds. However, they can infect a range of mammal species as well. There are nine recognized subtypes of H5 viruses, which include H5N1, H5N3, H5N2, H5N5, H5N4, H5N7, H5N6, H5N9 and H5N8. Among these subtypes, Worldwide, the viruses known as Low Pathogenic Avian Influenza (LPAI) are the most often seen in poultry and wild birds. Nevertheless, Highly Pathogenic Avian Influenza (HPAI) viruses are sporadically identified as well. The very first known instance of a highly pathogenic H5N1 virus infection in humans was reported by Yuen et al. 1998 [4]. These H5N1 viruses have significantly spread across regions in Asia, and the world. They have undergone rapid evolution, giving rise to ten distinct clades, labeled as clades 0 through 9. Since 2007, H5N1 viruses have been identified to be a significant hazard to men, wild birds, as well as poultry. Specifically, clade 2.2.2 was prevalent from 2007 to 2011, and from 2011 onwards, clade 2.3.2.1a has been in circulation [5].
The genotypes of H5N1 are determined by eight single-stranded RNA segments that encode a total of 11 proteins, among them surface glycoproteins known as neuraminidase (NA) and hemagglutinin (HA) [6]. Both HA and NA play essential roles that revolve around their contact with sialic acid. A terminal structure seen on glycoproteins or glycolipids that are expressed on the surface of cells is sialic acid [7]. The mechanism known as clathrin-mediated endocytosis, which is facilitated by HA's attachment to sialic acids on cellular receptors, allows the virus to enter cells. It's worth noting that the virus may also employ alternative endocytic pathways, like micropinocytosis, for cell entry [8].
NA is important in the final phases of infection. In particular, it functions to eliminate sialic acids from both cellular receptors and the freshly produced NA and HA present upon developing virions. The glycosylation activities of the host cell include the addition of these sialic acids [9, 10]. Moreover, NA's cleavage of sialic acids inhibits virions from clumping together and from attaching themselves to dying host cells via HA. This, in turn, facilitates the efficient release of virions and their spread to new target cells [9]. Given their crucial roles in the virus's pathogenicity, In the realm of vaccine research and development, HA and NA have become the main targets to neutralize avian influenza viruses (AIV) [10–12].
At present, there isn’t a vaccine strain on the market that can successfully produce protective immunity against the various clades and subclades of highly pathogenic avian influenza viruses (HPAIV) in the H5 lineage [13]. Nonetheless, Previously, research has indicated that vaccination against seasonal influenza A strains like H1N1 and H3N2 may improve cross-type cellular and humoral protection against the highly pathogenic avian influenza virus subtype H5N1 [14]. Both our research and the previously mentioned findings underscore the significant public health threat posed by H5N1 viruses, even though the number of human cases may be relatively small.
Consequently, the development of vaccines holds promise in helping to mitigate the impact of disease outbreaks. Reverse vaccinology is a recent breakthrough that, in addition to the conventional and laborious vaccine creation approaches, integrates immunogenomics and bioinformatics to quickly generate unique multi-epitope-based subunit vaccines. This approach offers the potential to expedite the vaccine development process [15, 16]. Today, there are extensive studies based on the use of bioinformatics and immunoinformatics tools to design new-generation vaccines [17–23].
Therefore, the current research aimed to produce a chimeric vaccine versus avian influenza A virus subtype H5N1 that is non-allergenic and immunogenic. This was achieved by utilizing a vaccinia’s approach, and It is advised that model animals be used in a laboratory environment to carry out the experimental validation of the suggested vaccination candidate.
Material and method
Antigen retrieval
The Influenza NCBI Research Database included the HA and NA protein sequences from several influenza H5N1 virus strains as described by Squires et al. 2012 [24].
Liner B cell epitopes and discontinues B cell epitopes prediction
To identify B cell antigenicity, Researchers used the Immune Epitope Database (IEDB) and BepiPred-2.0, a tool available at http://www.cbs.dtu.dk/services/BepiPred/, to detect linear B cell epitopes within the sequences of HA and NA proteins to predict B cell epitopes with 75% accuracy, 0.49 sensitivity, and 0.75 specificity, as detailed in the studies by Saadi et al. (2017) and Saha and Raghava [25](2006a). These tools incorporated several algorithms such as Emini surface accessibility prediction, and Bepipred linear epitope prediction 2.0 as well as Kolaskar and Tongaonkar antigenicity scale, which can be accessed at http://tools.iedb.org/bcell/. The semi-empirical Kolaskar and Tongaonkar technique takes into account the physicochemical characteristics of amino acid residues and their frequency in experimentally established segmental epitopes to predict antigenic determinants on proteins [26].
We also used the ElliPro server to forecast the discontinuous B cell epitopes' three-dimensional shape, which can be accessed at http://tools.iedb.org/ellipro/. ElliPro is a program that takes the three-dimensional structure of the protein into account to help anticipate conformational or discontinuous B cell epitopes. This is crucial for understanding how these epitopes are identified via the immunity system as well as for designing effective vaccines or therapeutic antibodies.
MHC-I epitope prediction
The T cell epitopes’ prediction, specifically cytotoxic T cell epitopes, has been carried out utilizing an enhanced neural network methodology offered via the NetMHC-4.0 server, accessible at https://services.healthtech.dtu.dk/service.php?NetMHC-4.0. In this analysis, all epitopes have been regarded to be 9-mers in length, as recommended by the server. These 9-mer epitopes are recognized by HLA Class I molecules, including HLA-A, HLA-C as well as HLA-B.
MHC-II epitope prediction
The epitope binding to MHC-II molecules has been predicted using the NetMHCII 2.3 server. This service predicts epitope affinity for a variety of HLA-II molecules, such as HLA-DR, HLA-DQ, and HLA-DP, using an artificial neural network. By using this prediction tool, Researchers can evaluate the probability that epitopes will attach to MHC-II molecules, which is critical for understanding and designing immune responses, particularly those involving helper T cells.
Population coverage determination
The Population Coverage instrument in the IEDB server, accessible at http://tools.iedb.org/population, was used to determine the population's coverage rate for particular epitopes. Determining the population coverage is essential to forecast the right epitopes for various HLA-binding interactions. Epitopes that exhibit strong binding to a variety of HLA alleles can provide broader coverage across different ethnicities, reducing the constraints of MHC limitations on T cell reactions. In such context, the evaluation of MHC-I HLA binding included a range of HLA alleles such as HLA-B5701, HLA-B5801. HLA-B5802, HLA-B2705, HLA-A0203, HLA-A2603, HLA-B8301, HLA-B4002, HLA-A3002, HLA-A2902, HLA-B4402, HLA-C0401, and HLA-C0303 for the NA and HA proteins. Additionally, MHC-II HLA binding assessments were performed for DRB1_0101, DPA10201-DPB10501, DPA10103-DPB10301, DQA10501-DQB10201, DRB1_0403, DPA10103-DPB10401, DRB1_0404, DRB1_0403, DRB1_0402, DQA10102-DQB10501, DRB1_0401, DRB1_1302, DQA10301-DQB10302 and DQA10401-DQB10402 for the NA and HA proteins. This comprehensive analysis helps in identifying epitopes that have the potential to generate immunity reactions across a broad range of human populations.
Analysis of epitopes conservation
The degree of conservation of particular epitopes was evaluated using the IEDB Conservancy Analysis tool as a monitoring tool about other sequences that were comparable. Epitopes can exhibit varying levels of conservation, ranging from a minimum of 0 percent to a maximum of 100 percent, depending on their specific length and sequence. This analysis is important in understanding how well-conserved an epitope is within a given protein or among related sequences. Highly conserved epitopes are more likely to be effective in stimulating immune responses because they are present in a broader range of strains or related proteins, making them potentially useful for vaccine design or therapeutic development. On the other hand, less conserved epitopes may be strain-specific and may have limited utility in generating immune responses across different variants or strains.
Physicochemical, antigenicity, allergenicity, toxicity properties of the vaccine
In the design of the vaccine construct, various linkers were strategically employed to connect different components and epitopes. These linkers serve as connectors between different functional elements and help optimize the vaccine’s overall design. AAY linkers, specific HEYGAEALERAG linkerGPGPG linkers, KK linkers and EAAAK linkers are used to bond Bcell, CLT and HLT epitopes. Also,HβD-3 and PADRE are used as adjuvants in the N-terminal of the construct.
The ProtParam instrument, available at https://web.expasy.org/protparam/, has been utilized for predicting the physicochemical characteristics of the final protein construct. It provides information about various protein parameters, such as molecular weight, and isoelectric point (pI), as well as amino acid composition.
The VaxiJen v2.0 server, accessible at http://www.ddg-pharmfac.net/vaxijen/VaxiJen.html, has been utilized for predicting the antigenic properties of the protein construct. It's considered an independent alignment method and is employed for antigen prediction. The server's accuracy typically ranges from 70 to 89%. AllerTOP v. 2.0, found at https://www.ddg-pharmfac.net/AllerTOP/, has been utilized for calculating the total protein sensitivity. This server uses an auto cross-covariance (ACC) as well as a k-nearest neighbor algorithm (kNN, k = 1) for predicting the proteins’ allergenicity. It considers various factors, including hydrophobicity, molecular weight, secondary structure characteristics, as well as the amino acids’ relative abundance. The ToxDL server, available at http://www.csbio.sjtu.edu.cn/bioinf/ToxDL/index.html, has been used to assess the protein's toxicity. This server employs an interpretable deep learning-based methodology for categorizing proteins into 2 categories: toxic and non-toxic. It uses a multi-modal approach like Convolutional Neural Networks (CNNs), and the InterProscan database. These tools and servers collectively provide valuable insights into physicochemical characteristics, antigenicity, allergenicity, along toxicity of protein construct, which are essential considerations in vaccine development and protein engineering.
The use of NetSurfP-2.0 online software, available at https://services.healthtech.dtu.dk/service.php?NetSurfP-2.0, has been implemented to predict the secondary structure of the protein. NetSurfP-2.0 is a sequence-based instrument that offers predictions regarding the secondary structure of amino acids within a protein.
3D modeling, refinement and validation of the H5N1 construct
The RoseTTAFold online software, accessible at https://robetta.bakerlab.org/, has been employed for constructing the three-dimensional (3D) structure of the protein. RoseTTAFold is considered a sophisticated “three-track” neural network, which means it regards multiple aspects simultaneously for predicting protein structures that have high accuracy. RoseTTAFold considers patterns in protein sequences, allowing it to understand the amino acid sequences that make up the protein.
To guarantee the precision and excellence of the protein structure simulation, PROCHECK and SAVES online servers, available at http://services.mbi.ucla.edu/PROCHECK and http://services.mbi.ucla.edu/SAVES, respectively, were used for structural validation.
The PROSA server, accessible at https://prosa.services.came.sbg.ac.at/prosa.php, has been utilized to identify the protein energy and Z-Score balance. This helps assess the quality and stability of the 3D structure of the protein.
The Ramachandran plot, which depicts the phi as well as psi angles of amino acids in protein structure, was generated using the online website http://molprobity.biochem.duke.edu/. It provides insight into the quality of the protein's backbone torsion angles.
The optimum spatial resolution for the study's optimal energy was found using the PyMol program. It is likely used for visualizing and analyzing the 3D protein structure. The website http://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE has been employed for protein structure refinement.
Docking analysis
The 3D structures of TLR7 (PDB code: 7CYN) and TLR8 (PDB code: 3w3g), were obtained from the RCSB server. The TLRs’ 3D structures have been adjusted for energy using the PyMOL v2.3.4 program. The vaccination and TLR PDB structures were cleared of water molecules. The prepared 3D structures were further processed within the Chimera V 1.13.1 program. H5N1 Influenza construct, TLR7 and TLR8 have been then submitted to the HDOCK server to identify interaction regions. Taking into account the complete molecular interface between H5N1-TLRs, the complexes with the lowest intermolecular binding energy have been ranked best. The choice was made with the lowest mean Root Mean Square Deviation (RMSD). H-bond formation has been evaluated using LigPlot+. These steps describe the process of retrieving, preparing, and analyzing the 3D structures of the components and their interactions to comprehend the binding and molecular contacts among the H5N1 influenza construct and TLRs.
Molecular dynamics and MM/PBSA analysis
Molecular dynamics simulations are a powerful tool for understanding the dynamic behavior of biomolecular complexes and are commonly used to investigate the interactions and stability of such complexes over time. The use of Gromacs and the CHARMM36 force field in this context helps provide valuable insights into the function of vaccine complexes in a simulated environment. Molecular dynamics calculations for the vaccine-m826 and vaccine-TLR7/8 complexes have been carried out using Gromacs v4.6.5 and the CHARMM36 all-atom force field. The protein–protein complexes have been solvated by adding water molecules, as well as the entire system has been then neutralized to ensure that it had an overall neutral charge. An energy minimization step was performed to optimize the geometry of the system and remove any steric clashes or unfavorable interactions. This helps to stabilize the system before proceeding to simulations. Optimization of the system was done for both NPT (constant number of particles, temperature and pressure) and NVT (constant number of particles, temperature and volume). These optimizations were carried out at 300 K (Kelvin), a 1 bar pressure, and with restraint forces set to 1000 kJ/mol. Following the optimization steps, molecular dynamics simulations were conducted for a total of 40,000 picoseconds (40 ns). During this simulation, the positions of all atoms in the system were updated over time to study the dynamics and behavior of the H5N1-TLR7 and H5N1-TLR8 complexes. To maintain stability during the simulation, all bonds have been constrained utilizing the LINCS (LINear Constraint Solver) algorithm, ensuring that bond lengths between atoms remain fixed.
