

Abstract
To evaluate the clinical impact of molecular tumor profiling (MTP) with targeted sequencing panel tests, pediatric patients with extracranial solid tumors were enrolled in a prospective observational cohort study at 12 institutions. In the 345-patient analytical population, median age at diagnosis was 12 years (range 0–27.5); 298 patients (86%) had 1 or more alterations with potential for impact on care. Genomic alterations with diagnostic, prognostic or therapeutic significance were present in 61, 16 and 65% of patients, respectively. After return of the results, impact on care included 17 patients with a clarified diagnostic classification and 240 patients with an MTP result that could be used to select molecularly targeted therapy matched to identified alterations (MTT). Of the 29 patients who received MTT, 24% had an objective response or experienced durable clinical benefit; all but 1 of these patients received targeted therapy matched to a gene fusion. Of the diagnostic variants identified in 209 patients, 77% were gene fusions. MTP with targeted panel tests that includes fusion detection has a substantial clinical impact for young patients with solid tumors.
In the United States, the Food & Drug Administration (FDA), which regulates medical devices including molecular testing, has fully approved five targeted next-generation sequencing (NGS) tests for advanced solid malignancies and approved the development of several additional targeted molecular tumor profiling (MTP) assays1–5. These FDA-approved and preapproved MTP tests for solid malignancies use paraffin-embedded tumor samples to sequence DNA and, in some cases, RNA, and issue reports on several hundred cancer-related gene mutations and copy number alterations and a more limited number of gene fusions. In addition, some academic laboratories have developed similar in-house targeted MTP assays generally referred to as laboratory-developed tests. Recommendations for use of these targeted NGS tests are included in national practice guidelines6,7 and national insurance coverage decisions8 for adult cancers. However, clinical trials supporting biomarker validation and subsequent targeted NGS testing and new therapeutic FDA approvals have not included children with solid malignancies, with the exception of the diagnosis- and age-agnostic trial that led to larotrectinib approval9. Thus, there are no national coverage determinations or practice guidelines regarding MTP for pediatric solid malignancies. The situation in the United States is not unique with regulatory approvals and consensus guidelines regarding multigene NGS panel tests focused on adult cancers also existing in Europe and Asia10,11.
Nevertheless, some pediatric patients with solid malignancies have targeted MTP performed. Pediatric cancer patients treated at academic institutions are undergoing targeted MTP, as evidenced by the fact that targeted sequencing data for over 2,000 patients aged 18 or younger with extracranial solid tumors have been deposited into the American Association of Cancer Research project ‘Genomics Evidence Neoplasia Information Exchange’ repository from 7 institutions12. Pediatric oncologists also utilize commercial targeted MTP, with Foundation Medicine reporting on 711 pediatric solid malignancies sequenced in 2017 (ref. 13). MTP of pediatric solid tumors is not universal; only one-third of National Cancer Institute (NCI)-Children’s Oncology Group (COG) Pediatric Molecular Analysis for Therapy Choice (MATCH) screening protocol participants had definitive evidence of MTP before enrolling on this national basket trial and one-third had definitive evidence of not having had MTP before enrollment14. The National Institutes of Health NCI Board of Scientific Advisors ad hoc working group in support of the Childhood Cancer Data Initiative report discusses the absence of a national standard of care for MTP and uneven access to modern molecular analyses in pediatric patients with cancer as important health equity and access to care issues15.
Several recently published studies demonstrated that comprehensive sequencing—including whole-genome, whole-exome and whole-transcriptome—of pediatric cancers can identify clinically relevant alterations in a substantial fraction of childhood patients with cancer and that some patients with actionable tumor alterations will receive and respond to matched targeted therapy (MTT)16,17. Importantly, fresh-frozen tumor samples have been used for these studies. In the United States, where targeted NGS tests are commonly performed on formalin-fixed paraffin-embedded (FFPE) tumor samples, these studies do not reflect typical clinical practice. In addition, these previous publications may be less relevant to patients with cancer and providers in other countries where resources do not permit this comprehensive sequencing approach. The multicenter observational prospective iCat2/Genomic Assessment Informs Novel Therapy Consortium (GAIN) Study follows a patient cohort, performs targeted NGS from FFPE tumor samples and collects clinical data to address the impact of MTP on patient outcomes. We report on 389 patients with extracranial solid tumors enrolled to facilitate data-driven insurance coverage decisions and testing guideline development in the United States and inform molecular testing practices in clinical settings where resource limitations prohibit acquisition, storage and transport of fresh-frozen tissue and comprehensive sequencing data generation, analysis and storage.
Results
Patients and molecular tumor profiling.
Patients consented to participation in the GAIN/iCat2 Study (NCT02520713), a multicenter prospective observational cohort study enrolling patients with relapsed/refractory or high-risk extracranial solid tumors aged ≤30 years at diagnosis at 12 pediatric oncology centers across the United States. All patients had MTP with the targeted DNA panel test OncoPanel and selected patients also had RNA sequencing (RNA-seq) performed, which included whole-transcriptome and targeted fusion panels, Laboratory for Molecular Pediatric Pathology (LaMPP) for most cases and a similar panel at the Massachusetts General Hospital. Patients who enrolled before 31 December 2018 and had at least 1 OncoPanel result completed by 4 April 2019 were included in the analytical population for this report. Of 389 patients enrolled between 1 November 2015 and 31 December 2018, 345 patients had 1 or more tumor samples successfully sequenced with OncoPanel (Extended Data Fig. 1). Patients in this 345-patient analytical population were newly diagnosed (n = 107) or had relapsed or refractory disease (n = 238) at enrollment. Patients had 59 distinct diagnoses, with the most common being osteosarcoma (19%), rhabdomyosarcoma (13%) and Ewing sarcoma (13%) (Table 1); 224 patients (65%) had a sarcoma. Median age at cancer diagnosis was 12 years (range 0–27.5 years), including 43 adults aged 18 years and older; median age at enrollment was 13 years (range 0.2–28.8 years) including 83 adults.
Table 1 |.
Baseline characteristics and sequencing performed for the analytical cohort (n = 345)
| Variable | |
|---|---|
| Age at diagnosis (years), median (range) | 12 (0–27.5) |
| Age at enrollment (years), median (range) | 13 (0.2–28.8) |
| Sex, n (%) | |
| Female | 151 (44) |
| Male | 194 (56) |
| Race, n (%) | |
| White | 244 (71) |
| Black or African-American | 25 (7) |
| Asian | 14 (4) |
| More than one race | 29 (8) |
| Other | 33 (10) |
| Ethnicity, n (%) | |
| Hispanic or Latino | 32 (9) |
| Non-Hispanic | 275 (80) |
| Unknown | 38 (11) |
| Cancer diagnosis, n (%) | |
| Osteosarcoma | 64 (19) |
| Rhabdomyosarcoma | 46 (13) |
| Ewing sarcoma | 44 (13) |
| Other sarcoma | 70 (20) |
| Renal tumor | 29 (8) |
| Neuroblastoma | 28 (8) |
| Liver tumor | 13 (4) |
| Carcinoma | 19 (6) |
| Other | 32 (9) |
| Sequencing method, n (%) | |
| Targeted DNA NGS only | 269 (78) |
| Targeted DNA NGS + RNA NGS | 32 (9) |
| Targeted DNA NGS + whole transcriptome | 26 (8) |
| Targeted DNA NGS + RNA NGS + whole transcriptome | 18 (5) |
Four hundred thirty-eight successful and 33 unsuccessful OncoPanel tests were run on 345 patients (failed OncoPanel testing rate 7%). Two hundred seventy-five patients had only 1 sample tested, 147 from initial diagnosis and 128 from recurrence. Seventy patients had >1 sample (range 2–6) tested with OncoPanel; 40 had paired diagnosis and recurrence samples tested. Forty-eight samples from 43 patients were run on OncoPanel V2, targeting 300 genes; 35 genes were targeted for fusions. Three hundred ninety samples from 311 patients were run on V3 targeting 447 genes; 60 genes were targeted for fusions. For these 438 OncoPanel cases, the mean target coverage was 314 reads, with an average of 98.3% of targeted bases with coverage greater than 30 reads. Mean turnaround time from sample accession to report was 42.4 d (median 40 d, range 7–321 d) using OncoPanel as a research assay. As point of reference, the current mean turnaround time for the clinical assay is 12.2 d.
An RNA fusion panel test was successfully run on 54 samples from 50 patients. Forty-eight patients had samples run on the LaMPP targeted fusion panel and 2 samples were evaluated with the similar targeted fusion panel at the Massachusetts General Hospital. Cases analyzed using the LaMPP fusion panel had an average of 238,469 unique reads. The mean turnaround time for the GAIN cases tested with the clinically validated fusion panel assay was 14.1 d; for other clinical cases, it is 10.3 d from sample receipt to report. Whole-transcriptome sequencing and fusion analysis was successful in 44 patients and unsuccessful in 9 patients (17% failure rate). Quality metrics for the successful cases include an average of >100 million reads aligned in pairs, with a mean turnaround time of 71 d. The Supplementary Data file contains all variants identified by OncoPanel and LaMPP.
Therapeutic impact.
Evidence identifying detected variants as biomarkers of potential response to MTT was evaluated according to the iCat evidence tiers18 (Supplementary Data). iCat tiers were assigned at the time the molecular profiling report was returned to the patient (February 2016–June 2019) and with updated evidence in May–June 2020, hereafter referred to as re-tiering. A molecular tumor board (MTB) discussion occurred when evidence was unclear. Two hundred forty of 345 patients (69%) in the analytical cohort had at least 1 gene variant for which an iCat therapeutic recommendation was made at the time the report was issued (Supplementary Data). Most of these patients had 1 (127 patients), 2 (71 patients) or 3 (34 patients) therapeutically actionable alterations resulting in an iCat recommendation, with few having 4–7 (Extended Data Fig. 2a). On a patient level, the highest-tier iCat recommendation at the time the report was issued was 1–2 (strong evidence from clinical studies) in 41% of patients (n = 140), 3–4 (evidence from preclinical studies) in 23% of patients (n = 79) and 5 (weaker evidence and study team consensus) in 6% of patients (n = 21; Extended Data Fig. 2b). After re-tiering the iCat recommendations with updated evidence, the proportions of iCat recommendations changed to 25% (tiers 1 and 2), 29% (tiers 3 and 4) and 12% (tier 5; Extended Data Fig. 3). The decrease in tier 1 and 2 alterations is largely attributed to a revised association between TP53 variants and WEE1 inhibitors. Initially this association was tier 2 based on a phase 1 clinical trial showing modest enrichment of TP53 variants in responders19. The association was not borne out in subsequent studies and MTB assigned this association a tier 5 because of ongoing biomarker selected clinical trials of WEE1 inhibitors in pediatric patients (NCT02813135). The iCat recommendations of 90 patients (26%) were discussed in the MTB. Of 408 alterations associated with iCat recommendations, 110 (27%) were fusions, 144 (35%) were copy number alterations and 154 (38%) were sequence variants (Extended Data Fig. 4a).
Tiers 1 and 2 iCat recommendations were most often made based on alterations in BRAF, SMARCB1, PIK3CA and for high tumor mutation burden (TMB). Alterations informing >5% of iCat recommendations were often lower tier: TP53 inactivating alterations (WEE1 inhibitor; tier 5); EWSR1-FLI1 fusions (EWSR1-FLI inhibitor TK-216; tier 3); MYC/MYCN amplification (BET inhibitor; tier 3 or 4); and CDKN2A/B deletions (CDK4 inhibitor; tier 5) (Fig. 1 and Extended Data Fig. 4). MTTs in iCat recommendations represented a wide range of therapeutic classes but were most often DNA damage response inhibitors (23%), epigenetic modifiers (18%), Ras-MAPK inhibitors (12%) and PI3K-Akt-MTOR inhibitors (11%). Three patients had tumors with TMB ≥ 10 mutations per Mb but <20 mutations per Mb; all 3 of these were tumors sequenced after exposure to chemotherapy. One patient with colorectal adenocarcinoma, previous history of astrocytoma, T cell lymphoblastic lymphoma and congenital mismatch repair deficiency (due to compound heterozygous MSH6 mutations) had TMB ≥ 20 mutations per Mb (GAIN183). Four patients had mismatch repair deficiency (MMRD) including the patient with high TMB.
Fig. 1 |. Relationship between genes containing actionable variants and the drug class of the iCat recommendation.
