

Abstract
Hereditary factors account for a significant proportion of breast cancer risk. Approximately 20% of hereditary breast cancers are attributable to pathogenic variants in the highly penetrant BRCA1 and BRCA2 genes. A proportion of the genetic risk is also explained by pathogenic variants in other breast cancer susceptibility genes, including ATM, CHEK2, PALB2, RAD51C, RAD51D and BARD1, as well as genes associated with breast cancer predisposition syndromes – TP53 (Li–Fraumeni syndrome), PTEN (Cowden syndrome), CDH1 (hereditary diffuse gastric cancer), STK11 (Peutz–Jeghers syndrome) and NF1 (neurofibromatosis type 1). Polygenic risk, the cumulative risk from carrying multiple low‐penetrance breast cancer susceptibility alleles, is also a well‐recognised contributor to risk. This review provides an overview of the established breast cancer susceptibility genes as well as breast cancer predisposition syndromes, highlights distinct genotype–phenotype correlations associated with germline mutation status and discusses molecular testing and therapeutic implications in the context of hereditary breast cancer.
Keywords: familial breast cancer, BRCA1, BRCA2, cancer syndromes, genotype–phenotype correlation

Introduction
Breast cancer is the leading cause of cancer death in women worldwide. 1 While most breast cancers are sporadic, approximately 5–10% are hereditary. 2 Pathogenic variants in the high‐penetrance genes BRCA1 and BRCA2 account for approximately 20% of heritable breast cancer risk. 3 , 4 BRCA1 and BRCA2 play an integral role in DNA damage repair by homologous recombination (HR), and germline variants in several other HR repair genes are also implicated in hereditary breast cancer risk, including CHEK2, PALB2, ATM, RAD51C, RAD51D and BARD1. 5 , 6 , 7 An additional proportion of patients with hereditary breast cancer have germline variants in genes associated with recognised cancer susceptibility syndromes, including TP53 (Li–Fraumeni syndrome), PTEN (Cowden syndrome), STK11 (Peutz–Jeghers syndrome), CDH1 (hereditary diffuse gastric cancer and hereditary lobular breast cancer) and NF1 (neurofibromatosis type 1). 8 An association between mutations in mismatch repair (MMR) genes (Lynch syndrome) and breast cancer has not been conclusively established. Furthermore, a substantial proportion of hereditary breast cancer risk is not explained by pathogenic variants in specific genes but is partly attributable to polygenic risk, referring to variable combinations of multiple low‐penetrance breast cancer susceptibility alleles. 9 Characteristic histopathological features and immunohistochemical phenotypes have been associated with germline variants in several breast cancer susceptibility genes. Recognition of these pathological features, together with known risk factors such as age and family history, informs patient risk prediction, guides genetic testing and facilitates the diagnosis of hereditary breast cancer, which influences patient management and surveillance. In this review we provide an overview of the established breast cancer susceptibility genes and syndromes, with an emphasis on distinct pathological and molecular associations and their clinical implications.
Historical background
A hereditary component to breast cancer predisposition was first proposed by the French surgeon, Pierre Paul Broca in 1866, when he described a familial cluster of breast cancer cases affecting multiple women across four generations. 10 Subsequent epidemiological studies firmly established family history as a major risk factor for developing breast cancer, 11 , 12 , 13 with the magnitude of risk further increased by the number of affected relatives, closer degree of relationship and younger age at diagnosis. 13 , 14 In 1988, segregation analysis of 1579 high‐risk breast cancer families with multiple affected individuals found that disease clustering was fully explained by an autosomal‐dominant model of transmission with a highly penetrant susceptibility allele. 15 Two years later, linkage studies revealed that the locus of the gene for early‐onset familial breast cancer was on chromosome 17. 16 In 1994, BRCA1 was discovered to be the causative gene at this locus. 17 In 1995, BRCA2 was identified as the second breast cancer susceptibility gene and localised to chromosome 13. 18 , 19 In 1997 the Breast Cancer Linkage Consortium reported distinct pathological characteristics of BRCA1/2‐associated familial breast cancers compared to sporadic cases,20 and key histological phenotypes associated with BRCA1/2 mutations were described in 1998. 21
Research into rare syndromes with a high incidence of breast cancer led to the recognition of several additional moderate‐ and high‐penetrance genes that contribute to hereditary breast cancer risk, including TP53, 22 CDH1, 23 PTEN, 24 STK11 25 and NF1. 26 Studies of non‐BRCA1/2 familial breast cancer patients implicated germline variants in multiple genes involved in DNA damage repair, including ATM, 27 CHEK2, 28 PALB2, 29 BARD1, 30 RAD51C 31 and RAD51D 32 in breast cancer susceptibility. In 2007, the first breast cancer genome‐wide association studies (GWAS) were published, 33 , 34 , 35 identifying several common low‐penetrance variants associated with genetic breast cancer risk, while the implementation of next‐generation sequencing (NGS) approaches in population‐based studies has defined rare genomic risk variants and quantified the magnitude of their associated risk. 6 , 7 Known germline variants in established high‐, moderate‐ and low‐penetrance genes collectively explain approximately 50% of familial breast cancer risk. 36 Ongoing research into the genetic basis of familial breast cancer will probably uncover novel risk variants, further elucidate the role of susceptibility genes in breast cancer aetiology and clarify how genetic factors interact with other risk factors, thereby improving individual risk prediction.
Heritable breast cancer risk
The genetic basis of breast cancer predisposition is a rapidly evolving topic and this is reflected in the substantially revised ‘genetic tumour syndromes of the breast’ chapter in the fifth edition of the World Health Organisation (WHO) classification of tumours of the breast (2019), 37 which incorporates a growing number of susceptibility genes and predisposition syndromes implicated in breast cancer risk (Table 1). The established and emerging breast cancer susceptibility genes and predisposition syndromes are summarised below.
Table 1.
Comparison of the topics included in the ‘genetic susceptibility: inherited syndromes’ chapter in the WHO classification, 4th edition (2012) and the ‘genetic tumour syndromes of the breast’ chapter in the WHO classification, 5th edition (2019). Of note, the association between Lynch syndrome and breast cancer is controversial, so the editorial board did not feel that there was sufficient evidence to include Lynch syndrome in the 2019 classification system. In the current classification system, Li–Fraumeni syndrome has also been split into two categories associated with variants in either TP53 or CHEK2. These classification systems may continue to evolve as more knowledge is accrued
| WHO classification 4th edition (2012) 199 | WHO classification 5th edition (2019) 37 |
|---|---|
| BRCA1 and BRCA2 syndromes | BRCA1/2‐associated hereditary breast and ovarian cancer syndrome |
| Li–Fraumeni syndrome | Li–Fraumeni syndrome, TP53‐associated |
| Li–Fraumeni syndrome, CHEK2‐associated | |
| Ataxia–telangiectasia | Ataxia–telangiectasia |
| Cowden syndrome | Cowden syndrome |
| Lynch syndrome | Not included in new edition |
| Other breast cancer‐predisposing genes | CDH1‐associated breast cancer |
| PALB2‐associated cancers | |
| Peutz–Jeghers syndrome | |
| Neurofibromatosis type 1 | |
| The polygenic component of breast cancer susceptibility |
Breast cancer predisposition syndromes
Hereditary breast and ovarian cancer syndrome
Hereditary breast and ovarian cancer syndrome is associated with germline mutations in the tumour suppressor genes BRCA1 and BRCA2 and is inherited in an autosomal‐dominant manner. BRCA1 and BRCA2 encode for BRCA1/2 proteins involved in the repair of double‐strand DNA breaks through HR. 38 , 39 Pathogenic variants in BRCA1/2 are highly penetrant and are associated with a significantly elevated lifetime risk of breast and ovarian cancer 40 , 41 , 42 and increased susceptibility to a number of other malignancies, particularly prostate (BRCA1/2) and pancreatic cancer (BRCA2). 43 , 44 , 45 , 46 In a large prospective cohort, the cumulative risk of breast cancer up to age 80 years was 72% for BRCA1 and 69% for BRCA2 mutation carriers. 41 In addition, BRCA1 and BRCA2 mutation carriers have an increased risk of contralateral breast cancer (40 and 26%, respectively),41 and germline mutations in BRCA2 are associated with male breast cancer. 47 , 48
Li–Fraumeni syndrome
Li–Fraumeni syndrome is an autosomal‐dominant syndrome associated with germline mutations in the tumour suppressor gene TP53. 22 TP53 encodes the protein p53, an important cell cycle regulator. 49 , 50 Germline mutations in TP53 are associated with an increased risk of early onset malignancy, including epithelial, mesenchymal and haematological malignancies. 51 , 52 , 53 , 54 Breast cancer is the most common epithelial malignancy in women, 55 with an 85% cumulative lifetime risk of breast cancer by age 60 years. 56 The median age of breast cancer diagnosis is 34 years 57 and germline TP53 mutations are identified in approximately 5–8% of women diagnosed with breast cancer before the age of 30 years. 58
Cowden syndrome
Cowden syndrome is an autosomal‐dominant syndrome associated with germline mutations in the tumour suppressor gene PTEN 24 and is the most common disorder in the PTEN hamartoma tumour syndrome spectrum. 59 The clinical manifestations of Cowden syndrome include the development of multiple hamartomas as well as an elevated risk of breast, thyroid, endometrial, renal and colorectal malignancies. 60 , 61 Breast cancer is the most common malignancy in Cowden syndrome, with an estimated lifetime risk of up to 85% 62 , 63 and an average age of diagnosis between 38 and 46 years. 64
Peutz–Jeghers syndrome
Peutz–Jeghers syndrome is an autosomal‐dominant syndrome attributable to mutations in the tumour suppressor gene STK11. 65 Peuts–Jeghers syndrome is characterised by the development of mucocutaneous pigmentation, hamartomatous gastrointestinal polyps and is associated with an increased risk of gastrointestinal, breast, lung, gynaecological and genitourinary malignancies. 66 , 67 , 68 , 69 , 70 Breast cancer is the second most common malignancy after gastrointestinal tumours, with a cumulative breast cancer risk of up to 54% at age 64 years and a mean age at diagnosis of 37 years. 67
