

Abstract
Background
Environmental factors can influence epigenetic regulation, including DNA methylation, potentially contributing to systemic lupus erythematosus (SLE) development and progression. We compared methylation of the B cell costimulatory CD70 gene, in persons with lupus and controls, and characterized associations with age.
Results
In 297 adults with SLE and 92 controls from the Michigan Lupus Epidemiology and Surveillance (MILES) Cohort, average CD70 methylation of CD4+ T cell DNA across 10 CpG sites based on pyrosequencing of the promoter region was higher for persons with SLE compared to controls, accounting for covariates [β=2.3, p=0.011]. Using Infinium MethylationEPIC array data at 18 CD70-annoted loci (CD4+ and CD8+ T cell DNA), sites within the promoter region tended to be hypomethylated in SLE, while those within the gene region were hypermethylated. In SLE but not controls, age was significantly associated with pyrosequencing-based CD70 methylation: for every year increase in age, methylation increased by 0.14 percentage points in SLE, accounting for covariates. Also within SLE, CD70 methylation approached a significantly higher level in Black persons compared to White persons (β=1.8, p=0.051).
Conclusions
We describe altered CD70 methylation patterns in T lymphocyte subsets in adults with SLE relative to controls, and report associations particular to SLE between methylation of this immune-relevant gene and both age and race, possibly a consequence of “weathering” or accelerated aging which may have implications for SLE pathogenesis and potential intervention strategies.
Keywords: epigenetics, autoimmunity, aging, epidemiology, immunoepidemiology, TNFSF7, CD4+, CD8+
INTRODUCTION
Systemic lupus erythematosus (SLE) is a prototypic autoimmune disease for which specific etiologic agents remain largely unclear. Several lines of evidence suggest that gene-environment interactions leading to inhibition of DNA methylation and overexpression of methylation-sensitive immune genes contribute to SLE development and progression (1,2). Although SLE has traditionally been referred to as a disease of reproductive-age women, recent epidemiologic evidence upends this perception as overly simplistic (3). Whereas Black women experience peak SLE incidence in their 20s, highest incidence for White women generally occurs in their 50s (around the average age of menopause) and for men in their 70s (4–7). One interpretation is that aging is an integral component of SLE risk, albeit unevenly expressed across groups. The accelerated development of SLE in Black women is compatible with the “weathering hypothesis”, which depicts the concept of faster, stress-related biological aging in historically marginalized groups (8,9).
DNA methylation is an epigenetic mechanism that silences gene expression by promoting a chromatin configuration inaccessible to transcription factors. In dividing T cells, suppression of DNA methylation—through direct inhibition of DNMT1 methyltransferase, or reduction of PkC-δ signaling by oxidative stress or agents associated with drug-induced lupus—converts normal CD4+ T cells into autoreactive, cytotoxic, and proinflammatory T cells that can cause lupus-like disease in animal models (10–12). CD70, a B cell costimulatory ligand encoded by the CD70 gene (also known as TNFSF7), has been depicted as hypomethylated and overexpressed in T cells both in lupus and when treated with lupus-inducing drugs (13,14). Its overexpression leads to increased B cell co-stimulation and concomitant immunoglobulin overproduction. Prior studies of DNA methylation in lupus have been limited by relatively small sample sizes in tertiary care settings and lack diversity reflective of the general population. While methylation of CD70 has not been detailed in association with age, CD70 gene expression in T lymphocyte subsets has been correlated with age in healthy persons, and CD70-expressing T cells from elderly persons demonstrate increased susceptibility to apoptosis (15).
We have performed a population-based, immunoepidemiology study characterizing CD70 DNA methylation patterns in T lymphocyte subsets in SLE and controls. Methylation, in contrast to gene expression, has the potential to serve as a more stable marker of environmental insults or alterations at this gene. As such, we have interrogated CD70 methylation associations with age and race, which may have implications related to weathering and SLE expression.
PARTICIPANTS AND METHODS
Study Population
This study was based on the Michigan Lupus Epidemiology and Surveillance (MILES) Cohort in southeastern Michigan. SLE cases were recruited from the MILES Surveillance Registry, one of the National US Lupus Registries in the Centers for Disease Control & Prevention network (6,16). Population-based controls were recruited from a random sample of households in the same geographic region as cases, detailed elsewhere (17,18). Briefly, controls were frequency-matched to cases with respect to race and age; male controls were over-sampled to achieve equivalent numbers of males per group, given the rarity of male lupus. Ethics approval was obtained from the Institutional Review Boards of the University of Michigan and Michigan Department of Health and Human Services. Study participants provided written, informed consent.
Data and specimen collection
Data from structured interviews and questionnaires, and biospecimens from the baseline visit were utilized. Sociodemographics, including race and ethnicity, were self-reported following methods similar to the National Health and Nutrition Examination Survey (19). Health insurance (past 12 months) was categorized as Medicaid, Medicare, private/other, or none, and household income in relation to US median income. Medication history (current/ever usage) included corticosteroids, antimalarials, methotrexate, azathioprine, and cyclophosphamide. Lupus activity and damage were assessed by validated instruments for epidemiologic settings—the Systemic Lupus Activity Questionnaire (SLAQ) (20) and Lupus Damage Index Questionnaire (LDIQ) (21), respectively. The SLAQ measures disease activity during the preceding three months; the LDIQ assesses SLE-related, irreversible organ damage. Higher scores indicate higher levels of activity or damage.
T lymphocyte purification and DNA isolation
From blood in lavender/EDTA vacutainers, peripheral blood mononuclear cells (PBMCs) were isolated by density gradient centrifugation using Ficoll-Paque Plus (GE Healthcare). T cell subsets were isolated using magnetic bead technology (Miltenyi Biotec, CD4+ and CD8+ isolation kits). Genomic DNA was purified by “ALL-Prep” kit (Qiagen). Extracted DNA concentration was measured by NanoDrop spectrophotometer (Thermo Scientific) and stored at −80°C until bisulfite conversion.
