

Abstract
The mitochondrion is a gatekeeper of apoptotic processes, and mediates drug resistance to several chemotherapy agents used to treat cancer. Neuroblastoma is a common solid cancer in young children with poor clinical outcomes following conventional chemotherapy. We sought druggable mitochondrial protein targets in neuroblastoma cells. Among mitochondria‐associated gene targets, we found that high expression of the mitochondrial adenine nucleotide translocase 2 (SLC25A5/ANT2), was a strong predictor of poor neuroblastoma patient prognosis and contributed to a more malignant phenotype in pre‐clinical models. Inhibiting this transporter with PENAO reduced cell viability in a panel of neuroblastoma cell lines in a TP53‐status‐dependant manner. We identified the histone deacetylase inhibitor, suberanilohydroxamic acid (SAHA), as the most effective drug in clinical use against mutant TP53 neuroblastoma cells. SAHA and PENAO synergistically reduced cell viability, and induced apoptosis, in neuroblastoma cells independent of TP53‐status. The SAHA and PENAO drug combination significantly delayed tumour progression in pre‐clinical neuroblastoma mouse models, suggesting that these clinically advanced inhibitors may be effective in treating the disease.
Keywords: neuroblastoma, PENAO, SAHA, SLC25A5
What's new?
Therapies that target the mitochondria can have a strong apoptotic effect and also increase the effectiveness of standard chemotherapy. Here, the authors went looking for druggable mitochondrial protein targets in neuroblastoma cells. They found that high expression of the mitochondrial adenine nucleotide translocase 2 (SLC25A5/ANT2) predicted poor prognosis in NB patients. Inhibiting this mitochondrial protein with phenylarsonous acid (PENAO) in combination with suberanilohydroxamic acid (SAHA) treatment delayed tumour progression of neuroblastoma in mouse models, regardless of TP53 mutation status. The two drugs worked synergistically against drug‐resistant, TP53‐mutant NB xenograft tumours.
Abbreviations
- CDDP
cisplatin
- CoxPH
cox proportional‐hazards
- CPA
cyclophosphamide
- DBD
DNA binding domain
- DHE
dihydroethidium
- DMEM
Dulbecco's modified eagle medium
- EFS
event free survival
- FBS
foetal bovine serum
- GDSC
genomics of drug sensitivity in cancer
- GSEA
gene set enrichment analysis
- GSH
glutathione
- HDACi
histone deacetylase inhibitor
- LBH589
Panobinostat
- MAGs
mitochondria‐associated genes
- MEM
minimum essential media
- MSigDB
molecular signatures database
- NB
neuroblastoma
- OS
overall survival
- PENAO
phenylarsonous acid
- ROS
reactive oxygen species
- RPMI
Roswell Park Memorial Institute 1640 medium
- SAHA
suberanilohydroxamic acid
- SLC25A5/ANT2
the mitochondrial adenine nucleotide translocase 2
- TH
tyrosine hydroxylase
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labelling
- VCR
vincristine
- VP16
etoposide
1. INTRODUCTION
Mitochondria‐targeted therapies are sought in the treatment of cancer as they produce a strong apoptotic phenotype and potentiate treatment responses to standard chemotherapy. 1 , 2 Successful small molecule inhibition of mitochondrial processes in neuroblastoma (NB) using U.S. FDA‐approved compounds such as venetoclax, arsenic trioxide, MIBG, metformin and phenformin provide a rationale for exploring potential drug targets among mitochondria‐associated genes. 3 , 4 One of the greatest barriers that mitochondrial‐targeted agents face is the dysregulation of the master tumour suppressor TP53, which is required for the efficient induction of apoptosis. 5 Two recent large‐scale pan‐cancer genome studies have revealed TP53 to be among the most common somatically mutated genes in paediatric cancers. 6 , 7
NB is the most common extracranial solid tumour of early childhood. 8 Approximately half of the children with high‐risk NB do not survive despite extensive therapeutic intervention. 9 At diagnosis, NB seldom presents with mutations in the TP53 pathway. 10 However, aberrations in the TP53 pathway have been observed at a higher frequency (2‐53%) following chemotherapy and at relapse in NB, suggesting clonal enrichment of minor subclones against therapy. 11 , 12 The mutational status of TP53 influences the drug sensitivity of many chemotherapy agents in NB. 13
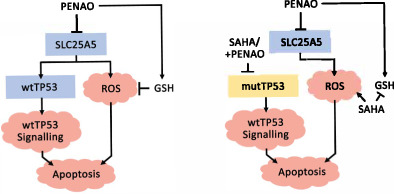
In NB, mitochondria are dysregulated by aberrant gene expression programs. Examples of this include the aberrant expression of the MYCN and BIRC5 oncogenes as well as the KIF1β tumour suppressor, owing to NB‐specific segmental copy number alterations (2p, 17q, 1p), which alter mitochondrial biogenesis and/or dynamics leading to drug resistance. 14 , 15 , 16 Here we identified the mitochondrial adenine nucleotide translocase 2 (SLC25A5 or ANT2) as a potential therapeutic target in NB. SLC25A5 is a key transporter of ATP between the outer and inner mitochondrial membrane, and regulates glycolysis. 17 The organo‐arsenical 4‐(N‐[S‐penicillaminylacetyl]amino) phenylarsonous acid (PENAO) is a mitochondria‐targeted agent that selectively reacts with and crosslinks cysteine residues (Cys57/257) of ANT homologues in tumour cells, resulting in apoptosis. 18 , 19 , 20 PENAO has efficacy against many cancer types 18 , 21 , 22 and is currently in early phase II clinical trials for a range of adult solid tumours. 23 Our studies, for the first time, identified SLC25A5 as a novel prognostic indicator and therapeutic target in NB cells. Moreover, the combination of PENAO (mitochondria inhibitor) and SAHA (chromatin modifier) had strong cytotoxic effects against both TP53 wild‐type and mutant NB cells, which are frequent in relapse/refractory NB cases.
2. METHODS
2.1. Tissue culture
The human neuroblastoma cell lines; SK‐N‐BE(2)‐C (CVCL_0529, BE(2)‐C), SH‐EP (CVCL_0524), SK‐N‐DZ (CVCL_1701), IMR‐32 (CVCL_0346), SK‐N‐F (CVCL_1702I), LAN‐1 (CVCL_1827), SK‐N‐AS (CVCL_1700), NB‐LS (CVCL_2136) and SH‐SY5Y (CVCL_0019) were maintained in Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies, Mulgrave, VIC, Australia) supplemented with 10% heat inactivated Foetal Bovine Serum (FBS) (Life Technologies). SH‐EP and SH‐SY5Y are subclones of the of the SK‐N‐SH cell line. KELLY (CVCL_2092) & CHP‐134 (CVCL_1124) human neuroblastoma cell lines were maintained in Roswell Park Memorial Institute 1640 medium (RPMI) (Life Technologies) supplemented with 10% heat inactivated FBS (Life Technologies). The human fibroblast cell lines MRC‐5 (CVCL_0440) and WI‐38 (CVCL_0579) were, respectively, maintained in Minimum Essential Media (MEM) (Life Technologies) and Alpha‐MEM (Life Technologies) supplemented with 10% heat inactivated FBS (Life Technologies). The cell line NBL‐S was kindly provided by Prof. Susan L. Cohn (Northwestern University, Chicago, IL). SH‐EP and SK‐N‐BE(2)‐C cell lines were kindly gifted from by Prof. June Biedler (Memorial Sloan Kettering Cancer Centre, New York, NY). SK‐N‐DZ and LAN‐1 cell lines were provided by Prof. John Maris. All other neuroblastoma cell lines were purchased from ATCC (Manassas, VA). MRC‐5 and WI38 human fibroblast cell lines were purchased from ATCC (Manassas, VA). All cell lines used were authenticated by Cell Bank Australia (Westmead, NSW, Australia) and were free from mycoplasma. All parental cell lines as well as the stable subclones have been authenticated using short tandem repeat profiling within the last 3 years. All cell lines were incubated at 37°C with 5% CO2 and were passaged at 80‐90% confluency to maintain an exponential growth phase. All experiments were performed with mycoplasma‐free cells.
