

Abstract
High-density lipoprotein (HDL) nanoparticles promote endothelial cell (EC) function and suppress inflammation, but their utility in treating EC dysfunction has not been fully explored. Here, we describe a fusion protein named ApoA1-ApoM (A1M) consisting of apolipoprotein A1 (ApoA1), the principal structural protein of HDL that forms lipid nanoparticles, and ApoM, a chaperone for the bioactive lipid sphingosine 1-phosphate (S1P). A1M forms HDL-like particles, binds to S1P, and is signaling competent. Molecular dynamics simulations showed that the S1P-bound ApoM moiety in A1M efficiently activated EC surface receptors. Treatment of human umbilical vein endothelial cells (HUVECs) with A1M-S1P stimulated barrier function either alone or cooperatively with other barrier-enhancing molecules, including the stable prostacyclin analog iloprost, and suppressed cytokine-induced inflammation. A1M-S1P injection into mice during sterile inflammation suppressed neutrophil influx and inflammatory mediator secretion. Moreover, systemic A1M administration led to a sustained increase in circulating HDL-bound S1P and suppressed inflammation in a murine model of LPS-induced endotoxemia. We propose that A1M administration may enhance vascular endothelial barrier function, suppress cytokine storm, and promote resilience of the vascular endothelium.
Introduction
Vascular endothelial dysfunction, which is induced by metabolic stress (such as diabetes or hypercholesterolemia), inflammatory and autoimmune states, is a major driver of chronic diseases, such as atherosclerosis, stroke, heart failure, rheumatoid arthritis, and systemic lupus erythematosus (1). Restoration of endothelial cell function attenuates inflammation-induced tissue damage, thrombosis, and chronic disease progression (2). However, therapeutic strategies to counter endothelial dysfunction are not available at present.
Circulating high-density lipoprotein (HDL) particles are important for cholesterol homeostasis and endothelial function. The major structural protein of HDL is apolipoprotein A1 (ApoA1), an amphipathic polypeptide that becomes lipidated after interaction with ABCA1/ABCG1 transporters and membranes (3). Nascent discoidal HDL particles transport cholesterol from tissues to the liver, a process termed reverse cholesterol transport (4). Although reverse cholesterol transport was originally thought to be solely responsible for the cardiovascular protective effects of HDL, the ability to attenuate endothelial dysfunction may be equally important (5, 6). HDL is associated with numerous proteins and lipids that confer cytoprotective and anti-inflammatory properties under normal conditions (7, 8). Among the HDL-associated proteins, the major structural protein ApoA1 decreases cytokine and endotoxin-induced NF-κB activation in myeloid cells (macrophages and neutrophils) (9) by interfering with inflammatory receptor signal transduction (10). ApoA1 also enhances endothelial-derived nitric oxide (NO) secretion, which promotes blood flow (11). In addition, HDL suppresses thrombosis by enhancing the activity of prostacyclin (PGI2) and attenuation of tissue factor expression (12, 13).
We have characterized apolipoprotein M (ApoM), a member of the lipocalin family of proteins associated with a subpopulation of HDL particles (14). We showed that ApoM is the key physiological chaperone for sphingosine 1-phosphate (S1P), a high affinity ligand for G-protein coupled S1P receptors (S1PRs). The S1P/S1PR1 axis is critical for vascular development, vascular barrier function, NO synthesis, and endothelial survival (15). HDL-bound S1P attenuates cytokine-induced NFκB activation and adhesion molecule expression by a mechanism that involves S1PR1 (16). This signaling axis suppresses lymphopoiesis in the bone marrow (17), endothelial injury in the lung (18) and liver fibrosis and promotes hepatocyte regeneration (19). In addition, S1PR1 signaling enables trans-endothelial passage of HDL particles into tissue parenchymal spaces (20). These studies suggest that HDL-bound S1P activation of endothelial S1PR1 may be therapeutically tractable to suppress endothelial dysfunction and suppress pathologic inflammation.
S1P bound to ApoM as well as other carrier proteins, such as albumin and ApoA4 can activate the S1PRs (21). However, mice that are deficient for ApoM exhibit increased vascular leak in lung microvessels (14, 22) and penetrating arterioles of the brain (23), suggesting that the ability of ApoM-bound S1P to promote vascular barrier function is physiologically critical. Solution binding as well as molecular dynamics (MD) simulation studies indicate that S1P dissociation from ApoM requires free energy to overcome the electrostatic interactions between the anionic head group of S1P and the cationic amino acid residues that form the lid of ApoM that blocks the of the ligand binding pocket (24). Thus, S1P transfer from ApoM to S1PRs may be fundamentally different from albumin-S1P/S1PR activation. This may explain the sustained and Gαi-biased signaling property of HDL-bound S1P (16).
We reported the development of an ApoM-Fc fusion protein that enhances endothelial barrier function, suppresses ischemia-reperfusion injury of the heart and the brain, and immune complex-mediated and acid-induced lung injury (18, 25, 26). However, we found that some of the endothelial protective effects of HDL-S1P were not mimicked by ApoM-Fc-S1P. To overcome this issue, we have designed and characterized ApoA1-ApoM (A1M) fusion protein, which forms spherical nano-sized lipoprotein particles, chaperones multiple bioactive lipids (S1P and PGI2 analogs), protects the endothelium, and suppresses platelet aggregation and inflammatory responses in vitro and in vivo.
Results
Design, expression and characterization of ApoA1-ApoM-S1P (A1M-S1P) nanoparticles
Because ApoM-Fc-S1P did not possess all HDL-S1P functions (19, 25, 26), we hypothesized that a fusion protein of ApoA1 and ApoM might confer additional endothelial protective properties. We designed and constructed an ApoA1-ApoM (A1M) fusion protein with a flexible linker domain (GGGGS) and 6X-Histidine tag at the carboxyl-terminus (Fig. S1) (27). We expressed and purified A1M protein from conditioned media from stably transfected CHO-S cells (Fig. 1A). A1M was loaded with S1P as described previously and purified by FPLC gel filtration chromatography (25). Unloaded and loaded A1M proteins had ~0.5 mol% and ~65 mol% of S1P, respectively (Fig. 1B).
Fig. 1. Production, purification, and characterization of S1P binding by the A1M fusion protein.
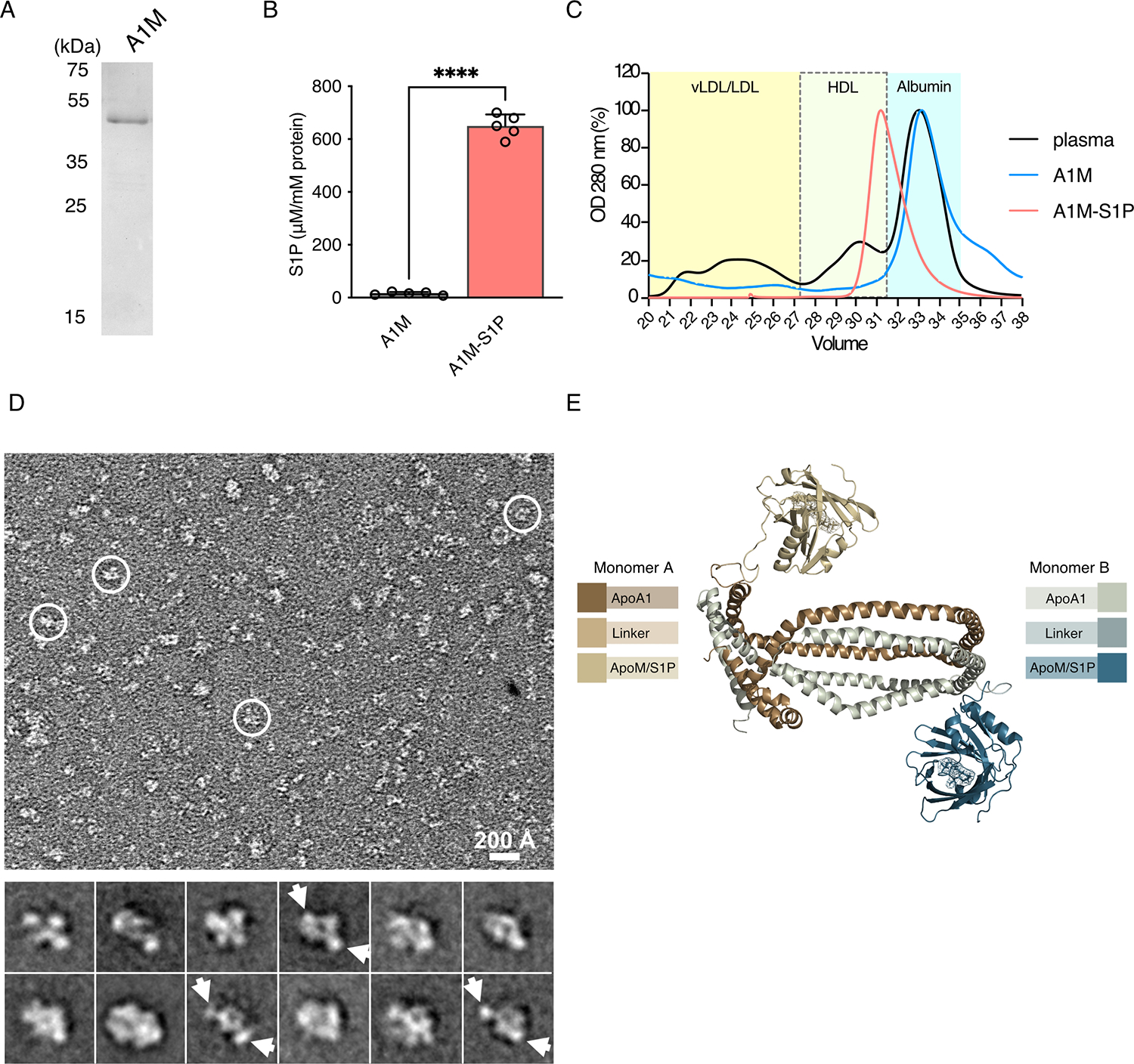
(A) Purified A1M (4 μg) from CHO cell conditioned media was separated by reducing 10% SDS-PAGE and stained with Coomassie brilliant blue. (B) Purified A1M was incubated with S1P for 24 hours, lipidated, and purified by gel filtration chromatography. S1P content of A1M-S1P was analyzed by electrospray ionization-MS/MS. The resulting data are the mean ± S.D.; N = 5 biological replicates per group. ****P < 0.0001 by unpaired t-test. (C) Representative FPLC elution profiles (OD 280 nm) of 200 μl of mouse plasma (black), purified recombinant A1M (blue), and lipidated A1M with S1P loading (A1M-S1P, red). FPLC elution of plasma standards: vLDL/LDL (21–26 mL), HDL (28–31 mL), and soluble protein (31–35 mL) fractions. Elution profiles are representative of 3 independent experiments. (D) Representative negative-stain transmission EM image of lipidated A1M-S1P complex with some particles highlighted in white circles (top) and 2D averages with potential ApoM densities indicated by arrows (bottom). Box dimension of each 2D average is 215 Å. Images are representative of 3 independent experiments. (E) Schematic model of A1M-S1P.
