

Abstract
Voltage‐gated L‐type Ca2+‐channels (LTCCs) are the target of Ca2+‐channel blockers (CCBs), which are in clinical use for the evidence‐based treatment of hypertension and angina. Their cardiovascular effects are largely mediated by the Cav1.2‐subtype. However, based on our current understanding of their physiological and pathophysiological roles, Cav1.3 LTCCs also appear as attractive drug targets for the therapy of various diseases, including treatment‐resistant hypertension, spasticity after spinal cord injury and neuroprotection in Parkinson's disease. Since CCBs inhibit both Cav1.2 and Cav1.3, Cav1.3‐selective inhibitors would be valuable tools to validate the therapeutic potential of Cav1.3 channel inhibition in preclinical models. Despite a number of publications reporting the discovery of Cav1.3‐selective blockers, their selectivity remains controversial. We conclude that at present no pharmacological tools exist that are suitable to confirm or refute a role of Cav1.3 channels in cellular responses. We also suggest essential criteria for a small molecule to be considered Cav1.3‐selective.
Keywords: Ca2+ channel blockers, Cav1.3 selective inhibitors, drug discovery, voltage‐gated Ca2+ channels
Abbreviations
- 6‐OHDA
6‐hydroxydopamine
- AP
aldosterone producing adenoma
- APCC
aldosterone‐producing cell cluster
- CICR
Ca2+‐induced Ca2+‐release
- CPP
conditioned place preference
- DA
dopamine
- DHN
dorsal horn neuron
- DHP
dihydropyridine
- LTCC
L‐type Ca2+‐channel
- LTP
long‐term potentiation
- MLI
molecular layer interneurons
- NMDAR
NMDA‐glutamate receptor
- SCI
spinal cord injury
- SN
Substantia nigra
- VTA
ventral tegmental area
1. INTRODUCTION
With the discovery of the new pharmacodynamic principle of ‘Ca2+ antagonism’ by substances such as verapamil and nifedipine, the German physiologist Albrecht Fleckenstein boosted research leading to the discovery of their molecular mechanism of action, the selective blockade of voltage‐gated L‐type Ca2+‐channels (LTCCs). LTCC inhibition in the heart and vascular smooth muscle explains their blood pressure‐lowering, anti‐anginal and antiarrhythmic actions. These clinical effects are largely mediated by Cav1.2, one of four members of the LTCC family. While Cav1.1 (skeletal muscle) and Cav1.4 (retina) have restricted functions, Cav1.3 LTCCs were found in most electrically excitable cells, and often together with Cav1.2 in the same cell (Figure 1). Over the years it became clear that the biophysical properties of Cav1.2 and Cav1.3 as well as their relative abundance in many cells differ, which allows them to serve distinct physiological functions. The clinically relevant pharmacological effects are mediated by Cav1.2 not only because of the preferential expression of Cav1.2 in the cardiovascular system but also because of the high sensitivity of Cav1.2 for inhibition by dihydropyridines (DHPs) in vascular smooth muscle cells due to the positive operation voltage range of these cells and the existence of splice variants favouring high sensitivity for DHPs (Liao et al., 2007; Ortner et al., 2017). Clinically available Ca2+‐channel blockers do not discriminate much between the two isoforms. An open questions still is whether the selective inhibition of either Cav1.2 (Harrison et al., 2019) or Cav1.3, which are both also widely expressed outside the cardiovascular system (including the brain and endocrine cells: Zamponi et al., 2015) has any therapeutic value. While this is unclear for Cav1.2, Cav1.3 appears as an attractive drug target as outlined below. Therefore, Cav1.3‐selective Ca2+‐channel blockers are of great interest as novel tools and lead compounds for further preclinical development.
FIGURE 1.
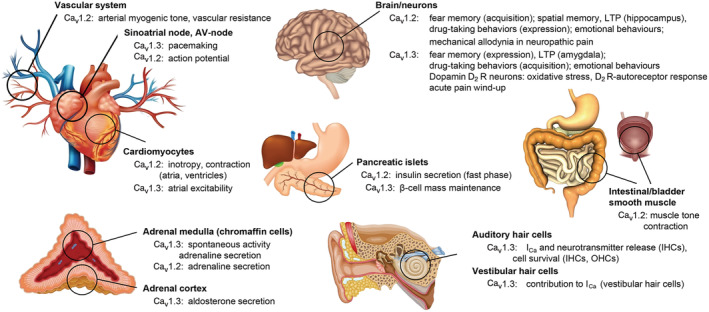
Physiological roles for Cav1.2 and Cav1.3 L‐type Ca2+‐channels (LTCCs). Cav1.2 and Cav1.3 are expressed in most excitable tissues and in most cases even together in the same cells. They can support different physiological functions due to their distinct gating properties (Cav1.3 channels are ‘low‐voltage‐activated LTCCs’), subcellular localization and/or protein‐interactions. Cav1.1 and Cav1.4 show more restricted expression in skeletal muscle and retinal cells and serve key functions for skeletal muscle contraction and photoreceptor signalling, respectively (Zamponi et al., 2015). AV‐node, atrioventricular node; ICa, inward Ca2+ current, IHC, inner hair cells; LTP, long‐term potentiation; OHC, outer hair cells. Adapted from Zamponi et al. (2015), redrawn with permission.
1.1. L‐type Ca2+‐channel drug binding domains
All LTCCs exhibit a uniquely high sensitivity for Ca2+‐channel blockers. High affinity drug binding domains exist on their pore‐forming α1‐subunits for the three major chemical classes of Ca2+‐channel blockers: dihydropyridines (DHPs, e.g., nifedipine, amlodipine and isradipine), phenylalkylamines (e.g., verapamil and gallopamil) and benzothiazepines (e.g., (+)‐cis‐diltiazem) (Striessnig, 2021a; Striessnig et al., 1998) (Figure 2b–d).
FIGURE 2.
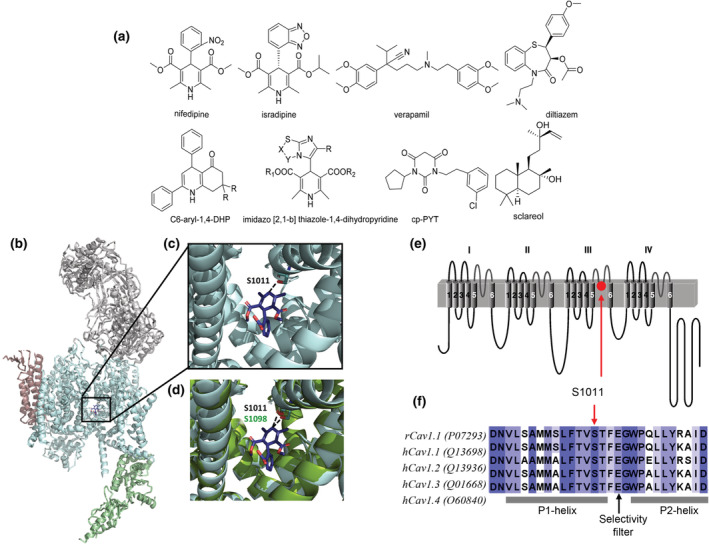
Chemical structures of compounds discussed in this article and L‐type Ca2+‐channel (LTCC) structure. (a) Isradipine (previously also referred to as PN200‐110) is shown as its active (S)‐(+)‐enantiomer, diltiazem as its active (+)‐cis‐diastereomer. Other names that can be found in the literature for cp‐PYT (1‐(3‐chlorophenethyl)‐3‐cyclopentylpyrimidine‐2,4,6‐(1H,3H,5H)‐trione) are compound 8 and BPN‐4689. (b) The structure of the rabbit Cav1.1 channel complex with nifedipine bound in the DHP binding pocket (PDB entry 6JP5 Zhao et al., 2019). α1, β, γ and α2δ1 subunits are coloured pale cyan, pale green, brown and grey, respectively. The DHP blocker nifedipine is shown in dark purple. Lipids facing the DHP binding‐site has been removed for clarity. (c) DHP‐binding site in the Cav1.1 structure with serine residue 1011 (numbering according to rabbit rCav1.1 α1‐sequence, Uniprot P07293) indicated. (d) Homology model of human hCav1.3 α1‐subunit (Uniprot Q01668) based on the rCav1.1 structure, generated with MOE (Molecular Operating Environment, version 2020.09, Molecular Computing Group Inc., Montreal, Canada). The hCav1.3 α1‐subunit (dark green) is shown in comparison to Cav1.1 (pale cyan). The serine residue (corresponding to S1098 in hCav1.3) represents the hydrogen‐bond acceptor partner of the hydrogen‐bond donor‐NH group in the DHP‐ring. This hydrogen bond is critical for the channel‐gating modifying activity of DHPs. (e) For orientation, the schematic transmembrane topology of Cav α1‐subunits is shown to highlight the approximate position of serine 1098 (red circle) in the P1 helix of repeat III in Cav1.3 α1. F. S1011 is highly conserved in all LTCCs and located close to one of the negative charges forming the channel's selectivity filter.
The three‐dimensional structure of the Cav1.1 LTCCs has recently been solved for the apo protein and in a complex with bound Ca2+‐channel blockers at high resolution (Gao & Yan, 2021; Wu et al., 2016; Zhao et al., 2019; for a detailed review on the structural details of voltage‐gated Ca2+‐ and Na+‐channels, see Catterall et al., 2020). An overview of the channel architecture in a complex with auxiliary subunits and with the DHP Ca2+‐channel blocker nifedipine is shown in Figure 2b. A serine residue in the P1 helix of repeat III forms the hydrogen‐bond interaction that is critical for DHP‐activity (Figure 2c), and it is highly conserved among all LTCCs including Cav1.3. (Figure 2d–f).
Upon binding, ligands induce conformational changes within the binding site (Wu et al., 2016; Zhao et al., 2019) due to the new interactions formed with the drug (Figure 3). The S4‐S5 linker of repeat III is shifted upwards and the S5‐helix of the same repeat is rotated towards nifedipine (Figure 3a). The S6‐helix rotates as exemplified by the movement of F1060 and in addition, Y1048 moves closer to Q939 to form a hydrogen bond that is not observed in the apo structure (Figure 3b) (Gao & Yan, 2021; Zhao et al., 2019).
FIGURE 3.
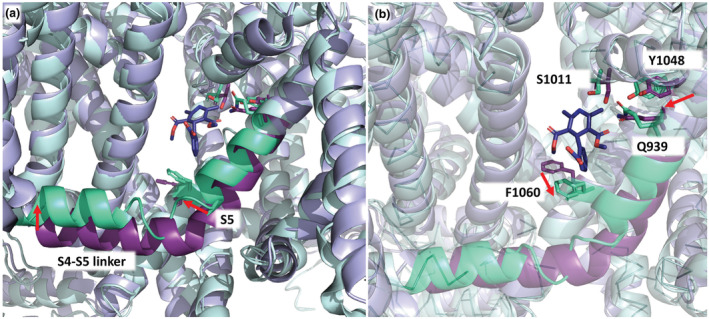
Comparison of the apo‐ and nifedipine‐bound structure of rabbit Cav1.1. The apo structure is shown in violet (PDB entry 5GJV, Wu et al., 2016), whereas the nifedipine‐bound structure (PDB entry 6JP5, Zhao et al., 2019) is depicted in pale cyan. (a) In the nifedipine‐bound Cav1.1 α1‐subunit, the IIIS4‐S5 linker is shifted upwards, whereas helix IIIS5 is rotated towards nifedipine. Red arrows indicate their movements. (b) S1011 of the repeat III pore segment, F1060 and Y1048 of IIIS6 and Q939 of IIIS5 are shown as solid sticks. They all change conformation upon ligand binding as indicated by red arrows. Please note that the S4‐S5 linker and S5 from both Cav1.1 structures are coloured with a darker shade (i.e., nifedipine‐bound Cav1.1 in green cyan, apo‐Cav1.1 in deep purple).