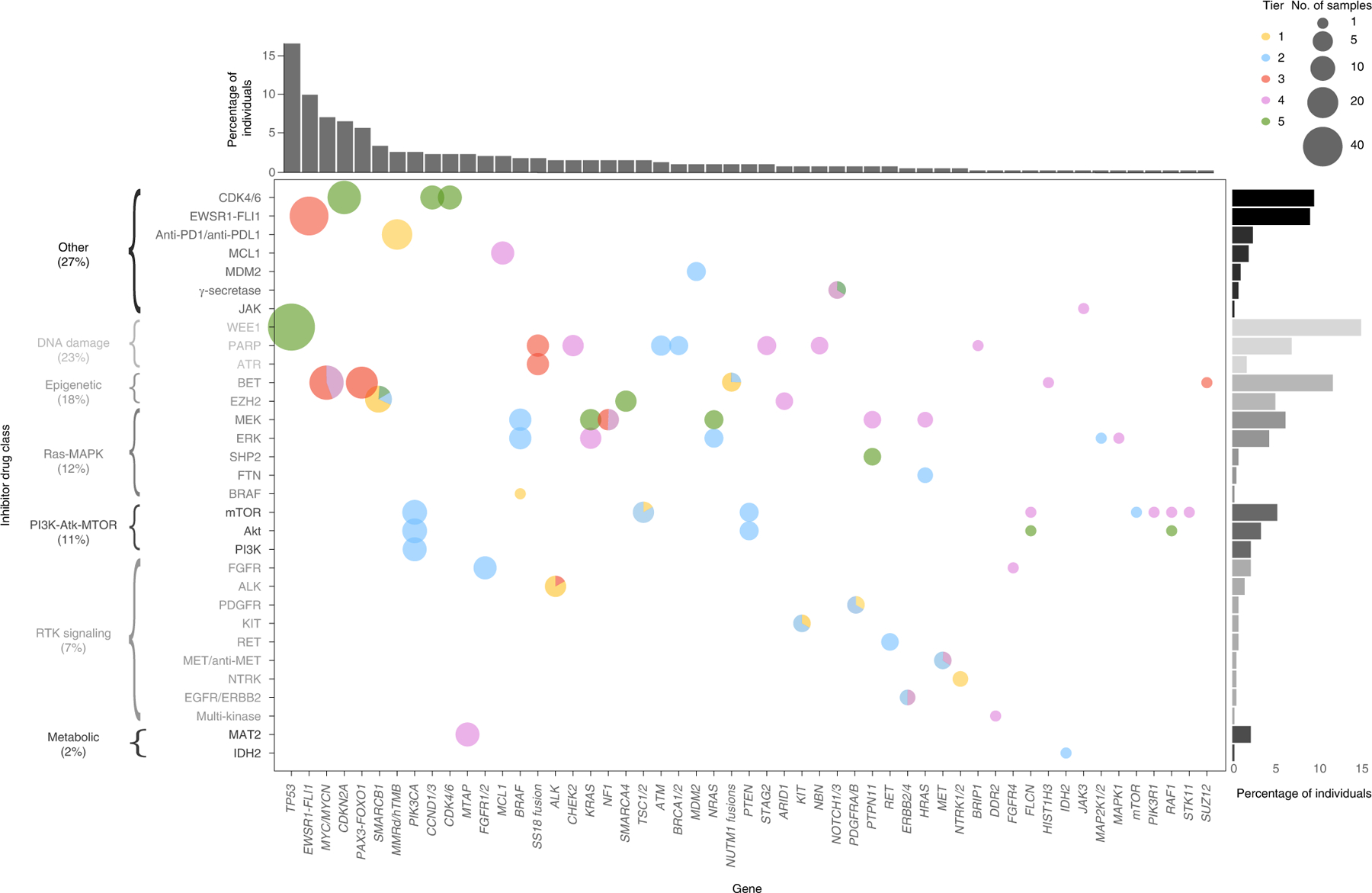
The size of each dot represents the number of patients who received iCat recommendations and the color represents the iCat recommendation tier.
In total, 52 of 345 patients (15%) had at least 1 gene variant that was an actionable mutation of interest (AMOI) of interest for the NCI-COG pediatric MATCH trial. This is lower than the 31.5% AMOI rate observed in the first 1,000 patients enrolled in the MATCH screening protocol, likely due to differences in the patient populations20. Specifically, sarcomas, representing two-thirds of the cases in this study, have a lower AMOI rate and brain tumors with an AMOI rate of 48% were excluded from this study. There were 46 actionable single-nucleotide variants (SNVs) in 39 patients, 19 actionable copy number variants (CNVs) in 17 patients and 1 actionable fusion in 1 patient. Six additional patients had fusions involving a gene in the pediatric MATCH AMOI list; however, the exact fusion was not on the AMOI list presumably because the fusion partner was new (CCDC6-ALK; CLIP2-RET; KHDRBS2-BRAF; MYH10-RET; PRMT7-RET; SEPTIN7-BRAF).
Treatment with MTT.
Two hundred patients were eligible for assessment of receipt and response to MTT, based on having an iCat recommendation (n = 240), being alive at the time of the return of the molecular report (n = 221) and having complete follow-up data (n = 200) (Extended Data Fig. 1). In these 200 patients, the median follow-up time was 10.6 months (range 0–41.0) from the date of relapsed or refractory disease status (n = 160) and 12 months (range 0–29.9) from the date of enrollment for newly diagnosed patients (n = 40). Ninety-six of 200 patients (48%) would not have been expected to consider MTT because they were either newly diagnosed and receiving frontline therapy (n = 29, 15%), received no cancer-directed systemic therapy during the follow-up period (n = 47, 24%) or no MTT drugs were available (n = 20, 10%). Of the remaining 104 who would have been expected to consider MTT, 29 (28%) received MTT (Fig. 2). Among the 104 expected to consider an MTT treatment option, patients who received MTT were more likely to have a tier 1 iCat recommendation (23%) than those who did not receive MTT (4%; P = 0.003), using the original tier provided at the time of the report.
Fig. 2 |. Summary infographic of the outcome for the 345 patients in the analytical cohort after return of genomic results with diagnostic or therapeutic significance.
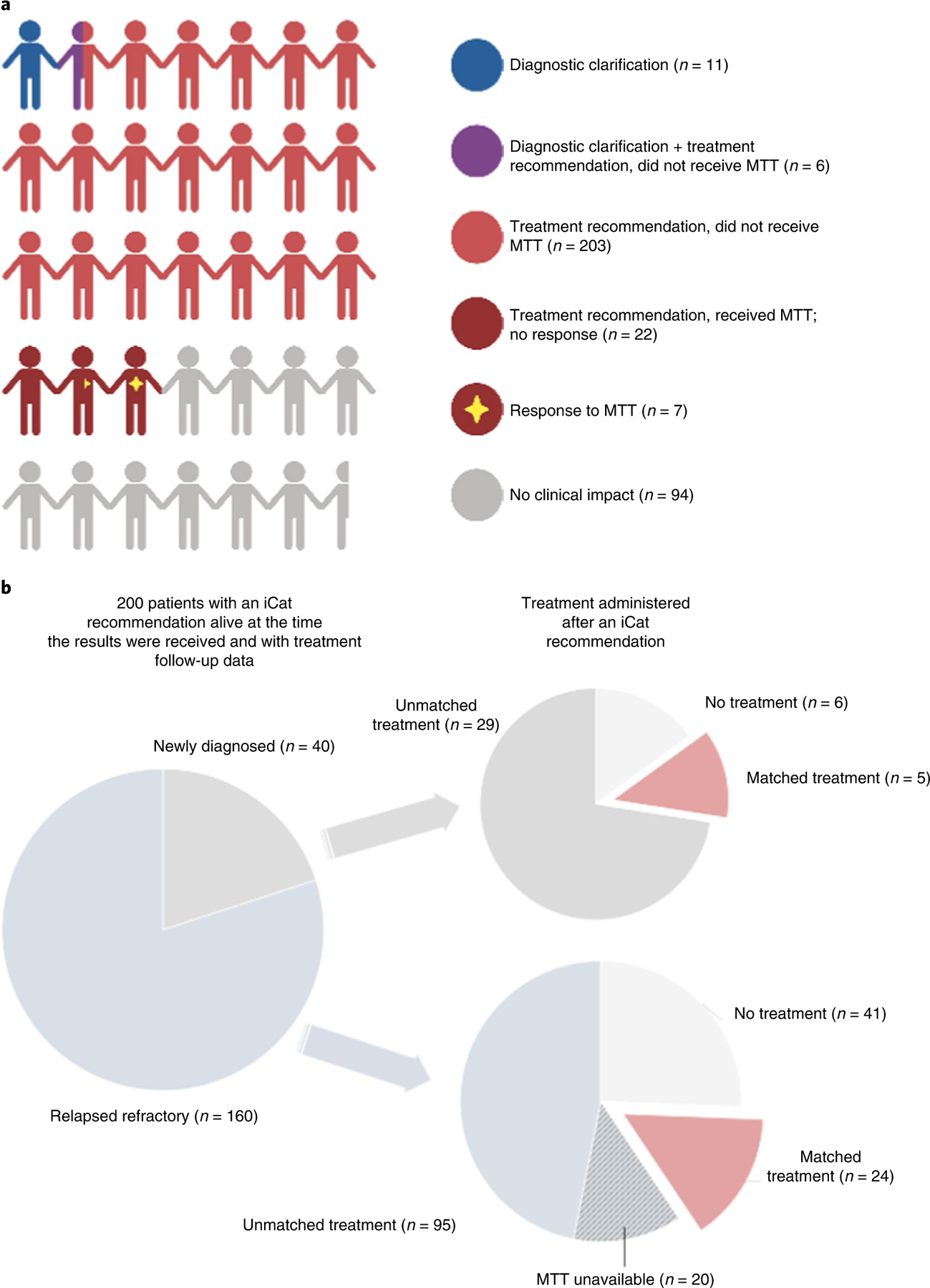
a, Each cartoon person represents 10 patients. Overall, 73% of patients had an impact on care, including 17 patients with a change in their diagnosis (blue and purple) and 29 patients receiving MTT (dark red) with 7 of these having a response (dark red with star). Created with Biorender. com. b, Treatment received by the 200 patients with therapeutically actionable alterations resulting in an iCat recommendation and sufficient treatment follow-up data to be eligible for assessment of MTT. Patients did not receive MTT because they either received no therapy (n = 46), were newly diagnosed and receiving initial treatment (n = 29) or MTT was not available (n = 23).
Of the 29 patients receiving MTT, 24% (n = 7) met criteria for a responder defined as a patient with RECIST measurable disease21 at the initiation of MTT without other concurrent tumor-directed therapy who either had a partial response or stable disease for ≥4 months (Fig. 3). All but one responder received targeted therapy matched to a fusion involving a gene that encodes a signaling protein. These fusions involved NTRK2, ALK, RET, NOTCH and BRAF (n = 2). Two cases have been reported previously22,23. The other responder (GAIN186), the patient with constitutional MMRD, enrolled with a newly diagnosed colorectal carcinoma but also had recurrent T lymphoblastic lymphoma and a distinct T cell lymphoma. This patient received nivolumab for more than four months with stable disease for the colorectal carcinoma sites; however, the lymphomas progressed. All responders had sarcomas or malignancies that are rare in pediatrics (Fig. 2 and Extended Data Fig. 5). Additional variants present in the tumors of patients who received MTT are shown (Extended Data Fig. 6).
Fig. 3 |. Swimmer plot of treatment response for 29 patients who received MTT.
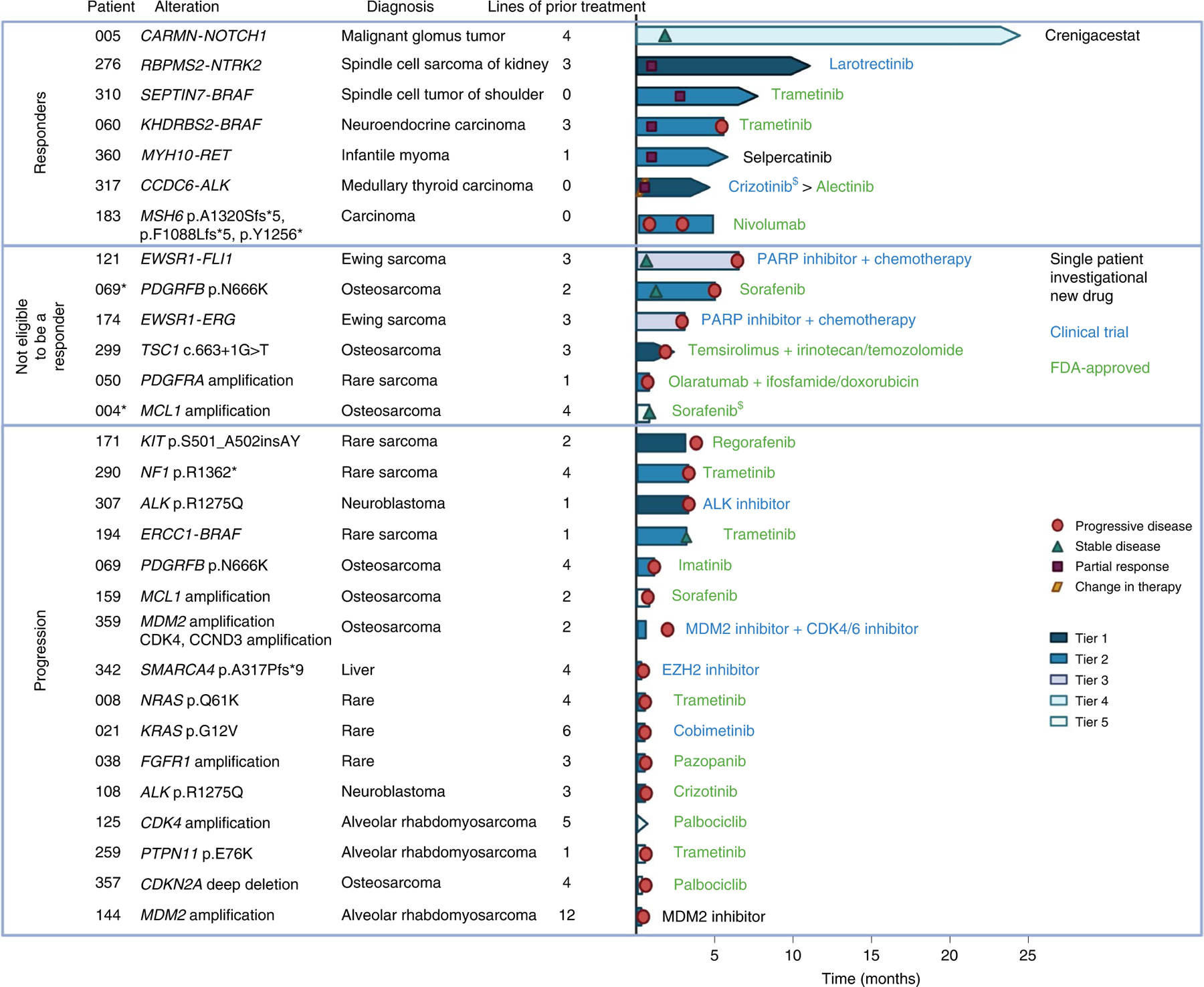
Additional details regarding responders are included in Supplementary Fig. 5. *Patient received treatment with evaluable disease. $Patient stopped treatment due to toxicity. Created with Biorender.com.