To prepare reconstituted HDL-like particles, A1M-S1P was lipidated with a mixture of phospholipids dimyristoyl phosphatidylcholine and dimyristoyl phosphatidyl glycerol (DMPC/DMPG) as described for ApoA1 (25, 28). FPLC elution profiles of the samples were compared to a reference of fractionated mouse plasma (Fig. 1C). Lipidated A1M-S1P eluted near the HDL fractions, suggesting conversion of nascent A1M to HDL-like particles. Lipidated A1M-S1P particles were further analyzed by negative staining and analyzed by transmission electron microscopy. The majority of the particles were ~ 8–12 nm in diameter with various shapes (Fig. 1D, top), in line with the conformational heterogeneity of A1M due to the flexible linker between ApoA1 and ApoM. Several two-dimensional (2D) averages showed two side lobes connecting to a central disc-shaped density (Fig. 1D, bottom), which is consistent with the flexibility of the linker domain that connects ApoA1 and ApoM domains (Fig. 1E). These studies suggested that lipidated A1M-S1P complex forms nanometer-sized lipoprotein particles that stably bind to S1P.
Molecular dynamic simulation study of the A1M-S1P complex
We next performed molecular dynamic (MD) simulation analyses of lipidated A1M-S1P using the known protein structure of ApoM-S1P complex (PDB 2YG21) (14) and the double belt structure of ApoA1 (29, 30). The HDL-like particle contained POPC:unesterified cholesterol:ApoA1 at a ratio of 160:24:2 in an antiparallel arrangement with LL5/5 registry (31) (Fig. 2A and Movie S1). To understand the structural stability of the lipidated A1M-S1P versus ApoM-S1P complexes at near physiological conditions, we calculated the root mean square deviation (RMSD) for all backbone residues. The time evolution of the RMSD of the conformation of ApoM-S1P reached a plateau around 2.3 ± 0.3 Å (ApoM-S1P interface: 1.7 ± 0.3 Å), suggesting the stability of this complex. However, the overall system of A1M-S1P exhibited a much higher degree of conformational variability with RMSD values of 18.4 ± 4.6 Å likely due to the presence of a flexible linker that connects ApoA1 and ApoM domains. Nevertheless, the ApoM-S1P moiety in the A1M-S1P particle displayed an RMSD of 2.4 ± 0.2 Å and 2.2 ± 0.3 Å in monomers A and B, respectively. Both S1P interfaces remained highly stable (RMSD – monomer A ApoM-S1P Interface: 1.5 ± 0.4 Å; monomer B ApoM-S1P Interface: 1.9 ± 0.3 Å). To better evaluate the conformational diversity, we performed principal component analysis (PCA) of sampled conformations in the A1M particle to allow the reduction of high-dimensional structural space and to understand main collective motions. The ensemble of structures was clustered by similarity into 5 representative clusters (Fig. 2B and Movie S2). The resulting eigenvalue contribution of PCA (Fig. 2C) showed that the first seven principal components were responsible for 86.4% of the total mean-square displacement of atom positional fluctuations (Movie S3). This capacity of rapid interconversions of conformations agreed with the electron microscopy data (Fig. 1D).
Fig. 2. Molecular dynamics simulation study of A1M-S1P complex.
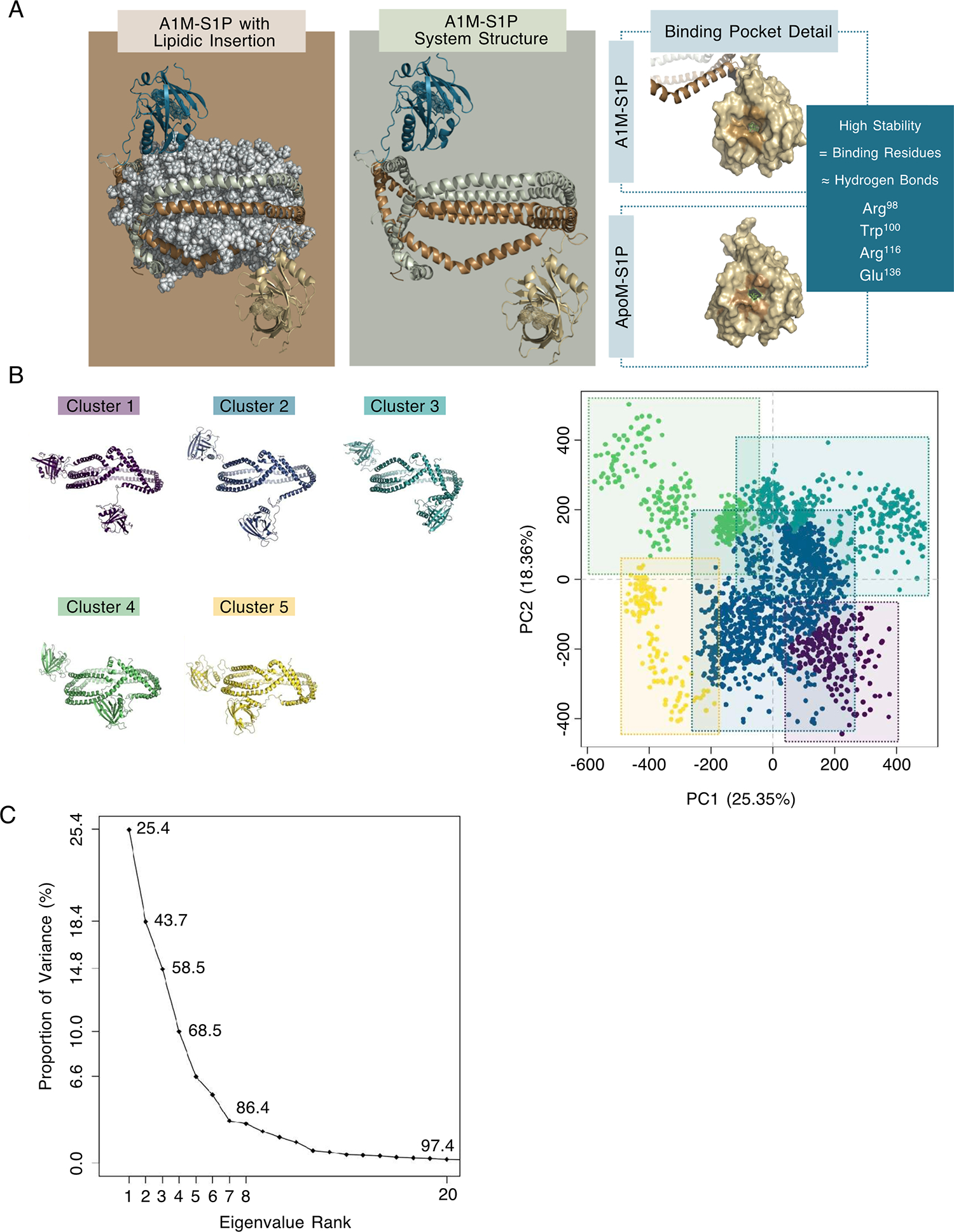
(A) Side view of A1M-S1P complex with and without the phospholipids, highlighting the S1P binding pocket in both the ApoM-S1P and ApoA1-fused systems. Monomer 1 - ApoA1 in sienna, linker in brown, and ApoM in beige. Monomer 2 - ApoA1 in sage, linker in turquoise, and ApoM in teal. Detail of the binding pocket in A1M-S1P and ApoM-S1P are shown in the right panel. (B) Projection of protein atoms along the top two dominant eigenvectors (eigenvector 1 - PC1 and eigenvector 2 – PC2) is responsible for over 40% of the dominant collective variances in the structure set, generating 5 clusters for which the three-dimensional structure of the most representative member of each cluster is shown. Permanence times: cluster 1 – 9.6%, cluster 2 – 52.9%, cluster 3 – 18.7%, cluster 4 – 12.2%, cluster 5 – 6.5%. (C) Distribution of the proportion of variance between the PCs.
Although we observed large-scale conformational changes of the fused A1M protein, the overall accessibility of the S1P binding interface was similar in both A1M-S1P and ApoM-S1P complexes. The ratio between the solvent accessible surface area (SASA) of interfacial residues on both A1M-S1P and ApoM-S1P was stable (close to 1, Fig. S2). Furthermore, the network of hydrogen bonds established between ApoM and S1P was also stably maintained in both complexes (Table S1). The more accessible conformational space explored by the fused protein led to greater movement of the polar S1P groups and replacement of the Arg143 contribution within this network by Arg98. Hydrogen network involvement in the S1P coupling is the main energetic barrier for S1P dissociation from the complex (24). Overall, analysis of the MD simulation data pointed to a stable binding of S1P to ApoM in both ApoM-S1P and lipidated A1M-S1P complexes. Further, the conformational diversity of ApoA1 and ApoM joined by a flexible linker in the A1M fusion protein may enable efficient action at cell surface S1P receptors.
A1M-S1P preferentially activates S1PR1 as a Gαi-biased agonist
The ability of the A1M-S1P complex to activate S1P receptors was studied using the real-time NanoBiT system that monitors receptor-induced heterotrimeric G protein dissociation and receptor-β-arrestin association (32). Temporal and dose-response analysis revealed A1M-S1P induced rapid and potent S1PR1-Gαi dissociation compared to ApoM-S1P and bovine serum albumin (BSA)-S1P, suggesting ApoM chaperone function was not impaired by fusion to ApoA1 or nanoparticle formation of the A1M-S1P particle (Fig. 3A and 3B). However, β-arrestin coupling to S1PR1 in response to A1M-S1P and ApoM-Fc-S1P was slower than BSA-S1P (Fig. 3C). Moreover, over a 4-log range of ligand concentrations, similar S1PR1-dependent activation of β-arrestin was observed for A1M-S1P, BSA-S1P, and ApoM-Fc-S1P (Fig. 3D). These data suggested that A1M-S1P, similar to ApoM-Fc-S1P, is a Gαi-biased activator of S1PR1. We also examined the ability of various chaperones to activate S1PR2 (Fig. 3E) and S1PR3 (Fig. 3F) by conducting β-arrestin coupling assays. We observed greater maximum stimulation of these receptors with BSA-S1P compared to either A1M-S1P and ApoM-Fc-S1P, suggesting that ApoM-bound S1P activates S1PR1 preferentially when compared to S1PR2 and S1PR3.
Fig. 3. A1M-S1P activates S1PRs in G-protein dissociation and β-arrestin coupling assays.