1.2. Cav1.3 Ca2+‐channels as potential drug targets
From the four mammalian LTCC family members only Cav1.2 and Cav1.3 are widely expressed in essentially all electrically excitable cells (Figure 1). In contrast, expression of Cav1.1 is largely restricted to skeletal muscle and Cav1.4 to the retina (Zamponi et al., 2015). The Cav1.2 and Cav1.3 α1‐subunits share high sequence homology within their voltage‐sensing and pore‐forming regions including their binding domains for the different Ca2+‐channel blockers. Despite this close structural relationship, they differ with respect to their biophysical properties. Of important physiological relevance is the finding that Cav1.3 channels activate at more negative voltages than Cav1.2. Therefore, Cav1.3 LTCCs underlie the ‘low‐voltage activated’ L‐type currents observed in various tissues (Avery & Johnston, 1996; Guzman et al., 2009;Mangoni et al., 2003; Marcantoni et al., 2010). Using Cav1.3‐deficient mice, low‐voltage activation of Cav1.3 has been confirmed in cochlea inner hair cells, adrenal chromaffin cells, sinoatrial node cells, and pancreatic β‐cells (Zamponi et al., 2015). With the exception of cochlear inner hair cells, Cav1.3 and Cav1.2 are expressed together in the same cell but their different biophysical properties allow them to serve distinct physiological functions. For example, in chromaffin cells or sinoatrial node cells, the negative activation voltage‐range enables subthreshold Cav1.3 (but not Cav1.2) currents supporting spontaneous pacemaking (Mangoni et al., 2003; Marcantoni et al., 2010). Alternative splicing affects the voltage‐dependent gating of LTCCs but so far, no Cav1.2 variant has been reported that activates at more negative voltages than Cav1.3 variants (Ortner et al., 2017; Zamponi et al., 2015).
The pacemaking function of Cav1.3 in the sinoatrial node and its absence in the ventricular myocardium predict Cav1.3‐selective inhibitors as bradycardic agents lacking relevant negative inotropy (Mangoni et al., 2003; Sinnegger‐Brauns et al., 2004a) with therapeutic potential in heart failure.
Low‐voltage‐activated LTCC‐mediated Ca2+‐influx has also been postulated to contribute to the selective vulnerability and degeneration of substantia nigra dopamine (SN DA) neurons in Parkinson's disease (PD) (Guzman et al., 2009; Liss & Striessnig, 2019; Surmeier, Obeso, & Halliday, 2017). In SN DA neurons, each action potential triggers large dendritic Ca2+‐transients. In these permanently active neurons, Ca2+‐load enhances mitochondrial stress and thus allows LTCCs to contribute to a synergistic network of toxicity pathways and PD stressors (Guzman et al., 2009; Liss & Striessnig, 2019; Surmeier, Obeso, & Halliday, 2017). Neuroprotection with DHP Ca2+‐channel blockers has been observed in some, but not all, in vivo mouse PD‐models (Liss & Striessnig, 2019; Ortner, 2021). The phase‐3 STEADY‐PD clinical trial investigated the neuroprotective potential of treatment with the DHP isradipine in early PD but it did not reach its primary endpoints (Parkinson Study Group STEADY‐PD III Investigators, 2020). One likely explanation for this finding is incomplete target engagement of Cav1.3 channels by the drug in the brain due to the low plasma levels reached in this study (Parkinson Study Group STEADY‐PD III Investigators, 2020) and the lower sensitivity of neuronal Cav1.3‐channels for DHPs as compared to vascular Cav1.2‐channels (Ortner et al., 2017). Higher dosing of isradipine or other DHPs is limited by hypotensive side effects mediated through inhibition of vascular Cav1.2‐channels. Cav1.3‐selective compounds may overcome this therapy‐limiting peripheral side effect.
As outlined below in this article, Cav1.3 inhibition may also reduce spasticity in patients after spinal cord injury (SCI) (Marcantoni et al., 2020).
In addition, rare human disorders have provided important insight into the pathogenic role of Cav1.3. Homozygous loss‐of‐function mutations in the human Cav1.3 α1‐subunit gene (CACNA1D) replicate the Cav1.3‐knockout phenotype observed in mice (Baig et al., 2011; Platzer et al., 2000). Affected individuals are congenitally deaf and exhibit an apparently benign sinoatrial node dysfunction. Notably, heterozygous loss‐of‐function in mice or humans is not associated with obvious disease symptoms (Baig et al., 2011; Platzer et al., 2000). In contrast, de novo (i.e., heterozygous) missense variants inducing gating changes, which also can promote Cav1.3 channel activity and Ca2+‐influx, are pathogenic in humans (Ortner, Kaserer, et al., 2020). Such CACNA1D missense mutations are found as somatic mutations in a substantial fraction of aldosterone‐producing adenomas (APAs) leading to hyperaldosteronism and treatment‐resistant hypertension (Azizan et al., 2013; Korah & Scholl, 2015; Scholl et al., 2013). Interestingly, such variants are even more frequently found in small so‐called aldosterone‐producing cell clusters (APCCs) (Nishimoto et al., 2015; Omata et al., 2017). Given this key role of Cav1.3‐channels in hyperaldosteronism, Cav1.3‐selective inhibitors may reduce aldosterone secretion and serve as specific antihypertensives in treatment–resistant hypertension associated with APAs or APCCs (Xie et al., 2016; Yang et al., 2020).
When present in the germline, such gating‐modifying missense mutations may lead to hyperaldosteronism and/or hyperinsulinemic hypoglycemia at birth (Cav1.3 channels are also expressed in pancreatic islet cells, Figure 1; Reinbothe et al., 2013; Zamponi et al., 2015). However, they are also associated with an usually severe neurodevelopmental syndrome. Symptoms include autistic behaviours, muscle hypotonia, hyperactivity, self‐aggressive behaviours, seizures and intellectual impairment (Ortner, Kaserer, et al., 2020; Striessnig, 2021b). These mutations provide unique insight into how increased Cav1.3 activity can also lead to neuronal dysfunction. Selective inhibition of hyperactive Cav1.3 signalling therefore appears as a potential treatment not only for the treatment of disorders with rare CACNA1D variants, but also for more frequent neuropsychiatric disorders, such as autism spectrum disorders or depression (Cav1.3‐deficient mice display reduced depression‐like behaviours, Sinnegger‐Brauns et al., 2004a).
Both channel isoforms also play an important role in central pain sensitization, including ‘wind‐up’ as a short‐term sensitization process and allodynia in neuropathic pain. In spinal dorsal horn neurons (DHNs), LTCCs postsynaptically modulate responses to physiological or pathological stimulation (Radwani et al., 2016). Dendritic Cav1.3‐channels mediate ‘wind‐up’, a progressive increase in spiking in response to nociceptive C‐fibre stimulation. They also induce plateau potentials that are followed by a sustained after‐discharge. In contrast, Cav1.2‐mediated changes in gene‐transcription participate in the expression of mechanical allodynia and DHN hyperexcitability (Fossat et al., 2010). It would therefore be interesting to determine to what extent Cav1.2 and/or Cav1.3 inhibition could prevent the development and expression of central sensitization in neuropathic pain (Radwani et al., 2016). Cav1.3‐selective inhibitors would be a valuable tool to help address this important question in preclinical pain models.
In conclusion, Cav1.3‐selective Ca2+‐channel blockers are predicted to have a number of interesting pharmacological properties of therapeutic potential. Together these findings represent a strong rationale for discovery of potent Cav1.3‐selective inhibitors.
1.3. Cav1.3–selective Ca2+‐channel blockers
The pharmacology of Cav1.3 Ca2+‐channels is complex. Among the existing drugs, DHPs are the best characterized with respect to their potential selectivity for Cav1.3. Independent reports found that DHPs, such as nifedipine (Wang et al., 2018), nimodipine (Huang et al., 2013; Xu & Lipscombe, 2001) and isradipine (Koschak et al., 2001; Ortner et al., 2017) inhibit heterologously expressed Cav1.3 channels with about 5–10‐fold higher IC50‐values than Cav1.2 when studied under identical experimental protocols. For isradipine this Cav1.2‐selectivity cannot easily be explained by differences in the affinity for the DHP binding pockets because in radioligand binding studies identical dissociation constants (KD values) were obtained for (+)‐[3H]isradipine binding to recombinant Cav1.3 and Cav1.2 channels (Koschak et al., 2001; Sinnegger‐Brauns et al., 2009). These findings are in excellent agreement with binding studies in brain membranes from a mouse model in which high DHP‐sensitivity had been removed selectively from Cav1.2‐channels by a single amino acid change (T1066Y, Sinnegger‐Brauns et al., 2004b; corresponding to T1056Y in human Cav1.2‐α1, Figure 4). This tyrosine causes a steric clash within the DHP binding site (Figure 4) and thereby prevents high affinity interaction with DHPs. In these mice, DHPs are ‘Cav1.3‐selective’ which allowed the detailed pharmacological analysis of the role of native Cav1.3 channels for many physiological functions (including behaviour, cardiac and endocrine functions) using DHPs without interference from Cav1.2‐inhibition (Zamponi et al., 2015). Radioligand binding in these mice found that Cav1.3‐channels comprise only about 10% of the total DHP binding sites in brain and confirmed essentially identical KD values for isradipine in brain membranes as compared to heterologously expressed Cav1.3 channels (Sinnegger‐Brauns et al., 2009). The differences in DHP‐sensitivity between Cav1.3 and Cav1.2 observed in functional studies must therefore involve conformational changes induced by changes of membrane‐voltage, protein interactions and/or alternative splicing. Cav1.2 splice variants (e.g., variants predominantly expressed in cardiac vs. smooth muscle) show considerable differences in DHP‐sensitivity (Liao et al., 2007). Likewise, C‐terminal splicing in the Cav1.3‐channel, which occurs outside the drug binding domains but causes profound changes in channel inactivation and voltage‐dependence of gating, strongly affects sensitivity for the DHP nimodipine (Huang et al., 2013), later also confirmed for isradipine (Ortner et al., 2017). In contrast to isradipine, some DHPs, such as nitrendipine and nifedipine, also have lower binding affinity for Cav1.3, indicating subtle differences in the coordination within the two binding pockets (Sinnegger‐Brauns et al., 2009).
FIGURE 4.
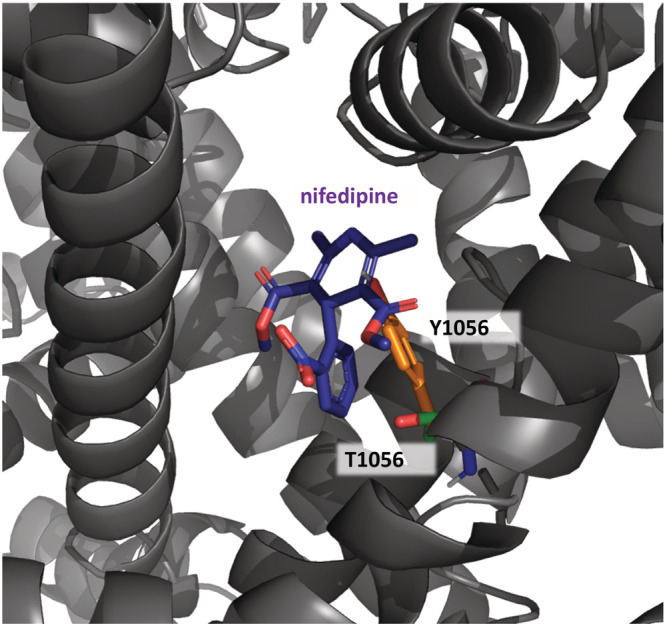
The T1056Y mutation causes a steric clash within the DHP binding site. In the model of hCav1.2 harbouring mutation T1056Y (corresponding to T1066Y in mouse Cav1.2; Sinnegger‐Brauns et al., 2009), the strongly reduced DHP‐sensitivity of the mutant is due to the steric clash between the Y1056 side chain and the DHP‐ring of DHPs (shown for nifedipine, blue), which prevents DHPs from accessing their binding site. This mutation has been used in in vitro and in vivo studies to prevent DHP effects through Cav1.2 channels making existing DHPs ‘selective’ for Cav1.3 in mutant cells. hCav1.2 is shown in grey and Y1056 in IIIS5 is shown as orange sticks. Green sticks represent corresponding T1056 belonging to the wild‐type hCav1.2. The homology model of hCav1.2 is based on PDB entry 6JP5 (Zhao et al., 2019). Model generation and insertion of the mutant residue were conducted with MOE (see legend to Figure 2).