Simulations of H5N1 immune response
The C-ImmSim server was used to model and forecast the immunological response to recombinant influenza structures. To anticipate immunological interactions, C-ImmSim combines an agent-based model with machine learning methods and immune epitope prediction. It does this by using a position-specific scoring matrix (PSSM). C-ImmSim replicates three different mammalian anatomical areas at the same time: the bone marrow (representing hematopoietic stem cells, lymphoid cells, and myeloid cells), the thymus (mimicking native T cell selection), as well as a third lymph node resembling typical lymph nodes. The Simulation Steps parameter has been set to 1050 to account for the vaccine's 4-week gap between doses one and two. Time-steps of 1, 336, and 672, each corresponding to eight hours in real life, have been considered for the three injections. Different parameters used in this study were applied as default settings.
Insilico cloning and vaccine optimization
The GeneScript program was used to optimize codons for the production of a recombinant construct in the pET-26b(+) vector. Because the expression host used in the investigation was different from the natural host for influenza, this optimization was required. To adapt the codon usage for the new host, based on the recommended codon use of influenza for production in Escherichia coli K12, the codon optimization has been modified. This ensures that the gene sequence is better suited for efficient expression in the new host. Two important metrics for evaluating codon optimization are the Codon Adaptation Index (CAI) score as well as the GC content. An ideal CAI score is 1.0, indicating a high level of adaptation to the host's codon usage, while a GC content of 30–70% is typically considered optimal for predicting protein expression levels in the host. To facilitate the cloning process, the locations of the NdeI as well as XhoI restriction enzyme sites have been recognized in the optimized nucleotide sequences. The insertion of the recombinant influenza gene sequence into the pET-26b(+) vector and effective target protein production in the selected host are made possible by these restriction sites. SnapGene has been utilized as a tool to aid in this identification and cloning process.
Results
Linear and conformational B cell epitope prediction
B cell epitopes have a crucial part in the H5N1 vaccine due to their ability to stimulate humoral immunity. To identify these epitopes, within the final design, both discontinuous and linear B cell epitopes have been predicted. Linear B cell epitopes have been determined utilizing BepiPred 2.0, while discontinuous epitopes were predicted with the IEDB servers. In total, 14 and 10 linear B cell epitopes have been detected for HA and NA, respectively, and detailed in Table 1. Additionally, Additional file 1: Tables S1 and S2 provide a summary of all B cell epitopes found within the NA and HA proteins.
Table 1.
Liner B-cell epitopes predicted from NA and HA antigens
| No | Start | End | IEDB | BepiPred 2.0 |
|---|---|---|---|---|
| HA protein | ||||
| 2 | 40 | 70 | VTVTHAQDILEKTHNGKLCDLDGVKPLILRD | VTVTHAQDILEKTHNGKLCDLDGVKPLILRD |
| 3 | 82 | 90 | MCDEFINVP | MCDEFINVP |
| 4 | 100 | 156 | NPNNDLCYPGSFNDYEELKHLLSRINHFEKIQIIPKNSWSDHEASSGVSAACPYLGS | NPNNDLCYPGSFNDYEELKHLLSRINHFEKIQIIPKNSWSDHEASSGVSAACPYLGS |
| 6 | 171 | 185 | STYPTIKKSYNNTNQ | STYPTIKKSYNNTNQ |
| 7 | 200 | 211 | AAEQTRLYQNPT | AAEQTRLYQNPT |
| 8 | 223 | 240 | QRLVPKIATRSKVNGQSG | QRLVPKIATRSKVNGQSG |
| 9 | 274 | 296 | KKGDSAIMKSELEYGNCNTKCQT | KKGDSAIMKSELEYGNCNTKCQT |
| 10 | 299 | 310 | GAINSSMPFHNI | GAINSSMPFHN |
| 11 | 330 | 346 | ATGLRNSPQRESRRKKR | ATGLRNSPQRESRRKKR |
| 12 | 359 | 397 | GWQGMVDGWYGYHHSNEQGSGYAADKESTQKAVDGVTNK | GWQGMVDGWYGYHHSNEQGSGYAADKESTQKAVDGVTNK |
| 16 | 410 | 420 | EAVGREFNNLE | EAVGREFNNLE |
| 18 | 452 | 460 | RTLDFHDSN | RTLDFHDSN |
| 20 | 476 | 481 | AKELGN | AKELGN |
| 21 | 495 | 527 | MESIRNGTYNYPQYSEEARLKREEINGVKLESV | MESIRNGTYNYPQYSEEARLKREEINGVKLESV |
| NA protein | ||||
| 1 | 38 | 55 | IQKGNQHQAESISNTNPL | QKGNQHQAESISNTNPL |
| 2 | 68 | 74 | NSSLCPI | NSSLCPI |
| 3 | 119 | 134 | LMNDKHSNGTVKDRSP | LMNDKHSNGTVKDRSP |
| 5 | 193 | 206 | TDTIKSWRNNILRT | TDTIKSWRNNILRT |
| 6 | 226 | 236 | PSNGQASYKIF | PSNGQASYKIF |
| 7 | 240 | 252 | KGKVVKSVELDAP | KGKVVKSVELDAP |
| 9 | 303 | 323 | GDNPRPNDGTGSCGPMSPNGA | GDNPRPNDGTGSCGPMSPNGA |
| 12 | 342 | 370 | TKSTNSRSGFEMIWDPNGWTGTDSSFSVK | TKSTNSRSGFEMIWDPNGWTGTDSSFSVK |
| 13 | 376 | 381 | ITDWSG | ITDWSG |
| 17 | 431 | 445 | DTVSWSWPDGAELPF | DTVSWSWPDGAE |
Selection of CTL epitopes prediction
To predict CTL (Cytotoxic T Lymphocyte) epitopes, the NetMHC4 server has been employed specifically for the NA and HA proteins. The selection of MHC class I epitopes was based on their high levels of immunogenicity and antigenicity. Additionally, epitopes with a strong binding score, as determined by the NetMHC4 server, were considered. Select MHC-I epitopes were merged when they overlapped with one another. Ultimately, a total of 21 MHC class I epitopes were chosen, comprising 9 from HA and 12 from NA, which were integrated into the influenza vaccine construct (as detailed in Table 2). There are all predicted CTL epitopes, those that bind to MHC-I, within Additional file 1: Tables S3 and S4.
Table 2.
MHC-I epitopes prediction. HA and NA epitopes have been chosen according to MHC-I HLAs binding affinity (nM), %rank and antigenicity rate
| pos | HLA | Peptide | Affinity(nM) | Allergenicity | Toxicity | Solubility | Antigenicity |
|---|---|---|---|---|---|---|---|
| HA protein | |||||||
| 14 | HLA-A0101 | KSDQICIGY | 38.08 | No | Non-Toxin | Good | 2.07 |
| 223 | HLA-A3101 | RLVPKIATR | 9.27 | No | Non-Toxin | Good | 1.009 |
| 240 | HLA-A3201 | RMEFFWTIL | 7.29 | No | Non-Toxin | Good | 1.195 |
| 304 | HLA-A3207 | MPFHNIHPL | 17.58 | No | Non-Toxin | Good | 1.263 |
| 304 | HLA-A3215 | MPFHNIHPL | 26.87 | No | Non-Toxin | Good | 1.263 |
| 304 | HLA-A6601 | MPFHNIHPL | 986.12 | No | Non-Toxin | Good | 1.2633 |
| 304 | HLA-A6823 | MPFHNIHPL | 9.68 | No | Non-Toxin | Good | 1.2633 |
| 304 | HLA-B0801 | MPFHNIHPL | 40.2 | No | Non-Toxin | Good | 1.2633 |
| 304 | HLA-B1402 | MPFHNIHPL | 980.92 | No | Non-Toxin | Good | 1.2633 |
| 239 | HLA-B2705 | GRMEFFWTI | 16.64 | No | Non-Toxin | Good | 1.2086 |
| 239 | HLA-B2720 | GRMEFFWTI | 8.67 | No | Non-Toxin | Good | 1.2086 |
| 304 | HLA-B3501 | MPFHNIHPL | 6.01 | No | Non-Toxin | Good | 1.2633 |
| 150 | HLA-B3503 | CPYLGSPSF | 1547.44 | No | Non-Toxin | Good | 1.1181 |
| 306 | HLA-B3801 | FHNIHPLTI | 370.07 | No | Non-Toxin | Good | 1.3066 |
| 304 | HLA-B3901 | MPFHNIHPL | 6.61 | No | Non-Toxin | Good | 1.2633 |
| 240 | HLA-B4013 | RMEFFWTIL | 140.81 | No | Non-Toxin | Good | 1.1954 |
| 304 | HLA-B4201 | MPFHNIHPL | 16.2 | No | Non-Toxin | Good | 1.2633 |
| 447 | HLA-B4402 | MENERTLDF | 12.92 | No | Non-Toxin | Good | 1.5311 |
| 447 | HLA-B4403 | MENERTLDF | 32.05 | No | Non-Toxin | Good | 1.5311 |
| 240 | HLA-B4801 | RMEFFWTIL | 244.69 | No | Non-Toxin | Good | 1.1954 |
| 150 | HLA-B5101 | CPYLGSPSF | 429.32 | No | Non-Toxin | Good | 1.1181 |
| 150 | HLA-B5301 | CPYLGSPSF | 22.92 | No | Non-Toxin | Good | 1.1181 |
| 304 | HLA-B5401 | MPFHNIHPL | 16.45 | No | Non-Toxin | Good | 1.2633 |
| 237 | HLA-B5801 | QSGRMEFFW | 3.97 | No | Non-Toxin | Good | 1.0528 |
| 237 | HLA-B5802 | QSGRMEFFW | 9413.88 | No | Non-Toxin | Good | 1.0528 |
| 304 | HLA-B8301 | MPFHNIHPL | 97.78 | No | Non-Toxin | Good | 1.2633 |
| NA protein | |||||||
| 105 | HLA-A0202 | HLECRIFFL | 25.01 | No | Non-Toxin | Good | 1.1819 |
| 23 | HLA-A0206 | LQIGNIISI | 7.02 | No | Non-Toxin | Good | 1.2621 |
| 393 | HLA-A0207 | GLDCIRPCF | 13,931.8 | No | Non-Toxin | Good | 1.4833 |
| 7 | HLA-A0211 | ITIGSICMV | 3.12 | No | Non-Toxin | Good | 2.0061 |
| 23 | HLA-A0212 | LQIGNIISI | 10.88 | No | Non-Toxin | Good | 1.2621 |
| 105 | HLA-A0217 | HLECRIFFL | 71.01 | No | Non-Toxin | Good | 1.1819 |
| 322 | HLA-A2301 | AYGVKGFSF | 99.25 | No | Non-Toxin | Good | 1.2975 |
| 322 | HLA-A2402 | AYGVKGFSF | 60.32 | No | Non-Toxin | Good | 1.2975 |
| 14 | HLA-A2601 | MVIGMVSLM | 5.23 | No | Non-Toxin | Good | 1.0720 |
| 14 | HLA-A2602 | MVIGMVSLM | 6.85 | No | Non-Toxin | Good | 1.0720 |
| 324 | HLA-A2902 | GVKGFSFKY | 22.73 | No | Non-Toxin | Good | 1.1111 |
| 324 | HLA-A3002 | GVKGFSFKY | 67.34 | No | Non-Toxin | Good | 1.1111 |
| 14 | HLA-A6601 | MVIGMVSLM | 2301.39 | No | Non-Toxin | Good | 1.0720 |
| 105 | HLA-B0802 | HLECRIFFL | 1975.27 | No | Non-Toxin | Good | 1.1819 |
| 105 | HLA-B0803 | HLECRIFFL | 5079.26 | No | Non-Toxin | Good | 1.1819 |
| 291 | HLA-B1501 | YQIGYICSG | 66.4 | No | Non-Toxin | Good | 1.3358 |
| 289 | HLA-B4001 | LEYQIGYIC | 158.53 | No | Non-Toxin | Good | 1.9916 |
| 289 | HLA-B4002 | LEYQIGYIC | 41.21 | No | Non-Toxin | Good | 1.9916 |
| 13 | HLA-B4801 | CMVIGMVSL | 1001.31 | No | Non-Toxin | Good | 1.9406 |
| 397 | HLA-C0702 | IRPCFWVEL | 644.03 | No | Non-Toxin | Good | 1.1888 |
| 328 | HLA-C0802 | FSFKYGNGV | 891.48 | No | Non-Toxin | Good | 1.3196 |
| 328 | HLA-C1203 | FSFKYGNGV | 12.1 | No | Non-Toxin | Good | 1.3196 |
Selection of HTL epitopes prediction
For the NA and HA antigens, the method used to predict how peptides will attach to MHC class II alleles has been the NetMHCII-2.3 server. In this process, all MHC class II epitopes have been thoroughly examined, and the epitopes have been chosen according to their strong immunogenicity, and antigenicity, as well as the highest binding score. Additionally, independent MHC class II epitopes were considered when they exhibited overlapping regions. Ultimately, an overall of 9 MHC class II epitopes have been selected for the construction of the H5N1 Vaccine, consisting of 6 HA epitopes and 3 NA epitopes, as outlined in Table 3. Thus, all the predicted HTL (Helper T Lymphocyte) epitopes, which bind to MHC class II, can be found in Additional file 1: Tables S5 and S6.
