

Abstract
Humans lacking heme oxygenase 1 (HMOX1) display growth retardation, haemolytic anaemia, and vulnerability to stress; however, cardiac function remains unclear. We aimed to explore the cardiac function of zebrafish lacking hmox1a at baseline and in response to stress. We generated zebrafish hmox1a mutants using CRISPR/Cas9 genome editing technology. Deletion of hmox1a increases cardiac output and further induces hypertrophy in adults. Adults lacking hmox1a develop myocardial interstitial fibrosis, restrain cardiomyocyte proliferation and downregulate renal haemoglobin and cardiac antioxidative genes. Larvae lacking hmox1a fail to respond to hypoxia, whereas adults are insensitive to isoproterenol stimulation in the heart, suggesting that hmox1a is necessary for cardiac response to stress. Haplodeficiency of hmox1a stimulates non‐mitochondrial respiration and cardiac cell proliferation, increases cardiac output in larvae in response to hypoxia, and deteriorates cardiac function and structure in adults upon isoproterenol treatment. Intriguingly, haplodeficiency of hmox1a upregulates cardiac hmox1a and hmox1b in response to isoproterenol. Collectively, deletion of hmox1a results in cardiac remodelling and abrogates cardiac response to hypoxia and isoproterenol. Haplodeficiency of hmox1a aggravates cardiac response to the stress, which could be associated with the upregulation of hmox1a and hmox1b. Our data suggests that HMOX1 homeostasis is essential for maintaining cardiac function and promoting cardioprotective effects.
Keywords: cardiac hypertrophy, cardiac output, heme oxygenase 1, interstitial fibrosis
1. INTRODUCTION
Heme oxygenase 1 (HMOX1), a stress‐inducible protein, degrades heme into bioactive signalling molecules carbon monoxide and biliverdin while simultaneously releasing iron. 1 Human HMOX1 deficiency is a rare autosomal recessive disorder with hallmark features of haemolytic anaemia, inflammation of multiple organs and vasculature, iron deposition, interstitial fibrosis and growth retardation. 2 , 3 , 4 , 5 An autopsy of a 6‐year‐old boy lacking HMOX1 revealed left ventricular hypertrophy. 6 In addition, a long GTn polymorphism in the promoter of HMOX1, exhibiting low transcriptional activity, is associated with cardiovascular diseases. 7 Consistent with HMOX1‐deficient patients, HMOX1‐deficient rodents displayed reduced body weight, anaemia, chronic inflammation and dysfunction of multiple organs. 8 , 9 , 10 Thus, the lack of HMOX1 appears to cause detrimental effects on mammalian tissues.
During the past two decades, the cardioprotective effect of HMOX1 induction has been extensively explored. 11 , 12 , 13 Overexpression of HMOX1 before ischemia/reperfusion (I/R) reduces cardiac ischaemic damage, whereas inhibition of HMOX1 activity deteriorates I/R injury. 14 , 15 In addition, mice with global and cardiac‐specific overexpression or activation of HMOX1 exhibit markedly lower infarct size and improved cardiac function after cardiac injury. 16 , 17 , 18 In contrast, the lack of HMOX1 in mice results in increased myocardial damage after I/R. 19 Cardioprotective effects of HMOX1 are mediated by diverse mechanisms, including reduction in oxidative stress, inhibition of apoptosis and modulation of mitochondrial function. 20 , 21
Although the cardioprotective effects of HMOX1 exist under diverse stress conditions, induction of HMOX1 has been associated with the development of chronic viral myocarditis. 22 Systemic overexpression of HMOX1 aggravates cardiac hypertrophy induced by pressure overload and fails to prevent arterial dysfunction upon injury in mice. 23 , 24 Despite the association of HMOX1 with protection against oxidative stress, 25 excess HMOX1 expression contributes to apoptosis mediated by oxidative stress. 22 HMOX1 appears to mediate pathological crosstalk between macrophages and cardiomyocytes, resulting in increased oxidative stress and cardiomyocyte apoptosis, consequently contributing to heart failure. 22 Thus, further studies are needed to elucidate the exact role of HMOX1 in various stressful events.
Zebrafish have emerged as a compelling vertebrate model for defining the conserved genetic and molecular basis of cardiac disease and for accessing the therapeutic potential of small molecules. Because of genome duplication events in teleosts, zebrafish have two paralogs of hmox1, hmox1a and hmox1b, showing significant conservation to human HMOX1. 26 Paralogs of hmox2, hmox2a and hmox2b, are closely related to human HMOX2 homologues. 27 Similar to human HMOX1, zebrafish hmox1a and hmox1b, but not hmox2a and hmox2b, display transcriptional responses to additional pro‐oxidant exposure. 26 , 27 Previous studies demonstrated a conserved role for hmox1a, but not hmox1b, in normal macrophage migration to the wound site and in protecting the host from M. marinum infection by generating a hmox1a mutant via TALENs. 28 , 29 In this study, we generated zebrafish hmox1a null mutants using CRISPR/Cas9 genome editing to explore the cardiac functional role of hmox1a at baseline and in response to stress.
2. MATERIALS AND METHODS
The detailed methods are provided in the Supplementary materials.
2.1. Generation of zebrafish hmox1a mutants
Zebrafish of the Turku line have been maintained in the zebrafish core facility at the University of Helsinki. Animal experiments were approved by the Regional Government Office of Southern Finland in agreement with the ethical guidelines of the European Union (ESAVI/4131/04.10.07/2017; ESAVI/16286/2020). The CRISPR/Cas9‐targeted mutagenesis was performed to generate zebrafish hmox1a (NM_001127516.1) mutants. The target site, 5′‐GGAGGCTCTGGGGCAGGACTTGG‐3′, was selected using ZiFiT Targeter software (http://zifit.partners.org/) without predicted off‐target site. Single‐guide RNA (sgRNA) was synthesised using the MAXIscript T7 kit (Life Technologies, Carlsbad, CA, USA). The sequence of hmox1a gRNA crRNA:tracrRNA was 5′‐gcgTAATACGACTCACTATAGGAGGCTCTGGGGCAGGACTGTTTTAGAGCTAGAAATAGC‐3′. Cas9 mRNA was synthesized with the plasmid pMLM3613 encoding Cas9 nuclease (Addgene, plasmid #42251) and the mMESSAGE T7 ULTRA kit (Life Technologies). Approximately 200 embryos were co‐injected with Cas9 mRNA and gRNA at the one‐cell stage. Mutated alleles were identified by high‐resolution melting (HRM) analysis (Roche Diagnostics GmbH, Mannheim, Germany) and Sanger sequencing. Ten founders (F0) carrying a 52‐bp deletion in exon 3 of hmox1a were outcrossed with the wild type to obtain F1 zebrafish heterozygous mutant. They were subsequently inbred to obtain F2 wild‐type (WT) (hmox1a +/+), heterozygous (HET) (hmox1a +/−) and homozygous (KO) (hmox1a −/−) zebrafish. HET mutants of F2 or later generations were inbred to generate WT, HET and KO embryos used in this study.
2.2. Genotyping
We genotyped either adult zebrafish or 3 dpf embryos at each generation as described previously 30 using the corresponding primers (Table S1). Each genotype displayed a distinct melting curve.
2.3. Cardiac function of zebrafish larvae
The cardiac function of larvae of mixed gender at 5–6 dpf was analysed as described earlier. 31 Ventricular volume was calculated based on ventricular width and length at the ends of diastole and systole. Ejection fraction (EF), stroke volume (SV) and cardiac output (CO) were calculated on the basis of ventricular volume. Five diastoles and systoles were analysed for each heart.
2.4. Echocardiography of adult zebrafish
Echocardiography of adult zebrafish of mixed gender was performed with a Vevo 2100® Image System (VisualSonics, Amsterdam, Netherlands) equipped with a high‐frequency transducer (MS700, 30–70 MHz) as described previously. 32 B‐mode videos were recorded in the longitudinal axis view, and pulsed‐wave Doppler (PWD) signals were obtained in the short axis view. EF, SV and CO were determined from B‐mode images. The maximal velocity of blood inflow across the atrioventricular valve during early diastole (E wave), atrial systole (A wave), and deceleration time of E wave were derived from PWD signals. HR was also obtained from the PWD image.
2.5. Hypoxia exposure in zebrafish larvae
At 4 dpf, larvae were exposed to hypoxia (3% O2) at 28°C in a Heracell VIOS 160i incubator (Thermo Fisher Scientific Inc., Waltham, MA, USA). Nitrogen gas was bubbled into the sealed incubator to maintain the oxygen conditions. Oxygen concentrations were measured with a Fibox 3 fibre optic oxygen probe and transmitter (PreSens Precision Sensing GmbH, Regensburg, Germany). Half of the embryos from each genotype were placed under hypoxic conditions, and half were placed under normoxic condition. After 24‐h exposure, larvae were immediately subjected to either cardiac function analysis or snap‐frozen.
2.6. Zn(II) Protoporphyrin IX treatment in adult zebrafish
Wild‐type adult zebrafish were anaesthetized with 0.02% tricaine in system water and i.p. injected with Zn(II) protoporphyrin IX (ZnPPIX, Frontier Scientific, Logan, UT, USA) at a dose of 5 mg/kg body weight on days 1 and 7. Control zebrafish received saline. Echocardiographic examinations were performed before and at 13 dpi.