1.4. Small molecule screens in native cells
Tenti and colleagues synthesized new C5‐unsubstituted C6‐aryl‐1,4‐DHPs (Figure 2a) for which they claimed improved Cav1.3 selectivity (Tenti et al., 2014). The pharmacological characterization of their compounds was based on a functional assay in native cells. Inhibition of Cav1.2 was quantified through inhibition of contraction of rat mesenteric resistance arteries, that were precontracted with 70 mM K+. Cav1.2 channels mediate contractility in arterial smooth muscle and the working myocardium (Zamponi et al., 2015), and the compound‐mediated inhibition can therefore report activity for this isoform. Inhibition of Cav1.3 was quantified in fluorescent Ca2+ imaging assays from the inhibition of cytosolic Ca2+‐transients induced by depolarization with 70 mM K+ in undifferentiated SH‐SY5Y human neuroblastoma cells. The assumption that this assay reports Cav1.3 activity, however, is not supported by sufficient evidence. Undifferentiated SH‐SY5Y cells express nifedipine‐sensitive L‐type and ω‐conotoxin GVIA‐sensitive N‐type currents (Reeve et al., 1994; Sousa et al., 2013). Both Cav1.3 (Sousa et al., 2013) and Cav1.2 α1‐subunit transcripts (Billingsley et al., 2018; Jiang et al., 2018; Sun et al., 2017; Wang et al., 2014) have been identified in these cells and their relative contribution to total L‐type current is unknown. It therefore remains unclear to what extent the inhibition of Cav1.3 currents contributes to the observed inhibition of Ca2+‐transients, which makes it impossible to interpret isoform‐selectivity of compounds using this assay. Moreover, the action of nifedipine, and perhaps also its DHP‐derivatives, is highly voltage‐dependent (Bean, 1984; Koschak et al., 2001; Lee & Tsien, 1983). DHPs preferentially bind to inactivated channel states. Therefore, they inhibit channels with much lower IC50 values when a cell is held at (or fires from) more depolarized membrane potentials, which favour inactivated channel states. In this respect, assay conditions were not comparable, because their compounds were tested for Cav1.2 effects in mesenteric arteries pre‐contracted (i.e., depolarized) with 70 mM K+, whereas they were preincubated in SH‐SY5Y cells before depolarization with 70 mM K+ to test for activity at Cav1.3 (Tenti et al., 2014). This can also explain their finding of a 950‐fold selectivity of nifedipine for Cav1.2, due to its low nanomolar IC50 for inhibition of mesenteric artery constriction as compared to micromolar concentrations required for half‐maximal inhibition of SH‐SY5Y Ca2+ transients. The very large selectivity for this Cav1.2‐mediated effect was reduced to between 2‐ and 8‐fold for some of their compounds due to a much lower potency for inhibition of arterial smooth muscle constriction. This reduction in selectivity was interpreted as drastically improved ‘selectivity’ towards Cav1.3 although these compounds were still, even if to a smaller extent, Cav1.2‐selective. No attempts were made to test if their compounds also inhibited non‐L‐type currents in SH‐SY5Y cells nor were they tested for potency or selectivity for Cav1.3 Ca2+‐channel currents in heterologous expression systems or in native cells, in which low‐voltage‐activated Cav1.3 currents can be isolated (e.g., mouse chromaffin or sinoatrial node cells; Zamponi et al., 2015).
A similar functional screen in native cells has also been performed for a large series of DHPs with different imidazo[2,1‐b]thiazole substituents in position C4 (Figure 2a) (Budriesi et al., 2008; Locatelli et al., 2013). These 4‐imidazo[2,1‐b]thiazole‐1,4‐DHPs were screened for their negative inotropic and chronotropic effects in guinea‐pig isolated left and right atria, and for inhibition of the contraction of K+‐depolarized (80 mM) guinea‐pig vascular (aortic strips) and nonvascular (ileum longitudinal) smooth muscle. In contrast to nifedipine, the tested compounds were only weak inhibitors of vascular smooth‐muscle contraction. They more efficiently inhibited ileal than aortic smooth muscle but in general with higher IC50‐values than those required for their negative inotropic effects. Some compounds showed selectivity for negative chronotropic effects measured in right atria. In addition, binding studies were performed in membranes prepared from guinea‐pig cardiac atria and ventricles or from rat brain cortex using the (unselective) DHP Ca2+‐channel blocker (+)‐[3H]isradipine (=PN200–110) (Budriesi et al., 2008; Locatelli et al., 2013). Some compounds partially inhibited (+)‐[3H]isradipine binding to guinea‐pig atrial membranes with IC50‐values about 10–50 times lower than to guinea‐pig ventricular membranes, whereas nifedipine showed similar affinity in both tissues (Locatelli et al., 2013). However, a methodological issue was that 100 μM nifedipine displaced only 72%–80% of (+)‐[3H]isradipine binding although even much lower concentrations (5 μM) were used to define nonspecific binding, questioning their results from binding studies. Most of the tested compounds also partially inhibited (+)‐[3H]isradipine binding to membranes prepared from rat brain cortex. From these studies, the authors concluded that 1,4‐DHP scaffolds bearing a imidazo[2,1‐b]thiazole ring at C4 can provide compounds with cardiac selectivity less affecting vascular smooth muscle and with different actions on different heart functions, including compounds with some selectivity for negative chronotropic effects. The authors discussed different actions of these molecules on ‘heterogeneously expressed Cav1.2 and Cav1.3 in heart and brain’ and that ‘the substituents at the imidazo[2,1‐b]thiazole ring can modulate the activity of compounds on different heart functions influenced by cardiac LTCCs isoforms Cav1.2 and Cav1.3’. However,whether their experiments can indeed differentiate between Cav1.2 and Cav1.3 remains unclear. One could speculate that they consider compounds with slight selectivity for negative chronotropic effects as Cav1.3‐selective because Cav1.3 supports pacemaking in the sinoatrial node (Mangoni et al., 2003; Zamponi et al., 2015). However, the authors did not test their compounds in the sinoatrial node and the negative chronotropic effects may also be due to inhibition of other ion channels including T‐type Ca2+ channels (Torrente et al., 2020). While the experimental design provides interesting structure–activity relationship data on imidazo[2,1‐b]thiazole–substituted DHPs and their effect on heart, smooth‐muscle and brain function, it can provide little evidence regarding the selectivity profiles of the tested compounds in the absence of binding or functional studies with identified Cav1.3 channels.
All of these studies included computational molecular modelling studies to explain the interaction of C5‐unsubstituted C6‐aryl‐ or imidazo[2,1‐b]thiazole – substituted DHPs (Budriesi et al., 2008; Locatelli et al., 2013; Tenti et al., 2014) with Cav1.2 and Cav1.3 binding pockets. However, these were based on preliminary models of the DHP binding domain constructed before the cryo‐electron microscopy structure of the Cav1.1 Ca2+‐channel complex in its apo‐ (Wu et al., 2016) and drug‐bound‐state (Zhao et al., 2019) became available. Consequently, the authors could not take the ligand‐induced conformational changes of the channel into account (Figure 3), which are required to fit the molecules in close proximity to the highly conserved serine and allow formation of the hydrogen bond that is critical for activity (Figure 2b–f).
Taken together, the concept of defining the selectivity of compounds for Cav1.2 or Cav1.3 channels by performing functional and binding studies in cells or tissues without clearly defined expression and a validated isoform‐specific functional readout is of limited value and even misleading for the discovery of Cav1.3‐selective inhibitors. Moreover, selectivity also needs to be confirmed in heterologous expression systems as well as in native cells with well‐characterized Cav1.2 and Cav1.3 current components.
1.5. Small molecule screens using recombinant channels
Based on the findings that LTCC inhibition by DHPs distinguishes to some extent between Cav1.2 and Cav1.3 (although with a preference for Cav1.2), Chang and colleagues (Chang et al., 2010) synthesized a library of 124 chemically diverse 1,4‐dihydropyridines in search of structures that are potent and selective inhibitors of Cav1.3. Channel inhibition was measured using tsA201 cells stably transfected with rabbit Cav1.2 or rat Cav1.3, either by testing the percent inhibition by a single concentration or by determining IC50 values in detailed concentration‐response curves using a FLIPR assay. Nifedipine served as the lead compound. Modifications included changes at the alkyl groups at the 2‐ and 6‐positions, at the 4‐position and at the amino group of the DHP ring. This should provide a detailed structure–activity relationship to identify structural features that increase potency and selectivity towards Cav1.3 channels. However, despite this effort, none of the more potent inhibitors of Cav1.3 exhibited relevant selectivity (<1.7‐fold). The two most selective analogues were only 2.2‐ and 2.4‐fold selective and both had only micromolar potencies.
In a follow‐up study (Kang et al., 2013a), the same authors explored diverse alternative scaffolds, including 1,4‐dihydropyrimidines and 4H‐pyrans, to introduce further structural variations. A library of 36 DHP‐mimetics was prepared, all with a 3‐nitrophenyl as the aromatic substituent (like in nitrendipine), but with modifications to the DHP‐scaffold (pyrimidinone, pyrimidinethione, and hydropyran), to each of the esters and to the alkyl side chain. The IC50 values for these compounds were determined by concentration–response curves in a FLIPR assay. Again, only modest improvements in selectivity could be achieved. The pure R‐enantiomer of the most potent compound inhibited Cav1.3 with an IC50 of 0.51 μM and Cav1.2 with an IC50 of 1.02 μM, corresponding to a difference of only about 2‐fold.
Despite the failure to discover Cav1.3‐selective lead compounds, these two studies (Chang et al., 2010; Kang et al., 2013) were very important, because they provided substantial evidence that relevant Cav1.3‐selectivity cannot be achieved by modifying the substitutions of the DHP scaffold.
Therefore, Kang et al. (2012) set out to discover novel scaffolds suitable for Cav1.3‐selective channel inhibition. In their FLIPR‐based high‐throughput screen they identified pyrimidine‐2,4,6‐triones, a class of compounds previously characterized as potential neuroprotective agents in amyotrophic lateral sclerosis and with favourable pharmacokinetic properties (Xia et al., 2011). Chemical modifications finally led to a Cav1.3‐selective compound, termed compound 8 (1‐(3‐chlorophenethyl)‐3‐cyclopentylpyrimidine‐2,4,6‐(1H,3H,5H)‐trione) (Figure 2a). In later publications it was also termed cp‐PYT or BPN‐4689. We will use cp‐PYT to refer to this compound throughout this article. In FLIPR‐assays it inhibited Cav1.3 with IC50‐values of 1.7–6.3 μM (Kang et al., 2012; Kang et al., 2013b) but it appeared as a weak inhibitor of Cav1.2 with an estimated 612‐fold selectivity for Cav1.3 (Kang et al., 2012). This was further confirmed in whole‐cell patch‐clamp experiments in HEK‐293 cells stably expressing Cav1.3 or Cav1.2. Cp‐PYT inhibited Cav1.3 with an IC50 of 24.3 μM and >100‐fold selectivity over Cav1.2 (Kang et al., 2012).
Unfortunately, these findings could not be reproduced by two other groups. Huang et al. (2014) studied the potency and selectivity of cp‐PYT using whole‐cell patch‐clamp recordings in transiently transfected HEK‐293 cells expressing C‐terminal long (Cav1.342) and short (Cav1.342a) rat Cav1.3 α1‐subunit splice variants or rat Cav1.2B15 (Tang et al., 2007) together with rat α2δ and the rat β‐subunit isoforms β1‐β4. Although recording conditions were similar (test pulses from −70 mV to 10 mV at 0.05 Hz) current inhibition by 50 μM of the compound was less than 50% for both Cav1.3‐splice variants and similar to Cav1.2 when β1‐, β3‐ and β4‐subunits formed part of the channel complex. However, upon co‐expression of β2a‐subunits (a palmitoylated β‐subunit that slows channel inactivation; Buraei & Yang, 2010; Ortner, Pinggera, et al., 2020 for review), cp‐PYT was even slightly Cav1.2‐selective (Huang et al., 2014).