Table 3.
MHC-II epitopes prediction from HA and NA proteins
| Pos | Allele | Peptide | Affinity (nM) | Antigenicity |
|---|---|---|---|---|
| HA protein | ||||
| 462–477 | DRB1_0103 | NLYDKVRLQLRDNAK | 2778.9 | 1.1390 |
| 443–457 | DRB1_0301 | ELLVLMENERTLDFH | 87.4 | 1.0452 |
| 210–224 | DRB1_0401 | PTTYISIGTSTLNQR | 19.7 | 1.0883 |
| 442–456 | DRB1_0405 | AELLVLMENERTLDF | 20.4 | 1.0504 |
| 464–476 | DRB4_0101 | LYDKVRLQLRDNAKE | 25.6 | 1.0144 |
| 210–224 | HLA-DQA10501-DQB10301 | PTTYISIGTSTLNQR | 29.9 | 1.0883 |
| NA protein | ||||
| 9–23 | DRB1_0403 | TIGSICMVIGMVSLM | 0.1 | 1.4483 |
| 13–27 | HLA-DQA10102-DQB10501 | ICMVIGMVSLMLQIG | 0.2 | 1.5671 |
| 10–24 | HLA-DQA10103-DQB10603 | GSICMVIGMVSLMLQ | 0.5 | 1.2727 |
Population coverage
The IEDB population coverage instrument, accessible at http://tools.iedb.org/population, has been employed to assess the extent of population coverage for each chosen epitope. As illustrated in Table 4, the greatest population coverage was found when both Class I and Class II epitopes were taken into account. This is noteworthy because NA and HA antigens linked to the [Influenza A virus (A/chicken/EastJava/UT551/2010(H5N1))] antigen originate in the Asian region. A comprehensive breakdown of population coverage in terms of Class I, and Class II, as well as the combined Class I and II epitopes associated with various continents has been presented in Table 4 and Fig. 1.
Table 4.
Population coverage (%) for specific epitopes containing HLA-binding alleles of MHC class I, class II, as well as combined class I and class II
| population/area | Class I | Class II | Class combined | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Coveragea | Average_hitb | pc90c | Coveragea | Average_hitb | pc90c | Coveragea | Average_hitb | pc90c | |
| Algeria | 0.0% | 0.0 | 0.0 | 83.55% | 1.42 | 0.61 | 83.55% | 1.42 | 0.61 |
| Argentina | 83.23% | 1.73 | 0.6 | 92.37% | 1.78 | 1.07 | 98.72% | 3.51 | 2.03 |
| Australia | 85.67% | 1.71 | 0.7 | 84.53% | 1.31 | 0.65 | 97.78% | 3.02 | 1.63 |
| Austria | 92.54% | 2.28 | 1.09 | 76.61% | 1.24 | 0.43 | 98.26% | 3.52 | 1.85 |
| Belgium | 85.65% | 2.06 | 0.7 | 77.25% | 1.25 | 0.44 | 96.74% | 3.3 | 1.52 |
| Brazil | 84.17% | 1.74 | 0.63 | 85.76% | 1.43 | 0.7 | 97.75% | 3.18 | 1.69 |
| Canada | 0.0% | 0.0 | 0.0 | 93.63% | 1.66 | 1.11 | 93.63% | 1.66 | 1.11 |
| Central Africa | 84.85% | 1.6 | 0.66 | 90.38% | 1.65 | 1.01 | 98.54% | 3.24 | 1.96 |
| Central America | 6.44% | 0.07 | 0.11 | 88.25% | 1.39 | 0.85 | 89.0% | 1.46 | 0.91 |
| Chile | 82.4% | 1.64 | 0.57 | 82.25% | 1.45 | 0.56 | 96.88% | 3.09 | 1.52 |
| China | 78.98% | 1.39 | 0.48 | 86.03% | 1.5 | 0.72 | 97.06% | 2.88 | 1.52 |
| Colombia | 2.84% | 0.03 | 0.1 | 84.34% | 1.45 | 0.64 | 84.79% | 1.48 | 0.66 |
| Croatia | 94.5% | 2.33 | 1.19 | 96.26% | 2.13 | 1.27 | 99.79% | 4.46 | 2.86 |
| Czech Republic | 87.79% | 2.07 | 0.82 | 94.04% | 2.05 | 1.15 | 99.27% | 4.12 | 2.29 |
| Denmark | 0.0% | 0.0 | 0.0 | 93.03% | 2.29 | 1.13 | 93.03% | 2.29 | 1.13 |
| East Africa | 87.8% | 1.9 | 0.82 | 95.27% | 1.96 | 1.2 | 99.42% | 3.86 | 2.31 |
| East Asia | 96.97% | 2.36 | 1.37 | 80.31% | 1.37 | 0.51 | 99.4% | 3.73 | 2.29 |
| England | 95.12% | 2.74 | 1.24 | 91.7% | 2.12 | 1.07 | 99.59% | 4.86 | 2.74 |
| Europe | 92.13% | 2.33 | 1.08 | 89.14% | 1.82 | 0.92 | 99.15% | 4.15 | 2.28 |
| France | 95.51% | 2.63 | 1.28 | 95.14% | 2.32 | 1.24 | 99.78% | 4.94 | 3.04 |
| Germany | 94.18% | 2.5 | 1.18 | 95.55% | 2.27 | 1.25 | 99.74% | 4.77 | 2.87 |
| Greece | 0.0% | 0.0 | 0.0 | 92.99% | 1.88 | 1.1 | 92.99% | 1.88 | 1.1 |
| Hong Kong | 86.36% | 1.59 | 0.73 | 0.0% | 0.0 | 0.0 | 86.36% | 1.59 | 0.73 |
| India | 80.13% | 1.61 | 0.5 | 86.55% | 1.58 | 0.74 | 97.33% | 3.19 | 1.61 |
| Indonesia | 65.87% | 1.02 | 0.29 | 84.82% | 1.31 | 0.66 | 94.82% | 2.33 | 1.22 |
| Iran | 83.59% | 1.46 | 0.61 | 88.19% | 1.62 | 0.85 | 98.06% | 3.08 | 1.77 |
| Ireland Northern | 95.02% | 2.69 | 1.23 | 81.01% | 1.46 | 0.53 | 99.05% | 4.15 | 2.21 |
| Israel | 81.19% | 1.75 | 0.53 | 93.51% | 1.94 | 1.13 | 98.78% | 3.69 | 2.08 |
| Italy | 94.7% | 2.53 | 1.2 | 93.46% | 1.84 | 1.11 | 99.65% | 4.37 | 2.58 |
| Japan | 97.56% | 2.39 | 1.44 | 82.55% | 1.4 | 0.57 | 99.57% | 3.79 | 2.4 |
| Korea; South | 95.86% | 2.26 | 1.27 | 74.06% | 1.16 | 0.39 | 98.93% | 3.42 | 2.08 |
| Lebanon | 0.0% | 0.0 | 0.0 | 84.42% | 1.38 | 0.64 | 84.42% | 1.38 | 0.64 |
| Malaysia | 55.3% | 0.78 | 0.22 | 65.45% | 0.85 | 0.29 | 84.56% | 1.63 | 0.65 |
| Mexico | 88.79% | 1.78 | 0.89 | 84.16% | 1.41 | 0.63 | 98.22% | 3.19 | 1.83 |
| Morocco | 91.75% | 2.32 | 1.07 | 87.3% | 1.61 | 0.79 | 98.95% | 3.93 | 2.19 |
| Netherlands | 0.0% | 0.0 | 0.0 | 91.94% | 2.0 | 1.07 | 91.94% | 2.0 | 1.07 |
| New Zealand | 0.0% | 0.0 | 0.0 | 73.31% | 1.09 | 0.37 | 73.31% | 1.09 | 0.37 |
| North Africa | 87.26% | 1.9 | 0.78 | 87.97% | 1.67 | 0.83 | 98.47% | 3.56 | 2.02 |
| North America | 91.84% | 2.14 | 1.07 | 92.53% | 2.0 | 1.09 | 99.39% | 4.13 | 2.39 |
| Northeast Asia | 80.08% | 1.41 | 0.5 | 86.03% | 1.5 | 0.72 | 97.22% | 2.91 | 1.55 |
| Norway | 0.0% | 0.0 | 0.0 | 94.51% | 2.34 | 1.2 | 94.51% | 2.34 | 1.2 |
| Oceania | 86.77% | 1.74 | 0.76 | 91.99% | 1.77 | 1.06 | 98.94% | 3.5 | 2.09 |
| Oman | 93.04% | 2.14 | 1.11 | 0.0% | 0.0 | 0.0 | 93.04% | 2.14 | 1.11 |
| Paraguay | 0.0% | 0.0 | 0.0 | 45.49% | 0.53 | 0.18 | 45.49% | 0.53 | 0.18 |
| Peru | 80.75% | 1.72 | 0.52 | 81.81% | 1.31 | 0.55 | 96.5% | 3.03 | 1.44 |
| Philippines | 86.69% | 1.73 | 0.75 | 19.14% | 0.2 | 0.12 | 89.24% | 1.92 | 0.93 |
| Portugal | 92.4% | 2.16 | 1.09 | 88.6% | 1.73 | 0.88 | 99.13% | 3.89 | 2.24 |
| Russia | 94.7% | 2.45 | 1.21 | 92.16% | 1.99 | 1.08 | 99.58% | 4.44 | 2.6 |
| Saudi Arabia | 86.23% | 1.96 | 0.73 | 58.23% | 0.8 | 0.24 | 94.25% | 2.76 | 1.23 |
| Serbia | 85.71% | 1.2 | 0.7 | 0.0% | 0.0 | 0.0 | 85.71% | 1.2 | 0.7 |
| Singapore | 79.61% | 1.4 | 0.49 | 74.83% | 0.92 | 0.4 | 94.87% | 2.31 | 1.22 |
| South Africa | 92.34% | 2.14 | 1.09 | 25.52% | 0.26 | 0.13 | 94.3% | 2.4 | 1.2 |
| South America | 86.48% | 1.71 | 0.74 | 84.11% | 1.42 | 0.63 | 97.85% | 3.13 | 1.7 |
| South Asia | 81.86% | 1.66 | 0.55 | 86.6% | 1.59 | 0.75 | 97.57% | 3.24 | 1.67 |
| Southeast Asia | 87.1% | 1.66 | 0.77 | 84.03% | 1.43 | 0.63 | 97.94% | 3.1 | 1.74 |
| Southwest Asia | 78.69% | 1.58 | 0.47 | 85.65% | 1.53 | 0.7 | 96.94% | 3.1 | 1.53 |
| Spain | 80.9% | 1.37 | 0.52 | 89.68% | 1.78 | 0.97 | 98.03% | 3.15 | 1.77 |
| Sweden | 96.98% | 2.44 | 1.33 | 89.41% | 1.85 | 0.94 | 99.68% | 4.3 | 2.57 |
| Thailand | 78.36% | 1.36 | 0.46 | 83.55% | 1.42 | 0.61 | 96.44% | 2.78 | 1.43 |
| Tunisia | 85.74% | 1.91 | 0.7 | 86.64% | 1.66 | 0.75 | 98.09% | 3.57 | 1.9 |
| Turkey | 1.99% | 0.02 | 0.1 | 95.32% | 2.07 | 1.21 | 95.41% | 2.09 | 1.22 |
| Ukraine | 0.0% | 0.0 | 0.0 | 21.22% | 0.42 | 0.25 | 21.22% | 0.42 | 0.25 |
| United Arab Emirates | 13.32% | 0.14 | 0.12 | 32.92% | 0.33 | 0.15 | 41.86% | 0.47 | 0.17 |
| United Kingdom | 0.0% | 0.0 | 0.0 | 38.38% | 0.38 | 0.16 | 38.38% | 0.38 | 0.16 |
| United States | 92.06% | 2.15 | 1.07 | 94.95% | 2.13 | 1.19 | 99.6% | 4.29 | 2.54 |
| Vietnam | 79.28% | 1.42 | 0.48 | 73.86% | 0.97 | 0.38 | 94.58% | 2.39 | 1.21 |
| West Africa | 90.38% | 1.88 | 1.01 | 98.42% | 2.36 | 1.55 | 99.85% | 4.24 | 2.85 |
| West Indies | 94.69% | 2.35 | 1.21 | 93.76% | 1.93 | 1.13 | 99.67% | 4.28 | 2.65 |
| Average | 68.12 | 1.49 | 0.66 | 77.89 | 1.45 | 0.73 | 92.24 | 2.94 | 1.63 |
| Standard deviation | 36.06 | 0.87 | 0.43 | 24.34 | 0.6 | 0.37 | 14.94 | 1.15 | 0.74 |
aProjected population coverage
bAverage number of epitope hits / HLA combinations recognized by the population
cMinimum number of epitope hits / HLA combinations recognized by 90% of the population
Fig. 1.

