

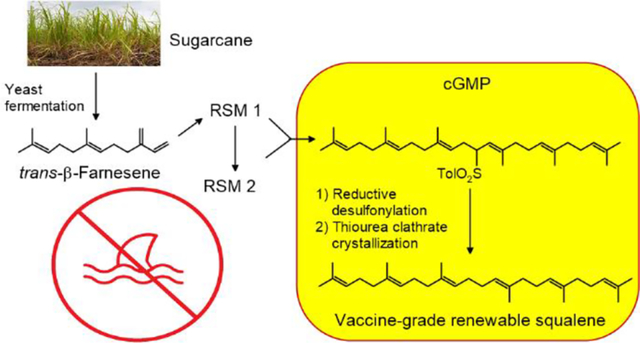
Abstract
Emulsions of the triterpene squalene ((6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene, CAS 111-02-4) have been used as adjuvants in influenza vaccines since the 1990s. Traditionally sourced from shark liver oil, the overfishing of sharks and concomitant reduction in the oceanic shark population raises sustainability issues for vaccine adjuvant grade squalene. We report a semisynthetic route to squalene meeting current pharmacopeial specifications for use in vaccines that leverages the ready availability of trans-β-farnesene ((6E)-7,11-dimethyl-3-methylene-1,6,10-dodecatriene, CAS 18794-84-8), manufactured from sustainable sugarcane via a yeast fermentation process. The scalability of the proposed route was verified by a kilo-scale GMP synthesis. We also report data demonstrating the synthesized semi-synthetic squalene’s physical stability and biological activity when used in a vaccine adjuvant formulation.
Keywords: Squalene, semi-synthesis, farnesene, allylic sulfone, reductive desulfonylation, thiourea inclusion complex crystallization, vaccine adjuvant
Graphical Abstract
INTRODUCTION:
Vaccine adjuvants are natural or synthetic substances that when incorporated in a vaccine formulation along with a suitable antigen, enhance the host antigen-specific immune response compared to the response resulting when the antigen is administered alone. The resultant beneficial properties imparted to adjuvanted vaccines, compared to non-adjuvanted vaccines, include dose sparing (i.e., a reduction in the amount of antigen needed to induce target antibody titers), enabling a more rapid immune response, antibody response broadening, and improved antibody response magnitude and functionality.1
Oil-in-water (o/w) emulsions of squalene have been used as vaccine adjuvants in hundreds of millions of doses of influenza vaccine, and are also under clinical evaluation for other indications, including tuberculosis, malaria, schistosomiasis, and leishmaniasis.2 Other than alumina salts, squalene-based emulsions are the most used vaccine adjuvants in licensed protein-based or inactivated virus vaccines.3 The source of the squalene used in licensed o/w emulsion vaccine adjuvants is shark liver oil, which poses a sustainability problem as shark populations have been subject to overfishing, and the global abundance of oceanic sharks and rays has declined by 71% since 1970, resulting in calls for precautionary catch limits and fishing bans to avert population collapse.4 An alternative, renewable, source of squalene with equivalent vaccine adjuvant properties to shark-derived squalene is therefore desirable to avert the population pressure on sharks and secure the long-term future of squalene emulsion vaccine adjuvants.
Metabolic engineering, using synthetic biology tools, has been used to develop industrial production of several sesquiterpenes and sesquiterpenoids by fermentation of yeast (Saccharomyces cerevisiae). An early example was the engineering of yeast to produce amorphadiene 5 or artemisinic acid 6, either of which can be chemically converted to the World Health Organization first-line antimalarial drug, artemisinin, though commercial production of semi-synthetic artemisinin uses fermentative production of artemisinic acid followed by photochemical conversion.7,8,9
A more recent example of industrial metabolic engineering is the development of yeast to produce the sesquiterpene trans-β-farnesene10, which is now produced commercially by fermentation of sustainably grown sugarcane.9 Amongst other products, trans-β-farnesene is used for the commercial semi-synthesis of squalane11, which is used as an emollient; it has also been used as the precursor for a series of squalene analogs, some of which display vaccine adjuvant properties equivalent or superior to squalene when appropriately formulated in o/w emulsions.12 While of great interest for the elucidation of squalene structure-function relationships relating to its vaccine adjuvant properties in o/w emulsions, these analogs have not undergone human testing, and would require extensive clinical trials before they could be considered as replacements for squalene in adjuvanted vaccines. As the need to replace shark squalene in o/w vaccine adjuvants remains pressing, we have developed a kilo-scale semi-synthesis of GMP vaccine-grade squalene with sustainable trans-β-farnesene as the feedstock, the details of which are reported herein.
A literature search reveals several synthetic routes to squalene dating back to the 1930s. The oldest example, associated with the confirmation at that time of the structure of squalene, involved homocoupling of farnesyl bromide in the presence of K or Mg and reportedly afforded in low yield a mixture of products which included, among others, squalene and a double bond isomer.13 Numerous examples using different chemistries have been reported in the subsequent years, including homocoupling of Grignard reagents14, alkylation of a heteroatom-stabilized carbanion15, the Claisen rearrangement16, the Wittig reaction17, copper-lithium dialkylamide complex-catalyzed allylic halide coupling18, cobalt (I)-catalyzed allylic halide coupling19, photocoupling of η3-allylpalladium complexes20, palladium/zinc-catalyzed allylic acetate coupling21, molybdenum(0)-catalyzed reductive allylic acetate coupling22, Ba-catalyzed allylic halide homocoupling23, Mn/Zr(IV)-catalyzed reductive coupling of allylic halides24, and Ti/Pd bimetallic system-catalyzed homocoupling25. Most of these routes take advantage of the symmetry of the squalene molecule and utilize coupling reactions of suitably functionalized C15 fragments (Scheme 1). Unfortunately, these approaches also share the features of poor overall yields and the formation of complex product mixtures, thus being generally unsuitable for scale-up, particularly those routes leading to significant amounts of squalene double bond isomers that would require extensive purification to meet the required purity criteria.
Scheme 1:
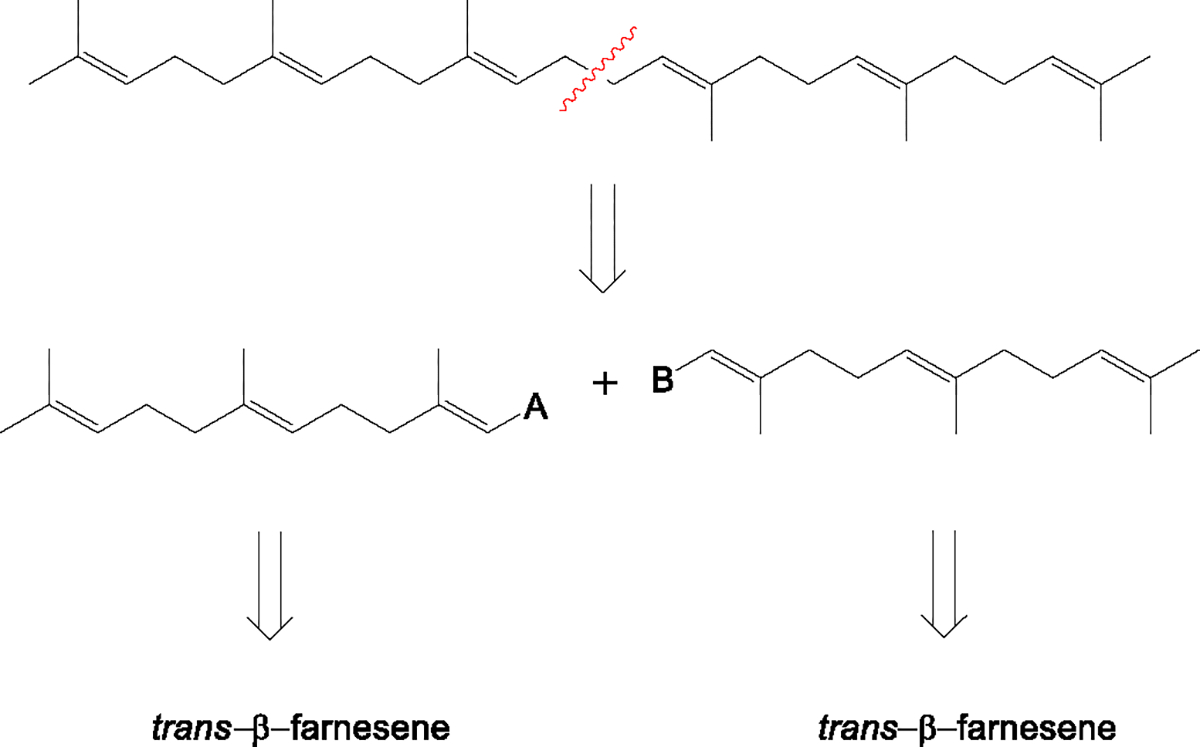
Retrosynthesis of squalene
Nevertheless, we also recognized that in some cases the nature of the particular impurities inherent to the chemistry used might provide handles to facilitate purification, and catalyst optimization might allow improving the squalene to by-product ratios and afford high yields. We have used these insights to develop a kilo-scale GMP semi-synthesis of squalene as described below.
RESULTS AND DISCUSSION
Chemistry
We have previously reported the synthesis of squalene using the coupling of farnesyl halide with a farnesyl sulfone26, followed by reductive cleavage of the resulting allylated sulfone.27 This route, first reported by Biellmann et al.28 and subsequently utilized by several other authors to synthesize squalene 29, 30, 31, met all our criteria, namely the use of well-established high-yielding reactions—several of which were already known to be amenable to scale up using readily available and relatively economical reagents—and from the sustainability perspective, by using our fermentation sourced trans-β-farnesene and carefully selecting the chemistry applied we could ensure that the carbon backbone of the resulting squalene would be 100% renewable as all those atoms would be sugarcane-derived (see the Supplementary Information section on the biological origination of trans-β-farnesene). The planned route is shown in Scheme 2. At the onset of the work the main unknowns were the overall yield of the reaction sequence and whether it would be possible to purify the squalene produced to meet the pharmacopeial specifications without resorting to chromatography. As envisioned, cGMP conditions would be applied to the last two steps in which the squalene skeleton is formed from the two regulatory starting materials (RSMs): farnesyl chloride (RSM 1, total area normalized (TAN) purity 82.3%) and farnesyl p-toylsulfone (RSM 2, made from RSM 1, TAN purity 98.4%). The discussion of the syntheses of RSM 1 and RSM 2 is presented in the Supporting Information and this section will focus on the two cGMP steps and the final product purification.
