

SUMMARY
Lactic acid has emerged as an important modulator of immune cell function. It can be produced by both gut microbiota and the host metabolism at homeostasis and during disease states. The production of lactic acid in the gut microenvironment is vital for tissue homeostasis. In the present study, we examined how lactic acid integrates cellular metabolism to shape the epigenome of macrophages during pro-inflammatory response. We found that lactic acid serves as a primary fuel source to promote histone H3K27 acetylation, which allows the expression of immunosuppressive gene program including Nr4a1. Consequently, macrophage pro-inflammatory function was transcriptionally repressed. Furthermore, the histone acetylation induced by lactic acid promotes a form of long-term immunosuppression (“trained immunosuppression”). Pre-exposure to lactic acid induces lipopolysaccharide tolerance. These findings thus indicate that lactic acid sensing and its effect on chromatin remodeling in macrophages represent a key homeostatic mechanism that can provide a tolerogenic tissue microenvironment.
Graphical Abstract
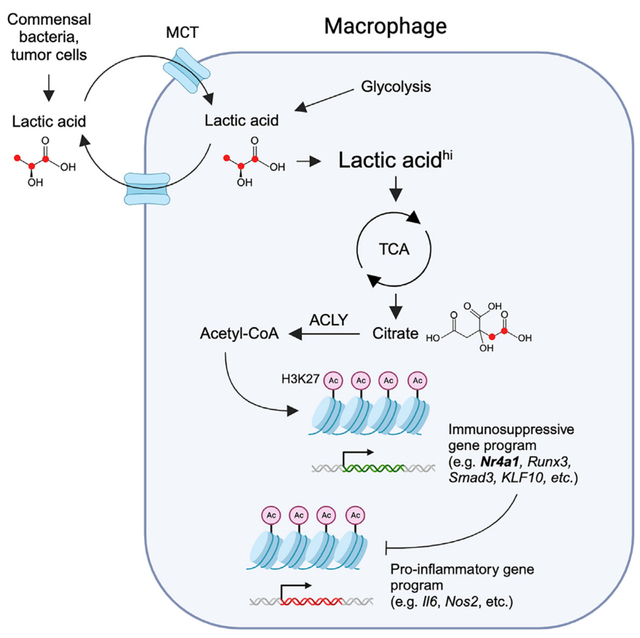
In brief
Lactic acid is emerging as a prominent immunosuppressive metabolite. Shi et al. reveal that lactic acid acts as a primary fuel source for the TCA cycle to promote H3K27 acetylation. This histone modification allows the expression of an immunosuppressive gene program including Nr4a1 that consequently inhibits macrophage pro-inflammatory response.
INTRODUCTION
Emerging evidence indicates that tissue microenvironments influence the functions of immune cells including tissue-resident macrophages.1 This interaction of immune cells with microenvironments is particularly essential in immune-privileged tissues where innate immune response is restricted in order to maintain immune tolerance and tissue homeostasis. Many of these tissues, including the gastrointestinal tract and placenta but also the tumor microenvironment, share overlapping metabolic characteristics and are enriched in lactic acid.2–4 Understanding the mechanisms of these overlapping immunosuppressive metabolic conditions has promise for improving anti-inflammatory treatments for chronic diseases such as inflammatory bowel disease (IBD). Lactic acid is an essential component of carbon metabolism and can be produced by lactic acid bacteria (i.e., Lactobacillus and Lactococcus species) and other commensal bacteria during the fermentation of carbohydrate and the host metabolism at homeostasis and during disease conditions, such as infection and cancer. Consequently, immune cells infiltrating or residing at different tissues are exposed to variable lactic acid levels. For example, at steady state, the level of lactic acid produced by commensal bacteria can be as high as 10 mM in the terminal ileum or colon,3,5 or even higher in the vagina,6 and it can reach up to 40 mM in the tumor microenvironment.4,7 Lactic acid has long been considered as a waste product of metabolism; however, it is becoming increasingly recognized as an essential metabolic signal responsible for regulating the effector functions of various immune cells, including macrophages and T cells.8,9
Pro-inflammatory response is generally triggered by innate immune receptors such as Toll-like receptors (TLRs) and essential for host defense against pathogens during infection and maintaining tissue homeostasis under noxious conditions.10 This innate immune response is mediated mainly by macrophages, which leads to the production of pro-inflammatory cytokines and chemokines. It has become clear that derangement of the processes involved in resolution or suppression of inflammation is an underlying feature of chronic inflammatory diseases such as type 2 diabetes, atherosclerosis, and IBD. In addition to pro-inflammatory function (i.e., M1-like macrophage), macrophages can also be polarized by interleukin (IL)-4 into M2 macrophages, which perform distinct functions, including anti-inflammatory response, tissue repair, and pro-tumor growth, by expressing M2-specific gene program (i.e., M2 genes).11 Recent studies on cellular metabolism by us and others have shown profound alterations in metabolic profiles and metabolite flux during macrophage activation.12–14 For example, M1 macrophages stimulated by TLR4 agonist lipopolysaccharide (LPS) undergo glycolytic switch by enhancing glucose influx while M2 macrophages stimulated by IL-4 increase the utilization of glucose, fatty acids, and glutamine.15–17 Shifts in metabolic state can orchestrate macrophage functions through its impact on epigenetic reprogramming, the reversible alterations of chromatin such as histone modifications that regulate access to the DNA and thus gene transcription. Tissue microenvironments therefore can exert their influence on macrophage through the input or generation of chromatin-modifying metabolites.18
Lactic acid sensing by macrophages modulates macrophage functional polarization. Much has been focused on the lactic acid effect on tumor-associated macrophages (TAMs) as lactic acid itself is sufficient to drives M2 gene expression or acts together with IL-4 to potentiate M2 macrophage polarization to promote tumor growth.8,19,20 However, how lactic acid controls macrophage pro-inflammatory response (i.e., M1 macrophages polarization) and maintain immune tolerance is largely unknown. Here, we report that, during LPS stimulation, lactic acid acts as a primary fuel source for tricarboxylic acid (TCA) cycle metabolism to promote epigenetic reprogramming via histone H3K27 acetylation, which allows the expression of lactic acid-specific immunosuppressive gene program including Nr4a1 that, in turn, inhibits macrophage pro-inflammatory response and promotes a long-term form of immunosuppression.
RESULTS
Lactic acid inhibits pro-inflammatory response but has no effect on the canonical pro-inflammatory signaling pathways in macrophages after LPS stimulation
Although lactic acid has been known to inhibit inflammatory response,21,22 mechanisms underlying the inhibitory effect remain poorly understood. To define this lactic acid effect, we used the activation by LPS of bone-marrow-derived macrophages (BMDMs) (Figure S1A) and found that lactic acid at concentrations as low as 10 mM significantly inhibited pro-inflammatory cytokine expression (Figure 1A) and production (Figures S1B–S1F). We then asked whether lactic acid inhibits LPS signaling (i.e., TLR4 signaling). However, lactic acid, even at higher concentrations (i.e., >10 mM), had no effect on the activation of nuclear factor κB (NF-κB) and p38 mitogen-activated protein (MAP) kinase pathways in macrophages as well as 293T cells overexpressing NF-κB reporter (Figures 1B and 1C). A kinetic experiment also showed that the inhibitory effect of lactic acid was found to occur as early as 1 h on the pro-inflammatory cytokine response, but not the LPS signaling (Figures 1D and 1E). These data suggest that the anti-inflammatory effect of lactic acid is not mediated via the inhibition of canonical LPS signaling.
Figure 1. Lactic acid inhibits pro-inflammatory response but has no effect on the canonical pro-inflammatory signaling pathways in macrophages after LPS stimulation.
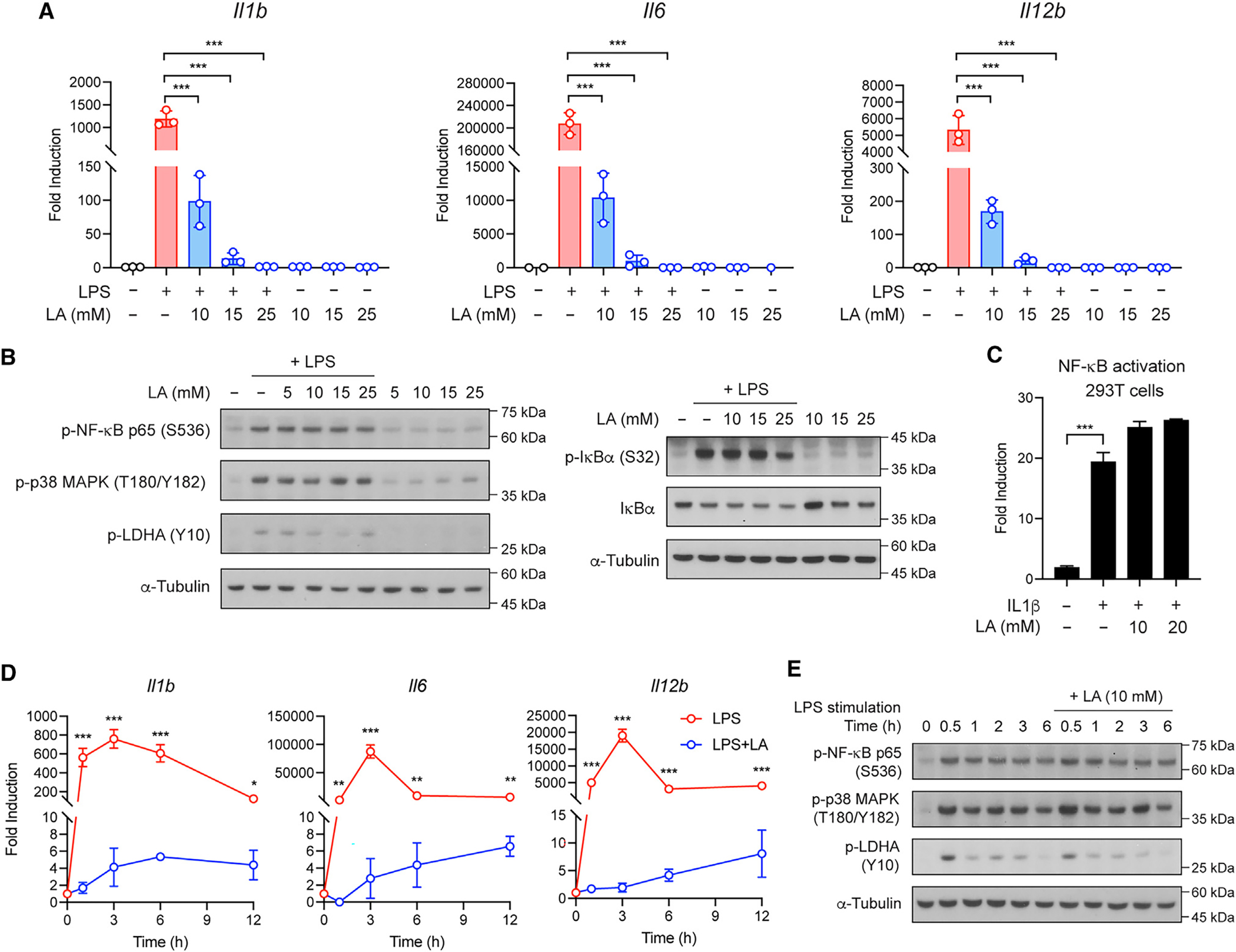
(A) Pro-inflammatory gene expression in BMDMs stimulated with LPS in the presence or absence of lactic acid (LA) at the indicated concentrations for 3 h.
(B) Activation of NF-κB and p38 MAP kinase pathways in BMDMs stimulated as in (A).
(C) NF-κB reporter assay in 293T cells stimulated with IL-1β in the presence or absence of LA at the indicated concentrations for 3 h.
(D) Pro-inflammatory gene expression in BMDMs stimulated with LPS in the presence or absence of LA at 10 mM for the indicated times.
(E) Activation of NF-κB and p38 MAP kinase pathways in BMDMs stimulated as in (D). Data are representative of at least three independent experiments. Data in (A), (C), and (D) are mean ± SD of triplicates. One-way ANOVA (A and C) or two-way ANOVA (D) followed by Tukey’s or Sidak’s post-test: **p < 0.01, ***p < 0.001.
Several G protein-coupled receptors (GPCRs), including GPR65 and GPR132, have been identified as extracellular pH sensors in acidic tumor microenvironment due to lactic acid secretion.23–25 To test whether GPCR signaling from the cell surface mediates the anti-inflammatory effect of lactic acid, we employed the adenylyl cyclase inhibitor MDL-12,330A hydrochloride (MDL-12). The inhibition of cyclic adenosine monophosphate (cAMP) de novo production by MDL-12, however, had no effect on lactic acid-suppressed pro-inflammatory response (Figure S2A). We therefore turned our focus to lactic acid transport across the plasma membrane. Our metabolomics analysis revealed that lactic acid together with LPS, but not lactic acid alone, significantly increased intracellular lactic acid level in BMDMs (Figure S2B), suggesting that LPS enhances lactic acid uptake, and intracellular lactic acid accumulation can act to suppress macrophage pro-inflammatory response. Consistent with this idea, BMDMs treated with 2-cyano-3-(4-hydroxyphenyl)-2-propenoic acid (CHC), an inhibitor of monocarboxylate transporter (MCT)-1 and MCT-4, which are known to transport lactic acid bidirectionally, resulted in a blockade of lactic acid secretion, accumulation of intracellular lactic acid, and, importantly, an inhibition of pro-inflammatory cytokine response during LPS stimulation (Figures S2C and S2D). Together, these observations suggest that lactic acid uptake and/or intracellular accumulation is essential for lactic acid immunosuppressive effect.
Lactic acid inhibits macrophage pro-inflammatory response via gene regulation
We then hypothesized that, after being taken up and accumulated intracellularly, lactic acid acts to regulate LPS-induced transcriptional response, leading to the inhibition of macrophage pro-inflammatory gene expression. To test this, we performed RNA sequencing (RNA-seq) analysis of BMDMs and examined the effect of lactic acid on macrophage gene expression after 3-h LPS stimulation. Principal-component analysis (PCA) of our RNA-seq data revealed distinct differences in transcriptomes of BMDMs stimulated with LPS in the presence or absence of lactic acid, or in an equivalent low-pH condition (i.e., pH 6.5) adjusted by hydrochloric acid (HCl) (Figure 2A), indicating an acid-independent effect of lactic acid. We focused on the 2,543 differentially expressed genes (DEGs) (LPS + LA vs. LPS, false discovery rate [FDR] <0.05, >2-fold differences in expression) (Figure 2B). Gene Ontology (GO) analysis revealed that DEGs upor downregulated by lactic acid are associated with distinct biological processes (Figure 2C). Consistent with the immunosuppressive effect of lactic acid, DEGs downregulated by lactic acid (1,358 genes) were involved in “immune system process” (GO: 0002376), “innate immune responses” (GO: 0045087), and “inflammatory processes” (GO: 0006954). In contrast, DEGs upregulated by lactic acid (1,185 genes) were associated with maintenance gene programs including the “cell cycle” (GO: 0007049), “regulation of DNA-templated transcription” (GO: 0006355), “negative regulation of gene transcription” (GO: 0000122 and GO: 0045892), and DNA damage/repair response (GO: 0006974 and GO: 0006281). In relation to macrophage pro-inflammatory polarization, lactic acid significantly suppressed pro-inflammatory gene (Il6, Il1b, Nos2, Ptgs2, etc.) and M1-related gene (Cd80, Cd86, Cd38, etc.) expression (Figure 2D). These RNA-seq results suggest that lactic acid broadly regulates macrophage transcriptional response to LPS and might induce gene programs that negatively regulate pro-inflammatory response.
Figure 2. LA inhibits macrophage pro-inflammatory response via gene regulation.