Hereditary diffuse gastric cancer and hereditary lobular breast cancer
Hereditary diffuse gastric cancer (HDGC) and hereditary lobular breast cancer (HLBC) are autosomal‐dominant syndromes associated with inactivating germline mutations in CDH1. 71 CDH1 encodes E‐cadherin, a transmembrane protein involved in cell‐to‐cell adhesion. 72 Both men and women with HDGC have an elevated lifetime risk of developing diffuse gastric cancer (70 and 56% by age 80 years, respectively) 73 and women also have a 42% lifetime risk of developing invasive lobular carcinoma (ILC). 73 , 74 ILC can be the first manifestation of HDGC, presenting as bilateral disease in women younger than 50 years of age. 75 HLBC includes families with germline CDH1 mutations who show a predisposition to ILC but do not have a family history of gastric cancer. 76 , 77 , 78 However, even without a family history of gastric cancer, patients with HLBC have a markedly elevated risk of developing occult gastric malignancy. 79 Breast cancer metastasis may need to be excluded in patients with diffuse gastric cancer who also have a history of ILC, as ILC is the most common breast cancer subtype to metastasise to the stomach and morphologically mimics diffuse‐type gastric cancer. 80 Differentiating metastatic breast from primary gastric carcinoma requires a panel of immunohistochemical markers; for example, GATA binding protein 3 (GATA3), oestrogen receptor (ER) and progesterone receptor (PR) positivity supports breast origin, while CK20 and CDX2 positivity is seen in a proportion of gastric carcinomas. 81 , 82
Neurofibromatosis type 1
Neurofibromatosis type 1 is associated with mutations in the tumour suppressor gene NF1, 83 which may be inherited in an autosomal‐dominant manner or occur sporadically in approximately half the cases. 84 Clinical manifestations include neurocutaneous lesions and an increased risk of malignancy, predominantly involving the nervous system and breast. 85 Women with neurofibromatosis type 1 have an increased risk of early‐onset breast cancer, with a four to 11‐fold increase in breast cancer risk up to the age of 50 years 84 and a 17% cumulative lifetime risk of breast cancer by age 70 years. 86
Lynch syndrome
Lynch syndrome is an autosomal‐dominant syndrome primarily attributable to germline mutations in the MMR genes MLH1, MSH2, MSH6 and PMS2. 87 The most common malignancies in Lynch syndrome involve the colon, endometrium, ovaries and stomach. 88 , 89 An association between Lynch syndrome and breast cancer has not been definitively established. Some studies have reported an increased risk of breast cancer 90 , 91 , 92 and a prospective cohort study of MMR gene mutation carriers identified a fourfold increase in breast cancer risk with a median follow‐up of 5 years. 88 However, data from the Prospective Lynch Syndrome Database showed that the risk of developing breast cancer in carriers of MLH1, MSH2 and MSH6 pathogenic germline variants was not significantly elevated among the general population. 89 , 93 , 94 In addition, microsatellite instability (MSI) as a result of MMR deficiency is uncommon in breast cancer and seen in fewer than 2% of cases. 95 In a cohort of 640 breast cancers, only 11 were found to have MMR deficiency using whole genome sequencing and more than 80% of the MMR‐deficient tumours did not have Lynch syndrome. 95 Furthermore, analysis of breast carcinomas arising in patients with Lynch syndrome found that only 51% of tumours demonstrated evidence of MMR deficiency on immunohistochemistry, 96 suggesting that half the cases had a different aetiology.
Homologous recombination deficiency and breast cancer risk
HR is a high‐fidelity DNA repair pathway utilised in the repair of double‐strand DNA breaks. 97 , 98 A defect in HR is termed homologous recombination deficiency (HRD) and is characterised by defective DNA repair, genomic instability and cancer predisposition. 99 In addition to BRCA1/2, several other genes involved in the HR repair pathway are associated with an increased risk of breast cancer, including PALB2, ATM, CHEK2, RAD51C, RAD51D and BARD1. Two recently published population‐based case–control studies BRIDGES 6 and CARRIERS 7 assessed sequencing data from almost 180 000 women, including unselected breast cancer patients as well as controls (unaffected individuals), to determine the prevalence of pathogenic variants in breast cancer predisposition genes in the general population and their associated breast cancer risk. Both studies identified an elevated breast cancer risk in women with germline pathogenic variants in eight genes involved in the HR repair pathway: BRCA1, BRCA2, PALB2, ATM, CHEK2, RAD51C, RAD51D and BARD1. Germline pathogenic variants in these genes, together with CDH1, NF1, PTEN and TP53, were detected in 5.03% of unselected breast cancer cases compared to 1.63% of controls. 7
PALB2
Partner and localiser of BRCA2 (PALB2) is a tumour suppressor gene involved in the HR repair pathway through its interaction with BRCA2. 100 , 101 , 102 Bi‐allelic germline PALB2 mutations are associated with Fanconi anaemia and a predisposition to childhood malignancies, 103 whereas mono‐allelic germline mutations confer an increased risk of breast, pancreatic and ovarian cancer. 29 , 104 , 105 , 106 , 107 , 108 Mono‐allelic germline PALB2 mutations are identified in up to ~5% of patients with hereditary breast cancer 104 , 109 and female PALB2 mutation carriers have a 2.3 to ninefold increased risk of breast cancer, 29 , 107 with an estimated cumulative lifetime risk of 35% by age 70 years. 107
ATM
Ataxia–telangiectasia‐mutated (ATM) gene is a tumour suppressor gene that encodes a serine/threonine protein kinase involved in the HR repair pathway, regulation of cell‐cycle check‐points and intracellular signalling pathways. 110 Bi‐allelic loss of function variants in ATM are associated with the development of ataxia–telangiectasia – a rare autosomal recessive neurodegenerative disorder – and confer increased cancer susceptibility. 27 , 111 , 112 Heterozygous carriers have a threefold increased risk of breast cancer and the risk is reported as sevenfold for women younger than 55 years. 112 The estimated cumulative lifetime risk of breast cancer in heterozygous mutation carriers is ~33% by age 80 years. 113
CHEK2
Check‐point kinase 2 (CHEK2) tumour suppressor gene encodes CHK2, a serine/threonine protein kinase involved in the HR repair pathway, cell cycle arrest and apoptosis in response to DNA damage. 114 Germline CHEK2 mutations are associated with an increased risk of breast, prostate, kidney, colon, thyroid and gastric cancer as well as sarcoma and non‐Hodgkin lymphoma, 115 , 116 , 117 , 118 in some cases manifesting in a Li–Fraumeni‐like phenotype (referred to as CHEK2‐associated Li–Fraumeni syndrome or Li–Fraumeni syndrome 2). 119 Pathogenic variants in CHEK2 are associated with a moderate breast cancer risk, with an odds ratio of 2.47 [95% confidence interval (CI) = 2.02–3.05] 7 The estimated lifetime breast cancer risk is 21% by age 70 years, 120 and the magnitude of this risk as much as doubles when both first‐ and second‐degree relatives have a history of breast cancer. 120 , 121
RAD51C, RAD51D and BARD1
RAD51C, RAD51D and BARD1 are tumour suppressor genes involved in the HR repair pathway and cell cycle progression. 122 Pathogenic variants in BARD1, RAD51C and RAD51D show a modest association with overall breast cancer risk, with reported odds ratios of 2.09 (95% CI = 1.35–3.23), 1.93 (95% CI = 1.20–3.11) and 1.80 (95% CI = 1.11–2.93), respectively;6 however, demonstrate a stronger association (odds ratios > 2) with ER‐negative and triple‐negative disease. 7 Carriers of protein‐truncating mutations in these genes are estimated to have a moderate absolute breast cancer risk (17–30%) by age 80 years. 6
Polygenic risk
Pathogenic variants in moderate‐ and high‐penetrance susceptibility genes explain approximately 25–30% of heritable breast cancer risk. 123 A further 18% is attributable to polygenic risk, pertaining to variable combinations of hundreds of common low‐penetrance breast cancer susceptibility alleles, identified using population‐based GWAS. 9 , 124 , 125 These low‐penetrance genetic variants are often located in non‐coding regions of DNA and individually confer a small risk of breast cancer (odds ratio < 1.5). 124 , 126 However, the magnitude of breast cancer risk can be substantial when the sum of individual risk is assessed, expressed as the polygenic risk score. 127 For women in the top centile of the polygenic risk score based on 313 confirmed risk loci, there was a ~ 33% lifetime risk of breast cancer, with a fourfold increased risk of developing ER‐positive breast cancer compared to women in the middle quintile. 9
Tumour pathology
Histopathology
Invasive carcinoma of no special type (IC‐NST) is the most common histological tumour subtype in hereditary breast cancer. BRCA1‐associated breast carcinomas are typically high‐grade and characteristically exhibit medullary pattern features, including a pushing margin, solid growth, necrosis and prominent lymphocytic infiltrate (Figure 1A); 21 , 128 however, low‐grade subtypes such as tubular carcinoma can rarely be seen. 129 BRCA2‐associated breast cancers are more heterogenous in terms of grade and spectrum of histological tumour subtypes, 130 , 131 , 132 with a larger proportion of tumours being low‐ and intermediate‐grade compared to BRCA1‐associated cancers. Germline CDH1 mutations are specifically associated with ILC (Figure 1B); 23 , 133 however, rare cases of IC‐NST have been described in CDH1 mutation carriers. 134 Distinct genotype–phenotype correlations are less well defined for other breast cancer risk genes. Most breast carcinomas described in the context of Li–Fraumeni syndrome 135 and neurofibromatosis type 1 136 as well as germline PALB2 137 and ATM 138 mutations are high‐grade IC‐NST. Tumours arising in PALB2 mutation carriers were also associated with minimal sclerosis, and this was reported to be predictive of PALB2 mutation status. 137 Breast carcinomas with MMR deficiency are typically high‐grade and are significantly more likely to show solid growth, necrosis, increased mitotic activity and a prominent lymphocytic infiltrate. 96 In contrast, breast carcinomas in Cowden syndrome are more commonly low‐ and intermediate‐grade IC‐NST, 139 , 140 with a proportion of cases exhibiting apocrine differentiation (Figure 1C). 141 Germline CHEK2‐associated breast cancer can be of any grade; the most prevalent c.1100delC variant is associated with IC‐NST142, 143, 144 while p.I157T carrier tumours show an association with lobular differentiation. 145 , 146 The polygenic risk score may also convey subtype‐specific risks; for example, an ILC‐specific predisposition polymorphism has been identified at 7q34 (rs11977670). 147
Figure 1.