Scheme 2:
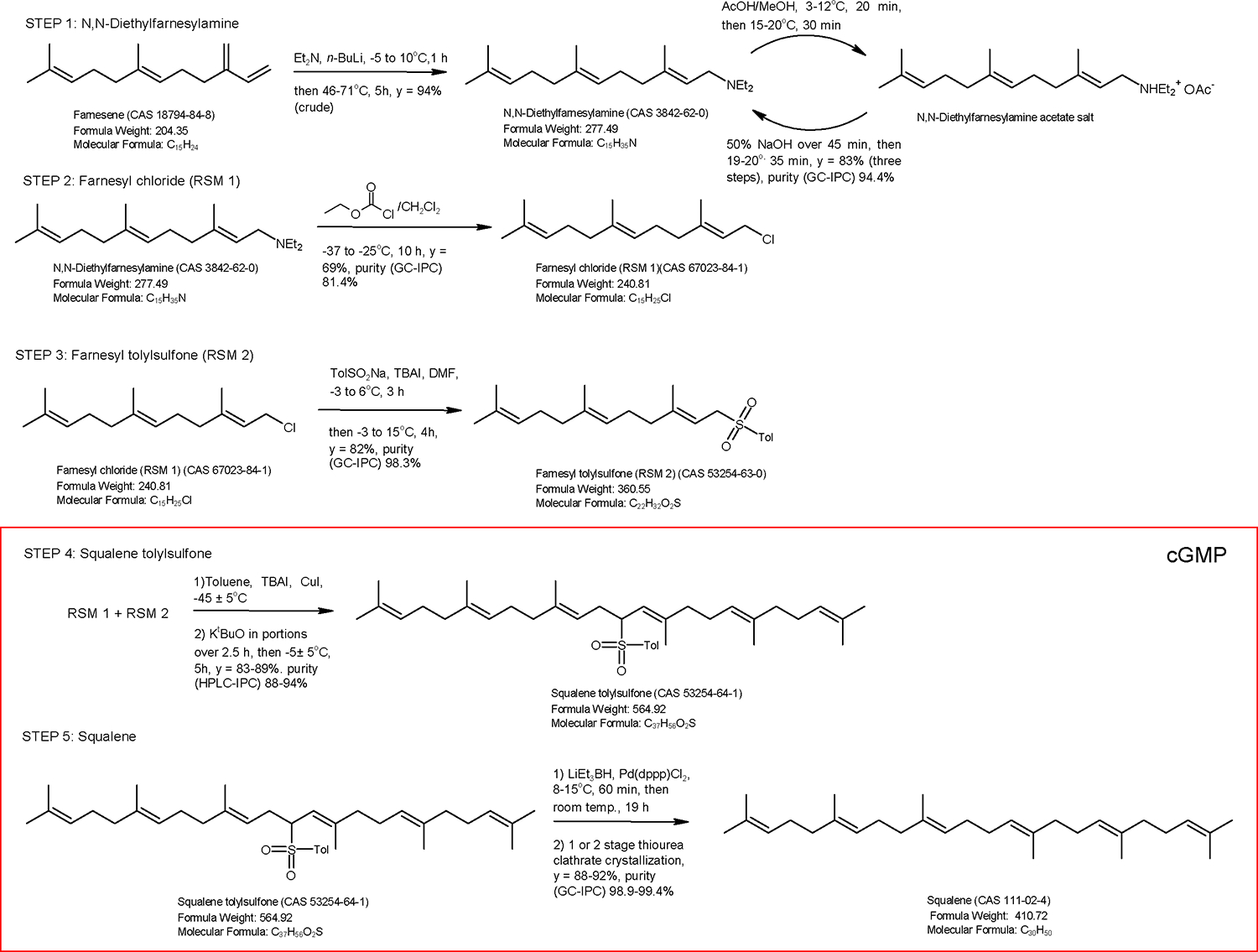
Planned route to vaccine-grade squalene. Glossary: Et2N = N,N-diethylamine; n-BuLi = n-butyllithium; AcOH = acetic acid; 50% NaOH = 50% aqueous sodium hydroxide; TolSo2Na = toluenesulfinic acid sodium salt; LiEt3BH = lithium triethylborohydride; Pd(dppp)Cl2 = palladium diphenylphosphino dichloride. cGMP = IPEC-PQG Joint Good Manufacturing Practices Guide for Pharmaceutical Excipients (v.5, 2002).
Analytical method development
Several new analytical test methods were developed for monitoring the synthesis steps and for the final characterization of the squalene target. A high-resolution gas chromatographic method using a flame ionization detector (FID) (henceforth identified as GC-IPC) was established for monitoring the formation and purity of the squalene product and of the C15 farnesyl amine and chloride reaction intermediates. This method was based on the European Pharmacopoeia (EP) method for the quantitation of squalene.32 The EP method itself was deemed unsuitable for the accurate analysis of all relevant compounds (e.g., intermediates, process-related impurities, and squalene isomers; for details see the Supporting Information). The adopted method, like the EP method, utilizes dimethylpolysiloxane (USP type G1) as the stationary phase. However, whereas the EP method indicates the use of a 30 m × 0.32 mm × 1 μm column, better resolution was achieved using a column with a higher phase-ratio (50 m x 0.2 mm × 0.11 μm). In addition, the EP method uses an isothermal oven program, however, much better resolution was obtained using an oven temperature gradient. In addition, a separate HPLC-DAD method (henceforth referred to as HPLC-IPC) had to be developed for the determination of the squalene tolyl sulfonate process intermediate. This compound was not amenable to gas chromatography due to either temperature instability and/or low volatility. Both methods were fully validated for GMP use (specificity, linear range and target concentration, limit of quantitation, precision, range, and stability) following standard protocols before their implementation33. Details of these analytical procedures are given in Supplementary Information.
Coupling of RSM 1 and RSM 2 (Scheme 2, Step 4) and Desulfonylation to Produce Squalene (Scheme 2, Step 5)
The low temperature (−40 to −45°C) coupling of RSM 1 and RSM 2 was performed in the presence of CuI/tetrabutylammonium iodide employing potassium tert-butoxide as the base. Screening experiments to optimize the RSM 1 + RSM 2 reaction parameters (Table 1) began with KOt-Bu (1.4 – 1.6 eq.), 1.0:1.1 ratios of RSM 1:RSM 2, varying amounts of CuI (0.1 – 0.5 eq.) and THF as the solvent at −78 °C. The reactions did not proceed very cleanly and significant amounts of an unknown by-product were formed (initially hypothesized to be the constitutional isomer, but later identified by LC-MS as the farnesyl sulfone trimer, presumably arising from of the reaction of the second activated hydrogen in the α position with respect to the RSM 2 sulfone moiety with another molecule of RSM 1). Additionally, −78 °C was not considered a viable temperature at scale. Further screening experiments at 0 – 10 °C, also showed large amounts of the impurity later identified as the trimer being formed. The thermal control units available for the process could hold −45 °C as the lowest consistent temperature, so this temperature was chosen for the addition. Both LiOt-Bu and NaOt-Bu were investigated as the choice of base, but neither gave acceptable conversions, with 10 – 20% unreacted starting materials remaining.
Table 1:
RSM1 + RSM2 reaction parameter screening
| RSM 1 (eq.) | RSM 2 (eq.) | CuI (eq.) | KOtBu (eq.) | TBAI (eq.) | Solvente | Isolated yield | Purity |
|---|---|---|---|---|---|---|---|
| 1.0 | 1.1 | 0.5 | 1.4 | NA | THF (6 vol) | 91% a | 87% |
| 1.0 | 1.1 | 0.5 | 1.8 | NA | THF (6 vol) | 93% | 75.5% |
| 1.0 | 1.1 | 0.25 | 1.4 | 0.1 | THF (6 vol) | 82% | 91% |
| 1.0 | 1.1 | 0.25 | 1.4 | 0.1 | 2MeTHF (6 vol) | 75% | 87% |
| 1.0 | 1.05 | 0.3 | 1.4 | 0.2 | Toluene (8 vol) | 81% | 89.5% |
| 1.03 | 1.0b | 0.35 | 1.6 | 0.25 | Toluene (5 vol) | 71% | 96% |
| 1.02 | 1.0 | 0.35 | 1.6c | 0.25 | Toluene (3 vol) | 91% | 91% |
| 1.05 | 1.0 | 0.35 | 1.6d | 0.25 | Toluene (1.5 vol) | 85% | 93% |
| 1.1 | 1.0 | 0.35 | 1.6f | 0.25 | Toluene (1.5 vol) | 92% | 94% |
| 0.96 | 1.0 | 0.25 | 1.5 d | 0.1 | Toluene (5 vol) | 87% | 92% |
| 1.0 | 1.0 | 0.25 | 1.4 | 0.1 | Toluene (4 vol) | 93% | 89%g |
Reaction performed at −78 °C
Recrystallized RSM 2
Added in 4 portions over 2 h
Added in 6 portions over 2 h
Volumes in relation to mass of RSM 2
Added in 2 portions over 1 h
6% RSM 2 remaining.
As the addition of the base is slightly exothermic, it was added in portions to maintain the reaction temperature around the −45 °C setpoint. On completion of the addition the reaction mixture was allowed to warm up to 0–5 °C over a period of 5−7 hours, while monitoring the consumption of farnesyl chloride (RSM 1). The reaction condition screening experiments indicated that warming the reaction mixture after the base addition was critical to obtain squalene of >99% purity after final purification by minimizing the formation of a C45 trifarnesyl sulfone that is not completely removed by the final purification procedure if present in amounts exceeding 2.5%, thus requiring additional purification in the form of a sequence of heptane/MeOH partitions before the sulfone group cleavage and generation of the squalene target. If the reaction mixture is kept at −45 °C for the duration of the coupling, up to 5–10% of RSM 2 may remain unreacted as RSM 1 is consumed by the undesired byproduct formation. The < 2.5% limit is determined by the fact that our smaller scale experiments indicated that if present in greater than this amount, the impurity is not completely removed by the final thiourea clathration, even if repeated twice, which is critical to achieve squalene of > 99% purity.