In addition, Ortner et al. (2014) could not observe Cav1.3 selectivity using whole‐cell patch‐clamp recordings with a pulse‐protocol and assay conditions similar to those in the original paper. TsA‐201 cells were transfected with the long splice variants of rat (rCav1.3L) or human Cav1.3 (hCav1.3L), rabbit rbCav1.2 (same construct as in the original study) or rbCav1.2S (truncated at amino acid position 1800 to account for proteolytically processed forms in heart and brain) together with rat β3 and rabbit α2δ1. In contrast to the other studies, 50 μM cp‐PYT strongly affected Cav1.3 and Cav1.2 current kinetics characterized by a slowing of the activation and inactivation time course as well as a pronounced slowing of deactivation upon repolarization. These changes resembled the kinetic changes induced by the LTCC activator FPL64176 (Kunze & Rampe, 1992). These effects on gating were absent only in a small minority of cells, in which cp‐PYT, however, blocked Cav1.3 and Cav1.2 weakly but to a similar extent. The absence of selective inhibition was not an artefact of heterologous expression because the slowing of tail current was further confirmed in native Cav1.2 and Cav1.3 LTCC current components in mouse chromaffin cells (Ortner et al., 2014). In summary, these results suggest, that cp‐PYT can behave as an activator rather than a blocking agent of LTCC currents. No evidence for selectivity was found for this compound.
In a more recent study (Cooper et al., 2020), the authors of the original findings tried to resolve these discrepancies by studying the molecular mechanism of cp‐PYT interaction within its binding pocket on Cav1.3‐channels in more detail. They confirmed the selectivity with an about 60% and 20% inhibition of Cav1.3 and Cav1.2 by 100 μM of the compound, respectively (again determined with whole‐cell patch‐clamp recordings as in the original study, Kang et al., 2012). A key observation was that a single amino acid exchange in Cav1.3 α1, T1081Y (which corresponds to T1066Y in ref. Sinnegger‐Brauns et al., 2004a, see Figure 4), which is known to eliminate high affinity for DHPs (see above), also prevented cp‐PYT inhibition (100 μM). This strongly suggested that cp‐PYT and DHPs bind to the same region. This was further supported by the observation that the binding of the tritiated cp‐PYT derivative [3H]‐D11 to recombinant Cav1.3 channels was partially displaced by the DHP isradipine (Cooper et al., 2020). The authors then constructed a homology model of Cav1.2 and Cav1.3 based on a cryo‐EM structure of Cav1.1 (skeletal muscle) Ca2+‐channels (Wu et al., 2016). From their model they predicted that the selectivity for Cav1.3 results from two residues within the DHP/cp‐PYT binding pocket that differ between Cav1.3 and Cav1.2 α1‐subunits (for sequence alignment see Figure 5): M1078 (in IIIS5), which forms the bottom of the binding pocket, and V1198 (located in S6 of domain III and not IVS6, as erroneously stated by the authors). Mutation of M1078 to valine, the corresponding amino acid in Cav1.2, reduced the sensitivity to cp‐PYT. Conversely, replacement of V1063 in Cav1.2, which corresponds to M1078 in Cav1.3, by methionine increased the sensitivity of Cav1.2 for the compound (about 50% inhibition by 100 μM cp‐PYT). Notably, mutation M1078V also strongly shifted the more negative current–voltage relationship of Cav1.3 to more positive voltages, indistinguishable from Cav1.2. Therefore, assuming state‐dependent effects of cp‐PYT (see below), the altered gating of this construct, rather than changes in its binding domain itself, may also account, at least in part, for its reduced sensitivity to cp‐PYT. Change of Cav1.3 V1198 to the corresponding isoleucine in Cav1.2 had no effect.
FIGURE 5.

Sequence alignment of rat Cav1.3 α1 subunit constructs used by different groups to study the selectivity of cp‐PYT. Alignments are shown for regions in homologous repeats III and IV with amino acid differences between the constructs. Note that residues M1078 and V1198 (orange) are not different (for details see text). The only difference is alternative splicing of exon 31 (Lieb et al., 2012), which was different in the construct used in the study by Ortner (Ortner et al., 2014) compared to those by the other laboratories (GenBank accession #: AF370009/AF370010; Cooper et al., 2020; Huang et al., 2014; Kang et al., 2012).
None of these mutations affected isradipine sensitivity, which is in contrast to an earlier report (Wang et al., 2018) demonstrating that M1078V in Cav1.3‐α1 (M1030V in Wang et al., 2018) increased, whereas the reciprocal Cav1.2 mutation decreased sensitivity for nifedipine.
The homology model generated by the authors (Cooper et al., 2020) provided a rational basis to predict which residues could account for the proposed selectivity of cp‐PYT towards Cav1.3 and was also used to predict a potential cp‐PYT binding mode. However, the conclusions derived thereof have to be interpreted with caution since the model was based on the apo‐structure of Cav1.1 (Wu et al., 2016) rather than on the structure of the drug‐bound conformation, which was already published at that time (Zhao et al., 2019), as outlined above (Figure 3). Therefore, their model predicted an isradipine‐docking pose unable to recapitulate the pose resolved for nifedipine in the cryo‐EM structure (Gao & Yan, 2021; Zhao et al., 2019).
As also shown for DHPs, cp‐PYT (10 μM) inhibition was voltage‐dependent and blocked Cav1.3 more efficiently (26% versus 46% inhibition) when cells were held at more depolarized voltages (−50 versus −80 mV, Cooper et al., 2020). In acutely isolated SN DA neurons, which express Cav1.3, Cav1.2, Cav2.3 and other voltage‐gated Ca2+ channels (Benkert et al., 2019; Poetschke et al., 2015), 50 μM cp‐PYT inhibited about 50% of the voltage‐gated Ca2+ current. Interestingly, the same concentration of cp‐PYT did not inhibit current in neurons prepared from Cav1.3‐deficient mice, interpreted as evidence for Cav1.3‐selectivity of the compound. However, the contribution of isradipine‐sensitive L‐type current to the overall Ca2+‐channel current in their SN DA neurons was not studied. Therefore, it remains unclear if a Cav1.2‐mediated L‐type current component even exists in these cells that, if not inhibited by cp‐PYT, would allow to prove its selectivity.
Application of cp‐PYT (50 μM) to ex vivo midbrain slices had no effect on pacemaking of SN DA neurons, but significantly reduced the dendritic Ca2+ oscillations, consistent with inhibition of LTCC activity. Similar to 1 μM isradipine, cp‐PYT (50 μM) also diminished mitochondrial oxidant stress in these neurons when compared to previously published historical controls (Figure 1h in Guzman et al., 2010).
Unlike observations by others in a previous publication (Ortner et al., 2014), only very minor gating changes were found by Cooper and colleagues (Cooper et al., 2020). A very small and non‐significant (at n = 3) slowing of deactivation (less than 2‐fold increase of the deactivation time constant) was evident at higher (50 μM) but not at lower (10 μM) concentrations of cp‐PYT. This was explained by the existence of an additional low affinity site on the channel, which could mediate slowing of deactivation but not channel inhibition (Cooper et al., 2020). Indeed, from their binding data using [3H]‐D11, a close structural analogue of cp‐PYT, the existence of a low affinity cp‐PYT binding site cannot be excluded. This is based on the observation that isradipine only displaced about 80% of specific binding. However, it is unclear if this partial inhibition resulted from partial displacement due to overlapping or negatively allosterically coupled binding sites for [3H]‐D11 and isradipine, or from occupation of an additional low affinity/high capacity site by [3H]‐D11 unrelated to LTCCs. The data do not allow conclusions about the overall binding capacity of [3H]‐D11 in their membrane preparation (the Scatchard plot revealed a Bmax of 0). Nevertheless, from their experiments the authors concluded that under their experimental conditions cp‐PYT is Cav1.3‐selective and that it shares a common binding pocket with DHPs.
At the moment, it is not clear why three independent laboratories obtained different results despite testing cp‐PYT under similar experimental recording conditions. It would therefore be important to elucidate the experimental and/or molecular mechanisms to evaluate under which conditions, cp‐PYT could behave as a Cav1.3 selective blocker. The use of different splice variants of the Cav1.3 and Cav1.2 α1‐subunits may contribute to the different observations. However, to date no experimental evidence has been reported to support this notion. In contrast to Cooper et al. (2020) and Huang et al. (2014), Ortner et al. (2014) employed a rat Cav1.3 α1‐subunit construct that was alternatively spliced to contain exon 31A instead of exon 31 (Figure 5 for sequence alignment) but, unlike stated in the paper (see published correction, Cooper et al., 2021) was otherwise identical to the other rat clones. Whether alternative splicing can account for the ‘agonistic’ effects of the compound has so far not been investigated. It should be kept in mind also for future studies that selectivity ratios are also affected by the nature of the Cav1.2 construct used as a reference. However, differences in the Cav1.2 constructs also appear unlikely in this case as Cooper et al. and Ortner et al. used the same rabbit construct (Uniprot accession #P15381). This corresponds to the cardiac splice variant of Cav1.2 (Mikami et al., 1989). It is important to note that the vascular smooth muscle splice variants of Cav1.2 inactivate at more negative voltages and, depending on resting membrane potential, are 6 to 10 times more sensitive to DHPs (Liao et al., 2007) than the cardiac splice variant used in these studies (Cooper et al., 2020; Kang et al., 2012). Given the fact that cp‐PYT is believed to bind within the DHP binding pocket and to also act in a voltage‐dependent manner no clear conclusions can be drawn regarding selectivity from experiments with the cardiac splice variant alone. Huang et al. employed a Cav1.2 splice variant preferentially expressed in aortic smooth muscle. It is possible (but not yet proven) that the different splicing also provided more robust block of Cav1.2 by cp‐PYT in their study, which could explain the lack of selectivity.
A recent cryo‐EM study tried to reveal the molecular basis of cp‐PYT binding to Cav1.3. While the structure of a cinnarizine‐Cav1.3 α1‐subunit complex could be solved, no electron density could be observed for cp‐PYT in the cryo‐EM structure of Cav1.3 incubated with 150 μM of cp‐PYT (Yao et al., 2022). The reason for this observation is unclear but may be due to the low affinity of this compound for the channel.
Given the current uncertainty regarding the Ca2+‐channel selectivity profile of cp‐PYT, the lack of reproducibility between different laboratories, and the absence of information on its action on other ion channel targets, we do not support its use or promote it as a Cav1.3‐selective Ca2+‐channel blocker, in contrast to other recommendations (Roca‐Lapirot et al., 2018).
1.6. Use of cp‐PYT as a Cav1.3‐selective tool in other studies
Until these discrepancies are resolved the evidence for cp‐PYT being a useful Cav1.3‐selective compound for further studies is limited and controversial. Data in native tissues are also not conclusive. Cp‐PYT modified gating similar to LTCC‐activators in mouse chromaffin cells (Ortner et al., 2014) but showed inhibitory effects in SN DA neurons (Cooper et al., 2020). As outlined above, it is unclear if Cav1.2 L‐type currents exist in these cells, therefore no conclusions about selectivity can be drawn from these experiments.
Despite these uncertainties, and the fact that cp‐PYT has not yet been investigated for its pharmacological effects on Cav2 and Cav3 (T‐type) Ca2+ channels, it has been used as a ‘Cav1.3‐selective’ tool in several studies. This is not surprising, given that the scientific community is eagerly awaiting such a compound to study the role of Cav1.3 as a drug target for a number of indications as outlined above.