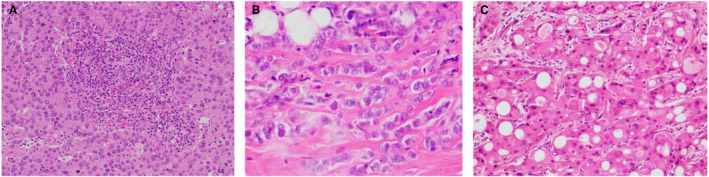
Examples of tumour pathology in hereditary breast cancer. A, BRCA1‐associated invasive carcinoma of no special type with medullary pattern; B, CDH1‐associated breast cancer is characteristically invasive lobular carcinoma; C, invasive carcinoma with apocrine differentiation can be associated with germline PTEN mutations (Cowden syndrome).
Immunohistochemical phenotypes and intrinsic breast cancer subtypes
Triple‐negative
BRCA1‐associated breast cancers are more likely to be ER‐, PR‐ and human epidermal growth factor receptor 2 (HER2)‐negative (‘triple‐negative’) 5 , 148 , 149 and the majority show a ‘basal’ phenotype (expression of high molecular weight cytokeratins such as CK5/6 and CK14 on immunohistochemistry). 150 , 151 , 152 Indeed, more than 60% of tumours arising in the context of BRCA1 mutations are triple‐negative 5 , 130 , 131 and a triple‐negative phenotype is highly predictive of BRCA1 mutation status. 153 Several other susceptibility genes involved in DNA damage repair are also associated with triple‐negative disease, including BRCA2, PALB2, RAD51C, RAD51D and BARD1 variants. 5 , 154 Analysis of the BRIDGES data set found that germline variants in nine of the breast cancer susceptibility genes (BRCA1, BRCA2, PALB2, ATM, CHEK2, RAD51C, RAD51D, BARD1 and TP53) accounted for 27.3% of all triple‐negative breast cancers in women aged 40 years or younger. 5 Breast carcinomas with MMR deficiency were significantly more likely to be ER‐/PR‐negative than MMR‐proficient tumours; however, the difference in HER2 expression was not statistically significant. 96
Hormone receptor‐positive
BRCA2‐associated breast cancers are more frequently ER‐positive and of luminal A or B intrinsic molecular subtype compared to BRCA1‐associated tumours. 151 , 155 Indeed, evaluation of the BRIDGES data set found that while BRCA2 pathogenic variants showed a strong association with the triple‐negative phenotype, they were most highly associated with hormone receptor‐positive, HER2‐negative disease. 5 Furthermore, high‐grade ER‐positive disease was found to be modestly predictive of BRCA2 mutation status, irrespective of age. 153 PALB2 variants are also highly associated with hormone receptor‐positive, HER2‐negative subtypes in addition to triple‐negative disease. 5 ATM variants are most commonly associated with ER‐/PR‐positive, HER2‐negative (luminal B) subtype. 5 , 138 , 156 , 157 CHEK2 variants are associated with all intrinsic breast cancer subtypes except for triple‐negative breast cancer. 5 , 143 , 144 , 145 , 146 Of the few reported breast cancer phenotypes in Peutz–Jeghers syndrome, most comprised ER‐positive, HER2‐negative disease. 158 The polygenic risk score may also inform intrinsic subtype‐specific risks corresponding to ER‐positive versus ER‐negative breast cancers. 9 , 124 , 126
HER2‐positive
TP53 pathogenic variants consistently show a strong association with HER2 positive breast cancer. 5 , 159 , 160 A study of 24 breast carcinomas arising in the context of neurofibromatosis type 1 demonstrated a higher prevalence of ER‐negative, HER2‐positive cases compared to age‐matched controls, particularly in women aged less than 50 years. 136 It should be noted that while BRCA1/2‐associated breast carcinomas were more significantly enriched for other subtypes, a lower but increased risk of HER2‐positive disease was also identified in mutation carriers, while CHEK2 variants showed similarly elevated odds ratios for HER2‐positive and hormone receptor‐positive/HER2‐negative disease. 5
Molecular testing and therapeutic implications
Germline testing
Germline testing detects heritable mutations present in the genome and is typically performed on a blood sample. Guidelines from expert groups such as the National Comprehensive Cancer Council (NCCN) outline criteria for germline testing based on factors such as age at breast cancer diagnosis, family history and tumour characteristics. 161 For example, according to current NCCN guidelines, germline testing is recommended for all women diagnosed with triple‐negative breast cancer as well as for women with ILC who have a personal or family history of diffuse gastric cancer. 161 In England, the National Genomic Test Directory outlines the eligibility criteria for genetic testing in suspected cases of inherited breast cancer. 162 According to the directory, patients who meet the testing criteria for inherited breast and ovarian cancer are eligible for germline BRCA1, BRCA2, PALB2, ATM and CHEK2 testing. In addition, several hereditary breast cancer risk assessment models have been devised, largely aimed at identifying patients with pathogenic variants in BRCA1/2, 163 , 164 , 165 although ongoing revisions of some models have allowed for a more broad assessment of genetic risk. 166 Nonetheless, genetic testing guidelines and risk assessment models may miss a significant proportion of patients with clinically actionable germline susceptibility variants167, 168, 169 and it was found that almost 50% of breast cancer patients with pathogenic or probably pathogenic variants in known susceptibility genes did not qualify for germline testing based on genetic testing guidelines. 167 In view of this, the American Society of Breast Surgeons has recently recommended that germline testing be offered to all breast cancer patients. 170 The increasing availability of NGS, combined with decreased cost and removal of gene patents, has allowed for routine implementation of multigene panel testing among multiple established breast cancer risk genes and the addition of the polygenic risk score, may further refine the accuracy of breast cancer risk stratification in hereditary breast cancer. 171 , 172 , 173 Germline testing results inform screening and risk reducing interventions and have implications for breast cancer management. For example, according to NCCN guidelines, there is currently sufficient evidence to consider risk reducing mastectomy in the setting of germline BRCA1/2, PALB2, TP53 and CDH1 mutations. 161 Furthermore, given that TP53 mutations are associated with an increased susceptibility to radiation‐induced malignancies, 174 identification of germline TP53 mutations modifies screening and management recommendations in order to minimise radiation exposure in this patient cohort. 55 , 175
Somatic testing
Molecular testing can be performed on the tumour to identify clinically actionable somatic driver mutations, treatment resistance‐associated mutations and to assess for gene expression profiles and mutational signatures that have prognostic and therapeutic implications. For example, the commercially available multigene expression assay Oncotype DX evaluates the expression of multiple cancer‐related genes to generate a recurrence risk score which is prognostic for recurrence risk and predictive of adjuvant chemotherapy benefit in early‐stage ER‐positive, HER2‐negative breast cancer. 176 , 177 In the advanced breast cancer setting, somatic testing can be utilised to identify clinically actionable mutations that may inform therapeutic options and clinical trial opportunities. Identification of PIK3CA mutations predicts response to adjuvant treatment with alpelisib in hormone receptor‐positive, HER2‐negative breast cancer, while detection of ESR1 mutations predicts resistance to hormone therapy. 178 Testing for MMR deficiency, MSI status and/or tumour mutation burden (TMB) may have a role in the advanced disease setting as the US Food and Drug Administration (FDA) has approved immunotherapy with pembrolizumab for patients with unresectable or metastatic solid tumours that are MMR‐deficient or TMB‐high and have progressed on initial treatment. 179 Immune check‐point inhibitors are also FDA‐approved for metastatic triple‐negative breast cancers that show evidence of programmed death ligand 1 (PD‐L1) expression on immunohistochemistry. 178 The predictive value of somatic BRCA1/2 mutations has not been definitively established in the breast cancer setting. 180
Homologous recombination deficiency and PARP inhibitors
Poly [adenosine diphosphate (ADP)‐ribose] polymerase (PARP) inhibitors, which inhibit the PARP enzymes from repairing single‐strand DNA breaks, induce synthetic lethality in the presence of HRD. 181 BRCA1/2 proteins are central to the HR repair pathway and impaired BRCA1/2 gene function renders tumour cells susceptible to PARP inhibitor therapy. 182 , 183 , 184 Clinically, adjuvant treatment with PARP inhibitors was associated with improved outcomes in patients with BRCA1/2‐associated breast cancer185, 186, 187, 188, 189 and hereditary breast cancer management guidelines have incorporated PARP inhibitors in the treatment of advanced BRCA1/2‐associated breast cancers and, more recently, in the setting of high‐risk early‐stage HER2‐negative disease. 175 , 190 Germline BRCA1/2 mutation testing is the recommended method of identifying breast cancer patients who may benefit from PARP inhibitor therapy, as there are currently few data on the clinical outcomes of patients with breast cancers harbouring somatic BRCA1/2 mutations. 191 However, a significant proportion of breast cancers may be associated with an HRD phenotype independent of BRCA germline status as a result of germline mutations in other pathway mediators (e.g. PALB2), epigenetic changes (e.g. somatic methylation of BRCA1 promoter), somatic mutational dysregulation of associated genes or, indeed, occurring through no obvious cause. 192 There is emerging evidence that this expanded patient cohort may also benefit from PARP inhibitor therapy. 193 , 194 Several testing methods have been developed to determine HRD status, including germline testing for mutations in HR pathway genes, detection of somatic genomic scars and mutational signatures associated with the HRD phenotype through various tumour‐based genomic analyses, as well as functional assessments of HR repair pathway deficiency. 195 , 196 , 197 , 198 However, it is not yet clear which of these assays is the most reliable method of determining HRD status or the predictive value of HRD biomarkers in identifying breast cancer patients who may benefit from PARP inhibitor therapy.
