

Abstract
Purpose
Since the discovery of IDH mutations in glioma over a decade ago, significant progress has been made in determining how these mutations affect epigenetic, transcriptomic, and metabolic programs in brain tumor cells. In this article, we summarize current understanding of how IDH mutations influence DNA damage in glioma and discuss clinical implications of these findings.
Methods
We performed a thorough review of peer-reviewed publications and provide an overview of key mechanisms by which IDH mutations impact response to DNA damage in gliomas, with an emphasis on clinical implications.
Results
The effects of mutant IDH on DNA damage largely fall into four overarching categories: Gene Expression, Sensitivity to Alkylating Agents, Homologous Recombination, and Oxidative Stress. From a mechanistic standpoint, we discuss how mutant IDH and the oncometabolite (R)-2HG affect each of these categories of DNA damage. We also contextualize these mechanisms with respect to ongoing clinical trials. Studies are underway that incorporate current standard-of-care therapies, including radiation and alkylating agents, in addition to novel therapeutic agents that exert genotoxic stress specifically in IDH-mutant gliomas. Lastly, we discuss key unanswered questions and emerging data in this field that have important implications for our understanding of glioma biology and for the development of new brain tumor therapies.
Conclusion
Mounting preclinical and clinical data suggest that IDH mutations alter DNA damage sensing and repair pathways through distinct mechanisms. Future studies are needed to deepen our understanding of these processes and provide additional mechanistic insights that can be leveraged for therapeutic benefit.
Keywords: DNA damage, IDH, Isocitrate dehydrogenase
Introduction
Mutations in IDH1 or IDH2 genes, which encode isocitrate dehydrogenase (IDH) enzymes, define diagnostic subsets of gliomas [1, 2]. The 2021 WHO classification now formally incorporates IDH mutation status as a component of glioma diagnoses, with the three primary adult-type diffuse gliomas being (1) astrocytoma, IDH-mutant, (2) oligodendroglioma, IDH-mutant, and 1p/19q-codeleted, and (3) glioblastoma, IDH-wildtype [3]. IDH-mutant gliomas are more often lower-grade (grade 2–3) and diagnosed in young adults. Wild-type IDH1/2 enzymes reversibly convert isocitrate to 2-oxoglutarate (2OG). In contrast, glioma-associated IDH mutants (the most common of which is IDH1R132H) are neomorphs that convert 2OG to (R)-2-hydroxyglutarate [(R)-2HG]. (R)-2HG accumulates to millimolar levels in IDH-mutant gliomas, constituting one of the most abundant metabolites in these tumors. Due to structural similarity between (R)-2HG and 2OG, (R)-2HG competitively inhibits 2OG-dependent enzymes [4–6] to promote tumorigenesis and is thus termed an “oncometabolite.”
In addition to oncogenic effects of (R)-2HG, (R)-2HG also confers bystander effects that may be therapeutically exploited. One of the most well-described of these so-called “collateral vulnerabilities” is an altered response to DNA damage. In this review, we discuss preclinical and clinical studies that illustrate mechanisms by which mutant IDH affects response to DNA damage. We describe molecular mechanisms underlying these effects, including gene expression changes, sensitivity to alkylating agents, homologous recombination defects, and response to oxidative stress. Lastly, we discuss key unanswered questions that inform translation of these findings to the clinic.
Gene expression
IDH mutations influence gene expression by regulating chromatin modifying enzymes. (R)-2HG competitively inhibits 2OG-dependent DNA and histone demethylases. One of the most well-established signatures of these epigenetic changes is the glioma CpG island methylator phenotype (G-CIMP) [7, 8]. Most promoter sites in the human genome are preceded by CpG islands, which are CG-rich regions that can be methylated by DNA methyltransferases to alter expression of downstream genes. Methylation of these CpG island sites leads to transcriptional repression and gene silencing (Fig. 1A). A glioma-specific G-CIMP phenotype was first discovered through profiling of mutations [8]. Mechanistically, this likely reflects (R)-2HG-mediated inhibition of the 2OG-dependent ten-eleven translocation (TET) family of DNA-modifying enzymes, in addition to other 2OG-dependent histone and DNA demethylases.
Fig. 1.