Population coverage (%) for specific epitopes containing HLA-binding alleles. For each chosen epitope, a global and average population coverage percentage is taken into account. The colors of MHC class I, and class II, as well as combined class I and class II are displayed in purple, green, and olive-green, correspondingly
Conservancy selected epitopes
The IEDB conservancy analysis instrument has been used to be a monitoring instrument for examining the level of conservation in chosen epitopes relative to other similar sequences. As shown in Table 5 epitopes exhibit varying lengths of minimum 0 and maximum 100 percent conservation.
Table 5.
Conservancy selected epitopes from HA and NA epitopes
| Allele | Epitope sequence | Epitope length | Percent of protein sequence m atches at identity < = 100% | Minim um identity (%) | Maxim um identity (%) |
|---|---|---|---|---|---|
| HA Protein | |||||
| HLA-A0101 | KSDQICIGY | 9 | 99.00% (99/100) | 88.89 | 100.00 |
| HLA-A3101 | RLVPKIATR | 9 | 82.00% (82/100) | 66.67 | 100.00 |
| HLA-A3201 | RMEFFWTIL | 9 | 96.00% (96/100) | 88.89 | 100.00 |
| HLA-A3207 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A3215 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A6601 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A6823 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B0801 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B1402 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B2705 | GRMEFFWTI | 9 | 95.00% (95/100) | 88.89 | 100.00 |
| HLA-B2720 | GRMEFFWTI | 9 | 95.00% (95/100) | 88.89 | 100.00 |
| HLA-B3501 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B3503 | CPYLGSPSF | 9 | 88.00% (88/100) | 77.78 | 100.00 |
| HLA-B3801 | FHNIHPLTI | 9 | 99.00% (99/100) | 88.89 | 100.00 |
| HLA-B3901 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B4013 | RMEFFWTIL | 9 | 96.00% (96/100) | 88.89 | 100.00 |
| HLA-B4201 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B4402 | MENERTLDF | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B4403 | MENERTLDF | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B4801 | RMEFFWTIL | 9 | 96.00% (96/100) | 88.89 | 100.00 |
| HLA-B5101 | CPYLGSPSF | 9 | 88.00% (88/100) | 77.78 | 100.00 |
| HLA-B5301 | CPYLGSPSF | 9 | 88.00% (88/100) | 77.78 | 100.00 |
| HLA-B5401 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-B5801 | QSGRMEFFW | 9 | 99.00% (99/100) | 77.78 | 100.00 |
| HLA-B5802 | QSGRMEFFW | 9 | 99.00% (99/100) | 77.78 | 100.00 |
| HLA-B8301 | MPFHNIHPL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| DRB1_0103 | NLYDKVRLQLRDNAK | 15 | 96.00% (96/100) | 93.33 | 100.00 |
| DRB1_0301 | ELLVLMENERTLDFH | 15 | 100.00% (100/100) | 100.00 | 100.00 |
| DRB1_0401 | PTTYISIGTSTLNQR | 15 | 83.00% (83/100) | 93.33 | 100.00 |
| DRB1_0405 | AELLVLMENERTLDF | 15 | 100.00% (100/100) | 100.00 | 100.00 |
| DRB4_0101 | LYDKVRLQLRDNAKE | 15 | 96.00% (96/100) | 93.33 | 100.00 |
| HLA-DQA10501-DQB10301 | PTTYISIGTSTLNQR | 15 | 83.00% (83/100) | 93.33 | 100.00 |
| NA Protein | |||||
| HLA-A0202 | HLECRIFFL | 9 | 2.00% (2/100) | 88.89 | 100.00 |
| HLA-A0206 | LQIGNIISI | 9 | 6.00% (6/100) | 66.67 | 100.00 |
| HLA-A0207 | GLDCIRPCF | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A0211 | ITIGSICMV | 9 | 95.00% (95/100) | 88.89 | 100.00 |
| HLA-A0212 | LQIGNIISI | 9 | 6.00% (6/100) | 66.67 | 100.00 |
| HLA-A0217 | HLECRIFFL | 9 | 2.00% (2/100) | 88.89 | 100.00 |
| HLA-A2301 | AYGVKGFSF | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A2402 | AYGVKGFSF | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A2601 | MVIGMVSLM | 9 | 2.00% (2/100) | 77.78 | 100.00 |
| HLA-A2602 | MVIGMVSLM | 9 | 2.00% (2/100) | 77.78 | 100.00 |
| HLA-A2902 | GVKGFSFKY | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A3002 | GVKGFSFKY | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-A6601 | MVIGMVSLM | 9 | 2.00% (2/100) | 77.78 | 100.00 |
| HLA-B0802 | HLECRIFFL | 9 | 2.00% (2/100) | 88.89 | 100.00 |
| HLA-B0803 | HLECRIFFL | 9 | 2.00% (2/100) | 88.89 | 100.00 |
| HLA-B1501 | YQIGYICSG | 9 | 99.00% (99/100) | 88.89 | 100.00 |
| HLA-B4001 | LEYQIGYIC | 9 | 99.00% (99/100) | 88.89 | 100.00 |
| HLA-B4002 | LEYQIGYIC | 9 | 99.00% (99/100) | 88.89 | 100.00 |
| HLA-B4801 | CMVIGMVSL | 9 | 2.00% (2/100) | 77.78 | 100.00 |
| HLA-C0702 | IRPCFWVEL | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-C0802 | FSFKYGNGV | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| HLA-C1203 | FSFKYGNGV | 9 | 100.00% (100/100) | 100.00 | 100.00 |
| DRB1_0403 | TIGSICMVIGMVSLM | 15 | 2.00% (2/100) | 86.67 | 100.00 |
| HLA-DQA10102-DQB10501 | ICMVIGMVSLMLQIG | 15 | 2.00% (2/100) | 86.67 | 100.00 |
| HLA-DQA10103-DQB10603 | GSICMVIGMVSLMLQ | 15 | 2.00% (2/100) | 86.67 | 100.00 |
Final epitopes selection, construction design and physicochemical properties
The nucleotide sequence of H5N1 can be found in Additional file 1: Table S7. Final selected epitopes and adjuvants are presented in Table 6.
Table 6.
Final selected epitopes. All overlapping sequences and epitopes are labeled as underlined and in bold form
| Epitopes/Adjuvants | Type |
|---|---|
| MRTLDFHDSN | B Cell |
| NSSLCPIK | B Cell |
| SDQICIGY | MHC-I |
| CPYLGSPSF | MHC-I |
| RLVPKIATR | MHC-I |
| QSGRMEFFWL | MHC-I |
| MPFHNIHPLTI | MHC-I |
| ITIGSICMV | MHC-I |
| MVIGMVSLM | MHC-I |
| LQIGNIISI | MHC-I |
| HLECRIFFL | MHC-I |
| LEYQIGYICSG | MHC-I |
| AYGVKGFSFKY | MHC-I |
| GLDCIRPCF | MHC-I |
| PTTYISIGTSTLNQR | MHC-II |
| AELLVLMENERTLDFH | MHC-II |
| NLYDKVRLQLRDNAKE | MHC-II |
| MRIHYLLFALLFLFLVPV | |
| PGHGGIINTLQKYYCRVR | |
| GGRCAVLSCLPKEEQIGK | |
| CSTRGRKCCRRKK | Adjuvant |
| AKFVAAWTLKAAA | Adjuvant |
The final selection of epitopes was interconnected using specific linkers. AAY linkers were chosen to link the MHC-I epitopes, and GPGPGPG linkers have been used for linking the MHC-II epitopes. Adjuvants such as HβD-3 and the PADRE sequence have been connected using EAAAK linkers at the C-terminal site. To bridge the final MHC-I epitope to the initial MHC-II epitope, the HEYGAEALERAG linker was employed. KK linkers were utilized for connecting B-cell epitopes. To complete the structure, a "Histidine Tag" has been included at the C-terminal and attached to the last construct as depicted in Fig. 2A. Also, the secondary structure of the H5N1 construct is shown in Fig. 2B.
Fig. 2.

A Amino acid sequence of H5N1 construct. Amino acids in gray are KK linkers, Amino acids in purple are AAY linkers, amino acids in blue are GPGPGPG linkers, amino acids in green are HEYGAEALERAG and amino acids in yellow are EAAAK linkers. B Secondary structure of H5N1 construct. Helixes are shown as orange color, Beta strands are shown as purple color and coils are shown as pink color and disorders are shown as grey color
The outcomes obtained from the ProtParam server revealed that the H5N1 vaccine amino acid sequence, which consists of 350 amino acids, is expected to produce a protein with a molecular weight of ~ 38.46 kDa. The theoretical isoelectric point (pI) and instability index (II) have been determined at 9.32 and 34.44, respectively, indicating that the influenza vaccine falls within the category of stable proteins. Moreover, this vaccine's half-life in a variety of prokaryotic and eukaryotic hosts is predicted to be between 10 and 30 h. The aliphatic index is 84.37 and the grand average of hydropathicity (GRAVY) to be 0.018.
Discontinues B cell from final H5N1 construct
Table 7 provides an overview of the conformational B cell epitopes from the final construct of the H5N1 vaccine. Each of the eight predicted scores for conformational B cell epitopes is shown in Fig. 3.
Table 7.
Predicted discontinuous epitope of final H5N1 construct
| No | Residues | Number of residues | Score |
|---|---|---|---|
| 1 | A:F169, A:G170, A:P171, A:G172, A:P173, A:G174, A:P175, A:G176, A:P177, A:T178, A:T179, A:Y180, A:I181, A:S182, A:I183, A:G184, A:T185, A:S186, A:T187, A:L188, A:N189, A:Q190, A:R191 | 23 | 0.813 |
| 2 | A:K315, A:K316, A:E317, A:A318, A:A319, A:A320, A:K321, A:A322, A:K323, A:F324, A:V325, A:A326, A:A327, A:W328, A:T329, A:L330, A:K331, A:A332, A:A333, A:A334, A:E335, A:A336, A:A337, A:A338, A:K339, A:E340, A:A341, A:A342, A:A343, A:K344, A:H346, A:H347 | 32 | 0.791 |
| 3 | A:R209, A:T210, A:L211, A:D212, A:F213, A:H214, A:G215, A:P216, A:G217, A:P218, A:G219, A:P220, A:G221, A:N222, A:L223, A:Y224, A:D225, A:K226, A:V227, A:R228, A:L229, A:Q230, A:L231, A:R232 | 24 | 0.777 |
| 4 | A:D233, A:N234, A:A235, A:K236, A:E237, A:E239, A:Y240 | 7 | 0.7 |
| 5 | A:P78, A:L79, A:T80, A:I81, A:A82, A:I85 | 6 | 0.695 |
| 6 | A:M250, A:R251, A:Y254 | 3 | 0.656 |
| 7 | A:G241, A:A242, A:E243, A:A244, A:E246, A:R247 | 6 | 0.638 |
| 8 | A:G161, A:L162, A:D163, A:C164, A:I165, A:R166 | 6 | 0.615 |
| 9 | A:V291, A:L292, A:S293, A:C294, A:L295, A:P296, A:K297, A:S305, A:T306, A:R307, A:G308, A:R309 | 12 | 0.566 |
Fig. 3.