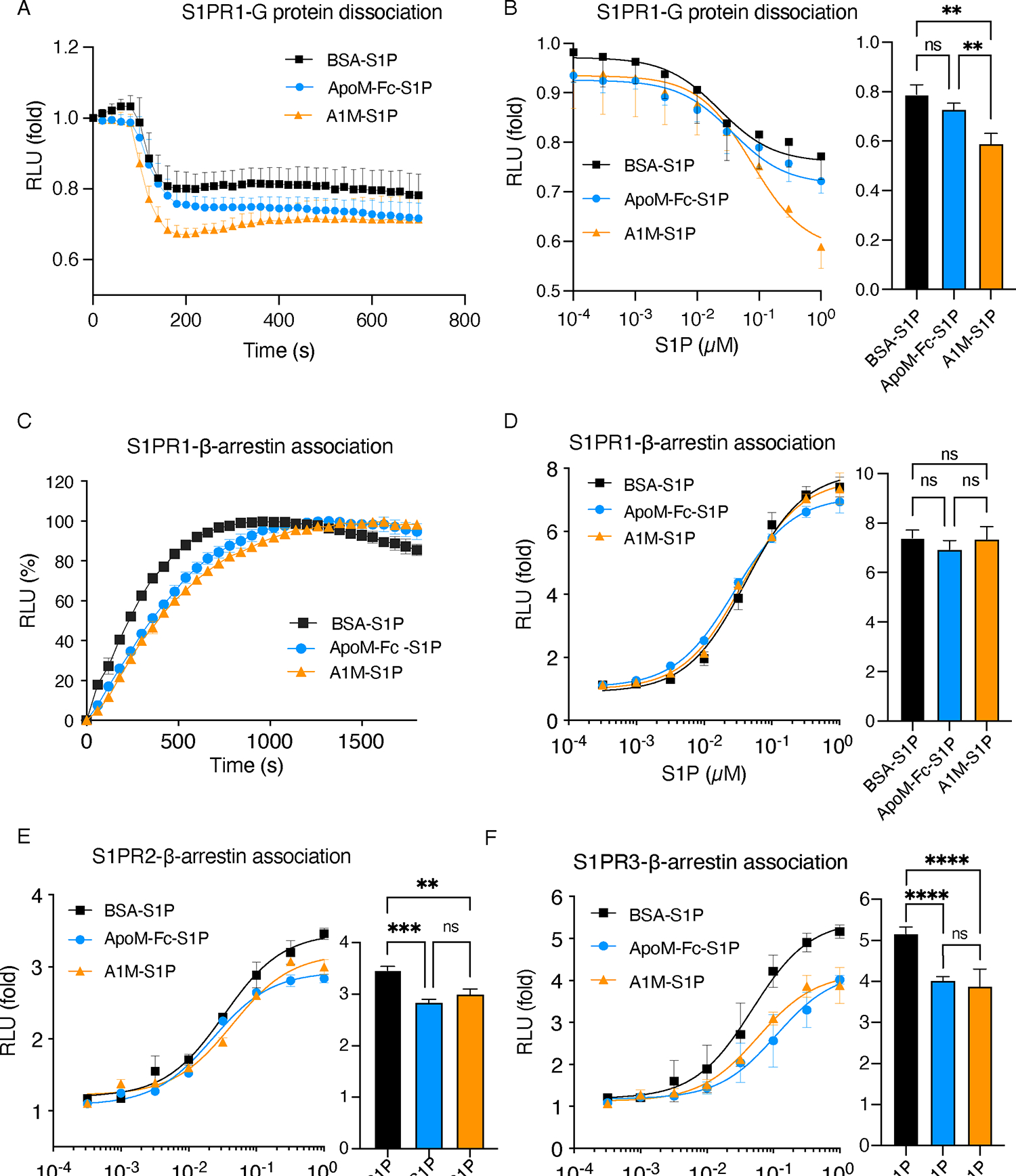
(A) Temporal analysis of S1P-induced S1PR1 activation in cells treated with 100 nM of S1P complexed with various chaperones through a NanoBiT G-protein dissociation assay (N = 3 biological replicates per group). (B) Dose-response analysis of S1PR1-dependent Gαi activation by bovine serum albumin-S1P (BSA-S1P), ApoM-Fc-S1P, or A1M-S1P by NanoBiT assay assessing G-protein dissociation (N = 3 biological replicates per group). (C) Temporal analysis of S1P-induced S1PR1 activation in cells treated with 100 nM of S1P complexed with various chaperones through a NanoBit β-arrestin association assay (N = 3 biological replicates per group). (D) Dose analysis of S1PR1-β-arrestin coupling induced by BSA-S1P, ApoM-Fc-S1P, or A1M-S1P by NanoBiT assay (N = 3 biological replicates per group). (E) Dose analysis of S1PR2-β-arrestin coupling induced by BSA-S1P, ApoM-Fc-S1P, or A1M-S1P by NanoBiT assay (N = 3 biological replicates per group). (F) Dose analysis of S1PR3-β-arrestin coupling induced by BSA-S1P, ApoM-Fc-S1P, or A1M-S1P by NanoBiT assay (N = 3 biological replicates per group). Data represent mean ± S.D. The bar graphs in (B), (D), (E), and (F) show the responses at 1 μM S1P. **P < 0.01, ***P < 0.001, ****P < 0.0001 by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
A1M-S1P stimulates endothelial barrier function
To determine the ability of lipidated A1M-S1P particles to regulate endothelial cell barrier function, trans-endothelial electrical resistance (TEER) analysis was performed using HUVECs (25). We performed a dose-response study of A1M-S1P (Fig. 4A), which showed similar potency of ApoM-Fc- and A1M-bound S1P. EC50 values of A1M-S1P and ApoM-Fc-S1P are 1.5 μg/mL and 2.2 μg/mL, respectively, which corresponded to ~ 21 and 29 nM S1P. Unloaded A1M and ApoM-Fc had no barrier stimulatory activity. This result suggests the equivalent functionality of ApoM as an S1P chaperone whether it is fused to the Fc or ApoA1 domains.
Fig. 4. A1M-S1P maintains endothelial barrier function in vitro and suppresses thrombin-induced barrier degradation.

(A) TEER analysis to measure barrier function was performed on HUVECs treated with A1M-S1P, ApoM-Fc-S1P or chaperone only (N = 3 biological replicates per group). (B) TEER analysis to measure barrier function was performed on HUVECs treated with Ang-1 (300 ng/mL), A1M-S1P (30 nM) or both (N = 3 biological replicates per group). (C and D) TEER analysis to measure barrier function was performed on HUVECs treated with APC (5 μg/mL) alone or with A1M-S1P (30 nM) (C) or ApoM-Fc-S1P (30 nM) (D). After 1 hour pretreatment, thrombin was added for an additional 2 hours. N = 3 biological replicates per group. The EC barrier index (above the baseline) and EC barrier degradation index (below the baseline) were analyzed by calculating the area under the curve of TEER value and are presented as means ± SD. ***P < 0.001, ****P < 0.0001 by ordinary one-way ANOVA with Tukey’s multiple comparisons test.
Angiopoietin-1 (Ang-1) is a polypeptide that stimulates endothelial barrier function and attenuates vascular leak during inflammation and thrombosis (33, 34). A1M-S1P and Ang-1 enhanced endothelial barrier function in an additive manner (Fig. 4B). Thrombin, a protease produced during blood clotting, activates G protein-coupled protease-activated receptors (PARs) to disrupt endothelial barrier function. S1P suppressed thrombin-induced barrier breach by activating S1PRs (35). We found that ApoM-Fc-S1P treated EC blocked thrombin-induced barrier degradation in a dose-dependent manner (Fig. S3A). In addition, ApoM-Fc-S1P and Ang-1 cooperated to suppress thrombin-induced barrier breach (Fig. S3B). In contrast to thrombin, activated protein C (APC) interacts with endothelial cell surface receptors such as thrombomodulin and the endothelial protein C receptor (EPCR) to enhance EC barrier function (36). We combined sub-optimal concentrations of APC (5 μg/mL) and S1P (30 nM) either complexed with A1M-S1P (Fig. 4C) or ApoM-Fc-S1P (Fig. 4D) to test if these combinations blocked thrombin-induced HUVEC barrier breach. Combination of suboptimal APC and A1M-S1P or ApoM-Fc-S1P blocked thrombin-induced barrier breach ~50% in a synergistic manner (Fig 4C and 4D). Together these data reveal that A1M-S1P, Ang-1, and APC cooperate to protect the endothelial barrier.
A1M binds to the stable prostacyclin analog iloprost, which cooperates with S1P to enhance barrier function
Endothelial cell derived PGI2 acts on the prostacyclin receptor (IP) to activate the Gαs/adenylate cyclase/cAMP pathway to suppress platelet aggregation, vascular smooth muscle dilation and inflammatory responses (37). IP activators also suppress inflammation-induced vascular leak by activating the cAMP/EPAC/Rap1 pathway (38). Moreover, the lability of PGI2 due to autohydrolysis is inhibited by HDL association (13). Therefore, we tested if A1M supplemented with iloprost, a stable PGI2 analog, would possess EC protective effects (39). We found that lipidated A1M-iloprost induced an IP/cAMP-responsive CREB luciferase reporter activity (Fig. 5A). 6 nM of iloprost showed an equivalent response to 1 μg/mL of A1M-iloprost. These data suggest that A1M-bound iloprost is biologically active and activates the IP receptor potently.
Fig. 5. Production and characterization of A1M-iloprost.

(A) Cells expressing a CREB-luciferase reporter with prostacyclin receptor (IP) were stimulated with vehicle or A1M-iloprost for 8 hours, and cell lysates were assayed for luciferase activity (N ≥ 3 biological replicates per group). 0 nM was used as a reference control and was normalized to 1. Data are presented as means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by ordinary one-way ANOVA with Dunnett’s multiple comparisons test. (B) TEER analysis to measure barrier function was performed on HUVECs treated with A1M-S1P (8 μg/mL with 100 nM S1P), A1M-iloprost (25 μg/mL with 200 nM iloprost), or both (N = 4 biological replicates per group). Data are presented as means ± SD. ****P < 0.0001 by ordinary one-way ANOVA with Tukey’s multiple comparisons test. (C) Human platelets were assayed for aggregation in response to the thrombin receptor mimetic peptide SFLLRN in the presence of vehicle control, A1M-iloprost (10 nM), A1M-S1P (200 nM), or in combination. Representative data from 3 biological replicates per group.
Because the cAMP/EPAC/Rap1 pathway stimulates endothelial barrier function, we tested if A1M-iloprost stimulated endothelial barrier function to a similar degree as low dose A1M-S1P (30 nM S1P) (Fig. 5B). Although effective as a single agent, combining A1M-iloprost and A1M-S1P provided additive barrier protection (Fig. 5B). Similarly, free iloprost (200 nM) cooperated with HDL-S1P (Fig. S4A) and the S1PR1-selective agonist AUY954 (Fig. S4B) to enhance endothelial barrier function in an additive manner.
Prostacyclin signaling through the IP receptor inhibits platelet aggregation, thereby inhibiting thrombosis (40). A1M-iloprost potently inhibited platelet aggregation induced by the thrombin receptor activating peptide SFLLRN (Fig. 5C). In contrast, A1M-S1P did not inhibit platelet aggregation to the same degree. Dose-response studies indicated an EC50 of ~ 11 nM for free iloprost and ~9 nM for ApoA1-associated-iloprost (Fig. S5A and S5B), suggesting a complete retention of potency when iloprost is associated with HDL-like nanodiscs. These data suggest that A1M supplemented with iloprost can function both as an anti-thrombotic as well as an endothelial barrier protective agent.
A1M attenuates TNFα-induced NF-κB activity and ICAM-1 expression
Intercellular adhesion molecule 1 (ICAM-1) is a cell surface glycoprotein and an adhesion receptor that regulates leukocyte recruitment from circulation to sites of inflammation (41). HDL-bound S1P attenuates TNFα-induced ICAM-1 expression (16, 42). In contrast, albumin-bound S1P did not suppress ICAM-1 expression, suggesting that S1PR1 agonism alone is not sufficient to suppress cytokine inflammatory responses. The ApoA1 moiety of HDL has been suggested to engage endothelial cells to induce NO release (43) and suppress TLR4- and TNFα-induced activation of the proinflammatory transcription factor NF-κB in myeloid cells (10). Here, we showed that ApoA1, A1M, and lipidated A1M-S1P inhibited TNFα-induced NF-κB activity ~25% in a luciferase-based NF-κB transcriptional reporter assay (Fig. 6A). Moreover, A1M lipoprotein particles, regardless of whether the bound lipid mediator was S1P or iloprost, suppressed the TNF-α−induced increase in ICAM-1 expression in HUVECs (Fig. 6B). In contrast, ApoM-Fc-S1P did not inhibit NF-κB activity or ICAM-1 up-regulation in response to TNF-α. Moreover, A1M-S1P induced cholesterol efflux in PMA-treated THP-1 cells (Fig. 6C), suggesting that similar to HDL particles, A1M-S1P can efflux cholesterol from cells by interacting with ATP-binding cassette transporters ABCA1 and ABCG1. This property of A1M likely promotes suppression of ICAM-1 expression.