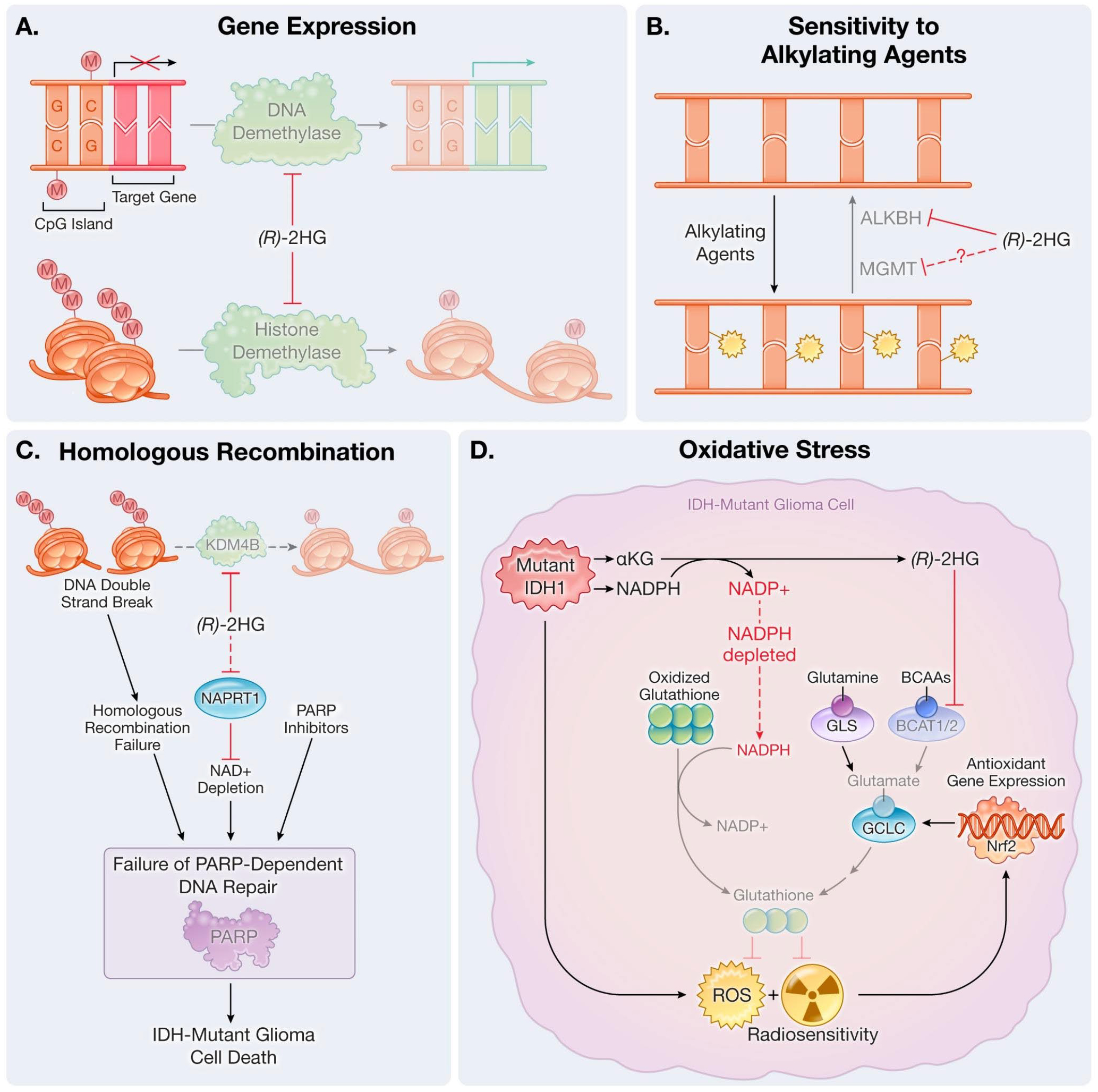
Altered responses to DNA damage in IDH-mutant gliomas. Examples of mechanisms related to (A) gene expression, (B) sensitivity to alkylating agents, (C) homologous recombination, and (D) oxidative stress. Dashed lines indicate indirect effects. BCAAs: branched-chain amino acids; ROS: reactive oxygen species
Epigenetic changes caused by mutant IDH have multiple functional effects, including maintenance of a stem-like state [8] and increased expression of glioma oncogenes [9]. However, much remains unknown regarding how mutant IDH functionally reprograms the glioma epigenome. Epigenetic-driven changes in gene expression by mutant IDH have been implicated as the cause of multiple “collateral vulnerabilities” that may be exploited therapeutically. One such example is highlighted in recent data from Liu and colleagues demonstrating increased expression of NRF2 pathway genes and reliance on this pathway for reactive oxygen species (ROS) homeostasis [10] (see “Oxidative Stress” below for further discussion). Importantly, the kinetics of these changes mirrored that of the long-term time course observed in mutant IDH-associated epigenetic reprogramming [8]
Mutant IDH-mediated epigenetic changes have also been implicated in directly controlling expression of genes that mediate DNA damage responses (DDR), with data supporting different mechanisms. Recent work from our group [11] revealed that mutant IDH sensitizes gliomas to inhibition of the de novo pyrimidine nucleotide synthesis pathway. Drugs that inhibit this pathway caused nucleotide pool imbalance, replication stress-dependent DNA damage, and cell death in multiple in vitro and in vivo models of IDH-mutant glioma. Mechanistically, these data support a model in which mutant IDH causes epigenetic and transcriptomic changes that silence DDR genes necessary to maintain genome integrity during nucleotide pool imbalance. In support of this hypothesis, an analysis of TCGA data demonstrated decreased expression of DDR-related genes in IDH-mutant as compared to IDH-WT gliomas. Similar findings of mutant IDH-driven DDR defects due to epigenetic changes have been made in IDH-mutant leukemia [12]. Additional data also suggest that mutant IDH increases replication stress by promoting heterochromatin formation, providing another mechanistic link between these mutations and susceptibility to DNA damage [13].
In contrast to these findings, data from Nuñez et al. [14] support an alternative hypothesis, in which mutant IDH acts as a tumor suppressor and increases DNA damage repair via epigenetic reprogramming. In the context of a genetically engineered mouse model harboring Trp53 loss, Atrx loss, and an Nras G12V mutation, mutant IDH1 enhanced histone H3K4 methylation and was associated with increased expression of DDR genes. These molecular changes were associated with radioresistance of IDH-mutant versus IDH-WT murine gliomas. These data raise interesting mechanistic questions regarding how genetic context may determine the impact of mutant IDH on expression of DDR genes.