Gene-specific DNA methylation via pyrosequencing
Assays were performed in blinded fashion with respect to participant characteristics. Pyrosequencing assessed methylation of 10 consecutive cytosine-phosphate-guanine (CpG) sites upstream of the CD70 transcription start site (TSS) in CD4+ T cell DNA (Supplementary Figure S1). Genomic DNA was bisulfite-converted using EpiTect Bisulfite Kit (Qiagen) per manufacturer protocol. HotStartTaq Master Mix (Qiagen) was used for amplification of the CD70 promoter by forward and reverse biotinylated primers, using approximately 40ng bisulfite-converted DNA. Primers were designed with PyroMark Assay Design Software v2.0 (Table S1). Percent methylated cells was quantified by pyrosequencing (22), using a PyroMark MD instrument (Qiagen) and calculated by Pyro Q-CpG Software (Qiagen). Each batch contained standard controls for quality control purposes, including no-DNA controls and controls with known methylation status. Additional quality control included ascertainment of complete bisulfite conversion and sufficient signal over background noise. Samples/CpG sites failing quality control were excluded. All samples were pyrosequenced in duplicate, with passing duplicate reads averaged. High precision was obtained; average % relative standard deviation (RSD) range was 1.3–4.8% across CpG sites. Duplicates with >15% RSD were excluded; one sample was dropped with this standard.
DNA Methylation via Infinium MethylationEPIC
DNA methylation at >800,000 CpG sites throughout the genome was measured via Infinium MethylationEPIC (‘EPIC’) array (Illumina) (23). We utilized EPIC data annotated to CD70 (18 CpG sites throughout the gene and its promoter region). After confirmatory DNA quantification by Picogreen, 250ng genomic DNA was bisulfite-converted at the UM Epigenomics Core using the Zymo EZ DNA Methylation kit. Samples were hybridized to EPIC chips and scanned at the UM Advanced Genomics Core. Raw image files were read into R using the minfi Bioconductor package (24). Quality control was performed via the ENmix Bioconductor package (25); three samples failing quality control were removed. After filtering failed samples and probes, probe intensities were background-corrected using out-of-band intensities and dye-corrected using the RELIC algorithm; inter-array normalization was performed by quantile normalizing methylated and unmethylated intensities (25). Probe-type biases were corrected by beta-mixture quantile normalization method (26). From this processed dataset, beta values (representing proportion methylated) from CD70-annotated sites were extracted and used in analyses. One of the 18 sites (probe cg22633597, chr19:6591674) was also covered in our pyrosequencing assay (pyrosequencing CpG site #1).
Statistical Analysis
Descriptive statistics
Continuous variables were summarized with means and standard deviations or medians and interquartile ranges as indicated by data distributions, and categorical variables as frequencies and percentages. Two sample t-tests were used to compare means between groups, and Pearson’s chi-squared test for categorical variable comparisons. CpG site-specific analyses included all participants with available data for the given site.
Summary measure of pyrosequencing-based intra-individual CD70 methylation status
As values from pyrosequencing data across CpG sites varied substantially, we centered and standardized each of the 10 sites using their respective means and variances, so that the measurements had the same scale (z-score): standardized CD70site(i) = [CD70site(i) – mean(CD70site(i))] / standard deviation(CD70site(i)), where i = CpG site 1,2,3…10. From the standardized data, the intra-person mean across sites was calculated. For participants with missing methylation values for ≥1 CpG site, the intra-person mean was calculated based on available sites. Primary analyses for mean methylation included all study participants; secondary analyses were restricted to the subset of participants with complete methylation data for all 10 sites.
Statistical analysis for pyrosequencing data
Univariate and multivariable linear regression were used to examine the association between age and methylation, and analyses were stratified according to case/control status. Separate univariate models were run for each of the 10 CpG sites as the outcome, as well as using intra-person mean methylation across all sites as the primary outcome for pyrosequencing data. Multivariable models included covariates for sex, race, health insurance, and income; for models specific to lupus cases, lupus disease activity and damage were additionally included as covariates.
Statistical analysis for EPIC Data
Beta values from 18 CpG sites annotated to the CD70 promoters, gene body, or introns were converted to percent methylation (0–100%). Independent t-tests were performed to compare DNA methylation levels between SLE cases and controls, separately for CD4+ and CD8+ samples. If Levene’s test for homogeneity of variances was significant (p<0.05), Welch’s two-sample t-test was used. Covariate-adjusted linear regression assessed associations between DNA methylation at each CpG site with case-control status and age. The interaction between age and case-control status was evaluated in secondary modelling.
REDCap electronic data capture tools hosted at the University of Michigan were used for data management (27). Stata v.15 (StataCorp, College Station, TX) and R v.3.5.1 (R Foundation) were utilized for statistical analyses.
RESULTS
Study population characteristics
This study included 389 participants (297 SLE, 92 controls) from the full cohort of 462 participants with SLE and 192 controls, who consented to having DNA isolated at baseline and who had available biospecimens. For the analyses based on pyrosequencing assays, CD4+ DNA from 386 participants (296 SLE cases, 90 controls) was available; five were excluded due to assays that did not pass quality control, resulting in a sample size of 381 (291 SLE, 90 controls). EPIC assays were completed on CD4+ T cell DNA for 388 participants (296 SLE, 92 controls), and on CD8+ T cell DNA for 185 participants (165 SLE, 20 controls). Participant characteristics are summarized in Table 1; sociodemographic composition for this study was similar to that for the full cohort, reported elsewhere (17,18).
Table 1.