Xie et al. (2016) used cp‐PYT to investigate the role of Cav1.3 on steroidogenesis in the human adrenocortical cell line, H295R, and in primary human adrenal cells. They transfected H295R cells with wild‐type or gain‐of‐function Cav1.3 channel mutants previously identified as disease‐causing in APAs (Azizan et al., 2013). These mutant channels promote higher aldosterone production compared to wild‐type‐transfected cells, which was inhibited only by very high concentrations (100 μM) of cp‐PYT. Interestingly, low concentrations of the compound almost doubled aldosterone secretion in wild‐type transfected H295R cells and also stimulated secretion in the mutants. This effect was not observed with nifedipine (Xie et al., 2016). These findings strongly suggest that at these lower concentrations cp‐PYT activated Ca2+‐current through Cav1.3, in agreement with the channel‐activating properties in one of the studies in tsA‐201 cells (Ortner et al., 2014). As mentioned by the authors, T‐type channels also control aldosterone secretion. Based on the unknown activity of the compound on T‐type channels, its usage is not suitable to unambiguously prove a role of Cav1.3 on steroidogenesis.
Cav1.3 has also been implicated as a therapeutic target for SCI. After SCI most individuals develop involuntary muscle contractions, including spasms, as a chronic complication (Jiang et al., 2021; Marcantoni et al., 2020). Altered excitatory and inhibitory synaptic input into motoneurons as well as hyperexcitabilty of motoneurons seem to underlie this phenomenon. In motoneurons LTCC activity can generate persistent inward currents supporting plateau potentials from which neurons can sustain firing. This enhanced activity of plateau potentials after SCI correlates with spasticity (Marcantoni et al., 2020). Due to their more negative activation‐range and higher activity at subthreshold potentials, Cav1.3 channels have been implicated in this process (Li & Bennett, 2003). This has recently been confirmed in an elegant study by showing that prolonged treatment of mice with the Ca2+‐channel blocker nimodipine, starting early after SCI, can prevent the development of increased muscle tone and spontaneous spasms. This is mainly due to the inhibition of Cav1.3 as demonstrated by a dramatic decrease of SCI‐induced aberrant muscle activity by neuron‐specific and constitutive Cav1.3‐knockout in mice (Marcantoni et al., 2020).
The involvement of LTCCs in SCI‐induced hyperexcitability was further explored in a mouse model of acute and chronic SCI (SCI) (Jiang et al., 2021). Cp‐PYT was employed in an attempt to demonstrate the specific involvement of Cav1.3. At high micromolar concentrations this compound inhibited in particular long‐lasting root reflexes and plateau potentials and reduced motoneuron firings evoked by intracellular current injection in a concentration‐dependent manner. While their data suggest that cp‐PYT (like in recombinant systems) can inhibit Cav1.3 channels at high concentrations, these experiments do not provide evidence for selectivity of this compound because the contribution of other L‐type currents (presumably Cav1.2) in motoneurons is small (Marcantoni et al., 2020). The authors misleadingly state that low selectivity of cp‐PYT reported in the study of Huang et al. (Huang et al., 2014) ‘is due to the existence of splice variants of Cav1.3 channels, which have various biophysical and pharmacological properties’. While the latter is true (see above), none of the references cited by the authors has ever investigated the effect of alternative splicing on cp‐PYT action. They also showed a strong inhibition of long‐lasting root reflexes by inhibiting NMDA‐receptors (NMDAR) with ketamine. Since no information about the activity of cp‐PYT on glutamate receptors is provided, a contribution through this mechanism cannot be excluded. In contrast to Cav1.3‐knockout (Marcantoni et al., 2020), the unknown selectivity profile of this compound does not allow conclusions about which LTCC is involved in spinal spasticity after SCI.
Degoulet et al. (2016) employed isradipine and cp‐PYT to study the role of LTCCs for ventral tegmental area (VTA) synaptic plasticity and for cocaine‐associated contextual memory. Their study built on previous observations that LTCC activation during postsynaptic depolarization can induce synaptic plasticity in multiple brain areas, including the cerebral cortex, hippocampus, amygdala and striatum. In addition, systemic administration of LTCC antagonists blocks the acquisition of drug‐induced conditioned place preference (CPP), a form of Pavlovian contextual cue learning dependent on NMDAR‐mediated signalling in the VTA (Degoulet et al., 2016). Using an elegant combination of ex vivo VTA slice‐recordings and behavioural studies in rats they found strong effects of LTCC‐inhibition on synaptic plasticity in the VTA. Isradipine blocked the induction of NMDAR mediated long‐term potentiation (LTP) but also facilitated the reversal of previously induced LTP in the VTA. In behavioural experiments isradipine injected into the VTA suppressed the acquisition of cocaine‐paired contextual cue memory assessed using a CPP‐paradigm and it abolished previously acquired cocaine and alcohol CPP. Like isradipine, cp‐PYT (20 μM) also mediated depotentiation of LTP in slice recordings and abolished previously acquired cocaine and alcohol CPP. While this elegant study demonstrates a novel role for LTCCs in the VTA and provides compelling evidence for a link between the observed changes in synaptic plasticity and the behavioural outcomes, the data cannot provide evidence for a selectivity of cp‐PYT. Even if Cav1.3 channels alone are involved in the observed drug effects and are inhibited by cp‐PYT, no implications for its selectivity can be made unless it is known to what extent cp‐PYT can spare Cav1.2‐mediated currents, if present at all.
In an elegant paper, Plotkin et al. (2013) employed two‐photon laser scanning microscopy and patch‐clamp electrophysiology in striatal ex vivo brain slices to study the regulation of dendritic Ca2+ release in striatal spiny projection neurons (SPNs), which underlies the induction of corticostriatal long‐term depression in these neurons. They found that theta‐burst‐stimulated back‐propagating action‐potentials induce a Ca2+‐transient in dendritic spines, which required opening of voltage‐gated Ca2+ channels and activation of ryanodine receptors and was thus attributed to Ca2+‐induced Ca2+‐release (CICR). Interestingly, activation of group I metabotropic glutamate receptor (mGluRs) by itself did not affect intraspine Ca2+‐concentration in the absence of somatic action potentials but increased the activity‐dependent intraspine Ca2+‐transient in proximal and distal dendrites of indirect pathway SPNs. This modulation was dependent on the activation of Cav1.3 LTCCs because it could be blocked by isradipine and was absent in neurons from Cav1.3 knockout‐mice (Plotkin et al., 2013). To test if CICR is dependent on Cav1.3 Ca2+‐channels also in the absence of mGluR activation they employed cp‐PYT at a concentration of 20 μM. Only a minimal inhibition was observed and attributed to the presence of other Ca2+‐channels, including Cav1.2 and Cav2.3 also expressed in these cells (Plotkin et al., 2013). However, the data with cp‐PYT are limited by the low potency (and unknown selectivity) of this compound. The authors considered 20 μM a ‘saturating concentration’. Yet in patch‐clamp studies with recombinant Cav1.3‐channels, the concentrations required for 50% inhibition of Cav1.3‐currents was 24 μM, and could be decreased to about 10 μM only at very positive holding potentials (Cooper et al., 2020). Again, these experiments allowed no conclusions regarding the drug's Cav1.3‐selectivity.
Sanchez‐Padilla and colleagues (Sanchez‐Padilla et al., 2014) studied mitochondrial oxidant stress in locus coeruleus neurons in ex vivo mouse brain slices using electrophysiological and optical techniques. Similar to previous findings in SN DA neurons they found that autonomous activity produced phase‐locked dendritic intracellular Ca2+‐oscillations. These Ca2+‐transients elevated mitochondrial oxidant stress and were due to the activation of LTCCs. To test for a role of Cav1.3, a single concentration of cp‐PYT (50 μM) was tested on both dendritic Ca2+‐spikes (observed in the presence of tetrodotoxin) and associated Ca2+‐transients. This concentration of cp‐PYT attenuated the amplitude and frequency of Ca2+‐spikes and of intracellular Ca2+‐oscillations by about 50%. The residual activity was eliminated by isradipine, believed to block additional Cav1.2 channels, which are even expressed at higher abundance than Cav1.3 in these cells (Sanchez‐Padilla et al., 2014). Interestingly, although amplitudes were decreased by cp‐PYT, spikes and transients became much wider, reminiscent of the data from one study (Ortner et al., 2014) showing that the compound can slow activation and inactivation kinetics of both Cav1.3 and Cav1.2 channels, which may have both been modulated by the compound in these cells.
Two publications (Bhandage et al., 2020; Kanatani et al., 2017) found that Toxoplasma gondii‐infection triggers a GABAA ‐dependent hypermigratory phenotype in dendritic cells that requires Cav1.3 Ca2+‐signalling. Cav1.3 was the most abundant Ca2+‐channel α1‐subunit expressed in these cells and further up‐regulated upon infection. The DHPs nifedipine and benidipine (10 μM) abolished this hypermigratory response, an effect seen with an even lower concentration (1 μM) of cp‐PYT. While inhibition of Cav1.3 may explain this effect, Cav1.2 expression in these cells is absent or low (Bhandage et al., 2020; Kanatani et al., 2017), again not permitting any conclusions about selectivity.
Rey et al. (2020) employed cp‐PYT to investigate the role of LTCCs in regulating GABA release from cerebellar molecular layer interneurons (MLIs) in rats. LTCC activation by BayK8644 increased peak Ca2+‐currents in MLIs, whereas nimodipine and cp‐PYT had inhibitory effects. cp‐PYT also inhibited action potential‐evoked presynaptic Ca2+‐transients and mIPSC frequency in MLIs and Purkinje cells, implicating a presynaptic function of LTCCs in the control of GABA‐release from MLIs. The authors acknowledged published evidence regarding the uncertainty about cp‐PYT selectivity and therefore implicated Cav1.2 or Cav1.3 or a combination of both as responsible for the observed effects.
Given the wide‐spread use of cp‐PYT as a tool compound, it would be of upmost importance to gain a better understanding of its pharmacological profile, not only regarding selectivity for Cav1.3 over Cav1.2, but also with respect to other potential targets.
1.7. Sclareol as a natural neuroprotective Cav1.3‐antagonist
A very recent study proposed sclareol (Figure 2a), a constituent of the Mediterranean herb Salvia sclarea, as a bioactive compound that inhibits Cav1.3 more strongly than Cav1.2 (Wang et al., 2022). It was discovered in an elegant new cell‐based high‐throughput screening assay. Cav1.2 and Cav1.3 channel complexes were expressed in HEK‐293 cells together with reporter proteins expressed under a synthetic, NFAT3‐based Ca2+‐sensitive promotor which responds to K+‐depolarization‐induced Ca2+ entry through these LTCCs. Surprisingly, screening of 42 essential oil products identified five oils that inhibited Cav1.3 activity with minimal effects on Cav1.2. Using in silico virtual screening and deep learning, 13 hits were identified as the most promising Cav1.3 inhibitors, including (6)‐gingerol and sclareol. However, in their assay no relevant selectivity for these compounds (about 2‐fold, at low micromolar IC50 values for both substances, Wang et al., 2022) was found. Although ‘sclareol inhibits Cav1.3 more strongly than Cav1.2’ this small difference cannot be regarded as Cav1.3‐selective. Even though functional Cav1.2 and Cav1.3 channel complexes were expressed in their HEK‐293 cell screening assay, no data from patch‐clamp studies were presented to validate the pharmacological properties of this compound on L‐type Ca2+ currents.
Due to earlier findings (see above) suggesting a neuroprotective effect of Cav1.3 inhibition in early PD (see Liss & Striessnig, 2019; Surmeier, Halliday, & Simuni, 2017, for reviews) the authors tested whether this compound has neuroprotective properties in a mouse 6‐hydroxydopamine (6‐OHDA) PD model. Indeed, daily i.p. injections of this compound in mice 2 days before a single unilateral 6‐OHDA injection above the substantia nigra (and not as in most other studies into the striatum) largely prevented the about 50% reduction of staining for the DA neuron‐specific marker tyrosine‐hydroxylase in the ipsilateral dorsal striatum (Wang et al., 2022). This represents indirect evidence for intact SN DA neuron innervation of the striatum and indicates a neuroprotective effect of the drug. A direct, Cav1.3‐dependent neuroprotective effect, evident as the prevention of the loss of vulnerable SN DA neurons, has not been assessed. However, evidence for a protective effect was obtained in behavioural experiments in which locomotor impairments induced by the unilateral 6‐OHDA injections in the vehicle‐treated group (rotational behaviour, hyperlocomotion) were essentially absent in the sclareol‐treated animals. Nevertheless, these findings cannot be taken as a proof for a contribution of Cav1.3 in neuroprotection or in vivo inhibition of Cav1.3. In ex vivo patch‐clamp recordings of putative SN DA neurons in midbrain slices, 10 μM of sclareol caused a large neuronal hyperpolarization (by about 18 mV) accompanied by an increased spiking threshold upon current injection. Such hyperpolarization has not been reported by inhibiting LTCCs with isradipine or in Cav1.3‐deficient SN DA neurons (Guzman et al., 2009; Puopolo et al., 2007). The experiments do not exclude the possibility that the effects of this compound are mediated by other signalling pathways affecting SN DA excitability and do not provide evidence for in vivo activity on Cav1.3.