The route described herein utilizes an aryl sulfone to stabilize an α-carbanion and facilitate the homocoupling of two farnesyl moieties. The use of sulfones for this type of reaction is an important synthetic strategy for the formation of carbon-carbon single and double bonds that facilitates the elaboration of molecules like our squalene target. Upon completion of the coupling, the sulfone auxiliary is usually removed reductively and replaced by hydrogen, although variants such as double bond formation (if a suitable β-substituted sulfone is used), sulfone replacement with an alkyl group, oxidative desulfonylations and nucleophilic displacement desulfonylations are all well-known.34 In our case a simple hydrogen replacement in an allylic sulfone is needed, and a long list of reducing agents can be used for this purpose, however, since it was critical to avoid the double bond migrations and isomerizations observed when other reducing agents are used, we resorted to the use of a hydride reagent in the presence of a Pd(0) complex catalyst, which is highly site- and stereoselective.31 Extensive screening experiments were performed to study the effects of different reducing agents and amounts and different catalyst levels. These are summarized in Table 2.
Table 2:
Reducing agent and catalyst loading screening experiments.
| Purity of squalene sulfone | Reductant | Pd catalyst | Purity of squalene at end of reaction | Isolated squalene yield |
|---|---|---|---|---|
| 92.5% | 1.2 eq Selectride | 5% Pd(dppf) | 77.5% | 63% |
| 89.5% | 1.2 eq Superhydride | 1% Pd(dppp) | 75.2% | 93% |
| 96% | 1.2 eq Superhydride | 5% Pd(dppp) | 83.7% | 81% |
| 93.3% | 1.2 eq Superhydride | 1.75% Pd(dppp) | 86.6% | 73% |
| 89.0% | 1.4 eq Selectride | 2.5 mol% Pd(dppp) | 77.9% | 93% |
| 89.6% | 1.2 eq Superhydride | 0.5 mol% Pd(dppp) | 88.0% | 79% |
| 89.6% | 1.2 eq Superhydride | 0.5 mol% Pd(dppb) | 89.1% | 87% |
| 89.6% | 1.2 eq Superhydride | 0.25 mol% Pd(dppb) | 91.0% | 94% |
| 89.6% | 1.2 eq Superhydride | 0.1 mol% Pd(dppb) | 89.0% | 92% |
The best conditions identified thus involved treatment of the squalene sulfone at 7–15 °C with Pd(dppb)Cl2 (0.0025 equivalents) and 1M LiEt3BH (Superhydride) in THF solution (1.2 eq.), followed by stirring at ~ 20°C until in-process testing by HPLC showed that < 3% of unreacted sulfone remained. At this time the reaction mixture was cooled again to 5°C and quenched by careful addition of water. The reaction mixture was partitioned with − 25% (wt/wt) aqueous NH4Cl and stirred for 1–2 h to quench any excess triethylborane. The aqueous layer was removed, and the organic layer was washed with 25% (wt/wt) NH4Cl and brine before stirring over activated charcoal (5% (wt/wt) with regards to the weight of squalene sulfone) for a minimum of 1 h. The suspension was filtered, washing with heptane. The solvent was removed under reduced pressure and then the solvent swapped with MeOH (2 × 1 vol in relation to the theoretical yield of squalene). These MeOH/heptane extractions were repeated at least three times to ensure the complete removal of any residual highly flammable borane byproducts (see the Safety Note below). Finally, the heptane was removed under reduced pressure until a constant mass of crude squalene was obtained for final purification.
Production of high-purity squalene by inclusion complex purification
While the described process readily provides ~90% (area) pure squalene, the required pharmacopeial purity specification (> 97.0 wt% by GC//FID)32, and the typical purity of commercial vaccine-grade squalene (>99%), indicated the need for a final purification method capable of robustly, routinely and economically providing a final product with this purity. As the literature suggested34, the formation of double bond isomers of squalene during the reductive removal of the sulfone group was anticipated to produce very closely related impurities that could prove hard to remove, for example, by fractional distillation, the method of choice for the purification of shark liver oil squalene that normally lacks such closely related contaminants. A literature survey of potential purification methods drew our attention to the potential of inclusion complex crystallization, discovered serendipitously by Bengen in the 1940s while studying the composition of pasteurized milk 35 when he observed that O-containing aliphatic compounds with six or more C atoms or straight-chain aliphatic hydrocarbons with six or more C atoms are separated from liquid organic mixtures containing them by addition of urea in the presence of small amounts of polar solvents such as water or low-molecular weight aliphatic alcohols, due to formation of what was later shown to be a crystalline inclusion complex or clathrate.36 Isolation and subsequent decomposition of the clathrate allows the separation of the included molecules from impurities (or in its reversed variant, of included impurities from a bulk compound to be purified). Soon thereafter the technique was extended to thiourea 37, which due to its different crystal unit size, shows a different substrate purification profile.38 The thiourea version figured prominently in early work to purify squalene and confirm its structure39 but since then had largely fallen in disuse in this context, although urea clathration is widely used nowadays in the industrial purification of fatty acids 40 and reportedly for the purification of squalene from plant sources.41, 42 Based on these literature protocols and extensive screening experiments to optimize the parameters such as squalene to thiourea ratio, solvent, and concentration, the temperature profiles of adduct formation and crystallization and ultimate complex decomposition and pure squalene isolation, we devised the protocol described in the Experimental which provided high purity squalene (> 99%) meeting the European Pharmacopoeia purity specification32 for use in vaccine adjuvants.
Formulation and characterization of semi-synthetic squalene formulated into vaccine adjuvant emulsion
With high purity and renewable semi-synthetic squalene in hand, all that remained was to formulate it in a vaccine adjuvant emulsion and prove its biological activity in comparison with standard shark squalene. For this purpose, semi-synthetic squalene at 4% v/v was emulsified by high-pressure homogenization with the stable emulsion (SE) composition, consisting of dimyristoyl phosphatidyl-choline, Poloxamer 188, α-tocopherol, glycerol, and ammonium phosphate buffer, as described previously.43 For comparison, an equivalent emulsion employing shark squalene was generated. Emulsion physicochemical stability was assessed using dynamic light scattering, pH measurement, and visual appearance. The droplet diameter, size polydispersity index, and pH of semi-synthetic squalene emulsion were comparable to those of shark squalene emulsion and within established specification limits over a 12-month storage period at 2–8°C (Figure 1). Likewise, visual appearance (milky-white liquid) remained unchanged for both emulsions over this period. Thus, the emulsion characteristics of semi-synthetic squalene were highly similar to those made using shark squalene.
Figure 1:
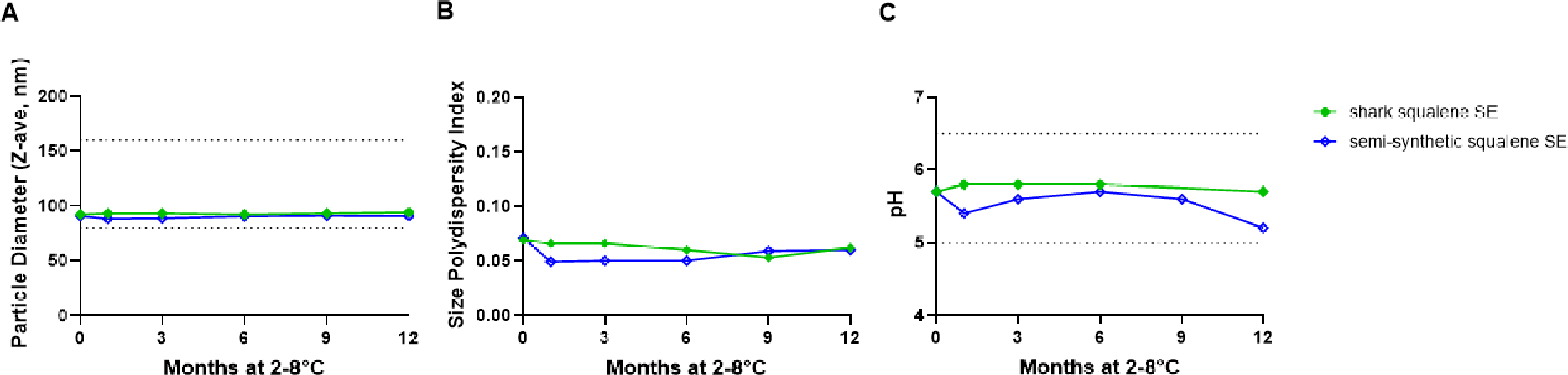
Emulsion physicochemical stability. Emulsions were stored at 2–8C for 12 months and monitored by dynamic light scattering and pH measurement. (A) Emulsion droplet diameter, (B) size polydispersity index, (C) pH. The dotted lines represent pre-established specification ranges.
Immunological activity of formulated semi-synthetic squalene adjuvant in a mouse model
Emulsions were mixed with split inactivated H5N1 vaccine antigen immediately before intramuscular immunization of C57BL/6 mice (n=8) with 0.01 μg antigen and 2% v/v oil.12 A control group of mice were administered antigen without emulsion. Mice were immunized twice, three weeks apart. Serum and splenocytes were collected three weeks after the second immunization for immune response analysis. Both emulsions elicited a highly similar immunogenicity profile, including enhanced antigen-specific serum IgG antibodies and increased numbers of antigen-stimulated IL-17-producing splenocytes compared to antigen alone (Figure 2). These results demonstrate that semi-synthetic squalene emulsion maintains humoral and cellular adjuvant activity compared to shark squalene emulsion in immunized mice.