2.2. Stable cell lines
Stable cell lines were previously generated from a panel of TP53 wild‐type neuroblastoma cell lines (IMR‐32, SH‐EP and SH‐SY5Y) using retroviral pBabe vectors containing short hairpin RNA (shRNA) sequences targeting either TP53 or GFP (control), which were transduced into cells using standard retroviral transduction protocols. 13 SH‐EP cells stably expressing mutant TP53 isoforms were also previously generated, by first introducing TP53 codon 135 mutations in a pCMV‐Neo vector containing the wild‐type TP53 cDNA sequence (C135R, C135P) and then transfecting SH‐EP cells with either the mutant TP53 constructs or pCMV‐Neo empty vector (control), using calcium phosphate transfection, followed by geneticin (G418) antibiotic selection and western blot validation of mutant p53 protein expression. 13
2.3. Drug treatments
For in vitro drug treatments, NB cell lines were seeded in either 96‐well, 6‐well plates or T25, T75 flasks (Corning) at densities that would reach 80‐90% confluency at the 24‐96‐h assay endpoints. Following an initial 24‐h incubation after seeding, drug treatments were made in fresh media. GSAO and PENAO were kindly provided by Prof. Philip J. Hogg (Australian Cancer Research Foundation, Centenary Cancer Research Centre, Charles Perkins Centre, USYD, NSW, Australia) and were constituted in saline (NaCl 0.9%). Vorinostat (SAHA) (#10009929, Cayman Chemical, MI) and Panobinostat (LBH589) (#13280, Cayman Chemical) were constituted in DMSO (Sigma‐Aldrich, Castle Hill, NSW, Australia). Glutathione ethyl ester (GSH‐EE) (G1404, Sigma‐Aldrich) and N‐Acetyl‐L‐Cysteine (NAC) (A9165, Sigma‐Aldrich) were constituted in Milli‐Q H2O. Cells were incubated at 37°C with 5% CO2 for 24 to 96 h, followed by various phenotypic assays.
For in vivo drug treatments, PENAO and GSAO were constituted in saline (NaCl 0.9%) prior to injection. The chemotherapy drugs; cisplatin (Hospira), vincristine (Tocris), etoposide (Sigma) and cyclophosphamide (Baxter) were diluted from their stock solution to 5% dextrose for injection. SAHA was diluted in a solution of 5% DMSO with saline (NaCl 0.9%) prior to injection.
2.4. Animal experiments
The efficacy of PENAO alone or in combination with chemotherapy agents was assessed in homozygous Th‐MYCN mice with established tumours. The Th‐MYCN model conditionally expresses the MYCN oncogene under the control of a tyrosine hydroxylase (TH) promoter. 24 This results in the formation of abdominal or thoracic tumours along the paraspinal ganglia which are histologically consistent with human NB. 24 Homozygous Th‐MYCN mice develop tumours at ~5.4 weeks of age with 100% tumour incidence. 25 Upon detection of an established tumour (5 mm in diameter) Th‐MYCN+/+ mice were separated into treatment cohorts, until the study endpoint (>10 mm tumour diameter by palpation or > 100 days survival). PENAO was administered at doses of either 10 mg/kg/day via oral gavage for up to 28 days, or 20 mg/kg/day via intravenous tail vail injections for only 5 days. Chemotherapy agents were administered alone or alongside PENAO treatments for only 5 days via intraperitoneal injections (I.P.) at the following doses; cisplatin (CDDP) 2 mg/kg/day, cyclophosphamide (CPA) 18 mg/kg/day, vincristine (VCR) 0.2 mg/kg/day and etoposide (VP‐16) 6 mg/kg/day.
Subcutaneous xenograft models of neuroblastoma were established by subcutaneous injection of 3.5 × 106 BE(2)‐C or 1 × 107 KELLY cells into the right flank of 5‐week old immunodeficient Balb/c nude mice. When tumours reached a volume of 50 mm3, as measured by callipers using the calculation; 0.5 × tumour length (mm) × (tumour width (mm)), 2 mice were assigned into treatment groups of either; 5% DMSO in saline, 17.5 mg/kg/day SAHA, 20 mg/kg/day PENAO or in combination. Mice were treated by I.P. injections on a 5 day on, 2 day off schedule for up to a total period of 42 days or until tumours reached a volume of 1000 mm3, at which point, the mice were sacrificed. The efficacy of PENAO combined with SAHA in Th‐MYCN homozygous mice was assessed by allocating 4‐week old mice with small palpable tumours (1‐2 mm in diameter), measured by abdominal palpations, into the aforementioned treatment groups. These mice were also treated by I.P. injections on a 5 day on, 2 day off schedule for up to a total period of 42 days or until tumours reached >10 mm in diameter by palpation, at which point, the mice were sacrificed.
2.5. Statistical analysis
Data obtained from all experiments were plotted and analysed using the software GraphPad Prism 7 (Applied BioSystems, Scoresby, VIC, Australia). Statistical analysis was completed using either; one‐way or two‐way ANOVA, with Tukey's multiple comparison tests, or unpaired t tests.
3. RESULTS
3.1. A subset of mitochondria‐associated genes associates with NB cell survival in vitro, chromatin accessibility and poor patient survival
We first sought to identify potential therapeutic targets among mitochondria‐associated genes (MAGs) expressed in human NB cell lines and tissues using publicly available datasets. A total of 28 gene sets with mitochondrial ontology from the Molecular Signatures Database (MSigDB) were integrated to generate a list of 1148 unique MAGs (Figure S1). To identify relationships among MAGs, the gene expression of each MAG in patient tumours of the SEQC NB RNA‐Seq cohort (GSE62564; n = 498) were scaled and hierarchically clustered, yielding six main gene clusters (C1‐C6) (Figure 1A). When patients were classified into each group using gene expression, we observed that patients in C5 & C6 groups had significantly poorer overall survival compared with other cluster groups (Figure 1B). MAG scores, which link the expression of each MAG with their ability to predict patient prognosis, were calculated and then also used to group patients, revealing a strong prognostic association between MAG expression and poor NB patient outcome (Figure S1). The predictive values of each MAG cluster were derived from univariate Cox Proportional‐Hazards (CoxPH) models of patient survival, using median gene expression to dichotomise the patient cohorts. We observed 2.66 to 7.21‐fold higher univariate hazard ratios in C5 & C6 compared with other clusters (Figure 1C).
FIGURE 1.
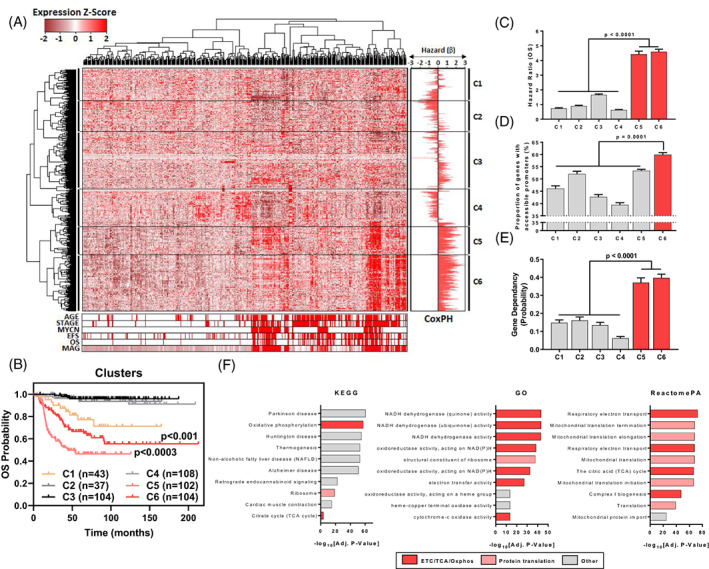
A subset of mitochondria‐associated genes associates with NB cell survival, high chromatin accessibility and poor patient survival. (A) 1148 Mitochondrial Associated Genes (MAGs) were derived from the integration of 28 mitochondrial gene sets from the MSigDB. Hierarchical clustering of gene expression (1148 MAGs) in the SEQC NB Cohort (RNA‐Seq; GSE62564) are represented as a heatmap with clinical annotations (Age [<18 Months vs >18 Months], Stage [1,2,3,4 s vs 4], MYCN Status [Non‐Amp vs Amp], EFS Event, OS Event & MAG scores for each patient). β‐coefficients from univariate Cox Proportional Hazards (CoxPH) models, using the median expression of each MAG (n = 1148) as a cut‐off, are shown as a bidirectional bar graph. (B) Patients from the SEQC NB cohort (n = 498) were subdivided based on cluster expression (RNA‐Seq) and their overall survival probability was plotted on a Kaplan‐Meier curve. Comparisons are made between C5 vs C1,2,3,4,6 and C6 vs C1,23,4. (C) Mean hazard ratios (HR = e[β]) of each cluster are shown with the error bars representing the SEM of HR's within a cluster. (D) The proportion of genes with accessible promoters in each cluster with error bars representing the SEM across all 7 NB cell lines (where an accessible promoter had significant enrichment of ATAC signal, FDR q‐value <0.05, −1000/+100 bp from TSS). (E) The mean gene dependency probability scores for each gene cluster derived from Project Achilles. Error bars represent the SE of the mean in each MAG cluster. (F) Gene set enrichment of C6 genes using KEGG, GO and ReactomePA databases, the top 10 genesets enriched for C6 genes are shown, with gensets being ranked by ‐log10[Adjusted P value]. Each gene set is also categorised by broad process involvement (ETC, electron transport chain; TCA, tricarboxylic acid cycle, Oxphos, oxidative phosphorylation). Error bars represent the SE of the mean in each MAG cluster
To discern factors governing MAG expression between clusters we assessed N‐Myc chromatin binding (GSE80151), as N‐Myc is a master transcription factor and strong prognostic factor in NB, as well as chromatin accessibility (GSE80152/GSE101294) at MAG promoter regions in NB cell lines (Figure S1). We observed no significant trends in the proportion of promoters enriched for N‐Myc in each cluster (Figure S1), however, we found that C6 genes had a higher proportion of promoters (6.50‐20.41%) that were accessible for gene transcription (Figure 1D). The importance of each MAG cluster in the growth of NB cells was also assessed using CRISPR interference screening data in NB cell lines (Project Achilles, n = 14). We found that NB cell lines had 2.30 to 6.35‐fold higher probability of being dependent on C5 & C6 MAGs for cell viability, compared with the other four clusters (Figure 1E). We then assessed the prognostic significance of each MAG against known predictors of NB patient outcome (MYCN amplification status, INSS disease stage & age at diagnosis) by multivariate CoxPH modelling in the SEQC NB patient cohort. We found that C5 & C6 MAGs had 1.60 to 2.20‐fold higher mean multivariate hazard ratios (mHR) compared with other clusters (Figure S1). Finally, we investigated cluster‐specific gene ontologies using the KEGG, GO, ReactomePA and Disease Ontology databases. We found that C5‐6 genes were enriched for pathways related to oxidative phosphorylation and protein translation, whilst C1‐4 were enriched for pathways related to cell metabolism, intracellular transport and cell death (Figures 1F and S1). We also found that C6 genes were uniquely enriched in mitochondrial metabolism diseases (Figure S1) suggesting that these genes were involved in vital functions in healthy cells. Taken together, C5‐6 MAGs had highly accessible promoters for gene transcription, were essential for cell viability, and were independent predictors of poor NB patient outcome. Hence, we next sought to identify therapeutic targets within the C5‐6 MAG subgroups.