1.8. Cav1.3‐selective toxins
At present no natural compounds, including animal toxins, with Cav1.3‐selectivity have been described. Calciseptine and the structurally highly related FS2 (from black mamba venom) and calcicludin (from green mamba venom) are known LTCC blockers (Wang et al., 2007; Zamponi et al., 2015). In functional studies in Langendorf‐perfused hearts, Barrere et al. (2020) have recently obtained evidence for Cav1.2‐selectivity of calciseptine.
2. CONCLUSIONS
Efforts have been made to build on DHP scaffolds to discover new drugs acting more potently on different cardiac functions or in neurons as compared to vascular smooth muscle. Although a number of interesting compounds have been characterized none of them were rigorously tested for Cav1.3‐selectivity. We propose that such selectivity testing should include heterologous systems and/or isolation of native Cav1.3‐mediated (sinoatrial node, inner hair cells and mouse chromaffin cells) or Cav1.2‐mediated (vascular smooth muscle, cardiomyocytes) current components. The concept of defining the selectivity of compounds for Cav1.2 or Cav1.3 channels by performing functional and binding studies on atrial, ventricular and arterial smooth muscle, rat brain cortex and SH‐SY5Y cells without providing evidence for a direct engagement of Cav1.3 channels (such as by demonstrating L‐type current inhibition) is misleading in the important drug discovery process towards potent and highly selective Cav1.3 inhibitors. Cp‐PYT is so far the only compound with some evidence for Cav1.3‐selectivity. However, it is at present unclear under which experimental conditions this selectivity can be observed and solid data obtained in independent laboratories reached different conclusions regarding its potency and selectivity. Overinterpretation of existing data and uncritical reporting cause a substantial overestimation of the suitability of this compound as a Cav1.3‐selective pharmacological tool. Until its activity on other major ionic currents (especially Cav2 and Cav3 Ca2+‐channels) has been systematically explored, it should not be used as a pharmacological tool to confirm or refute a role of Cav1.3 channels in cellular responses. This also holds true for sclareol. We therefore conclude that at present there is no published evidence for a validated Cav1.3‐selective compound.
Despite the clear therapeutic rationale and the need for a selective tool for pharmacological studies, it thus appears that the discovery of a compound with selectivity for Cav1.3 over Cav1.2 still remains a considerable challenge.
2.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in the IUPHAR/BPS Guide to PHARMACOLOGY http://www.guidetopharmacology.org and are permanently archived in the Concise Guide to PHARMACOLOGY 2021/22 (Alexander, Christopoulos et al., 2021; Alexander, Mathie, et al., 2021).
AUTHOR CONTRIBUTION
All authors wrote the manuscript and reviewed the final version; LF and TK performed molecular modelling for the illustrations used in this review.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
Filippini, L. , Ortner, N. J. , Kaserer, T. , & Striessnig, J. (2023). Cav1.3‐selective inhibitors of voltage‐gated L‐type Ca2+ channels: Fact or (still) fiction? British Journal of Pharmacology, 180(10), 1289–1303. 10.1111/bph.16060
Funding information This work was supported by the Austrian Science Fund (FWF P35722 to JS; P35087 to NJO), the Erika‐Cremer habilitation fellowship by the University of Innsbruck (to NJO) and the Tyrolean Science Fund (P7400‐037‐011 to LF).
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article because no new data were created or analysed in this study. Data for the molecular models used for illustration are available from the corresponding author upon request.
REFERENCES
- Alexander, S. P. , Christopoulos, A. , Davenport, A. P. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Abbracchio, M. P. , & CGTP Collaborators . (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: G protein‐coupled receptors. British Journal of Pharmacology, 178(S1), S27–S156. 10.1111/bph.15538 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. , Mathie, A. , Peters, J. A. , Veale, E. L. , Striessnig, J. , Kelly, E. , Armstrong, J. F. , Faccenda, E. , Harding, S. D. , Pawson, A. J. , Southan, C. , Davies, J. A. , Aldrich, R. W. , Attali, B. , Baggetta, A. M. , Becirovic, E. , Biel, M. , Bill, R. M. , Catterall, W. A. , … Zhu, M. (2021). THE CONCISE GUIDE TO PHARMACOLOGY 2021/22: Ion channels. British Journal of Pharmacology, 178(S1), S157–S245. 10.1111/bph.15539 [DOI] [PubMed] [Google Scholar]
- Avery, R. B. , & Johnston, D. (1996). Multiple channel types contribute to the low‐voltage‐activated calcium current in hippocampal CA3 pyramidal neurons. The Journal of Neuroscience, 16, 5567–5582. 10.1523/JNEUROSCI.16-18-05567.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azizan, E. A. B. , Poulsen, H. , Tuluc, P. , Zhou, J. , Clausen, M. V. , Lieb, A. , Maniero, C. , Garg, S. , Bochukova, E. G. , Zhao, W. , Shaikh, L. H. , Brighton, C. A. , Teo, A. E. D. , Davenport, A. P. , Dekkers, T. , Tops, B. , Küsters, B. , Ceral, J. , Yeo, G. S. H. , … Brown, M. J. (2013). Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nature Genetics, 45, 1055–1060. 10.1038/ng.2716 [DOI] [PubMed] [Google Scholar]
- Baig, S. M. , Koschak, A. , Lieb, A. , Gebhart, M. , Dafinger, C. , Nürnberg, G. , Ali, A. , Ahmad, I. , Sinnegger‐Brauns, M. J. , Brandt, N. , Engel, J. , Mangoni, M. E. , Farooq, M. , Khan, H. U. , Nürnberg, P. , Striessnig, J. , & Bolz, H. J. (2011). Loss of Cav1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nature Neuroscience, 14, 77–84. 10.1038/nn.2694 [DOI] [PubMed] [Google Scholar]
- Barrere, C. , Chinda, K. , Mesirca, P. , Aurélie, C. , Diochot, S. , Lazdunski, M. , Barrere‐Lemaire, S. , Mangoni, M. , & Nargeot, J. (2020). Evidence for a selective blockade of Cav1.2 versus Cav1.3 by the mamba toxin calciseptine in the mouse heart. Arch Cardiovasc Dis Suppl, 12, 259. [Google Scholar]
- Bean, B. P. (1984). Nitrendipine block of cardiac calcium channels: High‐affinity binding to the inactivated state. Proceedings of the National Academy of Sciences of the United States of America, 81, 6388–6392. 10.1073/pnas.81.20.6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benkert, J. , Hess, S. , Roy, S. , Beccano‐Kelly, D. , Wiederspohn, N. , Duda, J. , Simons, C. , Patil, K. , Gaifullina, A. , Mannal, N. , Dragicevic, E. , Spaich, D. , Müller, S. , Nemeth, J. , Hollmann, H. , Deuter, N. , Mousba, Y. , Kubisch, C. , Poetschke, C. , … Liss, B. (2019). Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson's disease. Nature Communications, 10, 5094. 10.1038/s41467-019-12834-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhandage, A. K. , Olivera, G. C. , Kanatani, S. , Thompson, E. , Loré, K. , Varas‐Godoy, M. , & Barragan, A. (2020). A motogenic GABAergic system of mononuclear phagocytes facilitates dissemination of coccidian parasites. eLife, 9, e60528. 10.7554/eLife.60528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billingsley, K. J. , Manca, M. , Gianfrancesco, O. , Collier, D. A. , Sharp, H. , Bubb, V. J. , & Quinn, J. P. (2018). Regulatory characterisation of the schizophrenia‐associated CACNA1C proximal promoter and the potential role for the transcription factor EZH2 in schizophrenia aetiology. Schizophrenia Research, 199, 168–175. 10.1016/j.schres.2018.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budriesi, R. , Ioan, P. , Locatelli, A. , Cosconati, S. , Leoni, A. , Ugenti, M. P. , Andreani, A. , di Toro, R. , Bedini, A. , Spampinato, S. , Marinelli, L. , Novellino, E. , & Chiarini, A. (2008). Imidazo[2,1‐b]thiazole system: A scaffold endowing dihydropyridines with selective cardiodepressant activity. Journal of Medicinal Chemistry, 51, 1592–1600. 10.1021/jm070681+ [DOI] [PubMed] [Google Scholar]
- Buraei, Z. , & Yang, J. (2010). The ß subunit of voltage‐gated Ca2+ channels. Physiological Reviews, 90, 1461–1506. 10.1152/physrev.00057.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall, W. A. , Lenaeus, M. J. , & Gamal El‐Din, T. M. (2020). Structure and pharmacology of voltage‐gated sodium and calcium channels. Annual Review of Pharmacology and Toxicology, 60, 133–154. 10.1146/annurev-pharmtox-010818-021757 [DOI] [PubMed] [Google Scholar]
- Chang, C. C. , Cao, S. , Kang, S. , Kai, L. , Tian, X. , Pandey, P. , Dunne, S. F. , Luan, C. H. , Surmeier, D. J. , & Silverman, R. B. (2010). Antagonism of 4‐substituted 1,4‐dihydropyridine‐3,5‐dicarboxylates toward voltage‐dependent L‐type ca2+ channels ca V 1.3 and Ca V 1.2. Bioorganic & Medicinal Chemistry, 18, 3147–3158. 10.1016/j.bmc.2010.03.038 [DOI] [PubMed] [Google Scholar]
- Cooper, G. , Kang, S. , Perez‐Rosello, T. , Guzman, J. N. , Galtieri, D. , Xie, Z. , Kondapalli, J. , Mordell, J. , Silverman, R. B. , & Surmeier, D. J. (2020). A single amino acid determines the selectivity and efficacy of selective negative allosteric modulators of Cav1.3 L‐type calcium channels. ACS Chemical Biology, 15, 2539–2550. 10.1021/acschembio.0c00577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper, G. , Kang, S. , Perez‐Rosello, T. , Guzman, J. N. , Galtieri, D. , Xie, Z. , Kondapalli, J. , Mordell, J. , Silverman, R. B. , & Surmeier, D. J. (2021). Correction to a single amino acid determines the selectivity and efficacy of selective negative allosteric modulators of Cav1.3 L‐type calcium channels. ACS Chemical Biology, 16, 1299–1300. 10.1021/acschembio.1c00368 [DOI] [PubMed] [Google Scholar]
- Degoulet, M. , Stelly, C. E. , Ahn, K. C. , & Morikawa, H. (2016). L‐type Ca2+ channel blockade with antihypertensive medication disrupts VTA synaptic plasticity and drug‐associated contextual memory. Molecular Psychiatry, 21, 394–402. 10.1038/mp.2015.84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fossat, P. , Dobremez, E. , Bouali‐Benazzouz, R. , Favereaux, A. , Bertrand, S. S. , Kilk, K. , Leger, C. , Cazalets, J. R. , Langel, U. , Landry, M. , & Nagy, F. (2010). Knockdown of L calcium channel subtypes: Differential effects in neuropathic pain. The Journal of Neuroscience, 30, 1073–1085. 10.1523/JNEUROSCI.3145-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. , & Yan, N. (2021). Structural basis of the modulation of the voltage‐gated calcium Ion Channel Cav 1.1 by Dihydropyridine compounds. Angewandte Chemie (International Ed. in English), 60, 3131–3137. 10.1002/anie.202011793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, J. N. , Sanchez‐Padilla, J. , Chan, C. S. , & Surmeier, D. J. (2009). Robust pacemaking in substantia nigra dopaminergic neurons. The Journal of Neuroscience, 29, 11011–11019. 10.1523/JNEUROSCI.2519-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman, J. N. , Sanchez‐Padilla, J. , Wokosin, D. , Kondapalli, J. , Ilijic, E. , Schumacker, P. T. , & Surmeier, D. J. (2010). Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ‐1. Nature, 468, 696–700. 10.1038/nature09536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison, P. J. , Tunbridge, E. M. , Dolphin, A. C. , & Hall, J. (2019). Voltage‐gated calcium channel blockers for psychiatric disorders: Genomic reappraisal. British Journal of Psychiatry, 216, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. , Ng, C. Y. , Yu, D. , Zhai, J. , Lam, Y. , & Soong, T. W. (2014). Modest Cav1.342‐selective inhibition by compound 8 is beta‐subunit dependent. Nature Communications, 5, 4481. 10.1038/ncomms5481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, H. , Yu, D. , & Soong, T. W. (2013). C‐terminal alternative splicing of Cav1.3 channels distinctively modulates their dihydropyridine sensitivity. Molecular Pharmacology, 84, 643–653. 10.1124/mol.113.087155 [DOI] [PubMed] [Google Scholar]