Summary
Hereditary breast cancer accounts for a substantial proportion of breast cancer cases. Some of the genetic risk can be explained by pathogenic variants in highly and moderately penetrant breast cancer susceptibility genes, several of which are also associated with recognisable cancer predisposition syndromes. Polygenic risk is also an established contributor to breast cancer risk. Characterisation of the genetic basis of breast cancer is central to the understanding of breast cancer biology and behaviour, which informs genetic testing recommendations and has significant implications for accurate risk prediction, patient management and surveillance.
Conflicts of interest
The authors have no conflicts of interest to disclose.
Acknowledgements
The authors are supported by funding from various organisations, including the National Health and Medical Research Council (1113867, 1164770). A.S. is funded by a fellowship from Breast Cancer Trials Australia and New Zealand. Open access publishing facilitated by The University of Queensland, as part of the Wiley ‐ The University of Queensland agreement via the Council of Australian University Librarians.
Data availability statement
Not applicable.
References
- 1. Sung H, Ferlay J, Siegel RL et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021; 71; 209–249. [DOI] [PubMed] [Google Scholar]
- 2. Claus EB, Schildkraut JM, Thompson WD, Risch NJ. The genetic attributable risk of breast and ovarian cancer. Cancer 1996; 77; 2318–2324. [DOI] [PubMed] [Google Scholar]
- 3. Melchor L, Benitez J. The complex genetic landscape of familial breast cancer. Hum. Genet. 2013; 132; 845–863. [DOI] [PubMed] [Google Scholar]
- 4. Stratton MR, Rahman N. The emerging landscape of breast cancer susceptibility. Nat. Genet. 2008; 40; 17–22. [DOI] [PubMed] [Google Scholar]
- 5. Breast Cancer Association C , Mavaddat N, Dorling L et al. Pathology of tumors associated with pathogenic germline variants in 9 breast cancer susceptibility genes. JAMA Oncologia 2022; 8; e216744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Breast Cancer Association C , Dorling L, Carvalho S et al. Breast cancer risk genes ‐ association analysis in more than 113,000 women. N. Engl. J. Med. 2021; 384; 428–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hu C, Hart SN, Gnanaolivu R et al. A population‐based study of genes previously implicated in breast cancer. N. Engl. J. Med. 2021; 384; 440–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Economopoulou P, Dimitriadis G, Psyrri A. Beyond BRCA: New hereditary breast cancer susceptibility genes. Cancer Treat. Rev. 2015; 41; 1–8. [DOI] [PubMed] [Google Scholar]
- 9. Mavaddat N, Michailidou K, Dennis J et al. Polygenic risk scores for prediction of breast cancer and breast cancer subtypes. Am. J. Hum. Genet. 2019; 104; 21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krush AJ. Contributions of Pierre Paul Broca to cancer genetics. Trans. Nebr. Acad. Sci. Affil. Soc. 1979; 316; 125–129. [Google Scholar]
- 11. Smithers DW. Family histories of 459 patients with cancer of the breast. Br. J. Cancer 1948; 2; 163–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Penrose LS, Mackenzie HJ, Karn MN. A genetical study of human mammary cancer. Br. J. Cancer 1948; 2; 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pharoah PD, Day NE, Duffy S, Easton DF, Ponder BA. Family history and the risk of breast cancer: a systematic review and meta‐analysis. Int. J. Cancer 1997; 71; 800–809. [DOI] [PubMed] [Google Scholar]
- 14. Nelson HD, Zakher B, Cantor A et al. Risk factors for breast cancer for women aged 40 to 49 years: a systematic review and meta‐analysis. Ann. Intern. Med. 2012; 156; 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman B, Austin MA, Lee M, King MC. Inheritance of human breast cancer: evidence for autosomal dominant transmission in high‐risk families. Proc. Natl. Acad. Sci. U. S. A. 1988; 85; 3044–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hall JM, Lee MK, Newman B et al. Linkage of early‐onset familial breast cancer to chromosome 17q21. Science 1990; 250; 1684–1689. [DOI] [PubMed] [Google Scholar]
- 17. Friedman LS, Ostermeyer EA, Szabo CI et al. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet. 1994; 8; 399–404. [DOI] [PubMed] [Google Scholar]
- 18. Wooster R, Bignell G, Lancaster J et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995; 378; 789–792. [DOI] [PubMed] [Google Scholar]
- 19. Wooster R, Neuhausen SL, Mangion J et al. Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12‐13. Science 1994; 265; 2088–2090. [DOI] [PubMed] [Google Scholar]
- 20. Pathology of familial breast cancer . Differences between breast cancers in carriers of BRCA1 or BRCA2 mutations and sporadic cases. Breast Cancer Linkage Consortium. Lancet 1997; 349; 1505–1510. [PubMed] [Google Scholar]
- 21. Lakhani SR, Jacquemier J, Sloane JP et al. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J. Natl. Cancer Inst. 1998; 90; 1138–1145. [DOI] [PubMed] [Google Scholar]
- 22. Malkin D, Li FP, Strong LC et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250; 1233–1238. [DOI] [PubMed] [Google Scholar]
- 23. Pharoah PD, Guilford P, Caldas C, International Gastric Cancer Linkage C . Incidence of gastric cancer and breast cancer in CDH1 (E‐cadherin) mutation carriers from hereditary diffuse gastric cancer families. Gastroenterology 2001; 121; 1348–1353. [DOI] [PubMed] [Google Scholar]
- 24. Liaw D, Marsh DJ, Li J et al. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat. Genet. 1997; 16; 64–67. [DOI] [PubMed] [Google Scholar]
- 25. Boardman LA, Thibodeau SN, Schaid DJ et al. Increased risk for cancer in patients with the Peutz‐Jeghers syndrome. Ann. Intern. Med. 1998; 128; 896–899. [DOI] [PubMed] [Google Scholar]
- 26. Uusitalo E, Rantanen M, Kallionpaa RA et al. Distinctive cancer associations in patients with Neurofibromatosis type 1. J. Clin. Oncol. 2016; 34; 1978–1986. [DOI] [PubMed] [Google Scholar]
- 27. Renwick A, Thompson D, Seal S et al. ATM mutations that cause ataxia‐telangiectasia are breast cancer susceptibility alleles. Nat. Genet. 2006; 38; 873–875. [DOI] [PubMed] [Google Scholar]
- 28. Meijers‐Heijboer H, van den Ouweland A, Klijn J et al. Low‐penetrance susceptibility to breast cancer due to CHEK2(*)1100delC in noncarriers of BRCA1 or BRCA2 mutations. Nat. Genet. 2002; 31; 55–59. [DOI] [PubMed] [Google Scholar]
- 29. Rahman N, Seal S, Thompson D et al. PALB2, which encodes a BRCA2‐interacting protein, is a breast cancer susceptibility gene. Nat. Genet. 2007; 39; 165–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ghimenti C, Sensi E, Presciuttini S et al. Germline mutations of the BRCA1‐associated ring domain (BARD1) gene in breast and breast/ovarian families negative for BRCA1 and BRCA2 alterations. Genes Chromosomes Cancer 2002; 33; 235–242. [DOI] [PubMed] [Google Scholar]
- 31. Meindl A, Hellebrand H, Wiek C et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat. Genet. 2010; 42; 410–414. [DOI] [PubMed] [Google Scholar]
- 32. Chen X, Li Y, Ouyang T et al. Associations between RAD51D germline mutations and breast cancer risk and survival in BRCA1/2‐negative breast cancers. Ann. Oncol. 2018; 29; 2046–2051. [DOI] [PubMed] [Google Scholar]
- 33. Stacey SN, Manolescu A, Sulem P et al. Common variants on chromosomes 2q35 and 16q12 confer susceptibility to estrogen receptor‐positive breast cancer. Nat. Genet. 2007; 39; 865–869. [DOI] [PubMed] [Google Scholar]
- 34. Easton DF, Pooley KA, Dunning AM et al. Genome‐wide association study identifies novel breast cancer susceptibility loci. Nature 2007; 447; 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hunter DJ, Kraft P, Jacobs KB et al. A genome‐wide association study identifies alleles in FGFR2 associated with risk of sporadic postmenopausal breast cancer. Nat. Genet. 2007; 39; 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wendt C, Margolin S. Identifying breast cancer susceptibility genes ‐ a review of the genetic background in familial breast cancer. Acta Oncol. 2019; 58; 135–146. [DOI] [PubMed] [Google Scholar]
- 37. WHO Classification of Tumours Editorial Board . WHO classification of Tumours, breast Tumours. 5th ed. Lyon: IARC Press, 2019. [Google Scholar]
- 38. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell 2002; 108; 171–182. [DOI] [PubMed] [Google Scholar]
