

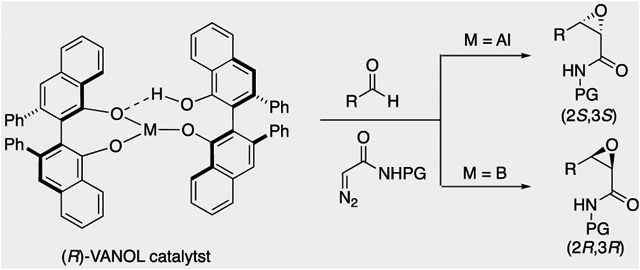
Abstract
Two epoxidation catalysts, one of which consists of two VANOL ligands and an aluminum and the other that consists of two VANOL ligands and a boron, were compared. Both catalysts are highly effective in the catalytic asymmetric epoxidation of a variety of aromatic and aliphatic aldehydes with diazoacetamides, giving high yields and excellent asymmetric inductions. The aluminum catalyst is effective at 0 °C and the boron catalyst at −40 °C. Although both the aluminum and boron catalysts of (R)-VANOL give very high asymmetric inductions (up to 99% ee), they give opposite enantiomers of the epoxide. The mechanism, rate- and enantioselectivity-determining step, and origin of enantiodivergence are evaluated using density functional theory calculations.
Keywords: asymmetric catalysis, epoxidation, enantiodivergence, DFT calculations
Graphical Abstract
Recently we reported an asymmetric catalytic method for the preparation of cis-α,β-epoxyamides from aldehydes and diazoacetamides with a chiral borate catalyst containing a VANOL ligand.1 With this, it was envisioned that access to various 3,4-epoxy-2-piperidone alkaloids could be achieved starting from the diazoacetamide 1 and aldehydes of the type 2 (Scheme 1). These would include (−)-tedanalactam 5,2 (+)-kausine 6,3 piplaroxide 7,2e,4 epoxydihydropiplartine 8,5 and epoxypipermethstine 9.6 The published syntheses of (−)-tedanalactam 5 have involved starting from the chiral pool (glucose),2d the use of a chiral auxiliary to effect resolution,2e and in the only asymmetric catalytic approach the use of the Sharpless AD reaction.2c The published syntheses of (+)-piplaroxide 72e and (−)-epoxypipermethystine 96b have both used a chiral auxiliary to effect resolution.2e No syntheses of (+)-kausine 6 or (−)-epoxydihydropiplartine 8 have been reported.
Scheme 1.
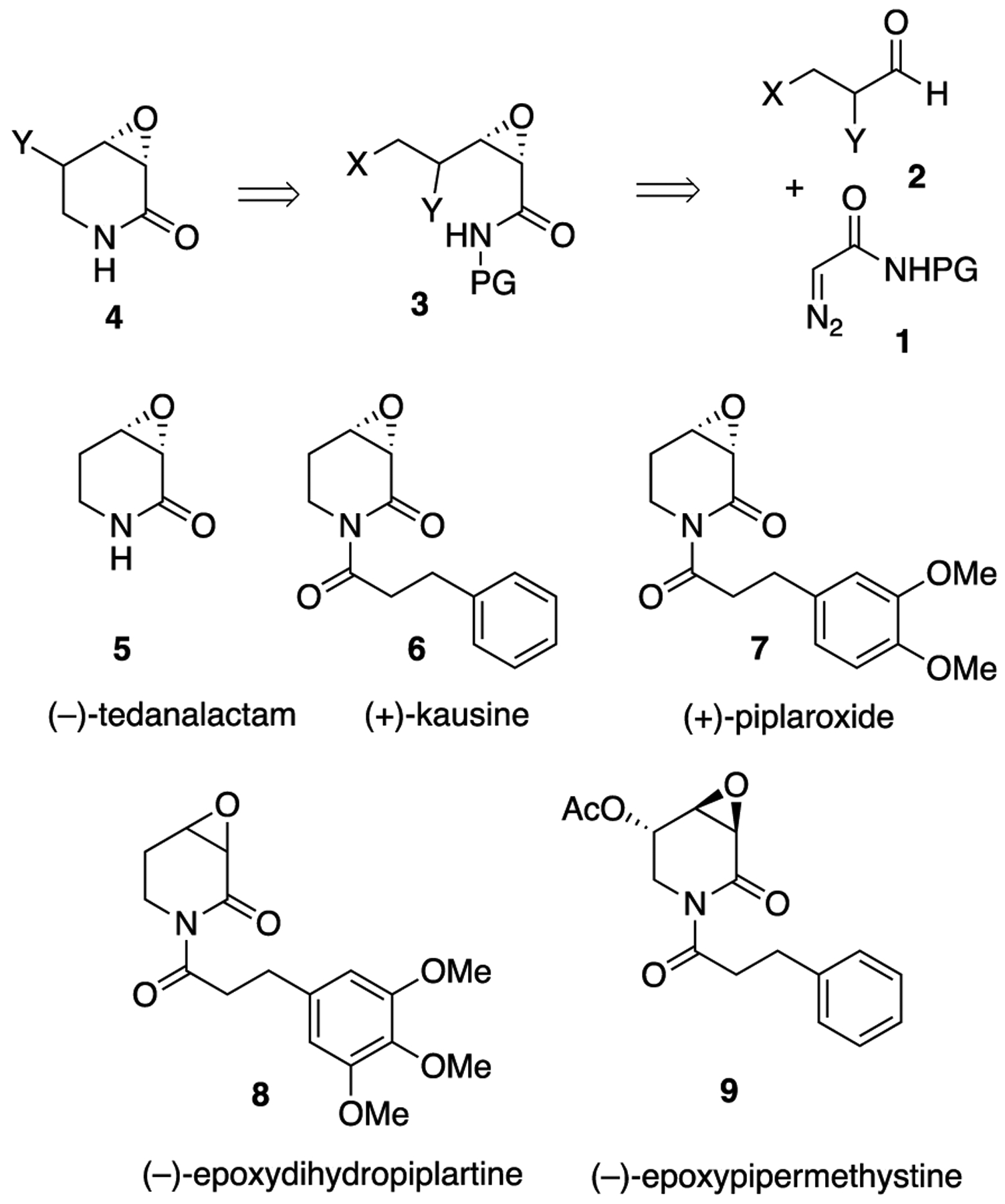
A Reterosynthetic Analysis for 3,4-Epoxy-2-piperidone Alkaloids
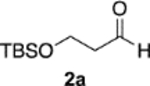
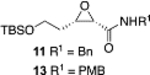
The synthesis of tedanalactam was to begin with the reaction of diazoacetamide 1a with the TBS-protected 3-hydroxypropanal 2a. When these two reagents were treated with the catalyst prepared as had been previously optimized for the epoxidation of aldehydes,1 none of the cis-epoxide 11 was observed and only 10% conversion was observed under the standard conditions (Scheme 2). This is in contrast to the reaction of diazoacetamide 1a with nonanal 2b, which gave the epoxide 12 in a 93% yield and >99% ee under the same conditions.1 The presence of DMSO in these reactions does not have a great deal of influence, typically giving slightly faster reactions and in some cases giving slightly higher asymmetric inductions.1 A repeat of the reaction of 1a and 2a without using DMSO in the preparation of the catalyst gave a 12% yield of 11 with a 15% conversion. This is again in contrast to the reaction of nonanal, which gave 12 in an 82% yield with >99% ee without DMSO.
Scheme 2.