2.7. Isoproterenol treatment in larval and adult zebrafish
Zebrafish embryos received 300 μM isoproterenol (ISO; Sigma‐Aldrich, Munich, Germany) at 2 dpf for 4 days. Larval cardiac function was monitored at 6 dpf. Adult zebrafish of mixed gender were anaesthetized with 0.02% tricaine in system water and i.p. injected with a single high dose of ISO at 150 mg/kg body weight. 33 Fish from each genotype were randomly divided into two groups based on body size: control and ISO. Control zebrafish received saline.
2.8. Primary cardiomyocyte isolation
Primary cardiomyocytes (CMs) were isolated and purified from adult ventricles of KO(52del), HET(52del) and WT zebrafish, as previously described. 34 Purified cardiomyocytes were seeded in poly l‐lysine‐coated cell culture plates (Agilent Seahorse XF96 Cell Culture Microplates, Santa Clara, CA, USA) at 10,000 cells/well and cultured at 28°C in 5% CO2.
2.9. Cardiomyocyte energy metabolism
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) in 4‐day primary CMs were measured with an XF Mito Stress Test Kit (Agilent) using a Seahorse XFe96 analyzer (Agilent) following the manufacturer's instructions. Prior to the assay, the culture medium was replaced with XF assay medium DMEM supplemented with 10 mM glucose, 2 mM glutamine and 1 mM pyruvate. During the assay, basal OCR and ECAR were measured, followed by sequential injections of 2 μM oligomycin, 3 μM carbonyl cyanide‐p‐trifluoromethoxyphenylhydrazone (FCCP), and 1 μM rotenone/antimycin A through drug injection ports. During the last injection step, cells were simultaneously stained with 2 μM Hoechst 33342 (Thermo Scientific, Rockford, IL, USA) to determine cell number using the Cytation 5 Cell Imaging Multi‐Mode Reader (Biotek, Agilent Technologies). For the ISO treatment experiment, cardiomyocytes isolated from HET(52del) and wild‐type adult hearts were treated with 10 μM ISO for 24 h prior to Seahorse assay. Data were analysed with Seahorse Wave software.
2.10. Western blotting
Pooled zebrafish larvae were lysed in chilled RIPA buffer supplemented with phosphatase and protease inhibitors (Roche) using a sonicator (Sonopuls HD2070, Bandelin, Berlin, Germany). Western blotting was performed as previously described. 35 The antibodies used are listed in Table S2.
2.11. RNA extraction and real‐time quantitative RT‐PCR
RNA extraction was performed with the miRNeasy Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer's instructions. Quantitative RT‐PCR was performed as described 35 at least three times with three technical replicates for each sample. The primer pairs used are listed in Table S1.
2.12. Histology
Acid fuchsin orange G (AFOG) staining (Sigma‐Aldrich, Saint Louis, MO, USA) was performed to detect fibrotic tissue according to the manufacturer's instructions. Slides were digitally scanned using a 3DHISTECH Pannoramic 250 FLASH II (3DHISTECH Ltd., Budapest, Hungary) and quantified with HistoQuant module (3DHISTECH Ltd.).
2.13. Immunohistochemistry
Paraffin sections from adult hearts were blocked and stained with antibodies against proliferating cell nuclear antigen (Pcna) and myocyte‐specific enhancer factor 2 (Mef2), followed by incubation with Alexa Fluor‐488‐ and Alexa Fluor‐594‐conjugated secondary antibodies. (Table S2). Nuclei were labelled with DAPI (4′,6‐Diamidino‐2‐Phenylindole, Dihydrochloride) (Molecular Probes, Eugene, OR, USA). Sections were mounted with ProLong Diamond antifade mountant (Molecular Probes) and imaged with a Zeiss LSM 780 confocal microscope (Carl Zeiss Microscopy GmbH, Germany). Quantitative identification of Collagen type I was performed using anti‐mouse Col1A1 antibody (Table S2) and EnVision™+ System‐HRP (Dako, Carpinteria, CA, USA) according to the manufacturer's instructions. Slides were digitally scanned using the 3DHISTECH Pannoramic 250 FLASH II and quantified with HistoQuant module. Two sections from each heart were stained, and the ventricular areas from each section were selected for quantification of Collagen type I relative to myocardium‐positive areas.
2.14. Statistical analysis
Data were analysed using Prism software (version 10.0; GraphPad, San Diego, CA, USA) and are presented as mean ± SD. One‐way ANOVA with Tukey's adjustment for multiple comparisons was used to calculate differences between more than two groups with normally distributed data. A two‐sample, unpaired, two‐tailed t‐test was performed to compare two groups with normally distributed data. Data that did not pass normality and lognormality tests were analysed with the Mann–Whitney test to compare the difference between two groups and with Kruskal‐Wallis followed by Dunn's test in the case of three groups. Statistical significance was set at p value <0.05.
3. RESULTS
3.1. Characterization of the CRISPR/Cas9‐generated zebrafish hmox1a mutant allele
We generated a zebrafish hmox1a mutant bearing a 52‐bp deletion in exon 3, hereinafter assigned as hmox1a (52del) (Figure 1A). The lesion resulted in a frameshift and a premature termination codon (Figure S1). We further investigated the expression of hmox1a in wild‐type (WT, hmox1a +/+), heterozygous (HET, hmox1a +/−), and knockout (KO, hmox1a −/−) offspring from F2 HET inbreeding by qRT‐PCR with a primer set spanning the exon 3 and 4 regions. As expected, the expression of hmox1a was almost undetectable in the KO(52del) larvae and markedly downregulated by approximately 50% in HET(52del) compared to WT (Figure 1B). To verify the possible genetic compensation response triggered by the deficiency of hmox1a, we quantitatively analysed the expression of the paralog hmox1b and the isoforms hmox2a and hmox2b. None of these related genes showed alteration at the transcriptional level in KO(52del) and HET(52del) compared to WT (Figure 1B), indicating that lack of hmox1a did not trigger a genetic compensation response. However, investigation of the protein level of Hmox1a was halted by the lack of an antibody specific to zebrafish. Nevertheless, the offspring of HET mutants followed Mendelian inheritance patterns. Both heterozygous and homozygous embryos displayed no gross morphological abnormalities (Figure 1C). They were adult viable and swam normally, but displayed a reduction in spawning frequency.
FIGURE 1.
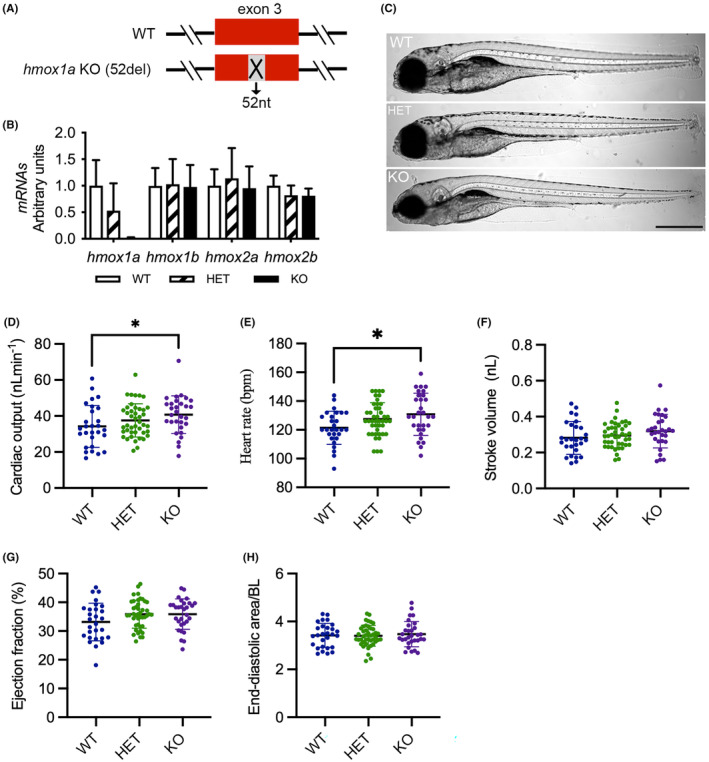
Deletion of hmox1a increases cardiac output in larvae. (A) Scheme of CRISPR/Cas9‐generated 52‐base pair deletion in exon 3 of hmox1a. (B) RT‐qPCR analysis of hmox1a, hmox1b, hmox2a and hmox2b in wild type (WT), heterozygous (HET) and homozygous (KO) hmox1a mutant at 5–6 dpf. The results are presented as fold change compared to WT. RNA was extracted from 5 to 10 pooled larvae. The number of RNA samples used for RT‐qPCR analyses as following: WT n = 5; HET(52del) n = 6; KO(52del) n = 6. (C) Representative images of hmox1a WT, HET(52del) and KO(52del) larvae at 5 dpf showing no obvious gross phenotype. Scale bar: 500 μm. (D–H) Analysis of cardiac function at 5–6 dpf larvae indicating an increase in cardiac output (D) and heart rate (E), and no significant change in stroke volume (F), ejection fraction (G) and end‐diastolic ventricular area normalized to body length (H) in hmox1a KO(52del). WT n = 28; HET(52del) n = 43; KO(52del) n = 30. Data are presented as mean ± SD. One‐way ANOVA with Tukey adjustment for multiple comparisons. *p < 0.05.
3.2. Deletion of hmox1a elevates cardiac output in larvae
We next evaluated the impact of hmox1a deficiency on cardiac function in zebrafish larvae. At 5–6 dpf, KO(52del) larvae displayed increased CO (p = 0.04) and HR (p = 0.03) in comparison with WT, whereas SV and EF showed no significant alteration in KO(52del) larvae (Figure 1D–G). EDA normalised to body length (BL) showed no obvious difference between the mutants and WT larvae (Figure 1H). Taken together, the deletion of hmox1a increases CO, which is ascribed to the enhanced HR in zebrafish larvae.