- 39. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: Different roles in a common pathway of genome protection. Nat. Rev. Cancer 2011; 12; 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chen S, Parmigiani G. Meta‐analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007; 25; 1329–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuchenbaecker KB, Hopper JL, Barnes DR et al. Risks of breast, ovarian, and contralateral breast cancer for BRCA1 and BRCA2 mutation carriers. JAMA 2017; 317; 2402–2416. [DOI] [PubMed] [Google Scholar]
- 42. Mavaddat N, Peock S, Frost D et al. Cancer risks for BRCA1 and BRCA2 mutation carriers: results from prospective analysis of EMBRACE. J. Natl. Cancer Inst. 2013; 105; 812–822. [DOI] [PubMed] [Google Scholar]
- 43. Mersch J, Jackson MA, Park M et al. Cancers associated with BRCA1 and BRCA2 mutations other than breast and ovarian. Cancer 2015; 121; 269–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Thompson D, Easton DF, Breast Cancer Linkage C . Cancer incidence in BRCA1 mutation carriers. J. Natl. Cancer Inst. 2002; 94; 1358–1365. [DOI] [PubMed] [Google Scholar]
- 45. Iqbal J, Ragone A, Lubinski J et al. The incidence of pancreatic cancer in BRCA1 and BRCA2 mutation carriers. Br. J. Cancer 2012; 107; 2005–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Breast Cancer Linkage C . Cancer risks in BRCA2 mutation carriers. J. Natl. Cancer Inst. 1999; 91; 1310–1316. [DOI] [PubMed] [Google Scholar]
- 47. Tai YC, Domchek S, Parmigiani G, Chen S. Breast cancer risk among male BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2007; 99; 1811–1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chamseddine RS, Wang C, Yin K et al. Penetrance of male breast cancer susceptibility genes: a systematic review. Breast Cancer Res. Treat. 2022; 191; 31–38. [DOI] [PubMed] [Google Scholar]
- 49. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408; 307–310. [DOI] [PubMed] [Google Scholar]
- 50. Engeland K. Cell cycle regulation: p53‐p21‐RB signaling. Cell Death Differ. 2022; 29; 946–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guha T, Malkin D. Inherited TP53 mutations and the Li‐Fraumeni syndrome. Cold Spring Harb. Perspect. Med. 2017; 7; a026187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Birch JM, Alston RD, McNally RJ et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene 2001; 20; 4621–4628. [DOI] [PubMed] [Google Scholar]
- 53. Bougeard G, Renaux‐Petel M, Flaman JM et al. Revisiting Li‐Fraumeni syndrome from TP53 mutation carriers. J. Clin. Oncol. 2015; 33; 2345–2352. [DOI] [PubMed] [Google Scholar]
- 54. Amadou A, Achatz MIW, Hainaut P. Revisiting tumor patterns and penetrance in germline TP53 mutation carriers: Temporal phases of Li–Fraumeni syndrome. Curr. Opin. Oncol. 2018; 30; 23–29. [DOI] [PubMed] [Google Scholar]
- 55. McBride KA, Ballinger ML, Killick E et al. Li–Fraumeni syndrome: cancer risk assessment and clinical management. Nat. Rev. Clin. Oncol. 2014; 11; 260–271. [DOI] [PubMed] [Google Scholar]
- 56. Mai PL, Best AF, Peters JA et al. Risks of first and subsequent cancers among TP53 mutation carriers in the National Cancer Institute Li–Fraumeni syndrome cohort. Cancer 2016; 122; 3673–3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Olivier M, Goldgar DE, Sodha N et al. Li‐Fraumeni and related syndromes: correlation between tumor type, family structure, and TP53 genotype. Cancer Res. 2003; 63; 6643–6650. [PubMed] [Google Scholar]
- 58. Schon K, Tischkowitz M. Clinical implications of germline mutations in breast cancer: TP53. Breast Cancer Res. Treat. 2018; 167; 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hobert JA, Eng C. PTEN hamartoma tumor syndrome: an overview. Genet. Med. 2009; 11; 687–694. [DOI] [PubMed] [Google Scholar]
- 60. Pilarski R, Burt R, Kohlman W, Pho L, Shannon KM, Swisher E. Cowden syndrome and the PTEN hamartoma tumor syndrome: systematic review and revised diagnostic criteria. J. Natl. Cancer Inst. 2013; 105; 1607–1616. [DOI] [PubMed] [Google Scholar]
- 61. Ngeow J, Sesock K, Eng C. Clinical implications for germline PTEN Spectrum disorders. Endocrinol. Metab. Clin. North Am. 2017; 46; 503–517. [DOI] [PubMed] [Google Scholar]
- 62. Ngeow J, Sesock K, Eng C. Breast cancer risk and clinical implications for germline PTEN mutation carriers. Breast Cancer Res. Treat. 2017; 165; 1–8. [DOI] [PubMed] [Google Scholar]
- 63. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. Lifetime cancer risks in individuals with germline PTEN mutations. Clin. Cancer Res. 2012; 18; 400–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pilarski R. Cowden syndrome: a critical review of the clinical literature. J. Genet. Couns. 2009; 18; 13–27. [DOI] [PubMed] [Google Scholar]
- 65. Hemminki A, Markie D, Tomlinson I et al. A serine/threonine kinase gene defective in Peutz‐Jeghers syndrome. Nature 1998; 391; 184–187. [DOI] [PubMed] [Google Scholar]
- 66. Beggs AD, Latchford AR, Vasen HF et al. Peutz‐Jeghers syndrome: a systematic review and recommendations for management. Gut 2010; 59; 975–986. [DOI] [PubMed] [Google Scholar]
- 67. Giardiello FM, Brensinger JD, Tersmette AC et al. Very high risk of cancer in familial Peutz‐Jeghers syndrome. Gastroenterology 2000; 119; 1447–1453. [DOI] [PubMed] [Google Scholar]
- 68. van Lier MG, Westerman AM, Wagner A et al. High cancer risk and increased mortality in patients with Peutz‐Jeghers syndrome. Gut 2011; 60; 141–147. [DOI] [PubMed] [Google Scholar]
- 69. Lim W, Olschwang S, Keller JJ et al. Relative frequency and morphology of cancers in STK11 mutation carriers. Gastroenterology 2004; 126; 1788–1794. [DOI] [PubMed] [Google Scholar]
- 70. Hearle N, Schumacher V, Menko FH et al. Frequency and spectrum of cancers in the Peutz‐Jeghers syndrome. Clin. Cancer Res. 2006; 12; 3209–3215. [DOI] [PubMed] [Google Scholar]
- 71. Guilford P, Hopkins J, Harraway J et al. E‐cadherin germline mutations in familial gastric cancer. Nature 1998; 392; 402–405. [DOI] [PubMed] [Google Scholar]
- 72. Berx G, Staes K, van Hengel J et al. Cloning and characterization of the human invasion suppressor gene E‐cadherin (CDH1). Genomics 1995; 26; 281–289. [DOI] [PubMed] [Google Scholar]
- 73. Hansford S, Kaurah P, Li‐Chang H et al. Hereditary diffuse gastric cancer syndrome: CDH1 mutations and beyond. JAMA Oncol. 2015; 1; 23–32. [DOI] [PubMed] [Google Scholar]
- 74. Xicola RM, Li S, Rodriguez N et al. Clinical features and cancer risk in families with pathogenic CDH1 variants irrespective of clinical criteria. J. Med. Genet. 2019; 56; 838–843. [DOI] [PubMed] [Google Scholar]
- 75. Benusiglio PR, Malka D, Rouleau E et al. CDH1 germline mutations and the hereditary diffuse gastric and lobular breast cancer syndrome: a multicentre study. J. Med. Genet. 2013; 50; 486–489. [DOI] [PubMed] [Google Scholar]
- 76. Corso G, De Scalzi A, Feroce I, Veronesi P, Bonanni B, Galimberti V. Clinical criteria revision for hereditary lobular breast cancer associated with E‐cadherin germline mutations. Per Med. 2018; 15; 153–155. [DOI] [PubMed] [Google Scholar]
- 77. Corso G. Pleiotropic cancer manifestations of germline CDH1 mutations: Risks and management. J. Surg. Oncol. 2022; 125; 1326–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Corso G, Figueiredo J, La Vecchia C et al. Hereditary lobular breast cancer with an emphasis on E‐cadherin genetic defect. J. Med. Genet. 2018; 55; 431–441. [DOI] [PubMed] [Google Scholar]
- 79. Gamble LA, Rossi A, Fasaye GA et al. Association between hereditary lobular breast cancer due to CDH1 variants and gastric cancer risk. JAMA Surg. 2022; 157; 18–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. D'Angelo FRA, Cardella S, Antolino L et al. Breast cancer metastasis to the stomach. J Cancer Metastasis Treat. 2019; 5; 30. [Google Scholar]
- 81. Chu PG, Weiss LM. Immunohistochemical characterization of signet‐ring cell carcinomas of the stomach, breast, and colon. Am. J. Clin. Pathol. 2004; 121; 884–892. [DOI] [PubMed] [Google Scholar]
- 82. Yim K, Ro SM, Lee J. Breast cancer metastasizing to the stomach mimicking primary gastric cancer: a case report. World J. Gastroenterol. 2017; 23; 2251–2257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Gutmann DH, Wood DL, Collins FS. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. U. S. A. 1991; 88; 9658–9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Howell SJ, Hockenhull K, Salih Z, Evans DG. Increased risk of breast cancer in neurofibromatosis type 1: current insights. Breast Cancer (Dove Med Press). 2017; 9; 531–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gutmann DH, Ferner RE, Listernick RH, Korf BR, Wolters PL, Johnson KJ. Neurofibromatosis type 1. Nat. Rev. Dis. Primers. 2017; 3; 17004. [DOI] [PubMed] [Google Scholar]