Fig. 6. A1M attenuates TNFα-dependent inflammation.

(A) HMEC-1 cells expressing an NF-κB-luciferase reporter were assayed for TNFα-induced NF-κB reporter activity in the presence of ApoA1, A1M, A1M-S1P, and ApoM-Fc-S1P (N = 3 biological replicates per group). Data are presented as means + S.D. **P < 0.01, ***P < 0.001, ****P < 0.0001 by ordinary two-way ANOVA with Tukey’s multiple comparisons test. TNFα is the reference group. (B) HUVECs were starved for 1 h, pre-treated for 10 minutes with ApoM-Fc-S1P (100 nM), iloprost (200 nM), both ApoM-Fc-S1P and iloprost, A1M (200 μg/mL), A1M-S1P (200 μg/mL), or A1M-iloprost (200 μg/mL) and induced with TNFα (10 ng/mL) for 5 hours. Lysates were subjected to immunoblot analysis for ICAM-1. Quantification of immunoblots was analyzed from 3 biological replicates per group. Data are presented as means ± S.D. *P < 0.05, **P < 0.01, ****P < 0.0001 by ANOVA with post-hoc Holm-Sidak’s multiple comparisons test. (C) Cholesterol efflux in response to human HDL (hHDL), human ApoA1 protein (hApoA1), and bacterial A1M (bA1M) in PMA-induced THP-1 cells (N ≥ 5 independent experiments). Data are presented as means + S.D. *P < 0.05, ***P < 0.001, ****P < 0.0001 by one-way ANOVA with Dunnett’s multiple comparisons test.
A1M suppresses inflammation in vivo
We next examined if A1M-S1P can impact the inflammatory responses in a murine sterile inflammation model. Thioglycolate injection into the peritoneum induces rapid neutrophil extravasation, resulting in acute inflammation (peritonitis) (44). We found that the intraperitoneal injection of lipidated A1M-S1P particle, but not non-S1P loaded A1M, markedly suppressed neutrophil influx into the peritoneum (Fig. 7A). Furthermore, peritoneal lavage fluid in A1M-S1P treated animals contained reduced amounts of inflammatory factors, including complement C5a, G-CSF, soluble ICAM-1, IL-6, IL-16, M-CSF and CCL2 (Fig. 7B). These data provide evidence for an anti-inflammatory and EC barrier protective functions of A1M-S1P in vivo.
Fig. 7. A1M-S1P suppresses inflammation in a murine peritonitis model.
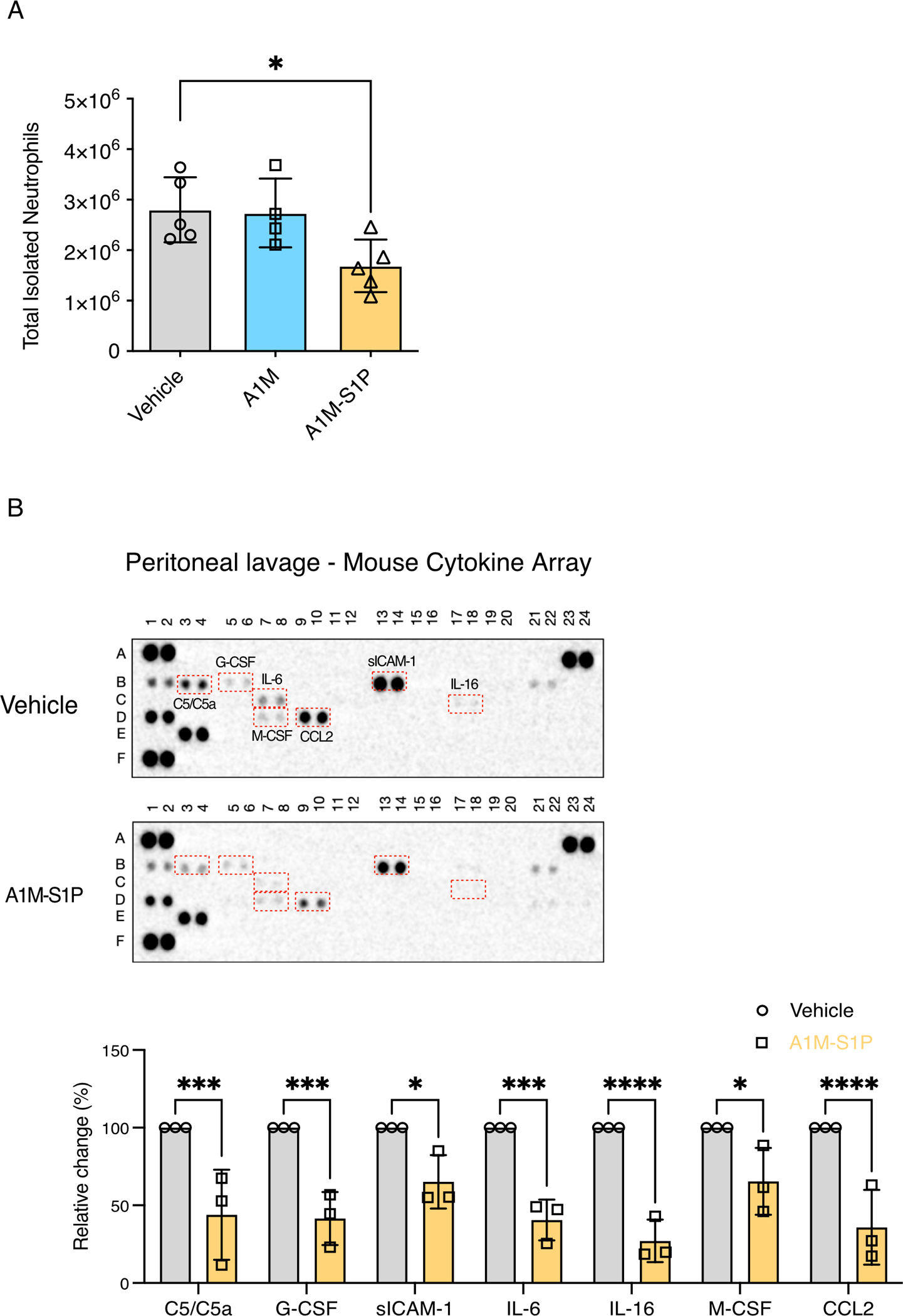
(A) Mice were treated intraperitoneally with thioglycolate and either PBS, A1M (0.2 mg/animal), or lipidated A1M-S1P (0.2 mg/animal). Peritoneal cells were collected at 4 h and analyzed by flow cytometry. N = 4–5 mice per group. Data are presented as means ± S.D. *P < 0.05 by one-way ANOVA with Dunnett’s multiple comparisons test. Vehicle was the reference group. (B) Peritoneal lavage supernatant from mice treated as described in (A) was assayed by cytokine array analysis. Statistical tests were done on analytes with changes in abundance after normalization (control was set at 100%). N = 3 mice per group. Data are presented as means ± S.D. *P < 0.05, ***P < 0.001, ****P < 0.0001 by ANOVA with Sidak’s multiple comparisons test.
To determine if systemic administration of nascent A1M suppresses the inflammatory response, we tested if intravenously injected recombinant A1M acquires S1P and gets lipidated into HDL particles in vivo. We constructed (Fig. S1) and purified a bacterial A1M fusion from E.coli (bA1M) (Fig. S6A). Nascent A1M was loaded with S1P and lipidated (bA1M-S1P) (Fig. S6B). Similar to CHO cell-derived A1M, bA1M-S1P induced barrier function in HUVECs in a dose-dependent manner (Fig. S6C). When bA1M was injected intravenously into mice, it was rapidly lipidated and appeared in the HDL fraction in plasma within 24 hours (Fig. S6D and S6E). In addition, S1P content of plasma and HDL fractions were increased by 2.5 and 4.5-fold, respectively (Fig. S6F and S6G), suggesting that intravenously injected nascent A1M protein picked up endogenous S1P and was lipidated into HDL particles.
We tested if systemic A1M administration influenced LPS-induced systemic inflammatory response in mice. C57Bl/6 mice were dosed intravenously with bA1M before being intraperitoneally injected with LPS. Although A1M treatment did not change the murine sepsis score (Fig. 8A), the LPS-induced decrease in body temperature was partially reversed by bA1M treatment (Fig. 8B). Similarly, plasma IL-6 levels were suppressed by bA1M (Fig. 8C). A1M-injected mice contained significant levels of circulating A1M and 2.13-fold more plasma S1P (Fig. 8D and 8E). These data suggest that the ApoA1 moiety of A1M suppressed systemic inflammatory responses induced by LPS in vivo.
Fig. 8. The ApoA1 moiety of bA1M suppresses the systemic inflammatory response induced by LPS.

(A to C) Mice were injected intravenously with 40 mg/Kg of bA1M or PBS (vehicle) for 1 hour to allow for A1M lipidation in vivo before being injected intraperitoneally with 10 mg/Kg of LPS or saline. Groups were saline (N ≥ 6 mice), LPS alone (N ≥ 14 mice), and LPS with A1M (N ≥ 10 mice). Mice were assessed for sepsis score (A), body temperature (B), and plasma IL-6 amounts (C) at 12 hours post-LPS injection. Data are presented as means ± S.D. ***P < 0.001, ****P < 0.0001 by one-way ANOVA with Tukey’s multiple comparisons test. (D) 0.5 μl of plasma from mice injected with LPS alone (10 mg/Kg) or LPS and bA1M (40 mg/Kg) at 12 hours post-injection were subjected to Western blot analysis for ApoM. Each lane represents an individual mouse. (E) Plasma S1P content in mice injected with saline, LPS alone, or LPS and bA1M at 12 hours post-injection were analyzed by LC-MS/MS (N = 4 mice per group). Data are presented as means ± S.D. ***P < 0.001 by one-way ANOVA with Tukey’s multiple comparisons test.
Discussion
Vascular endothelial dysfunction during infections, trauma, metabolic diseases and aging has been identified as a primary event that drives chronic pathology. Endothelial injury drives vascular leak and the subsequent activation of the innate immune response. Circulating HDL particles have vascular endothelial protective properties (6, 11). Specifically, ApoA1, the principal structural protein of HDL that carries out reverse cholesterol efflux, attenuates cytokine driven inflammatory responses (45). In addition, HDL binds to and stabilizes endothelial-derived prostacyclin, which inhibits platelet activation and thrombosis, blocks neutrophil adherence to the endothelium, and enhances endothelial barrier function (46). We previously found that the HDL-associated protein ApoM is the physiological chaperone for S1P, which enhances endothelial barrier function (14, 22) and protects from multiple organ injuries.
We used the vascular endothelial protective properties of HDL to create a recombinant A1M fusion protein. A1M had similar biochemical properties to HDL particles while retaining stable S1P binding, which is intrinsic to ApoM. Transmission electron microscopy analysis suggested that the A1M-S1P complex exhibited conformational heterogeneity of 10–12 nm size HDL-like particles. It is likely that the flexible linker between the ApoA1 and ApoM components enables variable orientations of those components in the A1M-S1P complex. Only 5% of endogenous plasma-borne HDL particles from human and mice contain ApoM (14). The ApoM content of HDL was needed for S1P association. Given that plasma ApoM levels are decreased by pathological conditions such as sepsis, diabetes, and viral infections, as well as normal aging (18, 47–50), A1M designer HDL particles that contain ~20x more ApoM than endogenous HDL may be able to replenish ApoM-S1P during pathological conditions.