Ongoing clinical trials will help clarify how IDH mutations impact DDR. An early-phase trial testing the de novo pyrimidine synthesis inhibitor BAY 2402234 in IDH-mutant glioma patients is forthcoming (Table 1). In addition, early data from the CODEL trial demonstrated a significant progression-free survival benefit in the radiation arms compared to temozolomide alone in IDH-mutant, 1p/19q codeleted patients [15], providing preliminary clinical evidence that IDH-mutant gliomas respond to radiation therapy. Given these early results, the CODEL trial is now undergoing redesign to omit the temozolomide-alone arm and will test radiation with either concurrent temozolomide or PCV.
Table 1.
Select completed and ongoing clinical trials related to DNA damage in IDH-mutant gliomas
| Patient population | Therapy | Phase | Status | Outcome | Reference / NCT |
|---|---|---|---|---|---|
| Grade 3, 1p/19q codeleted oligodendroglioma | Radiation, TMZ | III | Undergoing redesign | Improved PFS with RT | NCT00887146;[15] |
| Anaplastic gliomas, 1p/19q non-codeleted | Radiation, TMZ | III | Completed | Improved OS with adjuvant TMZ | NCT00626990; [1] |
| Recurrent grade 4 IDH-mutant glioma | BAY 2402234 (pyrimidine synthesis inhibitor) | 0 | In development | N/A | N/A |
| Grade 2–3 IDH-mutant glioma | Telaglenastat (glutaminase inhibitor) | I | Not accruing | N/A | NCT03528642 |
| Recurrent IDH-mutant glioma | Olaparib (PARP inhibitor) | II | Completed | Median PFS 2.3 months | NCT03561870; [30] |
| Recurrent grade 2–4 IDH-mutant astrocytoma | Niraparib (PARP inhibitor) | 0 | Accruing | N/A | NCT05076513 |
| Recurrent grade 2–4 IDH-mutant glioma | Pamiparib (PARP inhibitor) and metronomic TMZ | I/II | Not accruing | N/A | NCT03914742 |
| Recurrent IDH-mutant glioma | Olaparib and durvalumab | II | Accruing | N/A | NCT03991832 |
TMZ: Temozolomide
PFS: Progression-free survival
RT: Radiation therapy
OS: Overall survival
Sensitivity to alkylating agents
Alkylating agents are part of standard-of-care treatment for gliomas. These include temozolomide, CCNU, and procarbazine, the latter two of which comprise the three-agent regimen PCV along with vincristine. Given that alkylating agents are the primary systemic therapy class used for glioma treatment, multiple studies have deepened our understanding of how mutant IDH affects efficacy of alkylating chemotherapies.
Work from Wang et al. [16] and Chen et al. [17] explore the effect of (R)-2HG on the AlkB family of proteins, which are 2OG-dependent enzymes that repair alkylated DNA lesions [18, 19]. Wang et al. used engineered glioma and fibrosarcoma cell lines with or without mutant IDH1 to show that (R)-2HG inhibits the ALKBH2 and ALKBH3 enzymes, causing decreased repair of chemotherapy-induced DNA alkylation (Fig. 1B). Loss of function of ALKBH2 and ALKBH3 was due to inhibition of the catalytic activity of these enzymes as opposed to a decrease in expression. Furthermore, the catalytic activity of mutant IDH was necessary for mutant IDH-mediated sensitivity to alkylating agents, as demonstrated through experiments in which catalytically dead IDH1 double-mutants rescued the increased cell death observed in IDH1R132H single-mutant expressing cells. While response to temozolomide was not reported, mutant IDH expression sensitized cells to the alkylating agents CCNU and procarbazine, which was partially mitigated by exogenous expression of ALKBH2/3. These data suggest that inhibition of 2OG-dependent ALKBH dioxygenases by (R)-2HG may be functionally linked to sensitivity to alkylating agents. Additional characterization of the kinetics of inhibition under physiologic conditions has been described in work by Chen et al. [17].
In addition to ALKBH inhibition, mutant IDH confers metabolic vulnerabilities that may sensitize IDH-mutant glioma cells to alkylating agents. Work from Tateishi et al. demonstrate that mutant IDH1 decreases NAD + levels in glioma cells and increases susceptibility to therapeutics that deplete NAD + pools, such as NAMPT inhibitors [20]. This finding has important implications, as temozolomide-induced DNA damage can be repaired by the NAD+-dependent enzyme poly(ADP-ribose) polymerase (PARP). In follow-up work aiming to further exploit this dependency, they identified that temozolomide decreased NAD + levels, and that combination treatment with temozolomide and a NAMPT inhibitor enhanced efficacy of either treatment alone [21]. Similarly, NAD + depletion can also be exploited by use of temozolomide and inhibition of poly(ADP-ribose) glycohydrolase (PARG), the latter of which sequesters NAD + pools that are normally released following break-down of PAR chains after temozolomide-induced DNA damage [22]. These synergistic combinations may allow for use of reduced dose in patients, potentially mitigating dose-limiting toxicities encountered when using either treatment alone.