- 86. Wang X, Teer JK, Tousignant RN et al. Breast cancer risk and germline genomic profiling of women with neurofibromatosis type 1 who developed breast cancer. Genes Chromosomes Cancer 2018; 57; 19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Cerretelli G, Ager A, Arends MJ, Frayling IM. Molecular pathology of lynch syndrome. J. Pathol. 2020; 250; 518–531. [DOI] [PubMed] [Google Scholar]
- 88. Win AK, Young JP, Lindor NM et al. Colorectal and other cancer risks for carriers and noncarriers from families with a DNA mismatch repair gene mutation: a prospective cohort study. J. Clin. Oncol. 2012; 30; 958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Moller P, Seppala TT, Bernstein I et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the prospective lynch syndrome database. Gut 2018; 67; 1306–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Harkness EF, Barrow E, Newton K et al. Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: a cohort study. J. Med. Genet. 2015; 52; 553–556. [DOI] [PubMed] [Google Scholar]
- 91. Goldberg M, Bell K, Aronson M et al. Association between the lynch syndrome gene MSH2 and breast cancer susceptibility in a Canadian familial cancer registry. J. Med. Genet. 2017; 54; 742–746. [DOI] [PubMed] [Google Scholar]
- 92. Roberts ME, Jackson SA, Susswein LR et al. MSH6 and PMS2 germ‐line pathogenic variants implicated in lynch syndrome are associated with breast cancer. Genet. Med. 2018; 20; 1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Moller P, Seppala T, Bernstein I et al. Cancer incidence and survival in lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective lynch syndrome database. Gut 2017; 66; 464–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Moller P, Seppala T, Bernstein I et al. Incidence of and survival after subsequent cancers in carriers of pathogenic MMR variants with previous cancer: a report from the prospective lynch syndrome database. Gut 2017; 66; 1657–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Davies H, Morganella S, Purdie CA et al. Whole‐genome sequencing reveals breast cancers with mismatch repair deficiency. Cancer Res. 2017; 77; 4755–4762. [DOI] [PubMed] [Google Scholar]
- 96. Walsh MD, Buchanan DD, Cummings MC et al. Lynch syndrome‐associated breast cancers: clinicopathologic characteristics of a case series from the colon cancer family registry. Clin. Cancer Res. 2010; 16; 2214–2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Sung P, Klein H. Mechanism of homologous recombination: mediators and helicases take on regulatory functions. Nat. Rev. Mol. Cell Biol. 2006; 7; 739–750. [DOI] [PubMed] [Google Scholar]
- 98. Krejci L, Altmannova V, Spirek M, Zhao X. Homologous recombination and its regulation. Nucleic Acids Res. 2012; 40; 5795–5818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Stewart MD, Merino Vega D, Arend RC et al. Homologous recombination deficiency: concepts, definitions, and assays. Oncologist 2022; 27; 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Xia B, Sheng Q, Nakanishi K et al. Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2. Mol. Cell 2006; 22; 719–729. [DOI] [PubMed] [Google Scholar]
- 101. Sy SM, Huen MS, Chen J. PALB2 is an integral component of the BRCA complex required for homologous recombination repair. Proc. Natl. Acad. Sci. U. S. A. 2009; 106; 7155–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Zhang F, Ma J, Wu J et al. PALB2 links BRCA1 and BRCA2 in the DNA‐damage response. Curr. Biol. 2009; 19; 524–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Reid S, Schindler D, Hanenberg H et al. Biallelic mutations in PALB2 cause Fanconi anemia subtype FA‐N and predispose to childhood cancer. Nat. Genet. 2007; 39; 162–164. [DOI] [PubMed] [Google Scholar]
- 104. Casadei S, Norquist BM, Walsh T et al. Contribution of inherited mutations in the BRCA2‐interacting protein PALB2 to familial breast cancer. Cancer Res. 2011; 71; 2222–2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yang X, Leslie G, Doroszuk A et al. Cancer risks associated with germline PALB2 pathogenic variants: an international study of 524 families. J. Clin. Oncol. 2020; 38; 674–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Jones S, Hruban RH, Kamiyama M et al. Exomic sequencing identifies PALB2 as a pancreatic cancer susceptibility gene. Science 2009; 324; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Antoniou AC, Casadei S, Heikkinen T et al. Breast‐cancer risk in families with mutations in PALB2. N. Engl. J. Med. 2014; 371; 497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Norquist BM, Harrell MI, Brady MF et al. Inherited mutations in women with ovarian carcinoma. JAMA Oncol. 2016; 2; 482–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Janatova M, Kleibl Z, Stribrna J et al. The PALB2 gene is a strong candidate for clinical testing in BRCA1‐ and BRCA2‐negative hereditary breast cancer. Cancer Epidemiol. Biomarkers Prev. 2013; 22; 2323–2332. [DOI] [PubMed] [Google Scholar]
- 110. Ambrose M, Gatti RA. Pathogenesis of ataxia‐telangiectasia: the next generation of ATM functions. Blood 2013; 121; 4036–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Gumy‐Pause F, Wacker P, Sappino AP. ATM gene and lymphoid malignancies. Leukemia 2004; 18; 238–242. [DOI] [PubMed] [Google Scholar]
- 112. van Os NJ, Roeleveld N, Weemaes CM et al. Health risks for ataxia‐telangiectasia mutated heterozygotes: a systematic review, meta‐analysis and evidence‐based guideline. Clin. Genet. 2016; 90; 105–117. [DOI] [PubMed] [Google Scholar]
- 113. Marabelli M, Cheng SC, Parmigiani G. Penetrance of ATM gene mutations in breast cancer: a meta‐analysis of different measures of risk. Genet. Epidemiol. 2016; 40; 425–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Stracker TH, Usui T, Petrini JH. Taking the time to make important decisions: the checkpoint effector kinases Chk1 and Chk2 and the DNA damage response. DNA Repair (Amst). 2009; 8; 1047–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Naslund‐Koch C, Nordestgaard BG, Bojesen SE. Increased risk for other cancers in addition to breast cancer for CHEK2*1100delC heterozygotes estimated from the Copenhagen general population study. J. Clin. Oncol. 2016; 34; 1208–1216. [DOI] [PubMed] [Google Scholar]
- 116. Weischer M, Bojesen SE, Tybjaerg‐Hansen A, Axelsson CK, Nordestgaard BG. Increased risk of breast cancer associated with CHEK2*1100delC. J. Clin. Oncol. 2007; 25; 57–63. [DOI] [PubMed] [Google Scholar]
- 117. Cybulski C, Gorski B, Huzarski T et al. CHEK2 is a multiorgan cancer susceptibility gene. Am. J. Hum. Genet. 2004; 75; 1131–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Havranek O, Kleiblova P, Hojny J et al. Association of Germline CHEK2 gene variants with risk and prognosis of non‐Hodgkin lymphoma. PLoS One. 2015; 10; e0140819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Nevanlinna H, Bartek J. The CHEK2 gene and inherited breast cancer susceptibility. Oncogene 2006; 25; 5912–5919. [DOI] [PubMed] [Google Scholar]
- 120. Weischer M, Bojesen SE, Ellervik C, Tybjaerg‐Hansen A, Nordestgaard BG. CHEK2*1100delC genotyping for clinical assessment of breast cancer risk: Meta‐analyses of 26,000 patient cases and 27,000 controls. J. Clin. Oncol. 2008; 26; 542–548. [DOI] [PubMed] [Google Scholar]
- 121. Cybulski C, Wokolorczyk D, Jakubowska A et al. Risk of breast cancer in women with a CHEK2 mutation with and without a family history of breast cancer. J. Clin. Oncol. 2011; 29; 3747–3752. [DOI] [PubMed] [Google Scholar]
- 122. Stella S, Martorana F, Manzella L, Vigneri P. The other side of the coin: dissecting molecular mechanisms behind hereditary breast cancer in search of therapeutic opportunities. Transl Oncol. 2021; 14; 101104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Shiovitz S, Korde LA. Genetics of breast cancer: a topic in evolution. Ann. Oncol. 2015; 26; 1291–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Michailidou K, Lindstrom S, Dennis J et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017; 551; 92–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Michailidou K, Beesley J, Lindstrom S et al. Genome‐wide association analysis of more than 120,000 individuals identifies 15 new susceptibility loci for breast cancer. Nat. Genet. 2015; 47; 373–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Milne RL, Kuchenbaecker KB, Michailidou K et al. Identification of ten variants associated with risk of estrogen‐receptor‐negative breast cancer. Nat. Genet. 2017; 49; 1767–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sawyer S, Mitchell G, McKinley J et al. A role for common genomic variants in the assessment of familial breast cancer. J. Clin. Oncol. 2012; 30; 4330–4336. [DOI] [PubMed] [Google Scholar]