Figure 2:
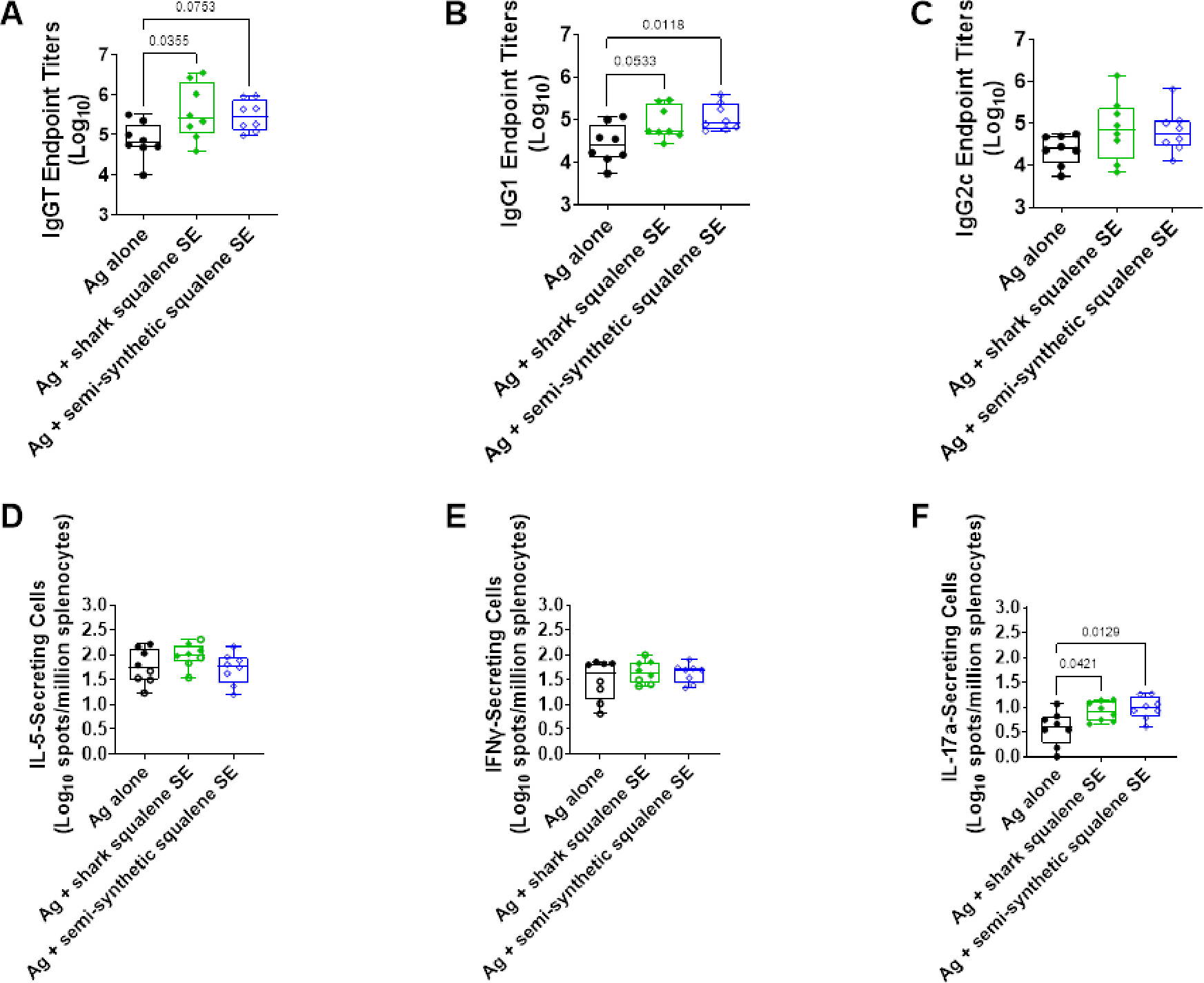
Emulsion adjuvant activity in mice immunized with split, inactivated H5N1 vaccine antigen. Vaccine antigen was mixed with emulsion immediately prior to intramuscular immunization of C57BL/6 (n = 8) mice. The control group was immunized with antigen alone. All mice were immunized twice, three weeks apart. Serum and splenocytes were collected three weeks after the 2nd immunization. Data are represented as box-whisker plots with bars representing median values, boxes representing 1st-3rd quartiles, and whiskers representing the maximum and minimum values. Statistical comparisons were performed by one-way ANOVA with Tukey’s correction for multiple comparisons, with p-values <0.1 reported on the plots. (A) Antigen-specific serum total IgG (IgGT) endpoint titer measured by ELISA. (B) Antigen-specific serum IgG1 endpoint titer measured by ELISA. (C) Antigen-specific serum IgG2c endpoint titer measured by ELISA. (D) Antigen-specific IL-5-secreting splenocytes measured by ELISpot. (E) Antigen-specific IFNγ-secreting splenocytes measured by ELISpot. (F) Antigen-specific IL-17a-secreting splenocytes measured by ELISpot.
CONCLUSION
The overfishing of oceanic sharks and rays has resulted in a significant decline in their populations (>70% since 1970) and threatens three-quarters of species comprising these groups with extinction4. Squalene, a triterpene used extensively as a vaccine adjuvant when appropriately formulated, is derived from shark liver, thus contributing to the overfishing of sharks for their squalene-rich liver oil. Clearly, an alternative source of vaccine-grade squalene is required to alleviate both the overfishing of sharks, and to safeguard the future supply of this pharmaceutically valuable compound.
The ready availability of trans-β-farnesene produced by fermentation of sustainable sugarcane has herein enabled us to develop a kilo-scale GMP synthesis of vaccine grade squalene (the biological origination of the trans-β-farnesene and the renewable nature of semi-synthetic squalene is described in the Supplementary Information). The well-established reactions used were selected with a view to further scale-up, and optimization of these reactions and the overall process allowed the synthesis of ~90% pure squalene, assessed by a GC-IPC procedure described here. Finally, the process features an efficient purification by thiourea clathrate crystallization that affords squalene meeting the pharmacopeial specifications (>97 weight% by GC-FID) and the customary sales specification purity (>99% by GC-IPC). Further testing showed that the resulting semi-synthetic squalene meets or exceeds all European Pharmacopeia monograph specifications (01/2020:2805) for squalene used as an adjuvant in vaccines.
Given that the ultimate usage of semi-synthetic squalene is to serve as a vaccine adjuvant when appropriately formulated, we assessed both its formulation characteristics and its immunological properties when incorporated into an influenza vaccine. Importantly, semi-synthetic squalene formed emulsions of ~100 nm diameter in a manner indistinguishable from shark squalene, and exhibited emulsion stability for at least one year, again indistinguishable from shark squalene. Immunological characterization of an emulsion formulated with semi-synthetic squalene in an established mouse model for vaccine adjuvanticity with split, inactivated H5N1 influenza virus12 demonstrated indistinguishable humoral and cellular adjuvant activity compared to emulsion formulated with shark squalene in immunized mice.
In summary, we have demonstrated a kilo-scale GMP synthesis of vaccine-grade semi-synthetic squalene that meets both European Pharmacopoeia specifications for squalene for use in vaccine adjuvants, and customary customer expectations of >99% purity. The resulting semi-synthetic squalene is functionally indistinguishable from shark squalene in both its formulation into emulsions and immunological properties in a mouse adjuvanted influenza model. The described semi-synthetic squalene has the potential to replace shark squalene in emulsion vaccine adjuvants, and the displacement of shark squalene should reduce the overfishing of sharks for harvesting of shark liver oil.
EXPERIMENTAL
General Information
Chemistry
Reagents and solvents were acquired from commercial suppliers and used as received, without further purification unless otherwise noted. Squalene reference material (Ph. Eur. Squalene CRS reference material, catalog number Y0002131, “as is” C30H50 content 99.8%) was purchased from the EQDM webstore (https://store.edqm.eu/index.html). trans-β-Farnesene produced by yeast fermentation was supplied by Amyris. 1H-and 13C-NMR spectra were recorded on Bruker Advance spectrometers operating at frequencies of 300, 500, or 600 and 75, 125 or 150 MHz, respectively. IR spectra were recorded in the 500–4000 cm−1 range on a JASCO FT-IR/4600 spectrometer equipped with an ATR Pro One accessory. Some of the Certificate of Analysis tests listed in Table 3 were performed at Galbraith Laboratories Inc. (Knoxville, TN, U.S.A.), MPL Laboratories (Sparta Township, NJ, U.S.A.) or Element Material Technologies (Santa Fe Springs, CA, U.S.A.), as indicated in the table footnotes.
Table 3:
Squalene sulfone GMP release specifications
| Attribute | Method | Acceptance Criteria | Results for described batch |
|---|---|---|---|
| Appearance | Visual | Yellow to red to red-brown transparent oil | Dark yellow transparent oil |
| Identification by RT | HPLC-IPC | RT of sample is within ± 5% of that of reference material | Conforms |
| Identification by FTIR | USP <197A> | Sample spectrum matches reference material spectrum | Conforms |
| Chromatographic Purity | HPLC-IPC | NLT 80 % | 90.9% |
| Water Content | Karl Fischer by USP <921> method Ic | Report Only | 100 ppm |
|
Residual Solvents Toluene (Class II) Methanol (Class II) Heptane (Class III) |
USP <467> Procedure C | Report Only | Conforms |
Biology
Shark-derived squalene was obtained from Sigma-Aldrich Fine Chemicals (St. Louis, MO, U.S.A.). DMPC was obtained from Lipoid (Newark, NJ, U.S.A.). Poloxamer 188, α-tocopherol, and glycerol were purchased from Spectrum Chemical (New Brunswick, NJ, U.S.A.). Buffer components were obtained from J.T. Baker/Avantor (Radnor, PA, U.S.A.). Split, inactivated H5N1 (A/Vietnam/1194/2004) was obtained from the National Institute for Biological Standards and Control (NIBSC, South Mimms, Hertfordshire, U.K.). C57BL/6 (B6) mice for animal experiments were purchased from The Jackson Laboratory (Bar Harbor, ME, U.S.A).