Clinically, temozolomide is part of standard-of-care treatment for gliomas as concurrent, adjuvant, or monotherapy treatment dependent on clinical scenario. Nevertheless, the CATNON trial in IDH-mutant, 1p/19q non-codeleted tumors suggest a benefit to adjuvant (but not concurrent) temozolomide [1], and the redesigned CODEL trial (enrolling IDH-mutant, 1p/19q codeleted patients) will test different alkylating chemotherapy regimens as concurrent treatment with radiotherapy [15]. Collectively, these data will help further clarify the role of IDH mutations in mediating sensitivity to alkylating agents and whether treatment timing impacts efficacy. Importantly, response to alkylating agents is not only affected by mutant IDH status, but also by known biomarkers such as O6-methylguanine methyltransferase (MGMT) promoter methylation in CpG islands [23]. While IDH mutations have been causally linked to CpG island hypermethylation (discussed above (in “Gene Expression”) [8]), not all IDH-mutant gliomas display MGMT promoter methylation [24, 25]. It thus remains unclear how the mechanisms of mutant IDH-driven response to alkylating agents highlighted above intersect with MGMT methylation status to control response to these therapies.
Homologous recombination
DNA damage can cause breaks that increase the potential for new mutations, thus representing an essential player in the development and progression of cancers. Homologous recombination (HR) is a high-fidelity mechanism that repairs various forms of DNA damage. HR promotes DNA repair through exchange of nucleotide sequences between similar or identical DNA strands, such as in double-strand break repair. Impairments in HR function can thus lead to genomic instability, which may render cancer cells sensitive to chemotherapies and radiation.
Tumor cells with IDH mutations have been shown to display HR defects driven by (R)-2HG accumulation. (R)-2HG inhibits 2OG-dependent dioxygenases [8, 26], including some enzymes that play important roles in the HR pathway. Work from Sulkowski et al. showed that (R)-2HG inhibits the 2OG-dependent dioxygenase KDM4B, leading to hypermethylation of histone 3 lysine 9 (H3K9). H3K9 methylation normally marks areas of DNA breaks and facilitates recruitment of HR machinery. In IDH-mutant tumor cells with high (R)-2HG and consequent H3K9 hypermethylation, H3K9 methylation-dependent activation of the Tip60 acetyltransferase-ATM kinase axis is impaired, and DNA double-strand break recognition is compromised. This prevents recruitment of downstream DNA repair factors like RPA, BRCA1, and RAD51 [27]. Thus, IDH-mutant tumor cells may display a “BRCAness” phenotype that confers sensitivity to PARP inhibitors [28] (Fig. 1C), though recent work has suggested an alternative mechanism that may contribute to this effect [13]. In addition to H3K9 hypermethylation, PARP inhibitor sensitivity is also driven by impaired NAD + metabolism in IDH-mutant glioma cells, as shown by Lu et al. [29]. Their work demonstrated that NAD + depletion in IDH-mutant glioma cells is associated with reduced PARP activity and hypersensitivity to combined PARP inhibitor and temozolomide treatment. This research builds on work from Tateishi et al. [20] and corroborates the mechanistic link between mutant IDH and perturbed NAD + homeostasis.
Like the variable impact of IDH mutations on DDR gene expression, there appear to be context-specific effects of IDH oncogenes on HR function. Contrasting the findings above, Ohba et al. found that IDH mutations increase HR-mediated DNA repair that is dependent on the recombinase RAD51. These findings were made in isogenic immortalized astrocyte cell lines harboring or lacking the IDH1-R132H oncogene, suggesting that IDH mutations increase HR activity when introduced in non-malignant cells. These findings are in agreement with those reported by Nuñez et al. [14], who found enhanced RAD51 expression in IDH-mutant glioma cells relative to IDH wild-type controls. Ohba and colleagues reported that increased HR efficiency driven by IDH mutations thus enhances resistance to temozolomide in their model systems.
Against the backdrop of an apparent complex relationship between IDH mutations and HR-dependent DNA damage repair, clinical trials testing PARP inhibitors in glioma patients are ongoing (Table 1). Olaparib as a monotherapy was investigated in the phase 2 OLAGLI clinical trial, which enrolled patients with recurrent IDH-mutant high-grade glioma. Olaparib was well tolerated, and median progression-free survival was 2.3 months [30]. Another phase 2 clinical trial reported that combination treatment with olaparib and the PD-L1 inhibitor durvalumab can be safely administered but displays limited efficacy [31]. Exploiting synthetic lethality between deficient HR DNA repair and IDH mutations through PARP inhibition may be enhanced by concurrent radiotherapy, as has been shown in preclinical models of glioma [32], and is being tested in ongoing clinical trials [33].
Oxidative stress
Oxidative stress can initiate cancers or prompt progression by chemically modifying DNA bases and causing mutations. IDH enzymes catalyze the conversion of isocitrate to 2OG while generating nicotinamide adenine dinucleotide phosphate (NADPH), a cofactor critical for maintaining cellular redox balance. IDH-mutant enzymes, in contrast, produce (R)-2HG through a catalytic mechanism that consumes NADPH [34]. IDH mutations have been demonstrated to reduce NADPH pools, which can inhibit recycling of oxidized glutathione to reduced glutathione and elevate ROS levels [35] (Fig. 1D). Reduced glutathione promotes elimination of free radicals, peroxides, lipid peroxides, and metals that may be harmful to DNA [36]. Moreover, reduced glutathione prevents oxidative DNA damage during radiotherapy [37].