MD simulation analysis of the ApoM-S1P and A1M-S1P complexes led to several insights that either added to or concurred with the experimental data. As previously reported, the ApoM-S1P complex is stable, with the ligand S1P buried in the internal binding pocket of the lipocalin fold of ApoM (14, 24, 25). The exterior lid of the ApoM moiety, which contains charged and polar residues, are thought to restrict dissociation of S1P from ApoM. Our MD simulation study showed that A1M-S1P complex maintains the stable S1P binding property of the ApoM protein. However, the flexible linker between ApoA1 and ApoM enables high conformational flexibility of the ApoM-S1P complex in the A1M molecule. These studies suggest that A1M-S1P should allow both ApoA1 and ApoM portions of this fusion protein to function independently to regulate cellular functions. We speculate that the ApoA1 interaction with its receptors on endothelial cells, such as scavenger receptor-B1 (51), would dock A1M-S1P complex at the endothelial cell surface. Conformational flexibility of the linker and the stability of the ApoM-S1P complex would then allow productive interaction with the S1P receptors on cell surface to induce intracellular signals.
HDL particles that contain ApoM exhibit anti-inflammatory properties of ApoA1 as well as the barrier protecting properties of S1P. Even though A1M-S1P can activate both Gαi and β-arrestin coupling, it shows a marked Gαi-biased signaling property. Although β-arrestin activation normally leads to termination of S1PR1 signaling, Gαi-biased A1M-S1P signaling is expected to activate EC protective pathways in a sustained manner, which we confirmed by TEER analysis. Additionally, we previously showed that A1M-S1P activates endothelial S1PR1, S1PR2, and S1PR3, but preferentially activated the barrier protective S1PR1 rather than the barrier disruptive S1PR2 (25). Both A1M and A1M-S1P were anti-inflammatory, reducing both TNFα-induced NF-κB activation in a reporter assay and inhibiting downstream inflammatory ICAM-1 expression in endothelium. We observed that other S1P chaperones ApoM-Fc and albumin had no inhibitory activity, suggesting that the ApoA1 moiety is the primary inhibitor of TNFR signaling and inflammatory responses.
Lipidated ApoA1 or A1M can bind to the stable PGI2 analog iloprost, consistent with the previous findings that PGI2 association with HDL or ApoA1 prolongs the half-life of this unstable lipid mediator by suppressing autohydrolysis (52, 53). We demonstrated that A1M/iloprost potently activated PKA-dependent CREB-signaling, promoted endothelial barrier function and inhibited human platelet aggregation. Our studies also show that A1M-bound iloprost cooperated with A1M-S1P to stimulate EC barrier function, suggesting that the IP/Gαs/cAMP/EPAC/Rap1 and S1PR1/Gαi/Rac1 pathways converge to assemble EC cell-cell junctions and enhance barrier function. Free iloprost has been previously suggested to be a protective factor in EC barrier integrity protection under endotoxic conditions (54). Indeed, we demonstrated the in vivo functionality of A1M-S1P in suppressing neutrophil infiltration into the peritoneum and inflammatory mediator secretion in a sterile peritonitis model of inflammation. The suppression of neutrophil influx by A1M-S1P but not A1M alone suggests that the ability of A1M-S1P to suppress inflammation-induced vascular leak is critical in the recruitment of neutrophils into the peritoneum. In the LPS model of endotoxemia, A1M suppressed cytokine storm in an ApoA1-dependent manner. These results suggest that A1M could be used not only to enhance endothelial barrier function but also to suppress inflammatory cytokine storm induction in vivo, which occurs in several pathological states.
Inflammatory activation of the endothelium leads to thrombin formation, which not only disrupts the endothelial barrier, but also induces thrombosis (11, 55). This is counteracted by the activated protein C (APC) pathway, which preserves the EC barrier and suppresses thrombin formation by the formation of APC and its cell surface functions. We demonstrated that A1M-S1P blocked thrombin-induced barrier disruption and enhanced the barrier-protective activity of APC. Moreover, we showed that A1M-iloprost inhibited thrombin-driven platelet aggregation in vitro. Thus, A1M-bound S1P and iloprost would be predicted to suppress pathologic vascular permeability and thrombosis.
Together, these results argue that recombinant A1M-bound S1P and prostacyclin provide a three-pronged approach to recapitulate the endothelial protective effects of HDL by enhancing endothelial barrier function through S1PR1 agonism, inhibiting inflammatory cytokine signaling with ApoA1, and dampening platelet activation and thrombotic responses by prostanoid receptor signaling. In contrast to small molecule S1PR functional antagonists, S1P chaperone-based receptor activation does not lead to inhibition of lymphocyte egress (25). Therefore, we suggest that A1M administration would not lead to lymphopenia and immune suppression. This approach has potential therapeutic utility in inflammatory and thrombotic states in which endothelial damage is driven by infectious pathogens, vascular reperfusion injury after trauma or in organ transplantation, as well as more chronic vasculopathies associated with atherosclerosis, diabetes, and autoimmunity.
Materials and Methods
Creation of the A1M fusion constructs
The A1M fusion was constructed using plasmids for murine ApoA1 (MR203500) and murine ApoM (MR201811) obtained from OriGene. The cDNA for ApoA1 was amplified to include the endogenous Kozak sequence and ORF of ApoA1 and to replace the stop codon with a codon for glycine. The cDNA for ApoM was amplified by sequential PCR to include a linker sequence encoding one copy of a five amino acid sequence linker (GGGGS) added to the 5’-end of ApoM, to remove the signal peptide (amino acids 1–20) and to insert a glycine codon and a 6 x histidine sequence before the stop codon. The two PCR products were linked after NOT1 digestion and the final fusion PCR product was cloned into the pCDH-puro (Invitrogen) expression vector. Primers used for ApoA1 cloning were 5’- TTTTCTAGAGAGGAGATCTGCCGCCGCGATCG-3’ (forward) and 5’-TTTGCGGCCGCGTACGTCTGGGCAGTCAGAG-3’ (reverse). Primers used for ApoM cloning were 5’-GGTGGAGGTGGATCTAATCAGTGCCCTGAGCACAGT-3’ (forward) and 5’GATGGTGATGTCCCTTGCTGGACAGCGGGCAGG-3’ (reverse) or 5’-TTTGCGGCCGCTTGGTGGAGGTGGATCTAATCAGTG-3’ (forward) and 5’-TTTGGATCCTCAGTGATGGTGATGGTGATGTCCCTTGCTGG-3’ (reverse).
Expression and purification of the A1M fusion protein from CHO-S cells
The resulting pA1M plasmid was transfected into adherent CHO-S cells (A11364–01, Invitrogen) using polyethylenimine (PEI, Sigma). Positive transfectants were obtained by selection in Puromycin (30 μg/mL, GIBCO) for 4 days. Cells were tested for expression of the fusion protein by immunoblot analysis for ApoA1 (Abcam), ApoM (Abcam), and His-tag (Santa Cruz). The drug-selected recombinant CHO-S cells were adapted to serum-free suspension culture using CD FortiCHO medium (Thermo Fisher). For large-scale cultures, cells were suspended at 3–5 × 105 cells per mL and maintained in culture to 2–4 × 106 cells/mL in a spinner flask (Corning). To collect secreted A1M protein from the conditioned medium, cells were removed from culture by centrifugation at 800 x g for 10 minutes at 4°C. The culture supernatant was further clarified by ultracentrifugation at >100,000 x g for 30 minutes at 4°C. The resulting supernatant was incubated with Ni-Sepharose beads (HisPur Ni-NTA resin, Thermo Fisher) at a final concentration of 2 mL of packed beads per 500 mL of culture overnight at 4°C. Next, beads were concentrated by centrifugation at 10,000 x g for 5 minutes at 4°C. Beads were washed with 50 volumes of His-wash buffer (20 mM Tris pH 8, 400 mM NaCl, 10 mM imidazole) until the flow through did not contain detectable protein. Protein was eluted from the column in the elution buffer (20 mM Tris pH 8, 400 mM NaCl, 300 mM imidazole). The resulting protein fractions were assayed using Bio-Rad protein reagent and were concentrated with Amicon Ultra-15 filters (Millipore Sigma). The final purified protein preparation was analyzed by SDS-PAGE and stained with Coomassie Brilliant Blue R-250 (Bio-Rad).
Preparation of S1P, loading S1P onto A1M, mass spectrometry and electron microscope (EM) analysis
S1P (1 mg) (Avanti Polar lipids) was resuspended in 13.4 mL of methanol and maintained at 37°C for 12 hours to achieve complete suspension. The solution was dispersed into 134 μl aliquots and dried under vacuum for 45 minutes to 1 hour at 37°C. Dried S1P was stored at −20°C until use. We previously described a method for loading S1P onto recombinant ApoM-Fc (25). Essentially, purified protein was suspended in PBS at 1 mg/mL (~20 μM) and mixed with 160 μM S1P by gentle pipetting. The sample was subjected to 3 × 30 s sonication in a bath sonicator and allowed to incubate for >24 hours by nutation. The resulting product was subjected to FPLC to separate the free S1P from the A1M-S1P protein complex and the resulting protein fractions were concentrated using Amicon Ultra-15 filters. A1M-S1P was lipidated as below. S1P was loaded onto albumin as described (56). Essentially, a 0.4% fatty acid free BSA (Sigma) solution in PBS was added to dried S1P and subjected to bath sonication for 3 cycles for 1 minute/cycle and maintained at 4°C for 24 hours. Mass spectroscopy-based quantitation of S1P was performed as described previously (57). S1P-bound ApoM or A1M proteins or plasma from mice injected with these proteins was extracted in methanol (20:1 vol/vol methanol:analyte). For EM analysis, A1M samples were diluted to 0.02 mg/mL and 2.5 μL was applied to glow-discharged copper EM grids covered with a thin layer of carbon film. Grids were stained with 1.5% (w/v) uranyl formate, blotted, and allowed to air dry. Negatively stained EM grids were imaged on a Tecnai T12 electron microscope (Thermo Fisher Scientific) operated at 120 kV at a nominal magnification of 67,000 x using a 4k x 4k CCD camera (UltraScan 4000, Gatan), corresponding to a pixel size of 1.68 Å. EM images were binned by two (3.36 Å per pixel), and particles were selected and subjected to two-dimensional classification (58).
Lipidation of A1M-S1P and A1M-iloprost
We employed a standard sonication and thermal cycling method for creating lipidated A1M as described (25, 28). A combination of 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) and 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG) (7:3 molar ratio) (Avanti) was resuspended in chloroform: methanol (95:5) and distributed into Eppendorf tubes and dried under vacuum. The lipid mixture was resuspended in PBS buffer, heated at 37°C for 5–10 minutes, and sonicated for 20 seconds to create a turbid lipid suspension. To generate lipidated A1M-S1P, S1P-loaded A1M was added to the lipid slurry (100:1 mol/mol lipid:protein). To generate A1M-iloprost, iloprost (Cayman Chemical) was dissolved in DMSO and added to the lipid solution at a final concentration of 1 mM. Purified A1M protein or human ApoA1 (Sigma) was resuspended in PBS and added to the lipid-iloprost slurry (100:1 mol/mol lipid:protein). Next, the solution was subjected to continuous bath sonication at 37°C for 30 min until the suspension exhibited clarification. The resulting lipidated solution was subjected to an FPLC purification (25), and relevant fractions corresponding to recombinant HDL nanoparticles were collected and concentrated by Amicon Filters.
TEER analysis of HUVEC
To study endothelial barrier function, transendothelial electrical resistance (TEER) on primary human umbilical vein endothelial cells (HUVECs; PCS-100–013, ATCC) was performed using a 96-well ECIS system (96W10idf PET array, Applied BioPhysics), as described previously (25). Essentially, HUVECs were maintained in HGM medium (M199 medium supplemented with 10% FCS, 1:100 penicillin-streptomycin, 8 mM glutamine, 2.5 U/mL heparin) and endothelial cell growth supplement (59) and on fibronectin (2 μg/mL in saline) coated plates. Wells were coated with fibronectin for 30 minutes at room temperature. HUVECs were harvested, resuspended in HGM medium at a cell density of 2.5–3 × 104 cells/well, and allowed to adhere overnight. Prior to analysis, culture media was removed and replaced with M199 media supplemented with 1% FCS and penicillin-streptomycin/glutamine for 30 minutes. For stimulation studies, either iloprost (Cayman Biochemicals), ApoA1-iloprost, A1M-iloprost, A1M-S1P, or ApoM-Fc-S1P were added to cultures, and TEER studies were performed from 3–24 hours. To study thrombin-induced endothelial barrier degradation, thrombin (Millipore Sigma) was used at a final concentration of 1 U/mL. Angiopoietin-1 (Ang-1, R&D systems) and activated protein C (APC, Enzyme Research laboratories) were used at a final concentration of 300 ng/mL and 5 μg/mL, respectively. For initial studies, Ang-1 was evaluated alone and in combination with ApoM-Fc-S1P. For thrombin studies, Ang-1, ApoM-Fc-S1P, and thrombin were co-added at the initiation of the study. For APC experiments, APC alone or in combination with A1M-S1P was added to the culture for 1 hour prior to the addition of thrombin, and TEER analysis was performed for an additional 2–8 hours. Data from TEER studies were analyzed using GraphPad Prism 7 (GraphPad Software, San Diego, CA). The endothelial barrier index (positive values) and endothelial barrier degradation (negative values) were measured by analyzing the area under the curve (AUC) of normalized TEER values for a given period of time.
Nanobit analysis of S1P receptor activation
The NanoBiT system employs split luciferase consisting of small and large fragments (SmBiT and LgBiT, respectively) and was previously utilized to monitor interactions of GPCRs with β-arrestin or specific Gα/βγ complexes in response to GPCR activation (32). This system was used to characterize A1M nanoparticles containing S1P. Briefly, HEK293A cells (R70507, Invitrogen) maintained in DMEM (GIBCO) supplemented with 10% FCS and penicillin-streptomycin, were dispersed into 6 well plates and allowed to adhere overnight. For functional studies, cells were transfected with the appropriate combinations of reporter plasmids. S1PR1-SmBiT and LgBiT-β-arrestin fusion proteins or Gαi-SmBiT combined with Gβ1 and LgBiT-Gγ1 were employed as described previously (32). After 24 hrs, cells were harvested, resuspended with the luciferase substrate coelenterazine (Cayman Chem; 50 μM), dispersed into white opaque-bottom 96-well plates (Greiner) and maintained at room temperature for 2 hours to quench the background. S1P-containing samples were added by multi-channel pipetting and the plate was immediately analyzed for luminescence for 30 minutes in a SpectraMax L 96-well plate reader (Molecular Devices). Data was integrated as described (32).
Inhibition of TNF-α-dependent NF-κB activation
TNFα-dependent NF-κB signaling was determined using an NFκB-Luciferase based reporter assay system (pGL4.32[luc2P/NF-κB-RE/Hygro]; Promega). We created a stable NFκB reporter cell line in human microvascular endothelial cells (HMEC-1; CRL-3243, ATCC). HMEC-1 cells were maintained in 10% FCS/penicillin-streptomycin MCDB media supplemented with 2 mM L-glutamine (Sigma), EGF (2 ng/mL; R&D systems) and hydrocortisone (1 ng/mL; Sigma). The 5 ×105 reporter cells were distributed to 12 well plates and allowed to adhere for 24 hours. Media was replaced with MCDB media supplemented with 1% FCS and L-glutamine (2 mM). Cells were pre-treated with either media alone, purified human ApoA1 (Sigma), A1M, A1M-S1P, ApoM-Fc-S1P, or BSA-S1P for 10 minutes and 2 ng/mL of TNFα (R&D Systems). Samples were extracted using cell lysis buffers (Promega), luciferin substrate was added, and the plates were measured for luminescence for 8–15 minutes in a SpectraMax L 96-well plate reader (Molecular Devices).
Inhibition of TNF-α-induced ICAM-1 expression in HUVECs
24 hours after plating, HUVECs were starved in M199 media supplemented with 1% FCS, 8 mM glutamine, and 1X penicillin-streptomycin. After 30 minutes, cells were pre-treated for 10 minutes with media, ApoM-Fc-S1P (100 nM S1P), iloprost (100 nM), A1M (200 μg/mL), A1M-iloprost (200 μg/mL with 100 nM iloprost) or A1M-S1P (200 μg/mL with 100 nM S1P) or in combinations and subsequently treated with TNF-α (10 ng/mL). After 5 hours, cells were lysed with cell lysis buffer (TBS-T; 20 mM Tris-pH 8, 160 mM NaCl, 1% Triton, 1X protease inhibitor cocktail (Sigma)). Cell lysates were collected and centrifuged at 20,000 x g for 5 minutes at 4°C. The supernatant extracts were analyzed by 10% SDS-PAGE, transferred to nitrocellulose membrane (Bio-Rad), blocked in 5% milk, and probed with antibodies to ICAM-1 (G-5, 1:1,000, sc-8439, Santa Cruz Biotech) and actin (C-2, 1:5,000, sc-8432, Santa Cruz Biotech). Blots were developed with appropriate secondary antibodies linked to HRP and visualized by chemiluminescence (Immobilon Western, EMD Millipore) using a ChemiDoc Imaging (Bio-Rad) or Azure Imaging Systems (AZI600–1, Azure Biosystems).
Iloprost-induced activation of the CREB–luciferase signaling
To evaluate the activity of lipoprotein-bound iloprost to activate the prostanoid receptor (IP), we used the downstream cAMP-dependent CREB-luciferase reporter system (pGL4.29 [luc2P/CRE/Hygro]; Promega). HEK293T (CRL-3216, ATCC) cells were maintained in DMEM supplemented with 10% FCS and penicillin-streptomycin. Cells were plated at 1 × 105 cells/well in 96-well plates and allowed to adhere for 24 hours. Media was replaced, and cells were transfected using PEI (0.5 μg/well) with either 0.1 μg/well of reporter plasmid alone or co-transfected with pCDH-IP receptor (0.1 μg/well). After 24 hours, media was replaced, and cells were incubated with media containing either vehicle, iloprost, or A1M-iloprost by titration for 8 hours at 37°C. Samples were lysed, luciferin was added, and luminescence was measured for 30 minutes in a SpectraMax L 96-well plate reader (Molecular Devices).
Inhibition of human platelet aggregation in vitro
Human studies were approved by the institutional review board of the Beth Israel Deaconess Medical Center (PI: R. Flaumenhaft). Whole blood samples were drawn from healthy donors in the presence of 10% sodium citrate (Sigma, S577–50mL) and were spun at 168 xg using Beckman centrifuge (GS-6K) for 20 min. The top platelet-rich plasma layer was collected and rested in a 37°C water bath for 30 min. The platelet-rich plasma layer was diluted with one-fifth volume of acid-citrate-dextrose buffer, and prostaglandin E1 (PGE1) was added to a final concentration of 0.15 μM. The mixture was spun in a conical-bottom tube at 2,000 g for 10 min, and the platelet pellet was collected and suspended in a pre-warmed HEPES-Tyrode-Glucose Buffer (HTG). The platelet concentration was determined and adjusted to ~2.5 × 105/μL with HTG on a Hematology cell counter System (Drew Scientific, 850FS). Light transmission aggregometry was used to evaluate platelet responses to agonists and antagonists. In a 4-channel aggregometer (Platelet ionized calcium aggregometer, Chronolog Corp Model 660), platelets were stirred in a cuvette at 37°C. Human platelets were incubated with A1M-iloprost, ApoA1-iloprost, or A1M-S1P alone or in combination for ~10 min before adding the thrombin receptor (PAR-1) agonist SFLLRN peptide (2 μM). Data were analyzed using AGGRO/LINK software package Ver 5.2.5 and Microsoft Office Professional Plus 2013.
Neutrophil Influx and cytokine content of peritoneal lavage fluid in thioglycolate-induced sterile peritonitis
C57Bl/6 mice were obtained from Jackson Labs (RRID: IMSR_JAX:000664, Bar Harbor, Maine). All in vivo experiments were performed according to an approved experimental protocol (1770) by IACUC at Boston Children’s Hospital. Mice were injected with 2 mL of 3% thioglycollate into the peritoneal cavity as described (44). One hour after thioglycolate induction, mice were intraperitoneally injected with either PBS (vehicle), or 200 μg of either A1M or A1M-S1P. After 4 hours, resident and infiltrating cells were isolated from peritoneal lavage by flushing with HBSS. Cells were washed, counted, and stained for flow cytometry using the neutrophil marker Ly6G-PE (1A8, 127608, BioLegend), gating for side-scatter and PE+ cells.
Approximately 3.5 mL of peritoneal lavage fluid was collected from each mouse after thioglycolate induction (see above). After cell clarification by centrifugation at 300 x g for 5 min, 1 mL aliquots of fluid were prepared and immediately frozen in liquid N2. 500 μL of peritoneal fluid was analyzed for cytokine expression using the Proteome Profiler Mouse Cytokine array Kit, Panel A (ARY006; R&D Systems), following the manufacturer’s instructions. After development, blots were subjected to ImageJ analysis, and data were analyzed using GraphPad Prism 7 as above.