Patients received MTT via a single patient protocol (3), enrolled on a clinical trial (8) or as an FDA-approved therapy (18). Two patients stopped treatment due to toxicity (GAIN004, GAIN317) one of whom transitioned to a different MTT (GAIN317). One patient (GAIN194) started trametinib with stable disease after chemotherapy; trametinib was stopped in the absence of response. All remaining patients who stopped MTT discontinued therapy due to progressive disease (Fig. 3).
Prognostic impact.
Fifty-six (16%) patients had 1 or more alterations identified that could impact prognosis based on having tier 1 or 2 evidence according to the Association for Molecular Pathology (AMP)/College of American Pathologists (CAP)/American Society for Clinical Oncology (ASCO) guidelines24, such as MYCN amplification or chromosome 11q loss in neuroblastoma, TP53 loss or chromosome 1q gain in Wilms tumor or STAG2 or TP53 inactivating alterations in Ewing sarcoma (Extended Data Fig. 7). None of these patients had a change in management based on these prognostic alterations and many were previously known. Unlike the diagnostic and therapeutic variants, none of the prognostic alterations were based on gene fusions (Extended Data Fig. 7 and Supplementary Data).
Diagnostic impact.
Of 345 GAIN patients in the analytical population, 209 (61%) had diagnostically significant alterations identified (Fig. 4) according to professional guidelines for the interpretation of clinical impact24. Most (80%, 168 of 209 patients) had structural variants, many resulting in fusions pathognomonic for specific rare pediatric cancers, such as EWSR1-FLI1 (Ewing sarcoma), PAX-FOXO1 (alveolar rhabdomyosarcoma), EWSR1-WT1 (desmoplastic small round cell tumor (DSRCT)), CIC-rearranged sarcoma, BCOR-altered sarcoma and NUTM1 fusions (NUT midline carcinomas). Sixteen percent had diagnostically significant SNVs with DICER1 variants being most common and 5% had diagnostically significant CNVs with SMARCB1 deficiency being the most common (Fig. 4, Extended Data Fig. 7 and Supplementary Data).
Fig. 4 |.
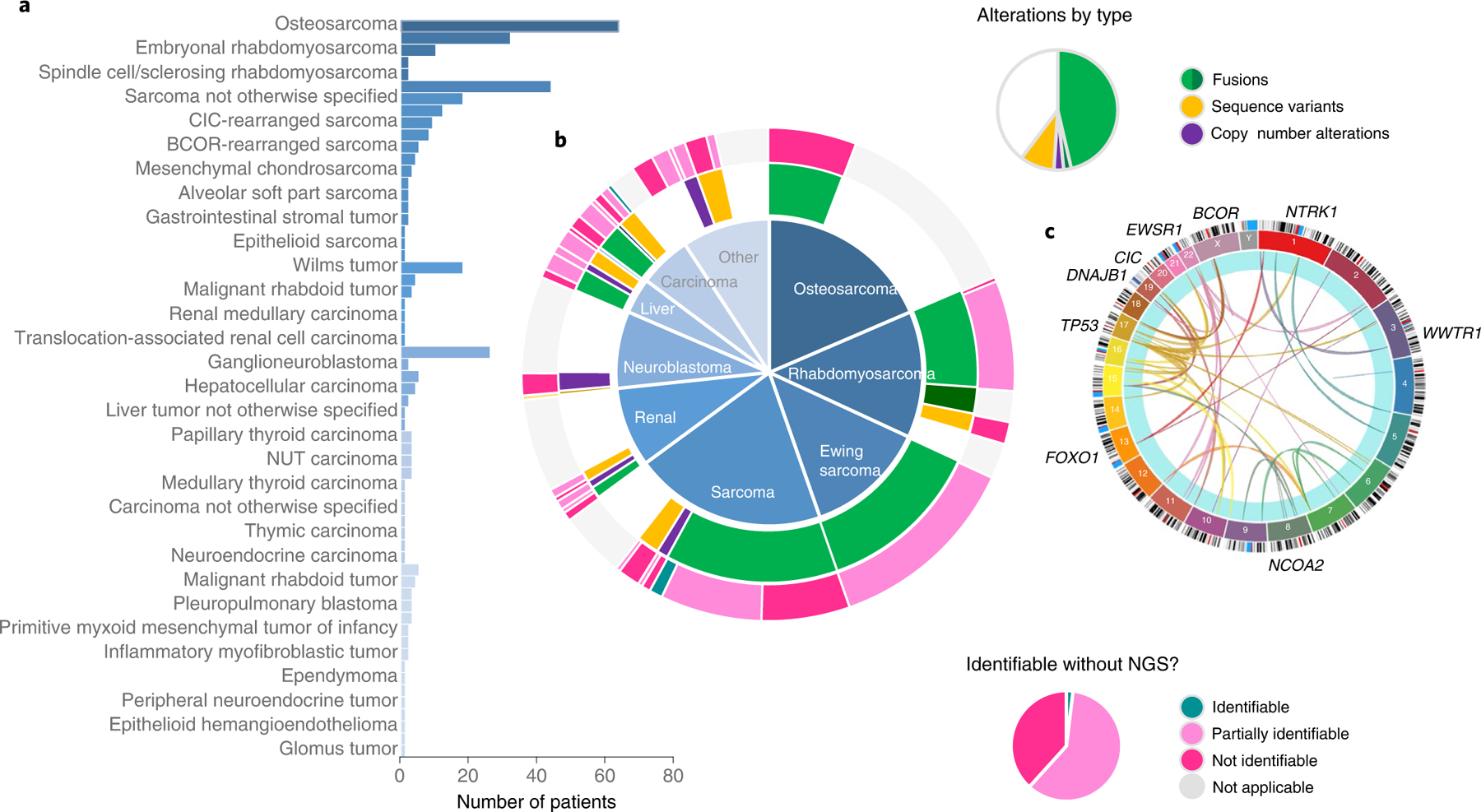
a,b, Diagnoses and diagnostically significant alterations. In 345 patients, 59 distinct solid tumor diagnoses were made, including many sarcomas and rare tumors, shown in the histogram (a), grouped by diagnostic bins in the central pie chart (b). Two hundred and eight patients had diagnostic alterations with tier 1 or 2 impact according to the AMP/CAP/ASCO guidelines. Diagnostic alterations for each patient are displayed in the inner ring and are grouped by diagnostic bins shown in the central pie chart. Fusions (green) consist of most of the diagnostically significant alterations. The outer ring shows patients whose alterations would have been identified using traditional techniques like FISH, PCR with reverse transcription or IHC: 4 patients (1.9%) would have had their diagnostic variant identified; 125 patients (60%) would have had their alterations partially identified; and the diagnostic alterations of 80 patients (38%) would have been completely missed using traditional assays. c, The Circos plot shows the wide variety of genes involved in diagnostic fusions. Circos plot created with Circa (http://omgenomics.com/circa).
MTP results clarified the diagnosis for 17 patients (5%; Table 2); 13 of these molecular alterations were fusions. In each case, the identified alteration(s) had a direct impact on the patient’s diagnostic classification and management by the clinical team. In five cases, traditional analyses including fluorescence in situ hybridization (FISH) failed to detect a fusion later identified with MTP. An illustrative real-time diagnosis impact example is GAIN318 (Extended Data Fig. 8). At age 14 the patient developed a distal tibia bone tumor with radiographic appearance suggestive of aneurysmal bone cyst (ABC), a benign tumor containing, in most cases, USP6 rearrangement25. The patient had removal with curettage, the appropriate treatment for ABC. Pathology demonstrated an ABC but with FISH testing negative for USP6 rearrangement. The mass recurred after a year. After biopsy, one pathology report was recurrent ABC and another pathology report was high-grade malignancy telangiectatic osteosarcoma requiring treatment with chemotherapy and radical resection. GAIN sequencing with OncoPanel, which can detect TP53 rearrangements, showed a fusion joining TP53 intron 1 to USP6 intron 7. Inactivating TP53 intron 1 rearrangements are recurrent in osteosarcoma26 and have not been described in any other malignancy but are not routinely tested for in bone tumors. The identified new TP53-USP6 fusion simultaneously inactivates TP53 and activates USP6 (Extended Data Fig. 8). A third pathology opinion, incorporating the TP53-USP6 fusion, diagnosed telangiectatic osteosarcoma; consequently, the patient’s oncologist and orthopedic surgeon treated the patient with chemotherapy and complete surgical resection of the primary tumor.
Table 2 |.
Details of 17 patients with a clarification in diagnosis after MTP
| GAIN ID | Diagnosis at enrollment | Pre-enrollment molecular testing | Alterations | Integrated diagnosis |
|---|---|---|---|---|
| 158 | Osteosarcoma | None | BCOR-CCNB3 | BCOR-CCNB3 sarcoma |
| 160 | Round cell malignant neoplasm | FOXO1 FISH, no rearrangement | EWSR1-WT1 | DSRCT |
| 193 | Primitive neuro-ectodermal tumor | EWSR1, FOXO1, SS18 FISH, no rearrangement, MYCN non-amplified | DICER1 p.C1197* DICER1 p.D1709N | DICER1-associated sarcoma |
| 234 | Round cell sarcoma consistent with DSRCT | EWSR1 FISH, no rearrangement | CTNNB1 p.S37F | Neoplasm, malignant |
| 276 | Malignant spindle cell neoplasm | SS18 FISH, no rearrangement, karyotype multiple abnormalities, no fusion | RBPMS-NTRK2 | NTRK-fusion malignancy |
| 028 | Round cell sarcoma, Ewing or Ewing-like | EWSR1 FISH, no rearrangement | EWSR1-FLI1 | Ewing sarcoma |
| 044 | Spindle cell tumor with rhabdomyosarcomatous differentiation | aCGH and ALK IHC | DICER1 c.904–1 G>A DICER1 p.D1709N | DICER1-associated sarcoma |
| 059 | Small round cell malignant neoplasm | EWSR1, FUS, BCOR, CIC FISH, no rearrangement | CIC-DUX4 | CIC-DUX4 sarcoma |
| 096 | Small round blue cell tumor | CCNB3 IHC | BCOR-CCNB3 | BCOR-CCNB3 |
| 154 | Small round blue cell tumor | EWSR1, CIC FISH, no rearrangement | EWSR1-CREB3L3 | Sclerosing epithelioid fibrosarcoma OR low-grade fibromyxoid sarcoma |
| 194 | Sarcoma arising in fibrous hamartoma of infancy | None | ERC1-BRAF | Fusion-positive spindle cell sarcoma |
| 199 | Sarcoma with features of ossifying fibromyxoid tumor | None | EWSR1-CREB3L1 | Sclerosing epithelioid fibrosarcoma OR low-grade fibromyxoid sarcoma |
| 284 | Renal cell carcinoma | None | TSC2 p.D1512Efs*9 | Eosinophilic granular subtype (indication for tuberous sclerosis evaluation) |
| 310 | Spindle cell process consistent with infantile fibromatosis | None | SEPTIN7-BRAF | Fusion-positive spindle cell sarcoma |
| 318 | Aneurysmal bone cyst | USP6 FISH, no rearrangement | TP53-USP6 | Osteosarcoma |
| 342 | Rhabdoid-like malignant neoplasm with retained INI1 expression | INI1 IHC | SMARCA4 p.A317Pfs*9 | Rhabdoid tumor |
| 371 | Congenital mesoblastic nephroma | None | PRMT7-RET | RET-fusion malignancy |
aCGH, array comparative genomic hybridization.
Comparing the diagnostic yield of the sequencing approach used in this study to traditional assays like FISH and immunohistochemistry (IHC), 80 of 209 patients (38%) with diagnostically significant results would have been missed with traditional assays, 125 patients (60%) would have had partial information such as the identification of an EWSR1 fusion but without the identification of the partner and 4 patients (1.9%) would have had their diagnostic alterations accurately identified. Approximately 63% (131 out of 209) of patients had diagnoses for which molecular testing is currently suggested as a potential method of analysis according to the National Comprehensive Cancer Network guidelines27–31 (Fig. 4).
Yield of RNA-seq.
Tumors were evaluated with whole transcriptome, a targeted RNA panel or both, according to the tissue triage strategy described in the Methods with additional detail in Extended Data Fig. 9b. The addition of whole-transcriptome and targeted RNA panels improved the MTP yield. Seventy-six cases underwent DNA- and RNA-seq (Table 1). Fusions were identified in 39 out of 76 cases with RNA-seq performed, with only 1 identified solely by whole-transcriptome sequencing. Only 11 out of 39 (28%) fusions were fully identified by the DNA panel test. Fusions were missed either because the variant was not assessed by OncoPanel (n = 13), because OncoPanel provided incomplete results (n = 7) or because it was not detected despite being targeted (n = 8; Supplementary Table 2). Whole-transcriptome sequencing identified one fusion, SEPTIN7-BRAF, which was initially missed by the targeted RNA panel test but later identified with an updated informatics pipeline (GAIN310). It also identified, in an inflammatory myofibroblastic tumor, a fusion not covered by the RNA panel test—VCAN-IL23R, reported only once before in this entity32. In three cases (two CIC-DUX4 fusions and one ASPSCR1-TFE3 fusion), whole-transcriptome sequencing missed fusions identified by the RNA panel test (Supplementary Table 2).
Discussion
This study is a comprehensive analysis of clinical impact of MTP in children with solid tumors, within a model and workflow that is directly applicable to the United States and other healthcare systems where there are regulatory approvals and consensus guidelines for targeted NGS panel tests. Tumor samples were collected at hospitals across the country according to their own procedures. MTP was conducted with targeted panel assays and using FFPE tissues. Treating oncologists received the MTP and clinical interpretation reports and made independent patient care decisions. Within this distributed delivery model, most patients had clinically significant MTP results. Summarizing the potential clinical impact of detected variants, 298 of 345 patients (86%) had 1 or more alteration with diagnostic, prognostic and/or therapeutic implications. After return of MTP results with diagnostic or therapeutic impact, 17 patients (5%) had a modification of their diagnosis, 240 patients (70%) received an iCat recommendation that could be used to select MTT and 29 of these patients received matched treatment. Of patients receiving MTT, seven responded to therapy (Fig. 2).
The results support the development of management guidelines and insurance reimbursement determinations addressing MTP with targeted panel tests in advanced pediatric solid malignancies. Given diagnostic significance in 61% of patients and real-time clarification of diagnostic classification in 5% of patients, performing MTP early in the disease course should be considered. As additional genetic associations with prognosis emerge, the importance of MTP for prognosis in pediatric solid tumors may increase, as it has for children with leukemia and brain tumors33,34.
Our findings highlight the importance of fusion detection for young patients with extracranial solid malignancies. All but one of the MTT responses involved a fusion. The majority of diagnostically relevant findings were fusions. These results are potentially impacted by a predominance of sarcomas in this cohort. Performing RNA-seq with a targeted RNA panel or with the whole transcriptome is needed because many DNA panel tests do not target fusions found in pediatric solid tumors. In addition, we showed that there can be false negative results for fusion detection with targeted DNA panel assays and with FISH. A wide variety of genes were involved in fusions in this cohort (Fig. 4c) and several patients, including responders to MTT, had unexpected fusions for the diagnosis, such as medullary thyroid carcinoma with an ALK fusion and a RET fusion in congenital mesoblastic nephroma. In such cases, iterative standard fusion testing can contribute to long delays in accurate diagnosis and initiation of MTT.
Three recent publications reported on pediatric cancer sequencing with more comprehensive approaches including whole-genome, whole-exome, whole-transcriptome and methylation profiling. These studies were conducted either at single well-resourced institutions or in a nationally funded genomics effort. The Australian Zero Childhood Cancer Program had similar findings: 32% of patients received MTT; 25% with extracranial solid malignancies who received MTT met our definition of a response17. The interim report from the INFORM Registry Study also showed a benefit, measured by prolonged progression-free survival, of receiving high-priority MTT in pediatric cancer16. INFORM found that a similar proportion of patients had a more precise diagnosis due to MTP. In the Genomes for Kids study, conducted by St. Jude Children’s Research Hospital, the proportion of patients with solid tumors with targetable alterations was lower (18%) as was the response to MTT (13%) but the sample size was small (84 patients) and skewed toward certain diagnoses35.
We did not aim to study the relative impact of targeted NGS compared to a more comprehensive sequencing approach. Our targeted NGS approach, without the whole transcriptome, detected 92% of targetable, prognostic or diagnostic variants in the Genomes for Kids patient population with solid tumors (Supplementary Data). Importantly, the RNA and DNA NGS assays used in this study were designed with input from pediatric oncologists and pathologists with molecular expertise. Some targeted DNA and RNA panels have a lower yield of clinically impactful results35. MTP with targeted NGS has important limitations. Targeted NGS limits genomic signature detection and research because discovery of previously unknown mechanisms of cancer development and treatment resistance is not possible. However, targeted NGS also has benefits including greater flexibility for tissue input, lower cost and lower staffing and computing requirements for data analysis and reporting. This makes targeted NGS a more viable option for molecular testing in clinical settings, where resources are more limited, such as low- and middle-income countries. Most targeted NGS assays are conducted without paired germline sequencing, limiting germline cancer risk mutation identification and confirmation of second hits in tumor suppressor genes. Unpaired germline sequencing was performed on this cohort and is reported separately36. Thirty-five of 160 (22%) patients had pathogenic or likely pathogenic germline variants, slightly higher than the rate reported in other studies37–39.
Another important study limitation is reporting on an interim patient population limiting patient numbers. Nevertheless, by focusing on solid malignancies, our study makes an important contribution to the previously published literature on the clinical impact of sequencing in this patient population16,17,35, increasing the number of reported cases by 60%. Solid tumors represent 42% of malignant cancers occurring in individuals aged <20 years40. Solid malignancies are also common in the relapsed and refractory population, representing 72% of the first 1,000 patients enrolled on the NCI-COG pediatric MATCH basket trial screening protocol20. There is a preponderance of sarcomas and of adolescent and young adult patients enrolled on this and the NCI-COG pediatric MATCH screening protocols41, highlighting adolescent and young adult sarcomas as unmet medical needs in pediatric oncology warranting intensified efforts in preclinical and clinical investigation.
The Ras/RAF/MAPK pathway was altered in 8% of patients. A related and similar finding in the NCI-COG pediatric MATCH is rapid accrual of patients with Ras/RAF/MAPK pathway alterations to the therapeutic subprotocol evaluating the MEK inhibitor selumetinib20. In light of FDA requirements mandated by the recent passage of the Research to Accelerate Cures and Equity Act42, new and more effective drugs targeting this pathway, such as covalent isoform-specific Ras inhibitors43 and allosteric SHP2 inhibitors44, should have pediatric study plans.
In summary, targeted NGS in 345 patients with advanced extracranial solid malignancies enrolled in the GAIN/iCat2 study had notable clinical impact for individual patients by providing more precise diagnostic classification and by resulting in responses to MTT. These results inform molecular testing approaches in this patient population and priorities for future pediatric oncology research efforts. The GAIN/iCat2 study is ongoing, with continued patient accrual. Further data collection will allow for additional analyses, such as frequency of rare genomic variants, and outcomes, such as event-free and overall survival.
Methods
Study design and objectives.
The GAIN/iCat2 study (NCT02520713) is a multicenter observational cohort study involving 12 pediatric oncology centers across the United States with remote consent available for patients cared for outside of the participating institutions. (see the study schema in Supplementary Fig. 9a). The study is conducted according to the principles of the Declaration of Helsinki (2013 version) and the International Conference on Harmonization Guidelines for Good Clinical Practice; it is approved by the institutional review board (IRB) of each of the participating institutions with the Dana-Farber/Harvard Cancer Center IRB serving as the lead IRB. The primary objective is to describe the overall survival of pediatric patients with advanced extracranial solid tumors who have or have not received molecularly targeted therapy matched to a genomic variant identified by MTP. Secondary and exploratory objectives include describing the frequency and range of molecular alterations in pediatric extracranial solid tumors and in-depth assessment of patients with response to MTT. In this interim analysis, we report on molecular alterations in an initial cohort characterized according to the impact on diagnosis, prognosis or response to MTT, as well as the intermediate clinical end points (response and prolonged stable disease) required to identify responders to MTT. Accrual is ongoing and data on the primary survival end point will be the focus of a future report.
Patients and tumor samples.
All patients provided informed written consent for participation. Patients are eligible if they have a high-risk newly diagnosed or relapsed/refractory solid malignancy outside of the central nervous system with an age at initial diagnosis of ≤30 years. High risk is defined as an expected 2-year progression-free survival of ≤50%. In addition, patients with extracranial solid tumors without a clear diagnosis after standard histological and molecular workup are eligible. Patients are required to have either a previously obtained sample available for sequencing, a procedure planned for clinical care expected to yield sufficient tumor for sequencing, or an MTP report from an accepted laboratory. Fresh-frozen or paraffin-embedded tumor samples obtained from procedures at diagnosis or recurrence in the course of clinical care were sequenced. Clinical and demographic data were collected by chart review at the time of enrollment including age, sex, diagnosis, date of diagnosis, stage at diagnosis, prior therapy and disease status (newly diagnosed or recurrent/refractory) and entered into the InForm v.6.2.1.0.21 clinical trial data capture system. Diagnoses were categorized using the World Health Organization’s International Classification of Diseases for Oncology ontology45. Patients who enrolled before 31 December 2018 and had at least 1 tumor sample OncoPanel sequencing test result completed by 4 April 2019 were included in the analytical population for this report. The few patients with only MTP reports from other laboratories were excluded from the analytical population.
MTP.
Patients had one or more tumor samples sequenced using the clinically validated OncoPanel assay at the Center for Advanced Molecular Diagnostics at Brigham and Women’s Hospital. OncoPanel is a DNA hybrid capture-based NGS assay that detects SNVs, insertions/deletions (indels) and CNVs in cancer genes (V2: 300; V3: 447) and rearrangements in cancer genes (V2: 35; V3: 60). The OncoPanel cancer gene list was determined with input from many clinicians including pediatric oncologists and pathologists. DNA was isolated using standard extraction methods (QIAGEN) and quantified with PicoGreen-based double-stranded DNA detection (Thermo Fisher Scientific). Indexed sequencing libraries were prepared from 50-ng sonically sheared DNA samples using Illumina TruSeq LT reagents (Illumina). Extracted DNA underwent targeted NGS using the KAPA HTP Library Preparation Kit (Roche), a custom RNA bait set (Agilent SureSelect) and sequenced with the Illumina HiSeq 2500 system. Pooled sample reads were deconvoluted (demultiplexed) and sorted with Picard v.1.92 and later (Broad Institute). Reads were aligned to the reference sequence b37 edition from the Human Genome Reference Consortium using the Burrows–Wheeler Aligner v.0.5.9 (Broad Institute). Duplicate reads were identified and removed with Picard. The median mean target coverage per sample after removal of duplicate reads was 169×. Alignments were further refined using the Genome Analysis Toolkit v.1.6 and later (Broad Institute) for localized realignment around indel sites. Recalibration of the quality scores was performed with the Genome Analysis Toolkit. Mutation analysis for SNVs was performed using MuTect v.1 0.27200 (Broad Institute). Indels were called using Indelocator (Broad Institute). Integrative Genomics Viewer v.2.0.16 or later (Broad Institute) was used for visualization and interpretation. Variants were filtered to exclude synonymous variants, known germline variants in the Single Nucleotide Polymorphism database, and variants that occur at a population frequency of >0.1% in the Exome Sequencing Project database. Copy number detection was performed by analysis of fractional coverage of a defined genomic interval compared with pooled normal samples. Structural variant analysis was performed using BreaKmer to detect larger indels. Analysis included the detection of TMB, microsatellite instability and mutational signatures46–49.
RNA-seq was performed and included the whole transcriptome and 64-gene Solid and Brain Tumor targeted fusion panel at the LaMPP at Boston Children’s Hospital. Because the samples used in this study have potential future clinical use, such as for clinical trial eligibility screening, a testing strategy was devised (described in Supplementary Fig. 10) that restricted transcriptome sequencing to a subset of cases with the highest probability of clinical utility. The LaMPP targeted RNA assay was developed by pathologists with pediatric and molecular expertise. For the targeted RNA assay, total nucleic acid or RNA alone was isolated, RNA was converted to cDNA by reverse transcriptase and library preparation was performed using a custom Archer FusionPlex kit (ArcherDX) using anchored multiplex PCR on an Illumina MiSeq sequencer. Sequencing reads were aligned, annotated and analyzed using the Archer Analysis bioinformatics software v6.2.7 system50. Whole-transcriptome sequencing, which was analyzed only for fusion detection, was performed at the Broad Institute, beginning with RNA extraction from frozen or FFPE samples. Illumina TruSeq Strand Specific Long Insert Whole Transcriptome Sequencing was used for the frozen samples. It combines poly(A) selection of messenger RNA transcripts with a strand-specific cDNA library preparation, resulting in fragments with a mean length of 550 base pairs (bp). The FFPE samples were sequenced using Illumina TCap, hybridization-based mRNA selection and transcriptome capture technology. The approach first prepares a stranded cDNA library from isolated RNA, then hybridizes the library to a set of DNA oligonucleotide probes to enrich the library for mRNA transcript fragments. It is a good option for RNA derived from potentially degraded samples. Libraries were sequenced on the latest Illumina sequencing platform to a minimum depth of 50 million reads. The paired-end reads were aligned and analyzed for gene fusions using our multi-caller fusion detection pipeline based in Google Cloud. The multi-caller fusion detection approach enabled us to address the high false positive rate typical for gene fusion calling in transcriptomic data while improving sensitivity to detect the more challenging fusions. STAR aligner and Bowtie were used to align the reads to the Hg19 human genome reference. STAR Fusion v1.2.0, Fusion Catcher v1.00, and Chime Pipe v0.9.5 were used to call fusions. All candidate fusions were analyzed with a custom algorithm, including filtering, annotation and prioritization of fusions for validation. The predicted fusion transcripts were inspected visually and fusions were selected based on relevance to diagnostic classification or therapy to be validated by an orthogonal method51,52. Genes targeted for the DNA and RNA panels tests, and all SNVs, CNVs and fusions identified and TMB and mismatch repair status determined by the DNA or RNA panel test are available in the Supplementary Data. R v.4.0.3 was used for iCatalog to analyze genomic data and create the manuscript figures.
Interpretation of evidence for clinical impact.
Professional guidelines from AMP, CAP and ASCO were used to classify variants of diagnostic and prognostic significance, with tier 1 alterations having strong evidence for diagnostic or prognostic impact and tier 2 alterations having moderate evidence for clinical impact24. Unexpected or discrepant results with diagnostic significance were delivered to the care teams at the time of the report, while comprehensive interpretations were rendered retrospectively for the whole cohort from January to July 2020.
Evidence identifying detected variants as biomarkers of potential response to MTT was evaluated according to the iCat evidence tiers18 (Supplementary Table 4). Tiers 1 and 2 were associated with evidence from clinical studies for response to MTT and tiers 3 and 4 were associated with evidence from preclinical studies, with tiers 1 and 3 used when data are in the same diagnosis and tiers 2 and 4 used when data are in a different diagnosis. Drugs with tissue-agnostic FDA approvals (larotrectinib and pembrolizumab) were considered to have clinical evidence in the same diagnosis (for example, tier 1). Tier 5 is associated with weaker evidence and consensus of the study team at the MTB described below. iCat tiers were assigned at the time of returning the molecular profiling report to the patient (February 2016–June 2019).
Patients with one or more detected variants with evidence supporting the variant as a biomarker of possible response to MTT received an iCat recommendation. The initial determination of therapeutic associations and iCat recommendations was performed by a study staff scientist. iCat recommendations and interpretations of diagnostic and prognostic significance were returned to the consenting provider or lead site investigator in a report. This report, which summarized the clinical interpretation, was drafted by a study staff scientist and then reviewed, edited and signed by one of the investigators. A knowledge base and report generating tool, iCatalog, developed for the study supported evidence interpretation and reporting (Supplementary Fig. 10). Reports contained information about the variant, a summary of evidence supporting the variant as a potential biomarker of response to MTT and a listing of specific drugs and clinical trials, including the ClinicalTrials.gov National Clinical Trial number. We also classified the availability of each of the iCat recommended drugs as: FDA-approved; clinical trial; and in clinical development but without an appropriate clinical trial for this patient (based on age and diagnosis).
An MTB meeting of an expert panel composed of molecular pathologists, investigators, study staff and genetic counselors from each of the participating sites occurred every other week. The treating oncologist for the patients being discussed was invited to attend. Cases were selected for discussion if the staff scientist preparing or investigator reviewing the clinical interpretation report identified conflicting or newly emerging evidence or if the iCat recommendation was tier 5. At the expert panel, a staff scientist or study investigator presented the sequencing results and evidence on which diagnostic and therapeutic assertions were based. The preliminarily assigned iCat tier was provided as were the issues for discussion. The final clinical interpretation report was generated after the MTB meeting.
Because evidence supporting genomic variants as biomarkers for response to MTT changes over time with emerging preclinical studies and clinical trial results, each iCat recommendation was systematically reevaluated with updated evidence in May–June 2020 (Supplementary Fig. 2) for this analysis. Systematic reevaluation, hereafter referred to as re-tiering, involved review of published literature and meeting abstracts and discussion in MTB meetings. Complete data on clinical interpretation of variants are available in the Supplementary Data file including the assignment of prognostic or diagnostic significance and associated AMP/CAP/ASCO tier and iCat recommendations. As an additional assessment of variants as biomarkers of response to MTT, we determined whether genomic variants were AMOIs allowing enrollment onto one of the treatment subprotocols of the NCI-COG pediatric MATCH clinical trial (NCT03155620).
Treatment and outcome data collection.
Receipt of MTT was defined as the patient having received at least one dose of a drug in the same therapeutic class as an iCat recommendation after receipt of the molecular profiling report. A responder to MTT was defined as a patient with RECIST measurable disease21 at the initiation of MTT given without other concurrent tumor-directed therapy who either had a partial response or stable disease on ≥4 months of MTT.
After receipt of a study-associated molecular report, patient vital status, disease status, treatment regimen and response data were entered into InForm on an ongoing basis (every three months for patients with recurrent/refractory disease and every six months for patients with newly diagnosed disease). SAS v.9.4 was used to analyze clinical data and Microsoft Excel was used to combine the analyzed clinical and sequencing data.
Extended Data
Extended Data Fig. 1 |. CONSORT diagram.
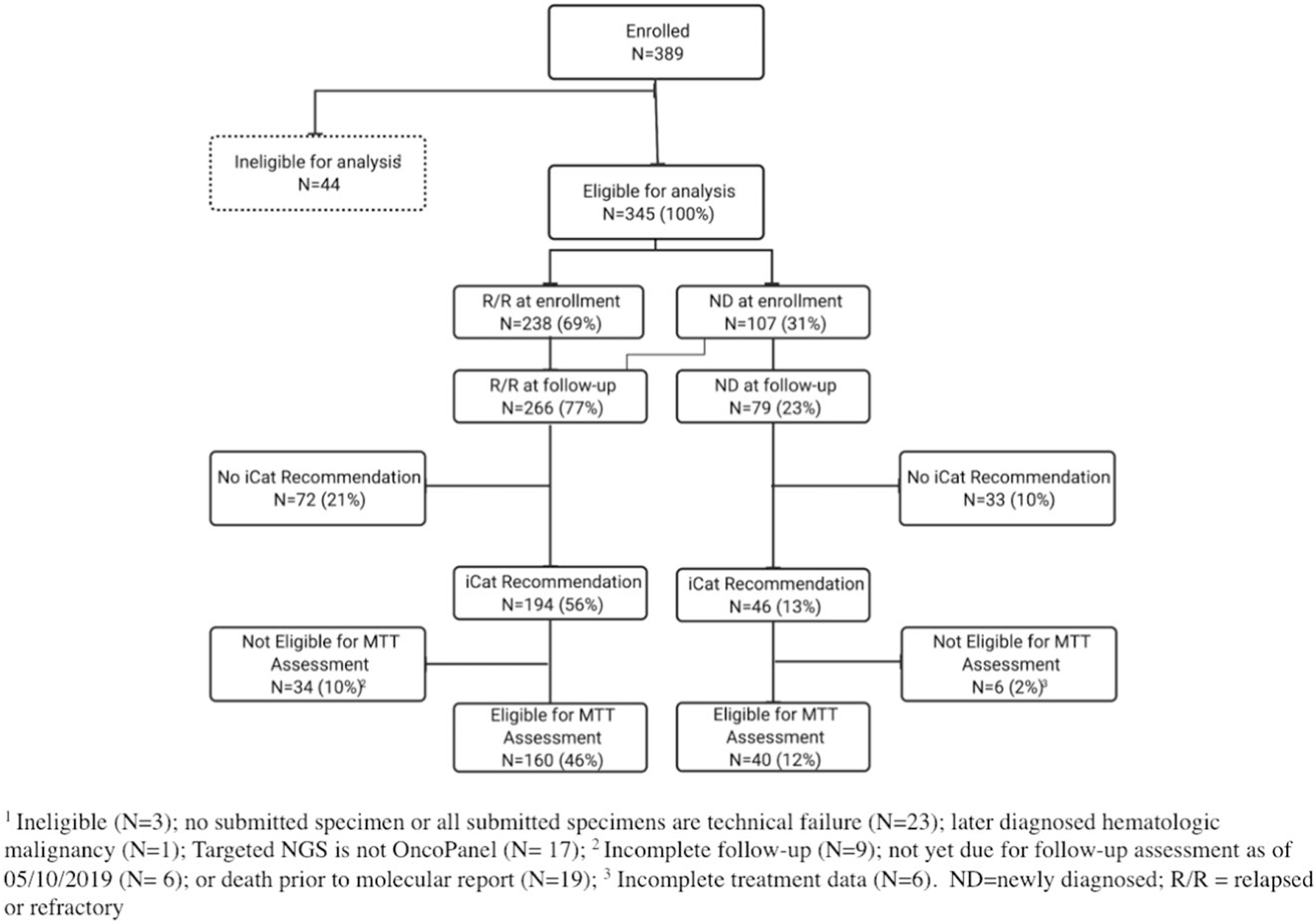
CONSORT diagram of 389 patients enrolled in the GAIN/iCat2 study between 11/2015 and 12/2018 identifying the analytic cohort. Percentages are based on the analytic population (n = 345).
Extended Data Fig. 2 |. iCat Recommendations.
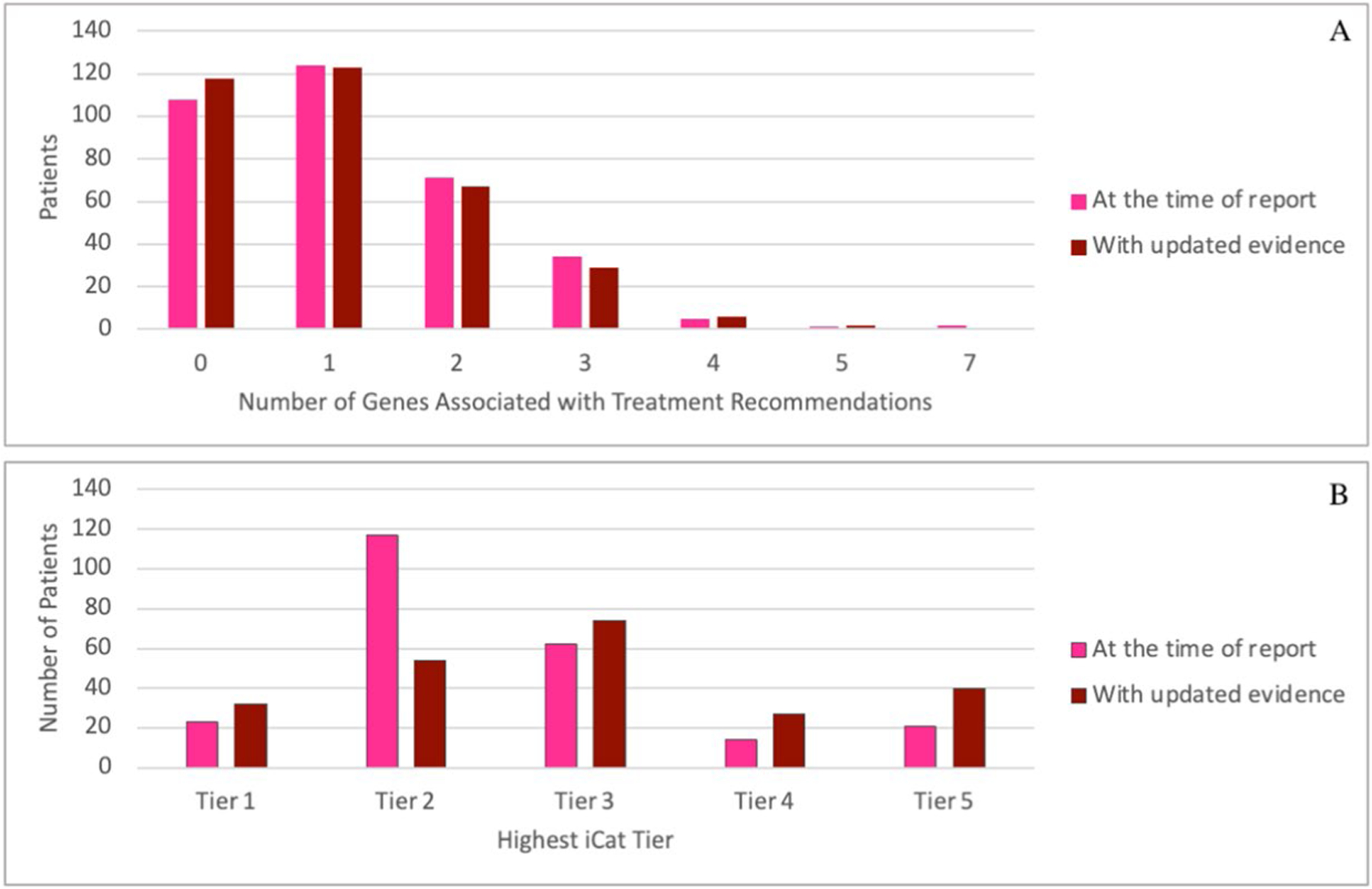
Number of genes with iCat recommendations per patient at the time of initial report (February, 2016 to June, 2019) and with updated evidence reviewed between May and June, 2020 (a). Highest tier of iCat therapeutic recommendation for each patient at the time of initial report (February, 2016 to June, 2019) and with updated evidence reviewed between May and June, 2020 (b).
Extended Data Fig. 3 |. Changes in iCat Recommendations.
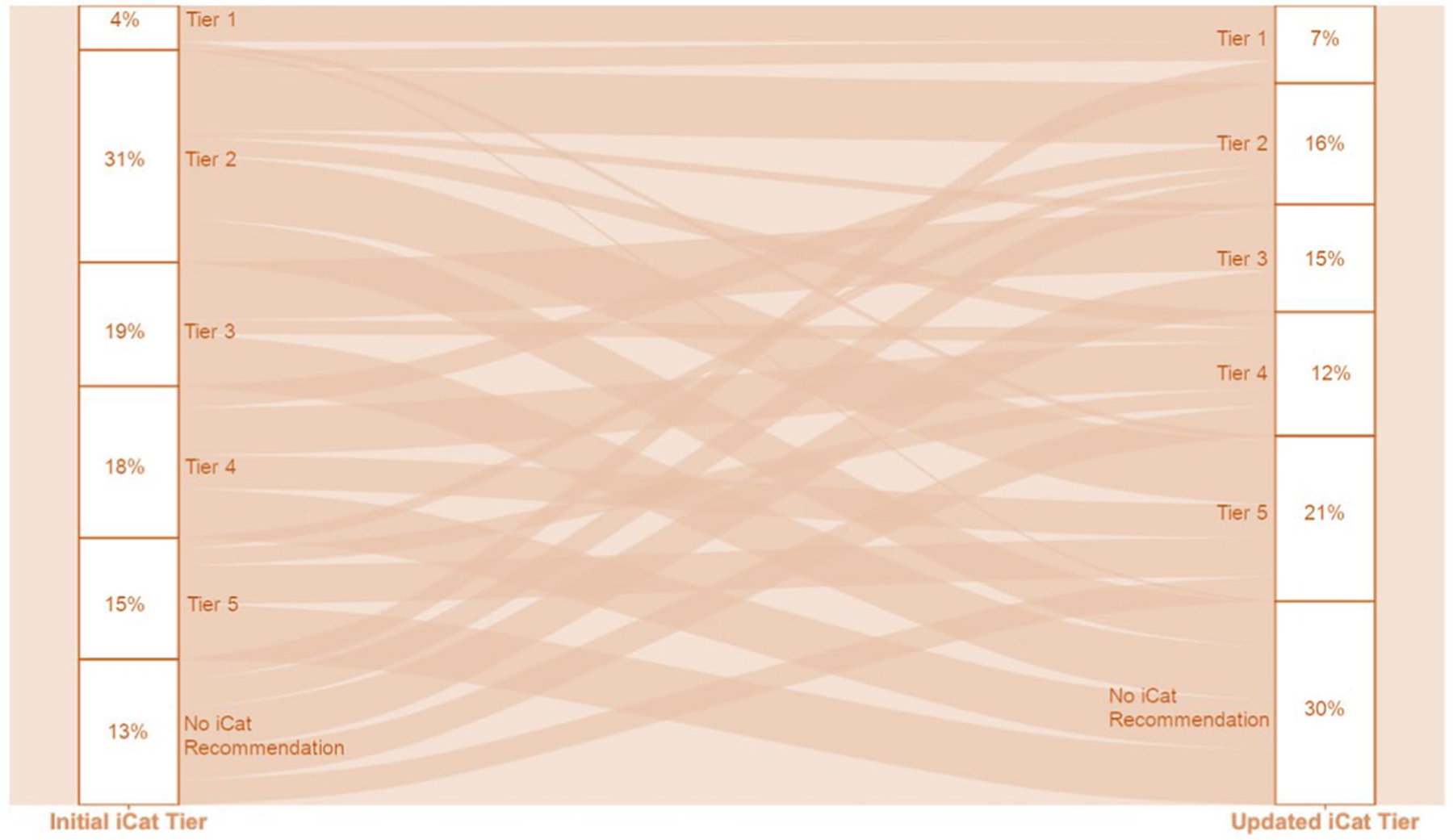
Changes in tiering of individual iCat recommendations from the time of initial report (February, 2016 to June, 2019) to re-tiering with updated evidence reviewed between May and June, 2020 upon reevaluation of evidence for expected response to matched targeted therapy.
Extended Data Fig. 4 |. Genes with iCat Recommendations.
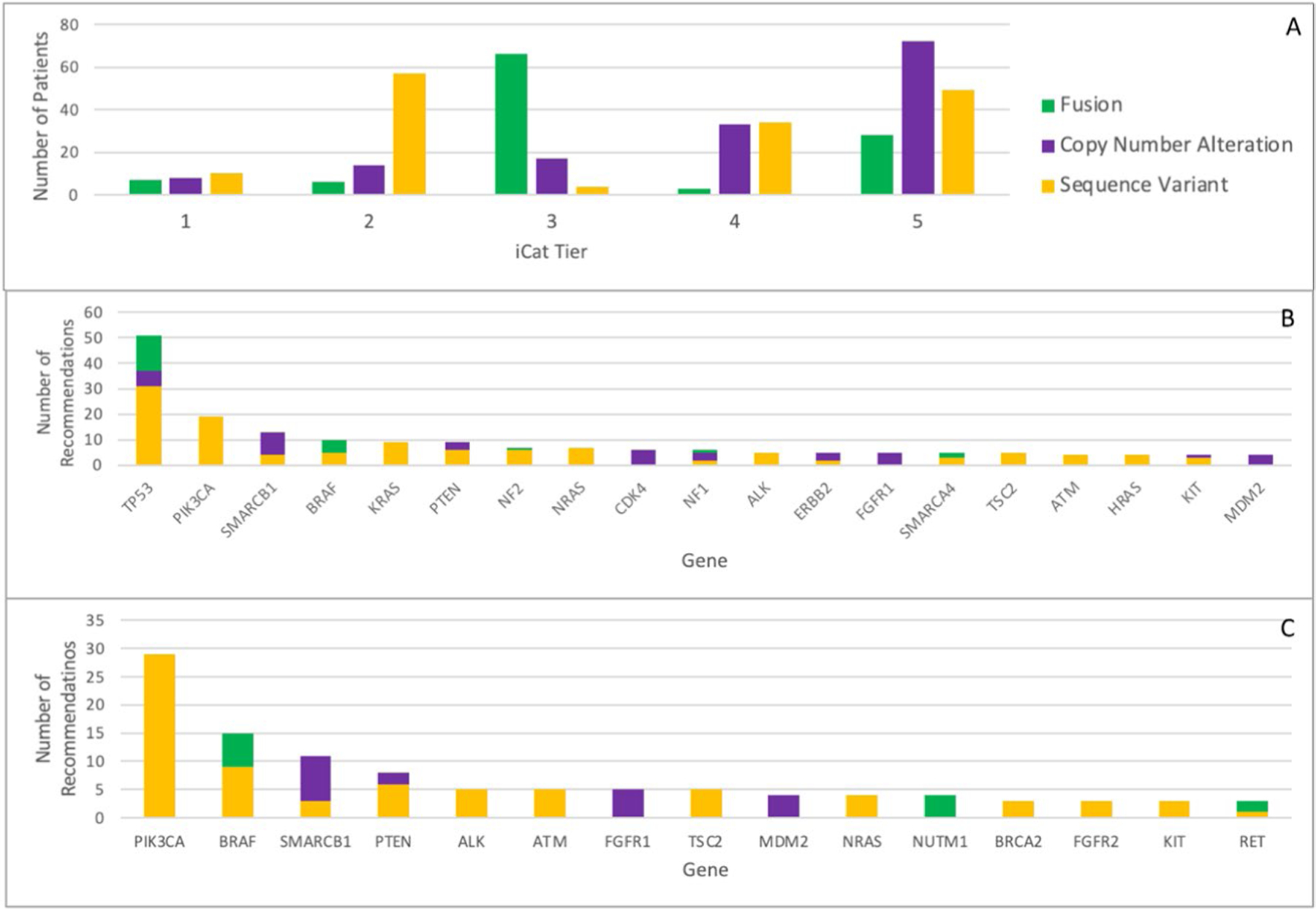
iCat tiers reported according to alteration type (a). Top genes with strong evidence for therapeutic impact (iCat tiers 1–2) at the time of report (February 2016 to June 2019) (b) and with updated evidence reviewed between May and June, 2020 (c).
Extended Data Fig. 5 |. Responders to Matched Targeted Therapy.
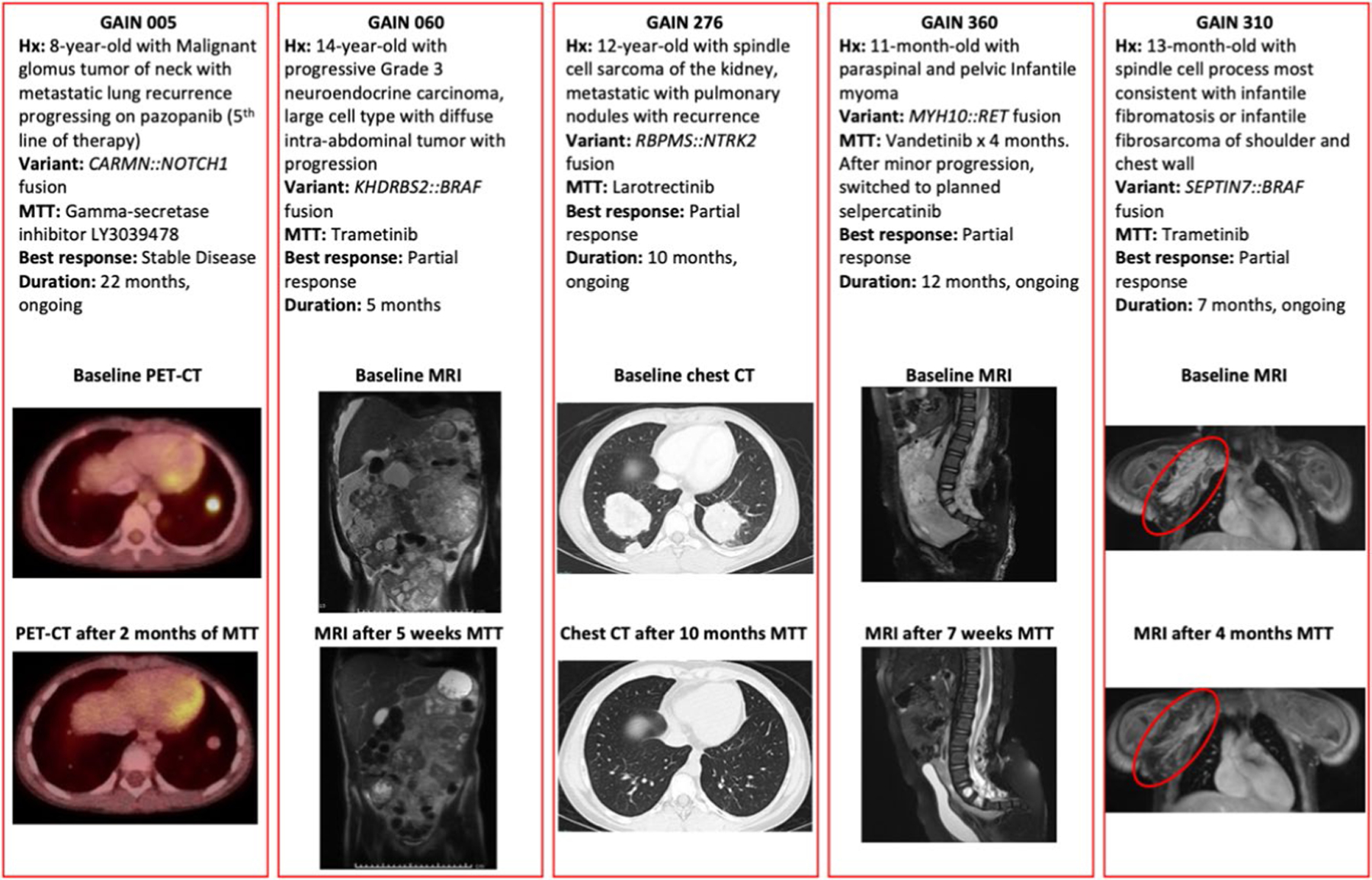
Details of 5 responders to matched targeted therapy. GAIN patient 317 with an ALK fusion in a medullary thyroid carcinoma also responded, with images available in the primary report (Hillier et al., 2019).
Extended Data Fig. 6 |. Oncoprint for Patients Receiving MTT.

Tumor mutational burden (TMB) and additional alterations in 29 patients who received MTT. Additional alterations are shown if they occurred in >1 case or were potentially targetable. (For the case with high TMB all potentially actionable variants are not shown).
Extended Data Fig. 7 |. Top Genes with Diagnostic and Prognostic Significance.
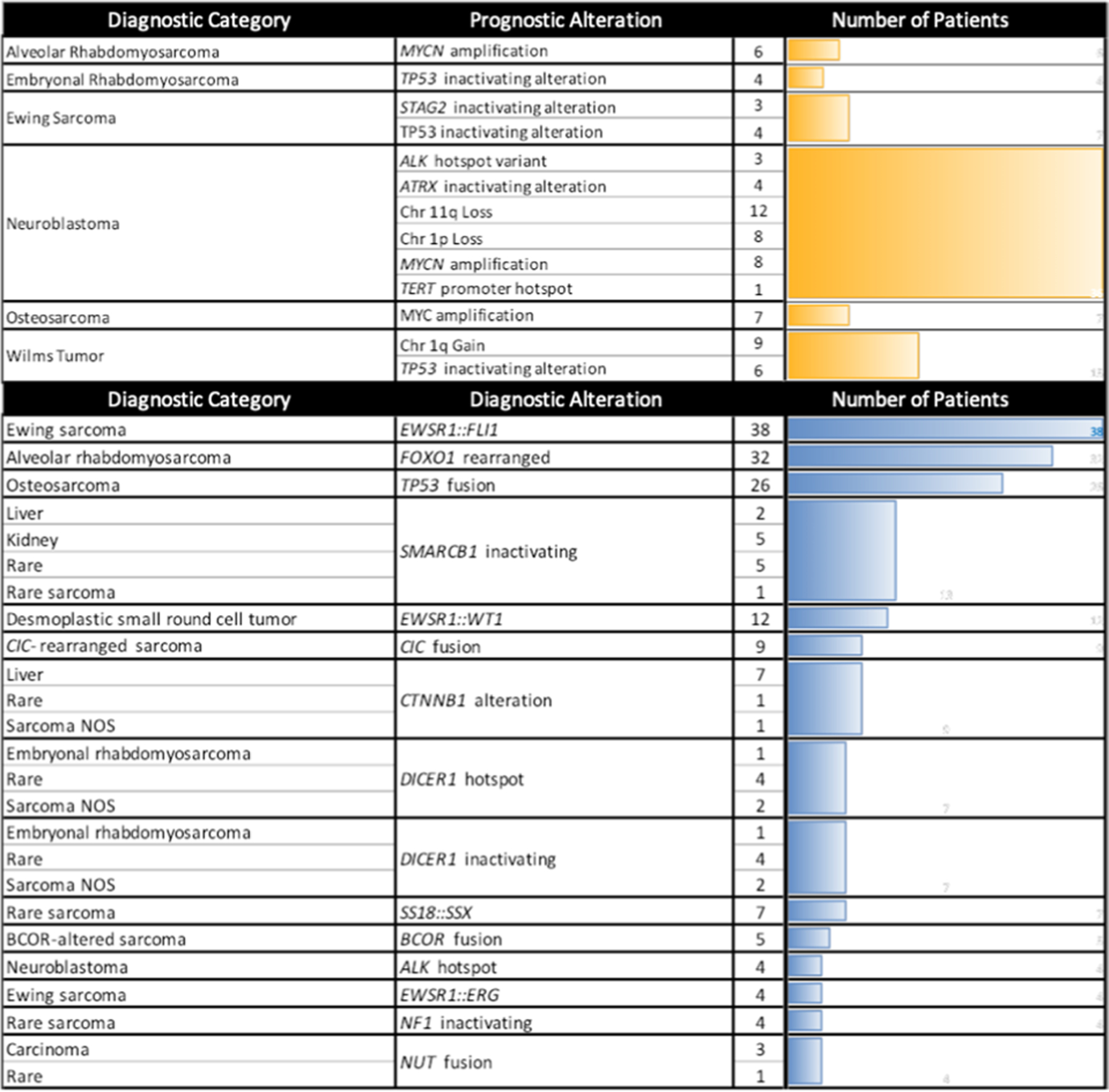
All genes or chromosomes arms with tier 1 or 2 AMP/CAP/ASCO guideline evidence for prognostic impact (top; yellow). Top alterations with tier 1 or 2 AMP/CAP/ASCO guideline evidence for diagnostic impact, representing 181 of 227 diagnostically significant alterations (bottom; blue).
Extended Data Fig. 8 |. Diagnostic Impact Case.
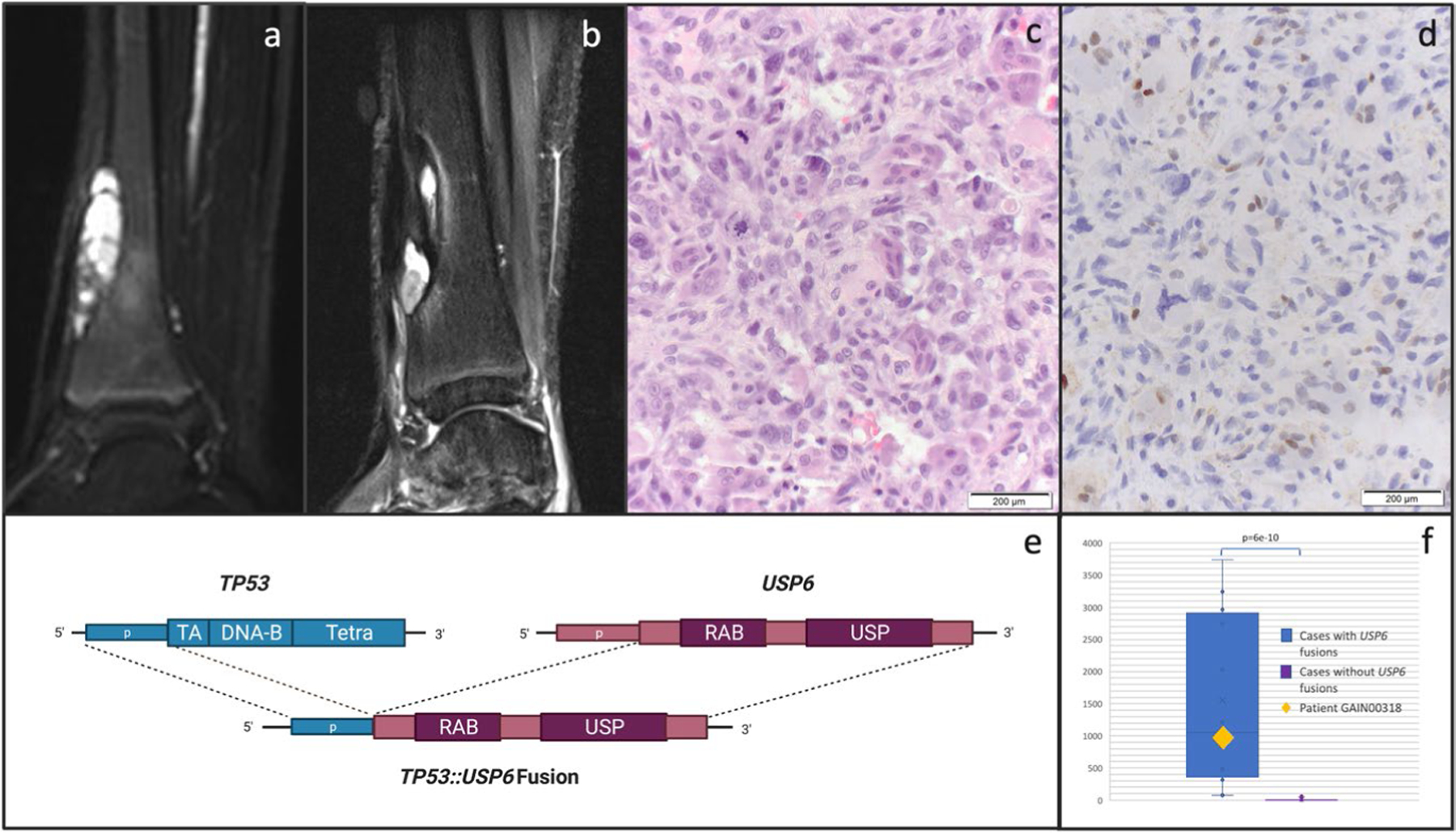
Details of an illustrative case (GAIN318) of diagnostic impact: MRI (sagittal short inversion time inversion recovery (STIR) sequence) of a distal tibia tumor at diagnosis (a) and one year later at recurrence (b). The diagnosis rendered in the pathology report at initial diagnosis was aneurysmal bone cyst (ABC) while biopsy of the recurrence demonstrated osteogenic cells with pleomorphism and atypical mitotic figures (c, H&E stain, single experiment, not repeated). p53 IHC (d) shows loss of p53 expression in tumor but not normal cells (single experiment, not repeated). GAIN sequencing identified a novel TP53::USP6 fusion connecting TP53 intron 1 to USP6 intron 7, supporting a diagnosis of osteosarcoma in which TP53 rearrangements are common (e, created with Biorender.com). RNA analysis shows high expression of USP6, as measured by the number of unique RNA reads across 4 USP6 target regions [chr17:5031701, chr17:5033235, chr17:5033666, chr17:5033937], shown in the context of 12 cases with USP6 fusions (left, average read count 1552 reads) compared to 20 control cases with no USP6 fusions (right, average read count 8.0). This represents a significant difference in expression (unpaired two-tail t-test, p = 6.3e-10). Box plots represent maximum and minimum values (whiskers), first and third quartiles (bounds of box) and median (center line) (f).
Extended Data Fig. 9 |. Overview of the iCat2/GAIN Study.
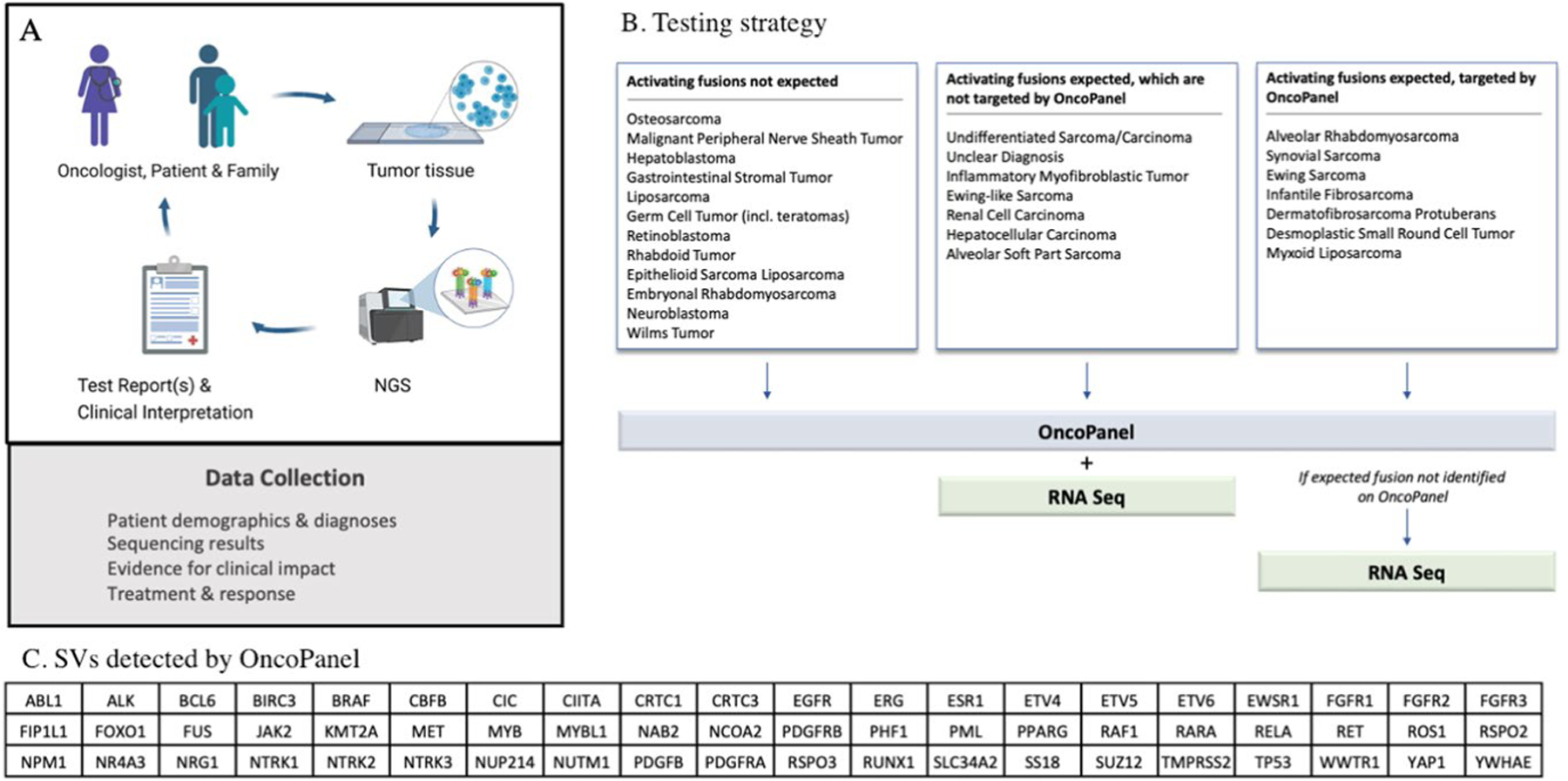
Overview of the iCat2/GAIN study (a, created with Biorender.com). Targeted DNA NGS is performed on one or more tumor samples from each patient. Selected patients also have tumors subjected to RNA sequencing. Test results are returned to the treating oncologist and follow-up treatment and response data are collected. Details of clinical interpretation of test reports including molecular tumor board are shown in extended data Fig. 3. Testing strategy (b) to select patients for additional sequencing with either whole transcriptome sequencing or targeted RNA fusion panel testing (RNASeq). RNASeq was not performed if it was unlikely to contribute to research findings or clinical care. In this study, transcriptome sequencing was analyzed only for structural variants (SVs) and OncoPanel detects rearrangements in 60 genes (c). The testing triage is based on several assumptions: 1) False positives for SV detection OncoPanel are uncommon; 2) If oncogenic fusions have not been described in a particular solid tumor in previous studies and typical oncogenic events for that diagnosis are present then novel oncogenic fusions are unlikely; and 3) very rare pediatric solid malignancies might harbor previously undescribed fusions because they are understudied.
Extended Data Fig. 10 |. Details of Clinical Interpretation.