In addition to consuming NADPH, IDH-mutant enzymes have also been shown to cause (R)-2HG-dependent inhibition of the 2OG-dependent transaminases BCAT1/2 [38]. Reduced BCAT activity decreases steady-state levels of glutamate and reduced glutathione, which is partly composed of glutamate. BCAT inhibition also increases reliance on the enzyme glutaminase (GLS) for glutamate and glutathione biosynthesis. These findings offer a potential mechanistic explanation for GLS hyperdependence displayed by IDH-mutant cells [39]. Notably, GLS inhibition is synthetic lethal with IDH mutations under conditions of oxidative stress or radiotherapy.
IDH-mutant glioma cells have been shown to display and respond to increased ROS levels by upregulating the Nrf2 antioxidant pathway [10]. Nrf2 is a transcription factor that regulates antioxidant gene expression and promotes redox homeostasis. In IDH1-mutant cells, elevated ROS leads to Nrf2 activation, which increases transcription of antioxidant genes, such as the gene encoding a key enzyme in the de novo glutathione biosynthesis pathway, glutamate-cysteine ligase (GCLC) [10]. The antioxidant response stimulated by Nrf2 can thus be seen as a compensatory response to the oxidative stress induced by IDH1-mutated cancer cells. Targeting the Nrf2 antioxidant pathway could prove to be a novel approach to the treatment of IDH1-mutant cancers.
Notably, work by Molenaar et al. demonstrated that the IDH1R132H inhibitor AGI-5198 protects IDH-mutant tumor cells from radiotherapy [40], consistent with its ability to restore NADPH, glutamate, and glutathione homeostasis. After exposure to radiation, higher levels of ROS, DNA double-strand breaks, and cell death were found in IDH1R132H-mutants as compared to IDH1 wild-type cells. Therefore, clinical trials of mutant IDH inhibitors are currently underway and, to date, do not involve combination with radiotherapy [41]. To exploit the radiosensitizing effects of IDH mutations, a trial is ongoing to test the addition of a GLS inhibitor, CB-839 (Telaglenastat), to standard-of-care radiation and temozolomide therapies for IDH-mutant glioma [42].
Discussion
IDH mutations induce profound metabolic, epigenomic, and transcriptomic reprogramming in glioma. Prior research has revealed multiple mechanisms through which the effects of IDH mutations converge on DNA damage repair pathways, leading to a number of clinical trials seeking to exploit these interactions for therapy (Table 1). Results from these ongoing studies will help resolve areas where data remain incomplete or conflicting, such as the role of mutant IDH in regulating radiosensitivity. Despite significant progress in understanding these mechanisms, important unanswered questions remain.
Given the multiple mechanisms through which IDH mutations impact DNA damage repair, disentangling the effects of any single mechanism in response to therapy remains challenging. However, this is an important goal in the field, considering that insights into these processes may inform rational design of combination therapy. For example, strategies that exploit DNA damage vulnerabilities caused by (R)-2HG would not be amenable to combination strategies involving a mutant IDH inhibitor. In contrast, therapies that exploit durable DNA damage repair deficits that are not altered by acute changes in (R)-2HG may permit use of concurrent mutant IDH inhibitors. The multiple downstream effectors of (R)-2HG can pose challenges for designing treatment strategies. For example, some data suggest that IDH-mutant gliomas develop alkylating agent-induced hypermutated phenotypes [43–45], possibly due to mutant-IDH driven DNA damage deficits. Exploiting this DNA damage deficit may therefore leverage increased tumor mutational burden and render these tumors susceptible to therapies such as immune checkpoint blockade. This is also supported by data suggesting that DNA damage can trigger an anti-tumor immune response [46]. However, other data suggest that (R)-2HG also suppresses the immune response and creates an immunosuppressive microenvironment [47–54] arguing for a mutant IDH inhibitor strategy to bolster immunotherapy response. Thus, it remains to be seen how to best optimize treatments that may exploit DNA damage (caused by mutant IDH and/or therapies such as alkylating agents or direct DDR inhibitors) in combination with mutant IDH inhibitors. Given the ongoing clinical testing of the efficacy of mutant IDH inhibitors in lower-grade gliomas [55], this question will likely have heightened relevance as outcomes are reported from this study in the future.
In addition to the therapeutic implications of mechanisms linking IDH mutations with DNA damage, emerging data suggest these mechanisms may also help define the genomic landscape of gliomas. For example, recent clinical data show that among IDH-mutant astrocytomas and oligodendrogliomas, increased glioma grade is correlated with increased copy number variation (CNV), increased chromothripsis, and gene expression signatures of chromosomal instability (CIN) [56–58]. These genomic features and processes are intimately linked to DNA damage. Importantly, IDH-mutant astrocytomas with high expression signatures of CIN display worse progression-free and overall survival, suggesting that this signature may have prognostic significance. Further work is needed to determine whether CNV and CIN alterations themselves cause DDR deficits in IDH-mutant glioma or whether these effects are the result of upstream DNA damaging processes driven by mutant IDH. Nevertheless, these findings may reveal additional mechanisms of interplay between DNA damage and IDH mutations distinct from those highlighted in this article. Presence of CNV/CIN may also serve as a biomarker to identify subsets of mutant IDH glioma patients who may benefit most from treatments targeting DDR deficits.