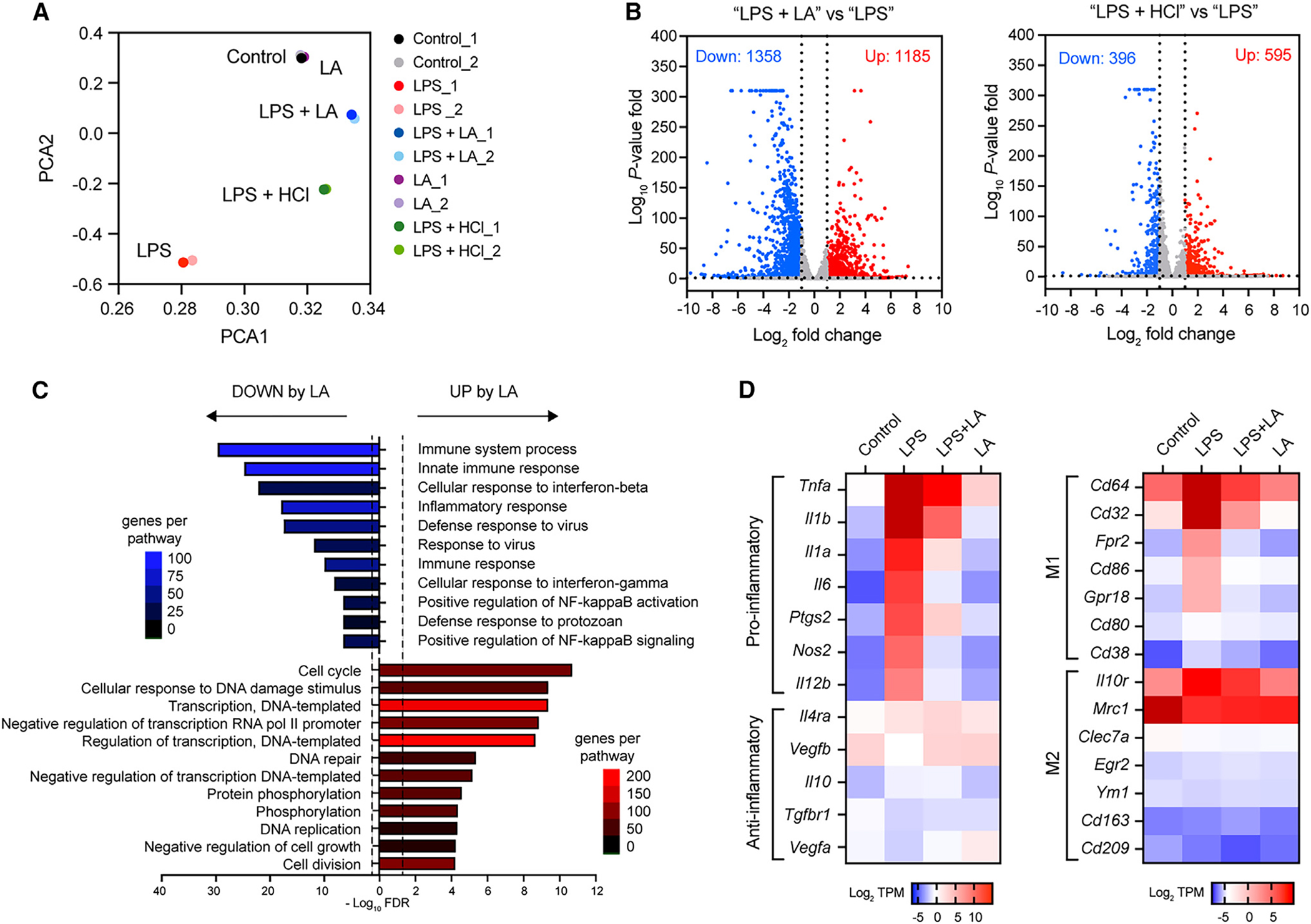
(A) PCA plot of RNA-seq data from BMDMs stimulated without (control) or with LPS in the presence or absence of LA at 10 mM or in low-pH condition with HCl for 3 h.
(B) Volcano plot of RNA-seq data showing cutoffs (log2 fold change ≥ or ≤ 1 and FDR ≤ 0.05) used to identify DEGs in BMDMs stimulated with LPS in the presence or absence of LA (LPS + LA vs. LPS) or LPS in low or normal pH condition (LPS + HCl vs. LPS).
(C) GO analysis of DEGs identified in (B) showing biological processes enriched in DEGs upregulated (red) or downregulated (blue) by LA in BMDMs stimulated with LPS.
(D) Heatmaps showing mRNA expression of selected gene subset between BMDMs stimulated as in (A). Colors on the heatmap represent scaled log2 of transcript count per million (TPM) from low (blue) to high (red) expression.
Previously studies have shown that lactic acid promotes M2-like macrophage polarization via the induction of M2-associated gene transcription (e.g., Arg1, Retnla, and Vegf).8,19,20 We next tested whether the M2-like polarization by lactic acid led to the inhibition of pro-inflammatory response. Our RNA-seq analysis of BMDMs stimulated with LPS for 3 h showed no significant induction of M2 genes by lactic acid (Figure 2D). A kinetic experiment by qPCR also revealed that the induction of M2 genes Arg1 and Retnla by lactic acid was a later event (>3–6 h) (Figure S2E) than that for the inhibition of pro-inflammatory genes by lactic acid (i.e., as early as 1 h) (Figure 1D), indicating that the transcriptional repression of pro-inflammatory genes by lactic acid occurs prior to the induction of M2 gene expression. Moreover, neither deletion of Retnla, among the earliest M2 gene induction by lactic acid, nor treatment of recombinant RELM-a (encoded by Retnla) in BMDMs had effect on LPS-induced pro-inflammatory gene expression (Figures S2F and S2G). These observations suggest that the inhibition of pro-inflammatory response by lactic acid is uncoupled from the M2 gene expression. Furthermore, our RNA-seq data showed that only less than 5% of DEGs upregulated by lactic acid (1,185 genes) (Figure 2B) were overlapping with genes induced by IL-4 (GEO: GSE181223),26 indicating that, rather than an M2-like phenotype, lactic acid polarizes macrophages into an immunosuppressive phenotype with a distinct gene expression profile.
Lactic acid modules cellular metabolism and promotes the production of citrate in TCA cycle
Recent studies by us and others have highlighted the impact of cellular metabolism on macrophage proand anti-inflammatory functions.12–14 A rapid switch from oxidative phosphorylation (OXPHOS) to glycolysis is the hallmark metabolic changes in macrophages following activation with LPS. We analyzed BMDMs for changes in the rate of extracellular acidification (ECAR) and the mitochondrial rate of oxygen consumption (OCR) as a measure of glycolysis and OXPHOS, respectively, after LPS stimulation. BMDMs stimulated with LPS in the presence of lactic acid became less glycolytic (i.e., lower basal ECAR) but more oxidative (i.e., higher basal OCR) as compared to cells stimulated with LPS alone (Figure 3A), suggesting that lactic acid opposes the glycolytic switch and promotes mitochondrial respiration and fitness. Consistent with this idea, the presence of lactic acid during LPS stimulation in BMDMs resulted in a higher maximal respiratory capacity (MRC) compared with the absence of lactic acid (Figures 3B and 3C). We then examined whether the inhibition of glycolysis mediates the anti-inflammatory effect of lactic acid. We used 2-deoxy-D-glucose (2-DG), an inhibitor of glycolysis (Figure S3A), or glucose-free medium to block glycolysis in BMDMs and found that, in the lack of glycolysis, lactic acid remained potent in suppressing pro-inflammatory gene expression (Figure S3B), suggesting that the inhibition of glycolysis is not required for the lactic acid effect.
Figure 3. LA modulates cellular metabolism and promotes the production of citrate in TCA cycle.
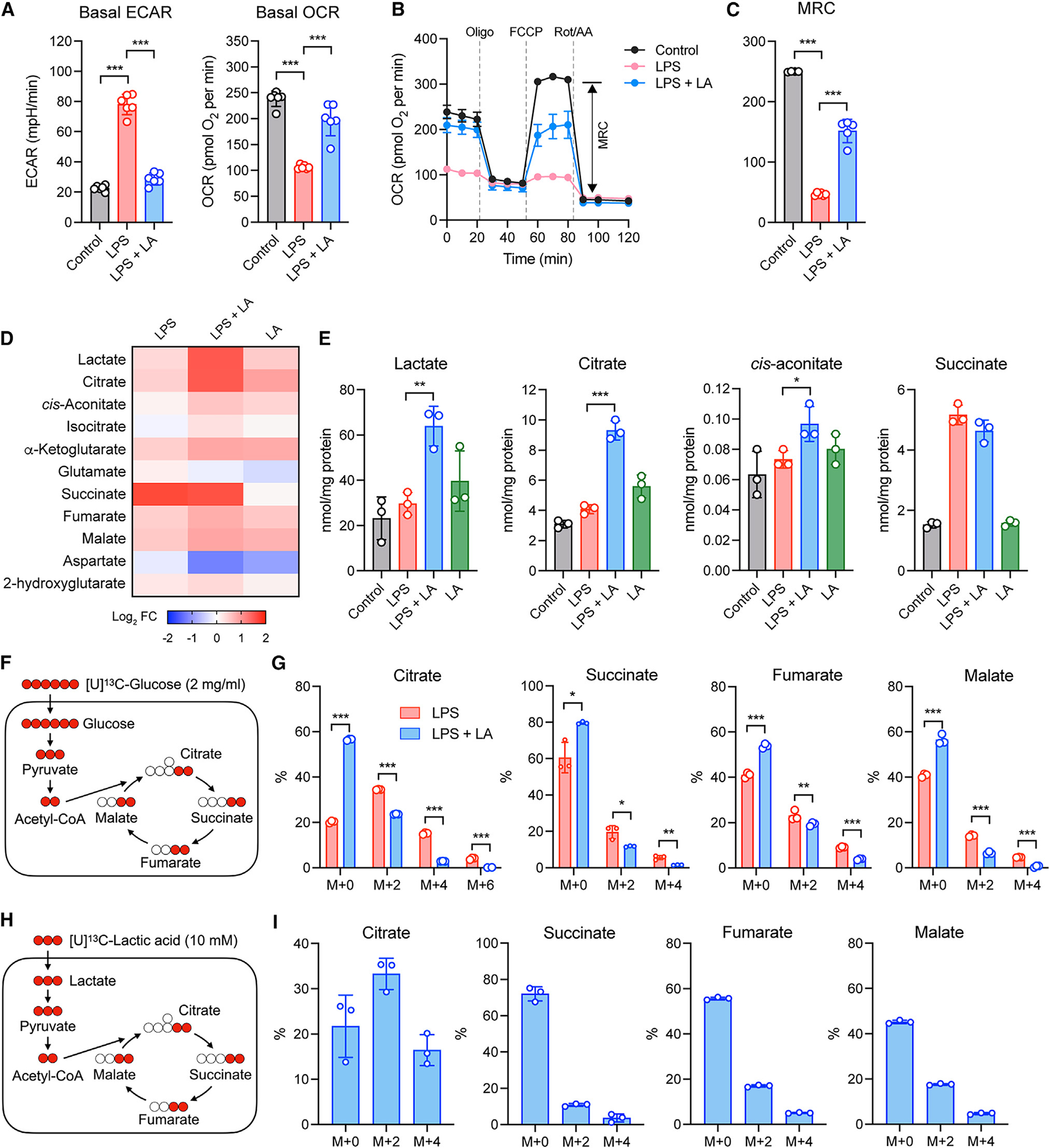
(A) Basal ECAR and OCR in BMDMs stimulated without (control) or with LPS in the presence or absence of LA at the indicated concentrations or 10 mM for 6 h.
(B) Real-time changes in the OCR of BMDMs stimulated as in (A) after treatment with oligomycin (Oligo), FCCP, and rotenone (Rot)/antimycin A (AA). MRC, maximal respiratory capacity (double-headed arrow; shown for the control).
(C) MRC of BMDMs measured by real-time changes in OCR as shown in (B).
(D) Heatmap showing TCA cycle metabolites in fold change, measured by targeted metabolomics in BMDMs stimulated as in (A).
(E) Shown are representative metabolites from (D).
(F and G) Isotopic flux analysis of [U]13C-glucose, illustrated in (F), in targeted TCA cycle metabolites (G) in BMDMs cultured in medium containing 2 mg/mL [U]13C-glucose for 3 h prior to LPS stimulation in the presence or absence of LA for 3 h.
(H and I) Isotopic flux analysis of [U]13C-LA, illustrated in (H), in targeted TCA cycle metabolites (I) in BMDMs cultured in medium containing 10 mM [U]13C-LA alone for 3 h. Data are mean ± SD of five replicates (A–C) or triplicates (E, G, I). Shown in (G) and (I) is the fractional enrichment of the indicated isotopologs. One-way ANOVA (A, C, and E) or two-way ANOVA (G) followed by Tukey’s or Sidak’s post-test: *p < 0.05, **p < 0.001, ***p ≤ 0.001.
To further gain insight into the mitochondrial metabolism promoted by lactic acid, we performed TCA cycle-targeted metabolomics analysis. Among the 11 targeted metabolites, five TCA cycle intermediates (citrate, cis-aconitate, isocitrate, α-ketoglutarate, malate) were affected by lactic acid (Figure 3D). Specifically, lactic acid significantly increased the production of citrate and cis-aconitate (Figure 3E). This enhanced TCA cycle metabolism could be due to two possibilities: lactic acid promotes metabolites derived from glucose metabolism (e.g., pyruvate) to enter TCA cycle and/or lactic acid serves as a primary fuel source for TCA cycle metabolism. To test the first possibility, we performed mass spectrometry of BMDMs pulsed with [U]13C-glucose during LPS stimulation in the presence or absence of lactic acid (Figure 3F). We observed that lactic acid significantly reduced the conversion of 13C-glucose into targeted TCA cycle intermediates (i.e., M + 2, M + 4, and M + 6), and a significant amount of the individual intermediates, including the fraction enhanced by lactic acid, were derived from a non-13C-glucose source (i.e., M + 0) (Figure 3G), suggesting that lactic acid inhibits the contribution of glucose metabolism to TCA cycle. We next tested the second possibility and pulsed BMDMs with [U]13C-lactic acid during LPS stimulation (Figure 3H). BMDMs significantly converted 13C-lactic acid into more than 50% of citrate (i.e., M + 2 and M + 4) but not into the other targeted intermediates (i.e., succinate, fumarate, malate) (Figure 3I). Together, these results suggest that lactic acid serves as a key fuel source for TCA cycle metabolism, especially for citrate production.
Lactic acid regulates histone H3K27 acetylation to inhibit pro-inflammatory response in macrophages
Lactic acid-derived citrate can result in enhanced cis-aconitate production in the TCA cycle as we observed (Figure 3E). Since cis-aconitate generates itaconate via aconitate decarboxylase (encoded by Acod1), and itaconate has been shown to play a role in limiting inflammatory response by activating nuclear factor erythroid 2-related factor 2 (Nrf2) and inhibiting succinate dehydrogenase,27,28 we next tested whether the cis-aconitate-itaconate axis mediates the anti-inflammatory effect of lactic acid. In line with the previous studies, deletion of Acod1 in BMDMs resulted in significantly increased pro-inflammatory response to LPS (Figure S3C). However, the anti-inflammatory effect of lactic acid remained largely intact in Acod1−/− cells (Figure S3C).