Characteristics of the study population, according to SLE or control status
| CD4+ T cell DNA analyses | CD8+ T cell DNA analyses | |||||
|---|---|---|---|---|---|---|
| SLE n=296 | Controls n=92 | p-value* | SLE n=165 | Controls n=20 | p-value* | |
|
|
|
|||||
| Age (years) | 53.8 (12.5) | 55.8 (12.9) | 0.2 | 54.5 (11.6) | 55.1 (14.7) | 0.9 |
| Sex | 0.05 | 0.9 | ||||
| female | 274 (92.6) | 79 (85.9) | 155 (93.9) | 19 (95) | ||
| male** | 22 (7.4) | 13 (14.1) | 10 (6.1) | 1 (5) | ||
| Race | 0.8 | 0.2 | ||||
| White | 156 (52.7) | 48 (52.2) | 84 (50.9) | 14 (70) | ||
| Black | 131 (44.3) | 40 (43.5) | 74 (44.8) | 6 (30) | ||
| other/unknown | 9 (3.0) | 4 (4.3) | 7 (4.2) | 0 (0) | ||
| Hispanic/Latino | 11 (3.7) | 1 (1.1) | 0.2 | 7 (4.2) | 0 (0) | 0.4 |
| Insurance | 0.4 | 0.7 | ||||
| private/other | 140 (47.3) | 39 (42.4) | 78 (47.3) | 9 (45.0) | ||
| Medicaid | 60 (20.3) | 26 (28.3) | 33 (20.0) | 4 (20.0) | ||
| Medicare | 89 (30.1) | 24 (26.1) | 52 (31.5) | 6 (30.0) | ||
| none | 7 (2.4) | 3 (3.3) | 2 (1.2) | 1 (5.0) | ||
| Lupus-specific variables | ||||||
| SLAQ total score | 12.1 ± 7.4 | -- | -- | 12.6 (7.4) | -- | -- |
| SLAQ flare (past 3 months) | ||||||
| no | 132 (45.2) | -- | 66 (41.0) | -- | ||
| mild | 92 (31.5) | -- | 56 (34.8) | -- | ||
| moderate | 56 (19.2) | -- | 34 (21.1) | -- | ||
| severe | 12 (4.1) | -- | 5 (3.1) | -- | ||
| LDIQ total score | 5.4 (4.0) | -- | 5.8 (4.2) | -- | ||
| SLE duration (years) (median, IQR) | 20 (14, 25) | -- | 20 (15, 26) | -- | ||
SLAQ=Systemic Lupus Activity Questionnaire; LDIQ = Lupus Damage Index Questionnaire; IQR = interquartile range
Data are expressed as mean (SD) for continuous data, unless otherwise noted, and frequency (percent) for categorical data.
p-values calculated by two sample t-tests (continuous variables) or Pearson's chi-squared test (categorical variables)
Males were over-sampled in controls
CD70 methylation in SLE compared to controls
When examining DNA methylation at individual CpG sites assessed by pyrosequencing, for both raw and standardized values, percent methylation was significantly lower in lupus compared to controls in one of the ten sites (site 3), higher in lupus than controls in four sites (sites 5–7, 9), and similar between groups for the remaining 5 sites; Figure 1. Overall, mean DNA methylation across the 10 sites was higher in SLE; Supplementary Figure S2. This held true both for the raw (SLE 50.1%, control 47.8%, p=0.01; S2a) and standardized (SLE 0.05, control −0.14, p=0.06; S2b) data. There was no evidence that medications commonly used in SLE (current or ever) were associated with methylation levels (data not shown). 80% of the study population had DNA methylation data for all 10 sites, which was similar between cases (80.1%) and controls (77.8%). In the subset of participants with complete data for the 10 sites, overall hypermethylation was again observed in SLE relative to controls (mean standardized methylation: SLE 0.04, control −0.18; p=0.04). Average methylation across sites was two percentage points higher for persons with SLE compared to controls in multivariable models adjusting for sex, race, and age [β=2.3 (95% CI 0.5, 4.0), p=0.011], and standardized DNA methylation likewise remained higher on average for SLE [β=0.2 (95% CI 0.0, 0.4), p=0.049].