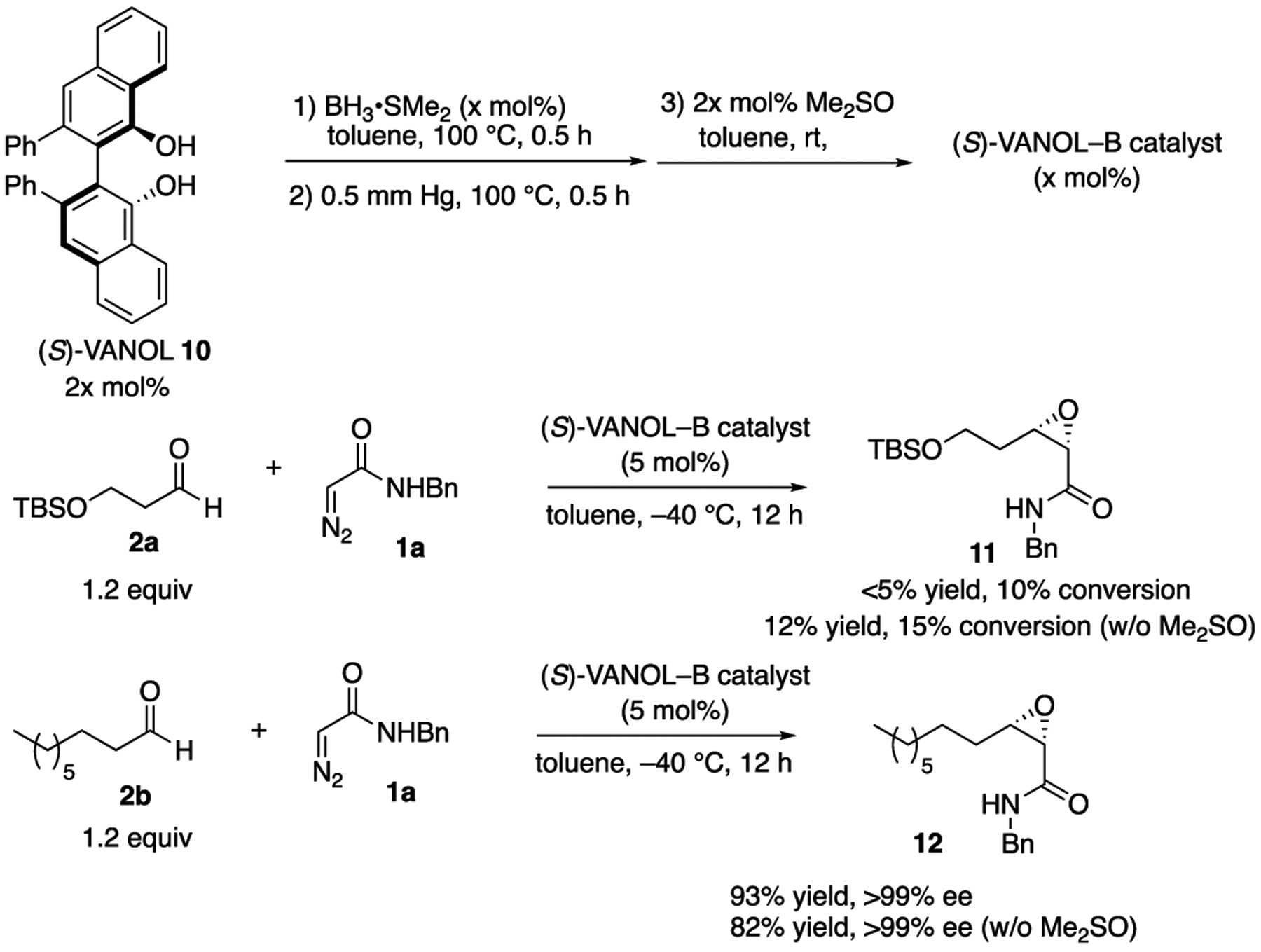
Failure of VANOL-borate Catalysts to Epoxidize Aldehyde 2a
SERENDIPITOUS DISCOVERY OF ENANTIODIVERGENCE IN ASYMMETRIC EPOXIDATION
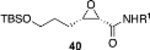
After several rounds of optimization (see Table S-I in the Supporting Information) involving variation of the catalyst loading, temperature, diazoacetamide, aldehyde silyl group, and amount of DMSO used in catalyst generation, the best results were obtained for the reaction of 2a and 1b using 20 mol % of the borate complex of VANOL prepared without any added DMSO. Under these conditions, the reaction conducted at 0 °C gives the cis-epoxide 13 in a 63% yield and 78% ee (Scheme 3). We then turned to the analogous heretofore unknown aluminate complexes of VANOL. The catalyst was generated by treating 2 equiv of the ligand with 1 equiv of trimethylaluminum in toluene at room temperature for 30 min. The findings reveal that the aluminum catalyst is superior to the boron catalyst. Using only 10 mol % aluminate (S)-VANOL complex as the catalyst at 0 °C, 13 was obtained in a 63% yield and 83% ee (Scheme 3). Incredibly, this slight increase in ee with the aluminate (S)-VANOL complex was accompanied by a reversal in the direction of the asymmetric induction relative to the reaction of the borate (S)-VANOL complex. Therefore, under otherwise identical reaction conditions and with the same enantiomer of the chiral ligand, a simple switch in the Lewis acid from boron to aluminum has led to the discovery of a perfectly enantiodivergent7 asymmetric epoxidation reaction. The discovery of conditions for the epoxidation of aldehyde 2a means that this methodogy for the synthesis of tedanalactam can go forward, but first we explored the scope of the epoxidation with the aluminum catalyst.
Scheme 3.
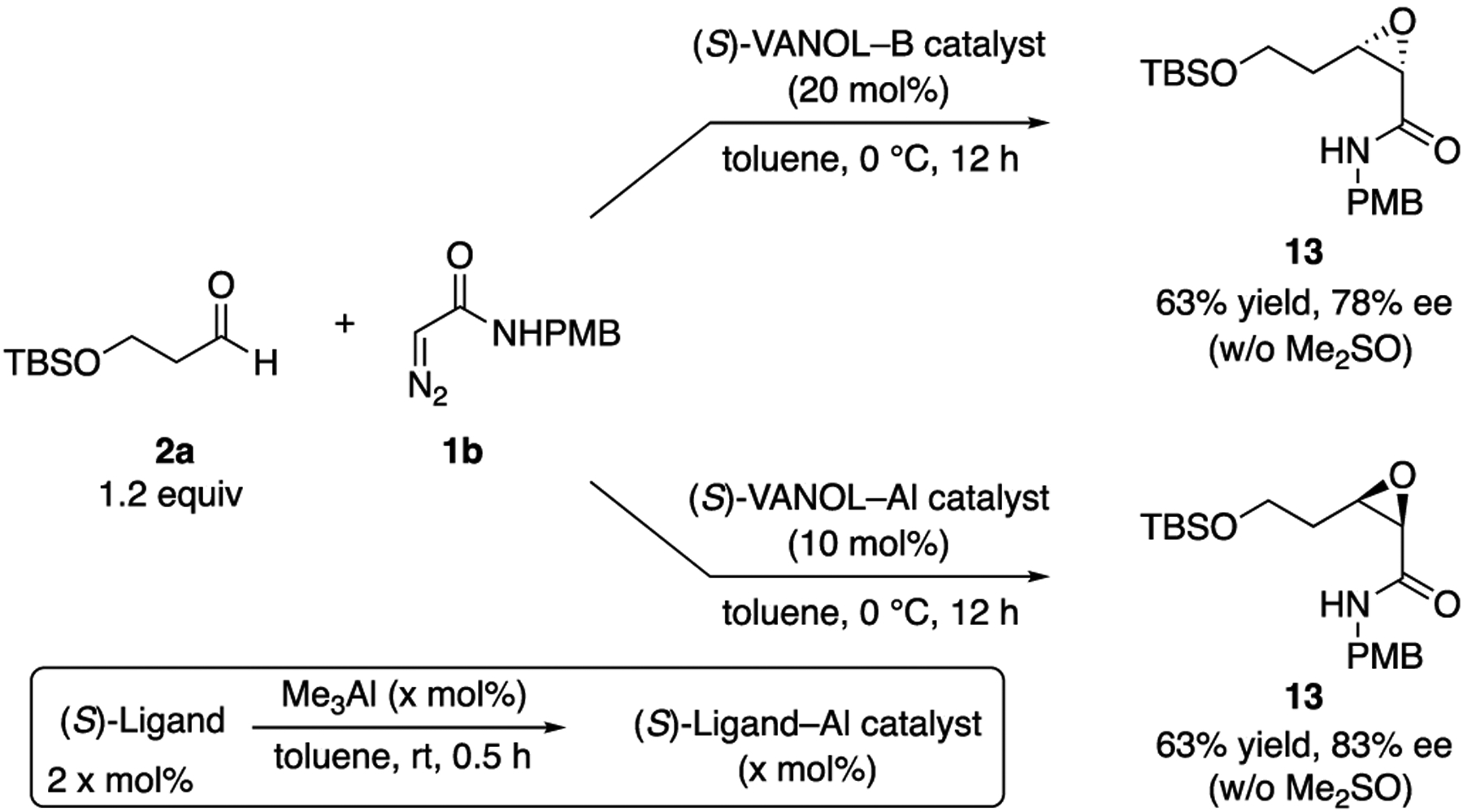
Unexpected Observation of Enantiodivergence upon Switching from Boron to Aluminum in the Asymmetric Epoxidation Reaction
Further optimization of the aluminate-catalyzed reaction (Table 1) by varying aldehyde (2a/2c), diazoacetamide (1a/1b), additive, and ligand led to the identification of the (S)-VAPOL (17) derived aluminate complex as the most efficient catalyst system; the reaction of 1a and 2a catalyzed by 10 mol % of the aluminate complex of (S)-VAPOL prepared without any added DMSO delivered the desired cis-epoxide 11 in an 80% yield and 96% ee (Table 1, entry 5). It is worth noting here that the borate complex of (S)-VAPOL did not give any useful reaction with aldehyde 2a (see the Supporting Information).
Table 1.
Epoxidation of 3-Siloxypropanal with the Aluminum Catalystsa
| ||||||||
|---|---|---|---|---|---|---|---|---|
| entry | ligand | R | R1 | catalyst (x mol %) | Me2SO (2x mol %) | epoxide | % yield epoxideb | % ee epoxidec |
| 1 | (S)-VANOL | TBS | Bn | 5 | 0 | 11 | 66 | 80 |
| 2 | (S)-VANOL | TBS | Bn | 10 | 0 | 13 | 63 | 83 |
| 3 | (S)-VANOL | TBS | Bn | 10 | 20 | 13 | 64 | 81 |
| 4 | (S)-VANOL | TBS | Bn | 5 | 0 | 11 | 85 | 91 |
| 5 | (S)-VANOL | TBS | Bn | 10 | 0 | 11 | 80 | 95 |
| 6 | (S)-VANOL | TBS | PMB | 5 | 0 | 13 | 76d | 92d |
| 7 | (R)-VANOL | TBS | PMB | 10 | 0 | 13 | 87e | −95e,f |
| 8 | (S)-VANOL | TBDPS | PMB | 10 | 0 | 14 | 77 | 94 |
| 9 | (S)-BINOL | TBS | Bn | 5 | 0 | 11 | (9) | nd |
Unless otherwise specified, all reactions were carried out in toluene at 0 °C for 12 h with 0.5 mmol diazo compound 1 at 0.1 M with 1.2 equiv of 2.
Isolated yields.
Determined by HPLC.
Average of two runs.
Average of four runs.
Enantiomer of epoxide 13 is formed.