- Jiang, M. C. , Birch, D. V. , Heckman, C. J. , & Tysseling, V. M. (2021). The involvement of Cav1.3 channels in prolonged root reflexes and its potential as a therapeutic target in spinal cord injury. Frontiers in Neural Circuits, 15, 642111. 10.3389/fncir.2021.642111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, Y. , Xu, B. , Chen, J. , Sui, Y. , Ren, L. , Li, J. , Zhang, H. , Guo, L. , & Sun, X. (2018). Micro‐RNA‐137 inhibits tau hyperphosphorylation in Alzheimer's disease and targets the CACNA1C gene in transgenic mice and human neuroblastoma SH‐SY5Y cells. Medical Science Monitor, 24, 5635–5644. 10.12659/MSM.908765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatani, S. , Fuks, J. M. , Olafsson, E. B. , Westermark, L. , Chambers, B. , Varas‐Godoy, M. , Uhlén, P. , & Barragan, A. (2017). Voltage‐dependent calcium channel signaling mediates GABAA receptor‐induced migratory activation of dendritic cells infected by toxoplasma gondii. PLoS Pathogens, 13, e1006739. 10.1371/journal.ppat.1006739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, S. , Cooper, G. , Dunne, S. F. , Dusel, B. , Luan, C.‐H. , Surmeier, D. J. , & Silverman, R. B. (2012). CaV1.3‐selective L‐type calcium channel antagonists as potential new therapeutics for Parkinson's disease. Nature Communications, 3, 1146. 10.1038/ncomms2149 [DOI] [PubMed] [Google Scholar]
- Kang, S. , Cooper, G. , Dunne, S. F. , Luan, C.‐H. , Surmeier, D. J. , & Silverman, R. B. (2013a). Antagonism of L‐type Ca2+ channels CaV1.3 and CaV1.2 by 1,4‐dihydropyrimidines and 4H‐pyrans as dihydropyridine mimics. Bioorganic & Medicinal Chemistry, 21, 4365–4373. 10.1016/j.bmc.2013.04.054 [DOI] [PubMed] [Google Scholar]
- Kang, S. , Cooper, G. , Dunne, S. F. , Luan, C. H. , Surmeier, D. J. , & Silverman, R. B. (2013b). Structure–activity relationship of N,N′‐Disubstituted Pyrimidinetriones as CaV1.3 Calcium Channel‐selective antagonists for Parkinson's disease. Journal of Medicinal Chemistry, 56, 4786–4797. 10.1021/jm4005048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korah, H. E. , & Scholl, U. I. (2015). An update on familial hyperaldosteronism. Hormone and Metabolic Research, 47, 941–946. 10.1055/s-0035-1564166 [DOI] [PubMed] [Google Scholar]
- Koschak, A. , Reimer, D. , Huber, I. , Grabner, M. , Glossmann, H. , Engel, J. , & Striessnig, J. (2001). α1D (Cav1.3) subunits can form L‐type calcium channels activating at negative voltages. The Journal of Biological Chemistry, 276, 22100–22106. 10.1074/jbc.M101469200 [DOI] [PubMed] [Google Scholar]
- Kunze, D. L. , & Rampe, D. (1992). Characterization of the effects of a new Ca2+ channel activator, FPL 64176, in GH3 cells. Molecular Pharmacology, 42, 666–670. [PubMed] [Google Scholar]
- Lee, K. S. , & Tsien, R. W. (1983). Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature, 302, 790–794. 10.1038/302790a0 [DOI] [PubMed] [Google Scholar]
- Li, Y. , & Bennett, D. J. (2003). Persistent sodium and calcium currents cause plateau potentials in Motoneurons of chronic spinal rats. Journal of Neurophysiology, 90, 857–869. 10.1152/jn.00236.2003 [DOI] [PubMed] [Google Scholar]
- Liao, P. , Yu, D. , Li, G. , Yong, T. F. , Soon, J. L. , Chua, Y. L. , & Soong, T. W. (2007). A smooth muscle Cav1.2 calcium channel splice variant underlies hyperpolarized window current and enhanced state‐dependent inhibition by nifedipine. The Journal of Biological Chemistry, 282, 35133–35142. 10.1074/jbc.M705478200 [DOI] [PubMed] [Google Scholar]
- Lieb, A. , Scharinger, A. , Sartori, S. B. , Sinnegger‐Brauns, M. J. , & Striessnig, J. (2012). Structural determinants of Cav 1.3 L‐type calcium channel gating. Channels, 6, 197–205. 10.4161/chan.21002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss, B. , & Striessnig, J. (2019). The potential of L‐type calcium channels as a drug target for neuroprotective therapy in Parkinson's disease. Annual Review of Pharmacology and Toxicology, 59, 263–289. 10.1146/annurev-pharmtox-010818-021214 [DOI] [PubMed] [Google Scholar]
- Locatelli, A. , Cosconati, S. , Micucci, M. , Leoni, A. , Marinelli, L. , Bedini, A. , Ioan, P. , Spampinato, S. M. , Novellino, E. , Chiarini, A. , & Budriesi, R. (2013). Ligand based approach to L‐type calcium channel by imidazo[2,1‐b]thiazole‐1,4‐dihydropyridines: From heart activity to brain affinity. Journal of Medicinal Chemistry, 56, 3866–3877. 10.1021/jm301839q [DOI] [PubMed] [Google Scholar]
- Mangoni, M. E. , Couette, B. , Bourinet, E. , Platzer, J. , Reimer, D. , Striessnig, J. , & Nargeot, J. (2003). Functional role of L‐type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proceedings of the National Academy of Sciences of the United States of America, 100, 5543–5548. 10.1073/pnas.0935295100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni, A. , Vandael, D. H. , Mahapatra, S. , Carabelli, V. , Sinnegger‐Brauns, M. J. , Striessnig, J. , & Carbone, E. (2010). Loss of Cav1.3 channels reveals the critical role of L‐type and BK channel coupling in pacemaking mouse adrenal chromaffin cells. The Journal of Neuroscience, 30, 491–504. 10.1523/JNEUROSCI.4961-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni, M. , Fuchs, A. , Low, P. , Bartsch, D. , Kiehn, O. , & Bellardita, C. (2020). Early delivery and prolonged treatment with nimodipine prevents the development of spasticity after spinal cord injury in mice. Science Translational Medicine, 12, eaay0167. 10.1126/scitranslmed.aay0167 [DOI] [PubMed] [Google Scholar]
- Mikami, A. , Imoto, K. , Tanabe, T. , Niidome, T. , Mori, Y. , Takeshima, H. , Narumiya, S. , & Numa, S. (1989). Primary structure and functional expression of the cardiac dihydropyridine‐sensitive calcium channel. Nature, 340, 230–233. 10.1038/340230a0 [DOI] [PubMed] [Google Scholar]
- Nishimoto, K. , Tomlins, S. A. , Kuick, R. , Cani, A. K. , Giordano, T. J. , Hovelson, D. H. , Liu, C. J. , Sanjanwala, A. R. , Edwards, M. A. , Gomez‐Sanchez, C. E. , Nanba, K. , & Rainey, W. E. (2015). Aldosterone‐stimulating somatic gene mutations are common in normal adrenal glands. Proceedings of the National Academy of Sciences of the United States of America, 112, E4591–E4599. 10.1073/pnas.1505529112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omata, K. , Anand, S. K. , Hovelson, D. H. , Liu, C.‐J. , Yamazaki, Y. , Nakamura, Y. , Ito, S. , Satoh, F. , Sasano, H. , Rainey, W. E. , & Tomlins, S. A. (2017). Aldosterone‐producing cell clusters frequently harbor somatic mutations and accumulate with age in Normal adrenals. Journal of the Endocrine Society, 1, 787–799. 10.1210/js.2017-00134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner, N. J. (2021). Voltage‐gated Ca2+ channels in dopaminergic substantia Nigra neurons: Therapeutic targets for neuroprotection in Parkinson's disease? Frontiers in Synaptic Neuroscience, 13, 636103. 10.3389/fnsyn.2021.636103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner, N. J. , Bock, G. , Dougalis, A. , Kharitonova, M. , Duda, J. , Hess, S. , Tuluc, P. , Pomberger, T. , Stefanova, N. , Pitterl, F. , Ciossek, T. , Oberacher, H. , Draheim, H. J. , Kloppenburg, P. , Liss, B. , & Striessnig, J. (2017). Lower affinity of Isradipine for L‐type Ca2+ channels during substantia Nigra dopamine neuron‐like activity: Implications for neuroprotection in Parkinson's disease. The Journal of Neuroscience, 37, 6761–6777. 10.1523/JNEUROSCI.2946-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner, N. J. , Bock, G. , Vandael, D. H. , Mauersberger, R. , Draheim, H. J. , Gust, R. , Carbone, E. , Tuluc, P. , & Striessnig, J. (2014). Pyrimidine‐2,4,6‐triones are a new class of voltage‐gated L‐type Ca2+ channel activators. Nature Communications, 5, 3897. 10.1038/ncomms4897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner, N. J. , Kaserer, T. , Copeland, J. N. , & Striessnig, J. (2020). De novo CACNA1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflügers Archiv, 472, 755–773. 10.1007/s00424-020-02418-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortner, N. J. , Pinggera, A. , Hofer, N. T. , Siller, A. , Brandt, N. , Raffeiner, A. , Vilusic, K. , Lang, I. , Blum, K. , Obermair, G. J. , Stefan, E. , Engel, J. , & Striessnig, J. (2020). RBP2 stabilizes slow Cav1.3 Ca2+ channel inactivation properties of cochlear inner hair cells. Pflügers Archiv, 472, 3–25. 10.1007/s00424-019-02338-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson Study Group STEADY‐PD III Investigators . (2020). Isradipine versus placebo in early Parkinson disease: A randomized trial. Annals of Internal Medicine, 172, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platzer, J. , Engel, J. , Schrott‐Fischer, A. , Stephan, K. , Bova, S. , Chen, H. , Zheng, H. , & Striessnig, J. (2000). Congenital deafness and sinoatrial node dysfunction in mice lacking class D L‐type Ca2+ channels. Cell, 102, 89–97. 10.1016/S0092-8674(00)00013-1 [DOI] [PubMed] [Google Scholar]