Figure 1. CD4+ T cell DNA methylation of CD70 at 10 CpG sites, based on pyrosequencing, for SLE cases compared to controls.
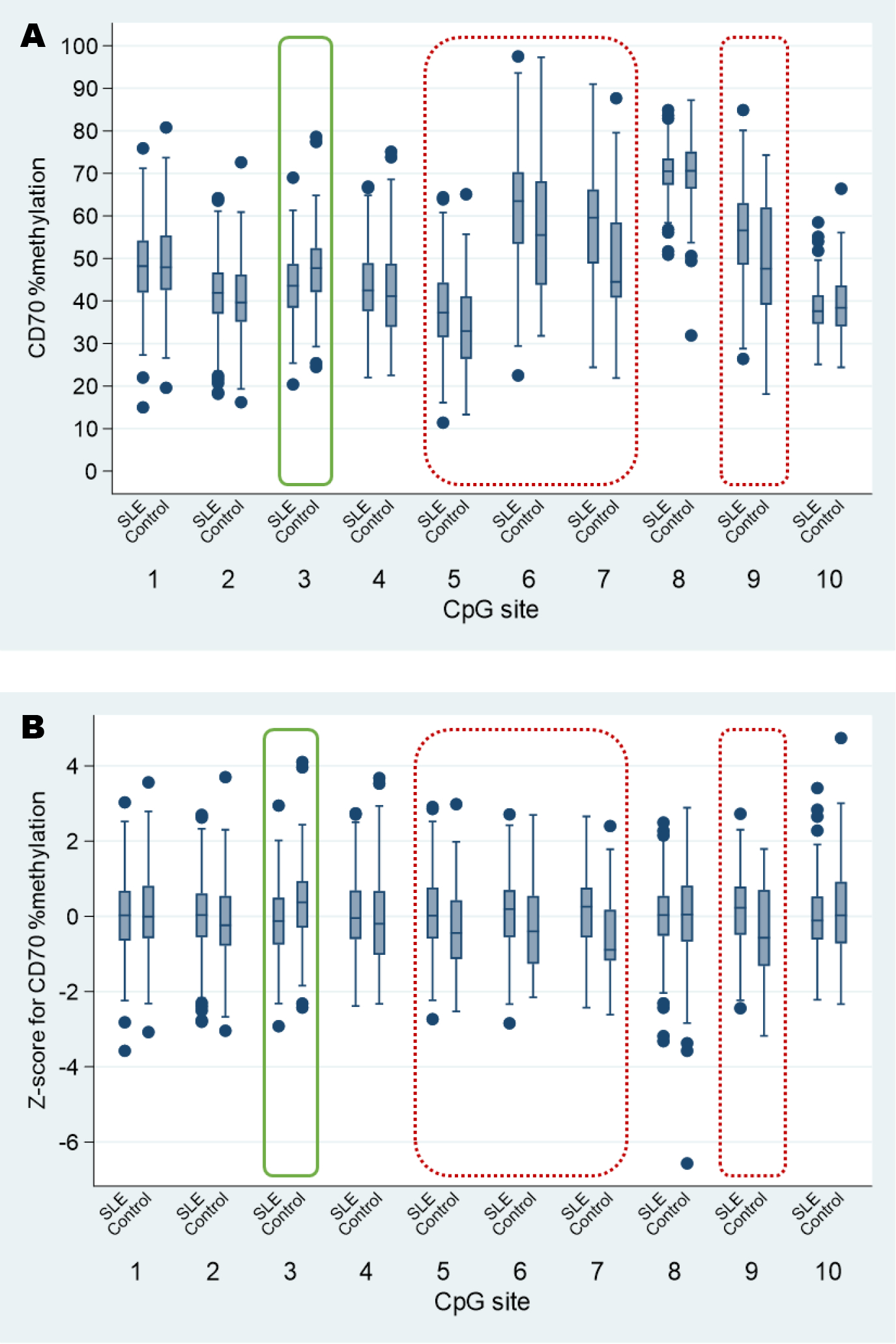
Data distributions differ across CpG sites for raw DNA methylation data (Panel A), expressed as % methylation; sites such as CD70site(8), which is much higher in relation to the other sites, will dominate the mean. Thus, we utilized standardized values expressed as a z-score for % methylation (Panel B) for further analyses. For both raw and standardized data, CD4+ T cell DNA from SLE was significantly hypomethylated relative to controls at site 3 (solid green box), and significantly hypermethylated at sites 5–7 and 9 (dashed red boxes); p-values ≤0.001 for these sites.
Boxplots: lower and upper hinges of box represent 25th & 75th percentiles; horizontal line within box represents median; navy dots represent outliers (outer 2.5 percentiles).
The EPIC array covered CpG sites interspersed throughout a 6090 bp long section of CD70 including part of the promoter (9 CpG sites) and gene body and introns (9 CpG sites). Among these sites, seven were significantly different in cases compared to controls in CD4+ T cells (Figure 2a), with 3 of these also significantly different in CD8+ T cells (Figure 2b). In adjusted models including sex, race, and age, eight CpG sites were significantly different between cases and controls in CD4+ T cells and three in CD8+ T cells. In general, the direction of association was the same whether in CD4+ or CD8+ cells. In persons with SLE compared to controls, CpG sites within the promoter region tended to be hypomethylated, while those within the gene were hypermethylated (Supplementary Table S2).
Figure 2. DNA methylation level at 18 CpG sites annotated to CD70 included on the MethylationEPIC Array in SLE cases and controls, assessed in CD4+ (Panel A) and CD8+ (Panel B) T cells.
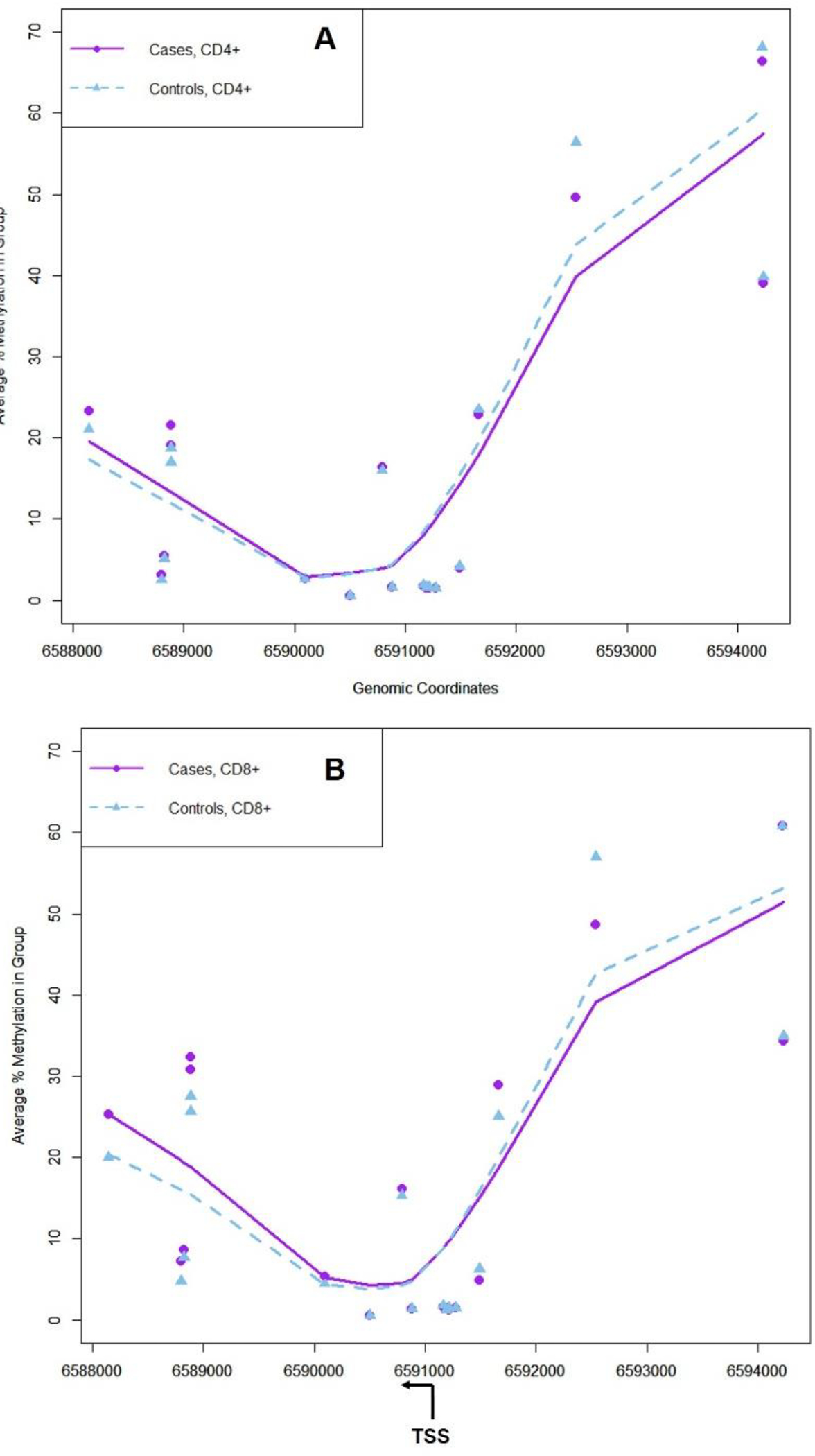
Average DNA methylation (%) in SLE case and control groups is shown for loci annotated to CD70 included on the Illumina Infinium EPIC array. Triangles are the average for all cases and squares for all controls. The location of the transcription start site (TSS) is shown along with direction of transcription. CpG sites that had significantly different DNA methylation between groups according to t-tests (p<0.05) are denoted with *. LOESS lines were fit to the scatterplot for each group. Genomic coordinates are according to GRCh38/hg38 build.