SYNTHETIC ELABORATION OF ALUMINATE-CATALYZED ASYMMETRIC EPOXIDATION OF ALDEHYDES
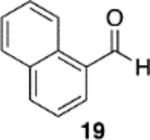
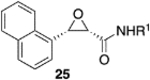
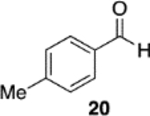
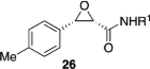
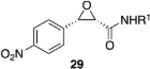
We were intrigued by the finding that boron and aluminum catalysts gave opposite enantiomers of 13, each from the same enantiomer of the ligand (Scheme 3). The first thought was that this could be somehow related to the nature of aldehyde 2a. To test this, a number of other aldehydes were examined (Table 2). The reaction of benzaldehyde 18 with diazoacetamide 1a gave the epoxide 24a in an 84% yield and 99% ee with the aluminum (S)-VAPOL catalyst and in an 89% yield and 81% ee with the aluminum (S)-VANOL catalyst (Table 2). Here, as with the aldehyde 2a (Table 1), the VAPOL aluminum catalyst gives higher asymmetric induction than the VANOL aluminum catalyst. Again, this is the opposite enantiomer of 24a that was seen with the VANOL-boron catalyst, which gave a a 98% yield of 24a in 97% ee (in this case the (S)-VANOL aluminum catalyst and the (R)-VANOL boron catalyst gave the same enantiomer of 24a).1 Four of the six aryl aldehydes in Table 2 are converted to the corresponding epoxide in 99% ee with 5 mol % of the aluminum VAPOL catalyst. The exceptions are 4-nitrobenzaldehyde 23, for which the induction in the epoxide 29a can be increased from 62% to 93% ee with 10 mol % catalyst, and 4-methoxybenzaldehyde, which was a very sluggish substrate, giving the epoxide 27a in a 11% yield at 50% conversion along with a 10% yield of the corresponding β-ketoamide. It is to be noted that the reaction of benzaldehyde is much faster with the boron catalyst than with the aluminum catalyst (Table 2). The reaction of diazoacetamide 1c and benzaldehyde with 5 mol % of the (S)-VANOL boron catalyst is complete in 2 h at −40 °C, giving an 86% yield of 24c with 93% ee, whereas the reaction with 10 mol % of the (S)-VANOL aluminum catalyst gives a 19% yield at −40 °C after 12 h.
Table 2.
Epoxidation of Aromatic Aldehydes with VANOL and VAPOL Aluminum Catalystsa
| ||||||
|---|---|---|---|---|---|---|
| aldehyde | epoxide | Ligand | catalyst (mol%) | R1 | % yield epoxideb | %ee epoxidec |
|
|
(S)-VAPOL | 5 | Bn | 97 | −99d |
| (S)-VAPOL | 5 | n-Bu | 86 | −97d | ||
| (S)-VAPOL | 10 | Bn | 84 | −99d | ||
| (S)-VANOL | 10 | Bn | 89 | −81d | ||
| (S)-VANOL | 10 | n-Bu | 19e,f | nd | ||
| (S)-VANOL | 5 | n-Bu | 86g | 93 | ||
|
|
(S)-VAPOL | 5 | Bn | 93 | −99d |
|
|
(S)-VAPOL | 5 | Bn | 76 | −99d |
| (S)-VAPOL | 5 | n-Bu | 66 | −93d | ||
|
|
(S)-VAPOL | 10 | Bn | 11e,h | nd |
|
|
(S)-VAPOL | 5 | Bn | 90 | 99d |
| (S)-VAPOL | 5 | n-Bu | 77 | −85d | ||
|
|
(S)-VAPOL | 5 | Bn | 55 | −62d |
| (S)-VAPOL | 5 | n- Bu | 36 | −71d | ||
| (R)-VAPOL | 10 | Bn | 70 | 93 | ||
Unless otherwise specified, all reactions were carried out in toluene at 0 °C for 12 h with 0.5 mmol diazo compound 1 at 0.1 M with 1.2 equiv of aldehyde. The catalyst was prepared as indicated in Table 1.
Isolated yields.
Determined by HPLC. nd = not determined.
The enantiomer of the epoxide was formed.
NMR yield.
This reaction as carried out at −40 °C for 12 h.
This reaction was with the boron catalyst prepared as in Scheme 2 without DMSO and was performed at −40 °C for 2 h. See ref 1.
Reaction went to 50% conversion and also gave a 10% yield of a β-keto amide.
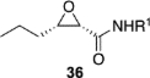
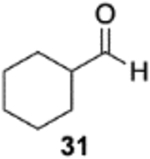
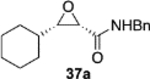
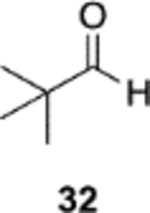
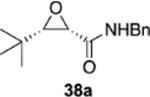
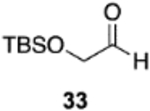
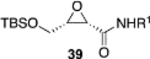
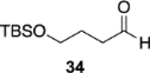
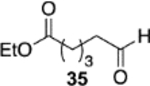
The reversal in enantioselectivity also extends to the series of aliphatic aldehydes shown in Table 3. The unbranched aldehyde butanal can be transformed to epoxide 36a in an 84% yield and 95% ee and to epoxide 36b in a 79% yield and 94% ee. The secondary carboxaldehyde 31 can be epoxidized to give 37a in an 81% yield with 94% ee. The epoxidation of the tertiary carboxaldehyde 32 by the VAPOL-Al catalyst gives the epoxide 38a in 99% ee but in only a 50% yield. The substrate scope was extended to t-butyldimethylsiloxy-substituted aldehydes 33 and 34, which have one less and one more methylene between the siloxy group and the aldehyde function than aldehyde 2a, respectively. While the latter gives epoxide 40a in an 88% yield and 92% ee, the former is not a good substrate, giving epoxide 39 at best in an 86% yield but only 54% ee. The aldehyde 35 was also examined in an effort to determine what other functionality might be tolerated in this reaction. Here the ester group is well tolerated, giving the epoxide 41a in a 74% yield and 96% ee with the (R)-VAPOL-Al catalyst. It is interesting to note that the (R)-VANOL-B catalyst (10 mol %) gives the enantiomer of the epoxide 41a in an 80% yield and 91% ee under the conditions shown in Scheme 2 without DMSO (Table 3).
Table 3.
Epoxidation of Aliphatic Aldehydes with VANOL and VAPOL Aluminum Catalystsa
| ||||||
|---|---|---|---|---|---|---|
| aldehyde | epoxide | Ligand | catalyst (mol%) | R1 | % yield epoxideb | %ee epoxidec |
|
|
(S)-VAPOL | 5 | Bn | 62 | −89 d |
| (R)-VANOL | 5 | Bn | 74 | 92 | ||
| (S)-VANOL | 10 | Bn | 84 | −95 d | ||
| (S)-VANOL | 10 | Bn | 24 | −86 d,e | ||
| (S)-VAPOL | 5 | PMB | 50 | −86 d | ||
| (R)-VAPOL | 10 | PMB | 79 | 94 | ||
|
|
(S)-VAPOL | 5 | Bn | 68 | −88 d |
| (R)-VAPOL | 10 | Bn | 81 | 94 | ||
|
|
(R)-VAPOL | 10 | Bn | 50 | 99 |
|
|
(S)-VAPOL | 5 | Bn | 78 | −56d |
| (R)-VAPOL | 10 | Bn | 73 | 58 | ||
| (S)-VAPOL | 5 | PMB | 73 | −50d | ||
| (R)-VAPOL | 10 | PMB | 86 | 54 | ||
|
|
(S)-VAPOL | 5 | Bn | 85 | −91d |
| (S)-VANOL | 5 | Bn | 66 | −80d | ||
| (R)-VAPOL | 10 | Bn | 80 | 96 | ||
| (S)-VAPOL | 5 | PMB | 76f | −92d,f | ||
| (R)-VAPOL | 10 | PMB | 87g | −95g | ||
| (S)-VANOL | 5 | PMB | 63 | −83d | ||
|
|
(S)-VAPOL | 5 | Bn | 84 | −50d |
| (R)-VAPOL | 10 | Bn | 88 | 92 | ||
| (S)-VAPOL | 5 | PMB | 67 | −51d | ||
| (R)-VAPOL | 10 | PMB | 78 | 88 | ||
|
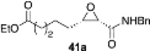
|
(S)-VAPOL | 5 | Bn | 75 | −76d |
| (R)-VAPOL | 10 | Bn | 74 | 96 | ||
| (R)-VANOL | 10 | Bn | 80 | −91d,h | ||
| (S)-VANOL | 10 | Bn | 99 | 95i | ||
Unless otherwise specified, all reactions were carried out in toluene at 0 °C for 12 h with 0.5 mmol diazo compound 1 at 0.1 M with 1.2 equiv of the aldehyde. The catalyst was prepared as indicated in Table 1.
Isolated yields.
Determined by HPLC.
The enantiomer of the epoxide is formed.
Reaction at −40 °C for 12 h.
Average of two runs.
Average of four runs.
This reaction was carried out with a boron catalyst as indicated in Scheme 3 in toluene at −40 °C for 12 h without DMSO.
Carried out as in h but with 20 mol % DMSO.