Synthesis of Squalene
Syntheses of p-tolylsulfonyl squalene from farnesyl chloride (RSM 1) and p-tolylfarnesyl sulfone (RSM 2) (Scheme 2, Step 4)
A 100 L reactor equipped with a temperature probe, mechanical stirrer and N2 inlet and connected to a Huber chiller (capable of holding −50 °C) was charged with farnesyl sulfone (RSM 2, 2.075 kg, 1.0 eq, TAN purity 98.4%), farnesyl chloride (RSM 1, 1.40 kg, 1.01 eq, TAN purity 82.3%) and toluene (7.13 kg, 4 vol.) and brought to an internal temperature of −45 ± 5 °C. Tetrabutylammonium iodide (TBAI, 0.54 kg, 0.25 equiv.) and copper (I) iodide (CuI, 0.39 kg, 0.35 equiv.) were added once the temperature was < 30 °C. Next potassium t-butoxide (KOtBu, 1.03 kg, 1.6 equiv.) was added in eight approximately equal portions at 30 – 45 minute intervals keeping the internal temperature −45 ± 5 °C. After base addition was complete (~2.5 h), the internal temperature was increased to −5 ± 5 °C over 5 hours, at which point an in-process test (HPLC-IPC) confirmed that the farnesyl chloride level was 0.3%. The reaction was warmed to 15 ± 5 °C and quenched with 37 (wt/wt)% sat. aq. ammonium chloride (NH4Cl, 4.01 kg, 2 volumes) and allowed to stir for 20 minutes. The biphasic suspension was filtered over Whatman™ #54 filter paper to remove insoluble particulates and washed with toluene (2 × 500 mL). The top organic layer was stirred over 37 (wt/wt)% sat. aq. NH4Cl (1 × 4.23 kg) for 12 minutes, settled, and the bottom layer was removed. The top organic layer was stirred over 20% (wt/wt) sodium thiosulfate pentahydrate (Na2S2O3·5H2O, 1 × 4.30 kg) for 10 minutes, allowed to settle, and the bottom layer removed. The top organic layer was stirred over water (1 × 4.22 kg) for 10 minutes, settled, and the bottom layer removed. Toluene was removed under reduced pressure in a 40 °C water bath until no more toluene was distilled. Residual toluene was solvent swapped with methanol (2 × 2.5 kg). After two solvent swaps the mass of crude squalene sulfone was 3.18 kg. An in-process check of the crude squalene sulfone (HPLC-IPC) determines the next order of operations. If the percentage of the trifarnesyl sulfone impurity with relative retention time (RRT) 1.067 min. < 2.5% the work-up is deemed complete and the squalene sulfone can be dried to constant mass (the < 2.5% limit for using one workup or the other is determined by the fact that the smaller scale R&D indicated that if present in greater than this amount, it is not completely removed by the final thiourea clathration, even if repeated twice, which is critical to achieve squalene of > 99% purity). If the percentage of this impurity is greater than 2.5%, as in the case of the cGMP batch described herein (4%), the following procedure should be followed: the crude squalene sulfone was transferred back to the cleaned 100 L reactor with heptane (2.0 L) and diluted with additional heptane (9.58 kg). The internal temperature was brought to 5.5 °C and the heptane layer was extracted with MeOH (1 × 7.55 kg, 1 × 4.98 kg). The internal temperature was brought to 0 °C and extracted with MeOH (5 × 2.50 kg). The combined MeOH extracts are concentrated under reduced pressure in a 30–50 °C water bath. Once distillation was complete MeOH was solvent swapped with heptane (2 × 2.30 kg) and concentrated until the mass of squalene sulfone was constant (2.33 kg, 71.7% yield). Average yields for this step range from 83–89%, with average sulfone purity of 88–94%. Intermediate release for the next synthesis step is contingent on the sulfone meeting the specifications listed in Table 3.
A sample of the sulfone was purified by silica gel chromatography using a 100% hexanes to 40% methylene chloride in hexanes step gradient to give 7.95 g of purified material which was 95.1% pure by HPLC-IPC. LC-MS: m/z 582 [M+NH3], which corresponds to MW = 564.
1H-NMR (600 MHz, CDCl3) δ 7.66 (d, J = 8.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 5.06 −4.96 (m, 4H), 4.95 −4.86 (m, 2H), 3.65 (td, J = 10.6, 3.3 Hz, 1H), 2.80 (ddd, J = 14.3, 7.4, 3.3 Hz, 1H), 2.37 (s, 3H), 2.27 (ddd, J = 14.3, 10.8, 7.1 Hz, 1H), 1.98 (m, J = 21.4, 7.3 Hz, 7H), 1.93 −1.84 (m, 10H), 1.61 (overlapping singlets, 6H), 1.54 (overlapping singlets, 6H), 1.53 (s, 6H), 1.51(s, 3H).
13C-NMR (151 MHz, CDCl3) δ 145.05, 144.17, 138.56, 135.57, 135.26, 135.17, 131.40, 131.31, 129.31, 129.19, 124.32, 124.25, 123.91, 123.53, 118.73, 117.16, 64.87, 39.80, 39.75, 39.73, 26.77, 26.73, 26.63, 26.61, 26.35, 25.70, 21.63, 17.70, 17.69, 16.66, 16.39, 15.97, 15.96.
Synthesis of Crude Squalene from Squalene Sulfone (Scheme 2, Step 5)
A 100 L reactor equipped with a mechanical stirrer, N2 inlet, Huber chiller, and temperature probe is charged with squalene sulfone (2.22 kg, 1.0 eq) and Me-THF (2.0 L, 2.34 kg) to ensure complete transfer of the liquid starting material. The reactor was charged with additional Me-THF (2.69 kg). The reactor was sparged with inert gas until the O2 level was ≤ 2.0% (as measured by an O2 meter, actual value = 1.3% O2) and the internal temperature was brought to 7.9 °C. Pd(dppb)Cl2 (6.00 g, 0.0025 eq, Strem) was added followed by immediate addition of lithium triethylborohydride (4.58 kg, 1M in THF, 1.3 equiv., Aldrich) at such a rate that the internal temperature remained below 15 °C (Taddn = 48 minutes, Thigh = 9.8 °C). Upon completion of addition, the internal temperature was brought to 21.5 °C (temperatures < 16 °C result in much slower conversion of squalene sulfone to squalene). The solution was allowed to react for 19 hours at which point IPC determined that % squalene sulfone was < 3.0% (% squalene sulfone for this batch = 0.5%). (NOTE: As observed during multiple experiments at different scales, the reaction is sensitive to the quality of reagents. For example, considerable variability is noted depending on the source of the triethylborohydride. Aldrich Super-Hydride® solution gave < 3.0% unreacted squalene sulfone after 16 h at 20 °C using 1.2 equivalents, whereas attaining the same result with Calselect™ Li triethylborohydride from Ascensus required 1.5 equivalents, as 1.2 equivalents were not enough to bring the reaction to completion). The internal temperature was brought to 7 ± 5 °C and the reaction was carefully quenched with water (410 mL, 0.2 vol.). The organic layer was stirred over 25% (wt/wt) aqueous NH4Cl (4.59 kg, 2 vol.) for 2 h. Upon settling, the bottom aqueous layer was removed, and the organic layer was stirred over 1 × 25% (wt/wt) aqueous NH4Cl 4.98 kg, 2 vol.) for 27 minutes. The layers were allowed to settle, and the bottom aqueous layer was removed. The organic layer was stirred over water (2 × 2.30 kg, 2 × 1 vol.). The crude squalene/Me-THF/THF solution was stirred over activated charcoal (DARCO®, Aldrich, 0.12 kg, 5 (wt/wt)% with respect to the weight of starting squalene sulfone) for 16 h at which point the charcoal was removed by filtration over glass fiber filter paper. The filter cake was washed with heptane (2 × 500 mL). The solvent mixture was removed under reduced pressure in a 25 °C water bath and then remaining solvent was azeotroped with MeOH (2 × 1.80 kg, 2 × 1 vol). Crude squalene was dissolved in heptane (2.25 kg, 2 vol.) and washed with MeOH (3 × 0.65 kg, 3 × ½ vol.) (SAFETY NOTE: These heptane/MeOH extractions are critical to ensure the removal of boron species produced during the reaction quench, such as dialkylboranes, which although less reactive than the original trialkylborane are nevertheless still pyrophoric; these are not completely removed, for example, by simple MeOH washes, a common workup for this kind of reaction. Although not implemented in our work, additional safety precautions can be taken, such as treatment with excess base followed by aqueous hydrogen peroxide to convert all boron species to the corresponding boronate and/or verification of the absence of reactive boron species using 11B-NMR). Heptane was removed under reduced pressure in a 35 °C water bath and then the residual heptane was azeotroped with MeOH (2 × 1.28 kg, 2 × 1 vol). Crude squalene was dried under reduced pressure in a 40 °C water bath to a constant mass (1.44 kg, 89.4% yield; typical yields are in the 80–87% range). Release for the next step is contingent on meeting the specifications listed in Table 4.
Table 4:
Crude squalene GMP release specifications
| Attribute | Method | Acceptance Criteria | Results for Described Batch |
|---|---|---|---|
| Appearance | Visual | Clear to yellow oil | Clear light-yellow oil |
| Identification by RT | GC-IPC | RT of sample is within + 5% of that of reference material | Conforms |
| Chromatographic Purity* | GC-IPC | Report | 91.1% |
Typical purity = 87–91 %
Purification of Squalene
A 100 L reactor equipped with a mechanical stirrer, N2 inlet, Huber chiller, and temperature probe was charged with MeOH (55.50 kg, 50 vol.; use of < 45 vol. does not reduce the sulfone trimer level below 0.5%). Thiourea (12.61 kg, 0.180 kg/L) is added, and the internal temperature is brought to 52 °C. The suspension was stirred until all thiourea was dissolved. Crude squalene (1.40 kg) was added, and the internal temperature is brought to 1.7 °C over 5–10 hours. The formed clathrate was recovered by filtration over #54 filter paper. The filter cake was not washed. Additionally, once all the clathrate was removed from the reaction, the remaining MeOH was not drained over the filter cake. The clathrate was dried on the Buchner funnel for 1.5 hours. The wet clathrate (10.07 kg) was broken by transferring the solid back to the reactor and suspending it in USP water (30.21 kg, 3 vol.) and heptane (6.89 kg, 1 vol.). The internal temperature was brought to 52 °C and stirred until the suspension became completely clear (extended stirring times do not appear to improve the squalene purity or reduce the trimer impurity levels). The bottom aqueous layer was removed, and the heptane layer was washed with USP water (2 × 1.40 kg, 2 × 1 vol.). The organic layer was dried with Na2SO4 (1.00 kg) and filtered over glass fiber filter paper. The heptane was removed under reduced pressure in a 33 °C water bath and residual heptane was azeotroped with MeOH (2 × 1.14 kg, 2 × 1 vol.) to afford semi-purified squalene (1.16 kg). The clathration process was repeated by adding MeOH (45.90 kg, 50 vol.) to the reactor. Thiourea (9.05 kg, 0.156 kg/L) was added, and the internal temperature was brought to 52 °C and the suspension was stirred until all thiourea was dissolved. Crude squalene (1.16 kg) was added and the internal temperature was brought to 4.8 °C over 5–10 hours. The formed clathrate was recovered by filtration over #54 filter paper. The filter cake was not washed. Additionally, once all the clathrate was removed from the reaction, any remaining MeOH was not drained over the filter cake. The clathrate was dried on the Buchner funnel for 1 hour. The wet clathrate (6.77 kg) was broken by transferring it back to the reactor and suspending it in USP water (20.31 kg, 3 vol.) and heptane (4.63 kg, 1 vol.). The internal temperature was brought to 52 °C and the mixture allowed to stir until the suspension became completely clear. The bottom aqueous layer was removed, and the heptane layer was washed with USP water (2 × 1.40 kg, 2 × 1 vol.). The heptane layer was dried with Na2SO4 (1.01 kg) and decolorized with DARCO® activated charcoal (76.1 g, 7 (wt/wt)%). The suspension was stirred for 16 h. The heptane suspension was filtered over glass fiber filter paper to remove activated charcoal. Heptane was removed under reduced pressure in a 45 °C water bath and residual heptane was azeotroped with MeOH (3 × 1.10 kg, 3 × 1 vol.). HPLC-IPC was performed to determine the % squalene sulfone (< LOD). Squalene was dried under reduced pressure in a 45 °C water bath until no change in the mass occurred. Residual solvents were determined (USP <467> Procedure C; MeOH < 1500 ppm, heptane < 2500 ppm). Squalene was isolated in 0.99 kg (70.7% recovery from crude squalene; typical recovery = 88–92%). Overall yield: From trans-β-farnesene to crude squalene = 33 %; from trans-β-farnesene to purified squalene = 23%. Typical purity by GG-IPC = 98.9 – 99.4%. %. Trimer by HPLC-IPC = < 0.05%. The purified semi-synthetic squalene was identical to a European Pharmacopeia shark squalene standard when compared by infrared absorption spectrophotometry (Figure 3).
Figure 3:
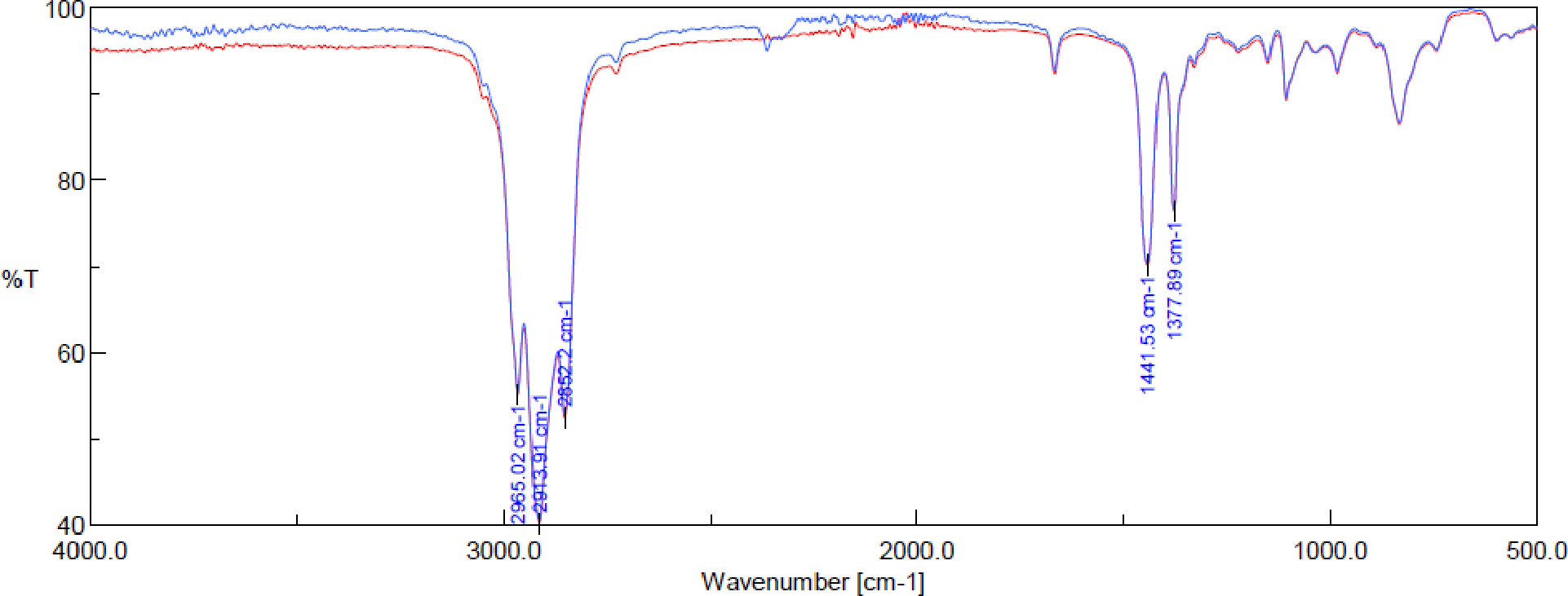
Infrared absorption spectrophotometry comparing semi-synthetic squalene (red) with European Pharmacopeia shark squalene standard (blue)
NMR data of semi-synthetic squalene are as follows: 1H-NMR: (600 MHz, CDCl3): 5.06–5.18 (m, 6H), 1.94–2.13 (m, 20H), 1.68 (bs, 6H), 1.60 (bs, 18H).
13C-NMR (150 MHz, CDCl3): 16.02, 16.06, 17.70, 25.72, 26.68, 26.79, 28.30, 39.75, 39.77, 124.29, 124.32, 124.43, 131.27, 134.92, 135,12 (as expected in light of the molecule’s symmetry, only 15 signals appear in this spectrum). These spectra are identical to those of a commercially available squalene sample.
The product meets all the EP monograph specifications for vaccine grade squalene. In addition to the pharmacopeial tests, residual solvents according to ICH Q3C Solvents, residual metals according to ICH Q3D Impurities and microbiological status according to ICH Q4B Annex 4C, Test for Microbiological Examination of Non-sterile Products, were determined to generate the product’s certificate of analysis, summarized in Table 5.
Table 5:
Semi-synthetic Squalene Certificate of Analysis
| Attribute | Method | Acceptance Criteria | Results |
|---|---|---|---|
| Appearance 1 | EP 2.21, 2.2.2 Method II | Examined to be clear and not more intensely colored than reference solution Y6 | Passes; clear and colorless |
| Identification by FTIR | USP <197A> (equivalent to EP 2.2.24) | Sample spectrum conforms to reference material spectrum | Conforms |
| Refractive lndex 1 | EP 2.2.6 | 1.491 to 1.499 | 1.4961 |
| Acid Value (mg KOH/g)1 | EP 2.5.1 | Maximum 1.0 | <0.03 mg KOH/g |
| Iodine Value 1 | EP 2.5.4 Method B (determined on 0.06 Q) | 350 to 450 | 386 g/100g |
| Peroxide Value 1 | EP 2.5.5 Method A | Maximum 5.0 | 1.4 mEq O2/kg |
| Saponification Value (mg/g) 1 | EP 2.5.6 | Maximum 5.0 | <1.0 mg/g |
| Water Content | USP <921> Method le (determined on 2.00 g, equivalent to EP 2.5.32) | NMT 0.2% | 26 ppm |
| Assay (wt%) | GC EP 2.2.28 | 97.0% to 103.0% | 97.3% |
| Chromatographic Purity | GC-IPC | NLT 99% | 99.4% |
| Individual Unspecified Impurities | GC-IPC | NMT 1% | Largest unspecified impurity: 0.2% |
| % Residual Tolyl Sulfonyl Squalene Intermediate | HPLC-IPC | NMT 0.05% (limit test) | < Detection Limit (0.025%) |
| Microbiological 2 | |||
| Microbial Limits | USP <61> | Total Aerobic Microbial Count: NMT 103 cfu/mL | <10 CFU/G |
| Total Combined Yeasts and Molds Count: NMT 102 cfu/mL | <10 CFU/G | ||
| Burkholderia cepacia | USP <60> | Absent | Negative |
| Elemental Impurities 3 | |||
| Cadmium | ICP-MS | NMT 0.2 ppm | ND |
| Lead | NMT 0.5 ppm | ND | |
| Arsenic | NMT 1.5 ppm | ND | |
| Mercury | NMT 0.3 ppm | ND | |
| Cobalt | NMT 0.5 ppm | ND | |
| Vanadium | NMT 1 ppm | ND | |
| Nickel | NMT 2 ppm | ND | |
| Lithium | NMT 25 ppm | ND | |
| Copper | NMT 30 ppm | ND | |
| Palladium | NMT 1 ppm | 0.004 | |
| Antimony | NMT 9 ppm | ND | |
| Residual Solvents 1 | |||
| Acetonitrile | GC-FID | NMT 410 ppm | <205 ppm |
| Methanol | NMT 3,000 ppm | <1500 ppm | |
| Tetrahydrofuran | NMT 720 ppm | <360 ppm | |
| Toluene | NMT 890 ppm | <445 ppm | |
| Dichloromethane | NMT 600 ppm | <300 ppm | |
| N,N’-Dimethylformamide | NMT 880 ppm | <440 ppm | |
| Heptane | NMT 5,000 ppm | <2500 ppm | |
| Ethyl Acetate | NMT 5,000 ppm | <2500 ppm | |
| Methyltetrahydrofuran | NMT 5,000 ppm | <2500 ppm | |
Tests performed at Galbraith Laboratories
Tests performed at MPL Laboratories
Tests performed at Element Material Technology
Adjuvanted vaccine formulation and testing
Squalene emulsion formulation
Squalene was formulated with a mixture of emulsifiers (DMPC and poloxamer 188), an antioxidant agent (α-tocopherol), a tonicity agent (glycerol), and a buffer system (25 mM ammonium phosphate, pH 5.8) and processed by high-shear mixing and high-pressure homogenization to generate 4% v/v oil-in-water nanoemulsions43.
Mice, immunizations, and sample collection
Experimental groups consisted of equal numbers of 6- to 8-week-old male and female mice. Mice were immunized by intramuscular injection of 100 μL total volume (50 μL in each hind leg) of vaccine containing 10 ng of split, inactivated H5N1 A/Vietnam/1194/2004 (NIBSC) and 2% v/v oil-in-water emulsion at Study Day 0 and Study Day 21. The animal experiment was performed in accordance with national and institutional guidelines for animal care of laboratory animals and approved by the Bloodworks Northwest Research Institute’s Institutional Animal Care and Use Committee. Results from the control groups were previously reported as part of a concurrent study12.
Blood and tissue collection and processing
Peripheral blood was collected at Study Day 21 by the retro-orbital route (prior to the boost immunization) under light sedation using isoflurane and at Study Day 42 via cardiac puncture after euthanasia. Blood was collected in Microvette® capillary blood collection tubes (Sarstedt, Nümbrecht, Germany) and centrifuged to separate the serum. Serum was stored at −20°C until analysis. On Study Day 42, mice were euthanized by controlled administration of carbon dioxide inhalation, followed by cervical dislocation, and spleens and bone marrow were removed aseptically. Lymphocytes were isolated from the spleen using manual disruption. Red blood cells contained in spleens were lysed using Red Blood Cell Lysis Buffer (eBioscience, San Diego, CA, U.S.A.). Lymphocytes were used for cytokine ELISpot (IFN-γ, IL-5, and IL-17a) assays as described below.
Serum ELISA
Antigen-specific total IgG (IgGT), IgG1, and IgG2c were measured in the serum samples from the immunized animals using antibodies purchased from Southern Biotech (Birmingham, AL, U.S.A.) that were diluted 1:5000, 1:3000, and 1:2000, respectively. Briefly, 384-well plates were coated with 1 μg/mL recombinant H5 A/Vietnam/1194/2004 antigen (Sino Biological, Wayne, PA, U.S.A.) overnight. The next day, plates were washed and blocked with phosphate-buffered saline (PBS) with 0.1% polysorbate 20 and 1% bovine serum albumin (BSA). After washing, plates were incubated with the serum followed by incubation with horseradish peroxidase (HRP)-conjugated antibodies and 3,3’,5,5’-tetramethylbenzidine (TMB) substrate. The reaction was stopped using 1N H2SO4 and read within 30 min using a Victor X4 (PerkinElmer, Waltham, MA, U.SA.) plate reader. Endpoint titers were interpolated using a sigmoidal curve fit and an arbitrary cutoff value of 0.5 or 0.75 depending on background signal. Endpoint titers below the standard curve range were assigned the value corresponding to 0.5x of the lowest standard curve dilution.
Splenocyte cytokine ELISpot
ELISpot plates were coated with IFN-γ (BD Biosciences, San Jose, CA, U.S.A.), IL-5 (BD Biosciences) or IL-17a (eBioscience) capture antibodies, diluted 1:200, overnight at 4°C. Plates were washed with PBST, blocked with complete RPMI medium for 2 h at ambient temperature, and then washed again. Splenocytes were plated at 2.0 × 105 cells per well and were stimulated with 10 μg/mL recombinant H5 A/Vietnam/1194/2004 antigen (Sino Biological) at 37°C with 5% CO2 for 48 h. The plates were washed with PBST, detection antibodies (BD Biosciences), diluted 1:250, were added, and the plates were then incubated overnight at 4°C. After incubation, plates were washed with PBST, and Avidin D (Av)-HRP (Invitrogen, Waltham, MA, U.S.A.), diluted 1:250, was added for a 45-min incubation at ambient temperature followed by a PBS wash. The plates were developed with AEC substrate kits according to the manufacturer’s protocol. The reaction was stopped by washing the plates with deionized water, plates were dried in the dark, and spots were counted using an automated ELISpot reader (CTL analyzer). Data were analyzed using ImmunoSpot version 7 professional software (Cellular Technology Ltd., Shaker Heights, OH, U.S.A.).
Statistical analysis
Adaptive immunity responses measured in vaccinated animals were log-transformed as indicated. Data were compared by one-way ANOVA with Tukey’s correction for multiple comparisons. Statistical analysis was performed using GraphPad Prism version 9.3.1. (GraphPad Software Inc., San Diego, CA, U.S.A.)
Supplementary Material
Acknowledgements
We thank Kathleen Valiasek and Annie Tsong for their support of this work, and Howard Fuller, Alicia Ng and Brett Harrison for QA/QC support. We are grateful to Julie Bakken and David Argilla for helpful technical assistance. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health (NIH) under grant award number R01AI135673 ($4.1 million). 6.7% of the total project cost was financed with federal money. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing Interests
All authors declare no competing non-financial interests, but the following declare competing financial interests: C.J.P., G.B., A.W.T. and D.J.M. own shares and possess stock options in Amyris, Inc. F.X.W. owns shares in Amyris, Inc. C.B.F. is an inventor on patents and/or patent applications involving terpenoid emulsion adjuvant formulations. K.J.F. and F.X.W. are inventors on a patent application involving the synthesis of squalene from farnesene.
Supporting Information
Biological origination of Amyris farnesene produced by fermentation and the renewable nature of semi-synthetic squalene, analytical method development, overview of syntheses of farnesyl chloride (RSM1) and farnesyl tolyl sulfone (RSM 2). Experimental section: Chemistry general information, analytical methods, chemistry step 1 (Synthesis of crude N,N-diethylfarnesylamine from farnesene), chemistry step 2 (Synthesis of farnesyl chloride (RSM1) from N,N-diethylfarnesylamine, chemistry step 3 (Synthesis of farnesyl tolyl sulfone (RSM2) from farnesyl chloride (RSM1) and sodium p-toluenesulfinate).
REFERENCES
- (1).Reed SG; Orr MT; Fox CB Key Roles of Adjuvants in Modern Vaccines. Nature Medicine (New York, NY, USA) 2013, 19 (12), 1597–1608. 10.1038/nm.3409. [DOI] [PubMed] [Google Scholar]
- (2).Fox CB; Carter D; Kramer RM; Beckmann AM; Reed SG Current Status of Toll-Like Receptor 4 Ligand Vaccine Adjuvants. In Immunopotentiators in Modern Vaccines (Second Edition); Schijns VEJC, O’Hagan DT, Eds.; Academic Press: San Diego, CA, USA, 2017; pp. 105–127. 10.1016/B978-0-12-804019-5.00006-2. [DOI] [Google Scholar]
- (3).Fox CB; Haensler J An Update on Safety and Immunogenicity of Vaccines Containing Emulsion-Based Adjuvants. Expert Rev. Vaccines 2013, 12 (7), 747–758. 10.1586/14760584.2013.811188. [DOI] [PubMed] [Google Scholar]
- (4).Pacoureau N; Rigby CL; Kyne PM; Sherley RB; Winker H; Carlson JK; Fordham SV; Barreto R; Fernando D; Francis MP; Jabado RW; Herman KB; Liu K-M; Marshall AD; Pollom RA; Romanov EV; Simpfendorfer CA; Yin JS; Kindsvater HK; Dulvy NK Half a Century of Global Decline in Oceanic Sharks and Rays. Nature 2021, 589 (7843), 567–571. 10.1038/s41586-020-03173-9. [DOI] [PubMed] [Google Scholar]
- (5).Westfall PJ; Pitera DJ; Lenihan JR; Eng D; Woolard FX; Regentin R; Horning T; Tsuruta H; Melis DJ; Owens A; Fickes S; Diola D; Benjamin KR; Keasling JD; Leavell MD; McPhee DJ; Renninger NS; Newman JD; Paddon CJ Production of Amorphadiene in Yeast, and Its Conversion to Dihydroartemisinic Acid, Precursor to the Antimalarial Agent Artemisinin. Proc. Nat. Acad. Sci. U.S.A 2012, 109 (3), E111–E118. 10.1073/pnas.1110740109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Paddon CJ; Westfall PJ; Pitera DJ; Benjamin K; Fisher K; McPhee D; Leavell MD; Tai A; Main A; Eng D; Polichuk DR; Teoh KH; Reed DW; Treynor T; Lenihan J; Fleck M; Bajad S; Dang G; Dengrove D; Diola D; Dorin G; Ellens KW; Fickes S; Galazzo J; Gaucher SP; Geistlinger T; Henry R; Hepp M; Horning T; Iqbal T; Jiang H; Kizer L; Lieu B; Melis D; Moss N; Regentin R; Secrest S; Tsuruta H; Vazquez R; Westblade LF; Xu L; Yu M; Zhang Y; Zhao L; Lievense J; Covello PS; Keasling JD; Reiling KK; Renninger NS; Newman JD High-Level Semi-Synthetic Production of the Potent Antimalarial Artemisinin. Nature 2013, 496 (7446), 528–532. 10.1038/nature12051. [DOI] [PubMed] [Google Scholar]
- (7).Singh D; McPhee D; Paddon CJ; Cherry J; Maurya G; Mahale G; Patel Y; Kumar N; Singh S; Sharma B; Kushwaha L; Singh S; Kumar A Amalgamation of Synthetic Biology and Chemistry for High-Throughput Nonconventional Synthesis of the Antimalarial Drug Artemisinin. Org. Process Res. Dev. 2017, 21 (4), 551–558. 10.1021/acs.oprd.6b00414. [DOI] [Google Scholar]
- (8).Burgard A; Gieshoff T; Peschl A; Hörstermann D; Keleschovsky C; Villa R; Michelis S; Feth MP Optimisation of the Photochemical Oxidation Step in the Industrial Synthesis of Artemisinin. Chem. Eng. J. (Amsterdam, Neth) 2016, 294, 83–96. 10.1016/j.cej.2016.02.085. [DOI] [Google Scholar]
- (9).Benjamin KR; Silva IR; Cherubim JP; McPhee D; Paddon CJ Developing Commercial Production of Semi-Synthetic Artemisinin, and of β-Farnesene, an Isoprenoid Produced by Fermentation of Brazilian Sugar. J. Braz. Chem. Soc. 2016, 27, 1339–1345. 10.5935/0103-5053.20160119. [DOI] [Google Scholar]
- (10).Meadows AL; Hawkins KM; Tsegaye Y; Antipov E; Kim Y; Raetz L; Dahl RH; Tai A; Mahatdejkul-Meadows T; Xu L; Zhao L; Dasika MS; Murarka A; Lenihan J; Eng D; Leng JS; Liu C-L; Wenger JW; Jiang H; Chao L; Westfall P; Lai J; Ganesan S; Jackson P; Mans R; Platt D; Reeves CD; Saija PR; Wichmann G; Holmes VF; Benjamin K; Hill PW; Gardner TS; Tsong AE Rewriting Yeast Central Carbon Metabolism for Industrial Isoprenoid Production. Nature 2016, 537 (7622), 694–697. 10.1038/nature19769. [DOI] [PubMed] [Google Scholar]
- (11).McPhee; Pin A; Kizer L; Perelman L Renewable Squalane from Sugarcane. Cosmet. Toiletries 2014, 129 (6), 20–25 [Google Scholar]
- (12).Fisher KJ; Kinsey R; Mohamath R; Phan T; Liang H; Orr MT; Lykins WR; Guderian JA; Bakken J; Argilla D; Ramer-Denisoff G; Larson E; Qi Y; Sivananthan S; Smolyar K; Carter D; Paddon CJ; Fox CB Semi-Synthetic Terpenoids with Differential Adjuvant Properties as Sustainable Replacements for Shark Squalene in Vaccine Emulsions. npj Vaccines 2023, 8 (1), 14. 10.1038/s41541-023-00608-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Karrer P; Helfenstein A Synthese Des Squalens. Helv. Chim. Acta 1931, 14 (1), 78–85. 10.1002/hlca.19310140107. [DOI] [Google Scholar]
- (14).Schmitt J Synthesen in Der Carotinoidreihe. II. Eine Neue Synthese Des Squalens. Justus Liebigs Ann. Chem. 1941, 547 (1), 115–122. 10.1002/jlac.19415470108. [DOI] [Google Scholar]
- (15).Biellmann JF; Ducep JB Synthese du squalene par couplage queue a queue. Tetrahedron Lett. 1969, 10 (42), 3707–3710. 10.1016/S0040-4039(01)88493-8. [DOI] [Google Scholar]
- (16).Werthemann L; Johnson WS Application of the Chloro Ketal Claisen Reaction to the Total Synthesis of Squalene*. Proc, Nat. Acad. Sci. U.S.A 1970, 67 (3), 1465–1467. 10.1073/pnas.67.3.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Axelrod E; Milne GM; Van Tamelen EE General 1,5-Diene Synthesis Involving Overall Allyl Alcohol Coupling with Geometrical and Positional Control. J. Am. Chem. Soc. 1970, 92 (7), 2139–2141. 10.1021/ja00710a061. [DOI] [Google Scholar]
- (18).Kitagawa Y; Oshima K; Yamamoto H; Nozaki H A New Stereospecific Synthesis of 1,5-Dienes. Tetrahedron Lett. 1975, 16 (22), 1859–1862. 10.1016/S0040-4039(00)75277-4. [DOI] [Google Scholar]
- (19).Momose D-I; Iguchi K; Sugiyama T; Yamada Y Reductive Coupling of Allylic Halides by Chlorotris(Triphenylphosphine)Cobalt(I). Tetrahedron Lett. 1983, 24 (9), 921–924. 10.1016/S0040-4039(00)81565-8. [DOI] [Google Scholar]
- (20).Muzart J; Pete JP Η3-Allylpalladium Complexes: Isomerization and Photolysis; Synthesis of Squalene. Bull. Soc. Chim. France, 1984, 56–58. [Google Scholar]
- (21).Sasaoka S; Yamamoto T; Kinoshita H; Inomata K; Kotake H Palladium-Catalyzed Coupling of Allylic Acetates with Zinc. Chem. Lett. 1985, 14 (3), 315–318. 10.1246/cl.1985.315. [DOI] [Google Scholar]
- (22).Masuyama Y; Otake k; Kurusu Y Hexacarbonylmolybdenum(0)-Catalyzed Reductive Coupling of Allylic Acetates. Bull. Chem. Soc. Jpn. 1987, 60, 1527–1528. [Google Scholar]
- (23).Yanagisawa A; Hibino H; Habaue S; Hisada Y; Yamamoto H Highly Selective Homocoupling Reaction of Allylic Halides Using Barium Metal. J. Org. Chem. 1992, 57 (24), 6386–6387. 10.1021/jo00050a006. [DOI] [Google Scholar]
- (24).Barrero AF; Herrador MM; Quílez del Moral JF; Arteaga P; Arteaga JF; Diéguez HR; Sánchez EM Mild TiIII- and Mn/ZrIV-Catalytic Reductive Coupling of Allylic Halides: Efficient Synthesis of Symmetric Terpenes. J. Org. Chem. 2007, 72 (8), 2988–2995. 10.1021/jo062630a. [DOI] [PubMed] [Google Scholar]
- (25).Millán A; Campaña AG; Bazdi B; Miguel D; Álvarez de Cienfuegos L; Echavarren AM; Cuerva JM Ti/Pd Bimetallic Systems for the Efficient Allylation of Carbonyl Compounds and Homocoupling Reactions. Chem. Eur. J. 2011, 17 (14), 3985–3994. 10.1002/chem.201003315. [DOI] [PubMed] [Google Scholar]
- (26).Fisher KJ; Woolard FX Synthesis of E,e-Farnesol, Farnesyl Acetate and Squalene from Farnesene Via Farnesyl Chloride. WO2019237005, December 12, 2019. https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2019237005 (accessed 2023-03-15). [Google Scholar]
- (27).Biellmann J-F; Ducep J-B Allylic and Benzylic Carbanions Substituted by Heteroatoms. In Organic Reactions; Wiley: Hoboken, NJ, USA, 1982; Vol. 27. [Google Scholar]
- (28).Biellmann JF; Ducep JB Synthèse du squalène et d’analogues. Tetrahedron 1971, 27 (23), 5861–5872. 10.1016/S0040-4020(01)91751-X. [DOI] [Google Scholar]
- (29).Grieco PA; Masaki Y General 1,5-Diene Synthesis. Application to the Synthesis of Squalene. J. Org. Chem. 1974, 39 (14), 2135–2136. 10.1021/jo00928a043. [DOI] [Google Scholar]
- (30).Terao S; Shiraishi M; Kato K Carbon-Carbon Bonding. Jpn. Kokai Tokkyo Koho 54030102, 1979. [Google Scholar]
- (31).Mohri M; Kinoshita H; Inomata K; Kotake H Palladium-Catalyzed Regio- and Stereoselective Desulfonylation Of Allylic Sulfones With LiHBEt3. Application to the Synthesis of Squalene. Chem. Lett. 1985, 14 (4), 451–454. 10.1246/cl.1985.451. [DOI] [Google Scholar]
- (32).European Pharmacopeia. Squalene; January/2020:2805. [Google Scholar]
- (33).International Conference on Harmonisation; Guideline on Validation of Analytical Procedures: Definitions and Terminology; Availability. Federal Register. https://www.federalregister.gov/documents/1995/03/01/95-4956/international-conference-on-harmonisation-guideline-on-validation-of-analytical-procedures (accessed 2023-04-04). [Google Scholar]
- (34).Alonso D; Nájera C Desulfonylation Reactions. In Organic Reactions; Denmark S, Ed.; Wiley: Hoboken, NJ, USA, 2008; Vol. 72, 367̵–656. [Google Scholar]
- (35).Bengen MF Mein Weg zu den neuen Harnstoff-Einschluß-Verbindungen. Angew. Chem. 1951, 63 (9), 207–208. 10.1002/ange.19510630903. [DOI] [Google Scholar]
- (36).Harris KDM Fundamental and Applied Aspects of Urea and Thiourea Inclusion Compounds. Supramol. Chem. 2007, 19 (1–2), 47–53. 10.1080/10610270600977706. [DOI] [Google Scholar]
- (37).Angla B Chimie Organique-Sur Les Complexes Moleculaires Fournis Par La Thiouree Avec Les Composes Organiques. Compt. Rend. 1947, 224, 402–404. [PubMed] [Google Scholar]
- (38).Schiessler RW; Flitter D Urea and Thiourea Adduction of C5—C42-Hydrocarbons. J. Am. Chem. Soc. 1952, 74 (7), 1720–1723. 10.1021/ja01127a033. [DOI] [Google Scholar]
- (39).Nicolaides N; Laves F On the crystal structure of squalene thiourea adducts and the stereochemistry of squalene. Z. Kristallogr. - Crystalline Materials 1965, 121 (1–6), 283–296. 10.1524/zkri.1965.121.16.283. [DOI] [Google Scholar]
- (40).Hayes DG; Bengtsson YC; Van Alstine JM; Setterwall F Urea Complexation for the Rapid, Ecologically Responsible Fractionation of Fatty Acids from Seed Oil. J. Am. Oil Chem. Soc. 1998, 75 (10), 1403–1409. 10.1007/s11746-998-0190-9. [DOI] [Google Scholar]
- (41).Toshiyuki T; Hidetada N; Mitsutoshi A Method for Manufacturing Refined Vegetable Squalene and Refined Vegetable Squalene. WO2012169443A1, December 13, 2012. https://patents.google.com/patent/WO2012169443A1/en (accessed 2023-03-21). [Google Scholar]
- (42).Masiá AS; Martínez Moreno JM Obtención de Escualeno a Partir de Las Oleínas de La Refinación Física Del Aceite de Oliva. Grasas Aceites (Sevilla, Sp) 1981, 32, 313–317. [Google Scholar]
- (43).Misquith A; Fung HWM; Dowling QM; Guderian JA; Vedvick TS; Fox CB In Vitro Evaluation of TLR4 Agonist Activity: Formulation Effects. Colloids Surf., B 2014, 113, 312–319. 10.1016/j.colsurfb.2013.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.