Age and CD70 DNA methylation
Linear regression results examining the marginal associations between age and pyrosequencing-based CD70 methylation at individual CpG sites are presented according to case and control status in Figure 3a. In SLE, positive associations between age and methylation were consistently detected for each of the 10 sites, whereby CpG site-specific methylation increased as age increased (p≤0.02 for all sites). In contrast, among controls, negative associations were generally observed, but none reached significance.
Figure 3. Associations between CD70 methylation (pyrosequencing) and age in CD4+ T cell DNA, for SLE cases and controls.
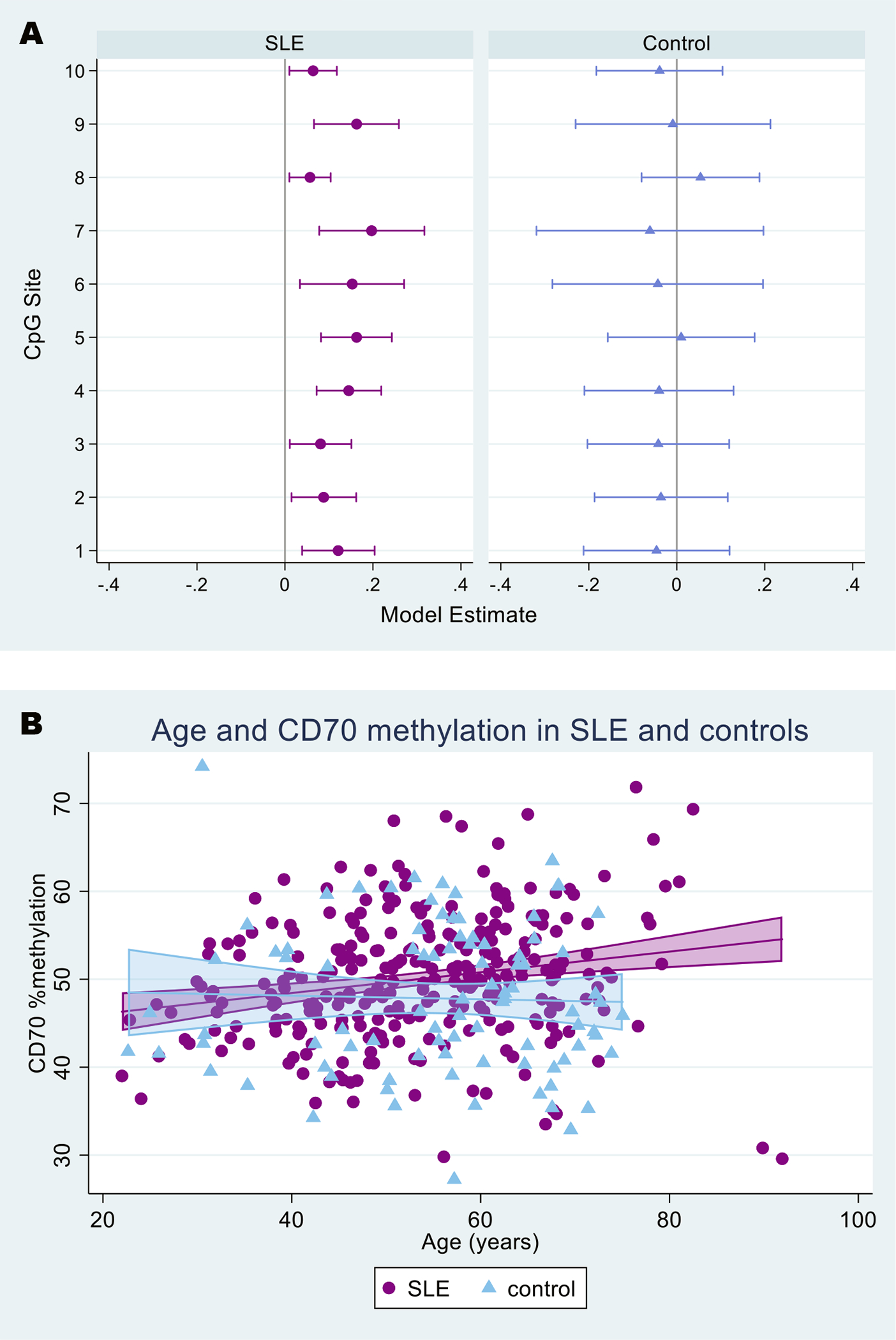
Increasing age was associated with increased % methylation in SLE but not controls, for each of the 10 CpG sites (p≤0.02 for all CpG sites among SLE) (Panel A), as well as on average across the 10 CpG sites (Panel B).