- Plotkin, J. L. , Shen, W. , Rafalovich, I. , Sebel, L. E. , Day, M. , Chan, C. S. , & Surmeier, D. J. (2013). Regulation of dendritic calcium release in striatal spiny projection neurons. Journal of Neurophysiology, 110, 2325–2336. 10.1152/jn.00422.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poetschke, C. , Dragicevic, E. , Duda, J. , Benkert, J. , Dougalis, A. , DeZio, R. , Snutch, T. P. , Striessnig, J. , & Liss, B. (2015). Compensatory T‐type Ca2+ channel activity alters D2‐autoreceptor responses of substantia nigra dopamine neurons from Cav1.3 L‐type Ca2+ channel knockout mice. Scientific Reports, 5, 13688. 10.1038/srep13688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo, M. , Raviola, E. , & Bean, B. P. (2007). Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. The Journal of Neuroscience, 27, 645–656. 10.1523/JNEUROSCI.4341-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwani, H. , Lopez‐Gonzalez, M. J. , Cattaert, D. , Roca‐Lapirot, O. , Dobremez, E. , Bouali‐Benazzouz, R. , Eiríksdóttir, E. , Langel, Ü. , Favereaux, A. , Errami, M. , Landry, M. , & Fossat, P. (2016). Cav1.2 and Cav1.3 L‐type calcium channels independently control short‐ and long‐term sensitization to pain. The Journal of Physiology, 594, 6607–6626. 10.1113/JP272725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve, H. L. , Vaughan, P. F. T. , & Peers, C. (1994). Calcium Channel currents in undifferentiated human neuroblastoma (SH‐SY5Y) cells: Actions and possible interactions of Dihydropyridines and ω‐Conotoxin. The European Journal of Neuroscience, 6, 943–952. 10.1111/j.1460-9568.1994.tb00588.x [DOI] [PubMed] [Google Scholar]
- Reinbothe, T. M. , Alkayyali, S. , Ahlqvist, E. , Tuomi, T. , Isomaa, B. , Lyssenko, V. , & Renström, E. (2013). The human L‐type calcium channel Cav1.3 regulates insulin release and polymorphisms in CACNA1D associate with type 2 diabetes. Diabetologia, 56, 340–349. 10.1007/s00125-012-2758-z [DOI] [PubMed] [Google Scholar]
- Rey, S. , Maton, G. , Satake, S. , Llano, I. , Kang, S. , Surmeier, D. J. , Silverman, R. B. , & Collin, T. (2020). Physiological involvement of presynaptic L‐type voltage‐dependent calcium channels in GABA release of cerebellar molecular layer interneurons. Journal of Neurochemistry, 155, 390–402. 10.1111/jnc.15100 [DOI] [PubMed] [Google Scholar]
- Roca‐Lapirot, O. , Radwani, H. , Aby, F. , Nagy, F. , Landry, M. , & Fossat, P. (2018). Calcium signalling through L‐type calcium channels: Role in pathophysiology of spinal nociceptive transmission: L‐type calcium channels and pain pathophysiology. British Journal of Pharmacology, 175, 2362–2374. 10.1111/bph.13747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez‐Padilla, J. , Guzman, J. N. , Ilijic, E. , Kondapalli, J. , Galtieri, D. J. , Yang, B. , Schieber, S. , Oertel, W. , Wokosin, D. , Schumacker, P. T. , & Surmeier, D. J. (2014). Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nature Neuroscience, 17, 832–840. 10.1038/nn.3717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholl, U. I. , Goh, G. , Stölting, G. , de Oliveira, R. C. , Choi, M. , Overton, J. D. , Fonseca, A. L. , Korah, R. , Starker, L. F. , Kunstman, J. W. , Prasad, M. L. , Hartung, E. A. , Mauras, N. , Benson, M. R. , Brady, T. , Shapiro, J. R. , Loring, E. , Nelson‐Williams, C. , Libutti, S. K. , … Lifton, R. P. (2013). Somatic and germline CACNA1D calcium channel mutations in aldosterone‐producing adenomas and primary aldosteronism. Nature Genetics, 45, 1050–1054. 10.1038/ng.2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns, M. J. , Hetzenauer, A. , Huber, I. G. , Renström, E. , Wietzorrek, G. , Berjukov, S. , Cavalli, M. , Walter, D. , Koschak, A. , Waldschütz, R. , Hering, S. , Bova, S. , Rorsman, P. , Pongs, O. , Singewald, N. , & Striessnig, J. (2004a). Isoform‐specific regulation of mood behavior and pancreatic beta cell and cardiovascular function by L‐type Ca2+ channels. The Journal of Clinical Investigation, 113, 1430–1439. 10.1172/JCI20208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns, M. J. , Hetzenauer, A. , Huber, I. G. , Renström, E. , Wietzorrek, G. , Berjukov, S. , Cavalli, M. , Walter, D. , Koschak, A. , Waldschütz, R. , Hering, S. , Bova, S. , Rorsman, P. , Pongs, O. , Singewald, N. , & Striessnig, J. (2004b). Isoform‐specific regulation of mood behavior and pancreatic β cell and cardiovascular function by L‐type Ca2+ channels. The Journal of Clinical Investigation, 113, 1430–1439. 10.1172/JCI20208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger‐Brauns, M. J. , Huber, I. G. , Koschak, A. , Wild, C. , Obermair, G. J. , Einzinger, U. , Hoda, J. C. , Sartori, S. B. , & Striessnig, J. (2009). Expression and 1,4‐dihydropyridine‐binding properties of brain L‐type calcium channel isoforms. Molecular Pharmacology, 75, 407–414. 10.1124/mol.108.049981 [DOI] [PubMed] [Google Scholar]
- Sousa, S. R. , Vetter, I. , Ragnarsson, L. , & Lewis, R. J. (2013). Expression and pharmacology of endogenous Cav channels in SH‐SY5Y human neuroblastoma cells. PLoS ONE, 8, e59293. 10.1371/journal.pone.0059293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig, J. (2021a). Calcium Channel Blockers. In Ortner N. J. (Ed.), Encycl Mol Pharmacol (pp. 375–383). Springer International Publishing. [Google Scholar]
- Striessnig, J. (2021b). Voltage‐gated Ca2+‐channel alpha1‐subunit de novo missense mutations: Gain or loss of function ‐ implications for potential therapies. Frontiers in Synaptic Neuroscience, 13, 634760. 10.3389/fnsyn.2021.634760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig, J. , Grabner, M. , Mitterdorfer, J. , Hering, S. , Sinnegger, M. J. , & Glossmann, H. (1998). Structural basis of drug binding to L Ca2+ channels. Trends in Pharmacological Sciences, 19, 108–115. 10.1016/S0165-6147(98)01171-7 [DOI] [PubMed] [Google Scholar]
- Sun, Y. , Zhang, H. , Selvaraj, S. , Sukumaran, P. , Lei, S. , Birnbaumer, L. , & Singh, B. B. (2017). Inhibition of L‐type Ca2+ channels by TRPC1‐STIM1 complex is essential for the protection of dopaminergic neurons. The Journal of Neuroscience, 37, 3364–3377. 10.1523/JNEUROSCI.3010-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. , Halliday, G. M. , & Simuni, T. (2017). Calcium, mitochondrial dysfunction and slowing the progression of Parkinson's disease. Experimental Neurology, 298, 202–209. 10.1016/j.expneurol.2017.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier, D. J. , Obeso, J. A. , & Halliday, G. M. (2017). Selective neuronal vulnerability in Parkinson disease. Nature Reviews. Neuroscience, 18, 101–113. 10.1038/nrn.2016.178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, Z. Z. , Hong, X. , Wang, J. , & Soong, T. W. (2007). Signature combinatorial splicing profiles of rat cardiac‐ and smooth‐muscle Cav1.2 channels with distinct biophysical properties. Cell Calcium, 41, 417–428. 10.1016/j.ceca.2006.08.002 [DOI] [PubMed] [Google Scholar]
- Tenti, G. , Parada, E. , León, R. , Egea, J. , Martínez‐Revelles, S. , Briones, A. M. , Sridharan, V. , López, M. G. , Ramos, M. T. , & Menéndez, J. C. (2014). New 5‐unsubstituted Dihydropyridines with improved Cav 1.3 selectivity as potential neuroprotective agents against ischemic injury. Journal of Medicinal Chemistry, 57, 4313–4323. 10.1021/jm500263v [DOI] [PubMed] [Google Scholar]
- Torrente, A. G. , Mesirca, P. , Bidaud, I. , & Mangoni, M. E. (2020). Channelopathies of voltage‐gated L‐type Cav1.3/alpha1D and T‐type Cav3.1/alpha1G Ca2+ channels in dysfunction of heart automaticity. Pflügers Archiv, 472, 817–830. 10.1007/s00424-020-02421-1 [DOI] [PubMed] [Google Scholar]
- Wang, H. , Xie, M. , Rizzi, G. , Li, X. , Tan, K. , & Fussenegger, M. (2022). Identification of Sclareol as a natural neuroprotective Cav 1.3‐antagonist using synthetic Parkinson‐mimetic gene circuits and computer‐aided drug discovery. Advanced Science (Weinheim, Baden‐Wurttemberg, Germany), 9, e2102855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Du, L. , & Peterson, B. Z. (2007). Calcicludine binding to the outer pore of L‐type calcium channels is allosterically coupled to dihydropyridine binding. Biochemistry, 46, 7590–7598. 10.1021/bi7001696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y. , Connors, R. , Fan, P. , Wang, X. , Wang, Z. , Liu, J. , Kayser, F. , Medina, J. C. , Johnstone, S. , Xu, H. , Thibault, S. , Walker, N. , Conn, M. , Zhang, Y. , Liu, Q. , Grillo, M. P. , Motani, A. , Coward, P. , & Wang, Z. (2014). Structure‐assisted discovery of the first non‐retinoid ligands for retinol‐binding protein 4. Bioorganic & Medicinal Chemistry Letters, 24, 2885–2891. 10.1016/j.bmcl.2014.04.089 [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Tang, S. , Harvey, K. E. , Salyer, A. E. , Li, T. A. , Rantz, E. K. , Lill, M. A. , & Hockerman, G. H. (2018). Molecular determinants of the differential modulation of Cav 1.2 and Cav 1.3 by Nifedipine and FPL 64176. Molecular Pharmacology, 94, 973–983. 10.1124/mol.118.112441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Yan, Z. , Li, Z. , Qian, X. , Lu, S. , Dong, M. , Zhou, Q. , & Yan, N. (2016). Structure of the voltage‐gated calcium channel Cav1.1 at 3.6 Å resolution. Nature, 537, 191–196. 10.1038/nature19321 [DOI] [PubMed] [Google Scholar]
- Xia, G. , Benmohamed, R. , Kim, J. , Arvanites, A. C. , Morimoto, R. I. , Ferrante, R. J. , Kirsch, D. R. , & Silverman, R. B. (2011). Pyrimidine‐2,4,6‐trione derivatives and their inhibition of mutant SOD1‐dependent protein aggregation. Toward a treatment for amyotrophic lateral sclerosis. Journal of Medicinal Chemistry, 54, 2409–2421. 10.1021/jm101549k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie, C. B. , Haris Shaikh, L. , Garg, S. , Tanriver, G. , Teo, A. E. D. , Zhou, J. , Maniero, C. , Zhao, W. , Kang, S. , Silverman, R. B. , Azizan, E. A. B. , & Brown, M. J. (2016). Regulation of aldosterone secretion by Cav1.3. Scientific Reports, 6, 24697. 10.1038/srep24697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , & Lipscombe, D. (2001). Neuronal Cav1.3α1 L‐type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. The Journal of Neuroscience, 21, 5944–5951. 10.1523/JNEUROSCI.21-16-05944.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, T. , He, M. , Zhang, H. , Barrett, P. Q. , & Hu, C. (2020). L‐ and T‐type calcium channels control aldosterone production from human adrenals. The Journal of Endocrinology, 244, 237–247. 10.1530/JOE-19-0259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao, X. , Gao, S. , & Yan, N. (2022). Structural basis for pore blockade of human voltage‐gated calcium channel Cav1.3 by motion sickness drug cinnarizine. Cell Research, 32, 946–948. 10.1038/s41422-022-00663-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi, G. W. , Striessnig, J. , Koschak, A. , & Dolphin, A. C. (2015). The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacological Reviews, 67, 821–870. 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Y. , Huang, G. , Wu, J. , Wu, Q. , Gao, S. , Yan, Z. , Lei, J. , & Yan, N. (2019). Molecular basis for ligand modulation of a mammalian voltage‐gated Ca2+ channel. Cell, 177, 1495–1506. 10.1016/j.cell.2019.04.043 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article because no new data were created or analysed in this study. Data for the molecular models used for illustration are available from the corresponding author upon request.