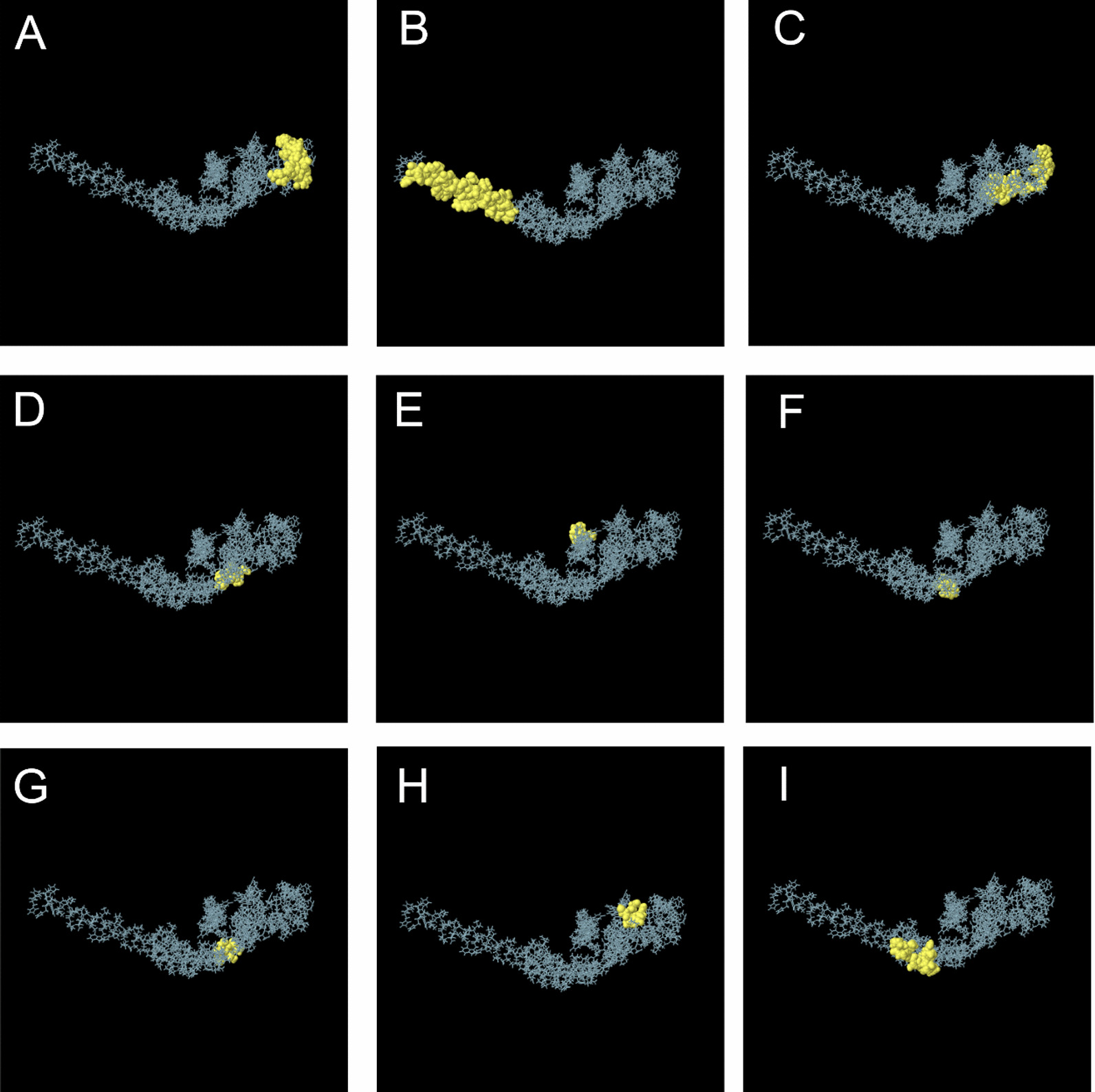
3D visualization of discontinuous B cell epitopes in H5N1 construct. Yellow regions are related to discontinuous amino acids in the final H5N1 construct. A Amino acids of F169, G170, P171, G172, P173, G174, P175, G176, P177, T178, T179, Y180, I181, S182, I183, G184, T185, S186, T187, L188, N189, Q190 and R191. B Amino acids of K315, K316, E317, A318, A319, A320, K321, A322, K323, F324, V325, A326, A327, W328, T329, L330, K331, A332, A333, A334, E335, A336, A337, A338, K339, E340, A341, A342, A343, K344, H346 and H347. C Amino acids of R209, T210, L211, D212, F213, H214, G215, P216, G217, P218, G219, P220, G221, N222, L223, Y224, D225, K226, V227, R228, L229, Q230, L231 and R232. DAmino acids of D233, N234, A235, K236, E237, E239, and Y240. E Amino acids of P78, L79, T80, I81, A82 and I85. F Amino acids of M250, R251, and Y254. G Amino acids of G241, A242, E243, A244, E246, and R247. H G161, L162, D163, C164, I165, and R166. (I)Amin acids of V291, L292, S293, C294, L295, P296, K297, S305, T306, R307, G308, and R309
Modeling of 3D construct, refinement and validation
The 3D structure of H5N1 has been modeled using the RoseTTAFold server, as illustrated in Fig. 4. Subsequently, the obtained H5N1 3D structure was subjected to the GalaxyRefine server for further refinement. Model validation was carried out using online software PROCHECK and PROSA. After refining, the findings showed a Z-Score of -0.87, which is used to evaluate the quality of the modeled protein based on NMR or X-ray methodologies (Fig. 5A). For proteins with less than 200 amino acids, the NMR methodology is usually utilized, however for proteins with more than 200 amino acids, the X-ray methodology is applied. The (blue-blue) NMR and (pale-blue) X-ray zones on the PROSA graph denote great simulation accuracy, the lowest error rate, as well as the strongest confidence in the simulated model, respectively, if the Z-Score dot falls inside them. Local model quality associated with the protein's structure is depicted in Fig. 5B.
Fig. 4.

3D structure of H5N1. Coils are shown as brown, α-Helixes are shown as blue and β-Sheets are shown as purple
Fig. 5.

A Z-Score validation of H5N1 3D homology modeling. A value of − 0.87 shows that the 3D H5N1 structure is located in X-ray crystallographic proteins. B The local model quality of H5N1 confirms that the energy of this structure is close to a stable manner. C Ramachandran plot associated with vaccine 3D structure. 98% of residues are modeled in favored regions and more than 99.8% of residues are modeled in allowed regions
Based on the Ramachandran graph analysis of the anticipated model, 336 out of 350 residues, or 96% of all residues, were found inside the preferred areas (with a 98% probability). In total, approximately 99.7% of all residues, or 349 out of 350, were within the allowed regions (with a likelihood exceeding 99.8%). The Ramachandran graph organizes amino acids based on the angles of phi and psi, as shown in Fig. 5C. This data reflects the overall quality and reliability of the model.
Protein–protein docking
The structures of H5N1 including TLR7 and TLR8 proteins have been submitted to the HDOCK server to determine the contact areas by protein–protein docking. The highest-ranking results for every complex have been chosen based on the least intermolecular binding energy among entire H5N1-Proteins complexes with the lowest docking score as shown in Tables 8 and 9.
Table 8.
An overview of the top ten H5N1-TLR7 models
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Docking Score | − 374.08 | − 373.18 | − 366.36 | − 361.21 | − 358.82 | − 357.58 | − 357.20 | − 349.83 | − 349.25 | − 341.88 |
| Confidence Score | 0.9888 | 0.9886 | 0.9870 | 0.9856 | 0.9849 | 0.9845 | 0.9844 | 0.9820 | 0.9817 | 0.9789 |
| Ligand rmsd (Å) | 92.47 | 70.15 | 71.18 | 68.52 | 80.36 | 122.48 | 69.70 | 54.50 | 68.29 | 88.55 |
| Interface residues | Model_1 | Model_2 | Model_3 | Model_4 | Model_5 | Model_6 | Model_7 | Model_8 | Model_9 | Model_10 |
Table 9.
An overview of the top ten H5N1-TLR8
| Rank | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 |
|---|---|---|---|---|---|---|---|---|---|---|
| Docking Score | − 414.39 | − 371.33 | − 351.41 | − 335.02 | − 331.52 | − 328.21 | − 318.14 | − 316.39 | − 313.14 | − 311.12 |
| Confidence Score | 0.9950 | 0.9882 | 0.9825 | 0.9759 | 0.9742 | 0.9725 | 0.9665 | 0.9654 | 0.9631 | 0.9617 |
| Ligand rmsd (Å) | 91.61 | 93.05 | 64.85 | 57.21 | 58.70 | 80.88 | 76.69 | 92.16 | 87.30 | 70.86 |
| Interface residues | Model_1 | Model_2 | Model_3 | Model_4 | Model_5 | Model_6 | Model_7 | Model_8 | Model_9 | Model_10 |
The docking results indicate that H5N1 is capable of forming interactions with the 3D structures of TLR7 and TLR8. Within the final construct, two sequences, PADRE and HβD-3, were selected and included as adjuvants at the C-terminal. As displayed in Figs. 6A and 7A, the initial amino acid regions of H5N1 were observed to be engaged with TLR7 and TLR8. Moreover, H5N1 directly established interactions with several amino acids within the binding sites of TLR7 and TLR8. This demonstrates the potential for a strong interaction between H5N1 and these immune-related receptors.
Fig. 6.

A Docking HADDOCK outcomes for the H5N1-TLR7 complex are visualized, B and the residues' interaction with the complex has been magnified. The H5N1 structure is shown as a green stick-filled carton. The blue cartons and sticks that make up the TLR7 structure are displayed. The dashed line in yellow represents hydrogen bonds
Fig. 7.

A Docking HADDOCK outcomes for the H5N1-TLR8 complex are visualized, B and the residues' contact with the complex has been magnified. An illustration of the H5N1 structure is a carton filled with green sticks. The cooper carton and sticks that make up the TLR8 structure are displayed. The dashed line in yellow represents hydrogen bonds
As illustrated in Figs. 6B and 7B, it was observed that specific amino acids in H5N1, including SER846, SER850, SER860 and PHE864 interacted with amino acids in the TLR7, such as ARG232, TYR224, PHE214 and LEU211. Also, GLN58, SER59 and ASN 75 amino acids from the H5N1 structure interacted with GLU438 and ARG628 amino acids of the TLR8 structure. This interaction demonstrates the potential binding between H5N1 and the TLR7 and TLR8.
The H5N1 exhibited interaction with TLR7 through 4 hydrogen bonds. Similarly, in the H5N1-TLR8 complex, interactions were facilitated by 3 hydrogen bonds to the interactions in this complex. These findings demonstrate the significance of hydrogen bond contacts in forming these complexes.
The 2D interactions are summarized and visualized, providing a detailed depiction of these interactions (H5N1-TLR7 in Fig. 8A and H5N1-TLR8 in Fig. 8B).
Fig. 8.

A Presentation of amino acids’ 2D interaction in H5N1-TLR7 complex. Green residues are presented as H5N1 structure and blue residues are presented as TLR7 structure. B Presentation of 2D interaction of amino acids in H5N1-TLR8 complex. Green residues are presented as H5N1 structure and blue residues are presented as TLR8 structure
Molecular dynamic and MM/PBSA analysis
The complexes underwent molecular dynamics (MD) simulations for a duration of 100 ns. The MD simulation results indicated the hydrogen bonds’ creation in H5N1-TLR7 and H5N1-TLR8 complexes with root mean square deviations (RMSD) of 2 and 3.5 nm, respectively, as shown in Figs. 9A and 10A. These RMSD values provide insights into the stability and behavior of the simulated complexes during the MD simulations.
Fig. 9.

MD simulation outcomes of H5N1 complex. A RMSD of H5N1-TLR7 in Pico seconds, B RMSD of H5N1-TLR7 in residues, C Gyration of H5N1-TLR7 in Pico seconds, D H-bond of H5N1-TLR7 in Pico seconds and E SASA graphs of H5N1-TLR7 complex
Fig. 10.

MD simulation outcomes of H5N1-TLR8 complex. A RMSD of H5N1-TLR8 in Pico seconds, B RMSD of H5N1-TLR8 in residues, C Gyration of H5N1-TLR8, Pico seconds (D) H-bond of H5N1-TLR8 in residues and E SASA graphs of H5N1-TLR8 complex
The root mean square fluctuation (RMSF) diagrams revealed that residues in the H5N1-TLR7 and H5N1-TLR8 complexes during MD simulation exhibited very weak volatility, as depicted in Figs. 9B and 10B.
These results suggest that the H5N1 structure is maintained in the binding interactions with TLR7 and TLR8 complexes. Additionally, it's worth noting that the H5N1-TLR7 complex returned to equilibrium after approximately 2 ns, while the and H5N1-TLR8 complex reached equilibrium around 3 ns after the start of the MD simulations. These findings indicate the stability and equilibration of the simulated complexes throughout the simulations.
The gyration radius data associated with H5N1-TLR7 and H5N1-TLR8 complexes can be found in Figs. 9C and 10C, providing insights into the compactness and overall structure of these complexes. Additionally, the H-bond and SASA overall results of H5N1-TLR7 and H5N1-TLR8 complexes are included in Figs. 9D and 10D as well as Figs. 9E and 10E, respectively, offering further details about the interactions within these complexes.