Panel A: Symbols designate point estimates (beta coefficients) from univariate linear regression (purple circles for SLE, blue triangles for controls); capped horizontal lines designate 95% confidence intervals (CIs). Estimates above zero represent positive associations (increased DNA methylation with increased age) and those below zero represent negative associations. Estimates where the 95% CIs do not overlap 0 are considered statistically significant (at the 0.05 level).
Panel B: Shaded areas include the line of best fit with 95% confidence intervals (SLE=purple, control=light blue).
Associations between age and intra-person average CD70 methylation across the 10 CpG sites were consistent with CpG site-specific results (Figure 3b). In univariate models, average methylation increased with age in SLE, but not in controls [SLE β=0.12 (95% CI 0.05, 0.18), p<0.001; control β=−0.02 (95% CI −0.16, 0.12), p=0.774]. In the multivariable regression (Table 2), the significant association between age and methylation in SLE persisted after adjusting for sex, race, health insurance, income, disease activity, and lupus damage. For every year increase in age within this adult population, CD70 % methylation increased by 0.14. In this multivariable model, methylation was 1.8 percentage points higher on average in Black persons compared to White persons with SLE, approaching statistical significance (p=0.051). None of the other covariates, including the lupus-specific variables, were associated with methylation. When we included SLE duration or SLE flare in the last three months in lieu of the continuous SLE activity variable, neither were associated with methylation. In contrast to findings within the SLE cases, none of the covariates was associated with CD70 methylation among controls. In analyses restricted to participants with complete data for all 10 CpG sites, results were similar, and the association between race and methylation in SLE was more pronounced and statistically significant [Black race β=2.23, 95% CI (0.24, 4.22), p=0.028].
Table 2. Associations with CD70 methylation of CD4+ T cell DNA within persons with SLE and controls.
Results based on multivariable regression models with average CD70 % DNA methylation across ten CpG sites assessed via pyrosequencing as the outcome, stratified by case-control status.
| SLE | Control | |
|---|---|---|
| β (95% CI) |
β (95% CI) |
|
| Age | 0.14 (0.06, 0.21)a | 0.02 (−0.15, 0.19) |
| Sex | 2.65 (−0.72, 5.94) | −1.16 (−6.62, 4.30) |
| Race (White referent) | ||
| Black | 1.77 (−0.005, 3.55)b | 1.62 (−2.73, 5.96) |
| all other or unknown | 1.77 (−3.57, 7.11) | −4.51 (−15.30, 6.29) |
| Insurance (private/other referent) | ||
| Medicaid | 0.17 (−2.47, 2.81) | −2.72 (−7.69, 2.25) |
| Medicare | −0.77 (−3.06, 1.51) | −3.79 (−9.14, 1.56) |
| none | 2.09 (−3.41, 7.58) | −2.21 (−13.48, 9.06) |
| Income below US median | −0.29 (−2.21, 1.63) | 3.05 (−1.55, 7.65) |
| SLE activity (SLAQ score) | −0.05 (−0.19, 0.08) | -- |
| SLE damage (LDIQ score) | 0.03 (−0.22, 0.27) | -- |
p=0.001
p=0.051
Based on EPIC array data, six of the CpG sites were significantly associated with age in CD4+ T cells when accounting for case-control status, sex, and race. These sites were all positively associated with age, except for a site (probe cg23263923) close to the TSS. In CD8+ T cells, 13 CpG sites were significantly associated with age. The direction of these associations varied by location and included positive associations with age among sites in the gene body (Figure 4). Evidence for an interaction between case-control status and age was limited, with only 1 CpG site (CD4+) having a p-value<0.05 for the interaction term (Supplementary Table S3).
Figure 4. Associations between age and methylation of CD4+ and CD8+ T cell DNA for CD70 CpG Sites included on the EPIC Array.
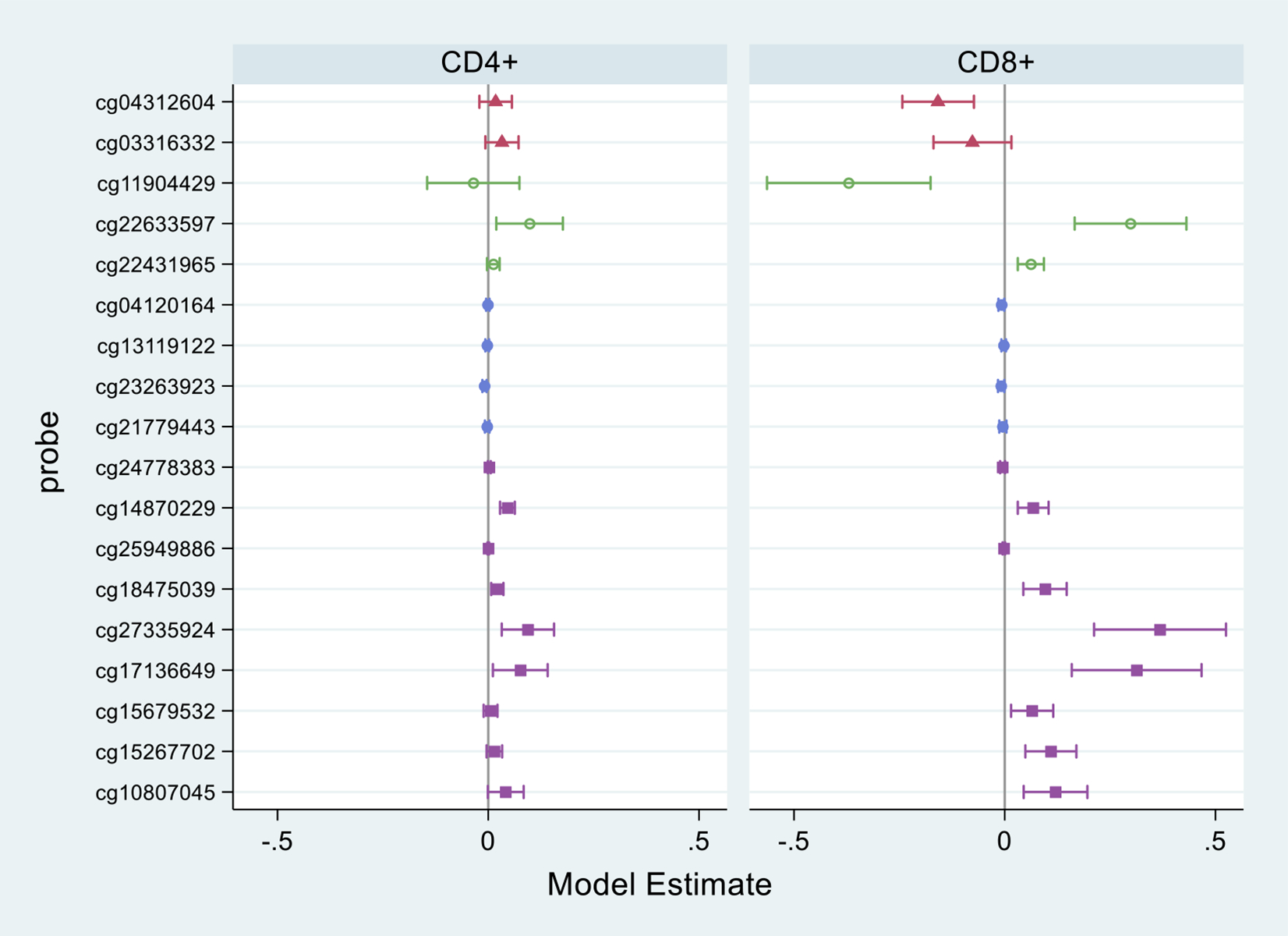
Symbols designate coefficients from multivariable regression models, which included adjustment for SLE/control status, sex, and race. Capped horizontal lines designate 95% confidence intervals (CIs). Estimates above zero represent positive associations (increased DNA methylation with increased age) and those below zero represent negative associations. Estimates where the 95% CIs do not overlap 0 are considered statistically significant (at the 0.05 level).
Regions relative to gene are represented by different marker colors/shapes: pink triangle=promoter/upstream; green hollow circle=1500 bp upstream of TSS; blue solid circle=200 bp upstream of TSS; purple square=in gene.
DISCUSSION
To our knowledge, this is the largest, population-based study of T cell DNA methylation in lupus and controls. Two main findings emerged. First, using two methods that covered DNA methylation at various regions of the CD70 (TNFSF7) gene, altered methylation was found in CD4+ T lymphocyte DNA for SLE cases relative to controls, with direction varying by genomic context. Similar results were found in CD8+ T lymphocyte DNA for the subset of participants with EPIC analysis performed on this cell type. Second, increasing age and Black race were both associated with increased CD70 promoter methylation among SLE cases.