MECHANISTIC INVESTIGATION
Having established the scope of this novel aluminate-catalyzed epoxidation reaction, we turned our attention to elucidating the mechanistic underpinnings of this reaction. In particular, we were interested in a comparison of the asymmetric epoxidation reaction enabled by the borate and aluminate complexes in order to understand the origin of the enantiodivergence induced by changing the Lewis acidic center from boron to aluminum. Accordingly, we modeled key transition states (TS) in the reactions of benzaldehyde 18 and diazoacetamide 1a resulting in the cis-epoxide 24a catalyzed by either the (R)-VANOL borate or the (R)-VANOL aluminate using B3LYP8-D39/6–31G(d)10 as implemented in Gaussian 16.11 High-level single point energy calculations were performed for all stationary points using M06–2x12/6–311++G** PCM13 (toluene). The Gibbs free energies reported in this article are obtained by adding the free energy correction from the optimizations to the high-level single point energy calculations. The free energies were corrected using Grimme’s quasi-rigid rotor -harmonic oscillator (qRRHO) approach.14 This approach is routinely utilized and is well-established to evaluate reactivity and selectivity in similar catalytic systems.15
Based on prior catalyst structure studies,1,16 we assumed that both the aluminum and boron catalysts have incorporated two molecules of the VANOL ligand. We envisioned three viable mechanistic pathways for aldehyde activation: Lewis acid activation (LA), Brønsted acid-assisted Lewis acid activation (BLA), and a Brønsted acid (BA) pathway, as shown in Scheme 4. In the BLA pathway, the aldehyde is activated by coordination to the Lewis acid (B/Al), which is flanked by two (R)-VANOL ligands. Three out of the four phenolic oxygen atoms of these two ligands are directly bonded to B/Al, and the free phenol unit of one of the ligands is H-bonded to an oxygen of the other ligand (BLA activation). It is this tricoordinated B/Al that serves as the chiral Lewis acid that activates the aldehyde in the BLA mechanism. The LA activation pathway has all the features of the BLA pathway except for the intramolecular H-bonding interaction. Finally, BA activation involves activation of the aldehyde by protonation with the spiro borate/aluminate serving as the chiral counterion that orchestrates the asymmetric transformation. In all three modes of aldehyde activation, the activated aldehyde 18 undergoes nucleophilic attack by 1a (TSCC) resulting in a betaine intermediate, which then undergoes an intramolecular SN2-like displacement of N2 by the aldehyde oxygen (TSRC) to form the epoxide product 24a. We initiated our DFT study by locating TSCC and TSRC steps for LA, BLA, and BA pathways for both the boron and aluminum catalyst systems. Our first goal was to identify the mode of catalysis in these reactions (LA/BLA/BA). The second objective was to identify the rate- and enantioselectivity-determining steps (TSCC or TSRC) within the operative mechanism for each catalyst system. Once we identify the enantioselectivity-determining step, our final goal was to understand the origin of enantioselectivity in each catalyst system and the origin of enantiodivergence observed across the two catalysts.
Scheme 4.
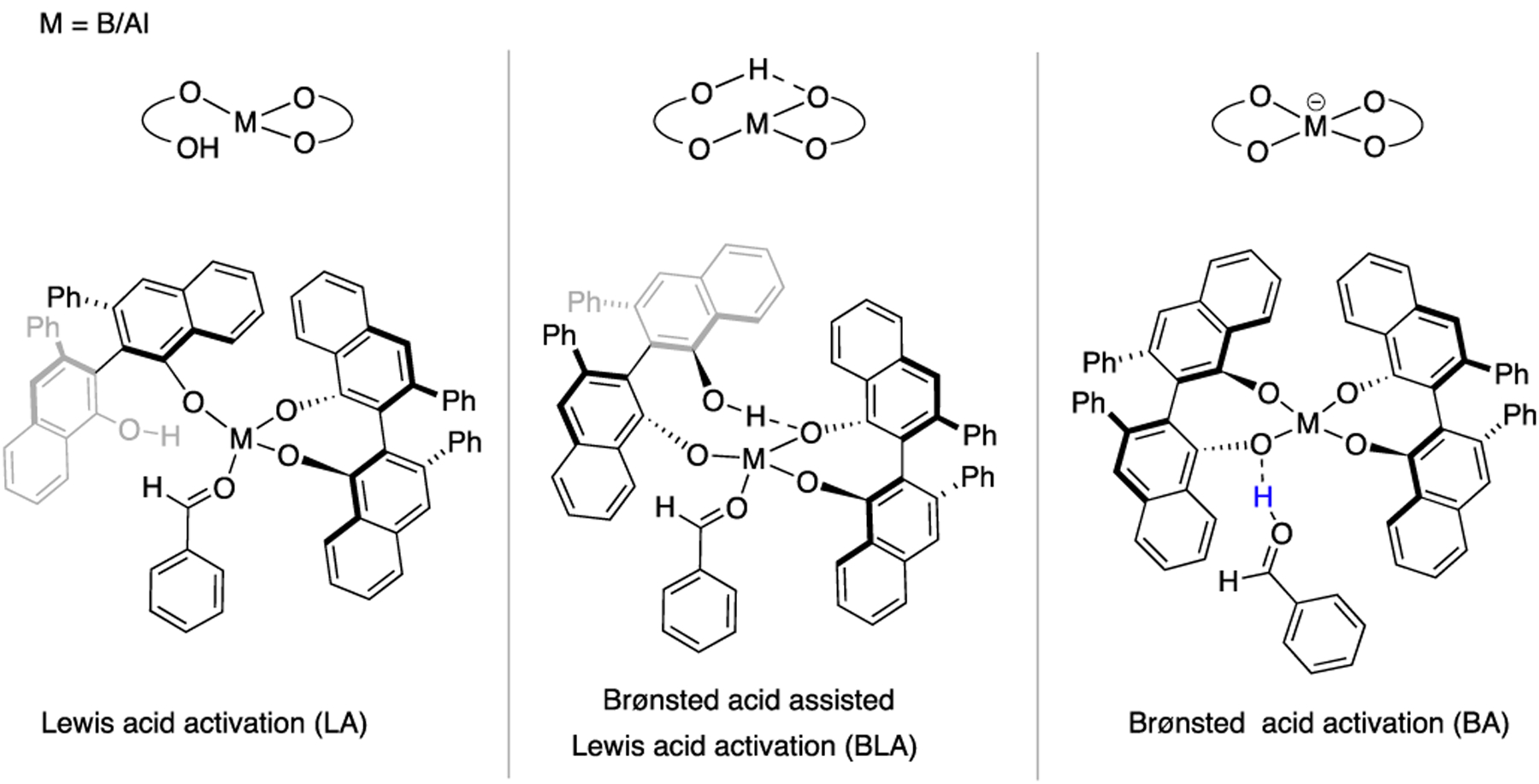
Three Distinct Pathways for Aldehyde Activation Investigated Using DFT Calculations for the (R)-VANOL-Lewis Acid-Catalyzed Asymmetric Epoxidation Reaction
In order to identify which of the three pathways is operative, we modeled each pathway leading to the major enantiomer of cis-epoxide 24a for both the boron and the aluminum catalysts. For both catalyst systems, between the two modes of Lewis acid activation (BLA and LA), it was found that the BLA pathway was favored over the LA pathway (see the SI for details). Therefore, our next step was to evaluate the relative energy of the BLA pathway to the BA pathway. Figure 1A shows the computed energy profile for the BLA and BA pathways for the boron catalyst relative to the lowest energy aldehyde-catalyst complex + 1a as reference. The key conclusions for the boron catalyst from this comparison are as follows: (a) For both BA and BLA pathways, initial C−C bond formation (TSCC-B-BLA-maj/TSCC-B-BA-maj) to form the betaine intermediate is lower in energy than the respective ring-closing steps that form the epoxide (TSRC-B-BLA-maj/TSRC-B-BA-maj), suggesting that betaine formation is reversible and ring-closure is the rate- and enantioselectivity-determining step of the reaction in both pathways. (b) The rate-determining step in the BLA pathway (TSRC-B-BLA-maj) is lower in energy than the rate-determining step in the BA pathway (TSRC-B-BA-maj), suggesting that the reaction most likely proceeds via the BLA pathway for the boron catalyst. Figure 1B shows the summary of an analogous analysis of the (R)-VANOL aluminate catalyst. The main conclusions from our investigation of the boron catalyst hold for the aluminum catalyst: the reaction proceeds via the BLA mechanism, and the ring-closure step (TSRC-Al-BLA-maj) is the rate- and enantioselectivity-determining step in the reaction for the aluminum catalyst. It is worth noting that a qualitatively similar free energy profile, where the ring-closing step was enantiodetermining, was previously established via experimental and computational studies of the Brønsted acid-catalyzed aziridination reaction of imines and diazoacetates developed in our lab.16,17
Figure 1.