3.2. High SLC25A5 gene expression is the strongest predictor of poor NB patient prognosis among MAGs
Multivariate analyses showed that the C6 MAG, SLC25A5, had the strongest independent prognostic effect among all MAGs (Figure 2A). We chose SLC25A5 for further study because of this strong prognostic value and the availability of an inhibitor in phase II clinical trials for advanced solid tumours; PENAO. 19 , 20 To determine the relationship between SLC25A5 expression and NB patient outcome, we dichotomised patient cohorts by median SLC25A5 gene expression and performed Kaplan‐Meier analyses. Higher SLC25A5 mRNA expression was significantly associated with poor event free survival (EFS) and overall survival (OS) in the SEQC cohort (Figures 2B and S2A). Multivariate CoxPH modelling revealed SLC25A5 gene expression to be a superior predictor of poor patient outcome compared with current clinical predictors (Figures 2C and S2B). We further assessed whether SLC25A5 gene expression correlated with these clinical predictors of poor outcome. Patients with MYCN amplification, INSS stage 4 of disease or diagnosed at >18 months of age, had significantly higher SLC25A5 gene expression compared with other patients (Figure 2D). All these findings were corroborated by similar analyses in the KOCAK NB patient cohort (n = 649) (Figure S2C‐F). These findings establish high SLC25A5 expression as a strong predictor of poor NB patient outcome. Additionally, the prognostic value of each of the three other ANT family members, SLC25A4, SLC25A6 and SLC25A31, were assessed using publicly available RNA‐Seq and gene microarray databases (SEQ Cohort GSE62564, and KOCAK cohort GSE45547, respectively) and compared with SLC25A5. This confirmed that SLC25A5 was the sole predictor of patient outcome (Figure S2I‐L).
FIGURE 2.
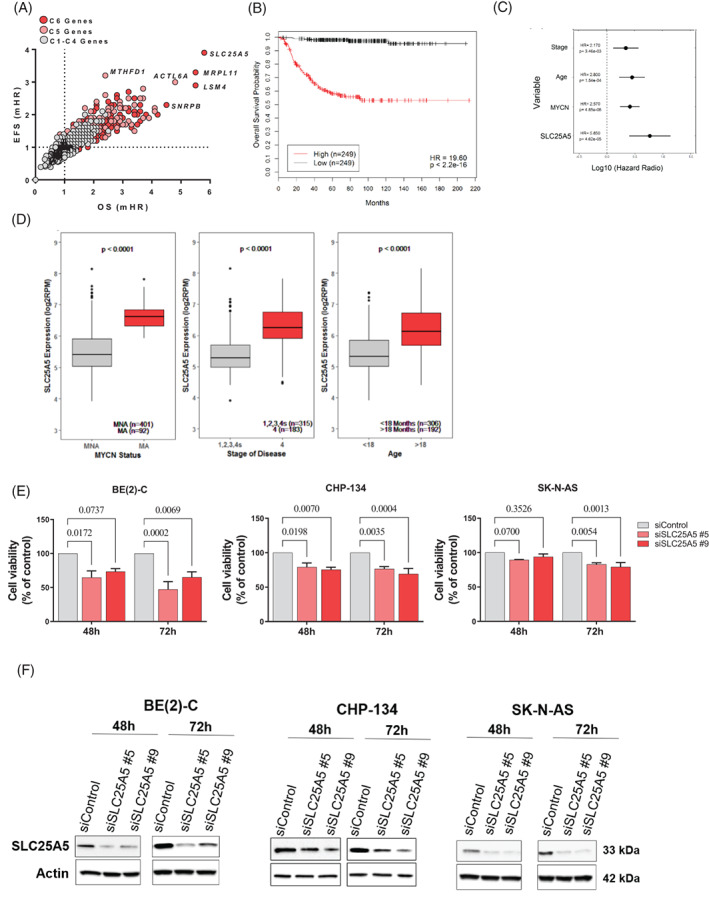
High SLC25A5 gene expression is predictive of poor NB patient prognosis. (A) The median gene expression (RNA‐Seq) of each MAG (n = 1148) was considered as a prognostic variable alongside classical predictors of NB patient outcome (MYCN Amplification Status [amplified vs non‐amplified], Stage of Disease [Stage 1,2,3,4 s vs Stage 4] & Age [>18 months vs < 18 months]) in iterative multivariate CoxPH models using the SEQC NB patient cohort (n = 498). The dot plot represents the multivariate hazards ratios from the CoxPH models for each gene with regard to event free survival (EFS)/overall survival (OS) probability of patients and cluster identity. (B) Kaplan‐Meier survival curves showing the OS probability of patients in the SEQC NB cohort (n = 498) when dichotomised by median SLC25A5 gene expression (RNA‐Seq). Hazard Ratio's (HR) and log‐rank P values are presented from a univariate CoxPH model. (C) Summary of the OS multivariate CoxPH models with SLC25A5 gene expression against classical prognostic predictors, dots represent the median HR whilst lines represent the 95% confidence interval. (D) Box plots of SLC25A5 gene expression subdivided using established prognostic predictors of poor NB patient outcome in the SEQC cohort (n = 498), patients are stratified by either; MYCN amplification status (MYCN non‐amplified; MNA, MYCN amplified; MA), stage of disease according to the International Neuroblastoma Staging System (INSS) (Stage 1,2,3,4 s vs 4) and age at diagnosis (<18 vs >18 months of age). (E) Cell viability of BE(2)‐C, CHP‐134 and SK‐N‐AS cell lines following siRNA mediated knockdown of SLC25A5 for 48‐72 h. SLC25A5 siRNA (#5 & #9) are compared with the non‐targeted siControl using a two‐way ANOVA with multiple comparison, reported P values are adjusted for multiple comparisons. (F) Representative western blot of SK‐N‐BE(2)‐C, CHP‐134 and SK‐N‐AS cells transfected with control or SLC25A5 siRNA for 72 h to confirm sufficient knockdown of SLC25A5 for cell viability assays
We then performed western blot analyses on the total cell protein lysates from NB cell lines and human fibroblast control cell lines and observed that SLC25A5 expression was on average 9.6‐fold higher in NB cell lines than in fibroblasts (Figure S2G‐H), however noted that SLC25A5 expression among NB cell lines varied. The phenotypic consequence of SLC25A5 ablation was then assessed using siRNA mediated interference, wherein knockdown of SLC25A5 mRNA resulted in significantly reduced cell viability between 48 and 72 h following transfection (Figure 2E) indicating that SLC25A5 expression was necessary for NB cell survival. Western blot of cell lysates following 72 h of transfection of SK‐N‐BE(2)‐C, CHP‐134 and SK‐N‐AS cells with control or two SLC25A5 siRNAs was performed, confirming the knockdown of SLC25A5 (Figure 2F).