- 128. Da Silva L, Lakhani SR. Pathology of hereditary breast cancer. Mod. Pathol. 2010; 23(Suppl 2); S46–S51. [DOI] [PubMed] [Google Scholar]
- 129. Rijnsburger AJ, Obdeijn IM, Kaas R et al. BRCA1‐associated breast cancers present differently from BRCA2‐associated and familial cases: long‐term follow‐up of the Dutch MRISC screening study. J. Clin. Oncol. 2010; 28; 5265–5273. [DOI] [PubMed] [Google Scholar]
- 130. Mavaddat N, Barrowdale D, Andrulis IL et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: results from the consortium of investigators of modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomarkers Prev. 2012; 21; 134–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Fountzilas E, Konstantopoulou I, Vagena A et al. Pathology of BRCA1‐ and BRCA2‐associated breast cancers: known and less known connections. Clin. Breast Cancer 2020; 20; 152–159. [DOI] [PubMed] [Google Scholar]
- 132. Palacios J, Honrado E, Osorio A et al. Immunohistochemical characteristics defined by tissue microarray of hereditary breast cancer not attributable to BRCA1 or BRCA2 mutations: differences from breast carcinomas arising in BRCA1 and BRCA2 mutation carriers. Clin. Cancer Res. 2003; 9(10 Pt 1); 3606–3614. [PubMed] [Google Scholar]
- 133. Adib E, El Zarif T, Nassar AH et al. CDH1 germline variants are enriched in patients with colorectal cancer, gastric cancer, and breast cancer. Br. J. Cancer 2022; 126; 797–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhu ZG, Yu YY, Zhang Y et al. Germline mutational analysis of CDH1 and pathologic features in familial cancer syndrome with diffuse gastric cancer/breast cancer proband in a Chinese family. Eur. J. Surg. Oncol. 2004; 30; 531–535. [DOI] [PubMed] [Google Scholar]
- 135. Masciari S, Dillon DA, Rath M et al. Breast cancer phenotype in women with TP53 germline mutations: a Li–Fraumeni syndrome consortium effort. Breast Cancer Res. Treat. 2012; 133; 1125–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Uusitalo E, Kallionpaa RA, Kurki S et al. Breast cancer in neurofibromatosis type 1: overrepresentation of unfavourable prognostic factors. Br. J. Cancer 2017; 116; 211–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Teo ZL, Provenzano E, Dite GS et al. Tumour morphology predicts PALB2 germline mutation status. Br. J. Cancer 2013; 109; 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Balleine RL, Murali R, Bilous AM et al. Histopathological features of breast cancer in carriers of ATM gene variants. Histopathology 2006; 49; 523–532. [DOI] [PubMed] [Google Scholar]
- 139. Schrager CA, Schneider D, Gruener AC, Tsou HC, Peacocke M. Clinical and pathological features of breast disease in Cowden's syndrome: an underrecognized syndrome with an increased risk of breast cancer. Hum. Pathol. 1998; 29; 47–53. [DOI] [PubMed] [Google Scholar]
- 140. Sabate JM, Gomez A, Torrubia S et al. Evaluation of breast involvement in relation to Cowden syndrome: a radiological and clinicopathological study of patients with PTEN germ‐line mutations. Eur. Radiol. 2006; 16; 702–706. [DOI] [PubMed] [Google Scholar]
- 141. Banneau G, Guedj M, MacGrogan G et al. Molecular apocrine differentiation is a common feature of breast cancer in patients with germline PTEN mutations. Breast Cancer Res. 2010; 12; R63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Weischer M, Nordestgaard BG, Pharoah P et al. CHEK2*1100delC heterozygosity in women with breast cancer associated with early death, breast cancer‐specific death, and increased risk of a second breast cancer. J. Clin. Oncol. 2012; 30; 4308–4316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Schmidt MK, Tollenaar RA, de Kemp SR et al. Breast cancer survival and tumor characteristics in premenopausal women carrying the CHEK2*1100delC germline mutation. J. Clin. Oncol. 2007; 25; 64–69. [DOI] [PubMed] [Google Scholar]
- 144. de Bock GH, Schutte M, Krol‐Warmerdam EM et al. Tumour characteristics and prognosis of breast cancer patients carrying the germline CHEK2*1100delC variant. J. Med. Genet. 2004; 41; 731–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Muranen TA, Blomqvist C, Dork T et al. Patient survival and tumor characteristics associated with CHEK2:P.I157T ‐ findings from the breast cancer association consortium. Breast Cancer Res. 2016; 18; 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Huzarski T, Cybulski C, Domagala W et al. Pathology of breast cancer in women with constitutional CHEK2 mutations. Breast Cancer Res. Treat. 2005; 90; 187–189. [DOI] [PubMed] [Google Scholar]
- 147. Sawyer E, Roylance R, Petridis C et al. Genetic predisposition to in situ and invasive lobular carcinoma of the breast. PLoS Genet. 2014; 10; e1004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Chen H, Wu J, Zhang Z et al. Association between BRCA status and triple‐negative breast cancer: a meta‐analysis. Front. Pharmacol. 2018; 9; 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Lakhani SR, Van De Vijver MJ, Jacquemier J et al. The pathology of familial breast cancer: predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER‐2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002; 20; 2310–2318. [DOI] [PubMed] [Google Scholar]
- 150. Foulkes WD, Stefansson IM, Chappuis PO et al. Germline BRCA1 mutations and a basal epithelial phenotype in breast cancer. J. Natl. Cancer Inst. 2003; 95; 1482–1485. [DOI] [PubMed] [Google Scholar]
- 151. Sonderstrup IMH, Jensen MR, Ejlertsen B et al. Subtypes in BRCA‐mutated breast cancer. Hum. Pathol. 2019; 84; 192–201. [DOI] [PubMed] [Google Scholar]
- 152. Lakhani SR, Reis‐Filho JS, Fulford L et al. Prediction of BRCA1 status in patients with breast cancer using estrogen receptor and basal phenotype. Clin. Cancer Res. 2005; 11; 5175–5180. [DOI] [PubMed] [Google Scholar]
- 153. Spurdle AB, Couch FJ, Parsons MT et al. Refined histopathological predictors of BRCA1 and BRCA2 mutation status: a large‐scale analysis of breast cancer characteristics from the BCAC, CIMBA, and ENIGMA consortia. Breast Cancer Res. 2014; 16; 3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Heikkinen T, Karkkainen H, Aaltonen K et al. The breast cancer susceptibility mutation PALB2 1592delT is associated with an aggressive tumor phenotype. Clin. Cancer Res. 2009; 15; 3214–3222. [DOI] [PubMed] [Google Scholar]
- 155. Waddell N, Arnold J, Cocciardi S et al. Subtypes of familial breast tumours revealed by expression and copy number profiling. Breast Cancer Res. Treat. 2010; 123; 661–677. [DOI] [PubMed] [Google Scholar]
- 156. Weigelt B, Bi R, Kumar R et al. The landscape of somatic genetic alterations in breast cancers from ATM germline mutation carriers. J. Natl. Cancer Inst. 2018; 110; 1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Renault AL, Mebirouk N, Fuhrmann L et al. Morphology and genomic hallmarks of breast tumours developed by ATM deleterious variant carriers. Breast Cancer Res. 2018; 20; 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Lipsa A, Kowtal P, Sarin R. Novel germline STK11 variants and breast cancer phenotype identified in an Indian cohort of Peutz‐Jeghers syndrome. Hum. Mol. Genet. 2019; 28; 1885–1893. [DOI] [PubMed] [Google Scholar]
- 159. Wilson JR, Bateman AC, Hanson H et al. A novel HER2‐positive breast cancer phenotype arising from germline TP53 mutations. J. Med. Genet. 2010; 47; 771–774. [DOI] [PubMed] [Google Scholar]
- 160. Melhem‐Bertrandt A, Bojadzieva J, Ready KJ et al. Early onset HER2‐positive breast cancer is associated with germline TP53 mutations. Cancer 2012; 118; 908–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology . Genetic/Familial High‐Risk Assessment: Breast, Ovarian, and Pancreatic. Version 2. Accessed 26 June 2022. Available from: https://www.nccn.org/professionals/physician_gls/pdf/genetics_bop.pdf. [DOI] [PubMed]
- 162. National Genomic Test Directory . Testing Criteria for Rare and Inherited Disease. Version 3.1. Accessed 29 August 2022. Available from: https://www.england.nhs.uk/wp‐content/uploads/2018/08/Rare‐and‐inherited‐disease‐eligibility‐criteria‐version‐3.1‐August‐2022.pdf.