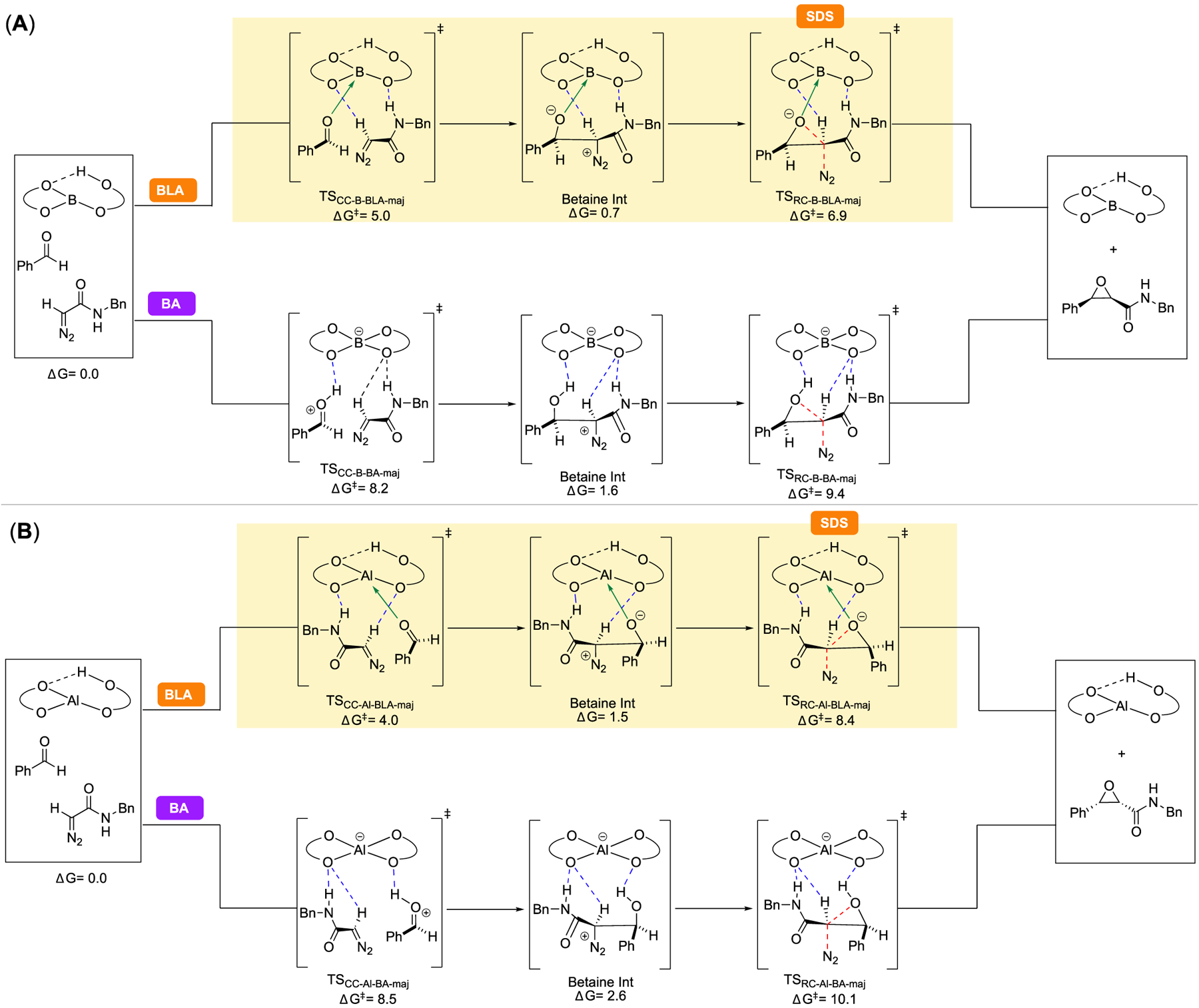
Free energies of key stationary points in the reaction coordinate leading to the major enantiomer of cis-epoxide 24a for the reaction catalyzed by the (A) (R)-VANOL boron-catalyst and (B) (R)-VANOL aluminum-catalyst, computed at M06–2x/6–311++G** PCM (toluene)//B3LYP-D3/6–31G*. Both Brønsted acid-assisted Lewis acid (BLA) and Brønsted acid (BA) pathways are shown, and the reported energies for each transition state and intermediate are relative to the prereactive complex of the respective catalyst with benzaldehyde (18) and diazoacetamide 1a in the BLA pathway, which is the lowest-energy stationary point in either pathway for both catalysts. The preferred pathway for each catalyst is highlighted in yellow, and the selectivity-determining step (SDS) is indicated.
SELECTIVITY-DETERMINING TRANSITION STRUCTURES FOR THE (R)-VANOL BORATE-CATALYZED EPOXIDATION REACTION
Based on our evaluation of the mechanism of the title reaction, in order to probe the origin of enantioselectivity and therefore the origin of enantiodivergence in these systems, we need to evaluate the relative energies of TSRC leading to both enantiomers of cis-24a for both catalyst systems. We first discuss our results from the reaction with the boron catalyst. We have already identified the lowest-energy TS leading to the major enantiomer (R,R) of cis-24a via the BLA mechanism (discussed in Figure 1A). The fine details of this TS, i.e., TSRC-B-BLA-maj, are shown Figure 2. It is worth noting here that it was during the extensive conformational search performed to locate TSRC-B-BLA-maj that we identified the existence of the LA pathway (discussed in Scheme 4). Also shown in Figure 2 is the lowest energy TS leading to (R,R)-24a in the LA pathway (TSRC-B-LA-maj), which is 4.9 kcal/mol higher in energy than TSRC-B-BLA-maj. Following this, we performed a thorough conformational search to locate TSRC leading to the minor enantiomer (S,S) of cis-24a to enable a theoretical prediction of the enantioselectivity observed in this reaction. Shown in Figure 2 are the lowest-energy transition structures leading to (S,S)-24 in the BLA (TSRC-B-BLA-min) and LA (TSRC-B-LA-min) pathways. The BLA pathway is found to be slightly favored over the LA pathway for the minor enantiomer by 1.8 kcal/mol. Therefore, the predicted enantioselectivity for the reaction of boron catalyst is obtained by comparing the relative energy of the lowest energy TSs leading to the major enantiomer (TSRC-B-BLA-maj resulting in (R,R)-24a highlighted in green in Figure 2) and the minor enantiomer (TSRC-B-BLA-min resulting in (S,S)-24a highlighted in red in Figure 2). The energy difference between these two TSs is 3.2 kcal/mol, a value that is in excellent agreement with the >99% ee observed experimentally for this reaction.1 Analysis of these highlighted TSs reveals that both of these TSs have identical bond-forming (rO−C = 2.12 Å) and bond-breaking (rC−N = 1.79 Å) distances for the SN2-like ring-closing event. Additionally, both these TSs are characterized by a short H-bonding interaction between the free phenolic OH of the VANOL ligand and one of the other oxygen atoms bound to boron, the interaction that is responsible for BLA activation. However, these TSs are fundamentally different in the nature of catalyst-substrate interactions that are involved in transition state stabilization. TSRC-B-BLA-maj is stabilized by a short, strong CH···O interaction (1.99 Å) between the acidic CH of 1a and one of the catalyst oxygen atoms and an intramolecular H-bonding interaction between the NH of 1a and the aldehyde oxygen (2.23 Å); on the other hand, TSRC-B-BLA-min is stabilized by a strong H-bonding interaction between the NH of 1a and one of the catalyst oxygen atoms (1.88 Å) and a possible CH−π interaction between the acidic CH of 1a and the aromatic rings of the VANOL ligand.
Figure 2.
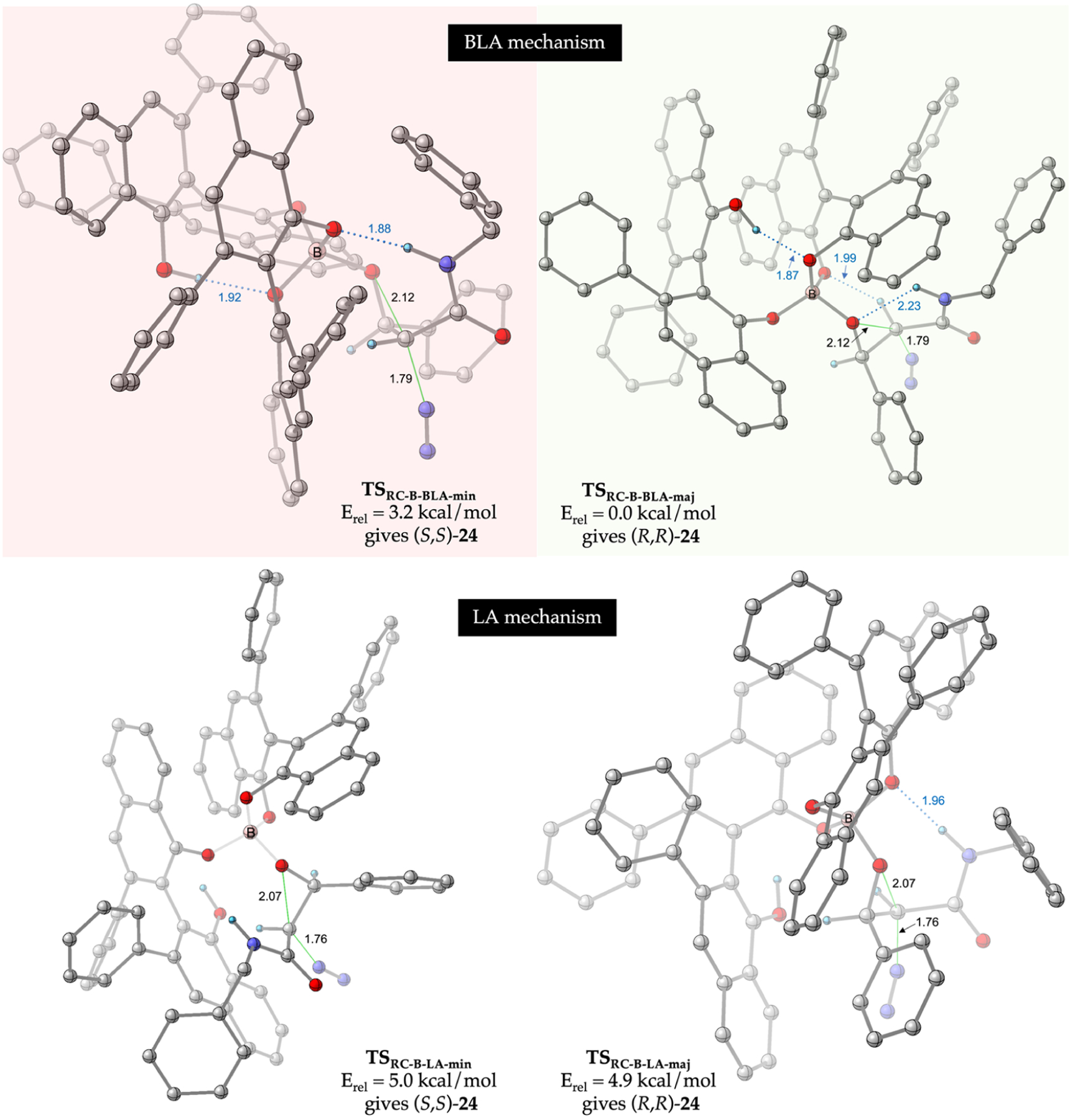
Transition state geometries of the rate- and enantioselectivity-determining ring-closure step leading to the major (R,R) and minor (S,S) enantiomer of the cis-epoxide 24a in the reaction of 18 and 1a catalyzed by the (R)-VANOL-B catalyst. The first two structures are for the BLA pathway, while the next two structures are for the LA pathway. The energies of all four structures, computed at M06–2x/6–311++G** PCM (toluene)//B3LYP-D3/6–31G*, are relative to the lowest-energy transition structure TSRC-B-BLA-maj. The lowest-energy transition structures leading to the major and minor enantiomers of cis-24a are highlighted in green and red boxes, respectively. All distances are in angstroms, and most hydrogen atoms have been removed for clarity.
SELECTIVITY-DETERMINING TRANSITION STRUCTURES FOR THE (R)-VANOL ALUMINATE-CATALYZED EPOXIDATION REACTION
For a direct comparison to the boron catalyst, we conducted a comprehensive conformational search for the rate- and enantioselectivity-determining ring-closing TS for both the BLA and LA pathways in the epoxidation reaction catalyzed by the aluminum catalyst. The lowest-energy transition structures leading to the experimentally observed major (S,S) and minor (R,R) enantiomers of cis-24a in both these pathways are shown in Figure 3. The four TSs shown in Figure 3 are very similar in geometry to the analogous TSs shown in Figure 2. However, remarkably and consistent with the experimentally observed enantiodivergence, our calculations show that TSRC-Al-BLA-maj (the ring-closing TS that gives (S,S)-24a) is now the lowest-energy TS for the reaction with the aluminum catalyst. Intriguingly, the lowest-energy TS leading to the minor (R,R) enantiomer proceeds via the LA activation pathway; TSRC-Al-LA-min is 1.8 kcal/mol higher in energy than TSRC-Al-BLA-maj, a value that is in good agreement with the 81% ee observed for this reaction (Table 2). Analysis of the key interactions stabilizing the aluminum TSs (Figure 3) reveals that they are very similar to the corresponding interactions in the analogous boron TSs (Figure 2). Therefore, the reversal in the sense of asymmetric induction upon switching from boron to aluminum must have its origin in the relative energetic contributions of these stabilizing interactions in the two catalytic systems.
Figure 3.
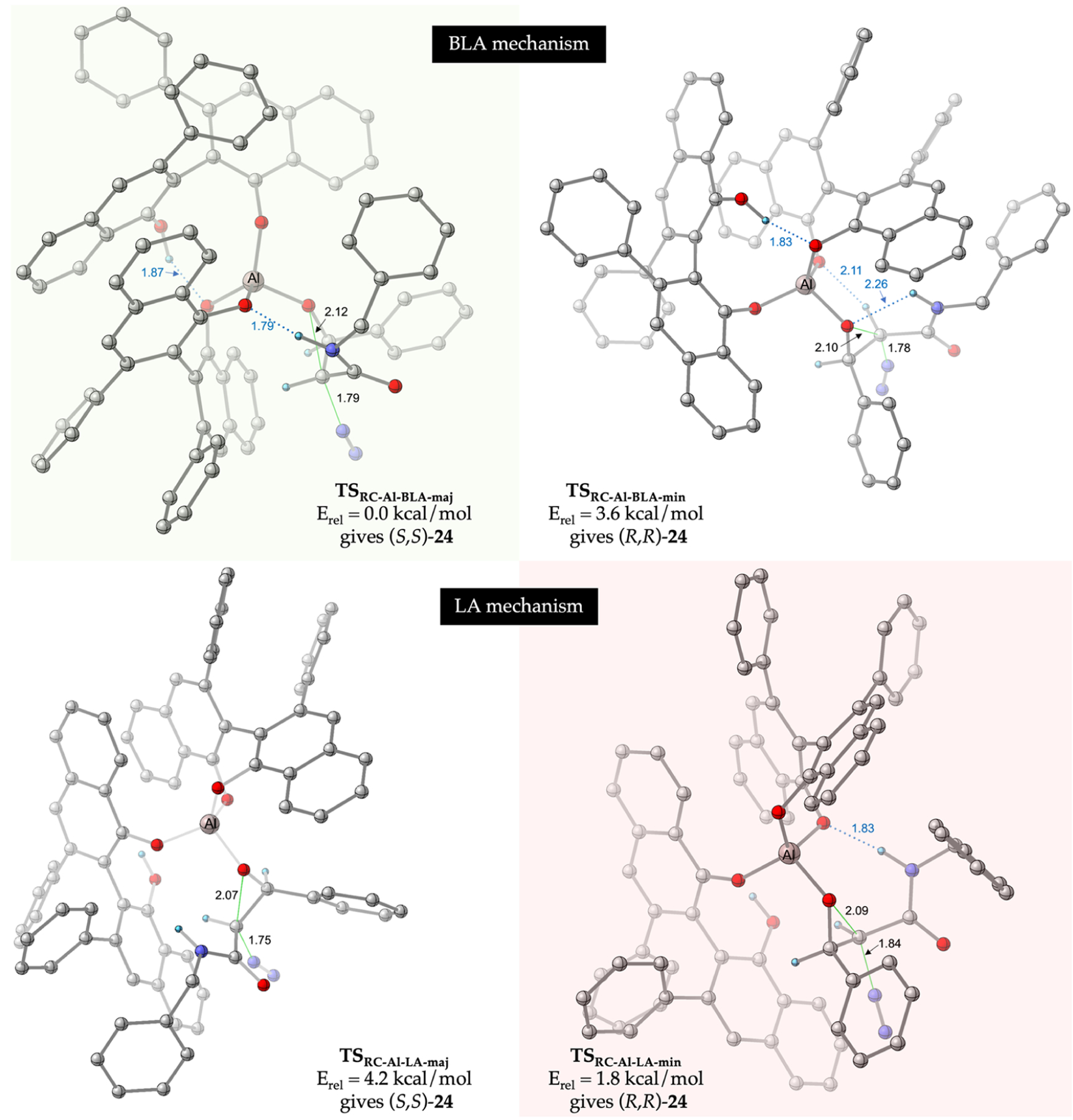
Transition state geometries of the rate- and enantioselectivity-determining ring-closure step leading to the major (S,S) and minor (R,R) enantiomers of the cis-epoxide 24a in the reaction of 18 and 1a catalyzed by the (R)-VANOL-Al catalyst. The first two structures are for the BLA pathway, while the next two structures are for the LA pathway. The energies of all four structures, computed at M06–2x/6–311++G** PCM (toluene)//B3LYP-D3/6–31G*, are relative to the lowest-energy transition structure TSRC-Al-BLA-maj. The lowest energy transition structures leading to the major and minor enantiomers of cis-24a are highlighted in green and red boxes, respectively. All distances are in angstroms, and most hydrogen atoms have been removed for clarity.
ORIGIN OF ENANTIOSELECTIVITY AND ENANTIODIVERGENCE OBSERVED IN THE (R)-VANOL ALUMINATE/BORATE-CATALYZED EPOXIDATION RECTION
A comparison of the transition structures leading to (S,S)-24a for the boron catalyst (Figure 2, TSRC-B-BLA-min) and the aluminum catalyst (Figure 3, TSRC-Al-BLA-maj) reveals that these structures are nearly identical in terms of the bond-forming/bond-breaking distances as well as the key stabilizing interactions. A similar conclusion can be drawn upon analysis of the corresponding transition structures leading to (R,R)-24a for the boron (Figure 2, TSRC-B-BLA-maj) and aluminum (Figure 3, TSRC-Al-BLA-maj) catalyst systems. Even though these pairs of analogous transition structures are near-identical, the calculations predict that (R,R)-24a is the major enantiomer for the boron catalyst and (S,S)-24a is the major enantiomer for the aluminum catalyst (Figures 2 and 3), consistent with the experimentally observed enantiodivergence observed across the two catalyst systems. Qualitatively, these observations suggest that the identity of the central Lewis acidic atom (B/Al), the difference in polarization, and bond lengths of the metal−oxygen bonds in the two metalate species likely play important roles in the observed enantiodivergence in these catalyst systems. The effects of these subtle differences in catalyst properties must stabilize the exact same transition state differently.
In order to evaluate the origin of enantioselectivity in each of these catalyst systems, we conducted an energy decomposition analysis of the transition structures leading to the major and minor enantiomers of cis-24a for the BLA mechanism (top panel, Figures 2 and 3).19 An energy decomposition analysis20 comparing TSs leading to the two enantiomers of product provides quantitative insight into the origin of the observed enantioselectivity by separating the effects of distortion (catalyst and substrate distortion from lowest-energy conformation) and interaction (H-bonding, dispersion etc.) energies to transition state stabilization.
The summary of this analysis is shown in Table 4. The key findings are as follows: (a) Comparison of the interaction energies reveals that both catalysts favor the (R,R) enantiomer (Table 4, middle column highlighted in green), boron by 1.0 kcal/mol and aluminum by 3.4 kcal/mol. This observation can be rationalized based on the fact that the (R,R)-TS in both catalyst systems is characterized by conventional H-bonding interactions (vide supra); with the more polarized Al−O bond providing better stabilization via the stronger H-bonds to the aluminate oxygen atoms relative to the borate oxygen atoms. (b) The major contributor to the observed enantiodivergence is the difference in the distortion energies at the transition states leading to the two enantiomers of cis-24; that is, the (R,R)-enantiomer pays a lower energetic penalty from distortion compared to the (S,S)-enantiomer (favored by 1.7 kcal/mol, Table 4, row 1, column 1 highlighted in green) for the boron catalyst, while the reverse is true for the aluminum catalyst, where the (S,S)-enantiomer is favored by 5.5 kcal/mol over the (R,R)-enantiomer (Table 4, row 2, column 1 highlighted in red). A possible explanation for this reversal in the trend of distortion interactions could be gleaned by visual analysis of the TS leading to the (S,S)-enantiomer in the boron (Figure 2, TSRC-B-BLA-min) and aluminum (Figure 3, TSRC-Al-BLA-maj). It is likely that the longer Al−O bond places the VANOL ligand farther away from the chiral pocket compared to its position with the boron catalyst and the shorter B−O bond. As a result, the diazoacetamide portion of the transition state has to distort less in TSRC-Al-BLA-maj compared to TSRC-B-BLA-min in order to avoid a deleterious steric interaction with the VANOL backbone. The combined effect of the distortion and interaction energies is that the (R,R) enantiomer is overall favored by 2.7 kcal/mol by the boron catalyst and the (S,S) enantiomer is overall favored by 2.1 kcal/mol (Table 4, column 3). In summary, the switch from boron to aluminum as the central metal results in a reversal of the sense of enantioselectivity because of changes in the steric features of the chiral pocket as well as the electronic properties of the H-bond acceptor oxygen atoms that flank the central atom (B/Al). The full details of the energy decomposition analysis can be found in the Supporting Information.
Table 4.
Summary of Key Findings from Energy Decomposition Analysis Performed on the Lowest Energy Transition Structures Leading to Either Enantiomer within the BLA Mechanism for Both Catalysts Using M06–2x-D3/6–311++G** PCM(toluene)a
| Favors (R,R)-24 | Favors (S,S)-24 | ||
|---|---|---|---|
| Distortion (kcal/mol) | Interaction (kcal/mol) | Overall (kcalmol) | |
| ΔΔE‡ (R)-VANOL-B catalyst | 1.7 | 1.0 | 2.7 |
| ΔΔE‡ (R)-VANOL- Al catalyst | 5.5 | 3.4 | 2.1 |
See ref 18. ΔΔE‡ represents the absolute value of the energy difference between the relevant transition structures for each component of the energy being compared (distortion and interaction). Numbers highlighted in green indicate an energy component that favors (R,R)-24, while those highlighted in red indicate the favorability for (S,S)-24.
SYNTHESIS OF (−)-TEDANALACTAM
With the initial success of the epoxidation of aldehyde 2a with the aluminum catalyst (Table 1), the epoxide 13 was then taken on to tedanalactam 5, which was when it was first realized that the boron and aluminum catalysts give opposite enantiomers in the epoxidation reaction: the rotation (αD = 7.1) did not match that reported for the natural product (αD = −7.6). This was of course rectified by switching to the (R)-VAPOL aluminum catalyst, as indicated in Scheme 5. The aldehyde 2a is commercially available but is very expensive and thus was prepared as indicated in Scheme 5 from 1,3-propanediol following the procedure of Allegretti and Ferreira.21 The epoxide 13 is then deprotected to give the 1°-alcohol 44, which is then converted to the 1°-tosylate and then directly cyclized with sodium hydride to give 3,4-epoxy-2-piperidone 45 in a 67% yield. If the tosylate is isolated first, the yield for this sequence drops to 47%. Deprotection of the lactam nitrogen in 45 can be effected with ceric ammonium nitate to give (−)-tedanalactam in a 75% yield (Scheme 5).
Scheme 5.
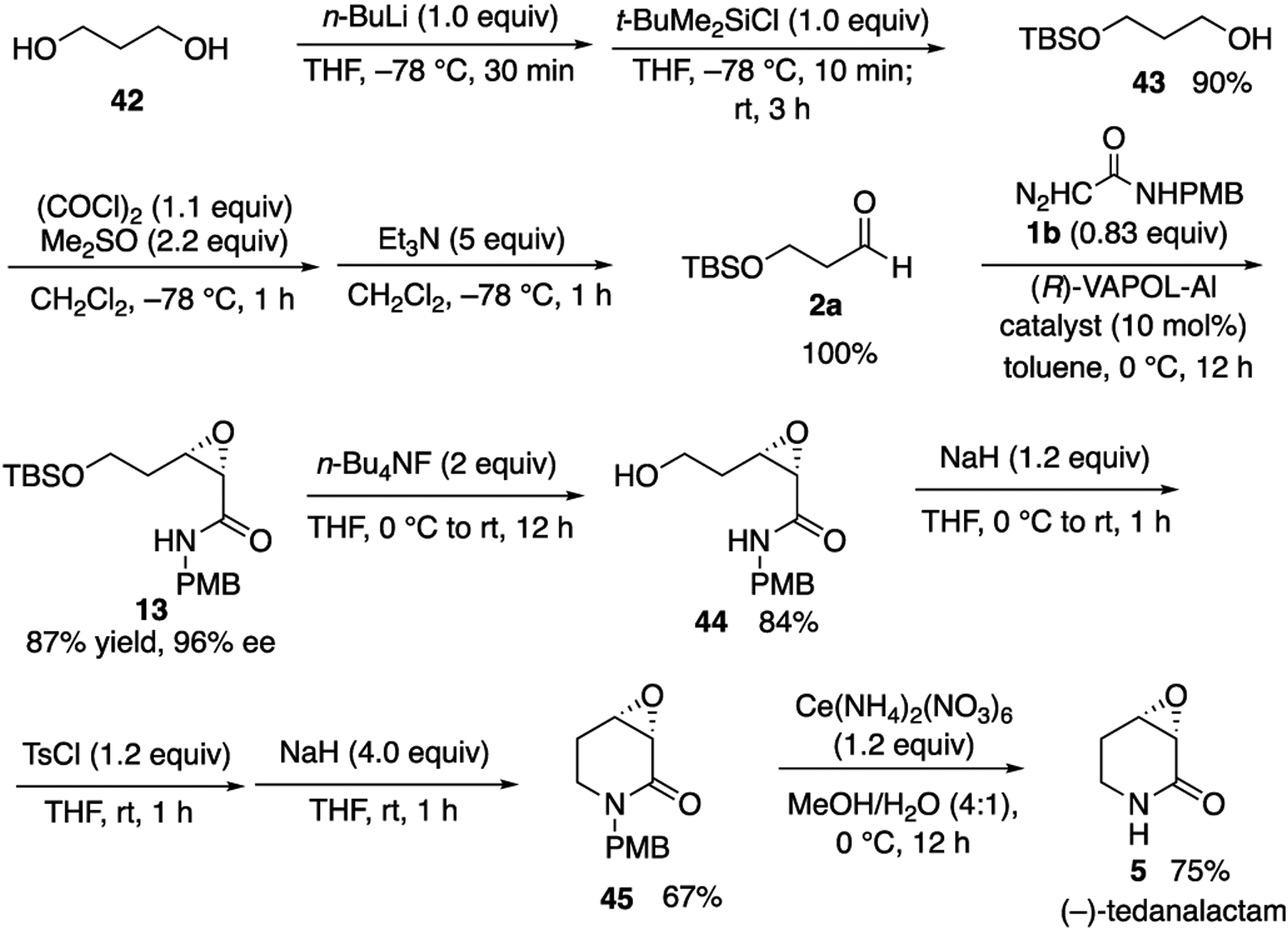
Synthesis of (−)-Tedanalactam 5
With the successful synthesis of (−)-tedanalactam, we have accomplished the original goal of our studies, the application of our asymmetric catalytic method for the preparation of cis-α,β-epoxyamides (from aldehydes and diazoacetamides using a chiral borate catalyst containing a VANOL ligand) to the synthesis of 3,4-epoxy-2-piperidone alkaloids. Critical failures in our exploratory studies toward achieving this goal led to the design and discovery of a new catalyst system involving a chiral aluminate catalyst containing two VANOL ligands that is structurally analogous to the borate catalyst. Synthetic studies have established the scope, and computational investigations have elucidated the origin of the enantiodivergence observed upon switching from boron to aluminum. We are currently evaluating the generality of enantiodivergence in other catalytic asymmetric reactions of aldehydes.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by a grant from the National Institutes of Health under R01 GM094478 (W.D.W.). Research reported in this publication was further supported by the National Institutes of Health under R35 GM145320 (M.J.V.). M.J.V. acknowledges support from the XSEDE Science Gateways Program (allocation ID CHE160009), which is supported by the National Science Foundation Grant ACI-1548562.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.3c03929.
Full experimental procedures and characterization data, copies of 1H and 13C NMR spectra for all new compounds, and full details of computational methods (PDF)
The coordinates of all computed structures can be found in the xyz coordinate file (XYZ)
Complete contact information is available at: https://pubs.acs.org/10.1021/acscatal.3c03929
The authors declare no competing financial interest.
Contributor Information
Aliakbar Mohammadlou, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Chetan Joshi, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States.
Brendyn P. Smith, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Li Zheng, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
Stephanie A. Corio, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States
Virginia M. Canestraight, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States
Saeedeh Torabi Kohlbouni, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States.
S. Maryamdokht Taimoory, Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109, United States.
Babak Borhan, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States;.
Richard Staples, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States;.
Mathew J. Vetticatt, Department of Chemistry, Binghamton University, Binghamton, New York 13902, United States;
William D. Wulff, Department of Chemistry, Michigan State University, East Lansing, Michigan 48824, United States;
REFERENCES
- (1).Gupta AK; Yin X; Mukherjee M; Desai AA; Mohammadlou A; Jurewicz K; Wulff WD Catalytic Asymmetric Epoxidation of Aldehydes with Two VANOL-Derived Chiral Borate Catalysts. Angew. Chem., Int. Ed 2019, 58, 3361–3367. [DOI] [PubMed] [Google Scholar]
- (2).(a) Cronan JM; Cardellina JH A Novel δ-Lactam from the Sponge Tedania ignis. Nat. Prod. Lett 1994, 5, 85–88. [Google Scholar]; (b) Lago JHG; Kato MJ 3α,4α-Epoxy-2-piperidone, a new minor derivative from leaves of Piper crassinervium Kunth (Piperaceae). Nat. Prod. Res 2007, 21, 910–914. [DOI] [PubMed] [Google Scholar]; (c) Majik MS; Parameswaran PS; Tilve SG Total Synthesis of (−)- and (+)-Tedanalactam. J. Org. Chem 2009, 74, 6378–6381. [DOI] [PubMed] [Google Scholar]; (d) Konda S; Kurva B; Nagarapu L; Dattatray AM A stereoselective approach for the total synthesis of (−)-tadanalactam from acetonide-d-glucose. Tetrahedron Lett. 2015, 56, 834–836. [Google Scholar]; (e) Romero-Ibanñez J; Xochicale-Santana L; Quintero L; Fuentes L; Sartillo-Piscil F Synthesis of the Enantiomers of Tedanalactam and the First Total Synthesis and Configurational Assignment of (+)-Piplaroxide. J. Nat. Prod 2016, 79, 1174–1178. [DOI] [PubMed] [Google Scholar]
- (3).Kaou AM; Mahiou-Leddet V; Canlet C; Debrauwer L; Hutter S; Azas N; Ollivier E New amide alkaloid from the aerial part of Piper capense L.f. (Piperaceae). Fitoterapia 2010, 81, 632–635. [DOI] [PubMed] [Google Scholar]
- (4).Capron MA; Wiemer DF Piplaroxide, an Ant-Repellent Piperidine Epoxide from Piper tuberculatum. J. Nat. Prod 1996, 59, 794–795. [Google Scholar]
- (5).Seeram NP; Lewis PA; Jacobs H; McLean S; Reynolds WF; Tay L-L; Yu M 3,4-Epoxy-8,9-dihydropiplartine. A New Imide from Piper verrucosum. J. Nat. Prod 1996, 59, 436–437. [Google Scholar]
- (6).(a) Dragull K; Yoshida WY; Tang C-S Piperidine alkaloids from Piper methysticum. Phytochemistry 2003, 63, 193–198. [DOI] [PubMed] [Google Scholar]; (b) Osorio-Nieto U; Vázquez-Amaya LY; Höpfl H; Quintero L; Sartillo-Piscil F The direct and highly diastereoselective synthesis of 3, 4-epoxy-2-piperidones. Application to the total synthesis and absolute configurational assignment of 3α, 4α-epoxy-5β-pipermethystine. Org. Biomol. Chem 2018, 16, 77–88. [DOI] [PubMed] [Google Scholar]
- (7).(a) Riehl PS; Richardson AD; Sakamoto T; Reid JP; Schindler CS Origin of enantioselectivity reversal in Lewis acidcatalysed Michael additions relying on the same chiral source. Chemical Science 2021, 12, 14133–14142. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rout S; Das A; Singh VK Metal-Controlled Switching of Enantioselectivity in the Mukaiyama−Michael Reaction of α, β-Unsaturated 2-Acyl Imidazoles Catalyzed by Chiral Metal−Pybox Complexes. J. Org. Chem 2018, 83, 5058–5071. [DOI] [PubMed] [Google Scholar]; (c) Mazumder S; Crandell DW; Lord RL; Baik M-H Switching the Enantioselectivity in Catalytic [4+ 1] Cycloadditions by Changing the Metal Center: Principles of Inverting the Stereochemical Preference of an Asymmetric Catalysis Revealed by DFT Calculations. J. Am. Chem. Soc 2014, 136, 9414–9423. [DOI] [PubMed] [Google Scholar]; (d) Riehl PS; Richardson AD; Sakamoto T; Schindler CS Eight-step enantiodivergent synthesis of (+)-and (−)-lingzhiol. Org. Lett 2020, 22, 290–294. [DOI] [PubMed] [Google Scholar]; (e) Zanoni G; Castronovo F; Franzini M; Vidari G; Giannini E Toggling enantioselective catalysis—a promising paradigm in the development of more efficient and versatile enantioselective synthetic methodologies. Chem. Soc. Rev 2003, 32, 115–129. [DOI] [PubMed] [Google Scholar]; (f) Macharia J; Wambua V; Hong Y; Harris L; Hirschi JS; Evans GB; Vetticatt MJ A Designed Approach to Enantiodivergent Enamine Catalysis. Angew. Chem., Int. Ed 2017, 56, 8756–8760. [DOI] [PubMed] [Google Scholar]
- (8).Becke A Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- (9).Grimme S; Ehrlich S; Goerigk L Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- (10).Hehre WJ; Stewart RF; Pople JA Self-Consistent Molecular-Orbital Methods. I. Use of Gaussian Expansions of Slater-Type Atomic Orbitals. J. Chem. Phys 1969, 51, 2657–2664. [Google Scholar]
- (11).Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams-Young D; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, rev. C.01; Gaussian, Inc., Wallingford, CT, 2016. [Google Scholar]
- (12).Zhao Y; Truhlar DG The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc 2008, 120, 215–241. [Google Scholar]
- (13).(a) Miertuš S; Scrocco E; Tomasi J Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys 1981, 55, 117–129. [Google Scholar]; (b) Tomasi J; Mennucci B; Cammi R Quantum mechanical continuum solvation models. Chem. Rev 2005, 105, 2999–3094. [DOI] [PubMed] [Google Scholar]
- (14).Grimme S Supramolecular binding thermodynamics by dispersion-corrected density functional theory. Eur. J. Chem 2012, 18, 9955–9964. [DOI] [PubMed] [Google Scholar]
- (15).(a) Vetticatt MJ; Singleton DA Isotope Effects and Heavy-Atom Tunneling in the Roush Allylboration of Aldehydes. Org. Lett 2012, 14, 2370–2373. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Duan M; Díaz-Oviedo CD; Zhou Y; Chen X; Yu P; List B; Houk KN; Lan Y Chiral Phosphoric Acid Catalyzed Conversion of Epoxides into Thiiranes: Mechanism, Stereochemical Model, and New Catalyst Design. Angew. Chem., Int. Ed 2022, 61, e202113204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hu G; Gupta AK; Huang L; Zhao W; Yin X; Osminski WEG; Huang RH; Wulff WD; Izzo JA; Vetticatt MJ Pyro-Borates, Spiro-Borates, and Boroxinates of BINOL—Assembly, Structures, and Reactivity. J. Am. Chem. Soc 2017, 139, 10267–10285. [DOI] [PubMed] [Google Scholar]
- (17).Vetticatt MJ; Desai AA; Wulff WD Isotope Effects and Mechanism of the Asymmetric BOROX Brønsted Acid Catalyzed Aziridination Reaction. J. Org. Chem 2013, 78, 5142–5152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).(a) Lockhart Z; Popescu M; Alegra-Requena J; Ahuja J; Paton R; Smith M A radical-polar crossover approach to complex nitrogen heterocycles via the triplet state. ChemRxiv, May 19, 2023, ver. 1. DOI: 10.26434/chemrxiv-2023-pcv6m. [DOI] [Google Scholar]; (b) Jones BA; Solon P; Popescu MV; Du J-Y; Paton R; Smith MD Catalytic Enantioselective 6π Photocyclization of Acrylanilides. J. Am. Chem. Soc 2023, 145, 171–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).We recognize that the minor enantiomer for the aluminum catalyst comes from the LA mechanism,but for this analysis we wanted to see how identical TSs were stabilized differently by the two catalysts. We have also performed this analysis by comparison to the highlighted TSs in Figure 3,and the results are in the Supporting Information.
- (20).(a) Bickelhaupt FM; Houk KN Analyzing Reaction Rates with the Distortion/Interaction-Activation Strain Model. Angew. Chem., Int. Ed. Engl 2017, 56, 10070–10086. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ess DH; Houk KN Distortion/interaction energy control of 1,3-dipolar cycloaddition reactivity. J. Am. Chem. Soc 2007, 129, 10646–7. [DOI] [PubMed] [Google Scholar]; (c) Maji R; Mallojjala SC; Wheeler SE Chiral phosphoric acid catalysis: from numbers to insights. Chem. Soc. Rev 2018, 47, 1142–1158. [DOI] [PubMed] [Google Scholar]
- (21).Allegretti PA; Ferreira EM Generation of α,β-Unsaturated Platinum Carbenes from Homopropargylic Alcohols: Rearrangements to Polysubstituted Furans. Org. Lett 2011, 13, 5924–5927. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.