Another key aspect of citrate is to undergo mitochondrial export and subsequent cleavage by ATP-citrate lyase (ACLY) to generate cytosolic acetyl-coenzyme A (acetyl-CoA) for histone acetylation in the nucleus.29 To investigate whether lactic acid regulates histone acetylation, we measured global histone acetylation in various lysine residues of histone H3 in BMDMs. Neither LPS nor lactic acid had any effect on acetylated H3K18, H3K14, or H3K9 (Figure 4A). However, LPS significantly decreased global H3K27 acetylation, and this reduction was restored to homeostatic levels by lactic acid in a dose-dependent manner (Figures 4A and 4B), but not by the low-pH condition with HCl (Figure 4C). Trichostatin A (TSA) is a well-characterized histone deacetylase (HDAC) inhibitor and known to exert anti-inflammatory effects on macrophages.30–33 We then compared the effect between lactic acid and TSA in BMDMs after LPS stimulation. Both lactic acid and TSA restored and promoted global H3K27 acetylation (Figure 4D), and they significantly inhibited pro-inflammatory gene expression (Figure 4E), suggesting that, similar to HDAC inhibitors, lactic acid acts to promote global H3K27 acetylation and suppress macrophage pro-inflammatory response. These data are consistent with a previous study where lactic acid alone (without LPS stimulation) can promote H3K27 acetylation.20 We next treated BMDMs with BMS (BMS-303141), an inhibitor of ACLY, to block acetyl-CoA production from citrate. BMDMs treated with BMS exhibited impaired H3K27 acetylation, as expected, and a partial but significant loss of lactic acid anti-inflammatory effect (Figures 4F and 4G). Together, these data suggest that lactic acid promotes H3K27 acetylation via acetyl-CoA production, resulting in transcriptional silencing of pro-inflammatory genes in macrophages.
Figure 4. LA regulates histone H3K27 acetylation to inhibit pro-inflammatory response in macrophages.
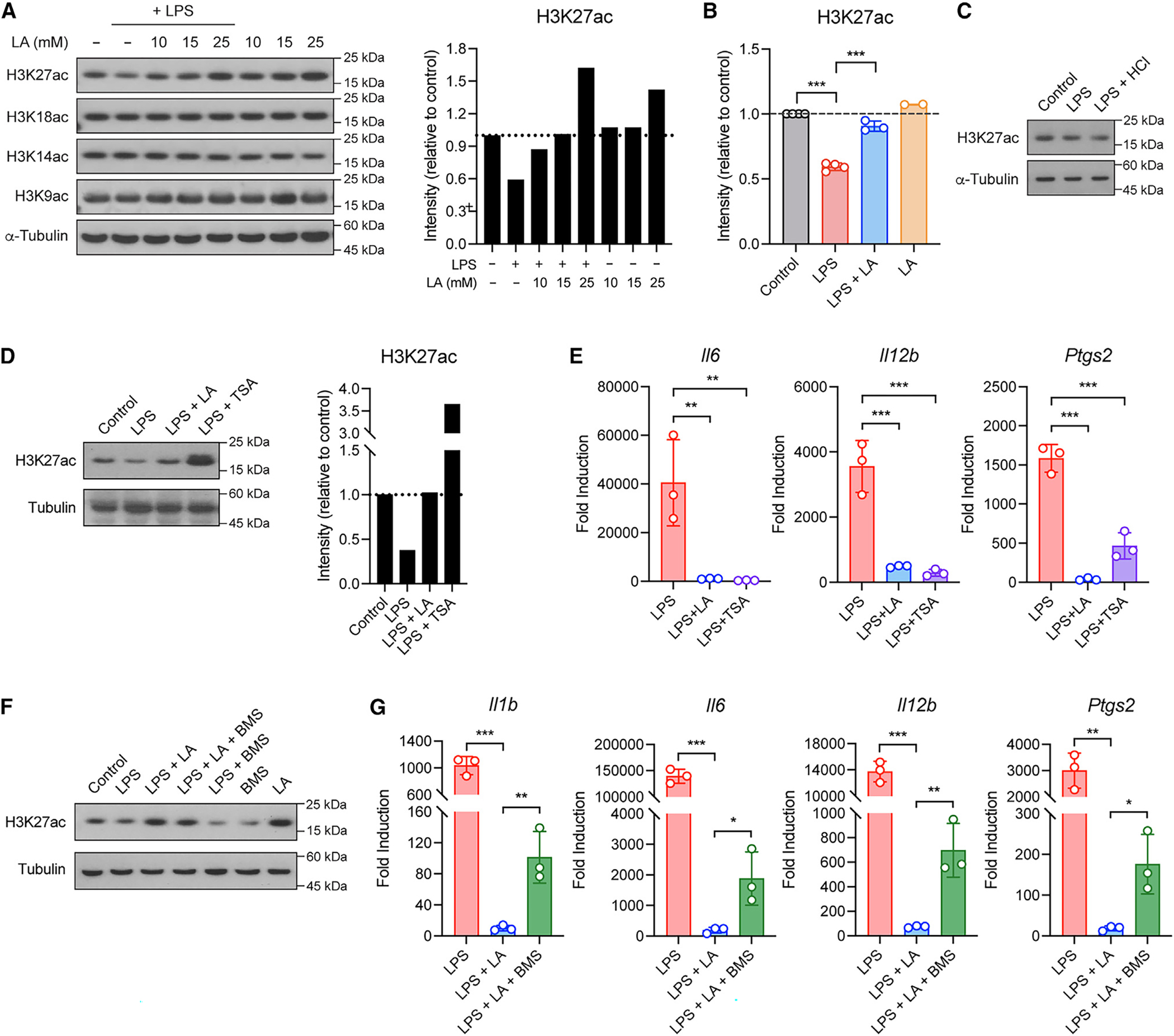
(A and B) Global histone acetylation in BMDMs stimulated without or with LPS in the presence or absence of LA at the indicated concentrations (A) or 10 mM (B). Shown in (A, right) and (B) are quantified signals for histone H3K27 acetylation.
(C) Global histone H3K27 acetylation in BMDMs stimulated without (control) or with LPS in normal or low-pH condition with HCl.
(D) Global histone H3K27 acetylation in BMDMs stimulated without (control) or with LPS in the presence or absence of TSA. Shown in the right are quantified signals for histone H3K27 acetylation.
(E) Pro-inflammatory gene expression in BMDMs stimulated as in (D).
(F) Global histone H3K27 acetylation in BMDMs stimulated without (control) or with LPS in the presence or absence of BMS.
(G) Pro-inflammatory gene expression in BMDMs stimulated as in (F). Data are mean ± SD of two to four independent experiments (B) or triplicates of representative experiments (E and G). One-way ANOVA (B, E, and G) followed by Tukey’s post-test: *p < 0.05, **p < 0.001, ***p < 0.001.
Alternatively, lactic acid might act directly as an HDAC inhibitor to regulate histone acetylation as shown by a previously study.34 It is also possible that other histone modifications might play a role in mediating lactic acid anti-inflammatory effect. A recent study has shown that lactic acid induces histone lysine lactylation (Kla) in a time-dependent fashion (e.g., 16–24 h), which is associated with the induction of M2-like genes including Arg1.35 However, we did not observe significant effect of lactic acid on lysine lactylation in all proteins (pan Kla) and H3K18la in BMDMs during the early phase of LPS stimulation (e.g., 0–3 h) (Figures S3D and S3E), suggesting that histone H3K27 acetylation promoted by lactic acid might precede histone lactylation. Lactic acid also had no effect on H3K27 trimethylation (Figures S3D and S3E).
Lactic acid modifies chromatin accessibility to pro-inflammatory gene loci
To assess the epigenetic basis for transcriptional changes induced by lactic acid in macrophages, we then performed an assay for transposase-accessible chromatin with high-throughput sequencing (ATAC-seq) to profile the chromatin landscape of BMDMs after LPS stimulation in the presence of lactic acid. The PCA of accessible chromatin regions indicated that BMDMs stimulated with LPS in the presence or absence of lactic acid form distinct clusters (Figure 5A; Figure S4A). Differential accessibility analysis revealed lactic acid-specific chromatin profiles (Figure 5B; Figure S4B). We identified 11,019 genomic regions (4,386 closed regions vs. 6,633 open regions) with altered chromatin accessibility induced by lactic acid in BMDMs during LPS stimulation (i.e., LPS + LA vs. LPS) (Figure 5B). This altered chromatin accessibility is significantly and positively correlated with gene expression induced by lactic acid (i.e., RNA-seq) (Figure S4C). GO analysis further showed that genes associated with the DNA regions closed with lactic acid during LPS stimulation are involved in innate immune processes including the “positive regulation of cytokine production” (GO: 0001819) and “regulation of inflammatory response” (GO: 00050727) (Figure 5C; Figure S4D), which is in line with the biological processes associated with lactic acid-downregulated genes from our RNA-seq data (Figure 2C), indicating that lactic acid inhibits pro-inflammatory response via reducing chromatin accessibility to the innate immune gene-associated regions. For example, Il6, Il12b, and Ptgs2 were among the top downregulated genes by lactic acid in LPS-stimulated BMDMs (Figure 5D), and the chromatin accessibility to DNA regions associated with these genes during LPS stimulation was significantly reduced by lactic acid (Figure 5E). Moreover, with motif enrichment analysis for the differentially accessible regions, we found that transcription factor (TF) binding sites for interferon regulatory factors (IRFs), NF-κB, and AP-1 were significantly depleted in the DNA regions closed with lactic acid (Figure 5F), including those regions associated with Il6 and Ptgs2 (Figure 5E). These results suggest that lactic acid mediates epigenetic reprogramming, leading to the reduction of pro-inflammatory gene expression.
Figure 5. LA modifies chromatin accessibility to pro-inflammatory gene loci.
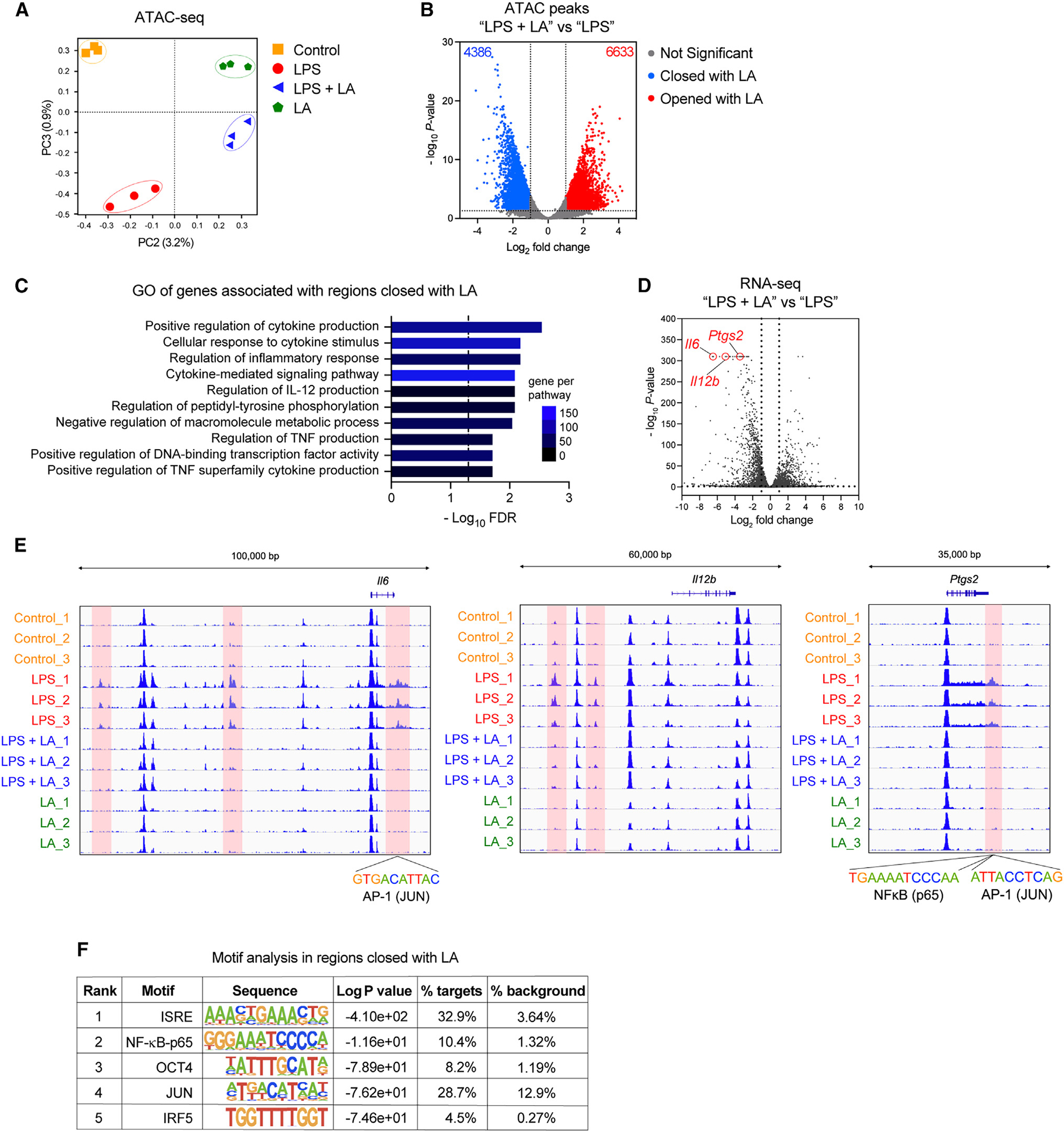
(A) PCA plot of ATAC-seq data from BMDMs stimulated without (control) or with LPS in the presence or absence of LA at 10 mM for 3 h.
(B) Volcano plot of ATAC-seq from (A) showing cutoffs (log2 fold change ≥ or ≤ 1 and FDR ≤ 0.05) used to identify differential accessibility regions in BMDMs stimulated with LPS in the presence or absence of LA (LPS + LA vs. LPS).
(C) GO analysis of differential accessibility regions identified in (B) showing biological processes enriched in DNA regions closed by LA in BMDMs stimulated with LPS.
(D) Volcano plot of RNA-seq data from Figure 2B indicating pro-inflammatory genes Il6, Il12b, and Ptgs2 (circled in red) highly suppressed by LA.
(E) Gene tracks of ATAC-seq data at Il6, Il12b, and Ptgs2 loci.
(F) Motif enrichment analysis of differential accessibility regions closed by LA.
Lactic acid regulates the enhancer repertoire and reduces pro-inflammatory gene-associated enhancer in BMDMs after LPS stimulation
Genomic annotations of the differential accessibility regions altered by lactic acid in BMDMs after LPS stimulation (LPS + LA vs. LPS) indicate a predominance of distal DNA regions from the transcription start site (TSS) of associated genes (i.e., >5 kb upstream or downstream of TSS) (Figure 6A), suggesting that those regions are enhancer regions. To examine this further, we carried out an enrichment analysis using public chromatin immunoprecipitation sequencing (ChIP-seq) data (ChIP-Atlas)36 and found that the ATAC-seq regions differentially altered by lactic acid (11,019) are significantly overlapping with enhancers marked by H3K27ac and H3K4me1 in macrophages and dendritic cells (Figure 6B). For example, our ATAC-seq regions closed by lactic acid in the Il6 locus are associated with H3K27ac and H3K4me1 enhancer regions previously reported (GEO: GSE38377)37 (Figure S5). To confirm these results with lactic acid, we performed H3K27ac ChIP-PCR for the ATAC-seq regions altered by lactic acid in BMDMs after LPS stimulation. Lactic acid-closed ATAC-seq regions in pro-inflammatory gene loci (i.e., Il6, Il12b, Nos2, and Ptgs2) were associated with reduced H3K27 acetylation (Figure 6C), strongly indicating that lactic acid represses pro-inflammatory genes by suppressing the function of their enhancers.
Figure 6. LA regulates the enhancer repertoire and reduces pro-inflammatory gene-associated enhancer in macrophages after LPS stimulation.