Significant progress has been made in our understanding of the complex interplay between DDR and IDH mutations in gliomas, with promising new data providing a foundation for additional investigation. Results from ongoing clinical trials, as well as future preclinical studies of this topic, will establish deeper mechanistic insights that may ultimately be leveraged to develop new treatment strategies for IDH-mutant glioma patients.
Acknowledgements
We thank Melissa Logies for providing illustrations accompanying this work.
Funding
D.D.S. is supported by the Holman Research Pathway, a Conquer Cancer Young Investigator Award, and the ASTRO Resident Research Biology Seed Grant. S.K.M. is supported by the Cancer Prevention and Research Institute of Texas (RR190034).
Financial interests
D.D.S. and S.A. have no financial interests to declare. S.K.M. has served as a paid advisor to Agios Pharmaceuticals. S.K.M. received research funding from Bayer Pharmaceuticals. Bayer had no influence over the design or execution of this work.
References
- 1.van den Bent MJ, Tesileanu CMS, Wick W et al. (2021) Adjuvant and concurrent temozolomide for 1p/19q non-co-deleted anaplastic glioma (CATNON; EORTC study 26053 – 22054): second interim analysis of a randomised, open-label, phase 3 study. Lancet Oncol 22:813–823. 10.1016/S1470-2045(21)00090-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cairncross JG, Wang M, Jenkins RB et al. (2014) Benefit from procarbazine, lomustine, and vincristine in oligodendroglial tumors is associated with mutation of IDH. J Clin Oncol 32:783–790. 10.1200/JCO.2013.49.3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Louis DN, Perry A, Wesseling P et al. (2021) The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 23:1231–1251. 10.1093/neuonc/noab106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Losman J-A, Looper RE, Koivunen P et al. (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Sci (New York NY) 339:1621–1625. 10.1126/science.1231677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu W, Yang H, Liu Y et al. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of α-ketoglutarate-dependent dioxygenases. Cancer Cell 19:17–30. 10.1016/j.ccr.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sulkowski PL, Oeck S, Dow J et al. (2020) Oncometabolites suppress DNA repair by disrupting local chromatin signalling. Nature 582:586–591. 10.1038/s41586-020-2363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Noushmehr H, Weisenberger DJ, Diefes K et al. (2010) Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell 17:510–522. 10.1016/j.ccr.2010.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turcan S, Rohle D, Goenka A et al. (2012) IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature 483:479–483. 10.1038/nature10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Flavahan WA, Drier Y, Liau BB et al. (2016) Insulator dysfunction and oncogene activation in IDH mutant gliomas. Nature 529:110–114. 10.1038/nature16490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y, Lu Y, Celiku O et al. (2019) Targeting IDH1-Mutated Malignancies with NRF2 Blockade. J Natl Cancer Inst 111:1033–1041. 10.1093/jnci/djy230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi DD, Savani MR, Levitt MM et al. (2022) De novo pyrimidine synthesis is a targetable vulnerability in IDH mutant glioma. Cancer Cell S. 10.1016/j.ccell.2022.07.011.1535-6108(22)00324-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Inoue S, Li WY, Tseng A et al. (2016) Mutant IDH1 Downregulates ATM and Alters DNA Repair and Sensitivity to DNA Damage Independent of TET2. Cancer Cell 30:337–348. 10.1016/j.ccell.2016.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schvartzman J-M, Forsyth G, Walch H et al. (2022) Oncogenic IDH mutations increase heterochromatin-related replication stress without impacting tumor mutation burden. 2022.08.04.502834 [DOI] [PMC free article] [PubMed]
- 14.Núñez FJ, Mendez FM, Kadiyala P et al. (2019) IDH1-R132H acts as a tumor suppressor in glioma via epigenetic upregulation of the DNA damage response. Sci Transl Med. 10.1126/scitranslmed.aaq1427.11: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaeckle KA, Ballman KV, van den Bent M et al. (2021) CODEL: phase III study of RT, RT + TMZ, or TMZ for newly diagnosed 1p/19q codeleted oligodendroglioma. Analysis from the initial study design. Neuro Oncol 23:457–467. 10.1093/neuonc/noaa168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang P, Wu J, Ma S et al. (2015) Oncometabolite D-2-Hydroxyglutarate Inhibits ALKBH DNA Repair Enzymes and Sensitizes IDH Mutant Cells to Alkylating Agents. Cell Rep 13:2353–2361. 10.1016/j.celrep.2015.11.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen F, Bian K, Tang Q et al. (2017) Oncometabolites d- and l-2-Hydroxyglutarate Inhibit the AlkB Family DNA Repair Enzymes under Physiological Conditions. Chem Res Toxicol 30:1102–1110. 10.1021/acs.chemrestox.7b00009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aas PA, Otterlei M, Falnes PO et al. (2003) Human and bacterial oxidative demethylases repair alkylation damage in both RNA and DNA. Nature 421:859–863. 10.1038/nature01363 [DOI] [PubMed] [Google Scholar]