3.3. Deletion of hmox1a induces cardiac remodelling in adults
We further investigated the cardiac function of adult hmox1a mutants at 6 months of age by echocardiography. A previous study indicated that normalising for body weight facilitates standardised comparison of derived ventricular volume between groups of zebrafish of different age and gender. 32 Accordingly, we normalised ventricular area and volume to body weight. KO(52del) zebrafish showed an increase in CO by 26.3% and in SV by 12.4% compared to WT, but neither of the increases was statistically significant (Figure 2A,B). However, KO(52del) zebrafish displays a significant increase in CO (33.1%, p = 0.03) and a trend of increase in SV (28%, p = 0.10) compared to HET siblings (Figure 2A,B), but unchanged HR and EF to their siblings (Figure 2C,D). We observed enlarged EDA in KO(52del) ventricles by 15.9% (p = 0.12) compared to WT and 21.5% (p = 0.02) to HET siblings (Figure 2E), suggestive of cardiac hypertrophy in KO(52del) adults. PWD signals displayed a heightened E wave (KO vs. WT p < 0.001; KO vs. HET p < 0.001; Figure 2F) and preserved A wave (Figure 2G) in KO(52del) hearts compared to their siblings, which ultimately led to an increase in E/A ratio (KO vs. WT p < 0.001; KO vs. HET p = 0.002; Figure 2H). However, the E‐wave deceleration times remained similar among the three genotypic groups (Figure 2I). The findings indicate that the increased CO could possibly be ascribed to increased SV resulted from enlarged ventricles in KO(52del). Notably, adult KO(52del) zebrafish displayed lower BW than their HET siblings (p = 0.03; Figure 2J). In line with KO(52del), inhibition of Hmox‐1 activity in WT zebrafish with ZnPPIX, a selective inhibitor of HMOX1, 36 induced an enlargement of EDA by 19.4% (p = 0.04; Figure 2K) at 13 dpi. In addition, treatment with ZnPPIX increased CO by 35.4% (Figure 2L), although the change did not reach statistical significance. Taken together, deficiencies in the expression and activity of Hmox‐1 lead to cardiac hypertrophy and increased CO in adult zebrafish.
FIGURE 2.
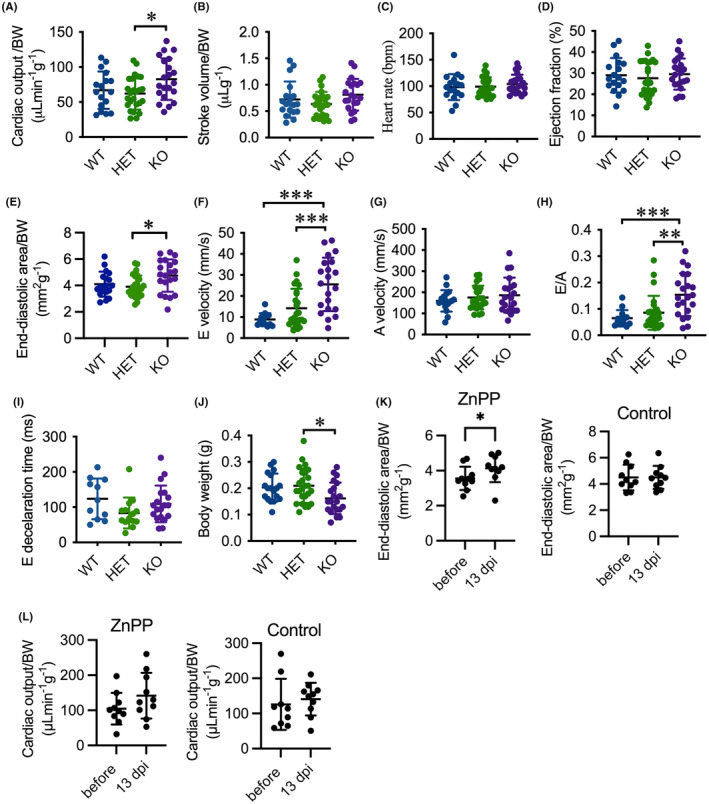
Deletion of hmox1a provokes cardiac output and hypertrophy in adults. (A–D) Echocardiography analysis depicting elevated cardiac output in KO(52del) zebrafish compared to their HET sibling (A) with a tendency toward increase in stroke volume (B) and no significant change in heart rate (C) and ejection fraction (D). (E) KO(52del) zebrafish exhibits enlarged end‐diastolic area compared to HET siblings. (F–H) PWD analysis indicating increased peak E wave velocity (F) and E/A ratio (H) without significant change on Peak A wave velocity (G). (I) Deceleration times of E wave from maximum velocity to baseline display no significant difference between the three genotypic groups. (J) KO(52del) zebrafish are lean in comparison with their HET siblings. (K) ZnPPIX treatment leads to enlarged end‐diastolic area in WT zebrafish at 13 dpi. Zebrafish treated with saline display no change at 13 dpi. (L) ZnPPIX treatment induces a tendency toward increase in cardiac output in WT zebrafish at 13 dpi. Zebrafish treated with saline display no change at 13 dpi. (A–J) WT n = 18; HET(52del) n = 25; KO(52del) n = 21. K, L ZnPPIX n = 10; Control n = 10. Data are presented as mean ± SD. One‐way ANOVA with Tukey adjustment for multiple comparisons in A–J. Two‐sample t‐test in K, L. *p < 0.05, **p < 0.01, ***p < 0.001.
Given that defects in the expression and activity of HMOX1 result in fibrosis and mitochondrial dysfunction in multiple metabolic tissues, 37 we therefore explored whether deletion of hmox1a induces cardiac fibrosis and mitochondrial impairment in adults. Indeed, histological staining revealed an abundant accumulation of collagen in the myocardium in KO(52del) zebrafish (p = 0.02) compared to WT siblings (Figure 3A,B). Furthermore, immunohistochemistry staining indicated an increase in the expression level of Collagen type I in the myocardium in KO(52del) zebrafish (p = 0.04; Figure 3C,D).
FIGURE 3.
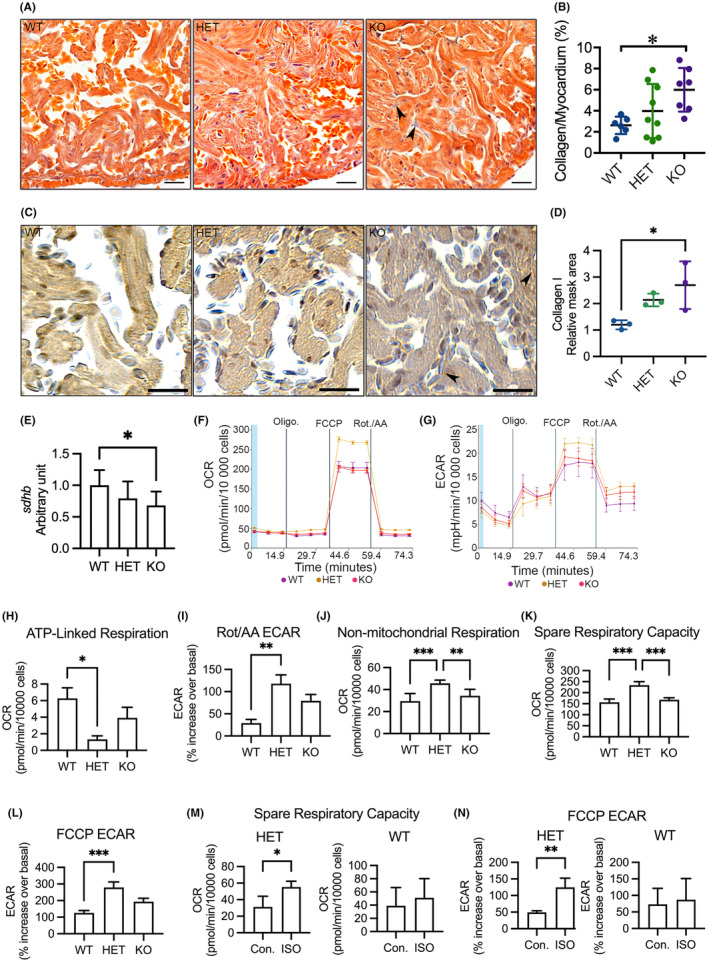
Analyses of myocardial interstitial fibrosis, cardiac OXPHOS gene expression and mitochondrial respiration in adult cardiomyocytes. (A) Representative images of acid fuchsin orange G (AFOG) staining of ventricular sections showing accumulation of collagen (blue) in the myocardium (orange). Arrowhead indicates accumulated collagen. (B) Quantification of AFOG staining indicating increased collagen accumulation in the myocardium in KO(52del). WT n = 6; HET(52del) n = 9; KO(52del) n = 7. (C) Representative images of immunohistochemical staining of ventricular sections for Collagen type I (brown). Arrowhead indicates Collagen type I‐positive signal. (D) Quantification of Collagen type I positive area relative to myocardium. WT n = 3; HET(52del) n = 3; KO(52del) n = 3. (E) RT‐qPCR analyses showing downregulation of OXPHOS complex II subunit sdhb in KO(52del) hearts compared to WT controls. sdhb, succinate dehydrogenase iron–sulfur subunit B; The graphs represent the quantification of two individual analyses of RNA extracts from pooled samples of two‐three hearts. Each analysis includes three replicates. WT n = 19; HET(52del) n = 19; KO(52del) n = 19. (F) Representative oxygen consumption (OCR) profile in zebrafish primary cardiomyocytes at basal respiration and after addition of oligomycin (Oligo.), carbonyl cyanide‐4 (trifluoromethoxy) phenylhydrazone (FCCP), followed by a combination of rotenone and antimycin A (Rot./AA). (G) Representative extracellular acidification (ECAR) profile in zebrafish primary cardiomyocytes at basal respiration and after addition of Oligo., FCCP and Rot./AA. (H) Cardiomyocytes from HET(52del) display a decline in the rate of respiration to drive mitochondrial ATP synthesis. (I) Cardiomyocytes from HET(52del) exhibit an increase in ECAR relative to the basal rate upon the addition of Rot./AA. (J) HET(52del) cardiomyocytes show increased non‐mitochondrial respiration. (K) HET(52del) cardiomyocytes show increased spare respiration capacity. (L) HET(52del) cardiomyocytes display increased ECAR relative to the basal rate upon the addition of FCCP. (M) Spare respiration capacity of HET(52del) and WT cardiomyocytes in response to ISO. (N) ECAR relative to the basal rate upon the addition of FCCP in HET(52del) and WT cardiomyocytes in response to ISO. (F–L) WT n = 15; HET(52del) n = 8; KO(52del) n = 7. M, N HET(52del) n = 15; WT n = 15. Data are presented as mean ± SD. One‐way ANOVA with Tukey adjustment for multiple comparisons in B, D, E and H–L. Two‐sample t‐test in M, N. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: 20 μm.