The H5N1-TLR7 and H5N1-TLR8 complexes' binding affinities were verified using the use of Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) modeling. Tables 10 and 11 provide an overview of the findings of these calculations, which identify the energy connected to the bonds that are created in each complex between the vaccine and the receptor. In the H5N1-TLR7 complex, the total binding energy was calculated to be − 29.97 kJ/mol, indicating a strong affinity between H5N1 and TLR7. On the other hand, in the H5N1-TLR8 complex, the total binding energy was found to be − 23.9 kJ/mol. These calculations provide insights into the thermodynamic stability and binding affinities of these complexes, with a notably high affinity observed in the case of H5N1-TLR7.
Table 10.
MMPB/SA calculation from H5N1-TLR7 complex
| Frames | VDWAALS | EEL | EPB | ENPOLAR | GGAS | GSOLV | TOTAL |
|---|---|---|---|---|---|---|---|
| Average | − 43.49 | − 100.18 | 120.04 | − 6.34 | − 143.67 | 113.7 | − 29.97 |
| SD | 3.48 | 15.79 | 16 | 0.57 | 17.15 | 15.75 | 2.5 |
| SEM | 1.05 | 4.76 | 4.83 | 0.17 | 5.17 | 4.75 | 0.75 |
Table 11.
MMPB/SA calculation from H5N1-TLR8 complex
| Frames | VDWAALS | EEL | EPB | ENPOLAR | GGAS | GSOLV | TOTAL |
|---|---|---|---|---|---|---|---|
| Average | − 38.06 | − 132.02 | 151.68 | − 5.49 | − 170.08 | 146.19 | − 23.9 |
| SD | 2.87 | 11.26 | 10.45 | 0.32 | 12.09 | 10.26 | 3.17 |
| SEM | 0.87 | 3.39 | 3.15 | 0.1 | 3.65 | 3.09 | 0.95 |
Additional investigations have shown that apart from total binding energy, van der Waals and electrostatic energies also have a substantial part in influencing complexes. In the H5N1-TLR7 complex, according to Table 10 and Fig. 11, the electrostatic energy has been determined to be − 100.18 kJ/mol, and the van der Waals energy to be − 43.49 kJ/mol. Conversely, in the H5N1-TLR8 complex, the van der Waals energy contribution was − 38.06 kJ/mol and the electrostatic energy was − 132.02 kJ/mol (Table 11 and Fig. 12).
Fig. 11.
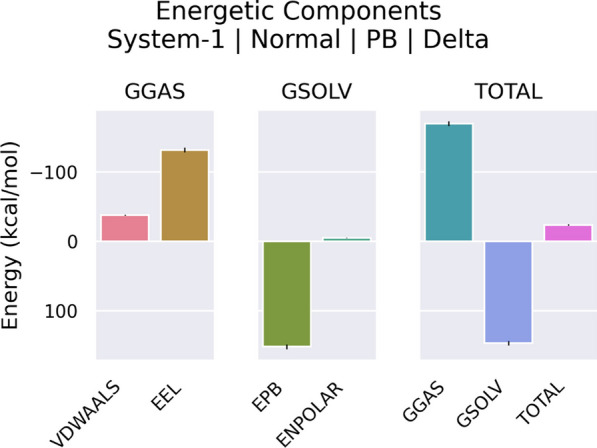
Graphical presentation of energy contribution of H5N1-TLR7 complex. Vander Waals (VDWAALS) energies are shown as pink, Electro static (EEL) energies are shown as brown, effective polarizable bonds (EPB) are as green, effective non-polarizable bonds (ENPOLAR) are shown as pale-green, gas phases (GGAS) are shown as blue, solvent phases (GSOLV) are shown as purple and total energies contribution (TOTAL) are shown as hot-pink
Fig. 12.
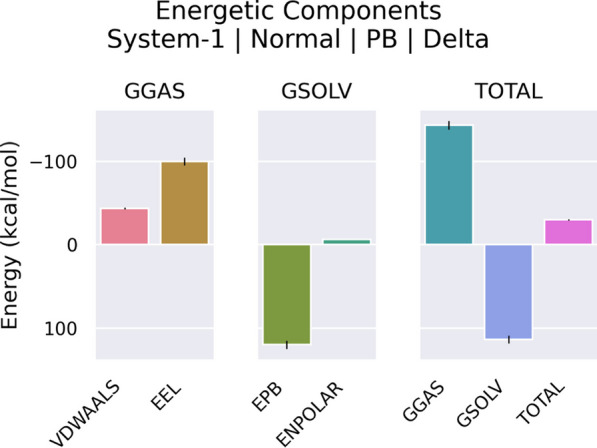
Graphical presentation of energy contribution of H5N1-TLR8 complex. Vander Waals (VDWAALS) energies are shown as pink, Electro static (EEL) energies are shown as brown, effective polarizable bonds (EPB) are as green, effective non-polarizable bonds (ENPOLAR) are shown as pale-green, gas phases (GGAS) are shown as blue, solvent phases (GSOLV) are shown as purple and total energies contribution (TOTAL) are shown as hot-pink
These energy components further characterize the nature of the contacts in complexes, with both van der Waals and electrostatic forces playing a role in influencing the binding affinities and stability of these complexes. The balance of van der Waals as well as electrostatic energies contributes to the overall stability of these interactions.
Immune simulation
The C-ImmSim web server's results showed a rise in secondary immune response generation that is consistent with the true immunological response. Notably, As the immunological response grew more intense, there was a rise in total IgM + IgG1 and IgG2 antibody levels, as depicted in Fig. 13A. Moreover, the quantity of B memory (y2) increased as the levels of IgG1 and IgG2 isotypes decreased, as depicted in Fig. 13B. Additionally, the outcomes demonstrated that an increase in TH memory (y2), as depicted in Fig. 13C, correlated with an increase in both TH and TC cell populations, as illustrated in Fig. 13D. Furthermore, there was an elevation in the focus of produced IFN-γ and an increase in TH cell population, as seen in Fig. 13E, F. Together, these results show that the C-ImmSim server's simulation of the immunological response to influenza H5N1 vaccination closely resembles the real immune response, underscoring the simulation's accuracy in simulating the activity of the immune system.
Fig. 13.

Immune response simulation against the H5N1 strain. A Immunoglobulin levels upon H5N1 construct. B Total B isotype (IgM, IgG1, IgG2) as the immunological response, and amount of B memory (y2). C CD4 T-helper cells as a result of the development of T-helpers. D Displaying both memory and total T-helper cells. E Cell populations of CD8 T-cytotoxic lymphocytes in each condition after being injected with antigen. F Generated IL-4, IL-6, and IL-12 concentrations together with IFN-γ
Insilico cloning and vaccine optimization
The nucleotide sequences of the finished construct were optimized using GenScript. The organism chosen for host expression was E. Coli K12. To prevent bacterial ribosome binding sites, cleavage sites for restriction enzymes, and rho-independent transcription terminators, certain settings were made in GenScript. GC-Content for the improved Influenza construct, which consists of 1032 nucleotides (excluding the His-Tag), was determined to be 54.46%. These values affirm the likelihood of successful protein expression. To facilitate intracellular expression, to exclude the PelB sequence from the final structure, Nde I and Xho I restriction enzyme sites have been introduced to the N- and C-terminals of the nucleotide sequence, respectively. Additionally, a stop codon has been included following the His-Tag sequence. Ultimately, SnapGene was used to clone the design into the pET-26b(+) plasmid, as shown in Fig. 14.
Fig. 14.

Cloned nucleotide sequence of H5N1 into pET-26b(+) plasmid. The H5N1 sequence is labeled as a green ribbon. NdeI and XhoI enzymes are located at N-terminal and C-terminal of H5N1 construct. His-Tag (6xHis) sequence is located at the C-terminal of H5N1 construct
Discussion
Influenza H5 viruses are responsible for respiratory infections in humans, and the severity of these infections can vary from no symptoms to being life-threatening. In the realm of infectious diseases, the influenza virus stands out as among the most substantial hazards to both human and avian populations worldwide. Virologists have issued a warning over the possibility of a brand-new, extremely damaging influenza pandemic, highlighting the pressing necessity of being ready and vigilant [27]. In the two decades following its initial emergence in China in 1996, approximately 60 nations have documented ongoing cases of H5 viruses affecting domestic poultry, wild bird populations, and human beings. This underscores the widespread and continuous nature of H5 virus incidents around the world [28, 29].
Recent progress in the fields of immunoinformatics, bioinformatics, and structural vaccinomics has led to a significant transformation in how antigens are identified. These advancements have also facilitated the development of the novel vaccines creation approach, enabling the vaccines targeting a broad spectrum of bacterial and viral infections. Our prior research, has demonstrated that multi-epitope peptide vaccines can use more diverse and higher effective factors against infectious agents compared to vaccines based on single epitopes [16, 30]. In line with this approach, the present study concentrated on multiepitope subunit vaccines. These vaccines are performed using multiple immunogenic factors of pathogens and possess the capability to direct the humoral immunity reaction towards particular antigenic epitopes, ultimately leading to a safer and more efficient immune response.
In this current work, we employed the NCBI database to obtain the sequences of two viral HA/NA proteins from AIV (H5N1) strain, following a comprehensive review of the existing literature. Two viral proteins, namely HA (GenBank: BAL61222.1) and NA (GenBank: BAL61230.1), serve as surface glycoproteins responsible for binding to host cells; are two of the proteins among the most abundant within the influenza A virus and play crucial functional and structural parts throughout life cycle of viruses [31].
Vaccines stimulate B cells to generate antibodies that, in turn, carry out effector functions by specifically interacting with pathogens or toxins. Due to their role in preserving memory cells, providing longer-term immunity, and defending versus reinfection, the humoral immunity reaction plays a vital part in safeguarding versus infections through vaccination. Numerous antigens and vaccinations also cause T-cell responses in addition to B-cell responses [32–34].
Well-known CD4+ T cell is another crucial type of immune cell capable of adopting Th1 or Th2 phenotypes as well as orchestrating immune reactions [35]. The Th1 response is responsible for activating cytotoxic CD8+ T lymphocytes natural killer cells, as well as macrophages. In contrast, the Th2 response has a crucial part in activating B cells, facilitating isotype switching, promoting affinity maturation, as well as generating antibodies that can target and neutralize external pathogens [36]. T cell epitope-based vaccination is therefore a unique strategy for generating a strong immune response against pathogenic pathogens [37]. Here, to found possible CTL and HTL immunogenic epitopes inside HA and NA protein, we used topological screening, the MHC-I and MHC-II binding predictions, and the VaxiJen server. These epitopes have been selected according to their ability to link to a wide range of HLA-A and HLA-B alleles having high binding affinity.
In this study, we utilized tools to evaluate the allergenicity of chosen epitopes. During the immune stimulation procedure, allergenicity, which poses a significant challenge in vaccine development, is detected in many vaccine candidates, potentially leading to an "allergic" response. According to a criterion for predicting allergenicity, a sequence is deemed possibly allergenic if, when compared to known allergens, it shares the identity of at least six consecutive amino acids within an 80-amino acid window [38]. In addition to that, one important factor in the reverse vaccinology technique is population coverage. As per the results, all the predicted T cell epitopes from HA and NA are capable of providing coverage for populations across the majority of geographic regions worldwide, with rates exceeding 90%. Moreover, since MHC superfamilies are important in the creation of vaccines and pharmaceuticals, it has become possible to ascertain the functional link among MHC variants by MHC cluster analysis.
In our study, we used a total of 18 B cell epitopes, CTL and HTL. We used two algorithms of Linear B-cell epitope prediction methodology, including IEDB server as well as BepiPred 2.0 for predicting amino acid likely B-cell epitopes. While over the past few decades, numerous B-cell epitope prediction methods have been developed, but they have mostly failed, the majority of experimental epitope determination efforts have concentrated on finding a linear B-cell epitope prediction technique [39]. Furthermore, accurate prediction and a quicker, less expensive vaccine design process can both be aided by linear B-cell epitope prediction [40].
Based on the Bepipred linear epitope prediction 2.0 for the AIV-A (H5N1) strain, the most effective B cell epitopes for each of the three proteins were selected as possible vaccination candidates. Linear epitopes from B cells, epitopes recognized by cytotoxic T cells (CTL) and helper T cells (HTL), along with Beta-defensin 2, as well as the PADRE peptide sequence, have been employed in the formulation of the ultimate vaccine proteins. In the past, adjuvants have been used as immunomodulators to increase the effectiveness of certain vaccination candidates [41–43]. The effectiveness of the Beta-defensin adjuvant as an immune booster has been demonstrated in numerous experiments against various organisms [44–46]. The VaxiJen server has been utilized to evaluate all of the acquired protein sequences, determining the most potential antigenic protein with the capacity to induce immunity.