- 19.Duncan T, Trewick SC, Koivisto P et al. (2002) Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci U S A 99:16660–16665. 10.1073/pnas.262589799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tateishi K, Wakimoto H, Iafrate AJ et al. (2015) Extreme Vulnerability of IDH1 Mutant Cancers to NAD + Depletion. Cancer Cell 28:773–784. 10.1016/j.ccell.2015.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tateishi K, Higuchi F, Miller JJ et al. (2017) The Alkylating Chemotherapeutic Temozolomide Induces Metabolic Stress in IDH1-Mutant Cancers and Potentiates NAD + Depletion-Mediated Cytotoxicity. Cancer Res 77:4102–4115. 10.1158/0008-5472.CAN-16-2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nagashima H, Lee CK, Tateishi K et al. (2020) Poly(ADP-ribose) Glycohydrolase Inhibition Sequesters NAD + to Potentiate the Metabolic Lethality of Alkylating Chemotherapy in IDH-Mutant Tumor Cells. Cancer Discov 10:1672–1689. 10.1158/2159-8290.CD-20-0226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hegi ME, Diserens A-C, Gorlia T et al. (2005) MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N Engl J Med 352:997–1003. 10.1056/NEJMoa043331 [DOI] [PubMed] [Google Scholar]
- 24.Chai R, Li G, Liu Y et al. (2021) Predictive value of MGMT promoter methylation on the survival of TMZ treated IDH-mutant glioblastoma. Cancer Biol Med 18:271–282. 10.20892/j.issn.2095-3941.2020.0179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulholland S, Pearson DM, Hamoudi RA et al. (2012) MGMT CpG island is invariably methylated in adult astrocytic and oligodendroglial tumors with IDH1 or IDH2 mutations. Int J Cancer 131:1104–1113. 10.1002/ijc.26499 [DOI] [PubMed] [Google Scholar]
- 26.Losman J-A, Kaelin WG (2013) What a difference a hydroxyl makes: mutant IDH, (R)-2-hydroxyglutarate, and cancer. Genes Dev 27:836–852. 10.1101/gad.217406.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sulkowski PL, Oeck S, Dow J et al. (2020) Oncometabolites suppress DNA repair by disrupting local chromatin signaling. Nature 582:586–591. 10.1038/s41586-020-2363-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sulkowski PL, Corso CD, Robinson ND et al. (2017) 2-Hydroxyglutarate produced by neomorphic IDH mutations suppresses homologous recombination and induces PARP inhibitor sensitivity. Sci Transl Med 9:eaal2463. 10.1126/scitranslmed.aal2463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu Y, Kwintkiewicz J, Liu Y et al. (2017) Chemosensitivity of IDH1-Mutated Gliomas Due to an Impairment in PARP1-Mediated DNA Repair. Cancer Res 77:1709–1718. 10.1158/0008-5472.CAN-16-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ducray F, Sanson M, Chinot OL et al. (2021) Olaparib in recurrent IDH-mutant high-grade glioma (OLAGLI). JCO 39:2007–2007. 10.1200/JCO.2021.39.15_suppl.2007 [DOI] [Google Scholar]
- 31.Ramos R, Climans SA, Adile A et al. (2021) Combination olaparib and durvalumab for patients with recurrent IDH-mutated gliomas. JCO 39:e14026–e14026. 10.1200/JCO.2021.39.15_suppl.e14026 [DOI] [Google Scholar]
- 32.Wang Y, Wild AT, Turcan S et al. (2020) Targeting therapeutic vulnerabilities with PARP inhibition and radiation in IDH-mutant gliomas and cholangiocarcinomas. Sci Adv 6:eaaz3221. 10.1126/sciadv.aaz3221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.The University of Hong Kong (2021) Combination Talazoparib - Carboplatin for Recurrent High-grade Glioma With DNA Damage Repair Deficiency (DDRd). clinicaltrials.gov
- 34.Dang L, White DW, Gross S et al. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462:739–744. 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi J, Sun B, Shi W et al. (2015) Decreasing GSH and increasing ROS in chemosensitivity gliomas with IDH1 mutation. Tumour Biol 36:655–662. 10.1007/s13277-014-2644-z [DOI] [PubMed] [Google Scholar]
- 36.Meister A (1994) Glutathione, ascorbate, and cellular protection. Cancer Res 54:1969s–1975s [PubMed] [Google Scholar]
- 37.Shimizu T, Iwanaga M, Yasunaga A et al. (1998) Protective role of glutathione synthesis on radiation-induced DNA damage in rabbit brain. Cell Mol Neurobiol 18:299–310. 10.1023/a:1022525214871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McBrayer SK, Mayers JR, DiNatale GJ et al. (2018) Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175:101–116e25. 10.1016/j.cell.2018.08.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seltzer MJ, Bennett BD, Joshi AD et al. (2010) Inhibition of glutaminase preferentially slows growth of glioma cells with mutant IDH1. Cancer Res 70:8981–8987. 10.1158/0008-5472.CAN-10-1666 [pii] 10.1158/0008-5472.CAN-10-1666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Molenaar RJ, Botman D, Smits MA et al. (2015) Radioprotection of IDH1-Mutated Cancer Cells by the IDH1-Mutant Inhibitor AGI-5198. Cancer Res 75:4790–4802. 10.1158/0008-5472.CAN-14-3603 [DOI] [PubMed] [Google Scholar]