Quantitative RT‐PCR indicated that the transcript of nuclear DNA‐encoded succinate dehydrogenase iron–sulfur subunit B (sdhb) of oxidative phosphorylation (OXPHOS) complex II markedly declined in hmox1a‐deficient hearts (KO vs. WT, p = 0.04; Figure 3E), whereas the mitochondrial DNA‐encoded cytochrome c oxidase subunit 1 (mt‐co1) of complex IV and NADH dehydrogenase subunit 1 (mt‐nd1) of complex I showed no significant alteration in KO(52del) compared to their siblings (Figure S2A,B). Concomitantly, mitochondrial DNA content was unchanged, suggesting that deletion of hmox1a has no major effect on mitochondrial abundance (Figure S2C). To further verify whether deletion of hmox1a could affect mitochondrial function, we isolated primary CMs from adult ventricles of each genotypic zebrafish and investigated real‐time cellular energy metabolism with Seahorse XF. Primary CMs isolated from WT and KO(52del) ventricles showed a subtle decrease in OCR upon addition of oligomycin to block mitochondrial ATP production, whereas CMs from HET(52del) were insensitive to oligomycin (Figure 3F), indicating a general low mitochondrial respiration in zebrafish primary CMs. However, OCR markedly increased when an artificial ATP demand was induced by FCCP and dropped when TCA cycle flux was shut down by rotenone and antimycin A (Figure 3F). In contrast, the primary CMs exhibited an increase in ECAR when oligomycin was added (Figure 3G), reflecting enhanced glycolytic turnover to meet energetic needs. ECAR was continually enhanced upon the addition of FCCP and decreased by rotenone and antimycin A (Figure 3G). Interestingly, CMs from HET(52del) exhibited a decrease in OCR (p = 0.02) devoted to ATP production (Figure 3H) and an increase in ECAR (p = 0.003) relative to basal rate upon addition of the respiratory inhibitors compared to WT controls (Figure 3I), indicating that haplodeficiency of hmox1a triggers non‐mitochondrial respiration for energy demand, possibly glycolysis, in cardiomyocytes. Consistently, non‐mitochondrial oxygen consumption was markedly increased in HET(52del) CMs compared with their siblings (HET vs. WT, p < 0.001; HET vs. KO, p = 0.006; Figure 3J). Furthermore, HET(52del) CMs displayed an increase in spare respiratory capacity (HET vs. WT, p < 0.001; HET vs. KO, p < 0.001; Figure 3K) and in ECAR (p = 0.001) relative to basal rate upon addition of FCCP (Figure 3L), suggesting a robust ability to respond to increased energy demand. Nevertheless, CMs from KO(52del) displayed no substantial alteration in mitochondrial respiration in comparison with WT controls. We further investigated the energy metabolism of HET(52del) CMs upon ISO treatment in comparison with WT CMs. ISO stimulated spare respiratory capacity (p = 0.02) and increased ECAR (p = 0.006) relative to basal rate upon addition of FCCP in HET(52del) CMs, but not in WT CMs (Figure 3M,N), suggesting a potential ability of HET(52del) CMs to meet energy demands possibly by increasing glycolysis in response to stress.
We next investigated cardiomyocyte proliferation. IHC indicated fewer Pcna+ cardiomyocytes in KO(52del) ventricles (p = 0.01) compared to their HET siblings, suggesting that deletion of hmox1a restrains cardiomyocyte proliferation (Figure 4A, B). Interestingly, HET(52del) exhibited increased cardiac cell proliferation compared to their KO(52del) siblings (p = 0.04; Figure 4C). Collectively, the data indicate that deletion of hmox1a increases cardiac output, induces cardiac hypertrophy and fibrosis, and restrains cardiomyocyte proliferation, suggestive of cardiac remodelling in adult KO(52del), whereas haplodeficiency of hmox1a triggers non‐mitochondrial respiration and stimulates cardiac cell proliferation.
FIGURE 4.
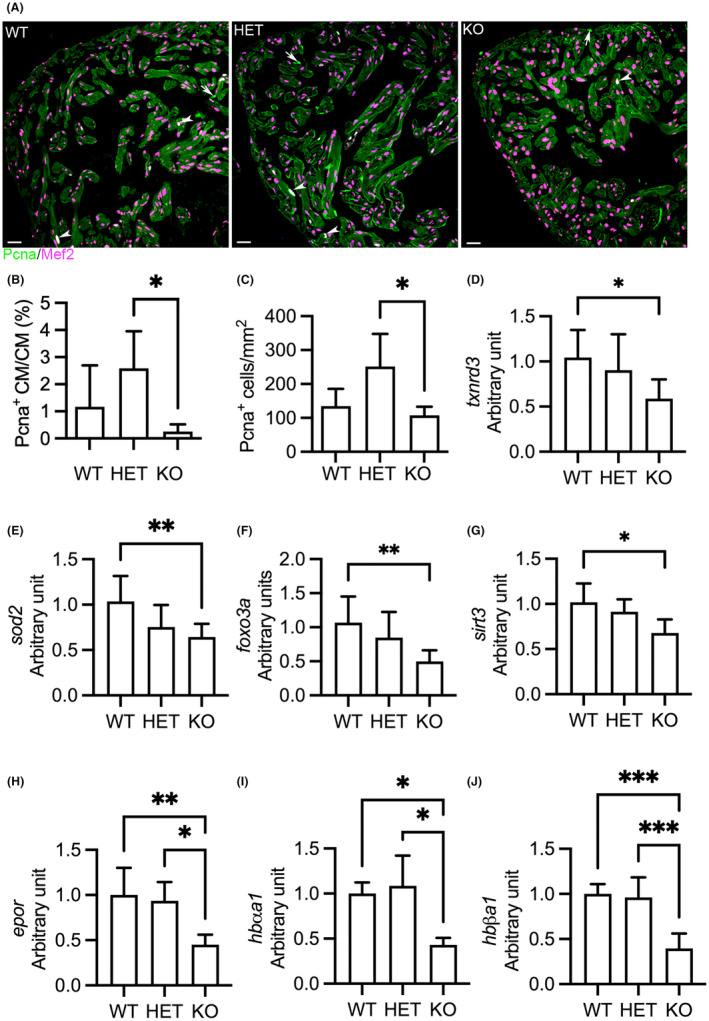
Deletion of hmox1a restrains cardiomyocyte proliferation and downregulates antioxidative genes and haemoglobin genes in adult zebrafish. (A) Representative immunohistochemical staining of Pcna (in green) and Mef2 (in meganne) in ventricular sections. Arrow indicates Pcna+ non‐cardiomyocytes. Arrowhead indicates Pcna+ cardiomyocytes. Pcna, proliferating cell nuclear antigen; Mef2, myocyte‐specific enhancer factor 2. (B) Quantification of Pcna+ cardiomyocytes relative to total cardiomyocytes. (C) Quantification of Pcna+ cardiac cells per mm2 ventricular area. Three hearts from each genotypic group were selected. Two sections from each heart were stained. The number of stain‐positive signals for Pcna and Mef2 from ventricular areas was quantified with ImageJ. (D–G) RT‐qPCR analyses indicating downregulation of antioxidative genes, txnrd3 (D) and sod2 (E), and the transcription regulators of antioxidative and antihypertrophic signalling, foxo3a (F) and sirt3 (G), in KO(52del) hearts. txnrd3, thioredoxin reductase 3; sod2, superoxide dismutase 2; foxo3a, forkhead box O3a; sirt3, sirtuin 3. The graphs represent the quantification of two individual analyses of RNA extracts from pooled samples of two‐three hearts. Each analysis includes three replicates. WT n = 19; HET(52del) n = 19; KO(52del) n = 19. (H–J) RT‐qPCR analyses showing downregulation of erythropoietin receptor epor (H), and adult haemoglobin genes hbαa1 (I) and hbβa1 (J) in kidney lacking hmox1a. epor, erythropoietin receptor; hbαa1, haemoglobin alpha adult‐1; hbβa1, haemoglobin beta adult‐1. The graphs represent the quantification of two individual analyses of RNA extracts from pooled samples of two kidneys. Each analysis includes three replicates. WT n = 10; HET(52del) n = 10; KO(52del) n = 10. Data are presented as mean ± SD. One‐way ANOVA with Tukey adjustment for multiple comparisons. *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars: 20 μm.