AAY linkers have been utilized to bond CTL (Cytotoxic T Lymphocyte) epitopes to each other. These linkers help separate and align CTL epitopes effectively. Specific HEYGAEALERAG linker serves as the interface among CTL epitopes as well as HTL (Helper T Lymphocyte) epitopes, ensuring a proper connection and interaction between these two critical components of the vaccine. GPGPG linkers act as connectors between different HTL epitopes. They help maintain structural integrity and spacing within the HTL epitope region. KK linkers have been used to connect B-cell epitopes, as well as have been utilized to link HβD-3 and PADRE adjuvants in the N-terminal of the construct. These linkers ensure that B-cell epitopes are properly connected, enhancing their accessibility for immune recognition. EAAAK was used to bond the vaccine construct to the HisTag sequence at the C-terminal. EAAAK Linkers help maintain flexibility and proper spacing between these components. The strategic use of these linkers is essential in vaccine design to ensure that different epitopes and components work together efficiently, promoting a robust immune response when administered.
Additionally, The ProtParam server has been utilized to examine the final construct's physicochemical characteristics. The molecular weight of the H5N1 vaccine construct is 38.46 kDa. This is deemed a suitable vaccine candidate, as proteins having a molecular weight of less than 110 kDa are generally regarded as more favorable for vaccine development because of their ease of purification [47]. After expression, the construct's heat stability was evaluated using the aliphatic index. The H5N1 protein is classified as stable by the estimated instability index since its value is less than 40. An instability index of less than 40 indicates stability, whereas a value above 40 suggests potential instability. The calculated GRAVY index of H5N1 serves as an indicator of its polar nature and its interaction with water. A greater solubility for this structure is indicated by a lower GRAVY score. Furthermore, the selected vaccine candidate exhibited a higher proportion of coil structure in its secondary structure. This suggests that random coils are significant in conferring greater protein flexibility and might contribute to an enhanced antibody binding capability [48].
Incorporating adjuvants into the subunit vaccine could enhance the immunity reaction it elicits. Adjuvants serve to be activators for Toll-like receptors (TLRs) and could be effectors to broaden antibody recognition [49]. It was observed that TLR7 and TLR8 are capable of recognizing Influenza Vaccine Antigen (IVA) due to their ability to couple with single-stranded RNAs (ssRNAs) [50]. TLR-7 was demonstrated to trigger both a humoral and long-lasting memory response in reaction to ssRNA-mediated infections as well as vaccinations, such as those involving Influenza and HIV [51, 52]. Additionally, It has been discovered that synthetic TLR7 and TLR8 agonists enhance both innate and adaptive immune responses [53].
The interaction between H5N1 3D structure and TLR7 and TLR8 structures has been examined through protein–protein docking. In the case of H5N1-TLR7 as well as H5N1-TLR8 complexes, it was observed that these interactions involved 4 and 6 hydrogen bonds, respectively.
After docking and determining how to connect complexes H5N1-TLR7 and H5N1-TLR8 we performed the MD test. The MD technique was performed to check the stability and behavior of the complexes per unit of time. Since it is possible that over time, the proteins in a complex move away from each other and the complex becomes unstable, MD can show the binding quality of two proteins resulting from their docking (within a certain time).
So in our study to do MD:
First, we ran each complex separately for 100 ps and analyzed the results through RMSD, RMSF, Gyration, H-bond and SASA graphs.
Then we went one step further and analyzed the MD results using MMPBSA to estimate the contribution of each of the energies involved in the complex such as electrostatics, van der Waals, etc.
The complexes of H5N1 with TLR7 and TLR8 demonstrated interactions between the proteins via the creation of hydrogen bonds. Analysis of RMSD diagrams for the H5N1-TLR7 and H5N1-TLR8 complexes revealed that the simulations were relatively stable. However, it's noteworthy that Compared to the H5N-TLR8 complex, which had a mean RMSD of 0.3 nm, the H5N1-TLR7 complex had a mean RMSD of 0.6 nm, indicating that its intermolecular energies are higher. The stability of the complex is confirmed by the gyration graph, which shows that the amino acids stay inside and don't go beyond their radial axis range during the simulation. Furthermore, the RMSF plot aligns with these outcomes, indicating that the amino acids within binding sites of H5N1-TLR7 and H5N1-TLR8 complexes keep their stability during simulation.
Overall, the results from the docking and molecular dynamics (MD) simulations suggest that these complexes are capable of maintaining their stability during the simulation. The outcomes from such a study indicate that recombinant construct may effectively stimulate TLR7 and TLR8. The fact that TLR7 and TLR8 attach to the residues in the vaccine structure suggests that the vaccination suggested in this study would probably cause the development of antibodies against it. This expectation stems from the importance of having the accurate 3D structure of antigen for it to be identified effectively via the humoral immunity mechanism. Consequently, all selected epitopes within the 3D-vaccine structure should closely mimic the correct fold found in the native structure of the antigen to elicit a comparable immunity response [54].
Additionally, the MM/PBSA outcomes revealed that, apart from the overall energy, the complexes are influenced by the energies arising from electrostatic and covalent bonds. Notably, the positive binding energy observed in the H5N1-TLR8 complex could be influenced by presence of salt bridges in contact between the TLR8 and vaccine structure [55].
Influenza viruses are ssRNA viruses that require the activation of T cell-dependent immune responses plus humoral immunity induced via B cells. This activation is crucial for preventing the proliferation and intracellular survival of the virus. According to predictions from the C-ImmSim Online server, the growth of both B cells and T cells leads to the establishment of long-term immune memory. Thus, IgG2 and IgG1 which are indicative of Th2 and Th1 responses to influenza antigens, respectively, play important roles in protecting against influenza infection [56, 57]. Past research has indicated that both avian and human hosts experience a reduction in their T-cell populations in reaction to avian influenza infection [58]. A decrease in T lymphocytes is frequently linked to proinflammatory cytokines’ upregulation, particularly interferons (IFNs), notably IFN-γ, and interleukins (ILs) like IL-6. These changes can, in the end, result in a condition called hypercytokinemia and trigger the cell apoptosis’ activation [59]. Increased levels of IL-12 and IFN-γ are indicative of a robust cellular immune response. While previous research has demonstrated that T epitopes from H5N1 strains designed to mimic human sequences may lead to weakened IFN responses in humans, the results from the ICM server indicate that this vaccine has an excellent capacity to stimulate IFN-γ, suggesting its potential to trigger a robust immune response [60–64]]. Nonetheless, the findings from the ICM server suggest that a rise in the count of Th1 cells is linked to the elevation in the number of cytotoxic T cells.
Supplementary Information
Acknowledgements
This study was supported by National Insititute for Medical Research Development, NIMAD.
Author contributions
FD, ZSR, JSN, FKG, SMAM, JZ, HT, SMRH, HM contributed to the conception, design, and drafting of the manuscript. AR, GP and FR have critically revised manuscript.
Funding
Hamed Mirzaei was supported by National Institute for Medical Research Development, NIMAD (Grant No. 4000478)
Availability of data and materials
Not applicable.
Declarations
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare no conflict of interest.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Hossein Tarrahimofrad, Email: h_tarrahimofrad@yahoo.com.
Seyed Mohammad Reza Hashemian, Email: smrhashemian@sbmu.ac.ir.
Hamed Mirzaei, Email: h.mirzaei2002@gmail.com.
References
- 1.Presti-Rachel M, Zhao G, Beatty-Wandy L, Mihindukulasuriya-Kathie A, Travassos da Rosa-Amelia PA, Popov-Vsevolod L, et al. Quaranfil, Johnston Atoll, and Lake chad viruses are novel members of the family orthomyxoviridae. J Virol. 2009;83(22):11599–11606. doi: 10.1128/JVI.00677-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Avian Influenza A. (H5N1) infection in humans. N Engl J Med. 2005;353(13):1374–1385. doi: 10.1056/NEJMra052211. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Xiao H, Lei F, Zhu Q, Qin K, Zhang XW, et al. Highly pathogenic h5n1 influenza virus infection in migratory birds. Science. 2005;309(5738):1206. doi: 10.1126/science.1115273. [DOI] [PubMed] [Google Scholar]
- 4.Yuen KY, Chan PKS, Peiris M, Tsang DNC, Que TL, Shortridge KF, et al. Clinical features and rapid viral diagnosis of human disease associated with avian influenza A H5N1 virus. Lancet. 1998;351(9101):467–471. doi: 10.1016/S0140-6736(98)01182-9. [DOI] [PubMed] [Google Scholar]
- 5.Kandeil A, Patton C, Jones JC, Jeevan T, Harrington WN, Trifkovic S, et al. Rapid evolution of A(H5N1) influenza viruses after intercontinental spread to North America. Nat Commun. 2023;14(1):3082. doi: 10.1038/s41467-023-38415-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar A, Meldgaard TS, Bertholet S. Novel platforms for the development of a universal influenza vaccine. Front Immunol. 2018;9:600. doi: 10.3389/fimmu.2018.00600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McAuley JL, Gilbertson BP, Trifkovic S, Brown LE, McKimm-Breschkin JL. Influenza virus neuraminidase structure and functions. Front Microbiol. 2019;10:39. doi: 10.3389/fmicb.2019.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lakadamyali M, Rust MJ, Zhuang X. Endocytosis of influenza viruses. Microbes Infect. 2004;6(10):929–936. doi: 10.1016/j.micinf.2004.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ilyushina Natalia A, Bovin Nicolai V, Webster RG. Decreased neuraminidase activity is important for the adaptation of H5N1 influenza virus to human airway epithelium. J Virol. 2012;86(9):4724–4733. doi: 10.1128/JVI.06774-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Kumari V, Kumbhar BV, Mukherjee A, Pandey R, Kondabagil K. Immunoinformatics approach for a novel multi-epitope subunit vaccine design against various subtypes of Influenza A virus. Immunobiology. 2021;226(2):152053. doi: 10.1016/j.imbio.2021.152053. [DOI] [PubMed] [Google Scholar]
- 11.Mia MM, Hasan M, Ahmed S, Rahman MN. Insight into the first multi-epitope-based peptide subunit vaccine against avian influenza A virus (H5N6): an immunoinformatics approach. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2022;104:105355. doi: 10.1016/j.meegid.2022.105355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Islam MSB, Miah M, Hossain ME, Kibria KMK. A conserved multi-epitope-based vaccine designed by targeting hemagglutinin protein of highly pathogenic avian H5 influenza viruses. 3 Biotech. 2020;10(12):546. doi: 10.1007/s13205-020-02544-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Criado Miria F, Sáe-Silva M, Lee D-H, de Salge-Carolina-Alves L, Spackman E, Donis R, et al. Cross-protection by inactivated H5 prepandemic vaccine seed strains against diverse goose/guangdong lineage H5N1 highly pathogenic avian influenza viruses. J Virol. 2020 doi: 10.1128/jvi.00720-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gioia C, Castilletti C, Tempestilli M, Piacentini P, Bordi L, Chiappini R, et al. Cross-subtype immunity against avian influenza in persons recently vaccinated for influenza. Emerg Infect Dis. 2008;14(1):121–128. doi: 10.3201/eid1401.061283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rahimnahal S, Yousefizadeh S, Mohammadi Y. Novel multi-epitope vaccine against bovine brucellosis: approach from immunoinformatics to expression. J Biomol Struct Dyn. 2023;41:15460. doi: 10.1080/07391102.2023.2188962. [DOI] [PubMed] [Google Scholar]
- 16.Tarrahimofrad H, Zamani J, Hamblin MR, Darvish M, Mirzaei H. A designed peptide-based vaccine to combat Brucella melitensis, B. suis and B. abortus: harnessing an epitope mapping and immunoinformatics approach. Biomed Pharmacother. 2022;155:113557. doi: 10.1016/j.biopha.2022.113557. [DOI] [PubMed] [Google Scholar]