DNA methylation defects in T lymphocytes have been linked to SLE pathogenesis; CD70 has been shown to be methylation-sensitive, with both promoter region hypomethylation and overexpression reported in adults with lupus, as well as in vitro in response to lupus-inducing drugs (13) and in autoimmune-prone mice (28). Further, its overexpression has been linked to autoreactivity, such as overstimulated B cell IgG production (29). Sustained CD70 overexpression has also been noted in a rheumatoid arthritis study; although DNA demethylation was not detected as an underlying mechanism, the authors reasoned that dysregulation of CD70 expression contributed to autoreactivity by lowering the threshold for activation of low avidity T cells (30). A small study of pediatric SLE did not detect differences in methylation of the CD70 promoter in DNA from unfractionated lymphocytes compared to healthy controls (31).
By providing data on CD70 methylation specific to T cell subsets in a diverse cohort, we extend observations from prior studies and provide added nuance. In particular, our finding of hypermethylation in a small region of the promoter in SLE compared to controls contrasts with prior findings of hypomethylation of CD70 in adult SLE (13). However, EPIC array results with coverage of individual sites throughout the gene show decreased promoter methylation and increased gene body methylation in lupus, both of which can contribute to increases in gene expression (32). Thus, CD70 may be poised for greater expression in the CD4+ T cells from persons with lupus. Broader coverage throughout the gene expands upon previous research that only focused on small regions within the promoter.
The larger size and diversity of the MILES population and genic regions analyzed could potentially account for findings that contrast with earlier studies which have been based primarily in academic, tertiary care settings, and included predominantly White females with lupus. Likewise, control groups have been small and largely recruited from convenience samples, such as hospital employees or clinic patients with other diseases. Our population-based control group was sampled from the same southeastern Michigan source population as the lupus cases, and is therefore not expected to be biased by the “healthy worker effect” (33).
The novel association between methylation of the CD70 gene and age detected in our adult lupus population, but not controls, suggests that regulation of CD70 may be part of lupus-specific “epigenetic aging.” While the control group was smaller and therefore had less statistical power than the SLE group to detect a significant age association, there was not even a trend observed in the same direction (Figure 3b). The premise that the age association was confined to lupus is reinforced by the CpG site-specific pyrosequencing results (Figure 3a), where all 10 sites corresponded to significant positive associations within SLE, in contrast to uniformly non-significant associations that were mostly in the opposite direction for the 10 sites in controls. Epigenetic aging is now a well-characterized phenomenon that is marked by two key features – decreased methylation when assessed at the global scale but locus-specific hypermethylation at certain genes (34,35), often at promoter regions. The locus-specific changes are hypothesized to contribute to the aging process (35). Increased DNA methylation at the CD70 promoter may thus be an indicator of accelerated epigenetic or biological aging. Whether this is caused by SLE disease progression or contributes to severity of the disease is yet to be determined. Data from the Women’s Health Initiative depicted higher levels of global epigenetic aging in post-menopausal, non-Hispanic Black compared to non-Hispanic White women, which was interpreted as partially mediating their increased mortality (36). In our study, Black race was associated with increased CD70 methylation in SLE cases. While a significant association between race and methylation was not detected in the smaller control group, the direction and magnitude of the point estimate for Black race were similar to that obtained in the SLE group. It is well recognized that lupus expression varies considerably by race, with Black persons experiencing increased incidence, prevalence, and younger onset compared to White persons (37,38), as well as higher levels of severity and involvement of certain organ systems, including the kidney. Our results point to the potential for epigenetics to aid our understanding of the interactions between age, race, and disease status.