In silico analyses of A1M-S1P and ApoM-S1P complexes
We have constructed two protein-ligand systems: ApoM with S1P (ApoM-S1P) and ApoA1 fused to ApoM with S1P (A1M-S1P). To construct ApoM-S1P, we used the crystal structure with Protein Databank ID 2YG2 (14). To ensure protonation states, we used the APBS-PDB2PQR software suite (29). We included all residues in their physiological protonation states (charged Glu, Asp, Lys, and Arg, all other residues neutral), except for His337, which we considered as double protonated/positively charged. We modeled His260, His277, and His386 as delta protonated and histidine317 as epsilon protonated. We fully optimized the substrate S1P, with a total charge of −1 (two negative charges on the phosphate and a positive ammonium group) using Open Babel (60) and ACPYPE (61) (or AnteChamber PYthon Parser interfacE), a wrapper script around the ANTECHAMBER software (62, 63). To construct the HDL complex, we fused ApoA1 to ApoM-S1P using the described linker. Due to its inherent high flexibility, ApoA1 exists in discoidal and spherical assemblies with varying lipid stoichiometries. Here, we used the double belt structure already modeled and stabilized by previously performed long molecular dynamics (MD) simulations (29, 30). Specifically, we used the double belt model for a nascent HDL of a disc containing 160:24:2 POPC:UC:ApoA1 (POPC: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine; UC: unesterified cholesterol) in an antiparallel arrangement with LL5/5 registry (31). To perform the MD simulations, we used GROMACS 2019.4 (64, 65) with the CHARMM36 force field (66). To generate the final structures and input files for the MD simulations, we utilized CHARMM-GUI (67, 68) using its Membrane/Bilayer builder. The ApoM-S1P and the A1M systems were composed of 38.082 and 408.528 atoms, respectively. We carried out the MD simulations in explicit solvent with the TIP3P water model, within the physiologic ionic concentration of 0.15 mM. To minimize electrostatic interactions between periodic images of the solute, we used a 10 Å separation between each edge of the box and the closest solute atom. We subjected the systems to an initial minimization using the steepest descent algorithm, followed by various production replicas. We ran six replicas of 0.5 μs for ApoM-S1P and 6 replicas of 0.35 μs for the A1M-S1P system due to its high size (11 times bigger than ApoM-S1P). To ensure temperature coupling, we used a Nose-Hoover (69) thermostat with a time constant of 1 ps and to ensure pressure coupling, we used the semi-isotropic Parrinello–Rahman barostat (70). In all MD simulations, we used the LINCS algorithm (71) to constrain the bond lengths involving hydrogens. To treat long-range electrostatic interactions, we used periodic boundary conditions using fast smooth Particle-Mesh Ewald (72). To investigate the stability of the complexes, we calculated the RMSDs for all MD simulations. To highlight the main collective movements, we reduced the observed variation within the 3D atomic coordinates of the protein structures, by identifying the significant PCs through PCA using the Bio3D package (73) of the R programming language. We subsequently subjected the retrieved PCs to agglomerative hierarchical cluster analysis, another unsupervised technique, using the complete-linkage algorithm. Cluster analysis allowed us to group the explored conformational space into subsets of similar structures. We calculated the binding interfaces using a 5 Å cut-off for the interatomic distance between protein residues and S1P. We chose to maintain the sampled residues with permanency times higher than 40% of the simulation time for further analysis. Because solvent accessible surface area (SASA) is a key factor for protein binding, we assessed it with FreeSASA within the vanddraabe R package (74). We measured the ratioSASA by residue at the binding pocket (ratioSASA = A1M-S1PSASA/ApoM-S1PSASA). We measured hydrogen bonds using the Visual Molecular Dynamics (VMD) package (75). All figures/videos were generated using the PYMOL, Affinity Designer, iMovie, and R packages.
Creation and characterization of the bacterial recombinant A1M (bA1M) fusion proteins
The A1M fusion for bacterial expression contains the pET15b derived translation start site sequence with an embedded 6x histidine tag for purification followed by the murine ApoA1 amino acids 25–264 of the open reading frame, a flexible linker region (GGGGS), and amino acids 21–190 of murine ApoM with a stop codon. The bA1M fusion was expressed in bacteria using the pET15b prokaryotic expression vector (Novagen). The open reading frame from the pCDH-A1M-puro plasmid described above was amplified by PCR using the following primers (ORF compatible sequence underlined): TTTCATATGGATGAACCCCAGTCCCAATGGGACA (for ApoA1) and TTTGGATCCTCACTTGCTGGACAGCGGGCAGGCCTCTT (for ApoM). The resulting PCR products and the pET15b vector were digested using NdeI and BamHI (NEB), purified and ligated using standard protocols. Positive clones were transformed into the BL21(DE3) protein expression strain (C2527I, NEB). Protein expression was performed using the standard IPTG induction protocols with several modifications. Overnight cultures were grown in terrific broth (TB) supplemented with 5 mM glucose and 100 μg/mL ampicillin. Cultures were diluted 1:30 in TB supplemented with 5 mM glucose and carbenicillin (Sigma) 100 μg/mL and maintained at 37°C at 180 rpm until OD600 reached 0.9. Cultures were diluted with 2 volumes of TB/carbenicillin for 4.5–5.5 hours at 180 rpm, pelleted by centrifugation, and frozen at −80°C. Pellets were resuspended in 50 mM Tris buffer pH8, 100 mM NaCl, 10 mM benzamidine (Sigma) supplemented with lysozyme and DNaseI and rocked at room temperature for 1 hour. The suspension was subjected to polytron disruption and centrifuged at 100,000 x g for 30 minutes. The resulting pellet was thoroughly disrupted using the B-Per Reagent (ThermoScientific) supplemented with 10 mM benzamidine and centrifuged at 100,000 x g for 30 minutes. The final pellet was considered to be inclusion bodies. Inclusion bodies were disrupted in 6 M guanidine-HCl (Sigma)/50 mM Tris pH8, rocked for 30 minutes, and centrifuged at 100,000 x g for 30 minutes. The resulting supernatant was mixed with HisPur Ni-NTA resin (ThermoFisher; 4 mL beads/L culture) for overnight bulk incubation at 4°C. The beads were washed with 50 volumes of wash buffer (PBS, 250 mM NaCl, 5 mM imidazole, 10 mM benzamidine) and protein was eluted as described above. Eluted protein was refolded step-wise using a previously published dialysis protocol (76). The resulting protein was cleared of residual endotoxin using Pierce high-capacity endotoxin resin (ThermoFisher) according to the manufacturer’s protocol, concentrated with Amicon® Ultra-4 Centrifugal Filter Unit (Ultracel® - 10K) and protein concentration was determined using the Bio-Rad Protein Assay. Western blot analyses were performed on 0.5 μL of isolated plasma using protocols described above using a rabbit monoclonal antibody specific for ApoM (EPR2904, ab91656, Abcam).
Analysis of the effects of A1M on endotoxemia in vivo
C57Bl/6J mice were obtained from Jackson Labs (Bar Harbor, Maine). 12-week-old male mice were pre-treated for 1 hour with saline or 40 mg/Kg of purified bA1M by intravenous injection. Mice were administered LPS (E. coli O111:B4, L4130, Sigma-Aldrich) 10 mg/Kg by intraperitoneal injection. Body temperature was measured at 0, 4, 8, and 12 hours post-injection using Temperature Controllers (TCAT-2DF, Physitemp). Clinical assessment was performed using the murine sepsis score (MSS) system based on observational characteristics (77). At 12 hours, mice were sacrificed, and blood was collected by cardiac puncture and maintained in EDTA. Complete blood count (CBC) was performed on an aliquot of blood using the Hemavet 950 (Drew Scientific). Plasma was collected after centrifugation at 2,000 × g for 15 minutes at 4°C. Plasma was maintained at −80°C until analyzed by Western blot or ELISA. An IL-6 ELISA was performed on 10 μl of plasma using the mouse Duoset ELISA kit (DY406–05, R&D systems) and all data were derived by comparison to the supplied IL-6 standards.
Cholesterol efflux assay
The cholesterol efflux was performed with the Cholesterol Efflux Assay Kit (ab196985, Abcam) by tracing fluorescently labeled cholesterol. Briefly, 100 nM phorbol 12-myristate-13-acetate (PMA) for 72 h was used to differentiate THP-1 cells (TIB-202, ATCC) to macrophage-like phenotype. Cells were harvested and plated 1 × 105 cells/well in a 96-well plate. After 4 h incubation, adherent cells were washed with serum-free medium and cultured in labeling medium in the dark for 1 h. After removing the labeling medium, cells were treated with the equilibration medium and incubated overnight. Cells were treated with cholesterol acceptors such as HDL and lipidated A1M particles for 5 h in the dark. The supernatant and cell lysates were collected and measured for fluorescence by plate reader at Ex/Em 485/523 nm. Cholesterol efflux from the labeled cells to HDL-like particles was calculated by dividing the fluorescence intensity (RFU) obtained for the supernatant by the sum of the fluorescence intensity of the supernatant and cell lysate of the same treatment.
Statistical analysis
Data are presented as means ± SD. Statistical analysis was performed using Prism software (GraphPad). Multiple comparison testing was performed according to the reference (78). P values < 0.05 were considered statistically significant.
Supplementary Material
Funding:
This work is supported by NIH grants (R35-HL135821 and RO1-EY031715 to TH, R35-HL135775 to RF, and R33-CA263705–01 to ZHG). ZHG was also supported by start-up funds from the Department of Genetics and Genomics at the Icahn School of Medicine at Mount Sinai. ISM and NR-F were supported by the European Regional Development Fund through the COMPETE 2020 - Operational Programme for Competitiveness and Internationalisation and Portuguese national funds through Fundação para a Ciência e a Tecnologia (FCT) [LA/P/0058/2020, UIDB/04046/2020, DSAIPA/DS/0118/2020 and UIDP/04046/2020]. NR-F was also supported by FCT through Ph.D. scholarships PD/BD/135179/2017. Author contributions: YL, SS, and TH conceived the project ideas, designed the experiments, and wrote the manuscript. YL and SS performed in vitro purification and functional assays. SS, AG, and SG performed and collected TEER data. YL, SS, AC, and TS conducted mouse experiments. AK conducted mass spectrometry experiments. SG, SP, AS, and RF designed and performed human platelet aggregation assays. ISM, NR-F and ZHG performed molecular dynamic simulation analyses. AC and ML contributed to the transmission EM analysis. ML established the NF-ĸB reporter cell line. AI contributed to the NanoBiT assays. YL and DZ performed statistical analysis. GAF contributed to the IP receptor experiments. TH provided funding, directed the project and managed collaborations.
Footnotes
Competing interests: TH and SS are named as inventors on patent applications (#XX) that describe A1M. The other authors claim that they have no competing interests.
Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. Metadata are available on DRYAD (URL). Request for DNA constructs for recombinant proteins will be honored upon the execution of a materials transfer agreement with the Boston Children’s Hospital technology and innovation development office.