3.3. Small molecule inhibition of SLC25A5 with PENAO reduces NB cell viability
We treated 10 NB and two normal fibroblast cell lines with the selective mitochondrial adenine nucleotide translocase inhibitor PENAO (Figure S3A) using a dose range (0‐20 μM) and measured cell viability relative to a vehicle control (Figure 3A). We observed that NB cell lines were on average 3.58‐fold more sensitive to PENAO than normal fibroblast cell lines (Figure S3B, C), suggesting that there might be a therapeutic window for in vivo therapy in pre‐clinical models of NB. We observed no significant correlation between SLC25A5 protein levels and PENAO IC50 suggesting that there may be additional factors governing sensitivity to PENAO (Figure S3D).
FIGURE 3.
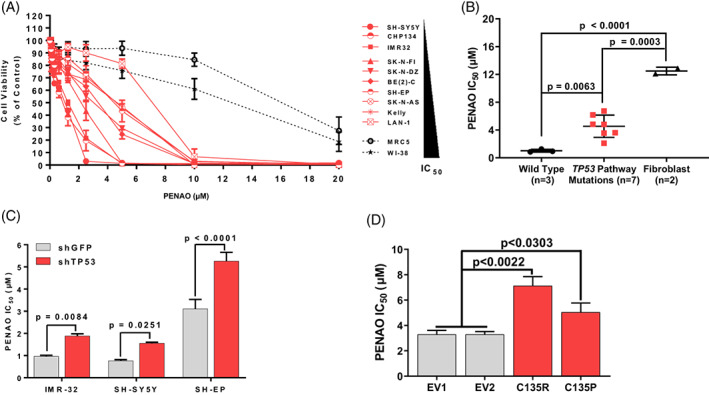
Direct targeting of SLC25A5 with the selective inhibitor PENAO significantly reduces NB cell viability. (A) Cell viability curves of NB and normal lung fibroblast cell lines upon PENAO treatment (0‐20 μM) for 72 h, cell lines are ranked according to IC50. (B) Average IC50's of cell lines subcategorised by mutations in the TP53 pathway (TP53, CDKN2A). (C) PENAO cytotoxicity (IC50) in neuroblastoma cell lines stably transduced with an shRNA construct targeting TP53 mRNA. (D) PENAO cytotoxicity (IC50) in SH‐EP clones stably expressing TP53 cDNA constructs with point mutations in the DNA‐binding domain (p.C135R & p.C135P mutants) or empty vector controls (EV1/EV2). Error bars represent the SE of the mean of at least three independent biological replicates
3.4. PENAO alone or in combination with cytotoxic chemotherapy has minimal efficacy against NB in vivo
We next assessed the efficacy of targeting SLC25A5 with PENAO in established NB tumours arising in homozygous Th‐MYCN+/+ NB transgenic mice. We found only small improvements in tumour‐free survival with PENAO treatment when compared with saline control mice (Figure S3E and Table S1). We further evaluated whether PENAO could potentiate induction chemotherapies in current clinical use for NB, such as cisplatin (CDDP), vincristine (VCR), cyclophosphamide (CPA) and etoposide (VP16) in Th‐MYCN+/+ mice (Figure S3F, G and Table S1). We observed either minor or no extension to tumour‐free survival in mice treated with PENAO and chemotherapy. The relatively poor efficacy of PENAO in vivo led us to seek more effective PENAO combination anti‐cancer therapies.
3.5. TP53 expression and mutational status are key determinants of PENAO sensitivity in NB cell lines
In order to develop a targeted combination therapy with PENAO, we first considered sensitivity/resistance factors for PENAO cytotoxicity. We grouped each cell line's PENAO IC50 by the presence or absence of TP53 pathway alterations, taking into consideration mutations in CDKN2A and TP53 as well as amplifications of MDM2. We found that cell lines with TP53 pathway alterations were on average 4.47‐fold more resistant to PENAO than those with no alterations (Figure 3B and Table S2).
We then validated TP53 expression as a sensitivity factor to PENAO, by assessing efficacy in NB cell lines with wild type TP53 stably expressing shRNA constructs against TP53 (shTP53) compared with vector controls. 13 We observed a significant increase in PENAO resistance in all three wild type TP53 cell lines following TP53 knockdown (Figure 3C and S3H, I). We then assessed the effect of TP53 mutation on resistance to PENAO. The NB SH‐EP cell line harbours a homozygous deletion of the CDKN2A gene, which results in supressed wild‐type TP53 signalling. 11 We utilised SH‐EP cells stably transfected with TP53 constructs harbouring point mutations in the DNA binding domain (DBD; p.C135R, p.C135P). 13 We observed a 1.53 to 2.17‐fold increased resistance to PENAO in the p.C135R/P mutant expressing cells compared with empty vector controls EV1/EV2 (Figures 3D and S3J, K). Taken together these findings suggest that wild type TP53 signal activation is required for a stronger PENAO sensitivity, whereas mutant TP53 mediates resistance to PENAO.
3.6. A combination of a histone deacetylase inhibitor and PENAO synergistically reduces cell viability in mutant TP53 NB cell lines
We next sought cytotoxic compounds whose activity was greater in TP53 mutant NB for combination therapy with PENAO. We examined publicly available drug sensitivity data (IC50) from the Genomics of Drug Sensitivity in Cancer (GDSC) database for drugs whose cytotoxic effects were greater in TP53 mutant, compared with TP53 wild type, NB cell lines. When we ranked 251 compounds for their cytotoxicity across all 32 available NB cell lines, we found 126 compounds with higher sensitivity (z‐score > 0) in TP53 mutant NB cell lines (Figure 4A, B). Of the top 5 scoring compounds, the histone deacetylase inhibitor (HDACi), SAHA (Vorinostat), was the most clinically advanced, being FDA approved 26 and used in several NB paediatric clinical trials (Figure 4B). 27 , 28
FIGURE 4.
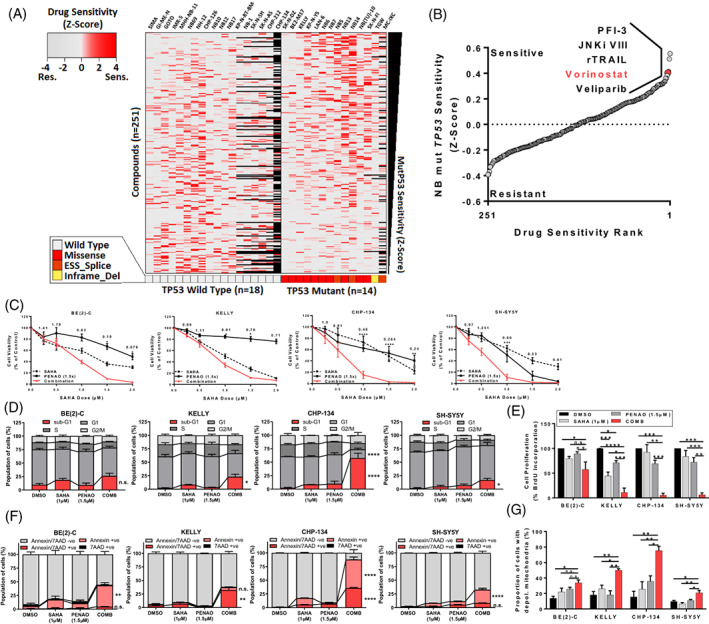
SAHA combined with PENAO synergistically induces apoptotic cell death in mutant TP53 NB cell lines. (A) Heatmap of drug sensitivity data (IC50) from the Genomics of Drug Sensitivity in Cancer database (GDSC) z‐scaled for 251 compounds across 32 NB cell lines, TP53 mutations identified by whole exome sequencing (WES) are annotated at the bottom. Black squares on the heatmap represent missing drug screening data. (ESS_Splice = mutation in the exonic splicing silencer site or at donor/acceptor splice sites, Inframe_Del = inframe deletion). (B) 251 compounds ranked by average drug sensitivity (z‐score) in mutant TP53 NB cell lines, the top five compounds are annotated. (C) Cell viability curves of SK‐N‐BE(2)‐C, Kelly, CHP134 & SH‐SY5Y neuroblastoma cells 72 h post treatment with SAHA (0‐2.0 μM) and/or PENAO (0‐3.0 μM) in a constant 1:1.5 ratio. Combination Index (CI) Values generated from CalcuSyn at the corresponding doses are also displayed. (D) Propidium iodide cell cycle assays represented in stacked column graphs indicating the proportion of cells in each cell cycle phase (including sub‐G1) after 72 h of treatment. (E) BrdU cell proliferation assays 72 h posttreatment in NB cell lines. (F) Annexin‐V/7‐AAD apoptosis assays in the same cell lines 72 h post treatment, represented in stacked column graphs indicating the proportion of apoptotic/dead cells. (G) JC‐1 assays representing the proportion of cells with depolarised mitochondria in each of the four cell lines after 48 h of treatment. *P < .05, **P < .01, ***P < .001, ****P < .0001, Reported P‐values are from one way ANOVA's, error bars represent the SE of the mean of at least three independent biological replicates
In cell viability assays in six NB and two normal human fibroblast cell lines we demonstrated the SAHA and PENAO combination was synergistic (Figures 4C and S4A). We confirmed similar synergy between the third generation HDACi, Panobinostat (LBH589) and PENAO (Figure S4B) as well as between PENAO's precursor, GSAO and SAHA (Figure S4C). We observed moderate to strong synergy (Combination Index, CI < 0.8) in DNA‐binding domain (DBD) TP53 mutant NB cell lines as well as in wild type TP53 NB cell lines (Figure S4D and Table S3). However, we found that there was an additive to antagonistic (CI > 0.9) interaction between PENAO and SAHA in c‐terminal deleted TP53 mutant NB cell lines (Figure S4D and Table S3). These results suggested there was broad synergism between histone deacetylase and SLC25A5 inhibitors in NB cell lines.
