

ABSTRACT
Cell-mediated immunity seems to be critical for prevention and resolution of invasive S. aureus infections, but an imbalance in this immunity may also produce SIRS and death or an inadequate protective response with prolonged bacteremia and death. This dysregulation is likely at the heart of mortality and severe disease in humans. Anti-toxin antibodies may also come into play in reducing the severity of S. aureus infections, but these antibodies might also address superantigen-induced immune dysregulation. Thus, while changing intrinsic T cell responses may be therapeutically difficult, monoclonal antibodies against superantigens may have utility in addressing dysfunctional immune responses to S. aureus. The models above are hypotheses for examining, and potentially dramatically improving immune response to and safety of S. aureus vaccines.
INTRODUCTION
Most adult humans have high levels of circulating antibodies against many staphylococcal antigens, indicative of prior subclinical infections, but these antibodies are generally not protective, and clinically significant infection with S. aureus fails to provide protective immunity. Multiple vaccines have been developed for the prevention of S. aureus infections, but none were proven efficacious in the human trials reviewed in references 1–4. All of the vaccine candidates functioned well in animal models, mostly murine models, but also in rabbits and primates. The reliance on murine models can be related to the extensive data available about murine immunity. Based on the large number of failures, a reasonable conclusion is that murine immunity and human preventive immunity against S. aureus are significantly different. The divergence of human and murine immunity has been detailed in the recent literature (1–6).
Clearly, one major hurdle in producing an S. aureus vaccine is the lack of detailed information about the human protective immune response to S. aureus (1–4). Several vaccine candidates that target surface antigens and produce opsonizing antibodies have reached clinical trials, but they have failed to protect or attenuate infections in humans. All of these antigens produced robust humoral immunity that provided protection in animal models. These data indicate that antibodies based on opsonophagoctyic activity are not protective in humans. While these results were very disappointing, they were not entirely surprising, because patients with B cell defects do not show increased frequency or severity of S. aureus diseases (2–4). Information from immune defects, clinical trials, and studies of human sepsis are providing important information about the immune response to S. aureus in humans, underlining the importance of cellular immunity. This information is examined here.
IMMUNE DEFECTS AND RECURRENT S. AUREUS INFECTIONS
The inborn and acquired immune defects that increase the incidence of S. aureus help to define the cells and pathways that provide protective immunity, and these are summarized in Table 1. Overall, evidence is mounting that helper T (TH) cells plus polymorphonuclear neutrophils (PMNs), dendritic cells, and macrophages are important for a protective immune response (7–14).
TABLE 1.
Immune defects that increase the incidence of S. aureus infectionsa
| Immune defect | Biological basis | Clinical presentation | References |
|---|---|---|---|
| Failure of dendritic cells | Vitamin D required for activation | Recurrent and more severe infections | 15, 16 |
| Failure to activate macrophages | Mutations in NGFβ and its receptor, NTRK1 | Recurrent and more severe S. aureus infections | 19 |
| Failure to activate PMNs | IRAK-4 or MyD88 deficiency | Failure of PMNs to kill S. aureus, S. pnumoniae, and P. aeruginosa despite Abs; invasive and skin infections; resolving in teenage years | 20–23 |
| Failure to differentiate naive T cells into TH17 cells | IL-6 autoantibodies or IL-6 deficiency | Recurrent S. aureus infections | 14 |
| Failure of TH17 cells to recruit PMNs | STAT-3 defect | Recurrent S. aureus skin and mucosal infections, starting in neonates | 8, 10, 13 |
| T-cell defects, e.g., with HIV or prednisone treatment | Reduced activation of T cells | Recurrent S. aureus infections, especially mucocutaneous | 34–36 |
| MAIT cell exhaustion(T cell exhaustion) | 10–45% of lymphocytesInactive after SAg exposure | Increased severity and frequency of infections in intensive care units with reduced MIAT cells | 39–44, 48 |
IRAK-4, interleukin-1 receptor-associated kinase-4; MAIT, mucosal-associated invariant T; MyD88, myeloid differentiation factor 88; NGFβ, nerve growth factorβ; STAT-3, signal transducer and activator of transcription 3; TH17, T helper cell-17.
Vitamin D Deficiency and Dendritic Cell Failure
Patients with vitamin D deficiency have increased numbers of S. aureus infections (15, 16). This may relate in part to decreased bactericidal activity of their dendritic cells (17). Indeed, some strains of S. aureus were able to reside within bone marrow dendritic cells and macrophages because they expressed high activity of the Agr quorum-sensing system and escaped from phagosomes by producing the lytic proteins phenol-soluble modulins (18). Therefore, having adequate vitamin D levels may help dendritic cells to express maximal bactericidal activity against S. aureus as well as to release mediators to activate the immune system.
Mutations in NGFβ and Its Receptor NTRK1 Reduce Phagocyte Activation
Mutations causing loss of nerve growth factor β (NGFβ) or its receptor have been associated with recurrent S. aureus infections in humans in skin, joints, bones, and the oral cavity (19). S. aureus factors such as peptidoglycan, protein A, α-hemolysin, and phenol-soluble modulins stimulate macrophages through nucleotide-binding oligomerization domain-like receptors to produce NGFβ (19). NGFβ is both a chemoattractant for and an activator of phagocytes. NGFβ activation of neutrophils and macrophages results in enhanced killing of S. aureus. Deletion of NGFβ or its receptor NTRK1 results in more severe S. aureus infections in animal models (19). Of note, the ability to activate macrophages to produce NGFβ is limited to S. aureus because not even Staphylococcus epidermidis causes the release of NGFβ. Patients deficient in NGFβ demonstrate the critical importance of phagocytes in the control of S. aureus infections.
IRAK-4/MyD88 Deficiency
Defects in interleukin-1 receptor-associated kinase-4 (IRAK-4) or myeloid differentiation factor 88 (MyD88) impair Toll-like receptor 2 (TLR-2)-mediated and interleukin-1 (IL-1) receptor-mediated immunity such that macrophages fail to produce inflammatory cytokines such as IL-1β, IL-6, IL-8, interferons (IFNs), and tumor necrosis factor alpha (TNF-α) (20–23). Without these cytokines, there is a failure to activate a specific subset of T-helper cells known as TH17 cells, which are critical for the activation of polymorphonuclear neutrophils (PMNs). These deficiencies manifest soon after birth with recurrent S. aureus, Streptococcus pneumoniae, and Pseudomonas aeruginosa infections. Prophylactic antibiotics and early aggressive antibiotic therapy are required to treat babies with these defects. While intravenous immunoglobulin is often used, with respect to pneumococcal infection, there is no correlation between the presence or absence of antipneumococcal antibodies and the occurrence of invasive pneumococcal disease in patients with these deficiencies. This demonstrates the critical importance of TH17 cells and PMNs in the outcome, even though it is well known that anticapsular antibodies are important in host defense against the pneumococcus (20–23). Those children that survive until their teenage years have a resolution of frequent invasive infections.
IL-6 Autoantibodies
A patient described by Puel et al. with recurrent S. aureus infections had normal immunoglobulins, suggesting that his infections could not have been the result of immunoglobulin deficiency. Instead, this patient was found to lack the proinflammatory cytokine IL-6 due to anti-IL-6 antibodies (14). The absence of IL-6 causes an immunodeficiency state, which can be assumed to be responsible for his recurrent S. aureus infections. IL-6 is normally produced in response to S. aureus interactions with macrophages, dendritic cells, and γδ-T cells. IL-6, in conjunction with transforming growth factor β (TGF-β), helps to direct the conversion of naive CD4 T cells to TH17 cells (24) and the proliferation of TH1 and TH2 cells, among many cell types (25). IL-6 is also important for blocking regulatory T cell (Treg) development and for stimulating B cells to produce antibodies (24). Overly active Tregs can produce immunosuppression by reducing TH1 and TH17 cellular activation of the bactericidal activity of phagocytes. On the one hand, activation of TH17 cells is critical for the control of S. aureus infections (26); on the other hand, the suppression of Tregs may allow for an overly exuberant inflammatory response to S. aureus infections (27, 28). Indeed, loss of Tregs can result in fatal inflammatory disease (29). Thus, IL-6 plays a key role in regulating TH17/Treg balance so as to control immunity and immunopathology.