The contradictory effect of HMOX1 on oxidative stress 22 , 25 prompts us to investigate the expression of cardiac genes involved in the antioxidative defence system. We found the downregulation of antioxidative genes, 38 thioredoxin reductase 3 (txnrd3) (p = 0.02; Figure 4D) and superoxide dismutase 2 (sod2) (p = 0.007; Figure 4E), in KO(52del) hearts. The expression level of forkhead box O3a (foxo3a) (p = 0.005; Figure 4F), a transcription regulator of antioxidative signalling 39 and a suppressor of myocardial hypertrophy, 38 was also decreased in KO(52del) hearts. We further observed the downregulation of sirtuin 3 (sirt3) (p = 0.02; Figure 4G), an activator of FOXO3, 40 in KO(52del) hearts. The data indicate that deletion of hmox1a diminishes antioxidative defences and downregulates the redox‐sensitive antihypertrophic regulators at adult age, which probably leading to increased oxidative stress and activated redox‐sensitive hypertrophic signalling.
As HMOX1‐deficient patients develop haemolytic anaemia, 41 we investigated erythropoiesis and haemoglobin synthesis in adults at the age of 5 months. Given that adult zebrafish erythropoiesis is maintained in the kidney marrow and regulated by erythroid transcription factors and erythropoietin signalling, 42 we examined the expression of genes essential for erythropoiesis in the kidney by qPCR. We observed significant downregulation of erythropoietin receptor (epor), which binds erythropoietin to stimulate erythropoiesis, in KO(52del) compared to their siblings (KO vs. WT p = 0.007; KO vs. HET p = 0.01; Figure 4H). In addition, the expression of haemoglobin alpha adult‐1 (hbαa1) (KO vs. WT, p = 0.05; KO vs. HET, p = 0.02) and beta adult‐1 (hbβa1) (KO vs. WT, p < 0.001; KO vs. HET, p < 0.001) was also reduced in KO(52del) zebrafish (Figure 4I, J). On the other hand, the expression of erythropoietin a (epoa) was unchanged (Figure S2D). Furthermore, the expression of transcription regulators in erythropoiesis, krüppel‐like factor d (klfd) and GATA binding protein 1a (gata1a), was not significantly different in KO(52del) compared to their siblings (Figure S2E,F). These results suggest that the deletion of hmox1a impairs haemoglobin synthesis in adults, which may contribute to cardiac remodelling.
3.4. HET(52del) larvae display increased cardiac output in response to hypoxia
HMOX1 exerts cardioprotective effects after injury. 11 Thus, we wondered whether disruption of hmox1a deteriorates cardiac dysfunction in response to injury in zebrafish. Hypoxia, which can occur in both physiological and pathological conditions, has significant implications for cardiovascular disease. 43 As Hmox1 plays a role in controlling cardiac function in response to hypoxia, 44 we exposed larvae to a low oxygen (hypoxia) environment (3% O2). Given that hypoxia induces ß‐tubulin gene expression mediated by hypoxia‐inducible factor (HIF), 45 we analysed the expression of ß‐tubulin in larvae exposed to normal oxygen (normoxia) or to 3% O2 for 24 h. The protein level of ß‐tubulin substantially increased in larvae exposed to 3% O2 (Figure 5A). We also observed the transcriptional upregulation of epoa (p < 0.001) and hmox1a (p < 0.001), which are target genes of Hif‐1α, in WT larvae exposed to 3% O2 (Figure 5B). The data indicate induction of hypoxia in vivo in larvae exposed to 3% O2. Notably, hypoxia had no substantial effect on the expression of hmox1b (Figure 5B).
FIGURE 5.
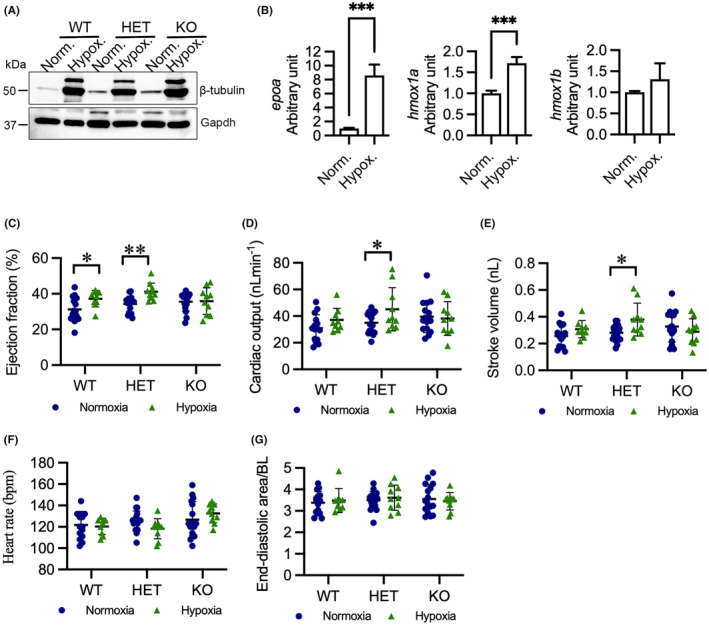
Hypoxia enhances cardiac output in HET(52del) larvae. (A) Representative immunoblots of β‐tubulin and Gapdh in WT, HET(52del), and KO(52del) at normoxic and hypoxic (3% O2 for 24 h) conditions. Gapdh serves as an internal control. Norm. normoxia; Hypox. Hypoxia. (B) RT‐qPCR analyses indicate that hypoxia induces upregulation of epoa and hmox1a, but not hmox1b, in WT larvae. (C–G) Hypoxia enhances ejection fraction (C) in WT and HET(52del) larvae, and cardiac output (D) and stroke volume (E) in HET(52del) larvae, but not in KO(52del), compared to normoxia. Hypoxia has no effect on heart rate (F) or diastolic area (G) in the three genotypic groups. WT, normoxia n = 15, hypoxia n = 9; HET(52del), normoxia n = 19, hypoxia n = 10; KO(52del), normoxia n = 16, hypoxia n = 10. Data are presented as mean ± SD. Two‐sample t‐test. *p < 0.05, **p < 0.01, ***p < 0.001.
We next evaluated cardiac function in normoxic and hypoxic larvae. Hypoxia caused elevated EF in WT and HET(52del) larvae (WT p = 0.05; HET p = 0.001), but not in KO(52del) larvae (Figure 5C). In addition, HET(52del) larvae exhibited enhanced CO (p = 0.02) and SV (p = 0.02) when exposed to hypoxia, whereas their WT and KO siblings showed no significant change (Figure 5D,E). Moreover, hypoxia had no impact on HR in either WT or KO but tended to reduce HR (p = 0.06) in HET (Figure 5F). Neither the mutants nor WT larvae exhibited changed EDA in response to hypoxia (Figure 5G). Collectively, the data suggest that deletion of hmox1a fails to induce cardiac compensation to overcome the lack of oxygen, whereas haplodeficiency of hmox1a boosts cardiac contractility and output in response to hypoxia.
3.5. HET(52del) adults deteriorate cardiac function in response to ISO treatment
A high dosage of ISO enhances myocardial function and eventually induces myocardial dysfunction in rats, representing an established ischaemic heart failure model. 33 Accordingly, we investigated whether deletion of hmox1a exacerbates cardiac dysfunction in response to ISO in zebrafish. ISO markedly boosts cardiac function in early larvae in all three genotypic groups (Figure S3). We treated WT adult zebrafish with ISO at a dosage of 150 mg/kg or 75 mg/kg and observed cardiac response to 150 mg/kg, but not 75 mg/kg, ISO (Figure S4). Thus, a high dosage of ISO (150 mg/kg) was administered to the adult zebrafish. We found reduced CO in WT (p = 0.05) and HET(52del) (p = 0.02) zebrafish in comparison with vehicle‐treated controls but had no effect in the KO(52del) siblings (Figure 6A). In addition, HET(52del) zebrafish also displayed reduced SV (p = 0.02) in response to ISO treatment, whereas WT and KO(52del) siblings showed no obvious alteration in SV (Figure 6B). ISO treatment had no substantial effect on HR and EF in either WT or the mutants (Figure 6C, D). However, ISO‐treated HET(52del) displayed smaller EDA (p < 0.001) and ESA (p = 0.001) compared to vehicle‐treated controls, while WT and KO siblings showed no obvious alteration in the cardiac area (Figure 6E, F). Taken together, the deletion of hmox1a fails to induce a cardiac response to ISO in adult zebrafish, but the haplodeficiency of hmox1a deteriorates cardiac function.
FIGURE 6.
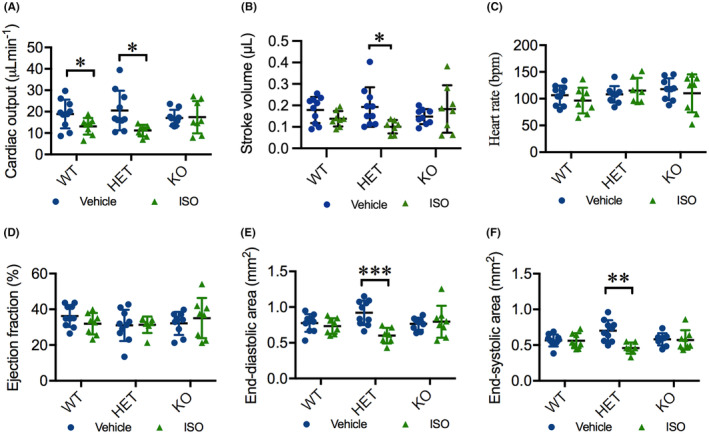
ISO deteriorates cardiac function in HET(52del) adults. (A) ISO treatment results in reduced cardiac output in WT and HET(52del), but not in KO(52del), compared to vehicle‐treated controls. (B) ISO treatment leads to reduced stroke volume in HET(52del), not in WT and KO(52del) compared to vehicle‐treated controls. (C, D) ISO treatment has no significant effect on heart rate (C) or ejection fraction (D) in the three genotypic groups compared to respective vehicle controls. (E, F) ISO treatment leads to reduced end‐diastolic (E) and end‐systolic area (F) in HET(52del), not in WT and KO(52del) compared to vehicle‐treated controls. WT, vehicle n = 10, ISO n = 8; HET(52del), vehicle n = 10, ISO n = 7; KO(52del), vehicle n = 9, ISO n = 8. Data are presented as mean ± SD. Two‐sample t‐test. * p < 0.05, ** p < 0.01, *** p < 0.001.