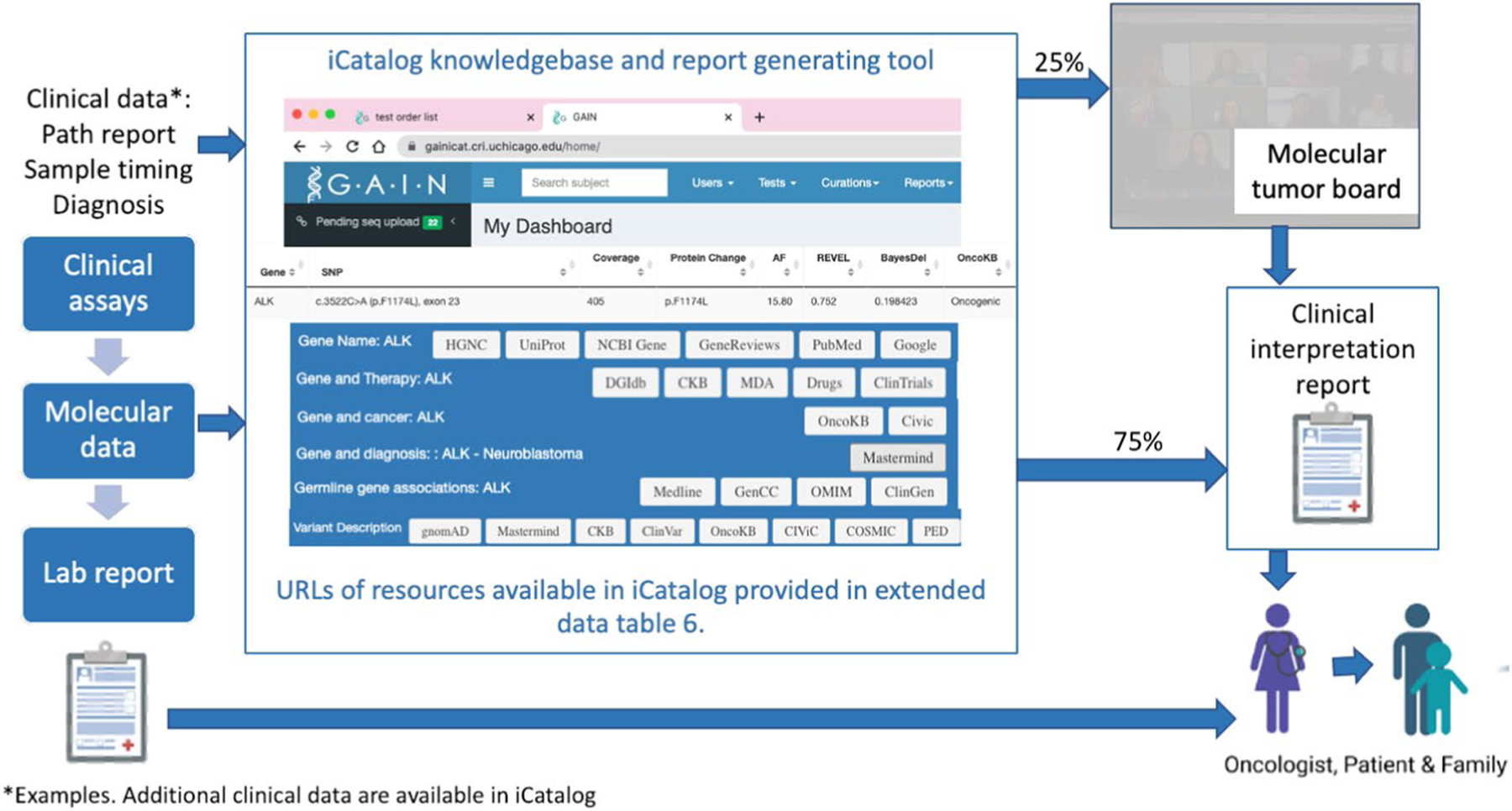
Details of clinical interpretation of test reports. A knowledgebase and report generation tool, iCatalog, was developed specifically for this study. iCatalog contains pediatric cancer specific knowledge on the gene and variant level including associated references (stored with PMID) and clinical trials (stored by NCT number). iCatalog knowledge is maintained by a staff scientist and research coordinator both at several scheduled times and when interpreting cases. iCatalog uses API to annotate variants. Resources available to the iCatalog user are shown below. Cases with Tier 5 iCat recommendations, previously undiscussed evidence or conflicting evidence are discussed at the molecular tumor board. Clinical interpretation reports are returned to the lead site investigator or enrolling oncologist.
Supplementary Material
Acknowledgements
Funding for this study was provided by the Precision For Kids Pan Mass Challenge Team, the 4C’s Fund, Lamb Family Fund, C&S Wholesale Grocers and C&S Charities and Alexandra Simpson Pediatric Research Fund. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. We also thank the study participants and their families and the research coordinators at each of the study sites.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41591-022-01856-6.
Reporting summary.
Further information on research design is available in the Nature Research Reporting summary linked to this article.
Code availability
There is no relevant code provided for this study.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s41591-022-01856-6.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41591-022-01856-6.
Peer review information Nature Medicine thanks Elaine Mardis and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling editor: Anna Maria Ranzoni, in collaboration with the Nature Medicine team.
Competing interests
A.J.C. sits on an advisory board for Bayer. L.B.C. is an employee at Sema4 and is a consultant for X-Chem and Biomatics. L.M. is a consultant for Jazz Pharmaceuticals. T.W.L. is an advisory board member for Bayer and has received honoraria from Bayer, Cellectis, Novartis, Deciphera, Jumo Health and Y-mAbs and has received research funding from Pfizer and Bayer. M.A.A. is an advisory board member for Fennec Pharmaceuticals. Y-C.L. is an advisory board member for Takeda Pharmaceutical Company. A.D.C. has received research support from Bayer and his spouse is employed by Labcorp. J.K.’s spouse has received consulting fees from ROME Therapeutics, Foundation Medicine, NanoString, Merck Millipore and Pfizer that are not related to this work. J.K.’s spouse is a founder and has equity in ROME Therapeutics, PanTher Therapeutics and TellBio, which is not related to this work. J.K.’s spouse receives research support from ACD Bio-Techne, PureTech Health and Ribon Therapeutics, which was not used in this work. S.L.V. is a consultant for CVS Accordant. E.V.A. has provided advisory/consulting work for Tango Therapeutics, Genome Medical, Invitae, Enara Bio, Janssen, Manifold Bio and Monte Rosa Therapeutics, has received research support from Novartis and Bristol Myers Squibb, holds equity in Tango Therapeutics, Genome Medical, Syapse, Enara Bio, Manifold Bio, Microsoft and Monte Rosa Therapeutics, has received travel reimbursement from Roche/Genentech and holds institutional patents filed on chromatin mutations and immunotherapy response and methods for clinical interpretation. S.G.D. has consulted for Bayer and received travel expenses from Loxo Oncology, Roche and Salarius. W.B.L. has served on the data safety monitoring boards for Merck Millipore and Jubilant Draximage. K.A.J. has consulted for Ipsen and Bayer, and has received honoraria from Foundation Medicine and Takeda Pharmaceutical Company. The other authors declare no competing interests.
Data availability
A Supplementary Data file contains all SNVs, CNVs and fusions identified and TMB and mismatch repair status determined by the DNA or RNA panel tests. The Supplementary Data file also includes clinical interpretation of variants determined to have prognostic or diagnostic significance and the associated AMP/CAP/ASCO tier. All iCat recommendations are provided in the Supplementary Data including the iCat tier at the time of the clinical interpretation report and at the time of re-tiering.
References
- 1.Brandão M, Caparica R, Eiger D & de Azambuja E Biomarkers of response and resistance to PI3K inhibitors in estrogen receptor-positive breast cancer patients and combination therapies involving PI3K inhibitors. Ann. Oncol 30, x27–x42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drilon A et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med 378, 731–739 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Havel JJ, Chowell D & Chan TA The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer 19, 133–150 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Le DT et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 357, 409–413 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lindeman NI et al. Updated molecular testing guideline for the selection of lung cancer patients for treatment with targeted tyrosine kinase inhibitors: guideline from the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology. J. Thorac. Oncol 13, 323–358 (2018). [DOI] [PubMed] [Google Scholar]
- 6.FDA Fact Sheet: CDRH’S Approach to Tumor Profiling Next Generation Sequencing Tests (US Food & Drug Administration, accessed May 23, 2022); https://www.fda.gov/media/109050/download
- 7.Guidelines Version 4.2021 Non-Small Cell Lung Cancer. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines) Vol. 2021 (National Comprehensive Cancer Network, 2021). [Google Scholar]
- 8.Next Generation Sequencing (NGS) for Medicare Beneficiaries with Advanced Cancer (CAG-00450N) (CMS.gov, 2018). [Google Scholar]
- 9.Laetsch TW & Hawkins DS Larotrectinib for the treatment of TRK fusion solid tumors. Expert Rev. Anticancer Ther 19, 1–10 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Mosele F et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann. Oncol 31, 1491–1505 (2020). [DOI] [PubMed] [Google Scholar]
- 11.Li W et al. Trends in molecular testing of lung cancer in mainland People’s Republic of China over the decade 2010 to 2019. JTO Clin. Res. Rep 2, 100163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.AACR Project GENIE: Data (American Association for Cancer Research, 2022). [Google Scholar]
- 13.Chmielecki J et al. Genomic profiling of a large set of diverse pediatric cancers identifies known and novel mutations across tumor spectra. Cancer Res 77, 509–519 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Parsons DW et al. Actionable tumor alterations and treatment protocol enrollment of pediatric and young adult patients with refractory cancers in the National Cancer Institute-Children’s Oncology Group Pediatric MATCH Trial. J. Clin. Oncol 10.1200/JCO.21.02838 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Data Sharing Opportunities in Childhood, Adolescent and Young Adult (AYA) Cancer Research for the National Cancer Institute: Report of the Board of Scientific Advisors on the Childhood Cancer Data Initiative (CCDI) (National Institutes of Health, National Cancer Institute Board of Scientific Advisors, 2020). [Google Scholar]
- 16.van Tilburg CM et al. The pediatric precision oncology INFORM registry: clinical outcome and benefit for patients with very high-evidence targets. Cancer Discov 11, 2764–2779 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wong M et al. Whole genome, transcriptome and methylome profiling enhances actionable target discovery in high-risk pediatric cancer. Nat. Med 26, 1742–1753 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Harris MH et al. Multicenter feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the Individualized Cancer Therapy (iCat) study. JAMA Oncol 2, 608–615 (2016). [DOI] [PubMed] [Google Scholar]
- 19.Leijen S et al. Phase I study evaluating WEE1 Inhibitor AZD1775 as monotherapy and in combination with gemcitabine, cisplatin, or carboplatin in patients with advanced solid tumors. J. Clin. Oncol 34, 4371–4380 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allen CE et al. Selumetinib in patients with tumors with MAPK pathway alterations: results from Arm E of the NCI-COG pediatric MATCH trial. J. Clin. Oncol 39, 10008 (2021). [Google Scholar]
- 21.Eisenhauer EA et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur. J. Cancer 45, 228–247 (2009). [DOI] [PubMed] [Google Scholar]
- 22.Ortiz MV et al. Activity of the highly specific RET inhibitor selpercatinib (LOXO-292) in pediatric patients with tumors harboring RET gene alterations. JCO Precis. Oncol 4, PO.19.00401 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hillier K et al. A novel ALK fusion in pediatric medullary thyroid carcinoma. Thyroid 29, 1704–1707 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Li MM et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J. Mol. Diagn 19, 4–23 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oliveira AM & Chou MM USP6-induced neoplasms: the biologic spectrum of aneurysmal bone cyst and nodular fasciitis. Hum. Pathol 45, 1–11 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Chen X et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep 7, 104–112 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thyroid Carcinoma, Version 3.2021. NCCN Practice Guidelines in Oncology (National Comprehensive Cancer Network, 2021). [Google Scholar]
- 28.Neuroendocrine and Adrenal Tumors, Version 4.2021. NCCN Clinical Practice Guidelines in Oncology (National Comprehensive Cancer Network, 2021). [DOI] [PubMed] [Google Scholar]
- 29.Bone Cancer, Version 2.2022. NCCN Practice Guidelines in Oncology (National Comprehensive Cancer Network, 2022). [Google Scholar]
- 30.Hepatobiliary Cancers, Version 5.2021. NCCN Practice Guidelines in Oncology (National Comprehensive Cancer Network, 2021). [DOI] [PubMed] [Google Scholar]
- 31.Soft Tissue Sarcoma, Version 3.2021. NCCN Practice Guidelines in Oncology (National Comprehensive Cancer Network, 2022). [Google Scholar]
- 32.Oberg JA et al. Implementation of next generation sequencing into pediatric hematology-oncology practice: moving beyond actionable alterations. Genome Med 8, 133 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Inaba H & Mullighan CG Pediatric acute lymphoblastic leukemia. Haematologica 105, 2524–2539 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ramaswamy V et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol 131, 821–831 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khater F et al. Molecular profiling of hard-to-treat childhood and adolescent cancers. JAMA Netw. Open 2, e192906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schienda J et al. Germline sequencing improves tumor-only sequencing interpretation in a precision genomic study of patients with pediatric solid tumor. JCO Precis. Oncol 5, PO.21.00281 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons DW et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol 2, 616–624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fiala EM et al. Prospective pan-cancer germline testing using MSK-IMPACT informs clinical translation in 751 patients with pediatric solid tumors. Nat. Cancer 2, 357–365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J et al. Germline mutations in predisposition genes in pediatric cancer. N. Engl. J. Med 373, 2336–2346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.NCCR*Explorer: An Interactive Website for NCCR Cancer Statistics (National Cancer Institute, 2021). [Google Scholar]
- 41.Parsons DW et al. Identification of targetable molecular alterations in the NCI-COG Pediatric MATCH trial. J. Clin. Oncol 37, 10011 (2019). [Google Scholar]
- 42.FDA Reauthorization Act of 2017 (FDARA) (US Food & Drug Administration, 2017). [Google Scholar]
- 43.Janes MR et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell 172, 578–589.e17 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Chen Y-N et al. Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152 (2016). [DOI] [PubMed] [Google Scholar]
- 45.International Classification of Diseases for Oncology (ICD-O) (World Health Organization, 2020). [Google Scholar]
- 46.Abo RP et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res 43, e19 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Garcia EP et al. Validation of OncoPanel: a targeted next-generation sequencing assay for the detection of somatic variants in cancer. Arch. Pathol. Lab. Med 141, 751–758 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Nowak JA et al. Detection of mismatch repair deficiency and microsatellite instability in colorectal adenocarcinoma by targeted next-generation sequencing. J. Mol. Diagn 19, 84–91 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sholl LM et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 1, e87062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Paulson VA et al. Recurrent and novel USP6 fusions in cranial fasciitis identified by targeted RNA sequencing. Mod. Pathol 33, 775–780 (2020). [DOI] [PubMed] [Google Scholar]
- 51.Haas BJ et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biol 20, 213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rodríguez-Martín B et al. ChimPipe: accurate detection of fusion genes and transcription-induced chimeras from RNA-seq data. BMC Genomics 18, 7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
A Supplementary Data file contains all SNVs, CNVs and fusions identified and TMB and mismatch repair status determined by the DNA or RNA panel tests. The Supplementary Data file also includes clinical interpretation of variants determined to have prognostic or diagnostic significance and the associated AMP/CAP/ASCO tier. All iCat recommendations are provided in the Supplementary Data including the iCat tier at the time of the clinical interpretation report and at the time of re-tiering.