Job’s Syndrome, a TH17/STAT3 Defect
Patients with autosomal-dominant hyper-IgE, or Job’s syndrome, have dominant-negative mutations in signal transducer and activator of transcription 3 (STAT3) (8, 10, 13), and mutations in STAT3 have a strong association with frequent and severe S. aureus mucocutaneous infections, including staphylococcal pneumonia, empyema, and “cold” skin abscesses. STAT3 is an intracellular signal transduction factor in TH17 cells, which links surface receptors to the activation of genes (30, 31). Activation by IL-6 of its receptor on TH17 cells plays a key role in the differentiation of naive T cells into TH17 cells. TH17 cells are critical for controlling S. aureus infections because they release IL-17, which recruits PMNs to the site of infection and helps to activate the PMNs for killing S. aureus (31). A key role for PMNs in the protection from S. aureus infections has also been established by the observation of recurrent S. aureus infections in patients with chronic granulomatous disease (primary defect in the production of reactive oxygen species by their PMNs) (32). However, the role of STAT3 may extend beyond PMNs because STAT3 is also involved in expression of host antimicrobial peptides in keratinocytes (31) and in the production of Reg3γ, an antimicrobial peptide produced by respiratory epithelial cells, whereby these respiratory mucosal cells become bactericidal for S. aureus (33). S. aureus interaction with respiratory epithelial cells activates them through STAT3 for the production of Reg3γ. Thus, while STAT3 is important for controlling systemic invasion by S. aureus by recruiting and activating PMNs, it also plays an important role in activating mucosal and skin cells. Hence, this defect is associated with recurrent S. aureus mucocutaneous infections.
Corticosteroids and AIDS
Patients with reduced cell-mediated immunity have an increased incidence of S. aureus infections. This can be due to an underlying disease such as systemic lupus erythematosus, AIDS infection, or the use of corticosteroids (34–37). Even moderate-dose prednisone therapy increases the risk of serious S. aureus infection (36). While we frequently think of viral infections as being associated with reduced cell-mediated immunity, the link to S. aureus infections comes from the reduced TH17 cell-PMN axis in prednisone-treated patients (38).
Mucosal-Associated Invariant T (MAIT) Cells, Superantigens, and T Cell Exhaustion
MAIT cells represent up to 10% of T cells in blood and up to 35% in liver and some mucosal sites (39–44), and they may be considered the first T cell responders to invading bacteria because they are activated during the early stages of bacterial infection and act as a bridge between the innate and adaptive immune systems by recognizing bacterial byproducts derived from riboflavin metabolism (42). However, unlike the situation with humans, MAIT cells are infrequent in laboratory mouse strains such as C57BL6, thereby limiting the utility of murine models without humanization (39). MAIT cells have been implicated in protecting the host from multiple species of bacteria (40–42, 44, 45). However, recent studies have shown that they also respond to superantigens (SAgs), such as staphylococcal enterotoxin B (SEB), and have been identified as extremely potent and fast-acting producers of proinflammatory cytokines (39). To quote from Taylor et al. (39), “We have identified a population of innate T-like cells, called MAIT cells, as the most powerful source of proinflammatory cytokines after exposure to SAgs.” In addition to being involved in immunopathogenic activity, MAIT cells may also be involved in “a novel mechanism of SAg-associated immunosuppression in humans” (39) wherein widespread activation of MAIT cells by SAgs results in their inability to respond to future challenges. Sandberg et al. (43) report that subsequent to such SAg activation, the MAIT cells remain unresponsive to stimulation with conventional bacterial antigens. This concept of immune exhaustion is supported by the observation that intensive care unit patients with reduced MAIT cells were more vulnerable to severe and subsequent bacterial infections during their stay in the intensive care unit (46). Equally, ill patients’ normal MAIT numbers did not show increased bacterial infections (46). Thus, SAg-producing S. aureus hijacks MAIT cells by inducing the cytokine storm and leaves them functionally impaired. A mechanism for this is based on the occurrence of anergy via upregulation of inhibitory receptors such as lymphocyte-activation gene 3 (39, 44, 47).
T Cell Exhaustion is Manifested in Humans by Low Levels of IL-2
SAgs induce other modifications to T cells (see Fig. 2). A marker of T cell exhaustion is low levels of IL-2 (39, 43, 48). IL-2 production increases dramatically in SEB-challenged MAIT cells compared to conventional T cells in peripheral blood mononuclear cells (39). This is in contrast to E. coli exposure to MAIT cells where IL-2 was not produced (49). These observations are consistent with previous findings where SEB and other SAgs delete or anergize T cells (50, 51). Moreover, exposure of human CD4+CD25− T cells to SEA induces proliferation and is followed by a switch to a CD25+FoxP3+ Treg phenotype that secretes IL-10 but not IL-2 (39). SEA also activates αβT cells (52). SEB activates αδT cells and stimulates the conversion of Treg to TH17-like cells (11, 39, 52–57). In summary, SAgs can induce a cytokine storm, but they are also potent immunomodulators of the T cell immune response. These effects would be anticipated to change during the course of S. aureus sepsis.
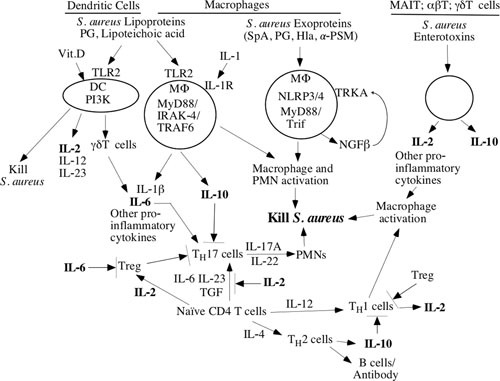
FIGURE 2.
S. aureus enterotoxin modifications of immune response.
Summary of Immune Defects and S. aureus Infections
In summary, the inborn and acquired immune defects that are associated with an increase in the incidence of S. aureus infections show that the macrophage-T cell axis is critical for the activation of phagocytes to control S. aureus invasive infections. Conspicuously absent is the need for opsonic antibodies for protection against S. aureus, because children with agammaglobulinemia do not show increased numbers of S. aureus infections (1–5). The innate activation of macrophages and dendritic cells, as well as the surface factors that activate complement, seem sufficient to control S. aureus without opsonization. On the other hand, antibodies against S. aureus toxins have been shown to reduce the severity and mortality of S. aureus toxin-mediated diseases (6, 58–61). As discussed below, staphylococcal SAgs can play a major role in the modulation of T cell development; therefore, antitoxin antibodies may be crucial in the maintenance of a balanced macrophage-T cell axis.
STAPHYLOCOCCAL SEPSIS AND CYTOKINE PRODUCTION
Some information is emerging about anti-S. aureus immunity that is based on studies of patients with sepsis, especially S. aureus bacteremia, and this is summarized in Table 2. In septic patients, the profile of cytokines produced during infection is a reflection of the cells that are activated (Table 2). Normally, interaction of invasive S. aureus with macrophages and dendritic cells causes the release of proinflammatory cytokines such as IL-1, IL-2, IL-6, IFN-γ, and TNF-α. This is later followed by IL-10, which downregulates the immune response. However, this normal pattern of immune response is not seen in patients that have worse outcomes from S. aureus sepsis. Thus, mortality is associated with dysregulation of the immune system in apparently normal individuals. As detailed in Table 2, mortality or more severe disease is associated with low IL-1, IFN-γ, and TNF-α but high IL-6 and IL-10.
TABLE 2.
Cytokines, sepsis, and survival in S. aureus infection
| Study | Type of infectiona | Survival or less complicated course | Death or complicated course |
|---|---|---|---|
| Söderquist et al. 1992 (84) | 65 patients with sepsis | Rapidly dropping IL-6 | Persistently elevated IL-6 |
| van Dissel et al. 1998 (67) | Sepsis | High IL-1 and TNF-α with low IL-10 | Low IL-1 and TNF-α with high IL-10 |
| Rose et al. 2012 (68), Rose et al. 2017 (69) | S. aureus bacteremia | High IL-1 and low IL-10 | Low IL-1β and IFN-ϒ; high IL-10 |
| V710 vaccine trailFowler 2013 (62); McNeely 2014 (63) | Invasive infections after cardiothoracic surgery | High IL-1, IL-2, and IL-17 | Low IL-2 and IL-17 preoperatively and all postoperative testing |
| McNicholas et al. 2014 (85) | 61 patients with SABa | Low levels of IL-6 predict better outcome | High levels of IL-6 predict worse outcome |
| Gupta et al. 2016 (65) | Posttrauma sepsis (mix of bacterial pathogens) | High IL-4, IL-10, TGF-β, TH17, and TH17/TregLow IL-2 and IFN-γ | |
| Minejima et al. 2016 (66) | 196 patients with SAB | Low TNF-α and IL-10; low IL-17A in all patients | High TNF-α and IL-10; low IL-17A in all patients |
| Chantratita et al. 2017 (86) | 327 patients with SAB | Low IL-6 and IL-8 had less respiratory failure | High IL-6 and IL-8 had more respiratory failure |
SAB, S. aureus bacteremia.
A dramatic example of immune dysfunction during S. aureus infection came from the V710 staphylococcal vaccine trial, where increased mortality occurred in vaccinated patients (62). V710 was a recombinant S. aureus surface protein, IsdB (iron surface determinant protein B), and was given to subjects prior to cardiovascular surgery. Patients that received the vaccine and subsequently developed invasive S. aureus infection during the postoperative period showed increased mortality. The cause of the increased mortality was systemic inflammatory response wherein control of the immune response was lost. Posttrial investigation found that low levels of IL-2 were observed not only during sepsis but even before patients received the V710 vaccine (63). There was a 5-fold increased mortality in vaccinees that had low prevaccination levels of IL-2. Thus, low baseline levels of IL-2 could predict a tragic outcome when vaccinees developed a postoperative infection. These patients also showed low levels of IL-17. These unanticipated results show that excess mortality was related to a dysregulated immune response even before the vaccine was given. Because IL-2 is critical for the survival of Tregs (64), one can postulate that the loss of these cells that modulate the immune response may have played a role in the mortality of these vaccinees. IL-2 is complex in that it produces both pro- and anti-inflammatory effects by downregulating TH17 but upregulating TH1 cells. Although increased numbers of TH17 cells were found in S. aureus sepsis in several studies (9, 65), this is usually not accompanied by an increased blood concentration of IL-17 (12, 62, 65, 66) and may help to explain why IL-17 levels were low in clinical studies when one would anticipate higher TH17 activity. It also raises the possibility that TH17 cells may act locally rather than systemically (Fig. 1).
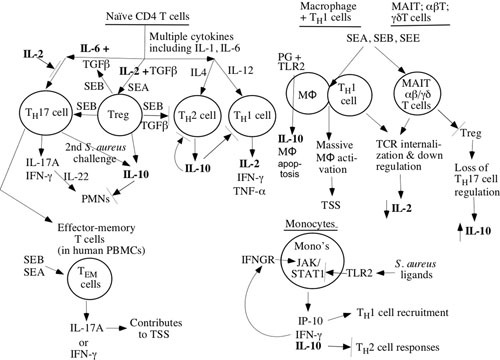
FIGURE 1.
Model for human immunity to S. aureus based on immune defects and cytokines in sepsis.
The data outlined in Table 2, suggesting that immune dysregulation correlates with more severe disease and mortality, are considered here. IL-1 is released from macrophages and dendritic cells (among other cells) when S. aureus is first met while invading tissues (12). However, patients that show low levels of IL-1 have worse outcomes during invasive S. aureus infections (67–69). Of the many functions that IL-1 has, perhaps a very important one for invasive S. aureus infections comes in directing TH17 differentiation and activation (70), because TH17 cells are critical for protection against S. aureus infections (10, 13). Therefore, it is striking that patients with S. aureus circulating in their bloodstream and interacting with macrophages would fail to produce IL-1, and this is an obvious sign of immune dysregulation.
Another cytokine released early in the course of S. aureus infection is IL-2 (71), and low levels have correlated with poor outcomes (62, 65). Broadly speaking, IL-2 ± TGF-β reduces inflammation by directing reduction of TH1, TH2, and TH17 differentiation and increasing Treg expansion (72). The importance of IL-2 in directing cells toward a moderated inflammatory response is underscored by the increased deaths in patients receiving the V710 S. aureus vaccine (62), wherein low prevaccination IL-2 levels correlated very strongly with a systemic inflammatory response syndrome (SIRS) response in these patients when an invasive S. aureus infection occurred. Moreover, IL-2 levels remained low during the infection. These observations are consistent with data from Gupta et al. (65), who studied sepsis in trauma patients, wherein death was associated with low level of IL-2 in culture supernatant of peripheral blood mononuclear cells from these patients. Of course, severe trauma itself causes changes in the immune response, so the immune dysregulation noted in this study is less clear cut than in the S. aureus vaccine study.
Also consistent with the IL-2 data for immune dysregulation are the observations from several studies that strongly correlate high levels of IL-10 with poor outcomes (65, 67–69). IL-10 downregulates the inflammatory response to S. aureus invasion. The increased mortality suggests that the balance between pro- and anti-inflammatory responses is incorrect. S. aureus itself may actively alter this balance by stimulating production of IL-10 directly. Quantitatively, S. aureus interactions with monocytes and macrophages via TLR-2 interactions produce 4- to 20-fold more IL-10 than interactions with dendritic cells (58). Thus, more IL-10 is produced during bacteremia than in cutaneous infections. While the downregulatory effects following interactions of S. aureus ligands with the TLR-2 on T cells, monocytes, macrophages, and dendritic cells may be critical for balancing the host response to SAgs (73–76), overly vigorous suppression of the immune response may lead to continued bacteremia, whereas inadequate suppression may result in death due to SIRS. Engagement of TLR-2 on macrophages can also produce the proinflammatory cytokine, IL-1, but in dendritic cells IL-2 is produced. IL-2 production is needed for the development and maintenance of Tregs, which suppress immune responses produced by TH17 cells (56). In some cases, TH17 cells, which are not thought of as IL-10 producers, can convert to IL-10 production when given a second S. aureus challenge (70), which might relate to the first challenge being vaccination and the second challenge being invasive infection (63). Hence, the inhibitory activity of IL-2 on TH2 and TH17 cells and stimulatory activity on Treg cells might be lost when IL-2 levels are low during sepsis and allow for increased IL-10 production. High IL-10 would decrease PMN activation and killing of S. aureus (70, 77–81), reduce TH17 proliferation (78, 82), and reduce Treg activity (78, 82, 83). A balance between too low and too high IL-10 levels in response to S. aureus infections has been suggested as needed to avoid an overly vigorous response (SIRS) or an incomplete activation (persistent bacteremia) (78, 82, 83).
Another consistent finding is that persistently elevated IL-6 correlates with death and/or with another worse outcome with S. aureus sepsis (84, 85, 86). IL-6 is produced by macrophages and dendritic cells shortly after S. aureus products bind to TLR-2 (14, 24, 27, 30, 81). IL-6 causes differentiation of naive T cells into TH17 and regular Tregs into proinflammatory Tregs that produce IL-17, IFN-γ, and TNF-α (14, 24, 27, 56). Thus, persistently elevated IL-6 may be linked to an overactive proinflammatory response that damages the host more than it protects it.
Finally, noninterleukins have also been examined during S. aureus sepsis in humans. Low levels of IFN-γ correlate with death (65, 68, 69). Early interactions of S. aureus with macrophages via TLR-2 lead to IFN-γ release (83). Similarly, when S. aureus interacts with dendritic cells via TLR-2, this results in dendritic-cell-mediated activation of TH1 and γδT cells to produce IFN-γ (18, 87). IFN-γ activates macrophages to kill S. aureus (83). MAIT cells can also produce IFN-γ after interactions with SEB (39). Clearly, low levels of IFN-γ might result in poor clearance of S. aureus by macrophages. Of interest, immunization of C57Bl/6 mice with killed S. aureus resulted in CD4 T-cell-dependent production of high IFN-γ levels with S. aureus bacteremia challenge that produced death (88). This provides another example where human and murine immunology diverge.
In contrast to IFN-γ levels, TNF-α levels yielded inconsistent results because high levels of TNF-α were associated with increased (66) or decreased (67) mortality from S. aureus bacteremia. TNF-α is important for activation of phagocytes, but it has also been associated with SIRS and death. More studies will be needed to sort this out. Nevertheless, the available studies of cytokine release during S. aureus sepsis provide strong evidence for immune dysfunction that leads eventually to death or more severe disease.
IMMUNE DYSREGULATION AND DEATH
Immune dysregulation, either over- or under-activation, of the immune response can be associated with mortality and/or more severe disease. This raises the question of why so many humans have a nonprotective response to S. aureus infections that is clearly related to immune dysregulation. We are familiar with Mycobacterium leprae and Trypanosoma cruzi as causing immunosuppression of the host, and hence one hypothesis is that the balance between the host and S. aureus has become lost. This might be due to the production of superantigens (SAgs) that can alter the responses of antigen-presenting cells and T cells (39, 43, 47, 50–53, 57, 74, 89–96). Observations from these papers are summarized in Fig. 2 and are offered as enterotoxins redirecting T cell differentiation. For example, SEB has many interactions with the human immune system. SEB can cause Tregs to develop a TH17 phenotype and to produce TGF-β (91). The TGF-β enhances naive T cells to differentiate into TH17 cells, and it inhibits TH2 cell activation. SEA, SEB, and SEE can activate TH1 and MAIT cells as superantigens that result in massive macrophage activation and toxic shock (39, 43, 46, 47, 49). After such activation, the T cells and MAIT cells downregulate and fail to respond to further stimulation, leaving the host immunocompromised for days to weeks (39, 46). In addition, SAgs can even inhibit the primary interactions between staphylococcal products and TLR-2 (89, 90). Thus, SAgs have a very large number of interactions with the human immune system, especially T cells and macrophages (Fig. 2). Notably, cutaneous infections and vaginal toxic shock syndrome with S. aureus do not reliably provoke durable, protective immune responses (58, 97), thereby opening the possibility for SAgs and other toxins to influence the immunity response; yet seemingly, they have provoked no immune response. These interactions may underlie failures of humans to respond with protective immunity against S. aureus because continued exposure to SAgs is not inhibited without the presence of antitoxin antibodies.
COMMENTS ON THE MODELS
Gaps exist in our knowledge of the construction of the models of human immunity against S. aureus presented in Fig. 1 and 2, but these generate testable hypotheses. A major breakthrough for the development of an S. aureus vaccine that could prevent disease and death, or at least reduce the severity of infections, would be the discovery of a biomarker. Examples of vaccine biomarkers are anti-hepatitis B viral surface antigen and antimeningococcal capsular antibodies whose presence correlates with protection from infection. Our lack of a biomarker that predicts protection from S. aureus infections relates directly to our lack of understanding of protective immunity against S. aureus. Many successful bacterial vaccines against S. pneumoniae, H. influenzae, Neisseria meningitidis, and Clostridium tetani have been developed when the mechanism of immune protection was known and a biomarker was followed during clinical trials.
While cytokines provide some indication of which cells are activated, the precise cells being activated are not defined because cytokines can be produced by multiple cells and pathways. There is limited information concerning the timing and precise pathways in S. aureus human infections, which are not well replicated by murine models (1, 4, 6, 59, 60, 88). Moreover, cytokines may be released just locally, not systemically, so blood levels are not helpful. Finally, the relative balance between T cell subsets may be critical to the outcome. For example, our current state of knowledge does not allow us to know whether T cell exhaustion, reduced action of Tregs, or overactivation of TH17 cells cause death with multiorgan failure during invasive S. aureus infections. In addition, the immune reactions to cutaneous/mucosal invasion versus systemic (bacteremic) infection are probably distinct, wherein γδT and TH17 cells (IL-17A) are more important for skin and respiratory infections, whereas TH1 (IFN-γ) responses are more important for survival during bacteremia (12, 98–100). We can see that sustained high levels of IL-10 correlate with death from S. aureus infections, yet it is not clear whether this is due to oversuppression of the immune response or a manifestation of the host attempt to reduce an overly exuberant immune response. When human peripheral blood mononuclear cells were differentiated into dendritic cells by culturing with granulocyte-macrophage colony-stimulating factor + IL-4, they produced a primarily TH1/TH17 stimulatory response, whereas when they were differentiated with macrophage colony stimulating factor, this triggered primarily an IL-10 response in response to TLR-2 activation by peptidoglycan or lipoteichoic acid (101). Hence, it is conceivable that the high levels of IL-10 during persistent S. aureus bacteremia come from continued activation of macrophages; however, it is also conceivable that the high levels of IL-10 downregulate the immune response needed to clear the bacteria.
Similarly, the data from the V710 vaccine trials, wherein low prevaccination IL-2 was strongly associated with SIRS, could be due to be overexpansion of or overly active TH17 cells, e.g., by reduced Treg activity and loss of suppressive effects on TH17 cells. Another hypothesis that springs out of Fig. 2 is that some patients may have T cell exhaustion and altered immune response due to colonization with superantigen-producing S. aureus strains (especially SEB), which can downregulate T cell receptors (92). For example, if superantigens led to MAIT cell downregulation, then low levels of IL-2 might prime the host for poor Treg and high TH17 responses when S. aureus invasion occurred. It would be anticipated that these patients might be colonized with SEB-producing strains, lack anti-SEB antibodies, and have low levels of IL-2. Low levels of IL-2 result in reduced Treg and increased TH17 activity, perhaps adding to an imbalanced immune response to invasive S. aureus infections as seen in the V710 clinical vaccine trial. Finally, patients with immunodeficiencies tend to have local infections that rarely develop spillover bacteremia (102), but we have little knowledge about the factors involved in protection from primary S. aureus bacteremia. Contrasting the immune responses to bacteremia to cutaneous infections may shed light on the context for T cell activation.
The models for human immune responses to S. aureus are really hypotheses (Fig. 1 and 2), and much more detail is needed about the types of T cells being activated, the balance between T cell types, the timing of T cell activation and inactivation, the impact of antibodies, and the correlations to outcomes. Given the multiple possible interpretations of changes in cytokines, a detailed analysis of the types of T cells produced, their state of activation, the time course of activation, and the relative balance between the different T cell subsets prior to and during S. aureus invasive infections should shed light on which pathway(s) are critical for immunity to S. aureus infections. In addition, prospective data on the types of antibodies being produced (opsonic versus toxin-neutralizing), especially during S. aureus bacteremia versus cutaneous infections, that are correlated with outcome will help to inform us about the relative value of these antibodies. Animal models do not substitute for human data. This basic knowledge will be critical as S. aureus vaccine trials are designed because patients should be stratified in future clinical trials to evaluate the impact of therapeutic interventions.
SUMMARY OF HUMAN IMMUNITY AGAINST S. AUREUS
Cell-mediated immunity seems to be critical for the prevention and resolution of invasive S. aureus infections, but an imbalance in this immunity may also produce SIRS and death or an inadequate protective response with prolonged bacteremia and death. This dysregulation is likely at the heart of mortality and severe disease in humans. Antitoxin antibodies may also come into play in reducing the severity of S. aureus infections, but these antibodies might also address superantigen-induced immune dysregulation. Thus, while changing intrinsic T cell responses may be therapeutically difficult, monoclonal antibodies against superantigens may have utility in addressing dysfunctional immune responses to S. aureus. The models above are hypotheses for examining, and potentially dramatically improving, the immune response to and safety of S. aureus vaccines.
REFERENCES
- 1.Fowler VG Jr, Proctor RA. 2014. Where does a Staphylococcus aureus vaccine stand? Clin Microbiol Infect 20(Suppl 5):66–75 10.1111/1469-0691.12570. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Proctor RA. 2012. Is there a future for a Staphylococcus aureus vaccine? Vaccine 30:2921–2927 10.1016/j.vaccine.2011.11.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 3.Proctor RA. 2012. Challenges for a universal Staphylococcus aureus vaccine. Clin Infect Dis 54:1179–1186 10.1093/cid/cis033. [PubMed] [DOI] [PubMed] [Google Scholar]
- 4.Proctor RA. 2015. Recent developments for Staphylococcus aureus vaccines: clinical and basic science challenges. Eur Cell Mater 30:315–326 10.22203/eCM.v030a22. [PubMed] [DOI] [PubMed] [Google Scholar]
- 5.Kolata JB, Kühbandner I, Link C, Normann N, Vu CH, Steil L, Weidenmaier C, Bröker BM. 2015. The fall of a dogma? Unexpected high T-cell memory response to Staphylococcus aureus in humans. J Infect Dis 212:830–838 10.1093/infdis/jiv128. [PubMed] [DOI] [PubMed] [Google Scholar]
- 6.Salgado-Pabón W, Schlievert PM. 2014. Models matter: the search for an effective Staphylococcus aureus vaccine. Nat Rev Microbiol 12:585–591 10.1038/nrmicro3308. [PubMed] [DOI] [PubMed] [Google Scholar]
- 7.Daum RS, Spellberg B. 2012. Progress toward a Staphylococcus aureus vaccine. Clin Infect Dis 54:560–567 10.1093/cid/cir828. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Jannière L, Fieschi C, Stéphan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gülle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL. 2008. Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 205:1543–1550 10.1084/jem.20080321. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Joshi A, Pancari G, Cope L, Bowman EP, Cua D, Proctor RA, McNeely T. 2012. Immunization with Staphylococcus aureus iron regulated surface determinant B (IsdB) confers protection via Th17/IL17 pathway in a murine sepsis model. Hum Vaccin Immunother 8:336–346 10.4161/hv.18946. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC. 2008. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205:1551–1557 10.1084/jem.20080218. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maher BM, Mulcahy ME, Murphy AG, Wilk M, O’Keeffe KM, Geoghegan JA, Lavelle EC, McLoughlin RM. 2013. Nlrp-3-driven interleukin 17 production by γδT cells controls infection outcomes during Staphylococcus aureus surgical site infection. Infect Immun 81:4478–4489 10.1128/IAI.01026-13. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miller LS, Cho JS. 2011. Immunity against Staphylococcus aureus cutaneous infections. Nat Rev Immunol 11:505–518 10.1038/nri3010. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Minegishi Y, Saito M, Nagasawa M, Takada H, Hara T, Tsuchiya S, Agematsu K, Yamada M, Kawamura N, Ariga T, Tsuge I, Karasuyama H. 2009. Molecular explanation for the contradiction between systemic Th17 defect and localized bacterial infection in hyper-IgE syndrome. J Exp Med 206:1291–1301 10.1084/jem.20082767. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Puel A, Picard C, Lorrot M, Pons C, Chrabieh M, Lorenzo L, Mamani-Matsuda M, Jouanguy E, Gendrel D, Casanova JL. 2008. Recurrent staphylococcal cellulitis and subcutaneous abscesses in a child with autoantibodies against IL-6. J Immunol 180:647–654 10.4049/jimmunol.180.1.647. [PubMed] [DOI] [PubMed] [Google Scholar]
- 15.Thomason J, Rentsch C, Stenehjem EA, Hidron AI, Rimland D. 2015. Association between vitamin D deficiency and methicillin-resistant Staphylococcus aureus infection. Infection 43:715–722 10.1007/s15010-015-0815-5. [PubMed] [DOI] [PubMed] [Google Scholar]
- 16.Wang JW, Hogan PG, Hunstad DA, Fritz SA. 2015. Vitamin D sufficiency and Staphylococcus aureus infection in children. Pediatr Infect Dis J 34:544–545 10.1097/INF.0000000000000667. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.van der Does AM, Kenne E, Koppelaar E, Agerberth B, Lindbom L. 2014. Vitamin D3 and phenylbutyrate promote development of a human dendritic cell subset displaying enhanced antimicrobial properties. J Leukoc Biol 95:883–891 10.1189/jlb.1013549. [PubMed] [DOI] [PubMed] [Google Scholar]
- 18.O’Keeffe KM, Wilk MM, Leech JM, Murphy AG, Laabei M, Monk IR, Massey RC, Lindsay JA, Foster TJ, Geoghegan JA, McLoughlin RM. 2015. Manipulation of autophagy in phagocytes facilitates Staphylococcus aureus bloodstream infection. Infect Immun 83:3445–3457 10.1128/IAI.00358-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hepburn L, Prajsnar TK, Klapholz C, Moreno P, Loynes CA, Ogryzko NV, Brown K, Schiebler M, Hegyi K, Antrobus R, Hammond KL, Connolly J, Ochoa B, Bryant C, Otto M, Surewaard B, Seneviratne SL, Grogono DM, Cachat J, Ny T, Kaser A, Török ME, Peacock SJ, Holden M, Blundell T, Wang L, Ligoxygakis P, Minichiello L, Woods CG, Foster SJ, Renshaw SA, Floto RA. 2014. Innate immunity. A Spaetzle-like role for nerve growth factor β in vertebrate immunity to Staphylococcus aureus. Science 346:641–646 10.1126/science.1258705. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pacquelet S, Johnson JL, Ellis BA, Brzezinska AA, Lane WS, Munafo DB, Catz SD. 2007. Cross-talk between IRAK-4 and the NADPH oxidase. Biochem J 403:451–461 10.1042/BJ20061184. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Picard C, von Bernuth H, Ghandil P, Chrabieh M, Levy O, Arkwright PD, McDonald D, Geha RS, Takada H, Krause JC, Creech CB, Ku CL, Ehl S, Maródi L, Al-Muhsen S, Al-Hajjar S, Al-Ghonaium A, Day-Good NK, Holland SM, Gallin JI, Chapel H, Speert DP, Rodriguez-Gallego C, Colino E, Garty BZ, Roifman C, Hara T, Yoshikawa H, Nonoyama S, Domachowske J, Issekutz AC, Tang M, Smart J, Zitnik SE, Hoarau C, Kumararatne DS, Thrasher AJ, Davies EG, Bethune C, Sirvent N, de Ricaud D, Camcioglu Y, Vasconcelos J, Guedes M, Vitor AB, Rodrigo C, Almazán F, Méndez M, Aróstegui JI, Alsina L, et al. 2010. Clinical features and outcome of patients with IRAK-4 and MyD88 deficiency. Medicine (Baltimore) 89:403–425 10.1097/MD.0b013e3181fd8ec3. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singh A, Zarember KA, Kuhns DB, Gallin JI. 2009. Impaired priming and activation of the neutrophil NADPH oxidase in patients with IRAK4 or NEMO deficiency. J Immunol 182:6410–6417 10.4049/jimmunol.0802512. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.van Bruggen R, Drewniak A, Tool AT, Jansen M, van Houdt M, Geissler J, van den Berg TK, Chapel H, Kuijpers TW. 2010. Toll-like receptor responses in IRAK-4-deficient neutrophils. J Innate Immun 2:280–287 10.1159/000268288. [PubMed] [DOI] [PubMed] [Google Scholar]
- 24.Tanaka T, Narazaki M, Kishimoto T. 2014. IL-6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol 6:a016295 10.1101/cshperspect.a016295. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hunter CA, Jones SA. 2015. IL-6 as a keystone cytokine in health and disease. Nat Immunol 16:448–457 10.1038/ni.3153. [PubMed] [DOI] [PubMed] [Google Scholar]
- 26.Lin L, Ibrahim AS, Xu X, Farber JM, Avanesian V, Baquir B, Fu Y, French SW, Edwards JE Jr, Spellberg B. 2009. Th1-Th17 cells mediate protective adaptive immunity against Staphylococcus aureus and Candida albicans infection in mice. PLoS Pathog 5:e1000703 10.1371/journal.ppat.1000703. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura A, Kishimoto T. 2010. IL-6: regulator of Treg/Th17 balance. Eur J Immunol 40:1830–1835 10.1002/eji.201040391. [PubMed] [DOI] [PubMed] [Google Scholar]
- 28.Smigiel KS, Srivastava S, Stolley JM, Campbell DJ. 2014. Regulatory T-cell homeostasis: steady-state maintenance and modulation during inflammation. Immunol Rev 259:40–59 10.1111/imr.12170. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lathrop SK, Bloom SM, Rao SM, Nutsch K, Lio CW, Santacruz N, Peterson DA, Stappenbeck TS, Hsieh CS. 2011. Peripheral education of the immune system by colonic commensal microbiota. Nature 478:250–254 10.1038/nature10434. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bustamante J, Boisson-Dupuis S, Jouanguy E, Picard C, Puel A, Abel L, Casanova JL. 2008. Novel primary immunodeficiencies revealed by the investigation of paediatric infectious diseases. Curr Opin Immunol 20:39–48 10.1016/j.coi.2007.10.005. [PubMed] [DOI] [PubMed] [Google Scholar]
- 31.Mogensen TH. 2013. STAT3 and the hyper-IgE syndrome: clinical presentation, genetic origin, pathogenesis, novel findings and remaining uncertainties. JAK-STAT 2:e23435 10.4161/jkst.23435. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heyworth PG, Cross AR, Curnutte JT. 2003. Chronic granulomatous disease. Curr Opin Immunol 15:578–584 10.1016/S0952-7915(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 33.Choi SM, McAleer JP, Zheng M, Pociask DA, Kaplan MH, Qin S, Reinhart TA, Kolls JK. 2013. Innate Stat3-mediated induction of the antimicrobial protein Reg3γ is required for host defense against MRSA pneumonia. J Exp Med 210:551–561 10.1084/jem.20120260. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crum-Cianflone N, Weekes J, Bavaro M. 2009. Recurrent community-associated methicillin-resistant Staphylococcus aureus infections among HIV-infected persons: incidence and risk factors. AIDS Patient Care STDS 23:499–502 10.1089/apc.2008.0240. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manfredi R, Calza L, Chiodo F. 2002. Epidemiology and microbiology of cellulitis and bacterial soft tissue infection during HIV disease: a 10-year survey. J Cutan Pathol 29:168–172 10.1034/j.1600-0560.2002.290307.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Ruiz-Irastorza G, Olivares N, Ruiz-Arruza I, Martinez-Berriotxoa A, Egurbide MV, Aguirre C. 2009. Predictors of major infections in systemic lupus erythematosus. Arthritis Res Ther 11:R109 10.1186/ar2764. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrera-Vargas A, Gómez-Martín D, Merayo-Chalico J, Ponce-de-León A, Alcocer-Varela J. 2014. Risk factors for drug-resistant bloodstream infections in patients with systemic lupus erythematosus. J Rheumatol 41:1311–1316 10.3899/jrheum.131261. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Espígol-Frigolé G, Corbera-Bellalta M, Planas-Rigol E, Lozano E, Segarra M, García-Martínez A, Prieto-González S, Hernández-Rodríguez J, Grau JM, Rahman MU, Cid MC. 2013. Increased IL-17A expression in temporal artery lesions is a predictor of sustained response to glucocorticoid treatment in patients with giant-cell arteritis. Ann Rheum Dis 72:1481–1487 10.1136/annrheumdis-2012-201836. [PubMed] [DOI] [PubMed] [Google Scholar]
- 39.Taylor AL, Llewelyn MJ, Rudak PT, Memarnejadian A, Szabo PA, Tun-Abraham ME, Rossjohn J, Corbett AJ, McCluskey J, McCormick JK, Lantz O, Hernandez-Alejandro R, Taylor AL, Llewelyn MJ. 2010. Superantigen-induced proliferation of human CD4+CD25- T cells is followed by a switch to a functional regulatory phenotype. J Immunol 185:6591–6598 10.4049/jimmunol.1002416. [PubMed] [DOI] [PubMed] [Google Scholar]
- 40.Ussher JE, Klenerman P, Willberg CB. 2014. Mucosal-associated invariant T-cells: new players in anti-bacterial immunity. Front Immunol 5:450 10.3389/fimmu.2014.00450. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao Y, Williams AP. 2015. Role of innate T cells in anti-bacterial immunity. Front Immunol 2:302. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Napier RJ, Adams EJ, Gold MC, Lewinsohn DM. 2015. The role of mucosal associated Invariant T cells in antimicrobial immunity. Front Immunol 6:344 10.3389/fimmu.2015.00344. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sandberg JK, Norrby-Teglund A, Leeansyah E. 2017. Bacterial deception of MAIT cells in a cloud of superantigen and cytokines. PLoS Biol 15:e2003167 10.1371/journal.pbio.2003167. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xiao X, Cai J. 2017. Mucosal-associated invariant T cells: new insights into antigen recognition and activation. Front Immunol 8:1540 10.3389/fimmu.2017.01540. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Reantragoon R, Boonpattanaporn N, Corbett AJ, McCluskey J. 2016. Mucosal-associated invariant T cells in clinical diseases. Asian Pac J Allergy Immunol 34:3–10. [PubMed] [Google Scholar]
- 46.Grimaldi D, Le Bourhis L, Sauneuf B, Dechartres A, Rousseau C, Ouaaz F, Milder M, Louis D, Chiche JD, Mira JP, Lantz O, Pène F. 2014. Specific MAIT cell behaviour among innate-like T lymphocytes in critically ill patients with severe infections. Intensive Care Med 40:192–201 10.1007/s00134-013-3163-x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 47.Shaler CR, Choi J, Rudak PT, Memarnejadian A, Szabo PA, Tun-Abraham ME, Rossjohn J, Corbett AJ, McCluskey J, McCormick JK, Lantz O, Hernandez-Alejandro R, Haeryfar SMM. 2017. MAIT cells launch a rapid, robust and distinct hyperinflammatory response to bacterial superantigens and quickly acquire an anergic phenotype that impedes their cognate antimicrobial function: defining a novel mechanism of superantigen-induced immunopathology and immunosuppression. PLoS Biol 15:e2001930 10.1371/journal.pbio.2001930. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balkhi MY, Ma Q, Ahmad S, Junghans RP. 2015. T cell exhaustion and interleukin 2 downregulation. Cytokine 71:339–347 10.1016/j.cyto.2014.11.024. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Dusseaux M, Martin E, Serriari N, Péguillet I, Premel V, Louis D, Milder M, Le Bourhis L, Soudais C, Treiner E, Lantz O. 2011. Human MAIT cells are xenobiotic-resistant, tissue-targeted, CD161hi IL-17-secreting T cells. Blood 117:1250–1259 10.1182/blood-2010-08-303339. [PubMed] [DOI] [PubMed] [Google Scholar]
- 50.Lussow AR, MacDonald HR. 1994. Differential effects of superantigen-induced “anergy” on priming and effector stages of a T cell-dependent antibody response. Eur J Immunol 24:445–449 10.1002/eji.1830240227. [PubMed] [DOI] [PubMed] [Google Scholar]
- 51.White J, Herman A, Pullen AM, Kubo R, Kappler JW, Marrack P. 1989. The V beta-specific superantigen staphylococcal enterotoxin B: stimulation of mature T cells and clonal deletion in neonatal mice. Cell 56:27–35 10.1016/0092-8674(89)90980-X. [DOI] [PubMed] [Google Scholar]
- 52.Ramesh N, Horner A, Ahern D, Geha RS. 1995. Bacterial superantigens induce the proliferation of resting gamma/delta receptor bearing T cells. Immunol Invest 24:713–724 10.3109/08820139509060700. [PubMed] [DOI] [PubMed] [Google Scholar]
- 53.Cho SN, Song CH, Jin J, Kim SH, Rha KS, Kim YM. 2014. Role of staphylococcal enterotoxin B on the differentiation of regulatory T cells in nasal polyposis. Am J Rhinol Allergy 28:e17–e24 10.2500/ajra.2014.28.3995. [PubMed] [DOI] [PubMed] [Google Scholar]
- 54.Gagliani N, Amezcua Vesely MC, Iseppon A, Brockmann L, Xu H, Palm NW, de Zoete MR, Licona-Limón P, Paiva RS, Ching T, Weaver C, Zi X, Pan X, Fan R, Garmire LX, Cotton MJ, Drier Y, Bernstein B, Geginat J, Stockinger B, Esplugues E, Huber S, Flavell RA. 2015. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 523:221–225 10.1038/nature14452. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ouyang W, Liao W, Luo CT, Yin N, Huse M, Kim MV, Peng M, Chan P, Ma Q, Mo Y, Meijer D, Zhao K, Rudensky AY, Atwal G, Zhang MQ, Li MO. 2012. Novel Foxo1-dependent transcriptional programs control T(reg) cell function. Nature 491:554–559 10.1038/nature11581. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pandiyan P, Zhu J. 2015. Origin and function of pro-inflammaory regulatory T cells. Cytokine 76:13–24 10.1016/j.cyto.2015.07.005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zeng WP, McFarland MM, Zhou B, Holtfreter S, Flesher S, Cheung A, Mallick A. 2017. Staphylococcal enterotoxin A-activated regulatory T cells promote allergen-specific TH2 response to intratracheal allergen inoculation. J Allergy Clin Immunol 139:508–518.e4 10.1016/j.jaci.2016.04.033. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Fritz SA, Tiemann KM, Hogan PG, Epplin EK, Rodriguez M, Al-Zubeidi DN, Bubeck Wardenburg J, Hunstad DA. 2013. A serologic correlate of protective immunity against community-onset Staphylococcus aureus infection. Clin Infect Dis 56:1554–1561 10.1093/cid/cit123. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG. 2013. In ammation and host response to injury, large scale collaborative research program. Genomic responses in mouse models poorly mimic human in ammatory diseases. Proc Natl Acad Sci U S A 110:3507–3512 10.1073/pnas.1222878110. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Spaulding AR, Lin YC, Merriman JA, Brosnahan AJ, Peterson ML, Schlievert PM. 2012. Immunity to Staphylococcus aureus secreted proteins protects rabbits from serious illnesses. Vaccine 30:5099–5109 10.1016/j.vaccine.2012.05.067. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rasigade JP, Sicot N, Laurent F, Lina G, Vandenesch F, Etienne J. 2011. A history of Panton-Valentine leukocidin (PVL)-associated infection protects against death in PVL-associated pneumonia. Vaccine 29:4185–4186 10.1016/j.vaccine.2011.04.033. [PubMed] [DOI] [PubMed] [Google Scholar]
- 62.Fowler VG, Allen KB, Moreira ED, Moustafa M, Isgro F, Boucher HW, Corey GR, Carmeli Y, Betts R, Hartzel JS, Chan ISF, McNeely TB, Kartsonis NA, Guris D, Onorato MT, Smugar SS, DiNubile MJ, Sobanjo-ter Meulen A. 2013. Effect of an investigational vaccine for preventing Staphylococcus aureus infections after cardiothoracic surgery. JAMA 309:1368–1378 10.1001/jama.2013.3010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 63.McNeely TB, Shah NA, Fridman A, Joshi A, Hartzel JS, Keshari RS, Lupu F, DiNubile MJ. 2014. Mortality among recipients of the Merck V710 Staphylococcus aureus vaccine after postoperative S. aureus infections: an analysis of possible contributing host factors. Hum Vaccin Immunother 10:3513–3516 10.4161/hv.34407. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Humrich JY, Riemekasten G. 2016. Restoring regulation: IL-2 therapy in systemic lupus erythematosus. Expert Rev Clin Immunol 12:1153–1160 10.1080/1744666X.2016.1199957. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Gupta DL, Bhoi S, Mohan T, Galwnkar S, Rao DN. 2016. Coexistence of Th1/Th2 and Th17/Treg imbalances in patients with post traumatic sepsis. Cytokine 88:214–221 10.1016/j.cyto.2016.09.010. [PubMed] [DOI] [PubMed] [Google Scholar]
- 66.Minejima E, Bensman J, She RC, Mack WJ, Tuan Tran M, Ny P, Lou M, Yamaki J, Nieberg P, Ho J, Wong-Beringer A. 2016. A dysregulated balance of proinflammatory and anti-inflammatory host cytokine response early during therapy predicts persistence and mortality in Staphylococcus aureus bacteremia. Crit Care Med 44:671–679. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.van Dissel JT, van Langevelde P, Westendorp RG, Kwappenberg K, Frölich M. 1998. Anti-inflammatory cytokine profile and mortality in febrile patients. Lancet 351:950–953 10.1016/S0140-6736(05)60606-X. [DOI] [PubMed] [Google Scholar]
- 68.Rose WE, Eickhoff JC, Shukla SK, Pantrangi M, Rooijakkers S, Cosgrove SE, Nizet V, Sakoulas G. 2012. Elevated serum interleukin-10 at time of hospital admission is predictive of mortality in patients with Staphylococcus aureus bacteremia. J Infect Dis 206:1604–1611 10.1093/infdis/jis552. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rose WE, Shukla SK, Berti AD, Hayney MS, Henriquez KM, Ranzoni A, Cooper MA, Proctor RA, Nizet V, Sakoulas G. 2017. Increased endovascular Staphylococcus aureus inoculum is the link between elevated serum interleukin 10 concentrations and mortality in patients with bacteremia. Clin Infect Dis 64:1406–1412 10.1093/cid/cix157. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zielinski CE, Mele F, Aschenbrenner D, Jarrossay D, Ronchi F, Gattorno M, Monticelli S, Lanzavecchia A, Sallusto F. 2012. Pathogen-induced human TH17 cells produce IFN-γ or IL-10 and are regulated by IL-1β. Nature 484:514–518 10.1038/nature10957. [PubMed] [DOI] [PubMed] [Google Scholar]
- 71.Crispín JC, Tsokos GC. 2009. Transcriptional regulation of IL-2 in health and autoimmunity. Autoimmun Rev 8:190–195 10.1016/j.autrev.2008.07.042. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Boyman O, Kolios AG, Raeber ME. 2015. Modulation of T cell responses by IL-2 and IL-2 complexes. Clin Exp Rheumatol 33(Suppl 92):S54–S57. [PubMed] [Google Scholar]
- 73.Chapman NM, Bilal MY, Cruz-Orcutt N, Knudson C, Madinaveitia S, Light J, Houtman JC. 2013. Distinct signaling pathways regulate TLR2 co-stimulatory function in human T cells. Cell Signal 25:639–650 10.1016/j.cellsig.2012.11.026. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chau TA, McCully ML, Brintnell W, An G, Kasper KJ, Vinés ED, Kubes P, Haeryfar SM, McCormick JK, Cairns E, Heinrichs DE, Madrenas J. 2009. Toll-like receptor 2 ligands on the staphylococcal cell wall downregulate superantigen-induced T cell activation and prevent toxic shock syndrome. Nat Med 15:641–648 10.1038/nm.1965. [PubMed] [DOI] [PubMed] [Google Scholar]
- 75.Li Z, Levast B, Madrenas J. 2017. Staphylococcus aureus downregulates IP-10 production and prevents Th1 cell recruitment. J Immunol 198:1865–1874 10.4049/jimmunol.1601336. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Li Z, Peres AG, Damian AC, Madrenas J. 2015. Immunomodulation and disease tolerance to Staphylococcus aureus. Pathogens 4:793–815 10.3390/pathogens4040793. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Andrade EB, Alves J, Madureira P, Oliveira L, Ribeiro A, Cordeiro-da-Silva A, Correia-Neves M, Trieu-Cuot P, Ferreira P. 2013. TLR2-induced IL-10 production impairs neutrophil recruitment to infected tissues during neonatal bacterial sepsis. J Immunol 191:4759–4768 10.4049/jimmunol.1301752. [PubMed] [DOI] [PubMed] [Google Scholar]
- 78.Leech JM, Lacey KA, Mulcahy ME, Medina E, McLoughlin RM. 2017. IL-10 plays opposing roles during Staphylococcus aureus systemic and localized infections. J Immunol 198:2352–2365 10.4049/jimmunol.1601018. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Martire-Greco D, Rodriguez-Rodrigues N, Landoni VI, Rearte B, Isturiz MA, Fernández GC. 2013. Interleukin-10 controls human peripheral PMN activation triggered by lipopolysaccharide. Cytokine 62:426–432 10.1016/j.cyto.2013.03.025. [PubMed] [DOI] [PubMed] [Google Scholar]
- 80.Shibata Y, Foster LA, Kurimoto M, Okamura H, Nakamura RM, Kawajiri K, Justice JP, Van Scott MR, Myrvik QN, Metzger WJ. 1998. Immunoregulatory roles of IL-10 in innate immunity: IL-10 inhibits macrophage production of IFN-gamma-inducing factors but enhances NK cell production of IFN-gamma. J Immunol 161:4283–4288. [PubMed] [Google Scholar]
- 81.Wang JE, Jørgensen PF, Almlöf M, Thiemermann C, Foster SJ, Aasen AO, Solberg R. 2000. Peptidoglycan and lipoteichoic acid from Staphylococcus aureus induce tumor necrosis factor alpha, interleukin 6 (IL-6), and IL-10 production in both T cells and monocytes in a human whole blood model. Infect Immun 68:3965–3970 10.1128/IAI.68.7.3965-3970.2000. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Brown AF, Leech JM, Rogers TR, McLoughlin RM. 2014. Staphylococcus aureus colonization: modulation of host immune response and impact on human vaccine design. Front Immunol 4:507 10.3389/fimmu.2013.00507. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brown AF, Murphy AG, Lalor SJ, Leech JM, O’Keeffe KM, Mac Aogáin M, O’Halloran DP, Lacey KA, Tavakol M, Hearnden CH, Fitzgerald-Hughes D, Humphreys H, Fennell JP, van Wamel WJ, Foster TJ, Geoghegan JA, Lavelle EC, Rogers TR, McLoughlin RM. 2015. Memory Th1 cells are protective in invasive Staphylococcus aureus infection. PLoS Pathog 11:e1005226 10.1371/journal.ppat.1005226. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Söderquist B, Sundqvist KG, Vikerfors T. 1992. Kinetics of serum levels of interleukin-6 in Staphylococcus aureus septicemia. Scand J Infect Dis 24:607–612 10.3109/00365549209054646. [PubMed] [DOI] [PubMed] [Google Scholar]
- 85.McNicholas S, Talento AF, O’Gorman J, Hannan MM, Lynch M, Greene CM, Humphreys H, Fitzgerald-Hughes D. 2014. Cytokine responses to Staphylococcus aureus bloodstream infection differ between patient cohorts that have different clinical courses of infection. BMC Infect Dis 14:580 10.1186/s12879-014-0580-6. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chantratita N, Tandhavanant S, Seal S, Wikraiphat C, Wongsuvan G, Ariyaprasert P, Suntornsut P, Teerawattanasook N, Jutrakul Y, Srisurat N, Chaimanee P, Mahavanakul W, Srisamang P, Phiphitaporn S, Mokchai M, Anukunananchai J, Wongratanacheewin S, Chetchotisakd P, Emond MJ, Peacock SJ, West TE. 2017. TLR4 genetic variation is associated with inflammatory responses in Gram-positive sepsis. Clin Microbiol Infect 23:47.e1–47.e10 10.1016/j.cmi.2016.08.028. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jin JO, Zhang W, Du JY, Yu Q. 2014. BDCA1-positive dendritic cells (DCs) represent a unique human myeloid DC subset that induces innate and adaptive immune responses to Staphylococcus aureus infection. Infect Immun 82:4466–4476 10.1128/IAI.01851-14. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Karauzum H, Haudenschild CC, Moore IN, Mahmoudieh M, Barber DL, Datta SK. 2017. Lethal CD4 T cell responses induced by vaccination against Staphylococcus aureus bacteremia. J Infect Dis 215:1231–1239 10.1093/infdis/jix096. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koymans KJ, Feitsma LJ, Brondijk TH, Aerts PC, Lukkien E, Lössl P, van Kessel KP, de Haas CJ, van Strijp JA, Huizinga EG. 2015. Structural basis for inhibition of TLR2 by staphylococcal superantigen-like protein 3 (SSL3). Proc Natl Acad Sci U S A 112:11018–11023 10.1073/pnas.1502026112. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Koymans KJ, Vrieling M, Gorham RD Jr, van Strijp JAG. 2017. Staphylococcal immune evasion proteins: structure, function, and host adaptation. Curr Top Microbiol Immunol 409:441–489 10.1007/82_2015_5017. [PubMed] [DOI] [PubMed] [Google Scholar]
- 91.Nagelkerken L, Gollob KJ, Tielemans M, Coffman RL. 1993. Role of transforming growth factor-beta in the preferential induction of T helper cells of type 1 by staphylococcal enterotoxin B. Eur J Immunol 23:2306–2310 10.1002/eji.1830230938. [PubMed] [DOI] [PubMed] [Google Scholar]
- 92.Niedergang F, Hémar A, Hewitt CR, Owen MJ, Dautry-Varsat A, Alcover A. 1995. The Staphylococcus aureus enterotoxin B superantigen induces specific T cell receptor down-regulation by increasing its internalization. J Biol Chem 270:12839–12845 10.1074/jbc.270.21.12839. [PubMed] [DOI] [PubMed] [Google Scholar]
- 93.Stach CS, Herrera A, Schlievert PM. 2014. Staphylococcal superantigens interact with multiple host receptors to cause serious diseases. Immunol Res 59:177–181 10.1007/s12026-014-8539-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Szabo PA, Goswami A, Mazzuca DM, Kim K, O’Gorman DB, Hess DA, Welch ID, Young HA, Singh B, McCormick JK, Haeryfar SM. 2017. Rapid and rigorous IL-17A production by a distinct subpopulation of effector memory T lymphocytes constitutes a novel mechanism of toxic shock syndrome immunopathology. J Immunol 198:2805–2818 10.4049/jimmunol.1601366. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Takahashi N, Hasegawa H, Komiyama M, Ohki T, Yada Y, Koike Y, Kawamata R, Honma Y, Momoi M. 2009. Selective excretion of anti-inflammatory cytokine interleukin-10 in a superantigen-inducing neonatal infectious disease. Cytokine 45:39–43 10.1016/j.cyto.2008.10.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 96.Tuffs SW, James DBA, Bestebroer J, Richards AC, Goncheva MI, O’Shea M, Wee BA, Seo KS, Schlievert PM, Lengeling A, van Strijp JA, Torres VJ, Fitzgerald JR. 2017. The Staphylococcus aureus superantigen SElX is a bifunctional toxin that inhibits neutrophil function. PLoS Pathog 13:e1006461 10.1371/journal.ppat.1006461. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stolz SJ, Davis JP, Vergeront JM, Crass BA, Chesney PJ, Wand PJ, Bergdoll MS. 1985. Development of serum antibody to toxic shock toxin among individuals with toxic shock syndrome in Wisconsin. J Infect Dis 151:883–889 10.1093/infdis/151.5.883. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Iwakura Y, Ishigame H, Saijo S, Nakae S. 2011. Functional specialization of interleukin-17 family members. Immunity 34:149–162 10.1016/j.immuni.2011.02.012. [PubMed] [DOI] [PubMed] [Google Scholar]
- 99.Iwasaka H, Noguchi T. 2004. Th1/Th2 balance in systemic inflammatory response syndrome (SIRS). Nihon Rinsho 62:2237–2243. (In Japanese.) [PubMed] [Google Scholar]
- 100.Lacey KA, Geoghegan JA, McLoughlin RM. 2016. The role of Staphylococcus aureus virulence factors in skin infection and their potential as vaccine antigens. Pathogens 17:E22. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Frodermann V, Chau TA, Sayedyahossein S, Toth JM, Heinrichs DE, Madrenas J. 2011. A modulatory interleukin-10 response to staphylococcal peptidoglycan prevents Th1/Th17 adaptive immunity to Staphylococcus aureus. J Infect Dis 204:253–262 10.1093/infdis/jir276. [PubMed] [DOI] [PubMed] [Google Scholar]
- 102.Donabedian H, Gallin JI. 1983. The hyperimmunoglobulin E recurrent-infection (Job’s) syndrome. A review of the NIH experience and the literature. Medicine (Baltimore) 62:195–208 10.1097/00005792-198307000-00001. [PubMed] [DOI] [PubMed] [Google Scholar]