We assessed whether cell‐cycle arrest (G0/1, G2) contributed to the synergistic reduction of cell viability in combination treated cells using propidium iodide (PI) staining (Figures 4D and S4E). We observed no changes in the proportion of cells in G0/1 or G2 phases of the cell‐cycle but saw increases in sub‐G1 cell proportions (Figure 4D). In contrast, human fibroblast cell lines treated with the combination did not display any significant increases in sub‐G1 cell proportions (Figure S4F). In BrdU cell proliferation assays, there were significant reductions in BrdU incorporation into cells treated with the combination, indicating a reduced proportion of cells in the proliferative S‐phase of the cell‐cycle (Figure 4E). Taken together these data suggested combination therapy primarily led to the activation of apoptosis and therefore a smaller proportion of cells available for cell replication.
We then used Annexin‐V/7‐AAD assays to measure apoptotic cell death in combination treated cells and found a significant increase in the proportion of cells undergoing early and/or late apoptosis in NB cell lines treated with the combination (Figures 4F and S4G). We observed comparatively less apoptosis in combination‐treated human fibroblast cell lines (Figure S4H). Terminal deoxynucleotidyl transferase dUTP nick end labelling (TUNEL) assays also indicated increased apoptosis in combination treated cells, albeit at later time points (Figure S4I, J). These data suggested that an apoptotic phenotype was being induced by the combination treatment, and that it was largely selective for NB cells.
We next assessed the activation of the intrinsic pathway of apoptosis by measuring characteristic changes in mitochondrial membrane potential via JC‐1 flow cytometric assays (Figures 4G and S4K). We observed a significant increase in the proportion of NB cells with depolarised mitochondrial membranes when treated with the combination (Figures 4G and S4L). This data confirmed that apoptosis which followed combination HDACi and PENAO treatment of NB cells was occurring via apoptotic intrinsic pathway mechanisms.
3.7. SAHA combined with PENAO induces oxidative stress and modulates glutathione levels in NB cell lines
Following our observations of intrinsically mediated apoptosis, we investigated reactive oxygen species (ROS) as a possible trigger of apoptosis in NB cells treated with the combination, since SAHA and PENAO were known to be potent ROS inducers. 21 , 29 , 30 , 31 We assessed mitochondrial superoxide formation using MitoSox Red (Figure S4M, N) and cytoplasmic superoxide formation using dihydroethidium (DHE) flow cytometric assays (Figure S4O, P). We showed that SAHA alone and the combination therapy had an equivalent effect in increasing both mitochondrial and whole cell superoxide production, indicating that SAHA treatment produced superoxide (Figure S4M, O). The superoxide scavenger N‐Acetyl Cysteine (NAC) was then used to ascertain if superoxide production was necessary for the efficacy of the combination. Cell viability assays following drug treatment, in the presence or absence of NAC, revealed a rescued phenotype in all cell lines treated with either PENAO or the combination (Figure S4Q), suggesting that ROS production was, in part, required for the action of PENAO.
We sought to understand whether changes in the key endogenous antioxidant, glutathione, contributed to ROS formation by quantifying levels of glutathione (GSH) following treatment (Figure S4R). We observed that PENAO alone increased total glutathione in NB cells, whilst SAHA alone depleted total glutathione and when used in combination only partially reversed PENAO‐mediated glutathione increase (Figure S4R). We introduced exogenous GSH in the form of glutathione mono‐ethyl ester (GSH‐EE) alongside treatments and found GSH‐EE rescued cell viability in cells treated with PENAO only (Figure S4S). This suggested that endogenous GSH levels were important for the efficacy of PENAO alone, and that the combination produced ROS which exceeded the antioxidant capacity of endogenous GSH pathways, enhanced by PENAO.
3.8. SAHA combined with PENAO synergistically reduces mutant p53 protein stability
To identify mechanisms of action in combination‐treated NB cells we subjected the TP53 wild type cell line SH‐SY5Y to either vehicle control, SAHA (1 μM), PENAO (1.5 μM) or combination treatments for 8 h, followed by mRNA microarray analysis. Differentially expressed genes between treatment conditions and the vehicle control were determined (Figure S5A, B and Table S4) and then subsequently ranked for a Gene Set Enrichment Analysis (GSEA) using the MSigDb hallmark database (Figure 5A). The TP53 pathway was the most significantly enriched pathway after combination treatment compared with the control (Figure 5A‐C and Table S5). This suggested that the TP53 pathway was the prime mechanism for apoptosis induction in a TP53 wild type setting. Hence, we investigated the effects of the combination therapy on mutant TP53 expressing NB cell lines.
FIGURE 5.
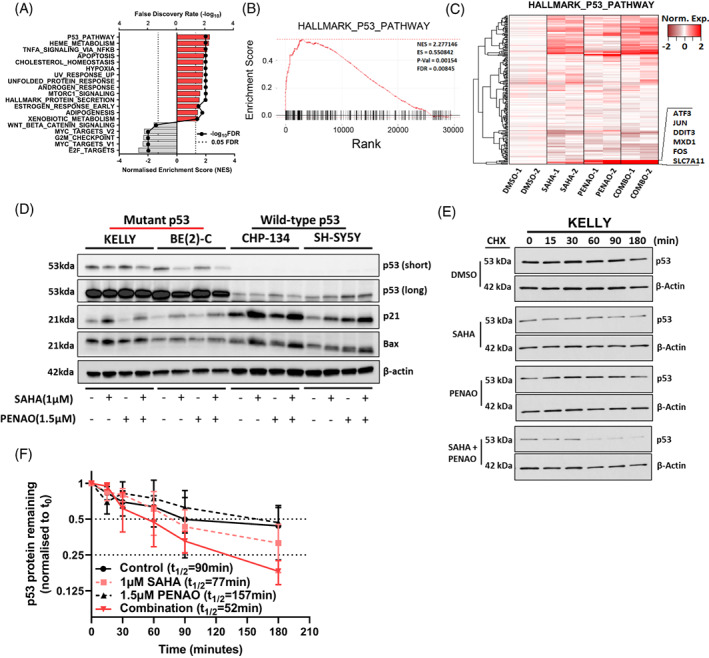
SAHA combined with PENAO synergistically reduces mutant p53 protein stability. (A) The TP53 wild type cell line SH‐SY5Y was subject to either vehicle control, SAHA (1 μM), PENAO (1.5 μM) or combination treatments for 8 h, followed by mRNA microarray analysis. Differentially expressed genes between conditions were determined and ranked for a Gene Set Enrichment Analysis (GSEA) using the MSigDb hallmark database. Significant pathways (FDR < 0.05) are displayed along with their normalised enrichment score (NES). (B) Enrichment plot displaying the relative enrichment and rank of differentially expressed genes present in the HALLMARK_P53_PATHWAY gene set (indicated by black lines on the x‐axis). Statistics from the GSEA are reported inside the plot. (C) Heatmap of gene expression among treatment conditions in genes within the HALLMARK_P53_PATHWAY gene set, highly expressed genes in the combination which are clustered tightly are highlighted and annotated on the side. (D) Immunoblot of NB cell lysates after 24 h of the indicated treatments, probed with either; anti‐p53, anti‐p21, anti‐Bax or anti‐β‐Actin antibodies. Approximate band sizes are shown to the left of the blots, cell lines used are indicated the top, treatments are indicated below. Both a short and long exposure of anti‐p53 probed membrane are provided. (E) Immunoblot of cycloheximide chases spanning 3 h in KELLY cells treated with DMSO, SAHA 1 μM, PENAO 1.5 μM or the combination for 24 h, probed with anti‐p53 and anti‐β‐Actin. (F) Densitometry of the cycloheximide chase immunoblots are provided, p53 levels are normalised to loading controls and then to the 0‐min time point, error bars represent the stand error of the mean from three independent experiments. The half‐life (t 1/2) of p53 in each condition is reported in the legend
We assessed the effect of the combination treatment on p53 protein expression by performing western blot analyses following treatments alongside other key TP53 pathway components (Figure 5D). We observed a decrease in mutant p53 protein levels in the TP53 mutant KELLY & BE(2)‐C cell lines when treated with the combination (Figures 5D and S5D, E). We observed no significant changes in the wild type TP53 CHP‐134 cell line but noted that the wild type TP53 cell line SH‐SY5Y exhibited an increase in p53 protein in response to combination treatment (Figures 5D and S5D‐G). We observed a broad induction of the p53 target genes, p21 and Bax, in each cell line by either SAHA or the combination treatments (Figures 5D and S5D‐G). The HDAC6‐HSP90 axis, responsible for mutant TP53 protection, 32 remained largely unchanged and the p53‐specific E3 ubiquitin ligases MDM2 and CHIP responsible for p53 degradation did not change (Figure S5C‐G). 33 Since the KELLY and BE(2)‐C cell lines possess TP53 mutations which increase the stability of p53 protein, 34 , 35 we assessed whether the reduction in protein levels could be attributed to loss of protein stability using cycloheximide chase (CHX) assays. We observed that KELLY cells treated with the combination exhibited lower p53 protein stability (t 1/2 = 52 min) than those treated with single agents or control alone (t 1/2 > 77 min) (Figure 5E, F). Taken together these findings indicated that SAHA combined with PENAO synergistically reduced mutant p53 protein stability, potentially triggering apoptotic cell death.
3.9. SAHA combined with PENAO significantly delays tumour growth and induces apoptosis in TP53 mutant NB xenograft models
As we observed synergy between PENAO and SAHA in the DBD TP53 mutant NB cell lines in vitro, we sought to assess efficacy in pre‐clinical xenograft models of these cell lines in vivo. Subcutaneous xenograft models of either SK‐N‐BE(2)‐C or KELLY cells were established in BALB/C nude mice. These mice were treated with either a saline control, SAHA (17.5 mg/kg), PENAO (20 mg/kg) or the combination of both via intraperitoneal administration until the study end‐points. In both SK‐N‐BE(2)‐C and KELLY xenograft models, combination treatments significantly improved median survival times by 7 to 19 days compared with individual agents (Figure 6A, B and Table S6). Tumour growth in these models was also delayed by combination treatments compared with all other treatments (Figure S6A, B and Table S6), which further indicated the anti‐tumour synergy between SAHA and PENAO in DBD mutant TP53 models of NB. We then assessed the efficacy of these treatments against the developing tumours of TP53 wild type Th‐MYCN+/+ mice. We observed significantly improved median survival times by 4‐10.5 days in combination treated mice compared with saline control, SAHA or PENAO treated mice (Figure S6C and Table S6). The body weight of treated mice did not differ from saline‐treated mice over the course of treatment and no signs of drug toxicity were observed (Figure S6D‐F).
FIGURE 6.
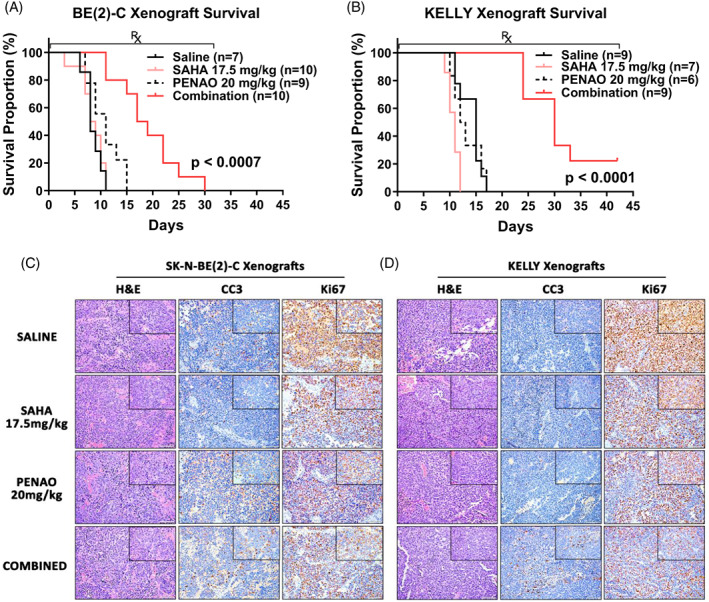
SAHA combined with PENAO significantly delays NB tumour growth and induces apoptosis in vivo. (A) Kaplan‐Meier survival curves of Balb/c nude mice xenografted with SK‐N‐BE(2)‐C or (B) KELLY cells, post tumour formation and during the administration of Saline, SAHA (17.5 mg/kg), PENAO (20 mg/kg I.P.) or a combination of both for up to 42 days on a 5 day on/2 day off treatment schedule. P values are from log‐rank statistic tests between the combination and all other treatment groups. (C) Representative ×20 images, with ×40 images inset (×10 objective lens), of treated SK‐N‐BE(2)‐C or (D) KELLY xenograft tumour sections stained with Haematoxylin & Eosin (H&E), anti‐Cleaved Caspase 3 (CC3) or anti‐Ki67. CC3 and Ki67 stained sections are counterstained with Haematoxylin. Scale bars represent 20 μM
To assess apoptosis and cell proliferation in vivo, we performed immunohistochemical analysis of xenograft tumour sections stained with the apoptotic marker, cleaved caspase 3 (CC3) and proliferative marker, Ki67 (Figure 6C‐D). We observed increased levels of CC3 and decreased levels of Ki67 staining in combination treated mice compared with single agents or saline treated mice (Figure 6C‐D), indicating that apoptosis and proliferative arrest were being induced by the combination therapy in vivo. Additionally, the in vivo inhibitory effect of SAHA when used in combination therapy with PENAO was confirmed using anti‐histone H3 antibody to measure the level of hyperacetylation of histone H3 (Figure S6G). Tumour xenograft samples treated with combination therapy showed the highest level of histone H3 hyperacetylation.
4. DISCUSSION
In cancer, mitochondria‐targeted therapies are sought to produce strong apoptotic phenotypes and potentiate chemotherapy. 2 , 36 However in NB, mitochondrial dysregulation is mediated by underlying genomic lesions, 14 , 15 , 16 changes in gene expression 37 , 38 or in response to chemotherapy. 15 , 39 Several mitochondria‐targeted agents have shown efficacy in NB in experimental systems but have achieved only limited clinical application, 4 , 37 suggesting the need for identifying more effective mitochondrial targets. Here we show that combined inhibition of mitochondrial translocases and histone deacetylation synergistically activated apoptotic signalling in NB cells with independent of TP53‐status, indicating a novel therapeutic strategy for neuroblastoma.
Mitochondrial gene signatures have previously contributed to patient prognosis prediction and target prioritisation in cancer. 40 Here, we identified a subset of mitochondria‐associated genes (MAGs) whose expression signified poor NB patient outcome. A recent study had shown similar prognostic value of MAG signatures in NB, such as those involved in the response to reactive oxygen species and mitophagy, 41 supporting the notion that MAG signatures correlate with NB patient prognosis and treatment resistance. Among this subset of MAGs, we identified the adenine nucleotide translocase, SLC25A5/ANT2, as having the highest prognostic value in determining poor NB patient outcome. High SLC25A5 expression has previously been associated with poor outcome in nonsmall cell lung cancer 42 and ovarian cancer patients, 43 which along with our findings in NB, encourages further exploration of SCL25A5 expression as a potential prognostic biomarker in other cancer types. There are four isoforms of mitochondrial adenine nucleotide translocases; SLC25A4, SCL25A5 SCL25A6 and SCL25A31 (ANT1‐4) sharing 80‐90% sequence homology. Each has a different tissue distribution, characteristics and expression in tumours, albeit, with SLC25A5 being most overexpressed in cancer, compared with the other isoforms. 44 , 45 Our experiments targeting SLC25A5 with PENAO revealed high cytotoxic selectivity against NB cell lines in vitro and preclinical efficacy in vivo in the absence of toxicities, which indicates that SLC25A5 may be an actionable target in children with NB.
The varying levels of PENAO sensitivity observed between the individual NB cell lines in our study indicated that there were cell‐intrinsic factors governing sensitivity, which has also been observed in past studies of other cancer types. 21 We observed no significant correlations between the IC50 of PENAO and the protein/gene expression of the target SLC25A5 in NB cell lines, which had been described previously in other cancers, 31 suggesting several factors mediating PENAO sensitivity that had yet to be determined. Reactive oxygen species (ROS) regulators and antioxidants such as glutathione (GSH) have been implicated in PENAO resistance. 19 Our studies utilising ROS/GSH modulators alongside PENAO treatment revealed that PENAO treatment efficacy was highly dependent on ROS induction and low endogenous GSH levels for efficacy. We have further shown that the expression and mutational status of the tumour suppressor TP53 are major determinants of PENAO sensitivity in NB. In our studies PENAO in combination with SAHA, produced potent anti‐tumour synergy in drug‐resistant, mutant TP53 NB xenograft models, highlighting the potential of this combination against a spectrum of other mutant TP53 cancers. The activation of wild‐type TP53 has not previously been suggested as a prerequisite for the anti‐cancer action of PENAO. Our work for the first time suggests a PENAO combination therapy which operates independent of TP53‐status and may have broad application across many cancer types.
The majority of somatic mutations in TP53 occur in the DNA binding domain (DBD) conferring either loss‐of‐function, gain‐of‐function or increased stability of the mutant p53 protein. 46 In the present study, we have shown that the combination of SAHA with PENAO reduces mutant p53 protein levels and stability in NB cell lines harbouring DBD TP53 mutations. Furthermore ATO, a precursor to PENAO, has also been shown to destabilise and degrade mutant p53 isoforms by upregulating the PIRH2 E3 ligase. 47 , 48 The combination of SAHA with ATO has been shown to act synergistically in eliciting cytotoxicity and mutant p53 degradation in cancer cell lines harbouring TP53 mutations, 48 which resonates with our findings on the combination of SAHA and PENAO. Our findings in this study are largely based on investigations in two cell line models of DBD mutant TP53 NB and would benefit from further ex vivo or in vivo testing in patient‐derived material with DBD TP53 mutations for further pre‐clinical validation.
5. CONCLUSION
PENAO has entered early phase II clinical trials for a range of solid tumours in adults 23 and SAHA is currently in early phase clinical trials for NB in paediatric patients, 28 but is already approved by U.S. FDA for use in adults with cutaneous T‐Cell lymphoma. 26 Our promising preclinical data on this combination therapy warrants further evaluation in an early phase clinical trial for NB. A recently completed phase I clinical trial in advanced adult solid tumours (NCT01339871) highlighted the profound clinical benefit of utilising a targeted drug combination of Pazopanib with SAHA in patients with TP53 hotspot mutations. 49 Thus, the combined inhibition of mitochondrial translocases and HDACs represents a novel therapeutic avenue against NB. There is an underlying importance in discovering novel actionable targets and understanding potential interplay with existing targets to better inform future combination therapy design. Our findings here, targeting the mitochondrial adenine nucleotide translocase SLC25A5 with PENAO and SAHA combination therapy, presents a novel therapeutic route that works independent of TP53‐status in neuroblastoma.
AUTHOR CONTRIBUTIONS
The work reported in the paper has been performed by the authors, unless clearly specified in the text. Conception and design: Janith A. Seneviratne, Daniel R. Carter, Belamy B. Cheung, Glenn M. Marshall. Development of methodology: Janith A. Seneviratne, Patrick Y. Kim, Aldwin S. Rahmanto, Jayne Murray, Chengyuan Xue, Sylvia A. Chung. Acquisition of data: Janith A. Seneviratne, Rituparna Mittra, Andrew Gifford, Patrick Y. Kim, Aldwin S. Rahmanto, Jie‐Si Luo, Jayne Murray, Ngan C. Cheng, Alice Salib, Zsuzsanna Nagy, Ane Kleynhans, Owen Tan, Selina K. Sutton, Yizhuo Zhang, Chengtao Sun, Li Zhang, Chelsea Mayoh, Qian Wang. Analysis and interpretation of data: Janith A. Seneviratne, Daniel R. Carter, Belamy B. Cheung, Glenn M. Marshall. Writing of the manuscript: Janith A. Seneviratne. Review, and/or revision of the manuscript: Janith A. Seneviratne, Daniel R. Carter, Rituparna Mittra, Belamy B. Cheung and Glenn M. Marshall. Administrative, technical, or material support: Chengyuan Xue, Pierre J. Dilda, Philip J. Hogg, Jamie I. Fletcher, Tao Liu, Michelle Haber, Murray D. Norris, Daniel R. Carter, Belamy B. Cheung, Glenn M. Marshall. Study supervision: Janith A. Seneviratne, Daniel R. Carter, Belamy B. Cheung, Glenn M. Marshall.
FUNDING INFORMATION
This work was supported by program grants (Glenn M. Marshall, Murray D. Norris, Michelle Haber) from the National Health and Medical Research Council (NHMRC) Australia (APP1016699), Cancer Institute NSW (10/TPG/1‐13), Cancer Council NSW (PG‐11‐06) and an Australia Research Training Program Scholarship, UNSW Sydney, Australia (Janith A. Seneviratne). This work was also supported by a NHMRC Project Grant (APP1125171; Glenn M. Marshall, Belamy B. Cheung) and Cancer Council NSW Project Grants (Belamy B. Cheung, RG21‐08 and Belamy B. Cheung, RG214491).
CONFLICT OF INTEREST
Pierre J. Dilda has a patent entitled “Organo‐arsenoxide compounds and use thereof” issued as well as a patent entitled “Pharmaceutical combinations of organo‐arsenoxide compounds and mTOR inhibitors” under examination. The other authors do not have a conflict of interest to declare.
ETHICS STATEMENT
These current animal studies were approved by the UNSW Animal Ethics Committee (ACEC Approval Numbers: 07/112A, 14/153B, 15/100B, 16/153B).
Supporting information
APPENDIX S1 Supporting Information
ACKNOWLEDGEMENT
The authors acknowledge the Steven Walter Children's Cancer Foundation, Saskia Loader's family and Neuroblastoma Australia for their continuous support. Open access publishing facilitated by University of New South Wales, as part of the Wiley ‐ University of New South Wales agreement via the Council of Australian University Librarians.
Seneviratne JA, Carter DR, Mittra R, et al. Inhibition of mitochondrial translocase SLC25A5 and histone deacetylation is an effective combination therapy in neuroblastoma. Int J Cancer. 2023;152(7):1399‐1413. doi: 10.1002/ijc.34349
Funding information Australia Research Training Program Scholarship; Cancer Council NSW, Grant/Award Number: PG‐11‐06; Cancer Council NSW Project Grant, Grant/Award Numbers: RG21‐08, RG214491; Cancer Institute NSW, Grant/Award Number: 10/TPG/1‐13; Neuroblastoma Australia; NHMRC Project Grant, Grant/Award Number: APP1125171; Steven Walter Children's Cancer Foundation; The National Health and Medical Research Council Australia, Grant/Award Number: APP1016699
Contributor Information
Belamy B. Cheung, Email: bcheung@ccia.unsw.edu.au.
Glenn M. Marshall, Email: glenn.marshall@health.nsw.gov.au.