- 41.Institut de Recherches Internationales Servier (2022) A Phase 1, Multicenter, Open-Label, Dose-Escalation and Expansion, Safety, Pharmacokinetic, Pharmacodynamic, and Clinical Activity Study of Orally Administered AG-120 in Subjects With Advanced Solid Tumors, Including Glioma, With an IDH1 Mutation. clinicaltrials.gov
- 42.Kizilbash SH, McBrayer S, Port J et al. (2019) A phase Ib trial of CB-839 (telaglenastat) in combination with radiation therapy and temozolomide in patients with IDH-mutated diffuse astrocytoma and anaplastic astrocytoma (NCT03528642). J Clin Oncol 37:TPS2075–TPS2075. 10.1200/JCO.2019.37.15_suppl.TPS2075 [DOI] [Google Scholar]
- 43.Johnson BE, Mazor T, Hong C et al. (2014) Mutational analysis reveals the origin and therapy-driven evolution of recurrent glioma. Science 343:189–193. 10.1126/science.1239947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Thuijl HF, Mazor T, Johnson BE et al. (2015) Evolution of DNA repair defects during malignant progression of low-grade gliomas after temozolomide treatment. Acta Neuropathol 129:597–607. 10.1007/s00401-015-1403-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Touat M, Li YY, Boynton AN et al. (2020) Mechanisms and therapeutic implications of hypermutation in gliomas. Nature 580:517–523. 10.1038/s41586-020-2209-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mender I, Zhang A, Ren Z et al. (2020) Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer Cell 38:400–411e6. 10.1016/j.ccell.2020.05.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kohanbash G, Carrera DA, Shrivastav S et al. Isocitrate dehydrogenase mutations suppress STAT1 and CD8 + T cell accumulation in gliomas. J Clin Invest 127:1425–1437. 10.1172/JCI90644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berghoff AS, Kiesel B, Widhalm G et al. (2017) Correlation of immune phenotype with IDH mutation in diffuse glioma. Neuro Oncol 19:1460–1468. 10.1093/neuonc/nox054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Friedrich M, Sankowski R, Bunse L et al. (2021) Tryptophan metabolism drives dynamic immunosuppressive myeloid states in IDH-mutant gliomas. Nat Cancer 2:723–740. 10.1038/s43018-021-00201-z [DOI] [PubMed] [Google Scholar]
- 50.Bunse L, Pusch S, Bunse T et al. (2018) Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat Med 24:1192–1203. 10.1038/s41591-018-0095-6 [DOI] [PubMed] [Google Scholar]
- 51.Lu M, Mellinghoff IK, Diaz A et al. (2020) Abstract 2046: Inhibiting IDH mutations in low-grade glioma alters cellular function and the immune environment. Cancer Res. 10.1158/1538-7445.AM2020-2046.80:2046 [DOI] [Google Scholar]
- 52.Notarangelo G, Spinelli JB, Perez EM et al. (2022) Oncometabolite d-2HG alters T cell metabolism to impair CD8 + T cell function. Science 377:1519–1529. 10.1126/science.abj5104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Amankulor NM, Kim Y, Arora S et al. (2017) Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev 31:774–786. 10.1101/gad.294991.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu M-J, Lei S, Dubrot J, Merritt J, Vijay V, Wei T-Y, Kessler E, Olander KE, Adil R, Pankaj A, Tummala KS, Weeresekara V, Zhen Y, Wu Q, Luo M, Shen W, García-Beccaria M, Fernández-Vaquero M, Hudson C, Ronseaux S, Sun Y, Saad-Berreta R, Jenkins RW, Wang T, Heikenwälder M, Ferrone CR, Goyal L, Nicolay B, Deshpande V, Kohli RM, Zheng H, Manguso RT, Bardeesy N (2022) Mutant IDH Inhibits IFNγ–TET2 Signaling to Promote Immunoevasion and Tumor Maintenance in Cholangiocarcinoma. Cancer Discovery 12(3):812–835. 10.1158/2159-8290.CD-21-1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.INDIGO : A global, randomized, double-blind, phase III study of vorasidenib (VOR; AG-881) vs placebo in patients (pts) with residual or recurrent grade II glioma with an isocitrate dehydrogenase 1/2 (IDH1/2) mutation. | Journal of Clinical Oncology. 10.1200/JCO.2020.38.15_suppl.TPS2574. Accessed 21 Aug 2022 [DOI] [Google Scholar]
- 56.Richardson TE, Sathe AA, Xing C et al. (2021) Molecular Signatures of Chromosomal Instability Correlate With Copy Number Variation Patterns and Patient Outcome in IDH-Mutant and IDH-Wildtype Astrocytomas. J Neuropathol Exp Neurol 80:354–365. 10.1093/jnen/nlab008 [DOI] [PubMed] [Google Scholar]
- 57.Mirchia K, Sathe AA, Walker JM et al. (2019) Total copy number variation as a prognostic factor in adult astrocytoma sub-types. Acta Neuropathol Commun 7:92. 10.1186/s40478-019-0746-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Richardson TE, Williams M, Galbraith K et al. (2020) Total copy number variation, somatic mutation burden, and histologic grade correlate with clinical outcome in oligodendroglioma. Clin Neuropathol 39:238–242. 10.5414/NP301260 [DOI] [PubMed] [Google Scholar]