- 163. Antoniou AC, Pharoah PD, McMullan G et al. A comprehensive model for familial breast cancer incorporating BRCA1, BRCA2 and other genes. Br. J. Cancer 2002; 86; 76–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Hoskins KF, Zwaagstra A, Ranz M. Validation of a tool for identifying women at high risk for hereditary breast cancer in population‐based screening. Cancer 2006; 107; 1769–1776. [DOI] [PubMed] [Google Scholar]
- 165. Bellcross CA, Lemke AA, Pape LS, Tess AL, Meisner LT. Evaluation of a breast/ovarian cancer genetics referral screening tool in a mammography population. Genet. Med. 2009; 11; 783–789. [DOI] [PubMed] [Google Scholar]
- 166. Carver T, Hartley S, Lee A et al. CanRisk tool‐a web Interface for the prediction of breast and ovarian cancer risk and the likelihood of carrying genetic pathogenic variants. Cancer Epidemiol. Biomarkers Prev. 2021; 30; 469–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Beitsch PD, Whitworth PW, Hughes K et al. Underdiagnosis of hereditary breast cancer: Are genetic testing guidelines a tool or an obstacle? J. Clin. Oncol. 2019; 37; 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. O'Leary E, Iacoboni D, Holle J et al. Expanded gene panel use for women with breast cancer: Identification and intervention beyond breast cancer risk. Ann. Surg. Oncol. 2017; 24; 3060–3066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Yang S, Axilbund JE, O'Leary E et al. Underdiagnosis of hereditary breast and ovarian cancer in Medicare patients: genetic testing criteria miss the mark. Ann. Surg. Oncol. 2018; 25; 2925–2931. [DOI] [PubMed] [Google Scholar]
- 170. Consensus Guideline on Genetic Testing for Hereditary Breast Cancer. Accessed 10 July 2022. Available from: https://www.breastsurgeons.org/docs/statements/Consensus‐Guideline‐on‐Genetic‐Testing‐for‐Hereditary‐Breast‐Cancer.pdf.
- 171. Kuchenbaecker KB, McGuffog L, Barrowdale D et al. Evaluation of polygenic risk scores for breast and ovarian cancer risk prediction in BRCA1 and BRCA2 mutation carriers. J. Natl. Cancer Inst. 2017; 109; djw302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Muranen TA, Greco D, Blomqvist C et al. Genetic modifiers of CHEK2*1100delC‐associated breast cancer risk. Genet. Med. 2017; 19; 599–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173. Mars N, Widen E, Kerminen S et al. The role of polygenic risk and susceptibility genes in breast cancer over the course of life. Nat. Commun. 2020; 11; 6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Heymann S, Delaloge S, Rahal A et al. Radio‐induced malignancies after breast cancer postoperative radiotherapy in patients with Li–Fraumeni syndrome. Radiat. Oncol. 2010; 5; 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Tung NM, Boughey JC, Pierce LJ et al. Management of Hereditary Breast Cancer: American Society of Clinical Oncology, American Society for Radiation Oncology, and Society of Surgical Oncology guideline. J. Clin. Oncol. 2020; 38; 2080–2106. [DOI] [PubMed] [Google Scholar]
- 176. Paik S, Shak S, Tang G et al. A multigene assay to predict recurrence of tamoxifen‐treated, node‐negative breast cancer. N. Engl. J. Med. 2004; 351; 2817–2826. [DOI] [PubMed] [Google Scholar]
- 177. Paik S, Tang G, Shak S et al. Gene expression and benefit of chemotherapy in women with node‐negative, estrogen receptor‐positive breast cancer. J. Clin. Oncol. 2006; 24; 3726–3734. [DOI] [PubMed] [Google Scholar]
- 178. Sun L, Wu A, Bean GR, Hagemann IS, Lin CY. Molecular testing in breast cancer: current status and future directions. J. Mol. Diagn. 2021; 23; 1422–1432. [DOI] [PubMed] [Google Scholar]
- 179. Chakravarty D, Johnson A, Sklar J et al. Somatic genomic testing in patients with metastatic or advanced cancer: ASCO provisional clinical opinion. J. Clin. Oncol. 2022; 40; 1231–1258. [DOI] [PubMed] [Google Scholar]
- 180. Kwon MJ. Predictive biomarkers for molecularly targeted therapies and immunotherapies in breast cancer. Arch. Pharm. Res. 2022; 45; 597–617. [DOI] [PubMed] [Google Scholar]
- 181. Lord CJ, Ashworth A. Targeted therapy for cancer using PARP inhibitors. Curr. Opin. Pharmacol. 2008; 8; 363–369. [DOI] [PubMed] [Google Scholar]
- 182. Fong PC, Boss DS, Yap TA et al. Inhibition of poly(ADP‐ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009; 361; 123–134. [DOI] [PubMed] [Google Scholar]
- 183. Farmer H, McCabe N, Lord CJ et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434; 917–921. [DOI] [PubMed] [Google Scholar]
- 184. Ashworth A. A synthetic lethal therapeutic approach: poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double‐strand break repair. J. Clin. Oncol. 2008; 26; 3785–3790. [DOI] [PubMed] [Google Scholar]
- 185. Tutt A, Robson M, Garber JE et al. Oral poly(ADP‐ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof‐of‐concept trial. Lancet 2010; 376; 235–244. [DOI] [PubMed] [Google Scholar]
- 186. Tutt ANJ, Garber JE, Kaufman B et al. Adjuvant Olaparib for patients with BRCA1‐ or BRCA2‐mutated breast cancer. N. Engl. J. Med. 2021; 384; 2394–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Kaufman B, Shapira‐Frommer R, Schmutzler RK et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015; 33; 244–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188. Litton JK, Rugo HS, Ettl J et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N. Engl. J. Med. 2018; 379; 753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Robson M, Im SA, Senkus E et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N. Engl. J. Med. 2017; 377; 523–533. [DOI] [PubMed] [Google Scholar]
- 190. Tung NM, Zakalik D, Somerfield MR, Hereditary Breast Cancer Guideline Expert P . Adjuvant PARP inhibitors in patients with high‐risk early‐stage HER2‐negative breast cancer and germline BRCA mutations: ASCO hereditary breast cancer guideline rapid recommendation update. J. Clin. Oncol. 2021; 39; 2959–2961. [DOI] [PubMed] [Google Scholar]
- 191. Condorelli R, Mosele F, Verret B et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO scale for clinical actionability of molecular targets (ESCAT). Ann. Oncol. 2019; 30; 365–373. [DOI] [PubMed] [Google Scholar]
- 192. Nones K, Johnson J, Newell F et al. Whole‐genome sequencing reveals clinically relevant insights into the aetiology of familial breast cancers. Ann. Oncol. 2019; 30; 1071–1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Tung NM, Robson ME, Ventz S et al. TBCRC 048: Phase II study of Olaparib for metastatic breast cancer and mutations in homologous recombination‐related genes. J. Clin. Oncol. 2020; 38; 4274–4282. [DOI] [PubMed] [Google Scholar]
- 194. Chopra N, Tovey H, Pearson A et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020; 11; 2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Pellegrino B, Herencia‐Ropero A, Llop‐Guevara A et al. Preclinical in vivo validation of the RAD51 test for identification of homologous recombination‐deficient tumors and patient stratification. Cancer Res. 2022; 82; 1646–1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Davies H, Glodzik D, Morganella S et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017; 23; 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Telli ML, Timms KM, Reid J et al. Homologous recombination deficiency (HRD) score predicts response to platinum‐containing neoadjuvant chemotherapy in patients with triple‐negative breast cancer. Clin. Cancer Res. 2016; 22; 3764–3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Paulet L, Trecourt A, Leary A et al. Cracking the homologous recombination deficiency code: how to identify responders to PARP inhibitors. Eur. J. Cancer 2022; 166; 87–99. [DOI] [PubMed] [Google Scholar]
- 199. Lakhani SR. WHO classification of tumours of the breast. 4. Ed. International Agency for Research on C, world health O, editors. International Agency for Research on Cancer: Lyon, 2012. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.