REFERENCES AND NOTES
- 1.Pober JS, Sessa WC, Evolving functions of endothelial cells in inflammation. Nat Rev Immunol 7, 803–815 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Kiseleva RY et al. , Targeting therapeutics to endothelium: are we there yet? Drug Deliv Transl Res 8, 883–902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yvan-Charvet L, Wang N, Tall AR, Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler Thromb Vasc Biol 30, 139–143 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ouimet M, Barrett TJ, Fisher EA, HDL and Reverse Cholesterol Transport. Circ Res 124, 1505–1518 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Remaley AT, Norata GD, Catapano AL, Novel concepts in HDL pharmacology. Cardiovasc Res 103, 423–428 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rohatgi A, Westerterp M, von Eckardstein A, Remaley A, Rye KA, HDL in the 21st Century: A Multifunctional Roadmap for Future HDL Research. Circulation 143, 2293–2309 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinecke JW, The protein cargo of HDL: implications for vascular wall biology and therapeutics. J Clin Lipidol 4, 371–375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shao B, Heinecke JW, Quantifying HDL proteins by mass spectrometry: how many proteins are there and what are their functions? Expert Rev Proteomics 15, 31–40 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Suzuki M et al. , High-density lipoprotein suppresses the type I interferon response, a family of potent antiviral immunoregulators, in macrophages challenged with lipopolysaccharide. Circulation 122, 1919–1927 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fotakis P et al. , Anti-Inflammatory Effects of HDL (High-Density Lipoprotein) in Macrophages Predominate Over Proinflammatory Effects in Atherosclerotic Plaques. Arterioscler Thromb Vasc Biol 39, e253–e272 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mineo C, Shaul PW, Regulation of signal transduction by HDL. J Lipid Res 54, 2315–2324 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mineo C, Deguchi H, Griffin JH, Shaul PW, Endothelial and antithrombotic actions of HDL. Circ Res 98, 1352–1364 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Morishita H, Yui Y, Hattori R, Aoyama T, Kawai C, Increased hydrolysis of cholesteryl ester with prostacyclin is potentiated by high density lipoprotein through the prostacyclin stabilization. J Clin Invest 86, 1885–1891 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Christoffersen C et al. , Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A 108, 9613–9618 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proia RL, Hla T, Emerging biology of sphingosine-1-phosphate: its role in pathogenesis and therapy. J Clin Invest 125, 1379–1387 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galvani S et al. , HDL-bound sphingosine 1-phosphate acts as a biased agonist for the endothelial cell receptor S1P1 to limit vascular inflammation. Sci Signal 8, ra79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blaho VA et al. , HDL-bound sphingosine-1-phosphate restrains lymphopoiesis and neuroinflammation. Nature 523, 342–346 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Z, Guo P, Yang D, Swendeman SL, Wang Z, Christoffersen C, Nielsen LB, Friedman SL, Powell CA, Hla T, Ding B, Aging attenuates interorgan protective signaling axis mediated by ApoM-bound sphingosine 1-phosphate. Dev Cell In Press (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ding BS et al. , HDL activation of endothelial sphingosine-1-phosphate receptor-1 (S1P1) promotes regeneration and suppresses fibrosis in the liver. JCI Insight 1, e87058 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Velagapudi S et al. , Apolipoprotein M and Sphingosine-1-Phosphate Receptor 1 Promote the Transendothelial Transport of High-Density Lipoprotein. Arterioscler Thromb Vasc Biol 41, e468–e479 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Obinata H et al. , Identification of ApoA4 as a sphingosine 1-phosphate chaperone in ApoM- and albumin-deficient mice. J Lipid Res 60, 1912–1921 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Christensen PM et al. , Impaired endothelial barrier function in apolipoprotein M-deficient mice is dependent on sphingosine-1-phosphate receptor 1. FASEB J 30, 2351–2359 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mathiesen Janiurek M, Soylu-Kucharz R, Christoffersen C, Kucharz K, Lauritzen M, Apolipoprotein M-bound sphingosine-1-phosphate regulates blood-brain barrier paracellular permeability and transcytosis. Elife 8 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang H, Pluhackova K, Jiang Z, Bockmann RA, Binding Characteristics of Sphingosine-1-Phosphate to ApoM hints to Assisted Release Mechanism via the ApoM Calyx-Opening. Sci Rep 6, 30655 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swendeman SL et al. , An engineered S1P chaperone attenuates hypertension and ischemic injury. Sci Signal 10 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Burg N, Swendeman S, Worgall S, Hla T, Salmon JE, Sphingosine 1-Phosphate Receptor 1 Signaling Maintains Endothelial Cell Barrier Function and Protects Against Immune Complex-Induced Vascular Injury. Arthritis Rheumatol 70, 1879–1889 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trinh R, Gurbaxani B, Morrison SL, Seyfzadeh M, Optimization of codon pair use within the (GGGGS)3 linker sequence results in enhanced protein expression. Mol Immunol 40, 717–722 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Oda MN et al. , Reconstituted high density lipoprotein enriched with the polyene antibiotic amphotericin B. J Lipid Res 47, 260–267 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Jurrus E et al. , Improvements to the APBS biomolecular solvation software suite. Protein Sci 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pourmousa M et al. , Tertiary structure of apolipoprotein A-I in nascent high-density lipoproteins. Proc Natl Acad Sci U S A 115, 5163–5168 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Segrest JP et al. , A detailed molecular belt model for apolipoprotein A-I in discoidal high density lipoprotein. J Biol Chem 274, 31755–31758 (1999). [DOI] [PubMed] [Google Scholar]
- 32.Hisano Y et al. , Lysolipid receptor cross-talk regulates lymphatic endothelial junctions in lymph nodes. J Exp Med 216, 1582–1598 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jeansson M et al. , Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J Clin Invest 121, 2278–2289 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parikh SM, Angiopoietins and Tie2 in vascular inflammation. Curr Opin Hematol 24, 432–438 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia JG et al. , Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest 108, 689–701 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Finigan JH et al. , Activated protein C mediates novel lung endothelial barrier enhancement: role of sphingosine 1-phosphate receptor transactivation. J Biol Chem 280, 17286–17293 (2005). [DOI] [PubMed] [Google Scholar]
- 37.Pluchart H, Khouri C, Blaise S, Roustit M, Cracowski JL, Targeting the Prostacyclin Pathway: Beyond Pulmonary Arterial Hypertension. Trends Pharmacol Sci 38, 512–523 (2017). [DOI] [PubMed] [Google Scholar]
- 38.Birukova AA et al. , Prostaglandins PGE(2) and PGI(2) promote endothelial barrier enhancement via PKA- and Epac1/Rap1-dependent Rac activation. Exp Cell Res 313, 2504–2520 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Skuballa W, Vorbruggen H, Synthesis of ciloprost (ZK 36 374): a chemically stable and biologically potent prostacyclin analog. Adv Prostaglandin Thromboxane Leukot Res 11, 299–305 (1983). [PubMed] [Google Scholar]
- 40.Moncada S, Higgs EA, Vane JR, Human arterial and venous tissues generate prostacyclin (prostaglandin x), a potent inhibitor of platelet aggregation. Lancet 1, 18–20 (1977). [DOI] [PubMed] [Google Scholar]
- 41.Bui TM, Wiesolek HL, Sumagin R, ICAM-1: A master regulator of cellular responses in inflammation, injury resolution, and tumorigenesis. J Leukoc Biol 108, 787–799 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cockerill GW, Rye KA, Gamble JR, Vadas MA, Barter PJ, High-density lipoproteins inhibit cytokine-induced expression of endothelial cell adhesion molecules. Arterioscler Thromb Vasc Biol 15, 1987–1994 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Yuhanna IS et al. , High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med 7, 853–857 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Michaud J, Kohno M, Proia RL, Hla T, Normal acute and chronic inflammatory responses in sphingosine kinase 1 knockout mice. FEBS Lett 580, 4607–4612 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Robert J, Osto E, von Eckardstein A, The Endothelium Is Both a Target and a Barrier of HDL’s Protective Functions. Cells 10 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Birukova AA et al. , Iloprost improves endothelial barrier function in lipopolysaccharide-induced lung injury. Eur Respir J 41, 165–176 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bisgaard LS, Christoffersen C, Apolipoprotein M/sphingosine-1-phosphate: novel effects on lipids, inflammation and kidney biology. Curr Opin Lipidol 30, 212–217 (2019). [DOI] [PubMed] [Google Scholar]
- 48.Frej C et al. , Sphingosine 1-phosphate and its carrier apolipoprotein M in human sepsis and in Escherichia coli sepsis in baboons. J Cell Mol Med 20, 1170–1181 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marfia G et al. , Decreased serum level of sphingosine-1-phosphate: a novel predictor of clinical severity in COVID-19. EMBO Mol Med 13, e13424 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winkler MS et al. , Erythrocytes increase endogenous sphingosine 1-phosphate levels as an adaptive response to SARS-CoV-2 infection. Clin Sci (Lond) 135, 2781–2791 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee MH et al. , S1P in HDL promotes interaction between SR-BI and S1PR1 and activates S1PR1-mediated biological functions: calcium flux and S1PR1 internalization. J Lipid Res 58, 325–338 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pirich C, Banyai M, Efthimiou Y, Sinzinger H, Lipoproteins and prostacyclin stability. Semin Thromb Hemost 19, 138–143 (1993). [DOI] [PubMed] [Google Scholar]
- 53.Yui Y et al. , Serum prostacyclin stabilizing factor is identical to apolipoprotein A-I (Apo A-I). A novel function of Apo A-I. J Clin Invest 82, 803–807 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Birukova AA et al. , Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochim Biophys Acta 1852, 778–791 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schmaier AA et al. , Tie2 activation protects against prothrombotic endothelial dysfunction in COVID-19. JCI Insight 10.1172/jci.insight.151527 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee MJ et al. , Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science 279, 1552–1555 (1998). [DOI] [PubMed] [Google Scholar]
- 57.Engelbrecht E et al. , Sphingosine 1-phosphate-regulated transcriptomes in heterogenous arterial and lymphatic endothelium of the aorta. Elife 9 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ru H et al. , Molecular Mechanism of V(D)J Recombination from Synaptic RAG1-RAG2 Complex Structures. Cell 163, 1138–1152 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hla T, Neilson K, Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A 89, 7384–7388 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.O’Boyle NM et al. , Open Babel: An open chemical toolbox. J Cheminform 3, 33 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sousa da Silva AW, Vranken WF, ACPYPE - AnteChamber PYthon Parser interfacE. BMC Res Notes 5, 367 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang J, Wang W, Kollman PA, Case DA, Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model 25, 247–260 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA, Development and testing of a general amber force field. J Comput Chem 25, 1157–1174 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Pronk S et al. , GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tan C et al. , Implementation of residue-level coarse-grained models in GENESIS for large-scale molecular dynamics simulations. PLoS Comput Biol 18, e1009578 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sippl MJ, Recognition of errors in three-dimensional structures of proteins. Proteins 17, 355–362 (1993). [DOI] [PubMed] [Google Scholar]
- 67.Jo S, Kim T, Iyer VG, Im W, CHARMM-GUI: a web-based graphical user interface for CHARMM. J Comput Chem 29, 1859–1865 (2008). [DOI] [PubMed] [Google Scholar]
- 68.Jo S et al. , CHARMM-GUI PDB manipulator for advanced modeling and simulations of proteins containing nonstandard residues. Adv Protein Chem Struct Biol 96, 235–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoover WG, Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A Gen Phys 31, 1695–1697 (1985). [DOI] [PubMed] [Google Scholar]
- 70.Aoki KM, Yonezawa F, Constant-pressure molecular-dynamics simulations of the crystal-smectic transition in systems of soft parallel spherocylinders. Phys Rev A 46, 6541–6549 (1992). [DOI] [PubMed] [Google Scholar]
- 71.Hess B, P-LINCS: A Parallel Linear Constraint Solver for Molecular Simulation. J Chem Theory Comput 4, 116–122 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Wang H, Gao X, Fang J, Multiple Staggered Mesh Ewald: Boosting the Accuracy of the Smooth Particle Mesh Ewald Method. J Chem Theory Comput 12, 5596–5608 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Grant BJ, Skjaerven L, Yao XQ, The Bio3D packages for structural bioinformatics. Protein Sci 30, 20–30 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Patel H, Gruning BA, Gunther S, Merfort I, PyWATER: a PyMOL plug-in to find conserved water molecules in proteins by clustering. Bioinformatics 30, 2978–2980 (2014). [DOI] [PubMed] [Google Scholar]
- 75.Humphrey W, Dalke A, Schulten K, VMD: visual molecular dynamics. J Mol Graph 14, 33–38, 27–38 (1996). [DOI] [PubMed] [Google Scholar]
- 76.Thomson CA, Olson M, Jackson LM, Schrader JW, A simplified method for the efficient refolding and purification of recombinant human GM-CSF. PLoS One 7, e49891 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shrum B et al. , A robust scoring system to evaluate sepsis severity in an animal model. BMC Res Notes 7, 233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Staffa SJ, Zurakowski D, Strategies in adjusting for multiple comparisons: A primer for pediatric surgeons. J Pediatr Surg 55, 1699–1705 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.