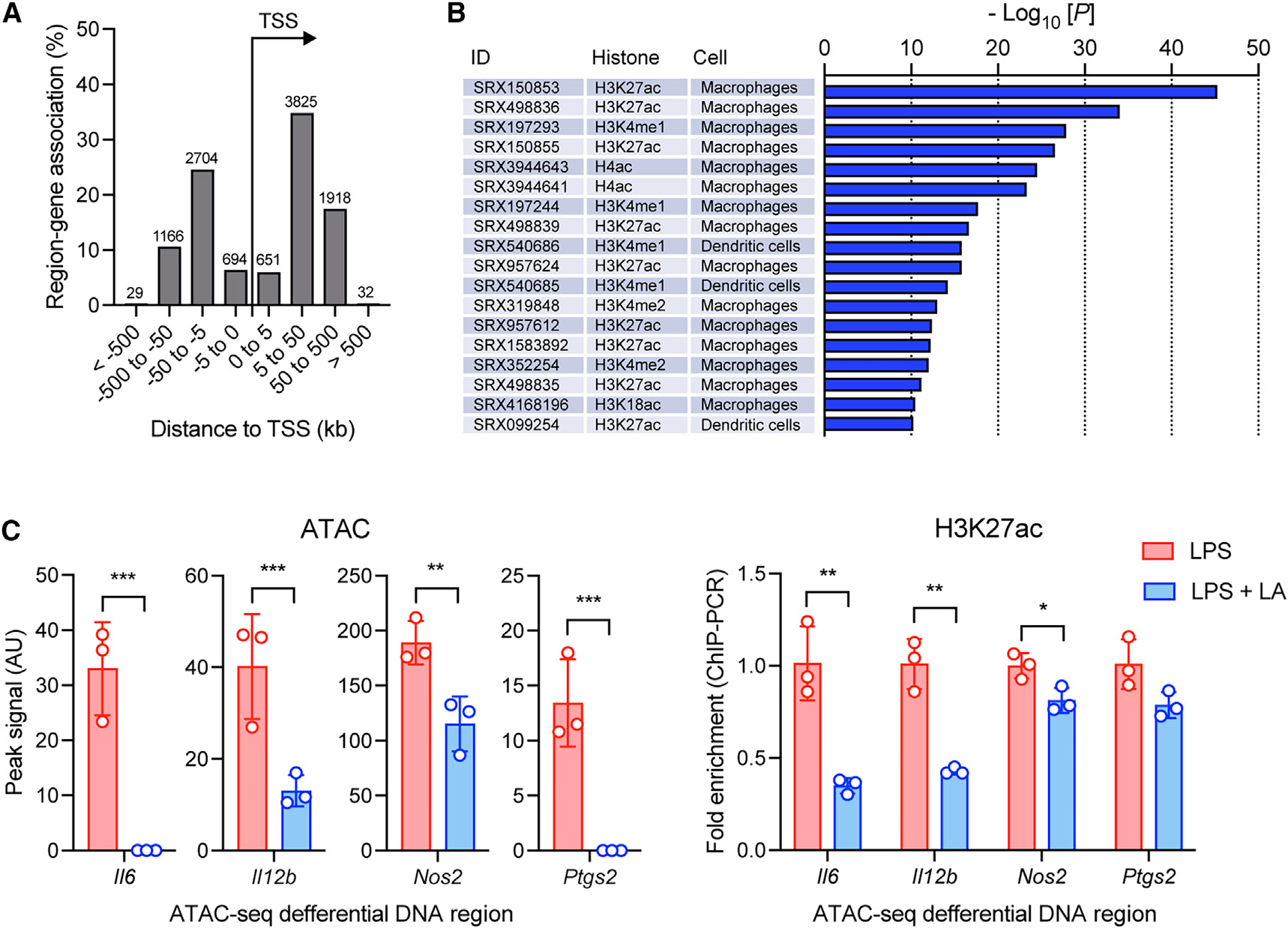
(A) Proximity of chromatin regions altered by LA in BMDMs stimulated with LPS to transcription start sites (TSSs).
(B) Enrichment analysis of chromatin regions altered by LA for enhancers using public chromatin immunoprecipitation sequencing (ChIP-seq) data (ChIP-Atlas).
(C) ChIP-qPCR analysis of H3K27 acetylation (right) in pro-inflammatory gene-associated ATAC-seq regions closed with LA (left). Data in (C) are mean ± SD of triplicates. Student’s t test (unpaired): *p < 0.05, **p < 0.001, ***p < 0.001.
Transcriptional repression of pro-inflammatory response by lactic acid and its long-term effect requires NR4A1
Both our RNA- and ATAC-seq data indicated that lactic acid positively regulated gene expression (i.e., 1,185 genes induced by lactic acid) and chromatin accessibility (i.e., 6,633 DNA regions opened with lactic acid) (Figures 2B and 5B). We therefore asked whether lactic acid-induced genes or enhancer regions are involved for the inhibition of pro-inflammatory response in macrophages. To test this, using RNA-seq analysis, we found that 198 out of 1,185 genes induced by lactic acid during LPS stimulation in BMDMs were involved in the “regulation of DNA-templated transcription” GO category (GO: 0006355) (Figure 2C). Within those genes, we identified Nr4a1, whose expression was one of the highest TF genes significantly induced by lactic acid (Figures 7A and 7B). This Nr4a1 induction was associated with increased chromatin accessibility and H3K27 acetylation in the gene locus (Figure 7C), suggesting that lactic acid promotes enhancer-dependent transcription at the Nr4a1 gene via H3K27 acetylation. Neither low-pH condition with HCl nor IL-4 could induce Nr4a1 (Figure 7D). In relation to the gene product of Nr4a1, motif enrichment analysis for ATAC-seq peaks also revealed that the TF binding site for NR4A1 is significantly enriched in the chromatin regions opened by lactic acid (Figure 7E). We therefore hypothesized that NR4A1 is required to mediate lactic acid anti-inflammatory effect. To tested this, we treated BMDMs with DIM-C-pPhOH, an inhibitor of NR4A1 (iNR4A1), together with LPS and lactic acid, and found that iNR4A1-treated BMDMs exhibited a significant loss of lactic acid inhibitory effect on pro-inflammatory Il1b, Ptgs2, Il6, and Il12b gene expression (Figure 7F). A partial loss of lactic acid inhibitory effect also occurred in macrophage cell line RAW 264.7 with small interfering RNA (siRNA) knockdown for Nr4a1 and BMDMs from NR4A1 knockout mice (Figures S6A and S6B). Together, these results suggest that lactic acid promotes the induction and activation of NR4A1, leading to the transcriptional repression of pro-inflammatory response in macrophages. Consistent with this, BMDMs treated with cytosporone B (Csn-B), an agonist that is known to enhance NR4A1 signaling,38 had significant reduced pro-inflammatory gene expression (Figure 7G), mimicking the lactic acid effect. A link between NR4A1 and the regulation of H3K27 acetylation has been previously reported.39,40 Our results thus suggest that NR4A1 might be required for mediating the epigenetic reprogramming by lactic acid.
Figure 7. Transcriptional repression of macrophage pro-inflammatory response by LA and its long-term effect requires NR4A1.
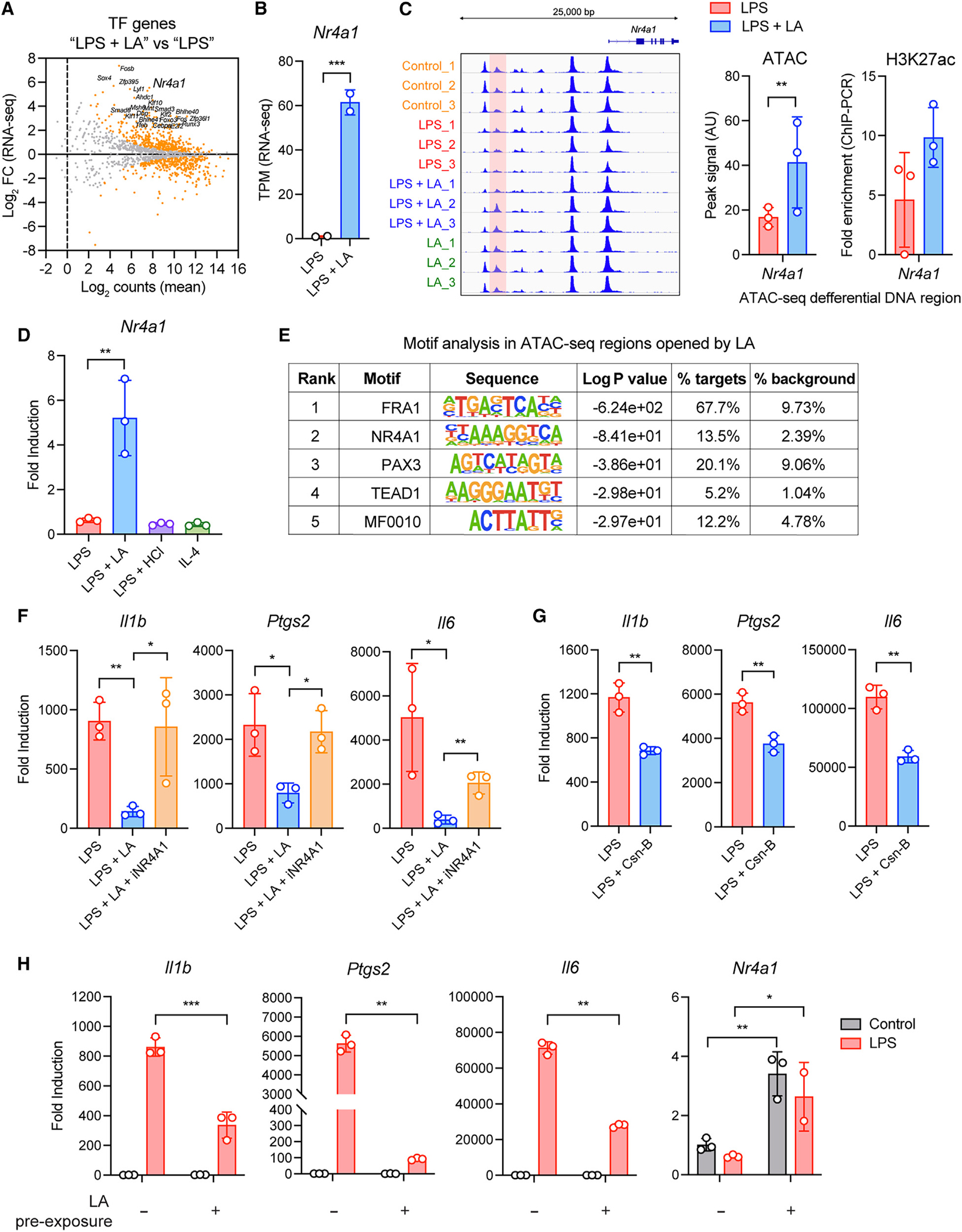
(A) MA plot of RNA-seq data showing TF gene expression in BMDMs stimulated with LPS in the presence or absence of LA at 10 mM for 3 h. Differentially expressed TF genes (FDR ≤ 0.05) are in orange.
(B) Nr4a1 expression from RNA-seq data as in (A).
(C) LA-altered chromatin accessibility region in Nr4a1 locus (left, middle) and its association with H3K27 acetylation measured by ChIP-qPCR (right).
(D) Nr4a1 expression in BMDMs stimulated with LPS as in (A) or in low-pH condition with HCl for 3 h or with IL-4 for 6 h.
(E) Motif enrichment analysis of differential accessibility regions opened by LA.
(F and G) Pro-inflammatory gene expression in BMDMs pre-treated with DIM-C-pPhOH (iNR4A1) and stimulated with LPS in the presence or absence of LA (F) or stimulated with LPS in the presence or absence of Csn-B (G).
(H) LPS tolerance in BMDMs pre-exposed to LA for 24 h. Data in (D) and (F–H) are mean ± SD of triplicates. Student’s t test (unpaired) (B, C, and G), one-way ANOVA (D and F), or two-way ANOVA (H) followed by Tukey’s or Sidak’s post-test: *p < 0.05, **p < 0.001, ***p < 0.001.
Functional annotation also revealed that the genes induced by lactic acid (i.e., 1,184 genes; Figure 2B) are significantly associated with the molecular function of repressors (KW:0678) and chromatin regulators (KW:0156) (Figure S6C). Several TF genes that are known to possess anti-inflammatory effect (e.g., Runx3, Smad3, Smad6, Klf10, Klf2, Hes1) were also highly induced by lactic acid (Figure S6D). It is therefore possible that, in addition to Nr4a1, other gene products induced by lactic acid might also participate in suppressing pro-inflammatory response epigenetically and/or transcriptionally.
Epigenetic reprogramming has been proposed as the molecular mechanism responsible for long-term functional modification of cells in the innate immune system (e.g., “trained immunity”).41 To test the long-term effect of lactic acid-mediated epigenetic reprogramming, we pre-exposed BMDMs to lactic acid 24 h prior to resting and subsequent LPS stimulation (Figure S7A). Lactic acid pre-exposure significantly inhibited subsequent pro-inflammatory response to LPS (Figure 7H), and the inhibitory effect was maintained and stable up to 24 h resting after initial lactic acid pre-exposure (Figure S7B). This long-term effect was also observed in BMDMs pre-exposed to Csn-B (Figure S7C). Together, these results suggest that lactic acid promotes a form of long-term immunosuppression (trained immunosuppression) on macrophage pro-inflammatory response via NR4A1. To test this immunosuppression in vivo, mice were administered with lactic acid enema for 3 days before and 3 days into dextran sodium sulfate (DSS)-induced colitis (Figure S7D). Lactic acid-treated mice exhibited substantial decrease in weight loss and inflammatory cytokine production and had reduced tumor necrosis factor (TNF)-α-producing macrophages and higher H3K27 acetylation in the colon lamina propria compared with control mice (Figures S7E–S7I), indicating that the pre-treatment with lactic acid exerts a long-term immunosuppression on intestinal inflammation.
DISCUSSION
Lactic acid has emerged as an important modulator of the immune system. High lactic acid concentrations in tissue microenvironments such as tumors and intestines are sufficient to exert immunosuppressive activity on both innate and adaptive immune cells.8,9 During infection or tissue injury, lactic acid regulates macrophage metabolism and prevents excessive inflammatory response; in tumors, lactic acid produced by tumor cells promotes the polarization of TAMs, which in turn promotes tumor progression. Several prior studies have shown that lactic acid drives M2 gene expression (i.e., IL-4-induced M2 macrophage-associated genes) to promote tumor growth and progression.8,19,20 However, how lactic acid suppresses macrophage pro-inflammatory response (i.e., M1 polarization) is largely unknown. Here, we provide evidence for a mechanism by which lactic acid exerts M2-gene-independent inhibitory effect on M1 macrophages and suggest that lactic acid acts as a primary fuel source for TCA cycle metabolism to promote histone H3K27 acetylation, which allows the expression of gene program including Nr4a1 that, in turn, inhibits macrophage pro-inflammatory response.
The anti-inflammatory effect of lactic acid on LPS-stimulated macrophages has been showed previously by others.21,22 Our RNA-seq data confirm this effect and further demonstrate that broader gene programs involved in inflammatory response but also other innate immune processes are suppressed by lactic acid, indicating a profound immunosuppressive activity exerted by lactic acid on macrophage functions. Furthermore, the gene expression profile by lactic acid is distinct from that by low-pH condition with HCl during LPS stimulation, indicating acid-independent gene regulation by lactic acid. While the expression of M2 genes might be responsible for the lactic acid effect, our data show that the transcriptional repression of pro-inflammatory response and other innate immune processes occur prior to the induction of M2 gene (e.g., Arg1, Mrc1, Ym1), suggesting that the immunosuppressive effect of lactic acid is uncoupled from the M2 gene expression. Instead of the M2 gene, lactic acid upregulates a set of genes (1,185 genes) associated with maintenance gene programs, including “cell cycle” and “regulation of gene transcription,” and only 5% of these genes are overlapping with genes induced by IL-4, indicating that, rather than a prototypic M2-like phenotype, lactic acid polarizes macrophages into a specific phenotype with a distinct gene expression profile. These lactic acid-specific gene inductions can be important in promoting immunosuppressive functions in macrophages.
The lactic acid effect on macrophages can be mediated via cell surface signaling receptors or intracellular metabolic processes. GPCRs have been shown to mediate the sensing of acidic pH environment due to lactic acid secretion by tumor cells.23–25 The lactic acid sensing by GPCRs can signal via cAMP to induce the expression of M2 genes, leading to the support of tumor growth.23,25 Here, we show that the inhibition of cAMP de novo synthesis by MDL-12, however, has no effect on lactic acid-suppressed pro-inflammatory response, suggesting that sensing by GPCR is not required for the lactic acid anti-inflammatory effect. Alternatively, the lactic acid effect can be mediated via intracellular lactic acid sensing. This is supported by our targeted metabolomics data showing that the presence of extracellular lactic acid promotes lactic acid uptake in macrophages during LPS stimulation. In addition, when carrier proteins MCT1 and MCT4, both important for bi-directionally transporting lactic acid across plasma membrane,42 are blocked by inhibitor CHC, accumulation of intracellular lactic acid occurs, leading to the inhibition of pro-inflammatory response. Together, our findings suggest that lactic acid influx or accumulation is required for mediating lactic acid anti-inflammatory effect. Consistent with this idea, Slc16a1, a gene encoding MCT1, is strongly upregulated by IL-10 according to our published RNA-seq data,13 indicating that controlling lactic acid flux in macrophages could be part of the processes mediating IL-10 anti-inflammatory effect. Future studies into the regulation of macrophage MCTs in vivo will help to better understand whether/how lactic acid transport in macrophages is critical in maintaining tissue homeostasis and controlling inflammation.
During LPS stimulation, macrophages undergo pseudo-hypoxia (also known as the Warburg effect) and switch their cellular metabolism from OXPHOS to anaerobic glycolysis.12 This glycolytic switch is required for M1 macrophage polarization as blocking glucose uptake by 2-DG reduces pro-inflammatory cytokine production (e.g., IL-1β) in LPS-stimulated macrophages.43 Our data showing the inhibition of glycolysis by lactic acid might therefore suggest that the anti-inflammatory effect of lactic acid is mediated via inhibiting glycolysis. However, the lactic acid effect remains largely intact even in glucose-free conditions or conditions where glycolysis is inhibited by 2-DG, indicating an alternative metabolic regulation is involved. Indeed, our findings from targeted metabolomics and isotype tracing using 13C-lactic acid demonstrate that lactic acid acts as a primary fuel source for the TCA cycle and citrate accumulation. This notion of lactic acid utilization in macrophages is similar to that in tumors where circulating/secreted lactic acid becomes a primary carbon source for the TCA cycle and thus of energy for tumor growth/progression under anaerobic conditions.44 Our further analysis on global histone modification shows that the citrate production from lactic acid is required for acetyl-CoA production and histone H3K27 acetylation. Blocking acetyl-CoA production from citrate by inhibiting ACLY results in reduced global H3K27 acetylation and loss of lactic acid anti-inflammatory effect, suggesting that the lactic acid utilization for citrate production in the TCA cycle is a pathway leading to the immunosuppression function of lactic acid. The requirement of ACLY-dependent histone acetylation is also observed in M2 polarization by IL-4 as well as lactic acid-potentiated M2 macrophages,17,20 indicating the importance of acetyl-CoA production in macrophage polarization for anti-inflammatory function and/or maintenance functions such as tissue repair and pro-tumor growth. How ACLY-dependent histone acetylation promotes the expression of specific gene programs between IL-4- and lactic acid-polarized macrophage remains to be addressed in future studies.
Recently, lactic acid has been shown to modify histones at lysine residues (i.e., histone lactylation or Kla) including H3K18, and increased Kla is associated with M2 gene expression such as Arg1 in macrophages during LPS stimulation.35 Here, while lactic acid is sufficient to suppress pro-inflammatory gene expression within 1–3 h of LPS stimulation, no Kla or H3K18la is induced by lactic acid during that period, suggesting that histone lactylation occurs rather in the later phase of the activation. This notion of time-dependent histone lactylation is supported by previous studies where Kla by lactic acid only occurs 16–24 h after LPS stimulation.35,45 Whether the citrate-to-acetyl-CoA pathway is required for histone lactylation (lactyl-CoA production) remains to be addressed. It is possible that earlier histone modification by lactic acid (H3K27 acetylation) is involved in controlling M1 polarization or pro-inflammatory response as we show in this study, and later histone modification (Kla) could be important for restoring steady state or promoting recovery from cell activation. A recent study also suggests that Kla is a consequence rather than a cause of macrophage activation but occurs under inflammatory duress (e.g., hypoxic lactic acid-rich inflammatory microenvironments).45 Our data implicating lactic acid in the epigenetic regulation via H3K27 acetylation are supported by our ATAC-seq results where differential accessible DNA regions induced by lactic acid are strongly associated with H3K27ac enhancer regions identified by others. Those enhancer regions in pro-inflammatory gene loci are significantly reduced by lactic acid. Together, these data support a model where lactic acid regulates the enhancer repertoire to control pro-inflammatory response in macrophages.
It is increasingly recognized that TF NR4A1, an orphan nuclear receptor of the NR4A family, is crucial in controlling pro-inflammatory response.46 Here, we show that NR4A1 expression is strongly upregulated by lactic acid, and the DNA-binding motif for NR4A1 is highly enriched in open chromatin regions associated with lactic acid, suggesting that NR4A1 is responsible for the lactic acid anti-inflammatory effect. In support of this, pharmaceutical inhibition of NR4A1 results in the loss of lactic acid anti-inflammatory effect. Conversely, agonistic activation of NR4A1 can mimic the lactic acid effect. However, macrophages from widely used NR4A1 knockout mice only exhibit partial loss of lactic acid effect, which could be due to other compensatory mechanisms or incomplete loss of NR4A1 activity. It has been shown that the expression of Nr4a2, a close homolog of Nr4a1, is highly increased following LPS challenge in the NR4A1 knockout mice,47 and the truncated NR4A1 peptide produced in the knockout mice remains capable of stabilizing and activating hypoxia-inducible factor-1α.48 Alternatively, other lactic acid-induced TF genes, in particular those with known anti-inflammatory effect, might also collectively contribute to the immunosuppressive effect of lactic acid. Our results demonstrating NR4A1 as a negative regulator for pro-inflammatory response are in line with previous studies showing that NR4A1 inhibits pro-inflammatory cytokine production in both human and mouse macrophages49–51 and protects from inflammatory diseases in mouse models such as DSS colitis and experimental autoimmune encephalomyelitis (EAE).52–54 In addition, NR4A1 has also been linked to epigenetic regulation. A recent study has found that NR4A1 is highly expressed in tolerant T cells, where it represses effector gene expression by inhibiting TF AP-1 function and promoting histone H3K27 acetylation, leading to activation of tolerance-related genes.40 In acute myeloid leukemia cells, NR4A1 has also been found to promote binding of p300 histone acetyltransferase to activate NR4A1-bound enhancers via histone H3K27 acetylation.39 Together with our findings on the regulation of histone H3K27 acetylation by lactic acid, it is therefore possible that lactic acid activates NR4A1 to promote lactic acid-specific enhancers, resulting in the expression of immunosuppressive genes. Further assessment is required to address the precise mechanism.
One strength of our study is that the lactic acid-induced epigenetic reprograming can result in a long-term form of immunosuppression. Macrophages pre-exposed to lactic acid exhibit long-lasting immunosuppressive function, resulting in the inhibition of LPS response, similar to that in LPS tolerance. This tolerance remains stable up to 24 h resting after initial lactic acid exposure. This long-lasting lactic acid effect is analogous to trained immunity, where histone modification is an essential mechanism for long-term memory of innate immunity.41 Pre-exposure to NR4A1 agonist has a similar effect to lactic acid, suggesting that not only the activation but also the expression of NR4A1 is critical to maintain a stable effect. The notion of trained immunosuppression by lactic acid is also supported by our in vivo data showing that mice pretreated with lactic acid enema are protected from DSS colitis. This protection is in line with the observations that dietary lactic acid supplementation or lactic acid bacteria strains (Lactobacillus) improve intestinal inflammation by modulating macrophage polarization.55–58
In summary, our study reveals a mechanism of lactic acid in suppressing macrophage pro-inflammatory response. We propose that lactic acid serves as a primary fuel source for TCA cycle metabolism to regulate H3K27ac enhancer repertoire, leading to the upregulation of lactic acid-specific gene program including Nr4a1, which in turn transcriptionally represses macrophage pro-inflammatory function. Note that this epigenetic reprogramming by lactic acid has a significant effect on promoting a form of long-term immunosuppression that leads to LPS tolerance in macrophages. These findings thus indicate that lactic acid sensing and its epigenetic reprogramming in macrophages represent a key homeostatic mechanism that promotes tolerogenic tissue microenvironment. Therapeutic targeting of the lactic acid-induced trained immunosuppression in macrophages therefore could be beneficial for treatment or prevention of inflammatory diseases.
Limitations of the study
Although our work reveals a regulatory role of lactic acid in epigenetic reprogramming, the extent to which this pathway controls gene transcription beyond suppressing pro-inflammatory response still needs to be assessed. Our RNA-seq data suggest that lactic acid induces a broad gene program including Nr4a1, but we were unable to determine if they collectively contribute to the anti-inflammatory effect of lactic acid, and if this lactic acid-specific gene program is mainly for immunosuppression or also involved in other cellular stress responses to protect macrophages from lactic acid-rich inflammatory microenvironments. It is possible that such a program is also induced in the tumor microenvironment via lactic acid sensing and might contribute to tumor growth, which requires further investigation.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, W. K. Eddie Ip (ip.eddie@mayo.edu).
Materials availability
Information and requests for resources and reagents may be directed to lead contact, W. K. Eddie Ip (ip.eddie@mayo.edu).
Data and code availability
RNA-seq and ATAC-seq data in this study have been deposited at GEO and are publicly available. The accession number is listed in the key resource table.
This study did not generate any unique code.
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Hamster monoclonal anti-mouse TNF-α | eBioscience | Cat # 14-7423-81; RRID: AB_2721314 |
| Rabbit polyclonal biotinylated anti-mouse TNF-α | eBioscience | Cat# 13-7341-81; RRID: AB_466950 |
| Rat monoclonal anti-mouse IL-6 | eBioscience | Cat# 14-7061-81; RRID: AB_468422 |
| Rat monoclonal biotinylated anti-mouse IL-6 | BioLegend | Cat# 504601; RRID: AB_2127458 |
| Hamster monoclonal anti-mouse IL-1β | eBioscience | Cat# 14-7012-81; RRID: AB_468396 |
| Rabbit polyclonal anti-mouse IL-1β | eBioscience | Cat# 13-7112-81; RRID: AB_466924 |
| Rat monoclonal anti-mouse IL-6, PerCP-eFluor 710 | Invitrogen | Cat# 46-7061-82; RRID: AB_2573829 |
| Rat monoclonal anti-mouse TNF-α, PE-efluor 610 | Invitrogen | Cat# 61-7321-82; RRID: AB_2574666 |
| Rat monoclonal anti-mouse IL-1β, PE-Cy7 | Invitrogen | Cat# 25-7114-82; RRID: AB_2573526 |
| Rat monoclonal anti-mouse iNOS, Alexa Fluor 700 | Invitrogen | Cat# 56-5920-80; RRID: AB_2848473 |
| Rat monoclonal anti-mouse F4/80, Super Bright 780 | Invitrogen | Cat# 78-4801-82; RRID: AB_2802477 |
| Mouse monoclonal anti-mouse CD64 (FcγRI), APC | BioLegend | Cat# 139305; RRID: AB_11219205 |
| Rat monoclonal anti-mouse CD11b, Super Bright 645 | Invitrogen | Cat# 64-0112-82; RRID: AB_2662387 |
| Rat monoclonal anti-mouse Ly6C, APC/Cyanine7 | BioLegend | Cat# 128025; RRID: AB_10643867 |
| Rat monoclonal anti-mouse MHC Class II (I-A), PE | Invitrogen | Cat# 12-5321-82; RRID: AB_465928 |
| Rat monoclonal anti-mouse CD45, Super Bright SB600 | Invitrogen | Cat# 63-0451-82; RRID: AB_2637149 |
| Mouse monoclonal anti-mouse NK1.1, Pacific Blue | BioLegend | Cat# 108721; RRID: AB_2234352 |
| Rat monoclonal anti-mouse Ly6G, Pacific Blue | BioLegend | Cat# 127611; RRID: AB_1877212 |
| Rat monoclonal anti-mouse TER119/Erythroid cells, Pacific Blue | BioLegend | Cat# 116231; RRID: AB_2149212 |
| Rat monoclonal anti-mouse CD3, Pacific Blue | BioLegend | Cat# 100213; RRID: AB_493644 |
| Rat monoclonal anti-mouse CD19, Pacific Blue | BioLegend | Cat# 115526; RRID: AB_493341 |
| Rabbit monoclonal anti-phospho-NF-kB p65 (Ser536) (93H1) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Rabbit anti-IκBα | Cell Signaling Technology | Cat# 9242; RRID: AB_331623 |
| Mouse monoclonal anti-phospho-IκBα (Ser32/36) (5A5) | Cell Signaling Technology | Cat# 9246; RRID: AB_2267145 |
| Mouse monoclonal anti-phospho-p38 MAPK (Thr180/Tyr182) (28B10) | Cell Signaling Technology | Cat# 9216; RRID: AB_331296 |
| Rabbit polyclonal anti-phospho-LDHA (Tyr10) | Cell Signaling Technology | Cat# 8176; RRID: AB_11220238 |
| Rabbit monoclonal anti-H3, acetyl (Lys27) (D5E4) XP | Cell Signaling Technology | Cat# 8173; RRID: AB_10949503 |
| Rabbit monoclonal anti-H3, acetyl (Lys18) (D8Z5H) | Cell Signaling Technology | Cat# 13998; RRID: AB_2783723 |
| Rabbit monoclonal anti-H3, acetyl (Lys14) (D4B9) | Cell Signaling Technology | Cat# 7627; RRID: AB_10839410 |
| Rabbit monoclonal anti-H3, acetyl (Lys9) (C5B11) | Cell Signaling Technology | Cat# 9649; RRID: AB_823528 |
| Rabbit monoclonal anti-H3, trimethyl (Lys27) (C36B11) | Cell Signaling Technology | Cat# 9733; RRID: AB_2616029 |
| Mouse monoclonal anti-α-tubulin (DM1A) | Cell Signaling Technology | Cat# 3873; RRID: AB_1904178 |
| Rabbit recombinant monoclonal anti-L-lactyl lysine | PTM BIO | Cat# PTM-1401RM; RRID: AB_2942013 |
| Rabbit recombinant monoclonal anti-H3, lactyl (Lys18) | PTM BIO | Cat# PTM-1406RM; RRID: AB_2909438 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Lipopolysaccharides from Escherichia coli O26:B6 | Sigma | L3755 |
| L-(+)-Lactic acid | Sigma | L6402 |
| ATP | Sigma | A6419 |
| L-Lactic acid-13C3 | Sigma | 746258 |
| cyano-4-hydroxycinnamic acid | Sigma | S8612 |
| MDL-12,330A hydrochloride | Sigma | M182 |
| Trichostatin A | Sigma | T8552 |
| BMS-303141 | Sigma | SML0783 |
| DIM-C-pPhOH | Selleck Chemicals | S6799 |
| Recombinant mouse TNF-α | PeproTech | 315-01A |
| Recombinant mouse IL-6 | PeproTech | 216-16 |
| Recombinant mouse IL-1β | PeproTech | 211-11B |
| Recombinant mouse RELM-α | PeproTech | 450-26 |
| 2-deoxy-D-glucose | Cayman Chemical | 14325 |
| D-glucose-13C6 | Cambridge Isotope | CLM-1396 |
| BD GolgiPlug | Fisher Scientific | BDB555029 |
| BD Cytofix/Cytoperm solution | Fisher Scientific | BDB554714 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | 13778030 |
| Cytosporone B | R&D Systems | 5459 |
| Dextran sulfate sodium (molecular mass 36–50 kDa) | MP Biomedicals | MFCD00081551 |
| Collagenase VIII | Sigma | C2139 |
| DNase I | ROCHE | 10104159001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Dual-Glo Luciferase Reporter Assay | Promega | E2920 |
| Prostaglandin E2 (PGE2) Parameter Assay Kit | R&D Systems | KGE004B |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | L34965 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq | This paper | GEO: GSE253050 |
| ATAC-seq | This paper | GEO: GSE253050 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| RAW264.7 | American Type Culture Collection | TIB-71 |
| 293T | American Type Culture Collection | CRL-3216 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 mice | Jackson Laboratory | 000664 |
| C57BL/6NJ mice (control mice for Acod1−/− mice) | Jackson Laboratory | 005304 |
| Acod1−/− mice | Jackson Laboratory | 029340 |
| Nr4a1wt control mice | Jackson Laboratory | 100903 |
| Nr4a1−/− mice | Jackson Laboratory | 06187 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer (qPCR): mouse Arg1 Forward: 5’-GTGAAGAACCCACGGTCTGT-3’ | This paper | N/A |
| Primer (qPCR): mouse Arg1 Reverse: 5’-CTGGTTGTCAGGGGAGTGTT-3’ | This paper | N/A |
| Primer (qPCR): mouse Gapdh Forward: 5’-GGTGCTGAGTATGTCGTGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Gapdh Reverse: 5’-CGGAGATGATGACCCTTTTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il1b Forward: 5’-CCCAAGCAATACCCAAAGAA-3’ | This paper | N/A |
| Primer (qPCR): mouse Il1b Reverse: 5’-GCTTGTGCTCTGCTTGTGAG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il6 Forward: 5’-TGTTCTCTGGGAAATCGTGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Il6 Reverse: 5’-AAGTGCATCATCGTTGTTCATACA-3’ |
This paper | N/A |
| Primer (qPCR): mouse Il12b Forward: 5’-AGGTCACACTGGACCAAAGG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il12b Reverse: 5’-TGGTTTGATGATGTCCCTGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Nos2 Forward: 5’-CACCTTGGAGTTCACCCAGT-3’ | This paper | N/A |
| Primer (qPCR): mouse Nos2 Reverse: 5’-ACCACTCGTACTTGGGATGC-3’ | This paper | N/A |
| Primer (qPCR): mouse Nr4a1 Forward: 5’-TTGAGTTCGGCAAGCCTACC-3’ | This paper | N/A |
| Primer (qPCR): mouse Nr4a1 Reverse: 5’-GTGTACCCGTCCATGAAGGTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Ptgs2 Forward: 5’-GCGAGCTAAGAGCTTCAGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Ptgs2 Reverse: 5’-TCATACATTCCCCACGGTTT-3’ | This paper | N/A |
| Primer (qPCR): mouse Retnla Forward: 5’-TGCTGGGATGACTGCTACTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Retnla Reverse: 5’-CTGGGTTCTCCACCTCTTCA-3’ | This paper | N/A |
| Primer (qPCR): mouse Tnf Forward: 5’-GCACAGAAAGCATGATCCG-3’ | This paper | N/A |
| Primer (qPCR): mouse Tnf, Reverse: 5’-GCCCCCCATCTTTTGGG-3’ | This paper | N/A |
| Primer (qPCR): mouse Vegf Forward: 5’-CGAGCTCATGGACGGGTGAG-3’ | This paper | N/A |
| Primer (qPCR): mouse Vegf Reverse: 5’-GCCTGGGACCACTTGGCAT-3’ | This paper | N/A |
| ChIP-qPCR primers (see Table S1) | This paper | N/A |
| NR4A1 (Nur77) siRNA | Santa Cruz Biotechnology | Sc36110 |
| Control siRNA | Santa Cruz Biotechnology | sc-37007 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 9 | GraphPad Software Inc. | RRID: SCR_002798 |
| FlowJo 10 | Becton Dickinson & Company | RRID: SCR_008520 |
| ImageJ | NIH | RRID: SCR_003070 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6 (000664), C57BL/6NJ (005304), and Acod1−/− mice (029340) were purchased from The Jackson Laboratory and maintained in our facilities. All mice were bred and housed in accordance with National Institutes of Health- and Mayo Clinic Department of Comparative Medicine-approved guidelines, and all animal procedures were performed in accordance with protocols approved by the Mayo clinic Institutional Animal Care and Use Committee.
Primary cells
For mouse bone-marrow derived macrophage (BMDM) preparation, bone marrow cells were flushed from femur and tibia bones of females 6–8 weeks old mice and were grown at 37°C in 5% CO2 (v/v) in a humidified incubator in RPMI-1640 medium (Thermo Fisher Scientific, 11875119) containing 10% fetal bovine serum (FBS; Gemini Bioproducts, A88G00KG), 1% sodium pyruvate (SigmaAldrich, S8636), 1% penicillin streptomycin (Thermo Fisher Scientific, 15070-063) and 30% L929 supernatant (BMDM growth medium) for 7–8 days. For Retnla−/− BMDMs, femur and tibia were isolated from Retnla−/− mice and agedand sex-matched control mice in the laboratory of Ruslan Medzhitov at Yale University School of Medicine and shipped at 4°C overnight to our laboratory at Yale University School of Medicine. Upon arrival bone marrow cells were prepared and grown as described above. Differentiated BMDMs were re-plated in BMDM growth media with 20% L929 supernatant 16–20 h prior to cell stimulation.
Cell culture
293T cells and RAW 264.7 cells were obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco modified essential medium (DMEM) (Thermo Fisher Scientific, 11995065) containing 10% FBS and 1% penicillin/streptomycin antibiotics at 37°C in 5% CO2 (v/v) in a humidified incubator.
METHOD DETAILS
Cell treatments
To stimulate macrophages, BMDMs were incubated with 100 ng/mL LPS in the presence or absence of L-(+)-lactic acid at 10 mM or indicated concentrations for 3 h or the indicated times. In some experiments, BMDMs were stimulated in the presence of absence of MDL-12 (20 μM), TSA (50 nM), CHC (5 mM), RELM-α (100 ng/mL), 2-DG (10 mM), or Csn-B (5 μg/mL), or pre-treated with BMS (10 μM) or DIM-C-pPhOH (15 μM) for 1 h prior to LPS ± lactic acid stimulation for 3 h or 1 h respectively. To control for the acidic pH due to lactic acid, BMDMs were stimulated with LPS in low pH condition, equivalent to that with lactic acid at 10 mM (i.e., pH 6.5), using hydrochloric acid (HCl). For IL-1β secretion, ATP (5 mM) was added for additional 30 min. For stimulation in the absence of glucose, BMDM growth media were replaced with glucose-free RPMI-1640 medium (Thermo Fisher Scientific, 11879020) containing 10% FBS for 3 h prior to LPS ± lactic acid stimulation. For lactic acid or Csn-B pre-exposure experiments, BMDMs were incubated with 10 mM L-(+)-lactic acid or 5 μg/mL Csn-B for 24 h, washed with warm PBS three times, and rested for the indicated times prior to stimulation with LPS for 3 h.
Real-time PCR
Total RNA was isolated from single-cell suspensions using RNeasy Mini Kit (QIAGEN, 74106) or RNA-Bee (Tel-Test). Poly(A)-tailed mRNA was reverse transcribed using SMART MMLV reverse transcriptase (Takara Bio, 639524). PCR was performed using intron-spanning gene-specific primers and SYBR green QPCR mix (Quantabio, 95056-02K) on BioRad real-time PCR machine. The expression levels of target genes were normalized to Gapdh. Sequences of the primers used in this study are listed in the key resource table.
Cytokine and prostaglandin E2 production
Cytokine levels in culture supernatants from BMDMs or mouse serum was measured by enzyme-linked immunosorbent assay (ELISA) using capture and detection antibodies against IL-6, IL-1β and TNF-α (eBioscience) according to manufacturer’s protocols. PGE2 in culture supernatants was measured by PGE2 Parameter Assay Kit (R&D Systems) according to the manufacturer’s instructions. Culture supernatants of BMDMs stimulated as described above were harvested after 3 h for TNF-α and IL-1β, 6 h for IL-6, or 24 h for PGE2. For intracellular cytokines in BMDMs, cells were stimulated as described for 1 h before adding GolgiPlug and incubated for another 3 h to accumulate intracellular cytokines. The cells were fixed and permeabilized in BD Cytofix/Cytoperm solution and stained with fluorochrome-conjugated antibodies against IL-1β, IL-6, IL-12β, iNOS and TNF-α. Intracellular cytokine production were analyzed by flow cytometry performed on Attune Flow Cytometer (ThermoFisher Scientific); analysis was performed with FlowJo (BD Biosciences).
Immunoblotting analysis
Electrophoresis of proteins was performed using NuPAGE system (Invitrogen) according to manufacturer’s protocol. Briefly, BMDMs stimulated as described above were lysed in lysis and loading buffer (250 mM Tri-HCl (pH 6.8), 10% SDS, 50% glycerol, 0.05% bromophenol blue, 5% b-mercaptoethanol, protease inhibitor cocktails (Roche, 4693159001) and phosphatase inhibitors (Roche, 4906845001)), followed by 10 s ultra-sonication and 5 min heating at 99°C, and stored at −20C until analyzed. Proteins were separated on 4%–12% NuPAGE gels (Invitrogen, NP0323BOX), transferred to Amersham Protran nitrocellulose membranes (Global Life Sciences Solutions) and blocked with 5% BSA (Gemini Bioproducts, 700-101P) in TBS-T buffer for at least 1 h at room temperature. All primary antibodies were incubated overnight at 4°C, whereas secondary antibodies were incubated for at least1 h at room temperature. Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific, 32106) was used to detect protein signals. ImageJ was used for quantifying the band intensity and signals for a-tubulin were used as loading control.
NF-κB reporter assay
Dual luciferase reporter assays for NF-κB activation were performed in 293T cells. In brief, 293T cells in 6-well tissue-culture plates were transfected with an NF-κB (2X) luciferase reporter construct and pRenilla (gift of Dr Ruslan Medzhitov) overnight using Lipofectamine 2000 (Invitrogen, 11668027) in accordance with the manufacturer’s instructions, and re-plated in 96-well tissue-culture plates. Transfected 293T cells were stimulated with 10 ng/mL human recombinant IL-1β (PeproTech) in the presence or absence of 10 mM L-(+)-lactic acid for 3 h at 37°C. Reporter gene activity was measured using the Dual-Glo luciferase assay system (Promega, E2920) according to the manufacturer’s instruction. Data were normalized for transfection efficiency with the control reporter activity (Renilla) from the same sample and presented as the mean NF-κB fold induction of control samples (without IL-1β ± lactic acid stimulation).
RNA-seq analysis
For bulk RNA sequencing, 1 × 106 BMDMs were stimulated as described above. Total RNA of BMDMs was prepared by using RNeasy kit (QIAGEN, 74106) with RNase free DNase set (QIAGEN, 79254). Quality and quantity of RNA was assessed using a NanoDrop One (Thermo Fisher Scientific). Library preparation, sequencing at 100 bp read length, and bioinformatics analysis including genome mapping, gene expression calculation, differentially expressed gene (DEG) detection and principal component analysis (PCA), were performed by BGI (Cambridge, MA). Gene ontology of DEG were performed using DAVID bioinformatics resource (https://david.ncifcrf.gov/).
Seahorse real-time cell metabolic analysis
Analysis of the extracellular acidification rate (ECAR) and oxygen consumption rate (OCR) was performed with a Seahorse XF96 Extracellular Flux Analyzer (Agilent) in BMDMs as a measure of lactic acid production (a surrogate for the glycolytic rate) and OXPHOS respectively. In brief, BMDMs were seeded overnight in quadruplicate at a density of 1 × 105 cells per well on a pretreated poly-D-lysine-coated 96-well polystyrene Seahorse plate in BMDM growth media with 20% L929 supernatant and stimulated as described above. Prior to starting the assay, cells were washed and incubated in Seahorse Assay Medium supplemented with 10 mM glucose and 1 mM sodium pyruvate in 37°C incubator without CO2 for 45 min. Oligomycin (1 μM), FCCP (0.2 μM), rotenone (0.5 μM) and antimycin (1 μM) were injected where indicated, and the ECAR (mpH/min) and OCR (pMoles O2/min) was measured in real time.
Targeted metabolomics
Three million BMDMs per sample were stimulated as described above. Plates containing cells were placed on ice and washed with cold PBS three times, and ice-cold 80% methanol was added. Cells were subsequently scraped and collected. Concentration of tricarboxylic acid (TCA) cycle metabolites (lactic acid, fumaric acid, succinic acid, ketoglutaric acid, malic acid, aspartic acid, glutamic acid, 2-hydorxyglutaric acid, cis-aconitic acid, citric acid and isocitric acid) was performed at Mayo Clinic Metabolomics Core by isotope dilution gas chromatograph mass spectrometry as previously described.59 Data were normalized to the total protein content of each sample.
13C-metabolic flux analysis
For 13C-glucose isotype tracing, 3 × 106 BMDMs were cultured for overnight, washed with pre-warmed PBS three time, and incubated with glucose-free RPMI medium (Invitrogen, 26400036) containing 10% dialyzed FBS (Invitrogen, 26400036) and 13C-glucose (2 mg/mL) for 3 h at 37°C prior to LPS stimulation as described above. For 13C-lactic acid tracing, 3 × 106 BMDMs were incubated with BMDM growth medium containing 13C-lactic acid (10 mM) for 3 h at 37°C. Plates containing cells were washed with cold PBS three times, transferred into −80°C freezer for 10 min, and placed on dry ice, followed by adding 80% methanol. Cells were subsequently scraped, collected, and centrifuged for 5 min at 2000 rpm at 4°C. Supernatants were transferred into glass tubes and dried with nitrogen gas. Dried metabolite samples were dissolved in 75 μL DMF first and then derivatized with 75 μL N-Methyl-N-(tertbutyldimethylsilyl)trifluoroacetamide (MTBSTFA) + 1% tertbutyldimetheylchlorosilane (TBDMCS) (270144; Regis Technologies) and incubated at room temperature for 30 min. Samples were analyzed using an Agilent 7890B GC coupled to a 5977A mass detector. 3 μL of derivatized sample were injected into an Agilent HP-5ms Ultra Inert column, and the GC oven temperature increased at 15°C/min up to 215°C, followed by 5°C/min up to 260°C, and finally at 25°C/min up to 325°C. The MS was operated in split-less mode with electron impact mode at 70 eV. Mass range of 50–800 was analyzed, recorded at 1,562 mass units/second. The following metabolites were detected as TBDMS derivatives: citrate (m/z 591), succinate (m/z 289), fumarate (m/z 287), and malate (m/z 419). Data was analyzed using Agilent MassHunter Workstation Analysis and Agilent MSD ChemStation Data Analysis softwares. IsoPat2 software was used to adjust for natural abundance as previously performed.60
ATAC-seq analysis
Fifty thousand BMDMs were stimulated as described above. ATAC-seq library preparation were performed by Mayo Clinic Epigenomic Development Laboratory and Recharge Center. The libraries were sequenced by Mayo Clinic Genome Analysis Core using Illumina HiSeq 4000 Next Generation DNA sequencers. The bioinformatics analysis of the ATAC sequencing data was performed using the HiChIP pipeline.61 Paired-end sequencing reads were mapped to the mouse genome (mm10/GRCm38) using Burrows-Wheeler Aligner (BWA, v0.5.9). Post-mapping, read pairs mapping to multiple locations in the genome were filtered out using in-house scripts and only those read pairs which mapped uniquely to the genome were retained for downstream analysis. In addition, reads arising from PCR amplifications were discarded as duplicate reads using Picard MarkDuplicates (v1.67; http://broadinstitute.github.io/picard/). For identifying accessible and enriched chromatin regions (“peaks”) against a genome-wide random background, the software package “Model-Based Analysis of ChIP-Seq” (MACS2, v2.0.10)62 was used. Statistically significant peaks were selected for analysis using a false discovery rate (FDR) cutoff of %1%. Genes nearest to the location of the peaks and the distance (in basepairs) of these peaks from the transcription start sites (TSS) of the nearest genes were annotated using the HOMER (v4.10.1) software’s “annotatePeaks” utility. Additionally, to generate visualization tracks compatible with the Integrative Genomics Viewer (IGV) software, normalized tag density profiles with a window size of 200bp and a step size of 20bp were generated using Bedtools (v2.16.2)63 and in-house Perl scripts. For comparison and identification of differential peaks between conditions, the software package DiffBind (v2.14.0) was used at statistical significance thresholds of p value <0.05 and an absolute log2 fold change >2. Identification of enriched transcription factor motifs within significant differentially accessible regions was performed using HOMER’s “findMotifsGenome” utility. Gene ontology of genes associated with differential peals were performed using DAVID bioinformatics resource. The correlation and directionality between open chromatin in promoters and gene expression was assessed using a scatterplot of log2 transformed fold change values between “LPS” and “LPS + LA” from both ATAC-seq and RNA-seq experiments. Specifically, for the 6114 differentially expressed genes identified between “LPS” and “LPS + LA” (adjusted p value <0.05), the log2 transformed fold changes from RNA-Seq (X axis) and log2 transformed fold changes of the ATAC peaks in the promoters of those genes (Y axis) were plotted. If multiple ATAC peaks were identified in the promoter of a gene, an average of the fold changes within those peaks was used to represent an overall ATAC fold change for that gene.
Chromatin immunoprecipitation
BMDMs were fixed with 1% formaldehyde for 10 min, and DNA-protein complexes were sonicated to obtain fragments of approximately 200–500 bp. 100 μg of chromatin complexes were incubated with anti-H3K27ac or IgG antibody for overnight and precipitated with protein A agarose (Thermo Fisher Scientific). Chromatin complexes were reversed cross links with at 65°C. DNA was purified using phenol:chloroform extractions. The genome locations of the selected ATAC-seq differential regions and the primers used for qPCR are listed in Table S1.
siRNA knockdown of Nr4a1
RAW 264.7 cells were plated in 24-well plate one day before transfection. Briefly, 20 pmol of Nr4a1 siRNA (sc36110, Santa Cruz Biotechnology Inc.) or control siRNA (sc-37007, Santa Cruz Biotechnology Inc.) was mixed with Lipofectamine RNAiMAX (13778030, ThermoFisher) in Opti-MEM (31985062, Life technology corporation) according to manufacturer’s protocols, and added to cells. Six hours after transfection, medium was replaced with fresh medium. Knockdown efficiency was determined with real-time PCR after 48 h.
Mouse colitis model
DSS (2%) was administrated in drinking water for 8 days, then removed. For lactic acid enema, 100 μL of 10 mM lactic acid or vehicle only (PBS) was administrated for 3 days before and 3 days into DSS treatment via rectal enema using a flexible polyurethane catheter (20 G) (Exelint International) inserted into the colon 2.5 cm proximally into the anal verge while mice were anesthetized with 2% isoflurane, and mice were held thereafter in a head-down position for 60 s. After DSS exposure, mice returned to standard drinking water for 2 days before being euthanized for serum and tissue collection. The weight of each mouse was determined daily during DSS treatment and the 2-day recovery with standard drinking water afterward. For gene expression in colon tissue, 5 mm distal colon tissue were collected into RNA-Bee solution (Tel-Test) and homogenized using a bead mill homogenizer (Bead Ruptor, Omni International). Total RNA was extracted according to the RNA Bee manufacturer’s instructions (Tel-Test).
Colonic lamina propria cell preparation and analysis for colon macrophages and intracellular TNF-α
To isolate colon lamina propria cells, whole colon tissue was dissociated from epithelial cells and digested in RPMI-1640 medium containing 10% FBS, collagenase VIII and DNase I with incubation in gentleMACS Tissue Dissociator (Miltenyi) for 25 min at 37°C. These cells were filtered through 40-μm strainers, washed in PBS, and resuspended in ice-cold RPMI-1640 medium with 10% FBS. To measure colon macrophage population and intracellular TNF-α, cells were stained at 4°C in the dark using LIVE/DEAD Fixable Aqua Dead Cell Stain and the antibodies against CD45, linage markers CD3, CD19, NK1.1, Ly6G, and TER119, and monocyte/macrophage markers CD64, CD11b, Ly6C and MHCII, intracellular TNF-α. Colon macrophage and intracellular TNF-a were analyzed by flow cytometry performed on Attune Flow Cytometer; analysis was performed with FlowJo. Single live cells were gated for CD45+ CD11b+ lineage− CD64+ CD11b+ for monocyte and macrophage populations. Newly recruited monocytes (P1), maturing monocytes (P2), and mature macrophages (P3) were further identified as Ly6C+ MHCII−, Ly6C+ MHCII+, and Ly6C− MHCII+ respectively.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
GraphPad Prism 9 software was used for data analysis. Data are represented as mean ± SD. Statistical significance was determined by unpaired two-tailed Student’s t-test for two-group comparisons, and one-way or two-way ANOVA followed by Tukey’s or Sidak’s post-test for comparing interactions between groups with two or more stimulus. p value <0.05 was considered statistically significant.
Supplementary Material
Highlights.
Lactic acid is metabolized by macrophages to fuel the TCA cycle
Citrate production from lactic acid promotes histone H3K27 acetylation
Histone acetylation by lactic acid leads to immunosuppressive gene expression
Nr4a1 expression facilitates lactic acid to develop long-term immunosuppression
ACKNOWLEDGMENTS
We thank Metabolomics Core and C-SiG Epigenomics Core (supported by C-SiG grant P30DK084567) at Mayo Clinic for targeted metabolomics and ATAC-seq analysis, all members of the Ip lab and the Faubion lab for their enthusiasm and insightful discussion, Ruslan Medzhitov (Yale University) for bone-marrow cells from Retnla−/− mice, Zeng Hu (Mayo Clinic) for the use of Seahorse Analyzer, Wale Bamidele (Mayo Clinic) for BMS-303141, and Virginia Shapiro for recombinant mouse IL-4. The authors also thank the BGI (Cambridge, MA) for RNA-seq and analysis. This project was supported by a startup fund from Mayo Foundation for Medical Education and Research (W.K.E.I.) and an NIH grant 1R01DK130854-01A1 (W.K.E.I.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113746.
REFERENCES
- 1.Lavin Y, Winter D, Blecher-Gonen R, David E, Keren-Shaul H, Merad M, Jung S, and Amit I (2014). Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell 159, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bax BE, and Bloxam DL (1997). Energy metabolism and glycolysis in human placental trophoblast cells during differentiation. Biochim. Biophys. Acta 1319, 283–292. [DOI] [PubMed] [Google Scholar]
- 3.Flint HJ, Duncan SH, Scott KP, and Louis P (2015). Links between diet, gut microbiota composition and gut metabolism. Proc. Nutr. Soc. 74, 13–22. [DOI] [PubMed] [Google Scholar]
- 4.Brizel DM, Schroeder T, Scher RL, Walenta S, Clough RW, Dewhirst MW, and Mueller-Klieser W (2001). Elevated tumor lactate concentrations predict for an increased risk of metastases in head-and-neck cancer. Int. J. Radiat. Oncol. Biol. Phys. 51, 349–353. [DOI] [PubMed] [Google Scholar]
- 5.Okada T, Fukuda S, Hase K, Nishiumi S, Izumi Y, Yoshida M, Hagiwara T, Kawashima R, Yamazaki M, Oshio T, et al. (2013). Microbiota-derived lactate accelerates colon epithelial cell turnover in starvation-refed mice. Nat. Commun. 4, 1654. [DOI] [PubMed] [Google Scholar]
- 6.O’Hanlon DE, Moench TR, and Cone RA (2013). Vaginal pH and microbicidal lactic acid when lactobacilli dominate the microbiota. PLoS One 8, e80074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walenta S, Wetterling M, Lehrke M, Schwickert G, Sundfør K, Rofstad EK, and Mueller-Klieser W (2000). High lactate levels predict likelihood of metastases, tumor recurrence, and restricted patient survival in human cervical cancers. Cancer Res. 60, 916–921. [PubMed] [Google Scholar]
- 8.Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Watson MJ, Vignali PDA, Mullett SJ, Overacre-Delgoffe AE, Peralta RM, Grebinoski S, Menk AV, Rittenhouse NL, DePeaux K, Whetstone RD, et al. (2021). Metabolic support of tumour-infiltrating regulatory T cells by lactic acid. Nature 591, 645–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi O, and Akira S (2010). Pattern recognition receptors and inflammation. Cell 140, 805–820. [DOI] [PubMed] [Google Scholar]
- 11.Murray PJ (2017). Macrophage Polarization. Annu. Rev. Physiol. 79, 541–566. [DOI] [PubMed] [Google Scholar]
- 12.Jung J, Zeng H, and Horng T (2019). Metabolism as a guiding force for immunity. Nat. Cell Biol. 21, 85–93. [DOI] [PubMed] [Google Scholar]
- 13.Ip WKE, Hoshi N, Shouval DS, Snapper S, and Medzhitov R (2017). Anti-inflammatory effect of IL-10 mediated by metabolic reprogramming of macrophages. Science 356, 513–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Teijlingen Bakker N, and Pearce EJ (2020). Cell-intrinsic metabolic regulation of mononuclear phagocyte activation: Findings from the tip of the iceberg. Immunol. Rev. 295, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu PS, Wang H, Li X, Chao T, Teav T, Christen S, Di Conza G, Cheng WC, Chou CH, Vavakova M, et al. (2017). alpha-ketoglutarate orchestrates macrophage activation through metabolic and epigenetic reprogramming. Nat. Immunol. 18, 985–994. [DOI] [PubMed] [Google Scholar]
- 16.Fukuzumi M, Shinomiya H, Shimizu Y, Ohishi K, and Utsumi S (1996). Endotoxin-induced enhancement of glucose influx into murine peritoneal macrophages via GLUT1. Infect. Immun. 64, 108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Covarrubias AJ, Aksoylar HI, Yu J, Snyder NW, Worth AJ, Iyer SS, Wang J, Ben-Sahra I, Byles V, Polynne-Stapornkul T, et al. (2016). Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. Elife 5, e11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai Z, Ramesh V, and Locasale JW (2020). The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 21, 737–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mu X, Shi W, Xu Y, Xu C, Zhao T, Geng B, Yang J, Pan J, Hu S, Zhang C, et al. (2018). Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 17, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noe JT, Rendon BE, Geller AE, Conroy LR, Morrissey SM, Young LEA, Bruntz RC, Kim EJ, Wise-Mitchell A, Barbosa de Souza Rizzo M., et al. (2021). Lactate supports a metabolic-epigenetic link in macrophage polarization. Sci. Adv. 7, eabi8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, Hellerbrand C, Kastenberger M, Kunz-Schughart LA, Oefner PJ, et al. (2010). Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J. Immunol. 184, 1200–1209. [DOI] [PubMed] [Google Scholar]
- 22.Peter K, Rehli M, Singer K, Renner-Sattler K, and Kreutz M (2015). Lactic acid delays the inflammatory response of human monocytes. Biochem. Biophys. Res. Commun. 457, 412–418. [DOI] [PubMed] [Google Scholar]
- 23.Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N, Aranda Lopez P, Hahlbrock J, Muth S, Endo S, et al. (2018). Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 19, 1319–1329. [DOI] [PubMed] [Google Scholar]
- 24.Chen X, Jaiswal A, Costliow Z, Herbst P, Creasey EA, Oshiro-Rapley N, Daly MJ, Carey KL, Graham DB, and Xavier RJ (2022). pH sensing controls tissue inflammation by modulating cellular metabolism and endo-lysosomal function of immune cells. Nat. Immunol. 23, 1063–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, Siegwart DJ, and Wan Y (2017). Gpr132 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proc Natl Acad Sci USA 114, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Czimmerer Z, Halasz L, Daniel B, Varga Z, Bene K, Domokos A, Hoeksema M, Shen Z, Berger WK, Cseh T, et al. (2022). The epigenetic state of IL-4-polarized macrophages enables inflammatory cistromic expansion and extended synergistic response to TLR ligands. Immunity 55, 2006–2026.e2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lampropoulou V, Sergushichev A, Bambouskova M, Nair S, Vincent EE, Loginicheva E, Cervantes-Barragan L, Ma X, Huang SC, Griss T, et al. (2016). Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab 24, 158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, and Thompson CB (2009). ATP-citrate lyase links cellular metabolism to histone acetylation. Science 324, 1076–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang PV, Hao L, Offermanns S, and Medzhitov R (2014). The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc Natl Acad Sci USA 111, 2247–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, and Rudensky AY (2013). Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 504, 451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han SB, and Lee JK (2009). Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch. Pharm. Res. 32, 613–624. [DOI] [PubMed] [Google Scholar]
- 33.Grabiec AM, Krausz S, de Jager W, Burakowski T, Groot D, Sanders ME, Prakken BJ, Maslinski W, Eldering E, Tak PP, and Reedquist KA (2010). Histone deacetylase inhibitors suppress inflammatory activation of rheumatoid arthritis patient synovial macrophages and tissue. J. Immunol. 184, 2718–2728. [DOI] [PubMed] [Google Scholar]
- 34.Latham T, Mackay L, Sproul D, Karim M, Culley J, Harrison DJ, Hayward L, Langridge-Smith P, Gilbert N, and Ramsahoye BH (2012). Lactate, a product of glycolytic metabolism, inhibits histone deacetylase activity and promotes changes in gene expression. Nucleic Acids Res. 40, 4794–4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oki S, Ohta T, Shioi G, Hatanaka H, Ogasawara O, Okuda Y, Kawaji H, Nakaki R, Sese J, and Meno C (2018). ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-seq data. EMBO Rep. 19, e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ostuni R, Piccolo V, Barozzi I, Polletti S, Termanini A, Bonifacio S, Curina A, Prosperini E, Ghisletti S, and Natoli G (2013). Latent enhancers activated by stimulation in differentiated cells. Cell 152, 157–171. [DOI] [PubMed] [Google Scholar]
- 38.Zhan Y, Du X, Chen H, Liu J, Zhao B, Huang D, Li G, Xu Q, Zhang M, Weimer BC, et al. (2008). Cytosporone B is an agonist for nuclear orphan receptor Nur77. Nat. Chem. Biol. 4, 548–556. [DOI] [PubMed] [Google Scholar]
- 39.Duren RP, Boudreaux SP, and Conneely OM (2016). Genome Wide Mapping of NR4A Binding Reveals Cooperativity with ETS Factors to Promote Epigenetic Activation of Distal Enhancers in Acute Myeloid Leukemia Cells. PLoS One 11, e0150450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu X, Wang Y, Lu H, Li J, Yan X, Xiao M, Hao J, Alekseev A, Khong H, Chen T, et al. (2019). Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Netea MG, Domínguez-Andrés J, Barreiro LB, Chavakis T, Divangahi M, Fuchs E, Joosten LAB, van der Meer JWM, Mhlanga MM, Mulder WJM, et al. (2020). Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Halestrap AP (2013). The SLC16 gene family - structure, role and regulation in health and disease. Mol. Aspects Med. 34, 337–349. [DOI] [PubMed] [Google Scholar]
- 43.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, et al. (2013). Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 496, 238–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dichtl S, Lindenthal L, Zeitler L, Behnke K, Schlösser D., Strobl B., Scheller J., El Kasmi KC., and Murray PJ. (2021). Lactate and IL6 define separable paths of inflammatory metabolic adaptation. Sci. Adv. 7, eabg3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lith SC, and de Vries CJM (2021). Nuclear receptor Nur77: its role in chronic inflammatory diseases. Essays Biochem. 65, 927–939. [DOI] [PubMed] [Google Scholar]
- 47.Crawford PA, Sadovsky Y, Woodson K, Lee SL, and Milbrandt J (1995). Adrenocortical function and regulation of the steroid 21-hydroxylase gene in NGFI-B-deficient mice. Mol. Cell. Biol. 15, 4331–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Koenis DS, Medzikovic L, Vos M, Beldman TJ, van Loenen PB, van Tiel CM, Hamers AAJ, Otermin Rubio I, de Waard V, and de Vries CJM (2018). Nur77 variants solely comprising the amino-terminal domain activate hypoxia-inducible factor-1alpha and affect bone marrow homeostasis in mice and humans. J. Biol. Chem. 293, 15070–15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Patino-Martinez E, Solis-Barbosa MA, Santana E, Gonzalez-Dominguez E, Segovia-Gamboa NC, Meraz-Rios MA, Cordova EJ, Valdes J, Corbi AL, and Sanchez-Torres C (2022). The Nurr7 agonist Cytosporone B differentially regulates inflammatory responses in human polarized macrophages. Immunobiology 227, 152299. [DOI] [PubMed] [Google Scholar]
- 50.Hanna RN, Shaked I, Hubbeling HG, Punt JA, Wu R, Herrley E, Zaugg C, Pei H, Geissmann F, Ley K, and Hedrick CC (2012). NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 110, 416–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koenis DS, Medzikovic L, van Loenen PB, van Weeghel M, Huveneers S, Vos M, Evers-van Gogh IJ, Van den Bossche J, Speijer D, Kim Y, et al. (2018). Nuclear Receptor Nur77 Limits the Macrophage Inflammatory Response through Transcriptional Reprogramming of Mitochondrial Metabolism. Cell Rep. 24, 2127–2140.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaked I, Hanna RN, Shaked H, Chodaczek G, Nowyhed HN, Tweet G, Tacke R, Basat AB, Mikulski Z, Togher S, et al. (2015). Transcription factor Nr4a1 couples sympathetic and inflammatory cues in CNS-recruited macrophages to limit neuroinflammation. Nat. Immunol. 16, 1228–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu H, Li XM, Wang JR, Gan WJ, Jiang FQ, Liu Y, Zhang XD, He XS, Zhao YY, Lu XX, et al. (2016). NUR77 exerts a protective effect against inflammatory bowel disease by negatively regulating the TRAF6/TLR-IL-1R signalling axis. J. Pathol. 238, 457–469. [DOI] [PubMed] [Google Scholar]
- 54.Hamers AA, van Dam L, Teixeira Duarte JM, Vos M, Marinkovic G, van Tiel CM, Meijer SL, van Stalborch AM, Huveneers S, Te Velde AA, et al. (2015). Deficiency of Nuclear Receptor Nur77 Aggravates Mouse Experimental Colitis by Increased NFkappaB Activity in Macrophages. PLoS One 10, e0133598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sun S, Xu X, Liang L, Wang X, Bai X, Zhu L, He Q, Liang H, Xin X, Wang L, et al. (2021). Lactic Acid-Producing Probiotic Saccharomyces cerevisiae Attenuates Ulcerative Colitis via Suppressing Macrophage Pyroptosis and Modulating Gut Microbiota. Front. Immunol. 12, 777665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rodríguez-Viso P, Domene A, Vélez D, Devesa V, Zúñiga M, and Monedero V (2023). Lactic acid bacteria strains reduce in vitro mercury toxicity on the intestinal mucosa. Food Chem. Toxicol. 173, 113631. [DOI] [PubMed] [Google Scholar]
- 57.Wang M, Fan Z, Chen D, Yu B, He J, Yu J, Mao X, Huang Z, Luo Y, Luo J, et al. (2023). Dietary lactate supplementation can alleviate DSS-induced colitis in piglets. Biomed. Pharmacother. 158, 114148. [DOI] [PubMed] [Google Scholar]
- 58.Yan Y, Li X, Yang Q, Zhang H, Hettinga K, Li H, and Chen W (2022). Dietary d-Lactate Intake Facilitates Inflammatory Resolution by Modulating M1 Macrophage Polarization. MolMol. Nutr. Food Res. 66, e2200196. [DOI] [PubMed] [Google Scholar]
- 59.Wilkins J, Sakrikar D, Petterson XM, Lanza IR, and Trushina E (2019). A comprehensive protocol for multiplatform metabolomics analysis in patient-derived skin fibroblasts. Metabolomics. 15, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiese EK, Hitosugi S, Loa ST, Sreedhar A, Andres-Beck LG, Kurmi K, Pang YP, Karnitz LM, Gonsalves WI, and Hitosugi T (2021). Enzymatic activation of pyruvate kinase increases cytosolic oxaloacetate to inhibit the Warburg effect. Nat. Metab. 3, 954–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yan H, Evans J, Kalmbach M, Moore R, Middha S, Luban S, Wang L, Bhagwate A, Li Y, Sun Z, et al. (2014). HiChIP: a high-throughput pipeline for integrative analysis of ChIP-Seq data. BMC Bioinf. 15, 280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ATAC-seq data in this study have been deposited at GEO and are publicly available. The accession number is listed in the key resource table.
This study did not generate any unique code.
Any additional information required to reanalyze the data reported in this paper is available from lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Hamster monoclonal anti-mouse TNF-α | eBioscience | Cat # 14-7423-81; RRID: AB_2721314 |
| Rabbit polyclonal biotinylated anti-mouse TNF-α | eBioscience | Cat# 13-7341-81; RRID: AB_466950 |
| Rat monoclonal anti-mouse IL-6 | eBioscience | Cat# 14-7061-81; RRID: AB_468422 |
| Rat monoclonal biotinylated anti-mouse IL-6 | BioLegend | Cat# 504601; RRID: AB_2127458 |
| Hamster monoclonal anti-mouse IL-1β | eBioscience | Cat# 14-7012-81; RRID: AB_468396 |
| Rabbit polyclonal anti-mouse IL-1β | eBioscience | Cat# 13-7112-81; RRID: AB_466924 |
| Rat monoclonal anti-mouse IL-6, PerCP-eFluor 710 | Invitrogen | Cat# 46-7061-82; RRID: AB_2573829 |
| Rat monoclonal anti-mouse TNF-α, PE-efluor 610 | Invitrogen | Cat# 61-7321-82; RRID: AB_2574666 |
| Rat monoclonal anti-mouse IL-1β, PE-Cy7 | Invitrogen | Cat# 25-7114-82; RRID: AB_2573526 |
| Rat monoclonal anti-mouse iNOS, Alexa Fluor 700 | Invitrogen | Cat# 56-5920-80; RRID: AB_2848473 |
| Rat monoclonal anti-mouse F4/80, Super Bright 780 | Invitrogen | Cat# 78-4801-82; RRID: AB_2802477 |
| Mouse monoclonal anti-mouse CD64 (FcγRI), APC | BioLegend | Cat# 139305; RRID: AB_11219205 |
| Rat monoclonal anti-mouse CD11b, Super Bright 645 | Invitrogen | Cat# 64-0112-82; RRID: AB_2662387 |
| Rat monoclonal anti-mouse Ly6C, APC/Cyanine7 | BioLegend | Cat# 128025; RRID: AB_10643867 |
| Rat monoclonal anti-mouse MHC Class II (I-A), PE | Invitrogen | Cat# 12-5321-82; RRID: AB_465928 |
| Rat monoclonal anti-mouse CD45, Super Bright SB600 | Invitrogen | Cat# 63-0451-82; RRID: AB_2637149 |
| Mouse monoclonal anti-mouse NK1.1, Pacific Blue | BioLegend | Cat# 108721; RRID: AB_2234352 |
| Rat monoclonal anti-mouse Ly6G, Pacific Blue | BioLegend | Cat# 127611; RRID: AB_1877212 |
| Rat monoclonal anti-mouse TER119/Erythroid cells, Pacific Blue | BioLegend | Cat# 116231; RRID: AB_2149212 |
| Rat monoclonal anti-mouse CD3, Pacific Blue | BioLegend | Cat# 100213; RRID: AB_493644 |
| Rat monoclonal anti-mouse CD19, Pacific Blue | BioLegend | Cat# 115526; RRID: AB_493341 |
| Rabbit monoclonal anti-phospho-NF-kB p65 (Ser536) (93H1) | Cell Signaling Technology | Cat# 3033; RRID: AB_331284 |
| Rabbit anti-IκBα | Cell Signaling Technology | Cat# 9242; RRID: AB_331623 |
| Mouse monoclonal anti-phospho-IκBα (Ser32/36) (5A5) | Cell Signaling Technology | Cat# 9246; RRID: AB_2267145 |
| Mouse monoclonal anti-phospho-p38 MAPK (Thr180/Tyr182) (28B10) | Cell Signaling Technology | Cat# 9216; RRID: AB_331296 |
| Rabbit polyclonal anti-phospho-LDHA (Tyr10) | Cell Signaling Technology | Cat# 8176; RRID: AB_11220238 |
| Rabbit monoclonal anti-H3, acetyl (Lys27) (D5E4) XP | Cell Signaling Technology | Cat# 8173; RRID: AB_10949503 |
| Rabbit monoclonal anti-H3, acetyl (Lys18) (D8Z5H) | Cell Signaling Technology | Cat# 13998; RRID: AB_2783723 |
| Rabbit monoclonal anti-H3, acetyl (Lys14) (D4B9) | Cell Signaling Technology | Cat# 7627; RRID: AB_10839410 |
| Rabbit monoclonal anti-H3, acetyl (Lys9) (C5B11) | Cell Signaling Technology | Cat# 9649; RRID: AB_823528 |
| Rabbit monoclonal anti-H3, trimethyl (Lys27) (C36B11) | Cell Signaling Technology | Cat# 9733; RRID: AB_2616029 |
| Mouse monoclonal anti-α-tubulin (DM1A) | Cell Signaling Technology | Cat# 3873; RRID: AB_1904178 |
| Rabbit recombinant monoclonal anti-L-lactyl lysine | PTM BIO | Cat# PTM-1401RM; RRID: AB_2942013 |
| Rabbit recombinant monoclonal anti-H3, lactyl (Lys18) | PTM BIO | Cat# PTM-1406RM; RRID: AB_2909438 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Lipopolysaccharides from Escherichia coli O26:B6 | Sigma | L3755 |
| L-(+)-Lactic acid | Sigma | L6402 |
| ATP | Sigma | A6419 |
| L-Lactic acid-13C3 | Sigma | 746258 |
| cyano-4-hydroxycinnamic acid | Sigma | S8612 |
| MDL-12,330A hydrochloride | Sigma | M182 |
| Trichostatin A | Sigma | T8552 |
| BMS-303141 | Sigma | SML0783 |
| DIM-C-pPhOH | Selleck Chemicals | S6799 |
| Recombinant mouse TNF-α | PeproTech | 315-01A |
| Recombinant mouse IL-6 | PeproTech | 216-16 |
| Recombinant mouse IL-1β | PeproTech | 211-11B |
| Recombinant mouse RELM-α | PeproTech | 450-26 |
| 2-deoxy-D-glucose | Cayman Chemical | 14325 |
| D-glucose-13C6 | Cambridge Isotope | CLM-1396 |
| BD GolgiPlug | Fisher Scientific | BDB555029 |
| BD Cytofix/Cytoperm solution | Fisher Scientific | BDB554714 |
| Lipofectamine RNAiMAX | Thermo Fisher Scientific | 13778030 |
| Cytosporone B | R&D Systems | 5459 |
| Dextran sulfate sodium (molecular mass 36–50 kDa) | MP Biomedicals | MFCD00081551 |
| Collagenase VIII | Sigma | C2139 |
| DNase I | ROCHE | 10104159001 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Dual-Glo Luciferase Reporter Assay | Promega | E2920 |
| Prostaglandin E2 (PGE2) Parameter Assay Kit | R&D Systems | KGE004B |
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermo Fisher Scientific | L34965 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq | This paper | GEO: GSE253050 |
| ATAC-seq | This paper | GEO: GSE253050 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| RAW264.7 | American Type Culture Collection | TIB-71 |
| 293T | American Type Culture Collection | CRL-3216 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6 mice | Jackson Laboratory | 000664 |
| C57BL/6NJ mice (control mice for Acod1−/− mice) | Jackson Laboratory | 005304 |
| Acod1−/− mice | Jackson Laboratory | 029340 |
| Nr4a1wt control mice | Jackson Laboratory | 100903 |
| Nr4a1−/− mice | Jackson Laboratory | 06187 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Primer (qPCR): mouse Arg1 Forward: 5’-GTGAAGAACCCACGGTCTGT-3’ | This paper | N/A |
| Primer (qPCR): mouse Arg1 Reverse: 5’-CTGGTTGTCAGGGGAGTGTT-3’ | This paper | N/A |
| Primer (qPCR): mouse Gapdh Forward: 5’-GGTGCTGAGTATGTCGTGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Gapdh Reverse: 5’-CGGAGATGATGACCCTTTTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il1b Forward: 5’-CCCAAGCAATACCCAAAGAA-3’ | This paper | N/A |
| Primer (qPCR): mouse Il1b Reverse: 5’-GCTTGTGCTCTGCTTGTGAG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il6 Forward: 5’-TGTTCTCTGGGAAATCGTGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Il6 Reverse: 5’-AAGTGCATCATCGTTGTTCATACA-3’ |
This paper | N/A |
| Primer (qPCR): mouse Il12b Forward: 5’-AGGTCACACTGGACCAAAGG-3’ | This paper | N/A |
| Primer (qPCR): mouse Il12b Reverse: 5’-TGGTTTGATGATGTCCCTGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Nos2 Forward: 5’-CACCTTGGAGTTCACCCAGT-3’ | This paper | N/A |
| Primer (qPCR): mouse Nos2 Reverse: 5’-ACCACTCGTACTTGGGATGC-3’ | This paper | N/A |
| Primer (qPCR): mouse Nr4a1 Forward: 5’-TTGAGTTCGGCAAGCCTACC-3’ | This paper | N/A |
| Primer (qPCR): mouse Nr4a1 Reverse: 5’-GTGTACCCGTCCATGAAGGTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Ptgs2 Forward: 5’-GCGAGCTAAGAGCTTCAGGA-3’ | This paper | N/A |
| Primer (qPCR): mouse Ptgs2 Reverse: 5’-TCATACATTCCCCACGGTTT-3’ | This paper | N/A |
| Primer (qPCR): mouse Retnla Forward: 5’-TGCTGGGATGACTGCTACTG-3’ | This paper | N/A |
| Primer (qPCR): mouse Retnla Reverse: 5’-CTGGGTTCTCCACCTCTTCA-3’ | This paper | N/A |
| Primer (qPCR): mouse Tnf Forward: 5’-GCACAGAAAGCATGATCCG-3’ | This paper | N/A |
| Primer (qPCR): mouse Tnf, Reverse: 5’-GCCCCCCATCTTTTGGG-3’ | This paper | N/A |
| Primer (qPCR): mouse Vegf Forward: 5’-CGAGCTCATGGACGGGTGAG-3’ | This paper | N/A |
| Primer (qPCR): mouse Vegf Reverse: 5’-GCCTGGGACCACTTGGCAT-3’ | This paper | N/A |
| ChIP-qPCR primers (see Table S1) | This paper | N/A |
| NR4A1 (Nur77) siRNA | Santa Cruz Biotechnology | Sc36110 |
| Control siRNA | Santa Cruz Biotechnology | sc-37007 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism 9 | GraphPad Software Inc. | RRID: SCR_002798 |
| FlowJo 10 | Becton Dickinson & Company | RRID: SCR_008520 |
| ImageJ | NIH | RRID: SCR_003070 |