To explore the potential mechanism underlying the distinct cardiac impact on heterozygous and homozygous hmox1a mutants in response to cardiac stress, we quantitatively analysed the expression of hmox1a in ISO‐treated larval and adult hearts compared with that in vehicle‐treated controls. As expected, the hmox1a transcript was absent in KO(52del) larvae treated either with ISO or vehicle (Figure S3F). In addition, ISO had no significant effect on the expression of hmox1a in either WT or HET(52del) larvae (Figure S3F). Intriguingly, ISO treatment resulted in profound upregulation of hmox1a in HET(52del) adult hearts (ISO vs. vehicle, p = 0.002), but not in WT controls (Figure 7A). We further observed a substantial upregulation of hmox1b in KO(52del) (ISO vs. vehicle, p = 0.002) and HET(52del) (ISO vs. vehicle, p = 0.004) hearts in response to ISO treatment, but no alteration in WT hearts (Figure 7B). However, ISO had no effect on the expression of hmox2a and hmox2b in either WT or mutant hearts (Figure 7C,D). These data suggest that upregulation of hmox1b exerts a counter‐protective effect in hmox1a KO hearts, whereas excessive expression of both HMOX1 paralogs elicits a detrimental effect in hmox1a HET hearts in response to ISO. In addition, IHC indicated that ISO treatment induced a significant increase in cell proliferation in the ventricles of all three genotypic groups (WT p = 0.01, HET p = 0.002, KO p = 0.02; Figure 7E–H). In contrast, the number of Pcna+ cardiomyocytes showed no significant difference upon ISO treatment in the three genotypic groups (Figure 7E–G,I). However, Pcna+ cardiac cells were mostly distributed in the compact layer of the ventricular wall in ISO‐treated WT (Figure 7E), whereas positive signals often appeared in the trabecular layer in ISO‐treated mutants (Figure 7F,G).
FIGURE 7.
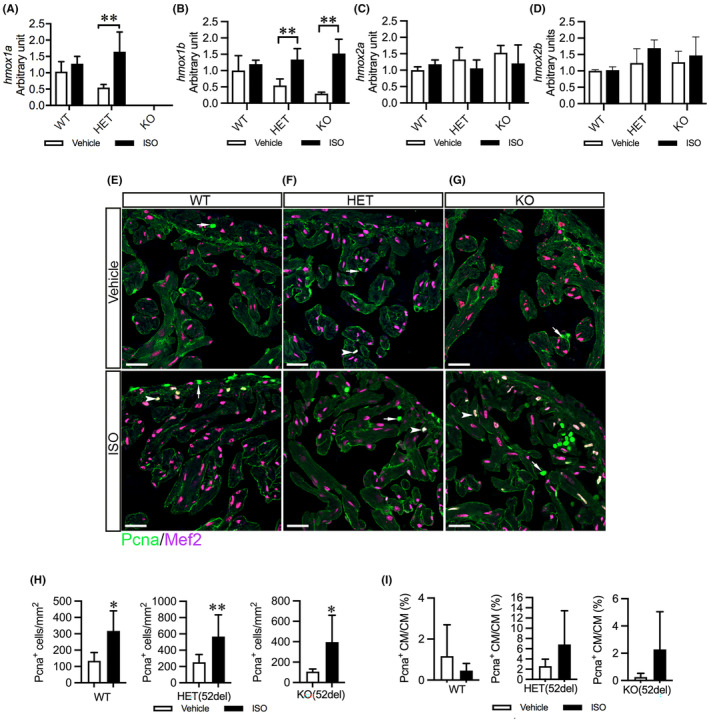
ISO upregulates cardiac hmox1 paralogs and increases cardiac cell proliferation in HET(52del) adults. (A–D) RT‐qPCR analyses of the expression of hmox1 homologues in ISO‐induced zebrafish hearts compared to vehicle‐treated controls. The results are presented as fold change compared to vehicle‐treated WT hearts. WT, vehicle n = 6, ISO n = 4; HET(52del), vehicle n = 6, ISO n = 4; KO(52del), vehicle n = 5, ISO n = 4. (E–G) Representative immunohistochemical staining of Pcna (in green) and Mef2 (in meganne) in ventricular sections from vehicle‐ or ISO‐treated zebrafish in each genotypic group. Arrow indicates Pcna+ non‐cardiomyocytes. Arrowhead indicates Pcna+ cardiomyocytes. (H, I) Quantification of Pcna+ cardiac cells per mm2 ventricular area (H) and Pcna+ cardiomyocytes relative to cardiomyocytes (I). Three hearts from each genotypic group were chosen. Two sections from each heart were stained. The number of stain‐positive signals for Pcna and Mef2 from ventricular areas was quantified with ImageJ. Data are presented as mean ± SD. Two‐sample t‐test. *p < 0.05, **p < 0.01.
4. DISCUSSION
We generated a mutation allele of hmox1a, hmox1a (52del), with a premature termination codon in exon 3 that possibly induces nonsense‐mediated mRNA decay using CRISPR. The deficiency of hmox1a did not induce the expression of its paralog hmox1b, or its isoforms, hmox2a and hmox2b. Neither homozygous nor heterozygous larvae displayed excess mortality or gross morphological abnormalities. Although they were adult viable, we noticed a short lifespan of homozygous mutants and a reduced spawning frequency in heterozygous adults. Unlike our findings, a previous study demonstrated that a minority of zebrafish hmox1a vcc42/vcc42 mutants displayed abnormal morphology at 3 dpf that progressed to lethality within 14 dpf. 28 However, hmox1a vcc42/vcc42 adult zebrafish showed no gross morphological abnormalities compared to their WT siblings. Genetic editing in the hmox1a locus led to a significant induction of hmox1b, which was even higher in abnormal embryos but not of hmox2a or hmox2b, in both hmox1a vcc42/vcc42 mutant and hmox1a crispants. 28 The mutant was generated by targeting the start code in exon 2 via TALENs, resulting in a deletion of exon 2 and a predicted mutant protein lacking 36 N‐terminal amino acid residues. The different phenotype between hmox1a (52del) and hmox1a vcc42/vcc42 mutant lines may be partially ascribed to the residual peptide translated from the hmox1a vcc42/vcc42 transcript and the upregulation of hmox1b.
Consistent with HMOX1 deficiency in mammals, hmox1a (52del) zebrafish developed interstitial fibrosis in the myocardium, dysregulated haemoglobin synthesis and reduced body weight compared to HET siblings. We demonstrate that deletion of hmox1a increases cardiac output in larval and adult zebrafish and induces cardiac hypertrophy in adult zebrafish. The different impact of the deletion of hmox1a in larvae versus adults could be because larvae are developing and can compensate for the deficiency of hmox1a, whereas the chronic absence of hmox1a leads to decompensation and remodelling with age. Similarly, patients with HMOX1 deficiency were initially normal until the onset of symptoms and rapid deterioration. 41 Similar to our findings, downregulation of Hmox1 with morpholinos results in a significant enlargement of the ventricle in 4 dpf zebrafish larvae. 44 However, unlike hmox1a mutant larvae, Hmox1 morphants exhibited no difference in CO despite significantly higher HR compared to controls. The discrepancy between hmox1a mutant and morphant phenotypes could be ascribed to a more severe effect of hmox1a deletion in comparison with the transient decline of Hmox1. An increase in cardiac output and enlargement of the ventricle have also been observed in phenylhydrazine hydrochloride‐induced anaemic rats and zebrafish. 46 , 47 Phenylhydrazine hydrochloride induces oxidative haemolysis of red blood cells and reduction of haemoglobin, ultimately resulting in anaemia. Anaemia consequently triggers cardiac remodelling by increasing cardiac output and ventricular stretch. 46 Indeed, we observed the downregulation of genes encoding haemoglobin in KO(52del). We also demonstrate that deletion of hmox1a downregulates the genes encoding antioxidative enzymes and transcription regulators that suppress cardiac hypertrophy in KO(52del). A previous study indicated that inhibition of HMOX1 activity increases oxidative stress, aggravating cardiac hypertrophy. 25 Accordingly, the elevated cardiac output and cardiac hypertrophy observed in hmox1a‐deficient zebrafish may be ascribed to anaemia, decreased antioxidative defences and impaired redox‐sensitive anti‐hypertrophic signalling.
