


Abstract
Ribosomal frameshifting refers to the process that ribosomes slip into +1 or −1 reading frame, thus produce chimeric trans-frame proteins. In viruses and bacteria, programmed ribosomal frameshifting can produce essential trans-frame proteins for viral replication or regulation of other biological processes. In humans, however, functional trans-frame protein derived from ribosomal frameshifting is scarcely documented. Combining multiple assays, we show that short codon repeats could act as cis-acting elements that stimulate ribosomal frameshifting in humans, abbreviated as CRFS hereafter. Using proteomic analyses, we identified many putative CRFS events from 32 normal human tissues supported by trans-frame peptides positioned at codon repeats. Finally, we show a CRFS-derived trans-frame protein (HDAC1-FS) functions by antagonizing the activities of HDAC1, thus affecting cell migration and apoptosis. These data suggest a novel type of translational recoding associated with codon repeats, which may expand the coding capacity of mRNA and diversify the regulation in human.
Graphical Abstract
Graphical Abstract.

Introduction
Canonical protein translation is a strictly regulated process, which guarantees that ribosomes initiate protein synthesis at a defined start codon, iteratively decode triplet codons during elongation, and terminate the synthesis at stop codons within the designated open reading frame (ORF) on mRNAs (1). However, in unconventional cases, translation elongation may breach this law, thus allowing ribosomes to produce multiple functional chimeric proteins from a given ORF. This process is termed translation recoding (2,3). Translational recoding consists of three types: translational readthrough (4,5), translational bypassing (6) and programmed ribosomal frameshifting (PRF) (7–9). PRF was first identified in Rous sarcoma virus and has been reported in all three domains of life, but detailed information comes mostly from studies on RNA viruses and a number of prokaryotic genes (9).
Ribosomal frameshifting refers to a process when ribosomes switch from the original reading frame (0 frame) into the +1 or −1 frame during the translation elongation. +1 ribosomal frameshifting (+1FS) refers to the slippage of the ribosome one nucleotide towards the 3′ end of the mRNA, while −1FS refers to the slippage of one nucleotide toward the 5′ end. The translational machinery is sophistically regulated to guarantee the ribosomes located in the correct reading frame. Studies on prokaryotic translation revealed that elongation factors play critical roles in maintaining the reading frame at ‘slippery sequences’ during the tRNA-mRNA translocation of ribosomes (10–12). Moreover, tRNA modifications, such as m1G37, have also been found to help stabilize certain codon-anticodon pairs during translocation (12–14). With these measures, spontaneous ribosomal frameshifting is maintained at a very low rate (10−5 per codon) (15,16). In contrast, PRF is a strictly controlled process, which occurs at a specific location with high efficiency varying between 1% and 80% (9,17). Viral and bacterial −1PRF is the best studied type, and it consists of two elements: a slippery site and a stimulator (18). The slippery site is a cis-element on mRNA characterized with a sequence motif, X XXY YYZ (X = A, U, C, G; Y = A or U; and Z ≠ G), which allows anticodons of tRNAs to pair with codons of the mRNA in an alternative frame (19–22). The stimulator is typically an RNA secondary structure, such as a stem loop (23) or pseudoknot (24,25), located immediately downstream of the slippery site. In some cases, proteins and miRNAs were also reported to participate in PRF regulation (26–28).
Recent biochemical and structural studies have shed light on the detailed mechanism of how frameshifting occurs inside the ribosome. For viral and bacterial −1PRF, when ribosomes are hindered by the downstream stimulator, the slippery site could allow anticodons of tRNAs to rematch codons in an alternative frame within the ribosome decoding center, thus leading to PRF (25,29–34). The bacteria Salmonella also features a special mechanism to induce +1FS by utilizing tRNAs with quadruplet anticodons, which could potentially expand the coding capacity of the genome (35). For example, SufB2, a tRNA with an extra nucleotide in its anticodon loop, initially pairs mRNA in the 0-frame with 3 bases but rematches the +1 frame with 3 or 4 bases at the late stage of the tRNA–mRNA translocation (36,37). In addition, environmental factors, such as amino acid starvation resulting in ‘hungry’ codons could also act as stimulator for PRF (34,38,39). The product of PRF, termed trans-frame proteins, usually have critical biological functions. In RNA viruses, trans-frame proteins are essential for viral replication (40,41). In prokaryotes, trans-frame proteins have also been well documented to participate in various biological processes, such as DNA replication (42), translation termination factor production (43) and copper transport (44).
In higher organisms, including humans, whether PRF widely exists in endogenous genes is under debate. Although viral-like −1PRF has been reported in the paternally expressed gene PEG10 derived from retrotransposons (45), one well-recognized case in eukaryotic genes is the +1PRF in ornithine decarboxylase antizyme 1 (OAZ1), which is highly conserved in eukaryotes from yeast to human (46,47). A typical OAZ1 mRNA contains two ORFs, ORF1 and ORF2 (47). When the ribosome translates to the stop codon of ORF1, +1PRF causes the ribosome to slip into ORF2 that is located in the +1 frame in regard to the ORF1(48). The occurrence and efficiency of +1PRF depends on the polyamine concentration and the OAZ nascent peptides (48,49), which produces a trans-frame protein essential for cell proliferation and development (50,51). In addition, computational analyses predicted that ∼10% of eukaryotic genes harbor viral-like −1PRF (52,53). The best evidence for this comes from human immunodeficiency virus-1 (HIV-1) co-receptor C-C chemokine receptor type 5 (CCR5), which harbors a −1PRF that can regulate the stability of CCR5 mRNA by miRNAs (26). However, the existence of viral-like −1PRF in eukaryotic genes remains controversial due to recent studies with a modified dual luciferase assay (54,55).
Other than PRF, recent evidence showed that in humans, under abnormal cellular conditions, ribosomal frameshifting could efficiently occur at select regions to produce aberrant trans-frame proteins. For example, in cancer cells, ‘sloppy’ ribosomal frameshifting could also occur at UGG codons upon tryptophan starvation, resulting in aberrant trans-frame peptides that are present on the cell surface (56,57). In addition, CAG and CGG codon repeat expansion, which has been noticed in a large category of neurodegenerative diseases (58), were reported to stimulate ribosomal frameshifting, thus producing aberrant trans-frame proteins that trigger protein aggregates (59–61). These studies raise an intriguing question whether in normal cells codon repeats could be the cis-acting elements that stimulate ribosomal frameshifting to produce beneficial trans-frame proteins.
Codon repeats refer to a string of identical codons, which widely represented in the human genes (Supplementary Figure S1). In this study, we combined various assays to show that normal codon repeats alone could trigger ribosomal frameshifting. Using proteome data from 32 normal human tissues, we could identify many putative ribosomal frameshifting events that occur on account of normal codon repeats. Through combining multiple assays, we show that the (UAC)3 codon repeat in the histone deacetylase 1 (HDAC1) mRNA is an important site for ribosomal frameshifting, which produces a small trans-frame protein (HDAC1-FS) that can affect cell migration and apoptosis.
Materials and methods
Human cell cultures and cell transfection
HEK293T, A549 and HeLa cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS, ExcellBio), 1 × penicillin–streptomycin (HyClone), at 37°C in a humidified incubator with 5% CO2.
HEK293T (1.7 × 105) cells were seeded in a 12-well plate at 70–80% coverage. After 16-h culturing, constructs were transiently transfected into cells using a calcium phosphate cell transfection kit (Beyotime, China). In brief, 1.5 μg plasmids were mixed with 40 μl CaCl2 solution. After 5 min incubation at room temperature, the DNA–CaCl2 mixture was added to 40 μl 2-[N,N-bis(N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES) buffer (known as BBS). After 10 min incubation at room temperature, the DNA–CaCl2–BBS mixture was added gently into the cell culture. The medium was changed after 8 h transfection at 37°C in a humidified incubator with 5% CO2. The HEK293T transfected cell line was selected with 50 mg/l hygromycin B (Amresco, K547). A549 and HeLa cells were transfected by Lipofectamine 2000 (Invitrogen). Briefly, 1.5 μg plasmids were mixed with 250 μl Opti-MEM medium. 3 μl of Lipofectamine 2000 were diluted with an additional 250 μl Opti-MEM medium for 5 min standing at room temperature. The diluted plasmids were mixed with diluted Lipofectamine 2000 and incubated for 20 min at room temperature. The DNA–lipid complex was then added to the cells. After 5 h transfection at 37°C in a humidified incubator with 5% CO2, the medium was refreshed with new DMEM including 10% FBS. Proteins were extracted 26 h after transfection.
Lentivirus generation and transduction
HEK293T cells were seeded onto 6-well plates for virus generation. The transfection was performed at 16 h after seeding at 70–80% confluency. 1 μg pMD2.G, 1.4 μg psPAX2, and 1.6 μg pcDNA3.1-HDAC1-FS (EV was used as a negative control) were mixed and diluted with 250 μl Opti-MEM medium. 8 μl of Lipofectamine 2000 (Invitrogen) was diluted into 250 μl Opti-MEM medium. After 5 min incubation at room temperature, the diluted plasmids were then added into the diluted Lipofectamine 2000 and the DNA–lipid complex was added into cells after 20 min incubation. Viruses were collected at 24, 48, 72 h and mixed. Viruses mixture were stored at −80°C after filtering through 0.45 μM syringe filters.
A549 cell lines were seeded onto 6-well plates at 80–90% coverage for viral transduction. The medium was evacuated after 16 h and replaced with 2 ml collected viruses. After 24 h, 300 mg/l hygromycin B was added for selection of stable cell lines.
Plasmid construction
Two dual-luciferase systems were used to detect the frameshifting ratio. First, using pcDNA3.1 as backbone, Renilla luciferase (RLuc) and firefly luciferase (FLuc) genes, separated by the P2A peptide sequence, were driven by the cytomegalovirus (CMV) promoter. This is termed the P2A system (sequence available in Supplementary Table S1). In this system, test sequences were inserted between P2A and FLuc (Figure 1A). Secondly, the F2A system was used as documented (55). In this system, RLuc and FLuc are driven by the CMV promoter and separated by two F2A peptides after translation. Here the test sequences were inserted between the two F2A peptides (Figure 1F).
Figure 1.

UUU codon repeats could trigger robust ribosomal frameshifting. (A) Schematic diagram of the P2A dual luciferase system (RLuc, Renilla luciferase; FLuc, firefly luciferase). 0F, +1F or −1F FLuc indicate the FLuc sequence is placed in the 0, +1 or −1 frame, respectively. 0, +1 and −1 UAA mean that a UAA stop codon was added in the 0, +1 or −1 frame, respectively. (B) Bar plot showing frameshifting ratio of HIV −1 PRF sequence in HEK293T cells by luciferase assays. SSm, slippery site mutation (UUUUUUA to CUUCUUA). SSd, slippery site deletion. (C) Bar plot showing Renilla luciferase (left) and firefly luciferase (right) activity of OAZ1 sequences. +1F and 0F means construct with FLuc in the +1 or 0 frame, as shown in (A) The cartoon on top showed the reporter structure. (D) Detection of ribosomal frameshifting induced by different number of UUU codon repeats with dual luciferase systems (top panel) and western blots (bottom panel). EV, empty vector. (E) Frameshifting ratio induced by (UUU)4 or (UUU)5 insertion (INS) with different vectors shown in (A). +1FLuc and −1FLuc means construct with FLuc in the +1 or −1 frame. (F) Schematic diagram of Stop-Go (SG) system. SG1 and SG2 indicate two F2A sequence. (G) Frameshifting ratios (top) and western blot results (bottom) from SG system with different number of UUU repeat insertion in HEK293T cells. EV, empty vector. (H) Schematic diagram of constructs (mC-E system) with OAZ1 (top) and (UUU)5 (bottom) insertion. 0, +1 and −1 mean the position of first stop codon in the corresponding frame after mCherry coding sequence. (I) Western blot analysis for the frameshifting proteins induced by OAZ1 sequence. NV, transfection without vector. (J) Western blot analysis the frameshifting induced by (UUU)5 codon in HEK293T cells. In this figure, FS stands for ribosomal frameshifting, EV for empty vector, error bars for standard deviation of three biological replicates. (*) P< 0.05, (**) P < 0.01 (Student's t-test for 2 data sets and one-way ANOVA for >2 data sets).
The frameshifting ratio of 61 types of codon repeats were detected using the P2A system. Codon repeats were inserted in front of the FLuc coding sequence (without AUG start codon) (Figure 2A). If the inserted codon sequence ended with U (NNU), a GGC sequence was added behind the codon repeats to avoid UGA stop codon in the −1 frame. One G/C was inserted for creating the +1 frame FLuc, while a G (the first base of the FLuc ORF) was deleted to create the −1 frame FLuc. The corresponding original plasmid (FLuc in 0-frame) was set as a normalization standard, that is, its firefly and Renilla luciferase activity were defined as 100%.
Figure 2.

Codon repeat-induced ribosomal frameshifting may widely occur in humans. (A) Frameshifting ratio determined by the P2A dual luciferase system from different types of codon repeat (5-time repeats) in HEK293T cells. Error bars, standard deviation of 3 biological repeats. (B) Detection of ribosomal frameshifting of (UGG)5 in the SPIDR gene. Diagram (top) shown the structure of the construct, with the dark green box indicating the position of (UGG)5 repeats. Middle and bottom panel showing the western blot result and luciferase activity assay. Three biological replicates were analyzed for each experiment. Error bars, standard deviation. (**) P < 0.01 (Student's t-test). (C) Diagram illustrating the definition of CRFS locus, trans-frame peptide and out-of-frame peptide in the trans-frame protein. Red and black asterisk mean the position of first stop codon in the +1 and 0 frame, respectively. (D) Pie chart showing the distribution of peptides detected in human tissue proteomes. Trans-frame or out-of-frame were defined in (C). Annotated protein means peptide sequences match unexpressed annotated proteins. Small peptide means sequences match the smProt database. Splice site means trans-frame peptides positioned at splicing junctions. (E) Venn diagram showing the number of CRFS loci supported by trans-frame, out-of-frame peptides or both. (F) Venn diagram showing the number of CRFS loci supported by trans-frame peptides detected in proteomic data of each biological repeats. (G) Diagram showing the number of CRFS loci supported by unique trans-frame and/or out-of-frame peptides from proteomic data of 32 human tissues. (H) Unique trans-frame peptides detected in two biological repeats and represented in > 16 human tissues. Red sequences are encoded by +1 frame, blue by −1 frame, and black by 0 frame.
The gene fragment insertions were defined as a region including the CRFS locus (excluding the stop codon) and 30 bp of upstream sequence (Figure 2C). Thus, the length of the inserted fragments will be (3n +1) bp or (3n − 1) bp, which set the FLuc ORF in −1 or +1 frame. A single nucleotide insertion or deletion was introduced to the 5′ end of the FLuc coding sequence (without AUG codon) so as to place the FLuc ORF in the 0-frame, which was used to normalize the frameshifting ratio of the inserted fragment. Primers used in this study are listed in Supplementary Table S1.
Luciferase activity measurement
Cell lysate production and luciferase activity measurements were done using the Dual-Luciferase® Reporter Assay System (Promega, E1960) according to the manufacture's protocol. Briefly, after transfection cells were cultured for 26 h and were lysed with 100 μl 1 × passive lysis buffer for 15 min at room temperature. The cell lysate was collected and centrifuged for 2 min at 4°C at 20 000 × g. Firefly and Renilla luciferase activity were individually measured with a Promega GloMax 20/20 Luminometer.
For a given inserted sequence, the +1 or −1 frameshifting ratio was calculated with following formula.
Insertion (test) sequence frameshifting ratio (%):
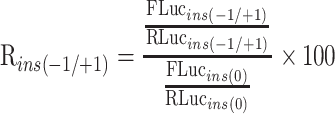 |
Here, FLucins(−1/+1) and RLucins(−1/+1) refers to the firefly or Renilla luciferase activities from the constructs with FLuc in alternative frames, whereas FLucins(0) and RLucins(0) refer to firefly or Renilla luciferase activities from the construct with FLuc in the 0 frame.
Accordingly, the frameshifting ratio of the empty vector (no insertion), which is used to evaluate the background frameshifting level of the reporter system, is calculated with formula below:
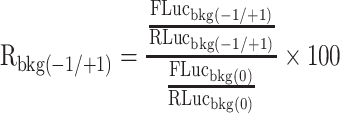 |
Wound healing assay and transwell assay
A549 cells were seeded to be 80% confluent with viral infection in a 6-well plate. To determine the function of HDAC1-FS, cells were infected with the collected viruses. 24 h after transfection the cells were scratched with 200 μl pipet tips and washed twice with PBS. The cells were maintained in DMEM supplemented with 1% FBS at 37°C in a humidified incubator with 5% CO2. Cell migration was photomicrographed (MF53, MSHOT) at 0, 24, 48 and 72 h after scratching. Wound width was measured for each transfection using ImageJ and used to calculate the wound closure rate. For HDAC1 knockdown, 113 pmol HDAC1-siRNA (synthesized by Sangon Biotech, si1: 5′-GAGAAAGACCCAGAGGAGAAG-3′, si2: 5′-UAGUAGUAACAGACUUUCCUC-3′) was transfected into A549 using 3 μl Lipofectamine RNAiMax (transfection procedure is the same as described above). Cells were collected and counted 24 h after transfection. 4 × 105 cells were seeded in 12-well plate for wound healing assay. The rest of the cells were resuspended in TRIzol reagent for reverse transcription quantitative PCR (RT-qPCR). Three biological replicates were performed for each experiment.
Transfected A549 cells (4 × 104) were resuspended in a transwell chamber (8 μm, PC membranes) in 200 μl DMEM. 600 μl DMEM containing 10% FBS was added outside of the transwell chamber. Cells were incubated for 12 h (siRNA transfected cells were incubated for 18 h) at 37°C in a humidified incubator with 5% CO2. The cells were swabbed on the top of the PC membranes and the membranes were stained with 0.2% crystal violet for 20 min and washed twice with PBS. The cells on the bottom of the PC membranes were photomicrographed (MF53, MSHOT) and counted. The cell number of the control group was set as 100% migration rate. Three biological replicates were performed for each experiment.
Protein extraction
HEK293T cells were washed with cold PBS and lysed with NP-40 lysis buffer (50 mM Tris–HCl [pH7.5], 150 mM NaCl, 0.5% NP-40, 1 mM DTT, 1 μg/ml pepstain A, 1 μg/ml leupeptin, 1 mM PMSF) for 15 min at 4°C with gently shaking. Cell lysates were collected and centrifuged for 10 min at 4°C at 20 000 × g. Supernatant was transferred to a new tube for further use or storage at −80°C.
Western blotting
Protein concentration was measured by Bradford assay (Pierce, 23246) and proteins were separated by 10% SDS PAGE in running buffer (3 g/l Tris, 14.4 g/l Glycine, and 1 g/l SDS), and then electro-transferred to a polyvinylidene fluoride (PVDF) membrane in transfer buffer (384 mM glycine, 50 mM Tris, 20% methanol). The PVDF membrane was immersed in 100% methanol for 1 min, air dried for 15 min, then incubated with 1 × PBS including 0.5% nonfat milk, 0.3% Tween-20 and corresponding antibodies for 2 h at room temperature or overnight at 4°C. After washing three times with PBS-Tween-20 (10 min each), the PVDF membrane was incubated with an HRP-linked secondary antibody. The protein bands were detected using ECL detection reagent (32106, Pierce) with a Tanon 4600SF Chemiluminescent Imaging system. Protein levels were quantified using Image J software. Three biological replicates were analyzed for each experiment.
Antibody information
Antibodies specific for H3K9ac (ab10812), Renilla luciferase (ab187338), and mCherry (ab183628) were purchased from Abcam. Anti-GAPDH (SC-32233) and anti-HDAC1 (sc-81598) were purchased from Santa Cruz. Anti-HA (SLAB0202) were purchased from Smart-Lifesciences. Anti-firefly luciferase (A18259), anti-Flag (AE024), HRP goat anti-rabbit (AS014) and HRP goat anti-mouse (AS003) were purchased from ABclonal. Anti-Flag M2 beads (M8823) were purchased from Sigma.
Apoptosis induction
Transfected HEK293T cells were changed with new medium containing DMSO (as control), 5 μM thapsigargin (TG), or 1 μg/ml tunicamycin (TM). For ChIP-qPCR and RT-qPCR assays, the cells were seeded in a 15-cm dish and treated for 4 h. For frameshifting ratio calculation, the cells were seeded in 12-well plates and treated for 8 h. For the apoptosis cell detection, the cells were seeded in 12-well plates and treated for 24 h. Three biological replicates were analyzed for each experiment.
Apoptosis cell detection
The annexin V/propidium iodide (PI) double staining assay were performed according to the manufacture's protocol (Proteintech, PF00005). Briefly, cells induced by DMSO or TG were digested with trypsin and washed with PBS twice. Then, cells were resuspended in 100 μl 1 × binding buffer. 5 μl annexin V and 5 μl PI were add into each tube and incubated at room temperature for 15 min in the dark. Next, 400 μl PBS was added to each tube and the stained cells were analyzed on a flow cytometer (FACS Aria SORP, BD). Annexin V was excited using a 488 nm laser and detected by the FITC channel. PI was detected using the PE-A channel. Data were analyzed using FlowJo 7.6.1. Cells with both Annexin V and Annexin V- PI positive were counted as apoptotic cells. Groups treated by DMSO were used for normalization. Relative apoptosis level was calculated by normalizing the apoptotic cell number from TG treatment with that from DMSO treatment. The relative apoptosis rate of cells transfected with EV were set as 0. Positive value of relative apoptosis rate indicates promote cell apoptosis compare to EV, negative value indicates suppress cell apoptosis to EV. Three biological replicates were analyzed for each experiment.
Chromatin immunoprecipitation assay (ChIP)
2 × 107 cells were cross-linked with 1% formaldehyde (Sigma-Aldrich, F8775) for 10 min at room temperature with gentle shaking. Glycine was then added to a final concentration of 0.125 M to stop the reaction. The cells were then washed with cold PBS twice, harvested in PBS, and centrifuged at 1300 rpm for 5 min at 4°C. The pellet was resuspended with 1 ml cold ChIP lysis buffer (20 mM Tris–HCl [pH 7.5], 50 mM NaCl, 0.1% SDS, 0.5% Triton X-100, 1 mM EDTA, 1 μg/ml pepstain A, 1 μg/ml leupeptin, 1 mM PMSF, 1 μg/ml Aprotinin, 1 mM DTT) for 20 min on ice, and then sonicated for 40 min in a Covaris M220 with the following settings: 10% duty factor, 75 W peak incident power, 200 cycles per burst. DNA fragments of 200–500 bp were produced. The sonicated sample was centrifuged at 20 000 × g for 10 min at 4°C and 400 ml supernatant (200 μl as Input) was transferred to a new tube and diluted to a final concentration of 0.1% SDS with ChIP lysis buffer. The chromatin immunoprecipitation (IP) was carried out with an antibody against H3K9ac at 4°C for 1 h followed by incubating with 30 μl Dynabead M-280 sheep anti-rabbit IgG (Invitrogen, 11203D) at 4°C overnight. The beads were washed twice with 1 ml cold Mixed Micelle Buffer (20 mM Tris–HCl [pH 8], 150 mM NaCl, 1% Triton X-100, 0.2% SDS, 5 mM EDTA, 65% sucrose), 1 ml cold Buffer 500 (25 mM HEPES, 500 mM NaCl, 1% Triton X-100, 0.1% Na deoxycholate, 10 mM Tris–HCl [pH 7.8], 1 mM EDTA), 1 ml cold LiCl/detergent wash (10 mM Tris–HCl [pH 8], 250 mM LiCl, 0.5% Na deoxycholate, 0.5% NP-40, 1 mM EDTA), and 1 ml cold TE buffer, respectively. Then, IP samples were eluted twice with 100 μl TE buffer set to a final concentration of 1% SDS at 65°C for 10 min at 1200 rpm. 200 μl IP sample was incubated at 65°C overnight for reverse cross-link. The IP sample was treated with RNase A at 37°C for 2 h, then 5 μl Proteinase K (Takara, #9034) was added into the IP sample for another 3 h at 55°C. DNA (IP DNA or input DNA) was precipitated with isopropyl alcohol and resuspended in 30 μl TE buffer. ChIP-seq libraries were produced using the VAHTS Universal DNA library prep kit for MGI (VAHTS, NDM607) according to the manufacture's protocol. Libraries were sequenced using the MGI seq2000 platform.
RNA isolation and quantitative PCR
For RT-qPCR analysis, apoptosis induced cells (in a 6-well plate) were collected in 600 μl TRIzol reagent, and transferred to a 1.5 ml tube. 200 μl chloroform was then added to each tube. All tubes were vortexed for 10 s, and incubated at room temperature for 15 min and then centrifuged at 15000 × g for 15 min at 4°C. The upper aqueous phase was removed (about 300 μl) and precipitated with 500 μl isopropyl alcohol at room temperature for 15 min. Samples were then centrifuged 20 000 × g for 15 min at 4°C. Pellets (RNA) were washed twice with 1 ml 80% cold ethanol, and air-dried at room temperature for 5 min. RNA pellets were resuspended in 200 μl of nuclease-free water. 500 ng RNA template were used for genome DNA wiping and cDNA synthesis using HiScript® II Reverse Transcriptase (R223-01, Vazyme). Each cDNA sample was diluted 1:10 in H2O and 2 μl of each diluted sample were used for each PCR. PCR (10 μl each) was performed in 384-well thermocycler plate using QuantStudio5 (Applied Biosystems). All PCR assays were performed in triplicate and were repeated using total RNA extracted from three independent samples. The primers used for RT-qPCR are listed in Supplementary Table S1.
For RT-PCR analysis in Supplementary Figures S2C, D and S4B, total RNA was isolated from corresponding transfected HEK293T cells. 500 ng RNA template was used for genome DNA wiping and cDNA synthesis using HiScript® II Reverse Transcriptase (R223-01, Vazyme). Nested PCR was performed, and primers used for PCR are listed in Supplementary Table S1. All PCR assays were performed in triplicate and were repeated using total RNA extracted from three independent samples.
For mRNA-seq, mRNA was isolated from 10 μg total RNA using NEBNext® Poly(A) mRNA Magnetic Isolation Module (E7490S) and dissolved in 30 μl nuclease-free water. 8 μl mRNA was pooled for library preparation using the VAHTS Universal V8 RNA-seq Library Prep Kit for MGI (VAHTS, NDM605) according to the manufacture's protocol. Libraries were sequenced using the MGI seq2000 platform.
For ChIP-qPCR analysis, HEK293T cells were seeded at 70% confluency prior to calcium phosphate-based transfection (described previously) in 15-cm plates. Cells were treated with DMSO or TG (described previously) at 24 h post transfection for 4 h. Next, chromatin was immunoprecipitated as described above. The immunoprecipitated DNA was dissolved in 30 μl H2O, the input DNA was dissolved in 100 μl H2O. 0.5 μl IP DNA was used for each PCR well. Input DNA was diluted 25-fold, and 1 μl was used for each PCR well. Three biological replicates were analyzed for each experiment.
Co-immunoprecipitation
Protein extracts (0.5 mg each sample) from transfected HEK293T cells (1 × 106) were diluted with 2 volumes cold TBS buffer (50 mM Tris–HCl, 150 mM NaCl, pH7.4). Anti-Flag M2 beads (20 μl each) were washed 3 times using TBS buffer before being added to the protein sample. All samples were gently rolled at 4°C for 4 h. The beads were then twice washed with cold TBS buffer. 30 μl TBS buffer and SDS PAGE loading buffer (50 mM Tris–HCl [pH6.8], 1 mM DTT, 20 g/l SDS, 0.1 g/l bromophenol blue, 50% glycerol) was added to beads and boiled for 10 min at 100°C. Samples were loaded and ran on a 10% SDS PAGE gel for 15 min at 100 V. All protein bands were cut out for mass spectrometry analysis. Mass spectrometry of the HDAC1 frameshifted peptide was performed without boiling and gel separation after IP.
Statistical analysis
Data from at least three biological replicates were used in the statistical analysis. The SPSS software was employed to compute the 95% (P< 0.05) or 99% (P< 0.01) confidence intervals of individual datasets and determine the statistically significant differences. Throughout the study, the comparison of 2 data sets was carried out with Student's t-tests, whereas the comparison of multiple data sets (>2) were carried out with one-way ANOVA.
Proteomic analyses
Genome-wide frameshifting peptide screening
Data description: The data contains the proteomes of 32 normal tissues from 14 normal individual humans (GTEx project with tandem mass tag (TMT) 10 plex/MS3 mass spectrometry strategy. Data resource: PXD016999). Specifically, the data contains two biological repeats. For each repeat, 28 MS runs were carried out to assess the proteomes of 32 organ samples, each of which contains organ samples from 8 individuals and mixture of the 8 samples (mega reference).
Reference making: We assume that CRFS events can only be detected from genes whose products can be identified by canonical translation. In the first-round searching, all annotated proteins (Uniport UP000005640) were set as reference for searching the expressed protein in different tissues. In the second-round searching, the reference contains all identified proteins of a given tissue (from first-round searching), as well as its corresponding +1 or −1 frameshifted protein, which are putatively frameshifted at all codon repeats (repeat number ≥ 3) of the expressed genes (Supplementary Table S2). The detected proteins from first-round searching was added as the background.
Searching method: All protein/trans-frame peptide identification were performed using MaxQuant (version 1.6.5.0) with settings (peptide false discovery rate (FDR) < 1% and minimum peptide length ≥ 7) as previously described (28), except that different proteome references were used (Supplementary Table S2). Raw files from 12 fractions of each sample were combined into a single searching. The searching criteria includes cysteine carbamidomethylation as a fixed modification. Peptide N-terminal and lysine TMT 10 plex modification, protein N-terminal acetylation and methionine oxidation were set as variable modifications. Up to two missed cleavages were allowed for trypsin digestion. Identified trans-frame or out-of-frame peptides were subject to whole annotate proteome (including unidentified proteins by first-round searching) to guarantee the sequence uniqueness. For a given biological repeat, which contains 28 MS runs with each run consisting of 8 samples and 2 mega references, if a trans-frame or out-of-frame peptide were detected in a given sample, it would be counted as 1 frameshifting event.
Frameshifting events were excluded in the following four conditions: (i) The detected frameshifting peptide is the same as the annotated protein (Uniport UP000005640). (ii) The intensity of trans-frame or out-of-frame peptides in tissue is lower than the average intensity of the two mega references. (iii) The trans-frame or out-of-frame peptides existed in the small peptide libraries (SmProt_MSdata, SmProt2_human_Ribo, and SmProt2_KnownDatabase). (iv) the frameshifting site (or codon repeat) of trans-frame peptide is on putative alternative splicing sites.
HDAC1 frameshifting peptide screening
Raw mass spectrometry files have been deposited to the ProteomeXchange Consortium, repository with the dataset identifier PXD043734. The reference proteomic databases contain HDAC1 in-frame proteins and trans-frame protein that is produced by +1FS at every codon of HDAC1 (Supplementary Table S3). Search analyses used MaxQuant (v2.2.0.0) (62) with built-in default parameters.
ChIP-seq data analysis
The raw reads (FASTQ) were mapped to the Homo Sapiens (GCRh38/hg38) genome using Bowtie2 (v2.3.5.1). Mapped reads were normalized by RPGC provided by command bamCoverage from deepTools (v3.5.1).
RNA-seq data analysis
The raw reads (FASTQ) were subjected to trim galore (version 0.6.7) and Bowtie2 (v2.3.5.1) for removing adaptors and ribosomal RNAs, respectively. The rest reads were mapped to Homo sapiens (GCRh38/hg38) reference genome using STAR (v2.7.9a). Read count extraction and normalization were performed using featureCounts (v2.0.1) to obtain gene expression levels.
Results
UUU codon repeats alone could trigger ribosomal frameshifting
To strictly evaluate ribosomal frameshifting events, we devised a dual luciferase reporter system with a self-cleaving peptide sequence P2A located in between the Renilla luciferase (RLuc) and the firefly luciferase (FLuc) gene (abbreviated as P2A system). Immediately in front of the test sequence we added 2 stop codons in the +1 and −1 frame, respectively, to cancel possible non-canonical translation occurring before the test sequence (Figure 1A). For each test sequence, to strictly normalize the luciferase signals so as to achieve accurate ribosomal frameshifting (FS) ratios, the control vector also harbors the same test sequence, except that the FLuc is placed in 0 frame (Supplementary Figure S2A). In HEK293T cells, the background FS ratios of this P2A system are 0.08% and 0.12% in the +1 and −1 frame, respectively (Supplementary Figure S2B). We next evaluated this system in HEK293T cells with the −1PRF site in HIV (40,63). As expected, robust −1FS ratio (9.64%) could be observed in the wild-type HIV sequence insertion, whereas either mutating the slippery site (SSm: UUUUUUA to CUUCUUA) or deleting the slippery site (SSd) abolished the −1FS (0.20% and 0.18%) (Figure 1B). For the human OAZ1 +1PRF site insertion (48,64), robust RLuc signals could be observed in both the control (0F, i.e. FLuc is in 0 frame) or the test construct (+1F, i.e. FLuc is in +1 frame) (Figure 1C, left). As expected, robust FLuc signal could only be detected from the test construct but not the control, since the +1PRF site harbors a 0-frame stop codon (UGA), which terminate the 0-frame translation before the FLuc (Figure 1C, right). Together, these data suggest that our P2A system is reliable to evaluate ribosomal frameshifting.
Given that pathological codon repeat expansion can cause random ribosomal frameshifting (59,60), we asked if short codon repeats, which are widely localized in human genome (Supplementary Figure S1), could also induce ribosomal frameshifting. We first tested the effect of UUU codon repeats in HEK293T cells, which have characteristics of a slippery site but lack of the downstream stimulator sequence. Surprisingly, (UUU)3 insertion showed modest but significant +1FS and −1FS (0.85% and 1.47%, P< 0.05) compared with the empty insertion, and the FS rate increased dramatically along with the increased number of UUU repeats (Figure 1D). Consistently, the frameshift product, FLuc, could also be robustly detected by western analyses in (UUU)4 and (UUU)5 insertion (Figure 1D).
To eliminate the possibility that the frameshifting signal was induced by uncanonical internal initiation or cryptic splicing events after UUU sequence insertion, a 0-frame stop codon (premature termination) was added in front of (UUU)4 or (UUU)5 insertion. We observed a substantial decrease of FS ratios in both insertions, indicating that there was no significant aberrant translational initiation in our P2A system (Figure 1E). Meanwhile, inserting a stop codon in the +1 frame or −1 frame after the insertions in the corresponding +1 or −1 frame also lead to substantial decrease of FS ratios, suggesting that detected FLuc signals should not be derived from cryptic splicing as previously described (54) (Figure 1E). Consistently, RT-PCR results with nested primers did not manifest cryptic splicing events in (UUU)4 and (UUU)5 constructs (Supplementary Figure S2C, D). Together, these data suggest that UUU codon repeats, as short as 3, might intrinsically trigger ribosomal frameshifting.
UUU repeats-induced ribosomal frameshifting could be recapitulated by different assays
As an important method to study ribosomal frameshifting, the fused dual luciferase reporter system has recently been questioned due to potential disturbances from cryptic splicing and/or altered luciferase activity (54,55). To rule out the possibility that fusion of inserted polyphenylalanine with FLuc might affect the luciferase activity and thus distort the frameshifting rate, we introduced UUU repeats into the dual StopGo (SG) reporter system as recently documented (55) (Figure 1F), which guarantee that both reporter proteins (RLuc and FLuc) are independent of the test sequence at the post-translational stage. As expected, the dual SG reporter system also showed robust ribosomal frameshifting as our P2A system (Figure 1G).
To rule out the possibility that the frameshifting is an artifact caused by the flanking luciferase gene sequences, we created another reporter system where the test sequence is inserted in between mCherry and EGFP, all of which are in 0 frame (Figure 1H). Since it is hard to evaluate the frameshifting ratio with the fluorescence intensity of GFP and mCherry, we turned to western analyses with mCherry antibody. For the control construct containing the +1PRF site of OAZ1 as the test sequence, canonical in a 30kD protein. If the +1PRF occurs, ribosomes will bypass the 0-frame stop codon, entering +1 frame (ORF2) and stop at the first +1-frame stop codon at the upstream EGFP, resulting in a 35 kDa protein (Figure 1H). Indeed, both the 30 kDa and 35 kDa bands were observed, with estimated frameshifting rate at ∼30% by normalizing the signals of 35 kDa (+1PRF) to 30 kDa bands (normal translation) (Figure 1I and Supplementary Figure S2E). Consistently, we did not detect the putative 45 kDa band that presumably derived from −1FS (Figure 1I), indicating that this assay could also accurately evaluate ribosomal frameshifting.
Using this assay, we tested the (UUU)5 insertion and observed 3 bands with expected sizes (Figure 1J). The 53 kDa band representing normal translation products (black arrow), as well as the 30 kDa (red arrow) and the 40 kDa (blue arrow) band representing +1FS and −1FS products, respectively (Figure 1H, J). Furthermore, to evaluate the FS ratios obtained from different reporter systems, we performed western analysis to evaluate the +1FS or −1FS ratios produced by the (UUU)5 insertion in the P2A system. After normalizing the level of RLuc and FLuc from the control (Fluc in 0 frame), the +1FS and −1FS ratio are 8.86% and 23%, respectively (Supplementary Figure S2F, G. Importantly, for both the +1FS and −1FS induced by the (UUU)5 insertion, all three different systems yield high FS ratios (Supplementary Figure S2G). Together, these data indicated that UUU codon repeats alone could trigger ribosomal frameshifting.
Short codon repeats might be slippery sites for ribosomal frameshifting in vivo
Compared with (UUU)5 repeats, the synonymous (UUC)5 repeats that feature no canonical slippery sequence also trigger significant +1FS (2.43%) and −1FS (2.91%) in the P2A system, respectively (Supplementary Figure S2J). We thus asked if other codon repeats might also trigger ribosomal frameshifting. Given that (UUU)5 manifests robust FS ratios in three different assays (Supplementary Figure S2G) and 5× codon repeats are common in the human genome (1402 sites in 707 genes) (Supplementary Figure S1), we preliminarily evaluated the frameshifting effects of all 61 codon types (excluding stop codons) by setting 5× repeat (GGG and CCC are 4× due to the limitation of primer syntheses) with our P2A dual luciferase assay. To our surprise, 46 types of codon repeats could significantly result in +1FS and/or −1FS (Supplementary Figure S2H, I and Supplementary Table S4). Among them, 13 types could induce robust +1 and/or −1 CRFS (>1%) (Figure 2A).
We next asked whether endogenous genes harboring codon repeats could also result in ribosomal frameshifting. Inspired by a recent finding that tryptophan starvation could induce ribosomal frameshifting in cancer cells at UGG codons (57), we choose the SPIDR gene, which harbors (UGG)5 repeats immediately before the stop codon (NM_001282919.1). If (UGG)5 could induce −1FS, translation will continue to stop at the −1 stop codon (stop codon located in −1 frame) which is 110 nt from the (UGG)5 repeats. To mimic the endogenous condition, a 149 nt fragment, consisting of (UGG)5 repeats as well as 24 nt upstream and 110 nt downstream sequence (to the nearest −1 stop codon), were inserted into the P2A luciferase reporter (Figure 2B, upper panel). As expected, we could observe the FLuc band by western analyses, suggesting that −1FS occurs. In contrast, deleting the (UGG)5 repeats from the 149 nt test sequence nearly abolished the FLuc signal (Figure 2B, lower panel). Analyses of the frameshifting ratios based on luciferase activities revealed a ∼2.5-fold higher (P< 0.01) level of FLuc in the wild-type (WT) SPIDR sequence, compared with that of the Δ(UGG)5 mutant sequence (Figure 2B). Together, these data suggest that codon repeats might serve as a mark to induce ribosomal frameshifting, hereafter termed codon repeat-induced ribosomal frameshifting or CRFS.
Codon repeats are commonly spread in the annotated human genome (GCRh38/hg38, Supplementary Figure S1). To quickly survey which genes might harbor CRFS, we assumed that translating ribosomes will shift at codon repeats, enter +1 and/or −1 frame and continue the translation until hitting the first stop codon in that frame, a region termed +1 or −1CRFS locus (Figure 2C). Consequently, this process will produce a chimeric protein, termed trans-frame protein, with a C-terminal sequence encoded by the +1 or −1 frame sequence. In proteomic analyses, trans-frame proteins were subject to trypsin digestion, resulting in two types of protein fragments bearing the information of ribosomal frameshifting. The trans-frame peptides are protein fragments with the N-terminus encoded by the 0 frame and C-terminus by the +1/−1 frame, whereas out-of-frame peptides are fully encoded by +1/−1 frame (Figure 2C). If these trans-frame proteins are relatively stable and abundant, the trans-frame and/or out-of-frame peptides should be detected from proteome data.
We first used the entire human proteome (UniPort UP000005640) to scan the public available tissue proteome data containing 32 normal human tissues collected from 14 individuals (65), which identified 12 690 annotated proteins when combining results of both biological repeats. We next created a trans-frame protein database based on the 26 749 codon repeats (≥3×) in the 12 690 proteins by assuming that frameshifting occurs at all positions of codon repeats, which resulted in a database containing 163 067 putative trans-frame proteins (Supplementary Table S2). We integrated this database with the expressed human proteome (12690 annotated proteins), then rescanned the proteome data. Collectively, 1890 trans-frame or out-of-frame peptides were obtained from the proteome of 32 human tissues, which in total belong to 706 unique frameshifted peptide sequences (Supplementary Table S5). To strictly evaluate these peptides, we carefully analyzed the sequences and excluded those sequences matching unexpressed annotated proteins (3%) or microproteins (smProt) (66) (17%). We also excluded those trans-frame peptide sequences matching the splicing junction sites (2%) (Figure 2D). Finally, we obtained 405 unique trans-frame peptides and 192 unique out-of-frame peptides.
These peptide sequences revealed 454 putative CRFS loci, of which 313 loci are directly supported by 405 trans-frame peptide sequences (Figure 2E). Twenty CRFS loci are supported by both trans-frame peptides and out-of-frame peptides (Figure 2E). Moreover, 91 CRFS loci could be supported by both biological repeats of proteome data, and CRFS-derived trans-frame peptides could be detected in all 32 tissues (Figure 2F, G and Supplementary Figure S3). Within them, 13 CRFS sites are supported by trans-frame peptides from more than half of the tissues (Figure 2H). Together, these data suggest that codon repeats, at least in some genes, might act as a cis-acting element that stimulates ribosomal frameshifting to produce stable trans-frame proteins.
Ribosomal frameshifting occurs in the upstream of HDAC1 mRNA
Within 13 identified CRFS candidates (Figure 2H), we noticed the +1CRFS in HDAC1, since its parental gene encodes the HDAC1, which is the first identified mammalian HDAC highly homologous to the yeast transcriptional regulator Rpd3 (67). In the past decades, the functional role of HDAC1 has been well-established in development and human diseases including cancers (68). Given that the putative trans-frame protein (termed HDAC1-FS, 29 a.a.) identified by mass spectrometry in HDAC1 is too small to detect by western analysis, we first designed a construct by fusing mCherry with full-length HDAC1 (1449 bp) and transfected HEK293T cells (Figure 3A). Western blot results reveal a major 82 kD band representing mCherry-HDAC1, as well as a small 30kD band whose size agrees with the putative +1CRFS product as identified in HDAC1 by mass spectrometry (26.73 kDa N-terminal mCherry +3.34 kDa HDAC1-FS) (Figure 3B). The ΔU43 of the HDAC1 plasmid was used as a positive control (Figure 3A, B). These data suggest that +1CRFS in HDAC1 mRNA might occur in the overexpression system.
Figure 3.

Ribosomal frameshifting efficiently occurs at the (UAC)3 repeat of HDAC1. (A) Cartoon showing constructs for mCherry-HDAC1 (mC-HDAC1) fusion protein. 4–88 nt mean 33 nt plus (UAC)3 repeat and 43 nt downstream of the (UAC)3 repeat. (*), in-frame stop codon. (B) Western blot detecting putative trans-frame proteins. The full length and trans-frame protein were marked by black and red arrows, respectively. (C) Frameshifting ratio of HDAC1 gene was detected by luciferase activity. INS, inserted HDAC1 fragment. 5′ ter and 3′ ter, premature stop at 5′ or 3′ of HDAC1 fragment, details in Figure S4A. (D) Diagrams of the truncated HDAC1 fusion constructs. BGH (Bovine growth hormone), transcription termination sequence. +1 and 0 stop codon mean the end of +1CRFS locus and end of protein, respectively. (E) Western blot detecting the HDAC1-FS trans-frame protein with constructs showed in this Figure (A and D) and Figure S4C. Protein produced by normal translation or +1 frameshifting were marked by black and red arrow, respectively. (F) Schematic diagram of dual fluorescent protein system with HDAC1 insert sequence and two stop codons in the +1 and −1 frame. (G) Flow cytometry analysis of HEK293T cells stably transformed with construct shown in (F). FITC-A and PE-Texas Red-A represent the fluorescence intensity of GFP and mCherry, respectively. (H) Diagram showing the construct for IP-MS and the experimental flow. (I) Table showing detected peptides related to CRFS. Relative positions shown in (H). (J) Diagram showing the frameshifting events at N-terminal 50 codons of HDAC1 from proteomic analyses. Pink and blue triangles represent +1 and −1 frameshifting positions, with number indicating the frequency of detected trans-frame peptides in all 32 tissues. The putative HDAC1-FS sequence resulted from +1CRFS, with representative mass spectrogram of trans-frame peptides is shown in bottom. (K) Heatmap showing the numbers of detected HDAC1 trans-frame peptides in proteomes of each 32 human tissues.
To verify the existence of +1CRFS in HDAC1, we cloned the 4–88 nt HDAC1 sequence consisting of a 33 nt upstream sequence (excluding the start codon) and 52 nt +1CRFS locus (from (UAC)3 repeats to the nearest stop codon in the +1 frame) (Figure 3A). We inserted this sequence into the P2A dual luciferase system and transfected this construct into HEK293T cells, which revealed a 16.9% frameshifting ratio based on luciferase activity (Figure 3C). Consistently, adding a premature stop codon before the insert sequence (5′ ter) or adding a +1 stop codon at the 3′ end of the CRFS locus (3′ ter) resulted in a sharp decrease of frameshifting ratios (Figure 3C and Supplementary Figure S4A). Meanwhile, no noticeable signals were detected other than the target band in RT-PCR results (Supplementary Figure S4B). These data suggest that the most detected FLuc signals should be derived from +1FS rather than aberrant translation initiation or cryptic splicing.
To further confirm this frameshifting event, we introduced a different system by using the CMV promoter to drive the expression of truncated HDAC1 fused with mCherry at the N-terminus. Here, the full-length fusion protein is 40.22 kDa, whereas +1 CRFS, if it occurs, will result in a 29.92 kDa protein (Figure 3D and Supplementary Figure S4C). As expected, we observed two bands in the western blots, with a frameshifting ratio estimated to be ∼30% (Figure 3E and Supplementary Figure S4D). Moreover, similar results could also be achieved from the same construct except using a weak human thymidine kinase (TK) promoter (Supplementary Figure S4E, F) (69). To rule out the possibility that such a ribosomal frameshifting might be an artifact from transient expression, the HDAC1 fragment (4–88nt, exclude start codon, Figure 3A) was inserted downstream of the P2A sequence in a HEK293T cell line that stably expresses a dual fluorescent reporter (EGFP in 0 frame and an mCherry (no start codon) in the +1 frame, Figure 3F). In this system, two stop codons individually at the +1 and −1 frame were inserted before the HDAC1 sequence to guarantee that the downstream mCherry reporter is only produced by +1FS (Figure 3F). By using fluorescence-activated cell sorting (FACS), our results showed that most of cells (97.5%, 82.6% and 69.9%, respectively) from three independently transformed cell lines have positive mCherry signals compared with the empty vector control (Figure 3G and Supplementary Figure S4G). Together, these data suggest that ribosomal frameshifting could efficiently occur within the inserted sequence of HDAC1.
(UAC)3 repeats are the dominant site for +1 ribosomal frameshifting in HDAC1
To pinpoint the cis-acting elements that stimulate ribosomal frameshifting in vivo, we designed another construct by introducing two stop codons individually in the +1 and −1 frame immediately after the start codon of HDAC1, and placed a Flag tag in the +1 frame after the HDAC1 (Figure 3H). Therefore, the Flag tag can only be expressed upon the occurrence of +1FS. After transfecting this construct into HEK293T cells, we performed immunoprecipitation with Flag antibodies and analyzed pulldown proteins with mass spectrometry (IP-MS) (Figure 3H). We identified three peptides resulting from +1FS with strong intensity, including one trans-frame peptide at (UAC)3 repeats (Figure 3I).
Limited by the depth of our mass spectrometry data, to further confirm if (UAC)3 codon repeats are the major site for +1FS, we specifically analyzed the first 50 codons of HDAC1 by building a database containing all putative trans-frame peptides resulting from +1FS and −1FS at every codon. We integrated this database with the entire expressed human proteome as described above and scanned the published proteome data containing normal human tissues (65). Indeed, 65% (97 out of 150) detected +1 trans-frame peptides are within or adjacent to (UAC)3 repeats in 25 out of 32 human tissues (Figure 3J,K). Consistently, deleting or mutating (UAC)3 repeats resulted in a significant drop of frameshifting ratios (Supplementary Figure S4H). Together, these results suggest that the (UAC)3 repeat should be a major site for +1FS in the HDAC1 mRNA.
HDAC1-FS protein might stably exist in the nucleus and cytoplasm
To further determine the existence of +1CRFS in HDAC1 and understand the subcellular localization of the HDAC1-FS trans-frame protein, we created a construct by fusing Flag tag and the N-terminus of HDAC1 (1–88nt) in the 0 frame with an AUG-free mCherry in the +1 frame (HDAC1-S-mC), thus allow the trans-frame protein to be visible upon the occurrence of +1CRFS (Figure 4A). To eliminate the possibility that the mCherry might be produced by alternative translation initiation from the +1 frame in front of the HDAC1, we inserted two +1 frame UAA stop codons in front of the HDAC1 fragment. As a control, we tagged the full-length HDAC1 with EGFP (HDAC1-EGFP) (Figure 4A).
Figure 4.

HDAC1-FS affects histone acetylation, cell migration and apoptosis. (A) Schematic diagrams of mCherry or EGFP constructs used to detect the protein localization. (B) Confocal microscopy showing subcellular localization of the HDAC1-FS and HDAC1 protein in HeLa cells with constructs shown in (A). (C) Metagene analyses of H3K9ac ChIP-seq with ectopic expression of 3× Flag-tagged HDAC1-FS, HDAC1 or empty vector (EV). (D) Wound closure assay showing the migration of A549 cells with ectopic expression of Flag-HDAC1-FS (left) and siRNA against HDAC1 (right). EV, empty vector. Error bars, standard deviation of three biological repeats. (**) P < 0.01 (one-way ANOVA). (E) Transwell assay showing the migration of A549 cells with ectopic expression of Flag-HDAC1-FS (left) and with siRNA against HDAC1 (right). EV, empty vector. Error bars, standard deviation of 3 biological repeats. (**) P < 0.01 (Student's t-test for 2 data sets and one-way ANOVA for >2 data sets). (F) Bar plot showing the changes of ribosomal frameshifting (FS) with thapsigargin (TG) or DMSO treatment of in HEK293T cells. The frameshifting ratios determined by P2A luciferase system. Error bars, standard deviation of 3 biological repeats. (**) P < 0.01(Student's t-test). (G) Apoptosis level measured by Annexin V-FITC/PI assay in thapsigargin (TG) treatment HEK293T cells. Three biological replicates were analyzed for each experiment in HEK293T cells. Error bars represent standard deviation. (**) P <0.01(Student's t-test). (H) Schematic diagram showing the experimental design for evaluating the effect of HDAC1-FS in cell apoptosis. (I) Representative western blot result detects HDAC1 protein after transformed with different constructs in wild-type (WT) HEK293T or HDAC1 KO HEK293T cells. EV, empty vector. NV, no transfection. (J) Flow cytometry analyses to TG-induced apoptosis in HEK293T cells transformed HDAC1(wt) and HDAC1(sm). Results are normalized with empty vector (EV). Error bars, standard deviation of three biological repeats. (**) P < 0.01 (Student's t-test).
We transfected these constructs into HeLa cells and observed mCherry signals in both nuclei and cytoplasm (Figure 4B). To better understand the subcellular localization, we co-transfected the HDAC1-EGFP plasmid with the HDAC1-S-mC plasmid. As expected, HDAC1-EGFP was shown to localize in the nucleus as previously described, due to the nuclear localization signal (NLS) sequence (Figure 4A) (70). However, for both constructs, no significant change of HDAC1-FS localization was observed (Figure 4B). In line with these observations, HDAC1-FS harbors no NLS sequence but could passively diffuse through the nuclear pore due to its small size (3.34 kDa) (71,72).
The strong HDAC1-FS-mCherry signals indicate that HDAC1-FS might be relatively stable in cells (Figure 4B). To further interrogate the stability of HDAC1-FS, we replaced the mCherry with a Hemagglutinin (HA) tag (9 a.a.) and transfected into HEK293T cells. As expected, with the treatment of cycloheximide (CHX), a protein synthesis inhibitor in eukaryotes, the decay rate of HDAC1-FS-HA is similar to that of a well-known reference protein, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (Supplementary Figure S4I, J). Together, these data suggest that the trans-frame protein HDAC1-FS could stably exist in both the nucleus and cytoplasm.
HDAC1-FS might act as an HDAC1 inhibitor
Given that a proportion of HDAC1-FS trans-frame proteins exist in the nucleus, to understand if HDAC1-FS protein might interact with HDAC1 in the nucleus, we overexpressed Flag-HDAC1-FS (ΔU43) in HEK293T cells and performed IP-MS assay with Flag antibodies, which identified 39 unique proteins (Supplementary Table S6). Among them, we noticed multiple HDAC-interacting proteins, especially HDAC1-interacting proteins (Supplementary Figure S5A, B), suggesting that HDAC1-FS might play a role in regulating deacetylation. However, the HDAC activity assay showed no noticeable difference between the mock (Flag) and synthesized HDAC1-FS peptides (Supplementary Figure S6A, B. We therefore hypothesized that HDAC1-FS might have more specific functions to regulate HDACs. Given that the HDAC1 has strong deacetylase activity against histone 3 lysine 9 acetylation (H3K9ac) (73), we ectopically expressed HDAC1-FS proteins in HEK293T cells and performed mRNA sequencing (RNA-seq) and chromatin immunoprecipitation sequencing (ChIP-seq) against H3K9ac. As expected, we observed a global increase of H3K9ac level (Figure 4C and Supplementary Figure S6C–E), as well as the increase of corresponding mRNA levels (Supplementary Figure S6F). These data suggest that at the molecular level, HDAC1-FS might act as an inhibitor against the histone deacetylation of HDAC1.
We next asked whether the inhibitory function of HDAC1-FS against HDAC1 could be observed at physiological level. The profound effect of HDAC1 on the regulation of gene transcription and many processes related to cell growth and apoptosis has been well-established in various cancers (74–77). Specifically, knockdown of HDAC1 could inhibit invasion and induces apoptosis in lung cancer cells (78). We selected A549 cells, an epithelial cell line derived from human lung cancer, and performed wound healing and transwell assays, two common approaches to investigate the cell migration in vitro. Specifically, the wound healing assay mimics cell migration during wound healing at 2D level (79), whereas the transwell assay mimics the process of cancer invasion by utilizing the chemotaxis of cells through a porous membrane (80). In line with previous results (78), knockdown of HDAC1 with siRNAs significantly inhibited cell migration in both assays (Supplementary Figure S7A, B and D). Surprisingly, cells overexpressing Flag-HDAC1-FS also resulted in significant suppressions of the cell migration, compared with the control (overexpressing Flag) (Figure 4D, E, and Supplementary Figure S7). In addition, similar inhibitory effect on cell migration could also be observed by using a weak TK promoter to drive the expression of Flag-HDAC1-FS (Supplementary Figure S8), further supporting the inhibitory role of HDAC-FS against HDAC1 in the cell migration.
We next interrogated the role of HDAC1-FS in the cell death. Gene ontology analyses also show that HDAC1-FS overexpression in HEK293T cells resulted in increased H3K9ac levels on apoptotic genes (Supplementary Figure S9A). We thus evaluated the frameshifting rate of the +1CRFS under a lethal concentration of thapsigargin (TG) treatment, known to trigger endoplasmic reticulum (ER) stress-induced apoptosis (81). As expected, both western analyses and the dual luciferase assay result manifested a significant increase of frameshifting under TG treatment (Figure 4F and Supplementary Figure S9B, C, indicating that HDAC1-FS level is increased under the stress conditions. In line with these results, compared with the control (EV), upon overexpressing HDAC1-FS we observed strong increases of apoptosis level in HEK293T cells (Figure 4G), as well as increased H3K9ac and mRNA levels in apoptotic genes (Supplementary Figure S9D, E. Together, these gain-of-function (overexpression) experiments suggest that HDAC1-FS likely acts as an inhibitor against the HDAC1.
To further confirms the function of HDAC1-FS in ER stress-induced apoptosis, we performed a loss-of-function experiment. We created a construct by introducing a synonymous mutation V19V (GUU to GUA) in the CRFS locus to generate a stop codon at the +1 frame. As such, the +1CRFS of HDAC1 here can only produce a truncated HDAC1-FS (18 a.a.), compared with full-length HDAC1-FS (29 a.a.) (Figure 4H). To best mimic the endogenous situation, we first knocked out the HDAC1 gene (HDAC1 KO) in HEK293T cell line by the CRISPR-Cas9 system. Then we selected the CMV promoter to drive the expression level of wild-type HDAC1, as well as that of a synonymous mutant HDAC1 (HDAC1sm), so as to keep the HDAC1 levels similar to that of wild type HEK293T cells (Figure 4H, I). We next tested the apoptosis level with flow cytometry. As expected, under TG treatment, compared with EV, we observed a 3-fold increase of apoptotic cells by ectopic expression of wild-type HDAC1. In contrast, compared with EV, we observed a 1.19-fold decrease of apoptotic cells upon ectopic expression of the mutant HDAC1sm (Figure 4J). Together, these data imply that HDAC1-FS may serve as a natural repressor to inhibit the function of HDAC1.
Discussion
Programmed ribosomal frameshifting has been well documented in viruses and bacteria, which produces trans-frame proteins essential for various biological processes (9). However, intriguingly in humans, except for retrotransposon-derived genes (45,82), only one case with strong evidence has been documented, i.e. +1PRF in OAZ1, which produces functional OAZ1 to down-regulate polyamine levels (47). Except for classic PRF, efficient ribosomal frameshifting could also occur within pathologically expanded codon repeats, which instead generate aberrant proteins (59–61). In this study, we unexpectedly found that normal codon repeats (≥3), widely existing in the human genome (Supplementary Figure S1), could trigger ribosomal frameshifting. We named this process as CRFS: codon repeats induced ribosomal frameshifting. Through multiple strictly controlled assays and using proteome data from normal human tissues, we uncovered many putative CRFS loci where codon repeats may serve as the frameshifting sites to produce trans-frame proteins. It implies that CRFS might represent a novel type of translational recoding and that it widely exists in human genes.
The dual luciferase assay has long stood as the cornerstone to quantify the efficiency of ribosomal frameshifting. However, recent studies with a modified structure (SG system, Figure 1F) generated controversial results (54,55). To investigate CRFS, we introduced dual luciferase reporters (the P2A and SG system) and dual fluorescent protein reporters (mC-E) to evaluate the frameshifting efficiency (Figure 1). Although 3 different assays manifest robust ribosomal frameshifting rates, the results are not equivalent (Supplementary Figure S2G). For example, for (UUU)5, the +1FS rate shown by the SG system is not only higher than the P2A system, but also ∼2-fold higher than quantifications of western blots. This discrepancy is likely derived from the background signals (no insertion), as the SG system appears to have a higher background than the P2A system in this case (Figure 1D, G, and Supplementary Figure S2B). In addition, different 2A sequences have different ‘cleavage’ activities (83), which might also contribute to the variation of frameshifting rates. In sum, these data suggest that for a given case, multiple methods should be applied to confirm the ribosomal frameshifting.
Different from viral-like −1PRF in humans (52), most of the identified codon repeats feature no canonical slippery sequence (Supplementary Table S5). In the −1CRFS locus of SPIDR, deleting the (UGG)5 codon repeat robustly reduces frameshifting efficiency (Figure 2B). Moreover, our IP-MS result also revealed that ribosomal frameshifting occurs at the (UAC)3 repeat in HDAC1 (Figure 3H, I). These data indicate that codon repeats might mark a new type of the cis-acting element that stimulates ribosomal frameshifting in human cells. Indeed, recent studies in yeasts revealed that certain codon combinations and poly(A) tracts could inhibit protein output by triggering ribosome stalling (84,85). Given that ribosome stalling is usually a prerequisite for ribosomal frameshifting (18,86), codon repeats might intrinsically cause stalling and hence induce frameshifting. However, since deletion or mutation of codon repeats does not totally abolish ribosomal frameshifting (Figure 2B and Supplementary Figure S4H), the mechanism of how CRFS is triggered warrants further investigation.
In this study, a large number of trans-frame peptides were identified from proteomes of normal human tissues, implying that at least some trans-frame proteins might be stable in tissues. In the case of HDAC1, trans-frame protein HDAC1-FS was identified in many tissues (Figure 3K). Considering that HDAC1-FS sequence is conserved in mammals (Supplementary Figure S9F), we further investigated the function of HDAC1-FS, which might serve as a natural repressor to inhibit the deacetylation function of HDAC1 at H3K9ac, thus suppressing cell migration and promoting apoptosis of lung cancer cells (Figure 4 and Supplementary Figures S6–S9). Given that many trans-frame proteins were identified from normal human tissues by mass spectrometry, it indicates that CRFS might play a role in many biological processes via producing trans-frame proteins with regulatory function.
Recently advances in sequencing technologies revealed that eukaryotic mRNAs could also be polycistronic, with one mRNA harboring multiple ORFs (87–91). Along with the main ORF, small open reading frames (smORFs) could be located upstream, downstream, and/or overlap with the annotated main ORFs. These alternative products were shown to positively or negatively regulate the activity of other proteins (92). Our results suggest that CRFS and its products might function as a new layer of translation regulation. Indeed, recent studies showed that synonymous mutations are non-neutral and associated with various human disease (93,94). Since the C-terminus of trans-frame protein is encoded by the +1 or −1 frame, synonymous mutation may result in silencing or nonsense mutation of trans-frame protein, and thus have profound effects during human development and diseases.
Supplementary Material
Acknowledgements
We acknowledge the Dang and Lai lab members for helping many sequencing works. We also thank Dr Xuna Wu (Yunnan University) for suggestions for mass spectrometry analysis. We thank Dr Leonard Krall (Yunnan University) for editing the manuscript.
Author contributions: Y. Dang and F. Lai conceived and supervised the overall project. G. Ren and X. Gu performed most of the experiments. L. Zhang, S. Gong, S. Song and Y. Dang performed bioinformatics analyses. G. Ren, X. Gu, Z. Chen, S. Chen, X. Wang, Y. Zhou and J. Yang prepared all constructs for frameshifing rate detection. Z. Li, G. Ren and L. Li prepared DNA and RNA libraries. S. Chen prepared samples for the high-throughput sequencing. Y. Dang, F. Lai, G. Ren, X. Gu and L. Zhang wrote the manuscript with critical feedback from all co-authors.
Contributor Information
Guiping Ren, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Xiaoqian Gu, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Lu Zhang, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Shimin Gong, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Shuang Song, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Shunkai Chen, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Zhenjing Chen, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Xiaoyan Wang, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Zhanbiao Li, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Yingshui Zhou, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Longxi Li, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Jiao Yang, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Fan Lai, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Yunkun Dang, State Key Laboratory for Conservation and Utilization of Bio-Resource in Yunnan, Key Laboratory for Southwest Microbial Diversity of the Ministry of Education, Yunnan Key Laboratory of Cell Metabolism and Diseases, Center for Life Science, School of Life Sciences, Yunnan University, Kunming 650021, China; Southwest United Graduate School, Kunming, 650092, China.
Data availability
All sequencing data generated in this study were deposited in NCBI GEO as accession number of GSE220678. Published proteomic data are available from ProteomeXchange Consortium PXD016999. Mass spectrometry data generated in this study have been deposited at ProteomeXchange Consortium (PXD043734). All customized codes and pipelines for bioinformatic analyses are available in GitHub (https://github.com/lu-1023). Data is also available in Zenodo: ChIP-seq (doi.org/10.5281/zenodo.10467767); RNA-seq (doi.org/10.5281/zenodo.10467769); and Mass spectrometry (doi.org/10.5281/zenodo.10464160).
Supplementary data
Supplementary Data are available at NAR Online.
Funding
National Natural Science Foundation of China [32171262, 31871254 to Y.D., 32070626 to F.L., 32002080 to G.R.]; Open Research Program of State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan [2023KF009 to Y.D., 2021KF002 to F.L.]; Yunnan Province Science and Technology Department [2019FY003020 to Y.D., 202201BF070001-015 to F.L., 202001BB050013 to G.R.]; startup funds from Yunnan University (to Y.D., F.L.). Funding for open access charge: National Natural Science Foundation of China [32171262, 31871254 to Y.D., 32070626 to F.L., 32002080 to G.R.]; Open Research Program of State Key Laboratory for Conservation and Utilization of Bio-Resources in Yunnan [2023KF009 to Y.D., 2021KF002 to F.L.]; startup funds from Yunnan University (to Y.D., F.L.)
Conflict of interest statement. None declared.
References
- 1. Jackson R.J., Hellen C.U.T., Pestova T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010; 11:113–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gesteland R.F., Atkins J.F. Recoding: dynamic reprogramming of translation. Annu. Rev. Biochem. 1996; 65:741–768. [DOI] [PubMed] [Google Scholar]
- 3. Rodnina M.V., Korniy N., Klimova M., Karki P., Peng B.Z., Senyushkina T., Belardinelli R., Maracci C., Wohlgemuth I., Samatova E. et al. Translational recoding: canonical translation mechanisms reinterpreted. Nucleic Acids Res. 2020; 48:1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Beznoskova P., Cuchalova L., Wagner S., Shoemaker C.J., Gunisova S., von der Haar T., Valasek L.S. Translation initiation factors eIF3 and HCR1 control translation termination and stop codon read-through in yeast cells. PLoS Genet. 2013; 9:e1003962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Namy O., Hatin I., Rousset J.P. Impact of the six nucleotides downstream of the stop codon on translation termination. EMBO Rep. 2001; 2:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang W.M., Ao S.Z., Casjens S., Orlandi R., Zeikus R., Weiss R., Winge D., Fang M. A persistent untranslated sequence within bacteriophage-T4 DNA topoisomerase gene-60. Science. 1988; 239:1005–1012. [DOI] [PubMed] [Google Scholar]
- 7. Jacks T., Varmus H.E. Expression of the Rous-Sarcoma Virus Pol gene by ribosomal frameshifting. Science. 1985; 230:1237–1242. [DOI] [PubMed] [Google Scholar]
- 8. Dinman J.D. Mechanisms and implications of programmed translational frameshifting. WIREs RNA. 2012; 3:661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Atkins J.F., Loughran G., Bhatt P.R., Firth A.E., Baranov P.V. Ribosomal frameshifting and transcriptional slippage: from genetic steganography and cryptography to adventitious use. Nucleic Acids Res. 2016; 44:7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Peng B.Z., Bock L.V., Belardinelli R., Peske F., Grubmüller H., Rodnina M.V. Active role of elongation factor G in maintaining the mRNA reading frame during translation. Sci. Adv. 2019; 5:eaax8030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Poulis P., Patel A., Rodnina M.V., Adio S. Altered tRNA dynamics during translocation on slippery mRNA as determinant of spontaneous ribosome frameshifting. Nat. Commun. 2022; 13:4231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gamper H.B., Masuda I., Frenkel-Morgenstern M., Hou Y.M. Maintenance of protein synthesis reading frame by EF-P and mG37-tRNA. Nat. Commun. 2015; 6:7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hoffer E.D., Hong S., Sunita S., Maehigashi T., Gonzalez R.L., Whitford P.C., Dunham C.M. Structural insights into mRNA reading frame regulation by tRNA modification and slippery codon-anticodon pairing. eLife. 2020; 9:e51898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gamper H.B., Masuda I., Frenkel-Morgenstern M., Hou Y.M. The UGG isoacceptor of tRNAPro is naturally prone to frameshifts. Int. J. Mol. Sci. 2015; 16:14866–14883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kurland C.G. Translational accuracy and the fitness of bacteria. Annu. Rev. Genet. 1992; 26:29–50. [DOI] [PubMed] [Google Scholar]
- 16. Parker J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989; 53:273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tsuchihashi Z., Brown P.O. Sequence requirements for efficient translational frameshifting in the Escherichia coli dnaX gene and the role of an unstable interaction between tRNA(Lys) and an AAG lysine codon. Genes Dev. 1992; 6:511–519. [DOI] [PubMed] [Google Scholar]
- 18. Korniy N., Samatova E., Anokhina M.M., Peske F., Rodnina M.V. Mechanisms and biomedical implications of −1 programmed ribosome frameshifting on viral and bacterial mRNAs. FEBS Lett. 2019; 593:1468–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brierley I., Jenner A.J., Inglis S.C. Mutational analysis of the slippery-sequence component of a coronavirus ribosomal frameshifting signal. J. Mol. Biol. 1992; 227:463–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Farabaugh P.J. Programmed translational frameshifting. Annu. Rev. Genet. 1996; 30:507–528. [DOI] [PubMed] [Google Scholar]
- 21. Plant E.P., Dinman J.D. Comparative study of the effects of heptameric slippery site composition on −1 frameshifting among different eukaryotic systems. RNA. 2006; 12:666–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Caliskan N., Peske F., Rodnina M.V. Changed in translation: mRNA recoding by −1 programmed ribosomal frameshifting. Trends Biochem. Sci. 2015; 40:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Larsen B., Gesteland R.F., Atkins J.F. Structural probing and mutagenic analysis of the stem-loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50%. J. Mol. Biol. 1997; 271:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Brierley I., Digard P., Inglis S.C. Characterization of an efficient coronavirus ribosomal frameshifting signal -requirement for an RNA pseudoknot. Cell. 1989; 57:537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Namy O., Moran S.J., Stuart D.I., Gilbert R.J.C., Brierley I. A mechanical explanation of RNA pseudoknot function in programmed ribosomal frameshifting. Nature. 2006; 441:244–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Belew A.T., Meskauskas A., Musalgaonkar S., Advani V.M., Sulima S.O., Kasprzak W.K., Shapiro B.A., Dinman J.D. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature. 2014; 512:265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang X., Xuan Y., Han Y., Ding X., Ye K., Yang F., Gao P., Goff S.P., Gao G. Regulation of HIV-1 gag-Pol Expression by shiftless, an inhibitor of programmed −1 ribosomal frameshifting. Cell. 2019; 176:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Napthine S., Treffers E.E., Bell S., Goodfellow I., Fang Y., Firth A.E., Snijder E.J., Brierley I. A novel role for poly(C) binding proteins in programmed ribosomal frameshifting. Nucleic Acids Res. 2016; 44:5491–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caliskan N., Katunin V.I., Belardinelli R., Peske F., Rodnina M.V. Programmed-1 frameshifting by kinetic partitioning during impeded translocation. Cell. 2014; 157:1619–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kim H.K., Liu F., Fei J., Bustamante C., Gonzalez R.L. Jr, Tinoco I. Jr A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U.S.A. 2014; 111:5538–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Choi J., O’Loughlin S., Atkins J.F., Puglisi J.D. The energy landscape of −1 ribosomal frameshifting. Sci. Adv. 2020; 6:eaax6969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen J., Petrov A., Johansson M., Tsai A., O’Leary S.E., Puglisi J.D. Dynamic pathways of −1 translational frameshifting. Nature. 2014; 512:328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bock L.V., Caliskan N., Korniy N., Peske F., Rodnina M.V., Grubmuller H. Thermodynamic control of −1 programmed ribosomal frameshifting. Nat. Commun. 2019; 10:4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Caliskan N., Wohlgemuth I., Korniy N., Pearson M., Peske F., Rodnina M.V. Conditional switch between frameshifting regimes upon translation of dnaX mRNA. Mol. Cell. 2017; 66:558–567. [DOI] [PubMed] [Google Scholar]
- 35. Gamper H., Masuda I., Hou Y.M. Genome expansion by tRNA+1 frameshifting at quadruplet codons. J. Mol. Biol. 2022; 434:167440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gamper H., Mao Y.J., Masuda I., McGuigan H., Blaha G., Wang Y.H., Xu S.J., Hou Y.M. Twice exploration of tRNA+1 frameshifting in an elongation cycle of protein synthesis. Nucleic Acids Res. 2021; 49:10046–10060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gamper H., Li H.X., Masuda I., Robkis D.M., Christian T., Conn A.B., Blaha G., Petersson E.J., Gonzalez R.L., Hou Y.M. Insights into genome recoding from the mechanism of a classic+1-frameshifting tRNA. Nat. Commun. 2021; 12:328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gallant J.A., Lindsley D Leftward ribosome frameshifting at a hungry codon. J. Mol. Biol. 1992; 223:31–40. [DOI] [PubMed] [Google Scholar]
- 39. Temperley R., Richter R., Dennerlein S., Lightowlers R.N., Chrzanowska-Lightowlers Z.M. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010; 327:301. [DOI] [PubMed] [Google Scholar]
- 40. Jacks T., Power M.D., Masiarz F.R., Luciw P.A., Barr P.J., Varmus H.E. Characterization of ribosomal frameshifting in HIV-1 Gag-Pol expression. Nature. 1988; 331:280–283. [DOI] [PubMed] [Google Scholar]
- 41. Kelly J.A., Woodside M.T., Dinman J.D. Programmed −1 ribosomal frameshifting in coronaviruses: a therapeutic target. Virology. 2021; 554:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tsuchihashi Z., Kornberg A. Translational frameshifting generates the gamma-subunit of dna polymerase-iii holoenzyme. Proc. Natl. Acad. Sci. U.S.A. 1990; 87:2516–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Devaraj A., Fredrick K. Short spacing between the shine-dalgarno sequence and P codon destabilizes codon-anticodon pairing in the P site to promote+1 programmed frameshifting. Mol. Microbiol. 2010; 78:1500–1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meydan S., Klepacki D., Karthikeyan S., Margus T., Thomas P., Jones J.E., Khan Y., Briggs J., Dinman J.D., Vazquez-Laslop N. et al. Programmed ribosomal frameshifting generates a copper transporter and a copper chaperone from the same gene. Mol. Cell. 2017; 65:207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Clark M.B., Janicke M., Gottesbuhren U., Kleffmann T., Legge M., Poole E.S., Tate W.P. Mammalian gene PEG10 expresses two reading frames by high efficiency-1 frameshifting in embryonic-associated tissues. J. Biol. Chem. 2007; 282:37359–37369. [DOI] [PubMed] [Google Scholar]
- 46. Ivanov I.P., Atkins J.F. Ribosomal frameshifting in decoding antizyme mRNAs from yeast and protists to humans: close to 300 cases reveal remarkable diversity despite underlying conservation. Nucleic Acids Res. 2007; 35:1842–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ivanov I.P., Gesteland R.F., Atkins J.F. Antizyme expression: a subversion of triplet decoding, which is remarkably conserved by evolution, is a sensor for an autoregulatory circuit. Nucleic Acids Res. 2000; 28:3185–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Matsufuji S., Matsufuji T., Miyazaki Y., Murakami Y., Atkins J.F., Gesteland R.F., Hayashi S. Autoregulatory frameshifting in decoding mammalian ornithine decarboxylase antizyme. Cell. 1995; 80:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kurian L., Palanimurugan R., Godderz D., Dohmen R.J. Polyamine sensing by nascent ornithine decarboxylase antizyme stimulates decoding of its mRNA. Nature. 2011; 477:490–494. [DOI] [PubMed] [Google Scholar]