- 17.Mahapatra SR, Sahoo S, Dehury B, Raina V, Patro S, Misra N, et al. Designing an efficient multi-epitope vaccine displaying interactions with diverse HLA molecules for an efficient humoral and cellular immune response to prevent COVID-19 infection. Expert Rev Vaccines. 2020;19(9):871–885. doi: 10.1080/14760584.2020.1811091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dey J, Mahapatra SR, Singh P, Patro S, Kushwaha GS, Misra N, et al. B and T cell epitope-based peptides predicted from clumping factor protein of Staphylococcus aureus as vaccine targets. Microb Pathog. 2021;160:105171. doi: 10.1016/j.micpath.2021.105171. [DOI] [PubMed] [Google Scholar]
- 19.Mahapatra SR, Dey J, Kaur T, Sarangi R, Bajoria AA, Kushwaha GS, et al. Immunoinformatics and molecular docking studies reveal a novel Multi-Epitope peptide vaccine against pneumonia infection. Vaccine. 2021;39(42):6221–6237. doi: 10.1016/j.vaccine.2021.09.025. [DOI] [PubMed] [Google Scholar]
- 20.Chatterjee R, Sahoo P, Mahapatra SR, Dey J, Ghosh M, Kushwaha GS, et al. Development of a conserved chimeric vaccine for induction of strong immune response against staphylococcus aureus using immunoinformatics approaches. Vaccines. 2021;9(9):1038. doi: 10.3390/vaccines9091038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mahapatra SR, Dey J, Kushwaha GS, Puhan P, Mohakud NK, Panda SK, et al. Immunoinformatic approach employing modeling and simulation to design a novel vaccine construct targeting MDR efflux pumps to confer wide protection against typhoidal Salmonella serovars. J Biomol Struct Dyn. 2022;40(22):11809–11821. doi: 10.1080/07391102.2021.1964600. [DOI] [PubMed] [Google Scholar]
- 22.Dey J, Mahapatra SR, Lata S, Patro S, Misra N, Suar M. Exploring Klebsiella pneumoniae capsule polysaccharide proteins to design multiepitope subunit vaccine to fight against pneumonia. Expert Rev Vaccines. 2022;21(4):569–587. doi: 10.1080/14760584.2022.2021882. [DOI] [PubMed] [Google Scholar]
- 23.Sahoo S, Mahapatra SR, Parida BK, Rath S, Dehury B, Raina V, et al. DBCOVP: a database of coronavirus virulent glycoproteins. Comput Biol Med. 2021;129:104131. doi: 10.1016/j.compbiomed.2020.104131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Y, Aevermann BD, Anderson TK, Burke DF, Dauphin G, Gu Z, et al. Influenza research database: an integrated bioinformatics resource for influenza virus research. Nucleic Acids Res. 2017;45(D1):D466–D474. doi: 10.1093/nar/gkw857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lundegaard C, Lund O, Nielsen M. Accurate approximation method for prediction of class I MHC affinities for peptides of length 8, 10 and 11 using prediction tools trained on 9mers. Bioinformatics. 2008;24(11):1397–1398. doi: 10.1093/bioinformatics/btn128. [DOI] [PubMed] [Google Scholar]
- 26.Kolaskar AS, Tongaonkar PC. A semi-empirical method for prediction of antigenic determinants on protein antigens. FEBS Lett. 1990;276(1–2):172–174. doi: 10.1016/0014-5793(90)80535-Q. [DOI] [PubMed] [Google Scholar]
- 27.Babon-Jenny-Aurielle B, Cruz J, Ennis-Francis A, Yin L, Terajima M. A Human CD4+ T cell epitope in the influenza hemagglutinin is cross-reactive to influenza A virus subtypes and to influenza B virus. J Virol. 2013;87(16):9396. doi: 10.1128/JVI.01387-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durand L, Glew P, Gross D, Kasper M, Trock S, Kim I, et al. Timing of influenza A(H5N1) in poultry and humans and seasonal influenza activity worldwide, 2004–2013. Emerg Infect Dis. 2015;21:202–208. doi: 10.3201/eid2102.140877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Olsen B, Munster VJ, Wallensten A, Waldenström J, Osterhaus ADME, Fouchier RAM. Global patterns of influenza A virus in wild birds. Science. 2006;312(5772):384–388. doi: 10.1126/science.1122438. [DOI] [PubMed] [Google Scholar]
- 30.Jafari-Najaf-Abadi MH, Abdi-Abyaneh F, Zare N, Zamani J, Abdoli A, Aslanbeigi F, et al. In silico design and immunoinformatics analysis of a chimeric vaccine construct based on Salmonella pathogenesis factors. Microb Pathogen. 2023;180:106130. doi: 10.1016/j.micpath.2023.106130. [DOI] [PubMed] [Google Scholar]
- 31.Nidom CA, Yamada S, Nidom RV, Rahmawati K, Alamudi MY, Kholik, et al. Genetic characterization of H5N1 influenza viruses isolated from chickens in Indonesia in 2010. Virus Genes. 2012;44(3):459–465. doi: 10.1007/s11262-012-0722-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frey SS, Versage E, Van Twuijver E, Hohenboken M. Antibody responses against heterologous H5N1 strains for an MF59-adjuvanted cell culture-derived H5N1 (aH5n1c) influenza vaccine in adults and older adults. Hum Vaccin Immunother. 2023;19(1):2193119. doi: 10.1080/21645515.2023.2193119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Honda-Okubo Y, Bart Tarbet E, Hurst BL, Petrovsky N. An Advax-CpG adjuvanted recombinant H5 hemagglutinin vaccine protects mice against lethal influenza infection. Vaccine. 2023;41(39):5730–5741. doi: 10.1016/j.vaccine.2023.08.009. [DOI] [PubMed] [Google Scholar]
- 34.Charostad J, Rezaei-Zadeh-Rukerd M, Mahmoudvand S, Bashash D, Hashemi SMA, Nakhaie M, et al. A comprehensive review of highly pathogenic avian influenza (HPAI) H5N1: an imminent threat at doorstep. Travel Med Infect Dis. 2023;55:102638. doi: 10.1016/j.tmaid.2023.102638. [DOI] [PubMed] [Google Scholar]
- 35.Khan MT, Islam R, Jerin TJ, Mahmud A, Khatun S, Kobir A, et al. Immunoinformatics and molecular dynamics approaches: next generation vaccine design against West Nile virus. PLoS ONE. 2021;16(6):e0253393. doi: 10.1371/journal.pone.0253393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ekkens MJ, Shedlock DJ, Jung E, Troy A, Pearce EL, Shen H, et al. Th1 and Th2 cells help CD8 T-cell responses. Infect Immun. 2007;75(5):2291–2296. doi: 10.1128/IAI.01328-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shrestha B, Diamond MS. Role of CD8+ T cells in control of west Nile virus infection. J Virol. 2004;78(15):8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKeever TM, Lewis SA, Smith C, Hubbard R. Vaccination and allergic disease: a birth cohort study. Am J Public Health. 2004;94(6):985–989. doi: 10.2105/AJPH.94.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.El-Manzalawy Y, Honavar V. Recent advances in B-cell epitope prediction methods. Immunome Res. 2010;6(2):S2. doi: 10.1186/1745-7580-6-S2-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galanis KA, Nastou KC, Papandreou NC, Petichakis GN, Pigis DG, Iconomidou VA. Linear B-cell epitope prediction for in silico vaccine design: a performance review of methods available via command-line interface. Int J Mol Sci. 2021;22(6):3210. doi: 10.3390/ijms22063210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Khan K, Khan SA, Jalal K, Ul-Haq Z, Uddin R. Immunoinformatic approach for the construction of multi-epitopes vaccine against omicron COVID-19 variant. Virology. 2022;572:28–43. doi: 10.1016/j.virol.2022.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Malonis RJ, Lai JR, Vergnolle O. Peptide-based vaccines: current progress and future challenges. Chem Rev. 2020;120(6):3210–3229. doi: 10.1021/acs.chemrev.9b00472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamley IW. Peptides for vaccine development. ACS Appl Bio Mater. 2022;5(3):905–944. doi: 10.1021/acsabm.1c01238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mei H-F, Jin X-B, Zhu J-Y, Zeng A-H, Wu Q, Lu X-M, et al. β-defensin 2 as an adjuvant promotes anti-melanoma immune responses and inhibits the growth of implanted murine melanoma in vivo. PLoS ONE. 2012;7(2):31328. doi: 10.1371/journal.pone.0031328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Yang YL, Jang SH, Jang YS. Human β-defensin 2 plays a regulatory role in innate antiviral immunity and is capable of potentiating the induction of antigen-specific immunity. Virol J. 2018;15(1):124. doi: 10.1186/s12985-018-1035-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng J, Yang J, Zhang Z, Liang X, Liu S, Pan Y, et al. An improved oral vaccine with molecular adjuvant β-defensin protects grouper against nervous necrosis virus infection. Fish Shellfish Immunol. 2023;136:108709. doi: 10.1016/j.fsi.2023.108709. [DOI] [PubMed] [Google Scholar]
- 47.Cao H, Xu H, Ning C, Xiang L, Ren Q, Zhang T, et al. Multi-omics approach reveals the potential core vaccine targets for the emerging foodborne pathogen campylobacter jejuni. Front Microbiol. 2021;12:665858. doi: 10.3389/fmicb.2021.665858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kovács D, Bodor A. The influence of random-coil chemical shifts on the assessment of structural propensities in folded proteins and IDPs. RSC Adv. 2023;13(15):10182–10203. doi: 10.1039/D3RA00977G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vogel FR. Improving vaccine performance with adjuvants. Clin Infect Dis. 2000;30(3):S266–S270. doi: 10.1086/313883. [DOI] [PubMed] [Google Scholar]
- 50.Pulendran B, Maddur MS. Innate immune sensing and response to influenza. Curr Top Microbiol Immunol. 2015;386:23–71. doi: 10.1007/82_2014_405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jeisy-Scott V, Kim Jin H, Davis William G, Cao W, Katz Jacqueline M, Sambhara S. TLR7 Recognition is dispensable for influenza virus A infection but important for the induction of hemagglutinin-specific antibodies in response to the 2009 pandemic split vaccine in mice. J Virol. 2012;86(20):10988–10998. doi: 10.1128/JVI.01064-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wille-Reece U, Wu CY, Flynn BJ, Kedl RM, Seder RA. Immunization with HIV-1 gag protein conjugated to a TLR7/8 agonist results in the generation of HIV-1 gag-specific Th1 and CD8+ T cell responses. J Immunol. 2005;174(12):7676–7683. doi: 10.4049/jimmunol.174.12.7676. [DOI] [PubMed] [Google Scholar]
- 53.Miller SM, Cybulski V, Walsh L, Livesay M, Bess L, Burkhart DJ, et al. Investigation of novel TLR7/8 ligands in combination with TLR4 ligands as adjuvants to drive cell mediated anti-influenza immunity. J Immunol. 2018;200(1):125.16–16. doi: 10.4049/jimmunol.200.Supp.125.16. [DOI] [Google Scholar]
- 54.Yu F, Song H, Wu Y, Chang SY, Wang L, Li W, et al. A potent germline-like human monoclonal antibody targets a ph-sensitive epitope on H7N9 influenza hemagglutinin. Cell Host Microbe. 2017;22(4):471–83.e5. doi: 10.1016/j.chom.2017.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu D, Tsai CJ, Nussinov R. Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng Des Sel. 1997;10(9):999–1012. doi: 10.1093/protein/10.9.999. [DOI] [PubMed] [Google Scholar]
- 56.Palladino G, Scherle PA, Gerhard W. Activity of CD4+ T-cell clones of type 1 and type 2 in generation of influenza virus-specific cytotoxic responses in vitro. J Virol. 1991;65(11):6071–6076. doi: 10.1128/jvi.65.11.6071-6076.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chen WH, Cross AS, Edelman R, Sztein MB, Blackwelder WC, Pasetti MF. Antibody and Th1-type cell-mediated immune responses in elderly and young adults immunized with the standard or a high dose influenza vaccine. Vaccine. 2011;29(16):2865–2873. doi: 10.1016/j.vaccine.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wang Z, Wan Y, Qiu C, Quiñones-Parra S, Zhu Z, Loh L, et al. Recovery from severe H7N9 disease is associated with diverse response mechanisms dominated by CD8+ T cells. Nat Commun. 2015;6(1):6833. doi: 10.1038/ncomms7833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang Z, Zhang A, Wan Y, Liu X, Qiu C, Xi X, et al. Early hypercytokinemia is associated with interferon-induced transmembrane protein-3 dysfunction and predictive of fatal H7N9 infection. Proc Natl Acad Sci. 2014;111(2):769–774. doi: 10.1073/pnas.1321748111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Noisumdaeng P, Roytrakul T, Prasertsopon J, Pooruk P, Lerdsamran H, Assanasen S, et al. T cell mediated immunity against influenza H5N1 nucleoprotein, matrix and hemagglutinin derived epitopes in H5N1 survivors and non-H5N1 subjects. PeerJ. 2021;9:e11021. doi: 10.7717/peerj.11021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gao W, Soloff AC, Lu X, Montecalvo A, Nguyen DC, Matsuoka Y, et al. Protection of mice and poultry from lethal H5N1 avian influenza virus through adenovirus-based immunization. J Virol. 2006;80(4):1959–1964. doi: 10.1128/JVI.80.4.1959-1964.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hoelscher MA, Garg S, Bangari DS, Belser JA, Lu X, Stephenson I, et al. Development of adenoviral-vector-based pandemic influenza vaccine against antigenically distinct human H5N1 strains in mice. Lancet. 2006;367(9509):475–481. doi: 10.1016/S0140-6736(06)68076-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Moris P, van der Most R, Leroux-Roels I, Clement F, Dramé M, Hanon E, et al. H5N1 influenza vaccine formulated with AS03 A induces strong cross-reactive and polyfunctional CD4 T-cell responses. J Clin Immunol. 2011;31(3):443–454. doi: 10.1007/s10875-010-9490-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Janssens Y, Joye J, Waerlop G, Clement F, Leroux-Roels G, Leroux-Roels I. The role of cell-mediated immunity against influenza and its implications for vaccine evaluation. Front Immunol. 2022;513:959379. doi: 10.3389/fimmu.2022.959379. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable.