

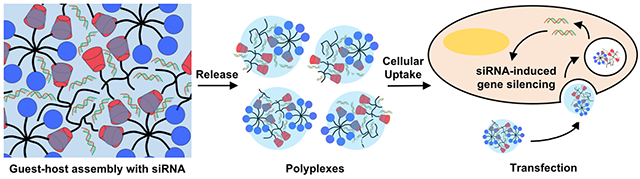
Abstract
While siRNA has tremendous potential for therapeutic applications, advancement is limited by poor delivery systems. Systemically, siRNAs are rapidly degraded, may have off-target silencing, and necessitate high working concentrations. To overcome this, we developed an injectable, guest-host assembled hydrogel between polyethylenimine (PEI) and polyethylene glycol (PEG) for local siRNA delivery. Guest-host modified polymers assembled with siRNAs to form polyplexes that had improved transfection and viability compared to PEI. At higher concentrations, these polymers assembled into shear-thinning hydrogels that rapidly self-healed. With siRNA encapsulation, the assemblies eroded as polyplexes which were active and transfected cells, observed by Cy3-siRNA uptake or GFP silencing in vitro. When injected into rat myocardium, the hydrogels localized polyplex release, observed by uptake of Cy5.5 siRNA and silencing of GFP for one week in a GFP-expressing rat. These results illustrate the potential for this system to be applied for therapeutic siRNA delivery, such as in cardiac pathologies.
Keywords: hydrogel, siRNA, polyethylenimine, cardiac, guest-host chemistry
Graphical Abstract
Introduction
Since the first report of RNA interference (RNAi) in 1998 by Fire et al., there has been great interest in applying RNAi therapeutically to silence the expression of pathologic genes.1 In this process, a double-stranded small RNA molecule, herein termed small interfering RNA (siRNA), enters a cell to interact with an RNA-induced silencing complex (RISC) and silence the expression of a complementary messenger RNA (mRNA) target.2,3 A single siRNA/RISC complex is able to silence cellular expression of gene targets specifically and efficiently. Particularly, siRNA therapeutics are able to target otherwise undruggable targets and have thus emerged as a potent therapeutic platform for various diseases including cancer and infection.4,5 However, despite the vast therapeutic potential, translation has been limited and there are currently no approved siRNA therapies clinically.6,7,8
Slow translation may be due to the numerous inherent challenges with siRNA delivery. siRNA is negatively charged and does not passively diffuse across cellular membranes, necessitating carriers like viruses, lipids or polymers for transfection.2,6,9 Moreover, siRNAs are particularly susceptible to nuclease-mediated hydrolysis and, when delivered systemically, are rapidly cleared from circulation (plasma half-life < 10 min).10 Other challenges include unfavorable aggregation with serum lipoproteins and erythrocytes, innate immune response induction, and off-target gene silencing.11,12 With systemic delivery, siRNA tends to accumulate in the lungs, kidneys, spleen and liver.10,13-15 Although this accumulation can be favorable for applications in these organs, attempts to deliver siRNA to other organs such as the heart necessitate higher loading concentrations or more frequent administrations, which can potentially increase costs, malignancy, inadvertent non-target organ toxicity, and potentially decreased patient adherence.
To overcome these limitations, siRNA can be delivered locally through a hydrogel. Hydrogels are water-swollen polymer networks crosslinked through physical or chemical bonds and have served as vehicles for the local and sustained delivery of various biomolecules, including siRNA.16-20 Release kinetics can be tuned through hydrogel properties such as polymer concentration, crosslink density, degradability, or drug-polymer affinity. siRNA is often delivered alone or in a cationic polyplex. By concentrating and eluting drugs locally, concentrations required for therapeutic efficacy are lowered by orders of magnitude, thereby additionally decreasing the potential for unintended adverse effects systemically.21,22 When compared to subcutaneous or intramuscular bolus injections, hydrogels further assist in local retention to limit off-target toxicity and promote sustained release.
While many hydrogels have been explored for this application, injectable hydrogels continue to gain interest for minimally invasive, or potentially non-invasive delivery.23-26 The design of injectable covalently crosslinked hydrogels is challenging, as rapid polymer diffusion from the injection site may occur if gelation is too slow,27 or clogging of the injection device may occur if gelation is too rapid.28 Hydrogels with physical crosslinks overcome these problems to an extent. In these systems, application of shear from a syringe can break physical bonds to permit flow until cessation of shear leads to reassembly. Many currently available systems exhibit long recovery times on the order of minutes to hours, leading to diffusion of polymers and associated biomolecules after injection.29,30 Thus, there is a need for a shear-thinning and self-healing hydrogel for local siRNA delivery that recovers rapidly upon injection.
We previously designed hyaluronic acid (HA) hydrogels based on guest-host chemistry between β-cyclodextrin and adamantane, which were shear-thinning, rapidly self-healing, and sustained the delivery of encapsulated biomolecules.14,23,31-34 Despite these properties, the use of anionic polymers like HA is not compatible with nucleic acids,35,36 which are typically delivered as cationic nanoparticles to permit transfection. To overcome this, we modified polyethylenimine (PEI), a widely used cationic polymer used for siRNA transfections,37-40 and polyethylene glycol, a neutral polymer, with guest-host chemistries. PEI is able to complex siRNA to form cationic polyplexes that can transfect cells and lead to gene silencing. In this system, we hypothesized that PEI would complex siRNA to promote siRNA transfection while also serving as one of the hydrogel components. PEI has been previously assembled into hydrogels for siRNA delivery for cancer.41,42 Our system expounds upon these previous reports as a shear-thinning, self-healing delivery system to improve injection towards minimally invasive delivery. Here, we explore our PEI guest-host assembly for the delivery of siRNA in vitro and upon injection into myocardium in vivo.
Experimental Section
CD-PEI synthesis:
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Branched PEI (25,000 g/mol) was modified with β-cyclodextrin according to a previously established protocol.43 Briefly, β-cyclodextrin (20.0 g, 17.6 mmol) was tosylated by reaction with p-toluenesulfonyl chloride (4.2 g, 22 mmol) in acetonitrile (10 mL) in de-ionized (DI) water. After two hours, sodium hydroxide (2.2 g, 53.6 mmol) was dissolved in DI water and added dropwise. The reaction was stirred another 30 minutes. pH was adjusted to 8.5 by addition of solid ammonium chloride. The solution was cooled on ice and the precipitate collected. The crude product was re-precipitated from cold DI water (3 × 200mL), washed by acetone (3 × 50mL) and dried under vacuum to afford the intermediate 6-o-monotosyl-6-deoxy-β-cyclodextrin as a white powder. Products were confirmed by 1H-NMR in DMSO-d6 (Bruker 360 MHz). To form cyclodextrin-PEI, 6-o-monotosyl-6-deoxy-β-cyclodextrin (740 mg, 0.57 mmol) was added dropwise to PEI (400 mg, 16.0 μmol) in 12 mL anhydrous DMSO via cannulation. Triethylamine (121 mg, 166 μL, 1.2 mmol) was added with a syringe. The reaction was stirred for 72 hours at 70°C and dialyzed against DI H2O for one week. Product was frozen and lyophilized to afford CD-PEI. Products were confirmed in 1H NMR in D2O. δ = 5.08 ppm (s, C1H of CD), 3.3-4.1 ppm (m, C2HC6H of CD), 2.5-3.2 ppm (m, CH2 of PEI). CD modification was obtained by normalizing C1H of CD (5.08 ppm) to [CH2CH2NH] repeats of PEI (2.5-3.2 ppm). CD-PEI obtained under these conditions has a modification of 25 CD molecules per PEI (~20% of primary amines) and has a re-calculated molecular weight of ~54,000 g/mol. Reaction times were decreased to achieve decreased CD modifications of 4, 7, 10, 12 CD/PEI.
Ad-PEG synthesis:
8-arm PEG-maleimide (20,000 g/mol, Creative PEGWorks) was modified with adamantane by reaction with 1-adamantanethiol through Michael addition. Briefly, 100 mg PEG-maleimide (5.0 μmol) was dissolved into 5 mL of DMSO. 20 mg (~100 μmol) of 1-adamantanethiol was dissolved into 20 mL of DMSO to react in excess. PEG-maleimide in DMSO was added slowly to the 1-adamantanethiol reaction dropwise. The reaction was stirred vigorously for hours at room temperature in a 50 mL round bottom flask. At reaction completion, the product was dialyzed against DI water for one week. By four days, unreacted adamantane precipitated out and was filtered from the product by vacuum filtration. After one week of dialysis, the product was frozen and lyophilized to afford Ad-PEG. 1H NMR (D2O) confirmed complete reaction of maleimides by disappearance of maleimide signal (δ = 6.8 ppm, d) and replacement by CH2 of Ad (δ = 1.5-2.2 ppm). Ad-PEG has a modification of 8 Ad per PEG and has a re-calculated molecular weight of ~21,000 g/mol.
CD-PEI and Ad-PEG fluorescent labeling:
To label CD-PEI, CD-PEI (~54 mg, 1.0 μmol) was reacted with Rhodamine Red™-X, Succinimidyl Ester, 5-isomer (Thermo Fisher Scientific) (~0.8 mg, 1.0 μmol) in DMSO to achieve an approximate functionalization of 1 rhodamine per PEI molecule. The reaction was carried out for 24 hours at 70°C. To label Ad-PEG, 8-arm-PEG-maleimide was reacted with a thiolated-FITC peptide (GCKK-FITC) based on a previously published protocol.44 Briefly, thiolated-FITC (893 g/mol, ~0.3 mg, 0.3 μmol) was dissolved in DMSO with PEG maleimide (~20 mg, 1.0 μmol) towards an approximate functionalization of 1 FITC molecule per PEG for two hours at room temperature, before being reacted with 1-adamantanethiol in excess. Synthesis was carried out in later steps as previously described.
Polyplex formation:
PEI (~25,000 g/mol), CD-PEI (~54,000 g/mol) and Ad-PEG (~21,000 g/mol) were resuspended in DEPC-treated H2O and filtered through a 0.2 μm filter before use to final concentrations of 1 mg/mL. For PEI/siRNA polyplex formation, 2.5 μl of 10 nM siRNA was combined with PEI, CD-PEI or Ad-PEG at various concentrations to form polyplexes in 500 μL DMEM or α-MEM and incubated at room temperature for 20 minutes under sterile conditions. Polyplexes were added dropwise to cells in serum-free conditions for 4 hours to avoid interactions with serum components and to parallel previous reports of nanoparticle/ nucleic acid delivery.20,45-47 After 4 hours, cells were washed in PBS and media was replaced with serum-containing media. For DLS and ζ-potential measurements, polyplexes were formed in DMEM as described above and measurements were made on a Zetasizer Nano (Malvern Instruments) for DLS or Delsa Nano C Particle Analyzer (Beckman Coulter). For ζ-potential measurements, polyplexes concentrations were scaled 80-fold. Sizes were reported from intensity distributions as z-average (d.nm). For heparin competition assays, polyplexes were formed as described above with Cy5.5-labeled siRNA. Heparin was added at varying concentrations and Cy5.5 fluorescence was measured at excitation of 670 nm and emission of 720 nm on a Tecan Infinite M200 plate reader.
siRNA sequences:
Cy3-labeled siRNA was purchased from SignalChem. All other siRNA purchases were from Dharmacon. Cy3-siCTRL: 5’-UUCUCCGAACGUGUCACGUTT-Cy3-3’ (Sense), 3’ TTAAGAGGCUUGCACAGUGCA 5’ (Antisense); Cy5.5-siGFP: 5’-GCAAGCUGACCCUGAAGUUCAU–Cy5.5-3’ (Sense), 3’-GCCGUUCGACUGGGACUUCAAG-5’ (Antisense); Cy5.5-siCTRL: 5’ UGGUUUACAUGUCGACUAAAU-Cy5.5- 3’ (Sense); 3’ GC ACCAAAUGUACAGCUGAUU 5’ (Antisense)
Hydrogel assembly:
Lyophilized polymers were sterilized under UV irradiation for 1 hour prior to resuspension in PBS matching CD to Ad. To form 100 μL gels at 20% wt, 8.9 mg of CD-PEI and 11.1 mg Ad-PEG were resuspended in PBS to match CD to Ad. To encapsulate siRNA, polymers were suspended in 50 μM siRNA solution. Before mixing, polymers were briefly sonicated, vortexed and incubated at room temperature with siRNA for 30 minutes. For release assays, gels were mixed in a 1.5 mL low-retention microcentrifuge tube (Fisher) and centrifuged and 500 μL of DMEM (HT1080), α-MEM (C166GFP) were added. Over two weeks, releasates were collected, stored at −20°C, and then thawed and added to cells under serum-free conditions for 24 hours for analysis. Cells were washed with PBS to remove polymer and siRNA polyplexes and media was replaced with serum-containing media after 24 hours.
Rheology characterization:
Rheological characterization was performed using an AR2000 stress-controlled rheometer (TA Instruments) fitted with a 20 mm diameter cone and plate geometry, 59 min 42 s cone angle, and 27 μm gap. Rheological properties were examined by oscillatory frequency sweeps (0.01–100 Hz; 1% strain), time sweeps (1.0 Hz; 1% strain), and continuous flow experiments (linearly ramped: 0–0.5 s−1 and returned). For shear recovery experiments, shear-thinning was performed at 250% strain with recovery at 0.5% strain, each at 1 Hz.
Characterization of hydrogel erosion, swelling, and siRNA release:
To quantify polymer erosion, CD-PEI-Rho, Ad-PEG-FITC, or Cy5.5-siRNA were assembled at 20 wt% in 1.5 mL Eppendorf tubes, with only one labeled component included in each formulation. Releasates were collected, solubilized in Triton X-100 and fluorescence was measured on a Tecan plate reader and normalized to a standard curve and to the final amount remaining. Rhodamine, FITC, and Cy5.5 fluorescence was measured at excitation/emission of 560/580 nm, 490/525 nm, and 670/720 nm. To quantify swelling, total gel volume increases at D1, D4, D7, and D14 were measured from releasate volume and normalized to beginning volume.
Cryogenic Transmission Electron Microscopy:
Gel/siRNA or control releasates were collected between D7 and D14. A droplet of 2 μL releasate solution was deposited onto a copper grid coated with a lacey carbon film, which was then loaded into a Gatan Cp3 cryoplunger. The sample was blotted by hand, plunged into liquefied ethane, and transferred to a Gatan CT3500TR cryoholder, which was then inserted into a JEOL2100 transmission electron microscope operating at an accelerating voltage of 200 kV. The images were recorded with low-dose mode in SerialEM using a Orius SC200 CCD camera.
Cell culture:
HT1080 human fibrosarcoma cells or C166GFP mouse endothelial cells were used for in vitro experiments. HT1080 cells were grown in α-MEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. C166GFP cells were grown in DMEM with 10% FBS and 0.2 mg/mL G418. All cells were seeded 24 hours prior to experiments at 1x105/mL in 1 mL of media in a 24-well plate a humidified CO2 (5%) atmosphere at 37°C.
Neonatal Rat Cardiomyocyte Isolation:
Neonatal rats were isolated between 0-36 hours after birth. Hearts were explanted and ventricles were minced and washed in Hank’s Balanced Salt Solution (HBSS) + 1% PSG solution. The tissue pieces were incubated in 0.1% trypsin in HBSS solution at 37°C while shaking. Supernatants were collected and placed on ice; this process was repeated 7 times. Supernatants were then centrifuged at 500 RCF for 5 min at 4°C and the cell pellet was resuspended in NRCM media (DMEM/M499 (4:1), 10% Horse serum, 5% FBS, 1% HEPES, and Medium 199). The cells were pre-plated on uncoated 10-cm dishes for 3 hours in the incubator; non-attached cells were plated on fibronectin-coated 96 well plates.
Cell viability assessment:
To HT1080 cells at 1x105 cells/mL, polymers were added drop-wise at various concentrations directly into the wells of the 24-well plate and incubated under a humidified CO2 (5%) atmosphere at 37°C for 24 hours in serum-containing media. Media was aspirated and replaced by 1 ml of 10% AlamarBlue® Cell Viability Reagent in serum-containing media according to Manufacturer’s protocols. Briefly, the plate was incubated for 4 hours under a humidified CO2 (5%) atmosphere at 37°C. After 4 hours, 250 μL of each sample were added to wells of a 96-well Costar® polystyrene assay plate (Corning). The fluorescence intensities were measured on a Tecan plate reader at 560 nm excitation/590 nm emission and normalized to untreated cells alone.
Flow cytometry:
Cells were detached from plates manually using a 220x11 mm cell scraper (CytoOne) and transferred into 1.5 mL microcentrifuge tubes. Tubes were centrifuged at 500 RCF for 5 minutes at room temperature. Cell pellets were washed three times in PBS and resuspended in 1 mL of PBS + 2% FBS and transferred to a polystyrene flow cytometry tubes (Falcon) while protected from light. Cells were analyzed in a BD FACSCalibur (BD Biosciences) to quantify Cy3 or GFP expression in the FL2 and FL1 channels, respectively. All conditions were normalized to untreated cells and scramble siRNA controls.
Injection into the myocardium:
For in vivo applications, gels were formed with siRNAs as previously described and manually transferred to a 27-Gx½” U-100 insulin syringe (Terumo) under sterile conditions on ice. Male rats (~300 g) were randomized to receive 60 μL total of gel or control injection (3 x 20 μL, 20% w/w, 50 μM siRNA). Briefly, rats were anesthetized with 3% isoflurane in an induction chamber (2L) and endotracheally intubated (Harvard Apparatus Regenerative Technology) with 1% isoflurane. A left lateral thoracotomy was performed at the fourth intercostal space to expose the heart in an established and highly reproducible model. Injections were made into the anterior and posterior left ventricular wall. For GFP knockdown studies, injections were made into Wistar-TgN(CAG-GFP)184Ys rats (Wistar GFP, Eiji Kobayashi, Jichi Medical School). Rats were genotyped prior to injection using real-time PCR, probing for eGFP (Transnetyx). Rats expressing an eGFP signal greater than 2.9, relative to a housekeeping gene, were selected for the study. Following injection, the chest was closed in 3 layers with a 3-0 polypropylene suture and animals were allowed to recover. After 24 hours or 7 days, the hearts were explanted, flushed with PBS, and distended with optimal cutting temperature embedding compound (Electron Microscopy Sciences) and frozen at −80°C. Hearts were sectioned along the short axis of the myocardium using Leica CM3050S cryostat at 10 μm thickness. Sections were washed 3 times in PBS, fixed in 4% paraformaldehyde for 10 minutes at room temperature, and blocked in 10% FBS for 1 hour at 37°C. Anti-GFP antibody (ab290, Abcam) was diluted at a ratio of 1:250 in PBS and incubated for 2 hours at 37°C. Alexa Fluor 488 secondary antibody (ab150065, Abcam) was diluted at a ratio of 1:1000 in PBS and incubated for 1 hour at 37°C. Nuclei were counterstained with 40,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories Inc, Burlingame, Calif). Sections were imaged using a Zeiss LSM 710 laser scanning confocal microscope (Oberkochen, Germany), with a 10x objective with the 405, 488, 514 and 651 nm laser lines. This project was approved by the University of Pennsylvania Institutional Animal Care and Use Committee and adhered to the National Institutes of Health guidelines on animal care and use.
Quantification of uptake and transfection:
To quantify GFP expression in images, GFP and Cy5.5 levels in separate channels were quantified along a horizontal axis using the plot profile feature of FIJI. To quantify GFP knockdown, GFP expression was measured in Cy5.5+ and Cy5.5− areas and compared between groups using thresholding. For GFP expression in Cy5.5+ areas, thresholding was used in the Cy5.5 channel to select a ROI that was analyzed in the GFP channel. For GFP expression in Cy5.5− areas, thresholding was done in the GFP channel to remove non-tissue areas and total GFP values were measured in the GFP+Cy5.5− area. Calculated total cryosection fluorescence (CTCF) was used to quantify total expression according to a previously established protocol to remove autofluorescence and background, where CTCF = Integrated Density – (Area of Selection x Mean Fluorescence of Background).48-50
Statistical Analysis:
All data are reported as means ± standard deviation (SD) and performed in triplicate unless otherwise indicated. Comparisons between two groups were performed by Students t-test with two-tailed criteria and significance determined at p < 0.05. For comparison between multiple groups, significance was determined by one-way ANOVA with post hoc testing. Bonferroni correction was used to account for multiple comparisons, with α = 0.05.
Results and Discussion
PEI modification for transfection and enhanced viability
Towards an injectable hydrogel for siRNA delivery, we conjugated guest-host pairs of β-cyclodextrin (CD) and adamantane (Ad) to branched polyethylenimine (PEI) and 8-arm poly(ethylene glycol) (PEG), respectively (Figure 1). CD is a macrocycle of 7D-glucose units that has an affinity for Ad through hydrophobic interactions to form inclusion complexes (Figure 1A). PEI was modified with CD to form CD-PEI through the nucleophilic tosylation reaction between the primary amines of PEI and mono-6-tosyl-β-cyclodextrin (Figure 1B).43 Neutrally charged 8-arm maleimide-functionalized PEG was modified with thiolated Ad to form Ad-PEG through a Michael Addition reaction. Synthesis was verified by 1H NMR (Figure S1). The modification of PEI with CD under these conditions led to ~25 CD/PEI. All maleimides on the PEG were modified with Ad (8 Ad/PEG). The guest-host interaction between CD and Ad permits the assembly of CD-PEI and Ad-PEG.
Figure 1. Schematic for modification of polyethylenimine (PEI) and polyethylene glycol (PEG) with guest host chemistries.
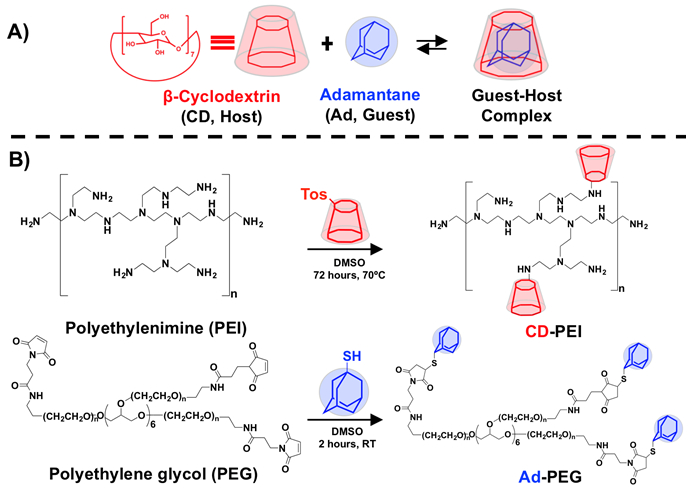
A) β-cyclodextrin and adamantane interact through supramolecular chemistry to form an inclusion complex (K = 1 x 10−5 M−1). B) Synthesis of guest-host modified CD-PEI and Ad-PEG polymers.
At low concentrations, CD-PEI and Ad-PEG were evaluated for their ability to complex siRNA into polyplexes and enable siRNA transfection in vitro (Figure 2A). The sizes of polyplexes were measured through dynamic light scattering (DLS). By DLS, CD-PEI/siRNA polyplexes were ~150 nm in size and increased in size to ~250 nm with addition of Ad-PEG at equimolar concentrations of CD and Ad; in contrast, PEI/siRNA polyplex controls were ~100 nm in size and did not increase in size with addition of Ad-PEG (Figure S2A). ζ-potential of CD-PEI polyplexes was ~7 mV, which was less than PEI polyplexes at ~11 mV to reflect the decrease in surface charge from amine modification (Figure S2B). Addition of Ad-PEG to CD-PEI decreased the ζ-potential to ~0 mV, reflecting a net neutral charge from coating by PEG. The co-localization of the various components was assessed through fluorescent microscopy, where siRNA, CD-PEI and Ad-PEG were labeled with Cy3, rhodamine and FITC, respectively. The three components were complexed together, where each was separately introduced with a fluorescent label in independent experiments, and were added to HT1080 cells. Fluorescent imaging after 24 hours of siRNA, CD-PEI and Ad-PEG confirmed localization of all three components within cells (Figure 2B). Compared to unmodified PEI, CD-PEI significantly enhanced transfection of Cy3-labeled siRNA into cells at 24 hours as measured by flow cytometry (Figure 2C). The uptake of siRNA also increased with increasing CD modification of PEI up to 25 CD/PEI (Figure S3A) at a single concentration (200 nM), where CD-PEI significantly enhanced siRNA uptake (Figure S3B). Furthermore, the addition of Ad-PEG to CD-PEI/siRNA polyplexes further increased siRNA uptake compared to PEI or CD-PEI alone. siRNA uptake increased with increasing Ad concentration until CD and Ad were matched in equimolar ratios, at which point uptake decreased. (Figure S3C).
Figure 2. Cyclodextrin modification and PEGylation through guest-host chemistry promotes siRNA transfection and decreases cytotoxicity.
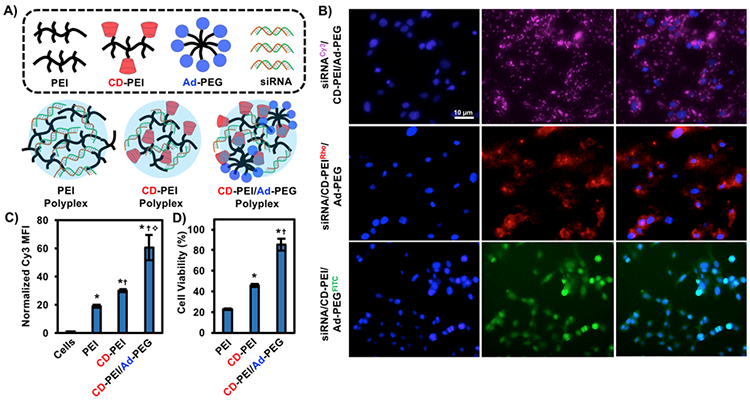
A) Schematic of PEI, CD-PEI and CD-PEI/Ad-PEG polyplexes with siRNA. B) Transfection of HT1080 cells with fluorescently-labeled siRNA and guest-host polymers, where each component was investigated individually (siRNA: Cy3, CD-PEI: Rhodamine, Ad-PEG: FITC). C) Flow cytometry of Cy3-siRNA uptake from PEI, CD-PEI and CD-PEI/Ad-PEG polyplexes compared to cells alone. *p<0.05 compared to cells alone, †p<0.05 compared to PEI, ✧p<0.05 compared to CD-PEI. D) Alamar Blue cell viability assay of PEI, CD-PEI and CD-PEI/Ad-PEG. *p<0.05 compared to PEI, †p<0.05 compared to CD-PEI.
To assess siRNA complexation efficiency, PEI and CD-PEI polyplexes were assembled with Cy5.5-labeled siRNAs in the presence of increasing concentrations of heparin, an anionic molecule known to displace siRNA from polyplexes. CD-PEI/siRNA complexes were as efficient as PEI in complexing siRNA at low concentrations of heparin. At high concentrations, heparin was able to displace siRNA from CD-PEI polyplexes (Figure S4). This was not surprising, as CD modification of primary amines reduced the charge and decreased the ability for PEI to complex with anionic phosphates of siRNA backbones.
Given previous reports of polycation-induced cytotoxicity from PEI,51,52 cell viability in the presence of the various polyplexes was also investigated. Towards this, HT1080 cells were plated with PEI, CD-PEI, or CD-PEI/Ad-PEG and toxicity was assessed through an Alamar Blue fluorometric assay, which measures metabolic activity. At cytotoxic concentrations of PEI, equimolar CD-PEI significantly enhanced cellular viability (Figure 2D), corroborating previous reports of cyclodextrin-modified PEIs in decreasing cytotoxicity.43,53 Cell viability was improved with as few as 4 CD/PEI (Figure S5A) and CD-PEI was significantly less cytotoxic than PEI at two different concentrations tested (Figure S5B). Moreover, the addition of Ad-PEG to CD-PEI improved cell viability in a dose-dependent manner (Figure S5C). Unsurprisingly, Ad-PEG alone had no significant effect on cell viability at any concentration tested (Figure S5D).
The ability for CD-PEI and Ad-PEG to improve transfection can be ascribed to a combination of improved cell viability to better facilitate endocytosis and improved condensation of polyplexes mediated by hydrophobic interactions between CD/Ad and CD/CD. Improvements in cell viability are most likely related to alterations in surface charge, wherein CD-modification of the primary amines of PEI directly decreases the amount of available free, protonated primary amines as suggested by the ζ-potential measurements. Addition of Ad-PEG through guest-host interactions further masks charge as PEG is neutrally charged to decrease cytotoxicity, while still allowing polyplexes to have some charge to traffic across cell membranes.43 This is also suggested by ζ-potential measurements. PEG may also play a role in enhancing salt stability of polyplexes to enhance transfection by coating the polyplex surface to limit undesired electrostatic interactions, which is supported by ζ-potential measurements in which CD-PEI/Ad-PEG polyplexes are approximately 0 mV.54,55 Lastly, cyclodextrin itself has been shown to be a viable siRNA transfection reagent in and of itself due to interactions with siRNA to promote endocytosis.6 Thus, there may be cooperativity between CD and PEI to enhance transfection in our modified polymers. This data corroborates previously published data, which had demonstrated the efficacy of both linear and branched CD-PEI polymers in plasmid DNA43,53 and siRNA delivery. 56,57
Guest-host polymer assembly into injectable, shear-thinning hydrogels
siRNA polyplexes formed from electrostatic interactions between siRNA and CD-PEI at low concentrations, where the polymer concentration was too dilute to assemble into 3-dimensional continuous networks. However, at higher concentrations (20 wt%) and equimolar amounts of CD and Ad, CD-PEI and Ad-PEG formed a supramolecular assembly through guest-host interactions between CD and Ad (Figure 3A). To demonstrate gelation qualitatively, individual CD-PEI and Ad-PEG polymers were dissolved in PBS to form viscous solutions that flowed with inversion. Conversely, when CD-PEI and Ad-PEG were combined and mixed manually, an assembly formed that was stable to flow within the first minute after during qualitative inversion (Figure 3B).
Figure 3. Guest-host CD-PEI/Ad-PEG assemblies exhibit frequency dependent moduli and shear-thinning with rapid recovery on oscillatory rheology.
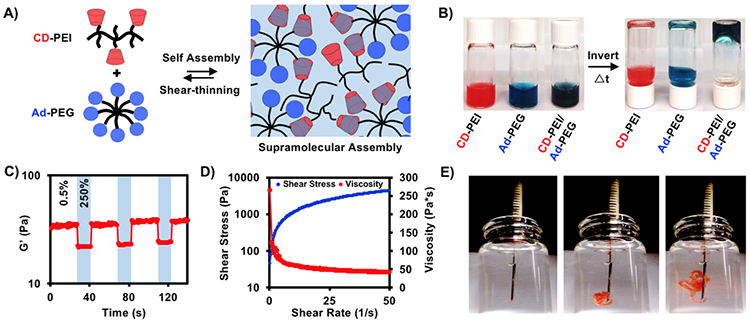
A) Schematic of CD-PEI and Ad-PEG assembly through guest-host complex formation – the reversible guest-host bond permits shear-thinning and rapid reassembly. B) Qualitative inversion after one minute of CD-PEI, Ad-PEG and CD-PEI/Ad-PEG. C) Storage modulus in response to cyclic strain (low = 0.5%, high = 250%) from oscillatory rheology. D) Shear stress and viscosity measurements from increasing shear rate by oscillatory rheology. E) Injection through 27Gx½” insulin syringes into water.
The viscoelastic properties of the assemblies were measured by oscillatory rheology. Given the viscoelastic nature of the material, moduli were frequency dependent. At 1 Hz, assemblies had a storage modulus (G’) of approximately 40 Pa, which increased several orders of magnitude as the frequency increased (Figure S6); notably, mechanics were not affected by encapsulation of siRNA. The relatively weak moduli of the assemblies can be ascribed to limitations in network formation as a result of low molecular weight and branching structures of the polymers (PEG:20k, PEI:25k) and the number of guest-host pairs possible between polymers (8 Ad/PEG, 25 CD/PEI). Whereas moduli can be improved by increasing polymer concentration and molecular weight, these changes compromise ease of injection and cell viability, respectively, as higher molecular weight PEI reduces viability (Figure S7). These assemblies are quite weak; however, they do form a continuous network and are stable over many weeks from in vitro erosion assays. However, their storage modulus (G’) is lower than their loss modulus (G”) across all frequencies tested rheologically; as such, these assemblies are more viscous than elastic.
Due to the dynamic reversibility of the guest-host complexes, assemblies were examined for shear-thinning and recovery. As the shear rate increased, the viscosity decreased with a corresponding increase in shear stress, demonstrating shear-thinning behavior (Figure 3C). To further explore this, assemblies were placed under alternating cycles of high and low amplitude oscillatory strain. Under high-amplitude strain (250%), the storage modulus decreased ~ 40%, which then rapidly recovered (<1 s) when returned to low-amplitude strain (0.5%), repeatedly (Figure 3D). These rheological measurements suggest that guest-host bonds can reverse under shear to permit flow with rapid bond reformation and reassembly upon cessation of shear, which will aid in injection and retention when introduced into a tissue. This was also observed empirically by injecting the assemblies through a 27-G insulin syringe into PBS, where both flow and rapid reassembly in the aqueous environment was observed without loss of material (Figure 3E). This confirms that these assemblies are both appropriately injectable and self-healing, as we have previously observed from hyaluronic acid systems.
siRNA release, transfection and gene silencing in vitro
Assemblies of siRNA/CD-PEI/Ad-PEG were hypothesized to encapsulate siRNA and then dissociate into polyplexes over time, that could then transfect cells (Figure 4A). This has previously been observed for gel/siRNA assemblies from cationic polymers, in which cationic polymers like PEI double as both the transfection reagent and network component.41,58-60 Unlike previously explored systems, our system takes advantage of guest-host chemistry to permit shear-thinning and rapid self-healing after injection to improve retention and delivery.
Figure 4. Guest-host assemblies with siRNA promote sustained polyplex release over two weeks.
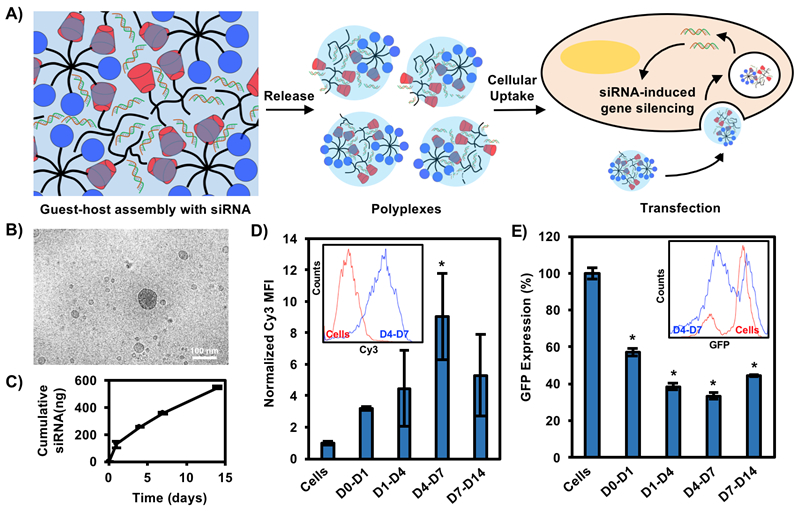
A) Schematic of assembly with siRNAs, which are sustained in the cationic gel through electrostatic interactions. siRNA is complexed to CD-PEI and Ad-PEG upon release, leading to transfection as polyplexes. B) Cryo-TEM of gel/siRNA assembly releasates. Scale bar = 100 nm. C) Cumulative siRNA release from assemblies over two weeks. D) Cy3-siRNA transfection from releasates collected over two weeks in HT1080 cells quantified by flow cytometry. *p<0.05 compared to cells alone. D) siGFP gene silencing from releasates collected over two weeks in GFP-expressing endothelial cells quantified by flow cytometry and normalized to siCTRL at each timepoint. *p<0.05 compared to cells alone.
Cy3-labeled siRNA was assembled with CD-PEI/Ad-PEG and releasates were collected over two weeks for siRNA polyplex imaging with cryo-TEM and quantification. Releasates from siRNA/CD-PEI/Ad-PEG assemblies showed spherical polyplexes that ranged from 20-300 nm in size in combination with small numbers of larger, micron-sized (>1000 nm) particles (Figure 4B). In contrast, releasates from CD-PEI/Ad-PEG assemblies without siRNA showed only micro-sized particles with no polyplexes (Figure S8), suggesting that free polymers alone did not assemble into polyplexes without siRNA. siRNA alone also did not form polyplexes without either CD-PEI or Ad-PEG. The formation of polyplexes is likely driven by the dynamic reversibility of the guest-host bond, in which CD-PEI and Ad-PEG are able to freely erode from the system via guest-host disassembly, either already in complex with siRNA or as free polymer chains that then interact with siRNA in releasates to form polyplexes.
siRNA release was sustained over this time period, with <10% of total siRNA released by two weeks (Figure 4C), suggesting the majority of the siRNA remained in the gel. The sustained release observed is most likely the result of electrostatic interactions with the CD-PEI and the anionic phosphates of the siRNA, limiting diffusion of siRNA. Erosion profiles of CD-PEI and Ad-PEG were sustained over the same time period where polymers achieved >60% erosion over the same window (Figure S9A), suggesting erosion as a primary mechanism for siRNA release. The disparate rates of release between siRNA and CD-PEI/Ad-PEG are most likely attributed to relative concentrations, as CD-PEI/Ad-PEG are present at much higher amounts in the network. The assemblies swelled to ~280% their original volume within the first week, suggesting swelling as a secondary mechanism for release (Figure S9B). It has been shown recently that these dynamic interactions can lead to a reorganization of the network, adding further complexity to release.61 Erosion and release profiles will also be greatly influenced by the environment and may be accelerated from these static in vitro cultures.
To confirm that released polyplexes were active, Cy3-siRNAs were included and releasates were added to HT1080 cultures. HT1080 cells are a fibrosarcoma line that represents a fibroblast lineage and potential in vivo target. Flow cytometry after 24 hours showed uptake of Cy3-siRNA over two weeks with maximal uptake of siRNA after one week, corresponding to ~10-fold increase in Cy3 fluorescence (Figure 4D, Figure S10A). To confirm that released polyplexes were functionally active, siRNA targeting green-fluorescent protein (siGFP) were used and releasates were collected and added to culture of GFP-expressing C166 murine endothelial cells. GFP knockdown was quantified through flow cytometry and normalized to a scramble control (siCTRL). Total GFP expression was decreased at all times by at least 30% with up to 55% knockdown of GFP expression for samples collected between days 4 and 7, coincident with maximal Cy3-siRNA uptake (Figure 4E, Figure S10B). Together, this suggests that released siRNA polyplexes are able to enter cells through endocytosis and escape the endosome to permit gene silencing. Releasates from D0-D1 were observed to be cytotoxic, which significantly improved at later collection timepoints (Figure S5E). This is likely due to burst release of high amounts of CD-PEI that do not adequately assemble into the network.
Taken together, this data supports a model in which siRNA is released in complex with CD-PEI and Ad-PEG as polyplexes that can transfect cells. Free PEI polymers that are not associated with siRNA can potentially improve transfection and enhance polyplex uptake; previous studies showed that increasing free PEI concentrations did not partake in polyplexes but still resulted in improvements in transfection.62,63 While this data confirms previously observed studies that cationic gel backbones can promote transfection of naked siRNAs, our system significantly improves injection potential towards use in biomedical applications.
siRNA uptake and gene silencing in vivo
Towards therapeutic siRNA delivery in vivo, assemblies were injected into the myocardium of the left ventricle of adult Wistar rats. The assemblies contained Cy3-labeled siRNA and rats were sacrificed after 24 hours for imaging. Cy3 signals were robust and localized to sites of injection, which were identified from areas of increased cellularity from nuclear stains. Imaging revealed pockets of hydrogel (DAPI negative, Cy3 positive) containing siRNA, suggesting that not all siRNA had been released by 24 hours. However, there was notably diffuse but punctate cytoplasmic Cy3 signals present, that mostly spared the nuclei (Figure 5A). This pattern of punctate, perinuclear fluorescence is characteristic of endosomal sequestration of Cy3-siRNAs,64,65 consistent with sustained endocytosis of siRNAs over the first 24 hours post injection. Moreover, the relative overexpression of siRNA in the cytoplasm compared to the nucleus observed is also characteristic of siRNA transfection,66 wherein siRNA, upon escaping the endosome, is unable to traffic across the nuclear membrane and leads to RNA interference in the cytoplasm.
Figure 5. Gel/Cy3-siRNA promotes Cy3-siRNA uptake in rat myocardium.
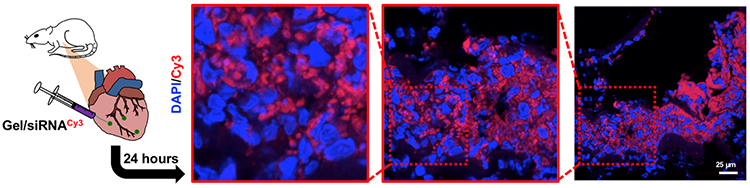
Immunohistochemistry of rat hearts 24 hours after Cy3-siRNA/gel injections with DAPI staining.
The assemblies were then investigated for gene silencing in the heart. In this model, Wistar-TgN(CAG-GFP)184Ys, transgenic GFP-expressing rats were employed to assess GFP knockdown. GFP is expressed downstream of a powerful CAG promoter to promote high levels of GFP expression. Animals were genotyped and only animals expressing high GFP levels (>2.9 normalized to housekeeping gene) were utilized for experiments. To confirm that such an approach to measure GFP knockdown was viable with siRNA to GFP (siGFP), primary neonatal rat cardiomyocytes from this animal model were plated in vitro and exposed to releasates from siGFP/CD-PEI/Ad-PEG. GFP knockdown was assessed through imaging and quantified in FIJI, which supported sustained GFP knockdown (Figure S10).
In this model, rats were injected with assemblies containing siGFP or control siRNA (siCTRL) and then sacrificed at 24 hours or 7 days. PBS/siGFP alone was also injected as a control. Importantly, siRNA was also labeled with Cy5.5, a near-IR fluorescent dye to minimize overlap with GFP emission and to co-localize GFP knockdown with Cy5.5 uptake. At 24 hours in gel/siGFP treated animals, Cy5.5 signal co-localized with a concomitant decrease in GFP expression, suggesting GFP gene silencing in cells taking up siRNA (Figure 6A). Cy5.5 signal co-localized with GFP in the cytoplasm, indicating uptake (Figure S11A). Using FIJI, pixel intensity of both GFP and Cy5.5 could be plotted against distance along a horizontal axis, which demonstrates that the strongest Cy5.5 signal occurs in the areas with the weakest GFP signal (Figure 6B). In contrast, in gel/siCTRL treated animals, siRNA did not affect GFP expression, supported by pixel intensity along the horizontal axis. To quantify knockdown in gel/siGFP compared to siCTRL, total GFP expression in Cy5.5-positive areas was measured and normalized to total GFP expression in Cy5.5-negative areas by thresholding in FIJI. By fluorescence, this corresponded to ~40% decrease in GFP expression at 24 hours with siGFP delivery (Figure 6C).
Figure 6. Gel/siGFP assemblies promote GFP gene silencing in vivo.
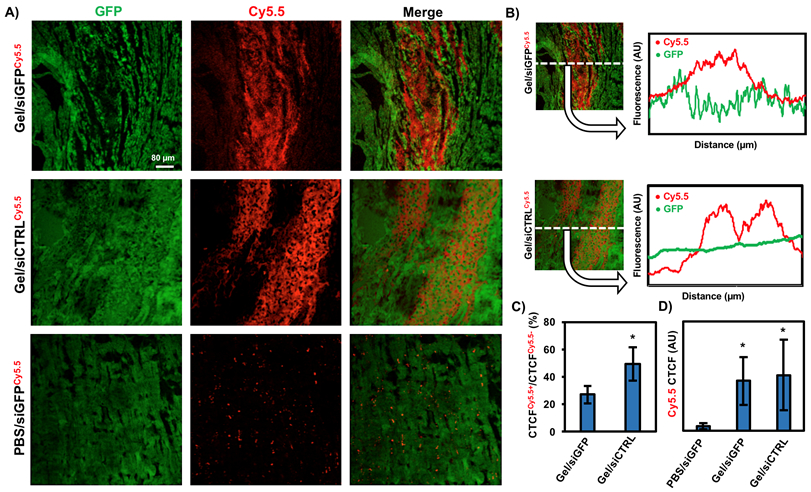
A) Immunohistochemistry of GFP expressing rat hearts 24 hours after Cy5.5-labeled siGFP injections either within a gel or alone, along with siCTRL within a gel control. B) FIJI analysis in the x-direction of Cy5.5 or GFP, where the pixel intensity was averaged across the entire y-direction of the image. C) Quantification of GFP in Cy5.5+ and Cy5.5− areas between groups from CTCF and FIJI threshold analysis to remove background and tissue tears. *p<0.05 compared to Gel/siGFP. D) Quantification of total Cy5.5 signal between groups after 24 hours from CTCF. *p<0.05 compared to PBS/siGFP.
GFP knockdown was observed out to 7 days in gel/siGFP treated groups, which co-localized with Cy5.5 (Figure S11B). However, the Cy5.5 signal at this timepoint was decreased compared to at 24 hours, suggesting hydrogel integration or dispersal into the tissue and potential siRNA degradation or exocytosis, especially because the Cy5.5 label is on the sense (passenger) strand that does not interact with RISC. In PBS/siGFP treated animals, there was no decrease in GFP knockdown. Moreover, the Cy5.5 signal was diffusely distributed into the tissue. Total Cy5.5 expression from PBS groups were compared to gel/siGFP or gel/siCTRL. In both cases, the hydrogel increased Cy5.5 expression significantly (Figure 6D). Notably, Cy5.5 signal from a PBS injection was not observed at 7 days (Figure S11C). Taken together, these data suggest that the injected assemblies promote gene silencing of GFP expression as early as 24 hours and up to one week and play an important role in localizing and retaining siRNA upon injection. The myocardium has previously been identified as a poor site of drug or therapeutic retention upon injection compared to other organs, owing largely to its contractile environment in combination with channel leakage and vascular drainage.67
The ability for this guest-host assembly to promote siRNA uptake and gene silencing of GFP in vivo corroborates previously observed in vitro data. Here, released polyplexes are likely uptaken into resident cells of the myocardium, for gene silencing. Moreover, free polymer chains from PEI can promote transfection, which can further facilitate the uptake of siRNA polyplexes.62,63 Whereas there have been previous reports of siRNA delivered to the heart through various mechanisms, this is the first ever report of siRNA delivery to the heart from a hydrogel towards local, sustained delivery. Future work will take this proof-of-concept into a therapy, through the silencing of genes that may influence outcomes after myocardial infarction. Such targets include ACE,68 RAGE,39 NSF,69 PHD2,70 and Nox2-NADPH oxidase,71 which have previously been explored for improving function after myocardial infarction.
Conclusions
In summary, we have developed an injectable polymer assembly from PEI and PEG to promote local and sustained siRNA delivery. This report builds on previous examples of biomaterials which enabled sustained siRNA release.41,72-74 In this report, guest-host modified polymers improved transfection and decreased cytotoxicity compared to PEI alone. Modified polymers formed injectable, shear-thinning assemblies that eroded into polyplexes that promoted siRNA uptake and gene silencing. Upon injection into the myocardium, these assemblies localized siRNA and sustained GFP silencing for up to one week. This generalizable approach can be used towards local, sustained applications for numerous applications such as cardiac pathologies, where an injectable hydrogel is useful for minimally invasive delivery and localized retention of siRNA cargo. It could also be tailored for other nucleic acid based therapeutics such as DNA or antisense oligonucleotide delivery.
Supplementary Material
Acknowledgments
The authors thank Dr. C. Rodell for helpful discussion concerning material synthesis and experimental design, and Patrick Dinh for assistance with implementation and analysis of the animal models. We thank the Penn Flow Cytometry and Cell Sorting Facility and the Nanoscale Characterization Facility in the Singh Center for Nanotechnology for use of flow cytometry and cryo-TEM equipment, respectively. This work was made possible by financial support from the American Heart Association through an established investigator award (JAB) and predoctoral fellowship (LLW), the National Institutes of Health (F30 HL134255), and the National Science Foundation through a MRSEC award to the University of Pennsylvania.
Footnotes
Supporting Information
1H NMR spectra of CD-PEI and AD-PEG, characterization of polyplex size and charge, additional cell viability and transfection data, heparin competition assays, oscillatory rheology frequency sweeps, cryo-TEM images and controls, gel erosion and swelling, flow cytometry dot plots, neonatal cardiomyocyte GFP quantification, and additional injection timepoints are available in the Supplementary Information document.
References
- (1).Fire A; Xu S; Montgomery MK; Kostas SA; Driver SE; Mello CC Nature 1998, 391 (6669), 806–811. [DOI] [PubMed] [Google Scholar]
- (2).Wang J; Lu Z; Wientjes MG; Au JL-S AAPS J. 2010, 12 (4), 492–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Milhavet O; Gary DS; Mattson MP Pharmacol. Rev 2003, 55 (4), 629–648. [DOI] [PubMed] [Google Scholar]
- (4).Wu SY; Lopez-Berestein G; Calin GA; Sood AK Sci. Transl. Med 2014, 6 (240), 240ps7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ozcan G; Ozpolat B; Coleman RL; Sood AK; Lopez-Berestein G Adv. Drug Deliv. Rev 2015, 87, 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kanasty R; Dorkin JR; Vegas A; Anderson D Nat. Mater 2013, 12 (11), 967–977. [DOI] [PubMed] [Google Scholar]
- (7).Wittrup A; Lieberman J Nat. Rev. Genet 2015, 16 (9), 543–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tiemann K; Rossi JJ EMBO Mol. Med 2009, 1 (3), 142–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tam YYC; Chen S; Cullis PR Pharmaceutics. 2013, pp 498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).van de Water FM; Boerman OC; Wouterse AC; Peters JGP; Russel FGM; Masereeuw R Drug Metab. Dispos 2006, 34 (8), 1393–1397. [DOI] [PubMed] [Google Scholar]
- (11).Kanasty RL; Whitehead KA; Vegas AJ; Anderson DG Mol. Ther 2012, 20 (3), 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Sarett SM; Nelson CE; Duvall CL J. Control. Release 2015, 218, 94–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tian Y; Liu Y; Wang T; Zhou N; Kong J; Chen L; Snitow M; Morley M; Li D; Petrenko N; Zhou S; Lu M; Gao E; Koch WJ; Stewart KM; Morrisey EE Sci. Transl. Med 2015, 7 (279). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rodell CB; MacArthur JW; Dorsey SM; Wade RJ; Wang LL; Woo YJ; Burdick JA Adv. Funct. Mater 2014, 25 (4), 636–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shi B; Keough E; Matter A; Leander K; Young S; Carlini E; Sachs AB; Tao W; Abrams M; Howell B; Sepp-Lorenzino LJ Histochem. Cytochem 2011, 59 (8), 727–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Huynh CT; Nguyen MK; Naris M; Tonga GY; Rotello VM; Alsberg E Nanomedicine (Lond). 2016, 11 (12), 1535–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Nguyen MK; Jeon O; Krebs MD; Schapira D; Alsberg E Biomaterials 2014, 35 (24), 6278–6286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Krebs MD; Jeon O; Alsberg EJ Am. Chem. Soc 2009, 131 (26), 9204–9206. [DOI] [PubMed] [Google Scholar]
- (19).Huynh CT; Nguyen MK; Tonga GY; Longé L; Rotello VM; Alsberg E Adv. Healthc. Mater 2016, 5 (3), 305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Segovia N; Pont M; Oliva N; Ramos V; Borrós S; Artzi N Adv. Healthc. Mater 2015, 4 (2), 271–280. [DOI] [PubMed] [Google Scholar]
- (21).Shea LD; Smiley E; Bonadio J; Mooney DJ Nat. Biotechnol 1999, 17 (6), 551–554. [DOI] [PubMed] [Google Scholar]
- (22).De Laporte L; Shea LD Adv. Drug Deliv. Rev 2007, 59 (4–5), 292–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mealy JE; Rodell CB; Burdick JA J. Mater. Chem. B 2015, 3 (40), 8010–8019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Purcell BP; Lobb D; Charati MB; Dorsey SM; Wade RJ; Zellars KN; Doviak H; Pettaway S; Logdon CB; Shuman JA; Freels PD; Gorman JH; Gorman RC; Spinale FG; Burdick JA Nat. Mater 2014, 13 (6), 653–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Soranno DE; Rodell CB; Altmann C; Duplantis J; Andres-Hernando A; Burdick JA; Faubel S Am. J. Physiol. Renal Physiol 2016, 311 (2), F362–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Soranno DE; Lu HD; Weber HM; Rai R; Burdick JA J. Biomed. Mater. Res. A 2014, 102 (7), 2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hillel AT; Unterman S; Nahas Z; Reid B; Coburn JM; Axelman J; Chae JJ; Guo Q; Trow R; Thomas A; Hou Z; Lichtsteiner S; Sutton D; Matheson C; Walker P; David N; Mori S; Taube JM; Elisseeff JH Sci. Transl. Med 2011, 3 (93), 93ra67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Martens TP; Godier AFG; Parks JJ; Wan LQ; Koeckert MS; Eng GM; Hudson BI; Sherman W; Vunjak-Novakovic G Cell Transplant. 2009, 18 (3), 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yan C; Altunbas A; Yucel T; Nagarkar RP; Schneider JP; Pochan DJ Soft Matter 2010, 6 (20), 5143–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Guvendiren M; Lu HD; Burdick JA Soft Matter 2012, 8 (2), 260–272. [Google Scholar]
- (31).Rodell CB; Mealy JE; Burdick JA Bioconjug. Chem 2015, 26 (12), 2279–2289. [DOI] [PubMed] [Google Scholar]
- (32).Rodell CB; Wade RJ; Purcell BP; Dusaj NN; Burdick JA ACS Biomater. Sci. Eng 2015, 1 (4), 277–286. [DOI] [PubMed] [Google Scholar]
- (33).Gaffey AC; Chen MH; Venkataraman CM; Trubelja A; Rodell CB; Dinh PV; Hung G; MacArthur JW; Soopan RV; Burdick JA; Atluri P J. Thorac. Cardiovasc. Surg 2015, 150 (5), 1268–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Rodell CB; Kaminski AL; Burdick JA Biomacromolecules 2013, 14 (11), 4125–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Lei Y; Huang S; Sharif-Kashani P; Chen Y; Kavehpour P; Segura T Biomaterials 2010, 31 (34), 9106–9116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lei Y; Rahim M; Ng Q; Segura TJ Control. Release 2011, 153 (3), 255–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Laroui H; Geem D; Xiao B; Viennois E; Rakhya P; Denning T; Merlin D Mol. Ther 2014, 22 (1), 69–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wu Y; Wang W; Chen Y; Huang K; Shuai X; Chen Q; Li X; Lian G Int. J. Nanomedicine 2010, 5, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hong J; Ku SH; Lee MS; Jeong JH; Mok H; Choi D; Kim SH Biomaterials 2014, 35 (26), 7562–7573. [DOI] [PubMed] [Google Scholar]
- (40).Zintchenko A; Philipp A; Dehshahri A; Wagner E Bioconjug. Chem 2008, 19 (7), 1448–1455. [DOI] [PubMed] [Google Scholar]
- (41).Kim Y-M; Park M-R; Song S-C ACS Nano 2012, 6 (7), 5757–5766. [DOI] [PubMed] [Google Scholar]
- (42).Kim Y-M; Park M-R; Song S-C Biomaterials 2013, 34 (18), 4493–4500. [DOI] [PubMed] [Google Scholar]
- (43).Pun SH; Bellocq NC; Liu A; Jensen G; Machemer T; Quijano E; Schluep T; Wen S; Engler H; Heidel J; Davis ME Bioconjug. Chem 2004, 15 (4), 831–840. [DOI] [PubMed] [Google Scholar]
- (44).Wade RJ; Bassin EJ; Rodell CB; Burdick JA Nat. Commun 2015, 6, 6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Godbey WT; Wu KK; Mikos AG Biomaterials 2001, 22 (5), 471–480. [DOI] [PubMed] [Google Scholar]
- (46).Xu Z; Shen G; Xia X; Zhao X; Zhang P; Wu H; Guo Q; Qian Z; Wei Y; Liang S J. Transl. Med 2011, 9, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yan M; Liang M; Wen J; Liu Y; Lu Y; Chen ISY J. Am. Chem. Soc 2012, 134 (33), 13542–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Potapova TA; Sivakumar S; Flynn JN; Li R; Gorbsky GJ Mol. Biol. Cell 2011, 22 (8), 1191–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Burgess A; Vigneron S; Brioudes E; Labbé J-C; Lorca T; Castro A Proc. Natl. Acad. Sci. U. S. A 2010, 107 (28), 12564–12569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Gavet O; Pines J Dev. Cell 2010, 18 (4), 533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Lv H; Zhang S; Wang B; Cui S; Yan JJ Control. Release 2006, 114 (1), 100–109. [DOI] [PubMed] [Google Scholar]
- (52).Cai J; Yue Y; Rui D; Zhang Y; Liu S; Wu C Macromolecules 2011, 44 (7), 2050–2057. [Google Scholar]
- (53).Forrest ML; Gabrielson N; Pack DW Biotechnol. Bioeng 2005, 89 (4), 416–423. [DOI] [PubMed] [Google Scholar]
- (54).Merdan T; Kunath K; Petersen H; Bakowsky U; Voigt KH; Kopecek J; Kissel T Bioconjug. Chem 16 (4), 785–792. [DOI] [PubMed] [Google Scholar]
- (55).Maurstad G; Stokke BT; Vårum KM; Strand SP Carbohydr. Polym 2013, 94 (1), 436–443. [DOI] [PubMed] [Google Scholar]
- (56).Ping Y; Liu C; Zhang Z; Liu KL; Chen J; Li J Biomaterials 2011, 32 (32), 8328–8341. [DOI] [PubMed] [Google Scholar]
- (57).Li J-M; Wang Y-Y; Zhang W; Su H; Ji L-N; Mao Z-W Int. J. Nanomedicine 2013, 8, 2101–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Kim Y-M; Song S-C Biomaterials 2014, 35 (27), 7970–7977. [DOI] [PubMed] [Google Scholar]
- (59).Han HD; Mora EM; Roh JW; Nishimura M; Lee SJ; Stone RL; Bar-Eli M; Lopez-Berestein G; Sood AK Cancer Biol. Ther 2011, 11 (9), 839–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ma Z; Yang C; Song W; Wang Q; Kjems J; Gao S J. Nanobiotechnology 2014, 12 (1), 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Rodell CB; Highley CB; Chen MH; Dusaj NN; Wang C; Han L; Burdick JA Soft Matter 2016, 309, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Zhao Q-Q; Chen J-L; Lv T-F; He C-X; Tang G-P; Liang W-Q; Tabata Y; Gao J-Q Biol. Pharm. Bull 2009, 32 (4), 706–710. [DOI] [PubMed] [Google Scholar]
- (63).Jin L; Zeng X; Liu M; Deng Y; He N Theranostics 2014, 4 (3), 240–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Shi B; Abrams M J. Histochem. Cytochem 2013, 61 (6), 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Erazo-Oliveras A; Muthukrishnan N; Baker R; Wang T-Y; Pellois J-P Pharmaceuticals (Basel). 2012, 5 (11), 1177–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Chiu Y-L; Ali A; Chu C-Y; Cao H; Rana TM Chem. Biol 2004, 11 (8), 1165–1175. [DOI] [PubMed] [Google Scholar]
- (67).Grossman PM; Han Z; Palasis M; Barry JJ; Lederman RJ Catheter. Cardiovasc. Interv 2002, 55 (3), 392–397. [DOI] [PubMed] [Google Scholar]
- (68).Wan W-G; Jiang X-J; Li X-Y; Zhang C; Yi X; Ren S; Zhang X-Z; W-g W; X-j J; X-y L; X-z Z J Biomed Mater Res Part A 2014, 102, 3452–3458. [DOI] [PubMed] [Google Scholar]
- (69).Zhou Y; Liu Y; Yang SX; Wang Z Genet. Mol. Res 2015, 14 (3), 9478–9485. [DOI] [PubMed] [Google Scholar]
- (70).Natarajan R; Salloum FN; Fisher BJ; Kukreja RC; Fowler AA Circ. Res 2006, 98 (1), 133–140. [DOI] [PubMed] [Google Scholar]
- (71).Somasuntharam I; Boopathy AV; Khan RS; Martinez MD; Brown ME; Murthy N; Davis ME Biomaterials 2013, 34 (31), 7790–7798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Nelson CE; Kim AJ; Adolph EJ; Gupta MK; Yu F; Hocking KM; Davidson JM; Guelcher SA; Duvall CL Adv. Mater 2014, 24 (4), 607–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Krebs MD; Alsberg E Chemistry 2011, 17 (11), 3054–3062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Segovia N; Pont M; Oliva N; Ramos V; Borrós S; Artzi N Adv. Healthc. Mater 2015, 4 (2), 271–280. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.