- 50. Ichiba T., Matsufuji S., Miyazaki Y., Murakami Y., Tanaka K., Ichihara A., Hayashi S. Functional regions of ornithine decarboxylase antizyme. Biochem. Biophys. Res. Commun. 1994; 200:1721–1727. [DOI] [PubMed] [Google Scholar]
- 51. Kang B., Jiang D.M., He H., Ma R., Yi Z.X., Chen Z.Y. Characterization of OAZ1 and its potential functions in goose follicular development. Electron. J. Biotechn. 2017; 26:1–6. [Google Scholar]
- 52. Belew A.T., Hepler N.L., Jacobs J.L., Dinman J.D. PRFdb: a database of computationally predicted eukaryotic programmed-1 ribosomal frameshift signals. Bmc Genomics [Electronic Resource]. 2008; 9:339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jacobs J.L., Belew A.T., Rakauskaite R., Dinman J.D. Identification of functional, endogenous programmed-1 ribosomal frameshift signals in the genome of Saccharomyces cerevisiae. Nucleic Acids Res. 2007; 35:165–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Khan Y.A., Loughran G., Steckelberg A.L., Brown K., Kiniry S.J., Stewart H., Baranov P.V., Kieft J.S., Firth A.E., Atkins J.F. Evaluating ribosomal frameshifting in CCR5 mRNA decoding. Nature. 2022; 604:E16–E23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Loughran G., Fedorova A.D., Khan Y.A., Atkins J.F., Baranov P.V. Lack of evidence for ribosomal frameshifting in ATP7B mRNA decoding. Mol. Cell. 2022; 82:3745–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Bartok O., Pataskar A., Nagel R., Laos M., Goldfarb E., Hayoun D., Levy R., Korner P.R., Kreuger I.Z.M., Champagne J. et al. Anti-tumour immunity induces aberrant peptide presentation in melanoma. Nature. 2021; 590:332–337. [DOI] [PubMed] [Google Scholar]
- 57. Champagne J., Pataskar A., Blommaert N., Nagel R., Wernaart D., Ramalho S., Kenski J., Bleijerveld O.B., Zaal E.A., Berkers C.R. et al. Oncogene-dependent sloppiness in mRNA translation. Mol. Cell. 2021; 81:4709–4721. [DOI] [PubMed] [Google Scholar]
- 58. Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007; 30:575–621. [DOI] [PubMed] [Google Scholar]
- 59. Girstmair H., Saffert P., Rode S., Czech A., Holland G., Bannert N., Ignatova Z. Depletion of cognate charged transfer RNA causes translational frameshifting within the expanded CAG stretch in huntingtin. Cell Rep. 2013; 3:148–159. [DOI] [PubMed] [Google Scholar]
- 60. Saffert P., Adamla F., Schieweck R., Atkins J.F., Ignatova Z. An expanded CAG repeat in huntingtin causes +1 frameshifting. J. Biol. Chem. 2016; 291:18505–18513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wright S.E., Rodriguez C.M., Monroe J., Xing J., Krans A., Flores B.N., Barsur V., Ivanova M.I., Koutmou K.S., Barmada S.J. et al. CGG repeats trigger translational frameshifts that generate aggregation-prone chimeric proteins. Nucleic Acids Res. 2022; 50:8674–8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tyanova S., Temu T., Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat. Protoc. 2016; 11:2301–2319. [DOI] [PubMed] [Google Scholar]
- 63. Horsfield J.A., Wilson D.N., Mannering S.A., Adamski F.M., Tate W.P. Prokaryotic ribosomes recode the HIV-1 Gag-Pol-1 frameshift sequence by an E/P site post-translocation simultaneous slippage mechanism. Nucleic Acids Res. 1995; 23:1487–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Rom E., Kahana C. Polyamines regulate the expression of ornithine decarboxylase antizyme in vitro by inducing ribosomal frame-shifting. Proc. Natl. Acad. Sci. U.S.A. 1994; 91:3959–3963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Jiang L., Wang M., Lin S., Jian R., Li X., Chan J., Dong G., Fang H., Robinson A.E., GTEx Consortium et al. A quantitative proteome map of the Human body. Cell. 2020; 183:269–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li Y., Zhou H., Chen X., Zheng Y., Kang Q., Hao D., Zhang L., Song T., Luo H., Hao Y. et al. SmProt: a reliable repository with comprehensive annotation of small proteins identified from ribosome profiling. Genomics Proteomics Bioinformatics. 2021; 19:602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Taunton J., Hassig C.A., Schreiber S.L. A mammalian histone deacetylase related to the yeast transcriptional regulator Rpd3p. Science. 1996; 272:408–411. [DOI] [PubMed] [Google Scholar]
- 68. Haberland M., Montgomery R.L., Olson E.N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 2009; 10:32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Arcot S.S., Flemington E.K., Deininger P.L. The Human thymidine kinase gene promoter-feletion analysis and specific protein-binding. J. Biol. Chem. 1989; 264:2343–2349. [PubMed] [Google Scholar]
- 70. Zhu Y.J., Vidaurre O.G., Adula K.P., Kezunovic N., Wentling M., Huntley G.W., Casaccia P. Subcellular distribution of HDAC1 in neurotoxic conditions is dependent on serine phosphorylation. J. Neurosci. 2017; 37:7547–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Bonner W.M. Protein migration into nuclei. II. frog oocyte nuclei accumulate a class of microinjected oocyte nuclear proteins and exclude a class of microinjected oocyte cytoplasmic proteins. J. Cell Biol. 1975; 64:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Paine P.L., Moore L.C., Horowitz S.B. Nuclear-envelope permeability. Nature. 1975; 254:109–114. [DOI] [PubMed] [Google Scholar]
- 73. Moreno-Yruela C., Zhang D., Wei W., Bæk M., Liu W.C., Gao J.J., Danková D., Nielsen A.L., Bolding J.E., Yang L. et al. Class I histone deacetylases (HDAC1-3) are histone lysine delactylases. Sci. Adv. 2022; 8:eabi6696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Falkenberg K.J., Johnstone R.W. Histone deacetylases and their inhibitors in cancer, neurological diseases and immune disorders. Nat. Rev. Drug Discov. 2015; 14:219–219. [DOI] [PubMed] [Google Scholar]
- 75. Lagger G., O’Carroll D., Rembold M., Khier H., Tischler J., Weitzer G., Schuettengruber B., Hauser C., Brunmeir R., Jenuwein T. et al. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002; 21:2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Senese S., Zaragoza K., Minardi S., Muradore I., Ronzoni S., Passafaro A., Bernard L., Draetta G.F., Alcalay M., Seiser C. et al. Role for histone deacetylase 1 in human tumor cell proliferation. Mol. Cell Biol. 2007; 27:4784–4795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bhaskara S. Histone deacetylases 1 and 2 regulate DNA replication and DNA repair: potential targets for genome stability-mechanism-based therapeutics for a subset of cancers. Cell Cycle. 2015; 14:1779–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zhang L., Bu L., Hu J., Xu Z., Ruan L., Fang Y., Wang P. HDAC1 knockdown inhibits invasion and induces apoptosis in non-small cell lung cancer cells. Biol. Chem. 2018; 399:603–610. [DOI] [PubMed] [Google Scholar]
- 79. Liang C.C., Park A.Y., Guan J.L. Scratch assay:: a convenient and inexpensive method for analysis of cell migration. Nat. Protoc. 2007; 2:329–333. [DOI] [PubMed] [Google Scholar]
- 80. Justus C.R., Marie M.A., Sanderlin E.J., Yang L.V. Transwell in vitro cell migration and invasion assays. Methods Mol. Biol. 2023; 2644:349–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Martinez T.F., Chu Q., Donaldson C., Tan D., Shokhirev M.N., Saghatelian A. Accurate annotation of human protein-coding small open reading frames. Nat. Chem. Biol. 2020; 16:458–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Manktelow E., Shigemoto K., Brierley I. Characterization of the frameshift signal of Edr, a mammalian example of programmed −1 ribosomal frameshifting. Nucleic Acids Res. 2005; 33:1553–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Vaseghi H.R., Qian L., Liu J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017; 7:2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Tesina P., Lessen L.N., Buschauer R., Cheng J.D., Wu C.C.C., Berninghausen O., Buskirk A.R., Becker T., Beckmann R., Green R. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 2020; 39:e103365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Gamble C.E., Brule C.E., Dean K.M., Fields S., Grayhack E.J. Adjacent codons act in concert to modulate translation efficiency in yeast. Cell. 2016; 166:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Rodnina M., Korniy N., Klimova M., Karki P., Peng B., Senyushkina T., Belardinelli R., Maracci C., Wohlgemuth I., Samatova E. et al. Translational recoding: canonical translation mechanisms reinterpreted. Nucleic Acids Res. 2020; 48:1056–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Chen J., Brunner A.D., Cogan J.Z., Nunez J.K., Fields A.P., Adamson B., Itzhak D.N., Li J.Y., Mann M., Leonetti M.D. et al. Pervasive functional translation of noncanonical human open reading frames. Science. 2020; 367:1140–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Cao X., Khitun A., Harold C.M., Bryant C.J., Zheng S.J., Baserga S.J., Slavoff S.A. Nascent alt-protein chemoproteomics reveals a pre-60S assembly checkpoint inhibitor. Nat. Chem. Biol. 2022; 18:643–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Cao X., Khitun A., Luo Y., Na Z., Phoodokmai T., Sappakhaw K., Olatunji E., Uttamapinant C., Slavoff S.A. Alt-RPL36 downregulates the PI3K-AKT-mTOR signaling pathway by interacting with TMEM24. Nat. Comm. 2021; 12:508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Na Z., Dai X., Zheng S.J., Bryant C.J., Loh K.H., Su H., Luo Y., Buhagiar A.F., Cao X., Baserga S.J. et al. Mapping subcellular localizations of unannotated microproteins and alternative proteins with MicroID. Mol. Cell. 2022; 82:2900–2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Prensner J.R., Enache O.M., Luria V., Krug K., Clauser K.R., Dempster J.M., Karger A., Wang L., Stumbraite K., Wang V.M. et al. Noncanonical open reading frames encode functional proteins essential for cancer cell survival. Nat. Biotechnol. 2021; 39:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Couso J.P., Patraquim P. Classification and function of small open reading frames. Nat. Rev. Mol. Cell Biol. 2017; 18:575–589. [DOI] [PubMed] [Google Scholar]
- 93. Sauna Z.E., Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat. Rev. Genet. 2011; 12:683–691. [DOI] [PubMed] [Google Scholar]
- 94. Shen X., Song S., Li C., Zhang J. Synonymous mutations in representative yeast genes are mostly strongly non-neutral. Nature. 2022; 606:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in this study were deposited in NCBI GEO as accession number of GSE220678. Published proteomic data are available from ProteomeXchange Consortium PXD016999. Mass spectrometry data generated in this study have been deposited at ProteomeXchange Consortium (PXD043734). All customized codes and pipelines for bioinformatic analyses are available in GitHub (https://github.com/lu-1023). Data is also available in Zenodo: ChIP-seq (doi.org/10.5281/zenodo.10467767); RNA-seq (doi.org/10.5281/zenodo.10467769); and Mass spectrometry (doi.org/10.5281/zenodo.10464160).