Given that HMOX‐1 exerts cardioprotective effects in response to injury, 11 we expected that the deletion of hmox1a would fail to protect against cardiac injury. We observed elevated cardiac contractility in WT and HET(52del) larvae when exposed to hypoxia. This could be ascribed to the less availability of oxygen in the environment, which triggers the cardiac response to overcome the demand for oxygen. This is in line with a previous study showing that genetically hypoxic larvae (vhl −/− ) 48 displayed higher cardiac output, especially at 6 dpf, than WT controls. 49 HET(52del) larvae exhibited significantly higher CO when exposed to hypoxia. This could be due to the robust ability of HET(52del) CMs to respond to the increased energy demand and stress. Similar to our finding, the downregulation of Hmox1 by morpholino also induced higher CO level in zebrafish larvae when exposed to hypoxia. 44 Strikingly, KO(52del) larvae failed to increase cardiac contractility and output when exposed to hypoxia. This suggests that hypoxia boosts cardiac function dependent on Hmox1a. Indeed, hypoxia induced the transcriptional upregulation of hmox1a in WT larvae.
Exposure to a high dosage of ISO caused a significant decrease in CO in WT and HET but not in KO(52del) adult zebrafish in the present study, suggesting that a lack of hmox1a prevented ISO from exerting its effect on specific sympathetic nerve stimulation of the heart. In support of our notion, previous studies demonstrated that ISO failed to elicit its beneficial effects on the treatment of sepsis when HMOX1 expression was suppressed by ZnPPIX, an HMOX1 inhibitor. 50 , 51 In addition, Hmox1 morphant larval hearts were insensitive to adrenaline under hypoxic conditions, whereas adrenaline increased HR in sham‐treated larvae. 44 We further found that ISO substantially stimulates the expression of hmox1a and hmox1b in HET(52del) zebrafish adult hearts. This is in good agreement with previous reports that ISO induced HMOX1 expression in mouse leukaemic monocyte‐macrophages and in heart and lung tissues in septic mice in a concentration‐dependent manner. 50 Induction of HMOX1 has been associated with chronic viral myocarditis and cardiac hypertrophy in mice. 22 Septic, critically ill patients carrying a short GTn allele in the HMOX1 promoter region have high plasma HMOX1 concentration and are associated with the development of acute kidney injury. 52 Excessive expression of hmox1a and hmox1b in HET(52del) hearts compared to WT controls may exert a detrimental effect on cardiac function in response to ISO.
Decrease of mitochondrial respiration or promotion of glycolytic metabolism is critical for cardiomyocyte proliferation and heart regeneration in zebrafish. 53 , 54 In line with this, heterozygous hmox1a(52del) exhibited increased non‐mitochondrial oxygen consumption in adult cardiomyocytes and heightened cardiac cell proliferation compared to their WT and KO siblings, indicating cardiac reprogramming from a high‐metabolic, non‐proliferative state to a low‐metabolic, proliferative state. Furthermore, they display a robust ability to the increased energy demand and in response to stress. Whether such cardiac reprogramming elicits a protective or adverse effect on cardiac function and structure needs to be studied further.
The presence of duplicate copies of mammalian HMOX1 and functional compensation in zebrafish can be a limitation of our study. Hmox1a appears to be necessary for normal development in zebrafish, and its expression is responsive to oxidative stress, suggesting a conserved physiological function to mammals. 28 Although knockout of hmox1a cannot completely delete Hmox1 protein, lowering the level of Hmox1 predisposes to cardiac remodelling with age. Thus, hmox1a knockout zebrafish could serve as a model resembling human long GTn HMOX1 polymorphism, which leads to lower HMOX1 protein levels and predisposes to cardiovascular disease.
In conclusion, hmox1a deficiency boosts cardiac output and induces cardiac hypertrophy in adult zebrafish. The changes in cardiac function and structure could be partially ascribed to restrained cardiomyocyte proliferation, myocardial interstitial fibrosis, impaired haemoglobin synthesis, decreased antioxidative defences and inactive anti‐hypertrophic signalling. Intriguingly, the haplodeficiency of hmox1a in adult zebrafish triggers upregulation of both hmox1a and hmox1b and aggravates cardiac dysfunction in response to ISO stimulation. Our data indicate that HMOX1 homeostasis is essential for maintaining cardiac function and prompting cardioprotective effects, suggesting that the induction of HMOX1 for cardiac therapeutics needs to be leveraged.
AUTHOR CONTRIBUTIONS
Hong Wang: Formal analysis (equal); investigation (equal); project administration (equal); visualization (lead); writing – original draft (lead); writing – review and editing (equal). Juuso Siren: Conceptualization (equal); formal analysis (equal); investigation (equal); project administration (equal). Sanni Perttunen: Formal analysis (equal); investigation (equal); project administration (equal). Katariina Immonen: Formal analysis (equal); investigation (equal). Yu‐chia Chen: Methodology (equal). Suneeta Narumanchi: Investigation (equal). Riikka Kosonen: Investigation (equal). Jere Paavola: Conceptualization (equal); writing – review and editing (equal). Mika Laine: Conceptualization (equal). Ilkka Tikkanen: Conceptualization (equal); funding acquisition (equal); supervision (equal). Päivi Lakkisto: Conceptualization (equal); funding acquisition (equal); supervision (equal); writing – review and editing (equal).
FUNDING INFORMATION
The work was supported by grants from the Finnish Cultural Foundation (HW, SN, PL), the Finnish Foundation for Cardiovascular Research (HW, IT, PL), the Aarne Koskelo Foundation (HW, SN, IT, PL), Ida Montin Foundation (SN), the Finnish Foundation for Laboratory Medicine (PL), Finska Läkaresällskapet (IT, PL), the Liv och Hälsa Foundation (IT, PL), the Finnish Society of Clinical Chemistry (PL), Päivikki and Sakari Sohlberg Foundation (PL), and Finnish state funding for university‐level research (IT, PL). Open access funding was provided by University of Helsinki.
CONFLICT OF INTEREST STATEMENT
The authors declare that there is no conflict of interest associated with this manuscript.
Supporting information
Data S1.
ACKNOWLEDGEMENTS
We thank Daria Blokhina and Elina Ojala for assistance with the hypoxia assays. We acknowledge the Biomedicum Imaging Unit (BIU), Zebrafish Unit at the University of Helsinki, Tissue Preparation and Histochemistry Unit, and Genome Biology Unit for excellent technical assistance.
Wang H, Siren J, Perttunen S, et al. Deficiency of heme oxygenase 1a causes detrimental effects on cardiac function. J Cell Mol Med. 2024;28:e18243. doi: 10.1111/jcmm.18243
DATA AVAILABILITY STATEMENT
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Maines MD. Heme oxygenase: function, multiplicity, regulatory mechanisms, and clinical applications. FASEB J. 1988;2:2557‐2568. [PubMed] [Google Scholar]
- 2. Chau AS, Cole BL, Debley JS, et al. Heme oxygenase‐1 deficiency presenting with interstitial lung disease and hemophagocytic flares. Pediatr Rheumatol Online J. 2020;18:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Radhakrishnan N, Yadav SP, Sachdeva A, et al. Human heme oxygenase‐1 deficiency presenting with hemolysis, nephritis, and asplenia. J Pediatr Hematol Oncol. 2011;33:74‐78. [DOI] [PubMed] [Google Scholar]
- 4. Tahghighi F, Parvaneh N, Ziaee V. Post‐mortem diagnosis of heme Oxygenase‐1 deficiency by whole exome sequencing in an Iranian child. Int J Mol Cell Med. 2019;8:300‐307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yachie A, Niida Y, Wada T, et al. Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase‐1 deficiency. J Clin Invest. 1999;103:129‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawashima A, Oda Y, Yachie A, Koizumi S, Nakanishi I. Heme oxygenase‐1 deficiency: the first autopsy case. Hum Pathol. 2002;33:125‐130. [DOI] [PubMed] [Google Scholar]
- 7. Daenen KEL, Martens P, Bammens B. Association of HO‐1 (GT)n promoter polymorphism and cardiovascular disease: a reanalysis of the literature. Can J Cardiol. 2016;32:160‐168. [DOI] [PubMed] [Google Scholar]
- 8. Atsaves V, Detsika MG, Poulaki E, Gakiopoulou H, Lianos EA. Phenotypic characterization of a novel HO‐1 depletion model in the rat. Transgenic Res. 2017;26:51‐64. [DOI] [PubMed] [Google Scholar]
- 9. Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre‐Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1‐deficient mice: effects on macrophage viability and tissue iron distribution. Blood. 2010;116:6054‐6062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Poss KD, Tonegawa S. Heme oxygenase 1 is required for mammalian iron reutilization. Proc Natl Acad Sci U S A. 1997;94:10919‐10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Otterbein LE, Foresti R, Motterlini R. Heme Oxygenase‐1 and carbon monoxide in the heart: the balancing act between danger signaling and pro‐survival. Circ Res. 2016;118:1940‐1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lakkisto P, Kyto V, Forsten H, et al. Heme oxygenase‐1 and carbon monoxide promote neovascularization after myocardial infarction by modulating the expression of HIF‐1alpha, SDF‐1alpha and VEGF‐B. Eur J Pharmacol. 2010;635:156‐164. [DOI] [PubMed] [Google Scholar]