DATA AVAILABILITY STATEMENT
Raw and processed microarray data generated in this study have been deposited in the gene expression omnibus under accession GSE196128 for public access. The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Sarosiek KA, Ni Chonghaile T, Letai A. Mitochondria: gatekeepers of response to chemotherapy. Trends Cell Biol. 2013;23:612‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol. 2015;11:9‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang SS, Hsiao R, Limpar MM, et al. Destabilization of MYC/MYCN by the mitochondrial inhibitors, metaiodobenzylguanidine, metformin and phenformin. Int J Mol Med. 2014;33:35‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bate‐Eya LT, den Hartog IJ, van der Ploeg I, et al. High efficacy of the BCL‐2 inhibitor ABT199 (venetoclax) in BCL‐2 high‐expressing neuroblastoma cell lines and xenografts and rational for combination with MCL‐1 inhibition. Oncotarget. 2016;7:27946‐27958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mihara M, Erster S, Zaika A, et al. p53 has a direct apoptogenic role at the mitochondria. Mol Cell. 2003;11:577‐590. [DOI] [PubMed] [Google Scholar]
- 6. Grobner SN, Worst BC, Weischenfeldt J, et al. The landscape of genomic alterations across childhood cancers. Nature. 2018;555:321‐327. [DOI] [PubMed] [Google Scholar]
- 7. Ma X, Liu Y, Liu Y, et al. Pan‐cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Howlader N, Noone A, Krapcho M, et al. SEER Cancer Statistics Review, 1975–2014. Bethesda, MD: National Cancer Institute; 2017:2018. [Google Scholar]
- 9. Pinto NR, Applebaum MA, Volchenboum SL, et al. Advances in risk classification and treatment strategies for neuroblastoma. J Clin Oncol. 2015;33:3008‐3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pugh TJ, Morozova O, Attiyeh EF, et al. The genetic landscape of high‐risk neuroblastoma. Nat Genet. 2013;45:279‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Carr J, Bell E, Pearson AD, et al. Increased frequency of aberrations in the p53/MDM2/p14(ARF) pathway in neuroblastoma cell lines established at relapse. Cancer Res. 2006;66:2138‐2145. [DOI] [PubMed] [Google Scholar]
- 12. Carr‐Wilkinson J, O'Toole K, Wood KM, et al. High frequency of p53/MDM2/p14ARF pathway abnormalities in relapsed neuroblastoma. Clin Cancer Res. 2010;16:1108‐1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xue C, Haber M, Flemming C, et al. p53 determines multidrug sensitivity of childhood neuroblastoma. Cancer Res. 2007;67:10351‐10360. [DOI] [PubMed] [Google Scholar]
- 14. Hagenbuchner J, Kuznetsov AV, Obexer P, Ausserlechner MJ. BIRC5/Survivin enhances aerobic glycolysis and drug resistance by altered regulation of the mitochondrial fusion/fission machinery. Oncogene. 2013;32:4748‐4757. [DOI] [PubMed] [Google Scholar]
- 15. Casinelli G, LaRosa J, Sharma M, et al. N‐Myc overexpression increases cisplatin resistance in neuroblastoma via deregulation of mitochondrial dynamics. Cell Death Discov. 2016;2:16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li S, Fell SM, Surova O, et al. The 1p36 tumor suppressor KIF 1Bbeta is required for calcineurin activation, controlling mitochondrial fission and apoptosis. Dev Cell. 2016;36:164‐178. [DOI] [PubMed] [Google Scholar]
- 17. Chevrollier A, Loiseau D, Chabi B, et al. ANT2 isoform required for cancer cell glycolysis. J Bioenerg Biomembr. 2005;37:307‐316. [DOI] [PubMed] [Google Scholar]
- 18. Don AS, Kisker O, Dilda P, et al. A peptide trivalent arsenical inhibits tumor angiogenesis by perturbing mitochondrial function in angiogenic endothelial cells. Cancer Cell. 2003;3:497‐509. [DOI] [PubMed] [Google Scholar]
- 19. Dilda PJ, Decollogne S, Weerakoon L, et al. Optimization of the antitumor efficacy of a synthetic mitochondrial toxin by increasing the residence time in the cytosol. J Med Chem. 2009;52:6209‐6216. [DOI] [PubMed] [Google Scholar]
- 20. Park D, Chiu J, Perrone GG, Dilda PJ, Hogg PJ. The tumour metabolism inhibitors GSAO and PENAO react with cysteines 57 and 257 of mitochondrial adenine nucleotide translocase. Cancer Cell Int. 2012;12:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gang BP, Dilda PJ, Hogg PJ, Blackburn AC. Targeting of two aspects of metabolism in breast cancer treatment. Cancer Biol Ther. 2014;15:1533‐1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shen H, Decollogne S, Dilda PJ, et al. Dual‐targeting of aberrant glucose metabolism in glioblastoma. J Exp Clin Cancer Res. 2015;34:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tran B, Hamilton AL, Horvath L, et al. First‐in‐man trial of 4‐(N‐[S‐penicillaminylacetyl]amino) phenylarsonous acid (PENAO) as a continuous intravenous infusion (CIVI), in patients (pt) with advanced solid tumours. J Clin Oncol. 2016;34:e14025‐e. [Google Scholar]
- 24. Weiss WA, Aldape K, Mohapatra G, Feuerstein BG, Bishop JM. Targeted expression of MYCN causes neuroblastoma in transgenic mice. EMBO J. 1997;16:2985‐2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rasmuson A, Segerström L, Nethander M, et al. Tumor development, growth characteristics and spectrum of genetic aberrations in the TH‐MYCN mouse model of neuroblastoma. PLoS One. 2012;7:e51297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mann BS, Johnson JR, Cohen MH, Justice R, Pazdur R. FDA approval summary: Vorinostat for treatment of advanced primary cutaneous T‐cell lymphoma. Oncologist. 2007;12:1247‐1252. [DOI] [PubMed] [Google Scholar]
- 27. DuBois SG, Groshen S, Park JR, et al. Phase I study of Vorinostat as a radiation sensitizer with 131I‐metaiodobenzylguanidine (131I‐MIBG) for patients with relapsed or refractory neuroblastoma. Clin Cancer Res. 2015;21:2715‐2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pinto N, DuBois SG, Marachelian A, et al. Phase I study of vorinostat in combination with isotretinoin in patients with refractory/recurrent neuroblastoma: a new approaches to Neuroblastoma therapy (NANT) trial. Pediatr Blood Cancer. 2018;65:e27023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petruccelli LA, Dupéré‐Richer D, Pettersson F, Retrouvey H, Skoulikas S, Miller WH Jr. Vorinostat induces reactive oxygen species and DNA damage in acute myeloid leukemia cells. PLoS One. 2011;6:e20987‐e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ruefli AA, Ausserlechner MJ, Bernhard D, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell‐death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci. 2001;98:10833‐10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsoli M, Liu J, Franshaw L, et al. Dual targeting of mitochondrial function and mTOR pathway as a therapeutic strategy for diffuse intrinsic pontine glioma. Oncotarget. 2018;9:7541‐7556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li D, Marchenko ND, Moll UM. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6‐Hsp90 chaperone axis. Cell Death Differ. 2011;18:1904‐1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li D, Marchenko ND, Schulz R, et al. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol Cancer Res. 2011;9:577‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Goldschneider D, Horvilleur E, Plassa L‐F, et al. Expression of C‐terminal deleted p53 isoforms in neuroblastoma. Nucleic Acids Res. 2006;34:5603‐5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meng J, Tagalakis AD, Hart SL. Silencing E3 Ubiqutin ligase ITCH as a potential therapy to enhance chemotherapy efficacy in p53 mutant neuroblastoma cells. Sci Rep. 2020;10:1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fulda S, Galluzzi L, Kroemer G. Targeting mitochondria for cancer therapy. Nat Rev Drug Discov. 2010;9:447‐464. [DOI] [PubMed] [Google Scholar]
- 37. Goldsmith KC, Gross M, Peirce S, et al. Mitochondrial Bcl‐2 family dynamics define therapy response and resistance in neuroblastoma. Cancer Res. 2012;72:2565‐2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Park SJ, Jo DS, Shin JH, et al. Suppression of Cpn10 increases mitochondrial fission and dysfunction in neuroblastoma cells. PLoS One. 2014;9:e112130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Santin G, Piccolini VM, Barni S, et al. Mitochondrial fusion: a mechanism of cisplatin‐induced resistance in neuroblastoma cells? Neurotoxicology. 2013;34:51‐60. [DOI] [PubMed] [Google Scholar]
- 40. Reznik E, Wang Q, La K, Schultz N, Sander C. Mitochondrial respiratory gene expression is suppressed in many cancers. Elife. 2017;6:e21592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Montemurro L, Raieli S, Angelucci S, et al. A novel MYCN‐specific antigene oligonucleotide deregulates mitochondria and inhibits tumor growth in MYCN‐amplified neuroblastoma. Cancer Res. 2019;79:6166‐6177. [DOI] [PubMed] [Google Scholar]
- 42. Jang JY, Kim YG, Nam SJ, et al. Targeting adenine nucleotide translocase‐2 (ANT2) to overcome resistance to epidermal growth factor receptor tyrosine kinase inhibitor in non‐small cell lung cancer. Mol Cancer Ther. 2016;15:1387‐1396. [DOI] [PubMed] [Google Scholar]
- 43. Sotgia F, Lisanti M. Mitochondrial mRNA transcripts predict overall survival, tumor recurrence and progression in serous ovarian cancer: companion diagnostics for cancer therapy. Oncotarget. 2015;8:66925‐66939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brenner C, Subramaniam K, Pertuiset C, Pervaiz S. Adenine nucleotide translocase family: four isoforms for apoptosis modulation in cancer. Oncogene. 2011;30:883‐895. [DOI] [PubMed] [Google Scholar]
- 45. Chevrollier A, Loiseau D, Reynier P, Stepien G. Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim Biophys Acta. 2011;1807:562‐567. [DOI] [PubMed] [Google Scholar]
- 46. Baugh EH, Ke H, Levine AJ, Bonneau RA, Chan CS. Why are there hotspot mutations in the TP53 gene in human cancers? Cell Death Differen. 2018;25:154‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yan W, Zhang Y, Zhang J, Liu S, Cho SJ, Chen X. Mutant p53 protein is targeted by arsenic for degradation and plays a role in arsenic‐mediated growth suppression. J Biol Chem. 2011;286:17478‐17486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yan W, Jung YS, Zhang Y, Chen X. Arsenic trioxide reactivates proteasome‐dependent degradation of mutant p53 protein in cancer cells in part via enhanced expression of Pirh2 E3 ligase. PLoS One. 2014;9:e103497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fu S, Hou MM, Naing A, et al. Phase I study of pazopanib and vorinostat: a therapeutic approach for inhibiting mutant p53‐mediated angiogenesis and facilitating mutant p53 degradation. Ann Oncol. 2015;26:1012‐1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
APPENDIX S1 Supporting Information
Data Availability Statement
Raw and processed microarray data generated in this study have been deposited in the gene expression omnibus under accession GSE196128 for public access. The data that support the findings of this study are available from the corresponding author upon reasonable request.