- 13. Segersvärd H, Lakkisto P, Hänninen M, et al. Carbon monoxide releasing molecule improves structural and functional cardiac recovery after myocardial injury. Eur J Pharmacol. 2018;818:57‐66. [DOI] [PubMed] [Google Scholar]
- 14. Akamatsu Y, Haga M, Tyagi S, et al. Heme oxygenase‐1‐derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J. 2004;18:771‐772. [DOI] [PubMed] [Google Scholar]
- 15. Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase‐1‐derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278:643‐H651. [DOI] [PubMed] [Google Scholar]
- 16. Kusmic C, Barsanti C, Matteucci M, et al. Up‐regulation of heme oxygenase‐1 after infarct initiation reduces mortality, infarct size and left ventricular remodeling: experimental evidence and proof of concept. J Transl Med. 2014;12:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li Q, Guo Y, Ou Q, et al. Gene transfer as a strategy to achieve permanent cardioprotection II: rAAV‐mediated gene therapy with heme oxygenase‐1 limits infarct size 1 year later without adverse functional consequences. Basic Res Cardiol. 2011;106:1367‐1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang G, Hamid T, Keith RJ, et al. Cardioprotective and antiapoptotic effects of heme oxygenase‐1 in the failing heart. Circulation. 2010;121:1912‐1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu X, Wei J, Peng DH, Layne MD, Yet SF. Absence of heme oxygenase‐1 exacerbates myocardial ischemia/reperfusion injury in diabetic mice. Diabetes. 2005;54:778‐784. [DOI] [PubMed] [Google Scholar]
- 20. Suliman HB, Carraway MS, Ali AS, Reynolds CM, Welty‐Wolf KE, Piantadosi CA. The CO/HO system reverses inhibition of mitochondrial biogenesis and prevents murine doxorubicin cardiomyopathy. J Clin Invest. 2007;117:3730‐3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yeh CH, Chen TP, Wang YC, et al. HO‐1 activation can attenuate cardiomyocytic apoptosis via inhibition of NF‐kappaB and AP‐1 translocation following cardiac global ischemia and reperfusion. J Surg Res. 2009;155:147‐156. [DOI] [PubMed] [Google Scholar]
- 22. Ursu ON, Sauter M, Ettischer N, Kandolf R, Klingel K. Heme oxygenase‐1 mediates oxidative stress and apoptosis in coxsackievirus B3‐induced myocarditis. Cell Physiol Biochem. 2014;33:52‐66. [DOI] [PubMed] [Google Scholar]
- 23. Chen C, Huo R, Tong Y, et al. Systemic heme oxygenase‐1 transgenic overexpression aggravates pressure overload‐induced cardiac hypertrophy in mice. Cell Physiol Biochem. 2011;28:25‐32. [DOI] [PubMed] [Google Scholar]
- 24. Gao X, Liu HB, Wu JC, et al. Heme oxygenase‐1 transgenic overexpression did not prevent artery injury induced by electric stimulation and pressure overload in mice. Eur J Pharmacol. 2011;659:199‐205. [DOI] [PubMed] [Google Scholar]
- 25. Waldman M, Nudelman V, Shainberg A, et al. The role of heme oxygenase 1 in the protective effect of caloric restriction against diabetic cardiomyopathy. Int J Mol Sci. 2019;20:E2427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Holowiecki A, O'Shields B, Jenny MJ. Characterization of heme oxygenase and biliverdin reductase gene expression in zebrafish (Danio rerio): basal expression and response to pro‐oxidant exposures. Toxicol Appl Pharmacol. 2016;311:74‐87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holowiecki A, O'Shields B, Jenny MJ. Spatiotemporal expression and transcriptional regulation of heme oxygenase and biliverdin reductase genes in zebrafish (Danio rerio) suggest novel roles during early developmental periods of heightened oxidative stress. Comp Biochem Physiol C Toxicol Pharmacol. 2017;191:138‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Luo K, Ogawa M, Ayer A, et al. Zebrafish heme oxygenase 1a is necessary for Normal development and macrophage migration. Zebrafish. 2022;19:7‐17. [DOI] [PubMed] [Google Scholar]
- 29. Luo K, Stocker R, Britton WJ, Kikuchi K, Oehlers SH. Haem oxygenase limits Mycobacterium marinum infection‐induced detrimental ferrostatin‐sensitive cell death in zebrafish. FEBS J. 2022;289:671‐681. [DOI] [PubMed] [Google Scholar]
- 30. Varshney GK, Carrington B, Pei W, et al. A high‐throughput functional genomics workflow based on CRISPR/Cas9‐mediated targeted mutagenesis in zebrafish. Nat Protoc. 2016;11:2357‐2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dash SN, Narumanchi S, Paavola J, et al. Sept7b is required for the subcellular organization of cardiomyocytes and cardiac function in zebrafish. Am J Physiol Heart Circ Physiol. 2017;312:H1085‐H1095. [DOI] [PubMed] [Google Scholar]
- 32. Wang LW, Huttner IG, Santiago CF, et al. Standardized echocardiographic assessment of cardiac function in normal adult zebrafish and heart disease models. Dis Model Mech. 2017;10:63‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Grimm D, Elsner D, Schunkert H, et al. Development of heart failure following isoproterenol administration in the rat: role of the renin‐angiotensin system. Cardiovasc Res. 1998;37:91‐100. [DOI] [PubMed] [Google Scholar]
- 34. Sander V, Suñe G, Jopling C, Morera C, Belmonte JCI. Isolation and in vitro culture of primary cardiomyocytes from adult zebrafish hearts. Nat Protoc. 2013;8:800‐809. [DOI] [PubMed] [Google Scholar]
- 35. Wang H, Segersvärd H, Siren J, et al. Tankyrase inhibition attenuates cardiac dilatation and dysfunction in ischemic heart failure. Int J Mol Sci. 2022;23:10059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nowis D, Bugajski M, Winiarska M, et al. Zinc protoporphyrin IX, a heme oxygenase‐1 inhibitor, demonstrates potent antitumor effects but is unable to potentiate antitumor effects of chemotherapeutics in mice. BMC Cancer. 2008;8:197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sasson A, Kristoferson E, Batista R, McClung JA, Abraham NG, Peterson SJ. The pivotal role of heme Oxygenase‐1 in reversing the pathophysiology and systemic complications of NAFLD. Arch Biochem Biophys. 2021;697:108679. [DOI] [PubMed] [Google Scholar]
- 38. Keceli G, Gupta A, Sourdon J, et al. Mitochondrial creatine kinase attenuates pathologic remodeling in heart failure. Circ Res. 2022;130:741‐759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klotz L‐O, Sánchez‐Ramos C, Prieto‐Arroyo I, Urbánek P, Steinbrenner H, Monsalve M. Redox regulation of FoxO transcription factors. Redox Biol. 2015;6:51‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rangarajan P, Karthikeyan A, Lu J, Ling EA, Dheen ST. Sirtuin 3 regulates Foxo3a‐mediated antioxidant pathway in microglia. Neuroscience. 2015;311:398‐414. [DOI] [PubMed] [Google Scholar]
- 41. Yachie A. Heme Oxygenase‐1 deficiency and oxidative stress: a review of 9 independent human cases and animal models. Int J Mol Sci. 2021;22:1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kulkeaw K, Sugiyama D. Zebrafish erythropoiesis and the utility of fish as models of anemia. Stem Cell Res Ther. 2012;3:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bilo G, Gatterer H, Torlasco C, Villafuerte FC, Parati G. Editorial: Hypoxia in cardiovascular disease. Front Cardiovasc Med. 2022;9:990013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tzaneva V, Perry SF. Evidence for a role of heme oxygenase‐1 in the control of cardiac function in zebrafish (Danio rerio) larvae exposed to hypoxia. J Exp Biol. 2016;219:1563‐1571. [DOI] [PubMed] [Google Scholar]
- 45. Raspaglio G, Filippetti F, Prislei S, et al. Hypoxia induces class III beta‐tubulin gene expression by HIF‐1alpha binding to its 3′ flanking region. Gene. 2008;409:100‐108. [DOI] [PubMed] [Google Scholar]
- 46. Fang Y, Sun Y, Luo C, et al. Evaluation of cardiac dysfunction in adult zebrafish using high frequency echocardiography. Life Sci. 2020;253:117732. [DOI] [PubMed] [Google Scholar]
- 47. Norman TD, McBroom RD. Cardiac hypertrophy in rats with phenylhydrazine anemia. Circ Res. 1958;6:765‐770. [DOI] [PubMed] [Google Scholar]
- 48. van Rooijen E, Voest EE, Logister I, et al. Zebrafish mutants in the von Hippel‐Lindau tumor suppressor display a hypoxic response and recapitulate key aspects of Chuvash polycythemia. Blood. 2009;113:6449‐6460. [DOI] [PubMed] [Google Scholar]
- 49. Yaqoob N, Schwerte T. Cardiovascular and respiratory developmental plasticity under oxygen depleted environment and in genetically hypoxic zebrafish (Danio rerio). Comp Biochem Physiol A Mol Integr Physiol. 2010;156:475‐484. [DOI] [PubMed] [Google Scholar]
- 50. Ha YM, Ham SA, Kim YM, et al. Beta(1)‐adrenergic receptor‐mediated HO‐1 induction, via PI3K and p38 MAPK, by isoproterenol in RAW 264.7 cells leads to inhibition of HMGB1 release in LPS‐activated RAW 264.7 cells and increases in survival rate of CLP‐induced septic mice. Biochem Pharmacol. 2011;82:769‐777. [DOI] [PubMed] [Google Scholar]
- 51. Sun J, Kim SJ, Park MK, et al. Selective activation of adrenergic beta1 receptors induces heme oxygenase 1 production in RAW264.7 cells. FEBS Lett. 2005;579:5494‐5500. [DOI] [PubMed] [Google Scholar]
- 52. Vilander LM, Vaara ST, Donner KM, et al. Heme oxygenase‐1 repeat polymorphism in septic acute kidney injury. PloS One. 2019;14:e0217291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Honkoop H, de Bakker DE, Aharonov A, et al. Single‐cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. 2019;8:e50163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fukuda R, Marín‐Juez R, El‐Sammak H, et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. 2020;21:e49752. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author upon reasonable request.