We cannot directly address the mechanisms underlying hypermethylation with age and Black race, or why the age association is particularly evident for SLE. Indirect exposure to racism has been associated with disease activity among Black women with SLE (39), and it has been suggested that psychological distress mediates the association between racism-related stress and SLE disease activity (40). Akin to the weathering hypothesis (8,9), it has been suggested that physical and emotional stress contribute to “inflammaging” (41,42). DNA methylation signatures have also been linked with chronic, low-grade inflammation in complex diseases, in both European and African American populations (43). Within this framework, we propose that stressors associated with structural racism may contribute to our finding that Black race was independently associated with CD70 methylation among persons with SLE.
It is known that genomic context (i.e., position in the gene, local genetic sequence/variation, transcription factor binding) contributes to extent of DNA methylation and its subsequent influence on gene expression. By using two laboratory methods, we were able to inspect a well-studied region of the promoter, as well as sites scattered throughout the gene. In the focused pyrosequencing analysis, the region that we interrogated contained CpG sites spanning 89 base pairs. The sequenced region includes putative binding sites for transcription factors with strong evidence for NF-kappa B, AP4, GATA1, GATA2, and GATA3. Results included one site (CpG site 3, or −508 bp 5’ from TSS) in which DNA from persons with SLE was hypomethylated, as hypothesized, compared to controls. However, four of the ten sites were hypermethylated in SLE. Notably, CpG site 8 (−439 bp 5’ from TSS) had the highest methylation levels of any of the ten sites in both cases and controls; this is compatible with the observation by Lu et al. that this same CpG site appears to be “reproducibly resistant to demethylation” (13). Another consistency with Lu et al. was the lack of association between methylation level and lupus disease activity; they likewise reported that unlike for other methylation-sensitive genes implicated in SLE pathogenesis, such as perforin, methylation of CD70 in CD4+ T cells was not related to physician-assessed lupus activity by SLEDAI (13,44). This could imply that CD70 methylation may be a more stable marker of immune dysregulation relevant to lupus development.
The EPIC analysis expands our scope beyond one section of the CD70 promoter to include sites scattered throughout the promoter, exons, and introns of the gene. Results of the EPIC analysis suggest that epigenetic regulation varies by case-control status and age in several regions of the gene. In general, the promoter region is hypomethylated in cases compared to controls, whereas the gene body is hypermethylated in both cell populations. Gene body methylation has been linked to increased expression of other genes (45). DNA methylation of this gene increases with age in various regions, except for a few sites in the promoter that decrease with age. Thus, regulation of CD70 DNA methylation varies by genomic context, and this may be important for dynamic regulation of CD70 by SLE and aging.
Our study has several strengths. To our knowledge, this is the first lupus cohort study to include lymphocyte fractionation and T cell subset-specific DNA isolation on a large scale. Epigenetic patterns are tissue- and cell-type specific, and cellular composition of whole blood can confound DNA methylation results (46). Cell separations are most reliably performed on fresh specimens prior to cryopreservation, which restricts the feasibility of banking lymphocyte subsets within the context of epidemiologic fieldwork and has limited investigation of epigenetics at the population level. Our design and implementation of the MILES Cohort and Biorepository addresses this critical gap, allowing for the advancement of epigenetic epidemiology related to lupus. We also assayed DNA methylation using two, complementary laboratory methods. Pyrosequencing allowed for directed interrogation of a specific candidate region, applying methodology extensively used in prior SLE studies which optimized comparability. The high-throughput EPIC array was advantageous in that it allowed for more expansive coverage. Utilization of a separate methodology on the same set of specimens was particularly valuable given the contrasting results of our pyrosequencing data with prior research. Both pyrosequencing and EPIC assays were performed in blinded fashion with respect to associated participant data and without regard to results of the other assay. The sociodemographic diversity of our study population, reflective of the underlying region encompassing one quarter of the Michigan population, increases generalizability of results. This is of particular importance given wide disparities in lupus risk and outcomes across racial and ethnic groups (38). Our study included 171 persons who self-identified their race as Black, a considerably higher number than included in many previous studies in the field. However, owing to the demographics of our geographic region, some racial and ethnic groups, including Asians and Hispanics, are not well-represented. Our study population also spans a broad age range across all stages of adulthood, allowing for age-related patterns to be effectively studied, with the exception of pediatric stages during which SLE is rare (36).
Our study also has limitations. First, while CD70 promoter hypomethylation has previously been shown to correspond to gene expression and functional consequences including increased antibody production, we did not directly measure CD70 gene or protein expression or other downstream endpoints in our study. Second, although we included a broad age range, we were unable in this cross-sectional study to assess longitudinal trajectories of methylation with aging, or to assess the temporality of epigenetic variations in relation to SLE onset. Third, lupus activity measurement by patient-reported outcome, while validated for epidemiologic research, may differ from physician-driven assessments applied in clinical settings. Fourth, we did not include medication exposures in the modeling, given the heterogeneity in medication usage and doses over time and collinearity with disease activity. Further, medications commonly used in lupus generally do not target epigenetic pathways and were not associated with methylation in univariate analyses. Fifth, the control group sample size was smaller than for SLE, lessening statistical power within controls. While it was not feasible to enroll a larger control group, this group of nearly 100 controls is substantially larger than typically included lupus epigenetics studies, particularly when dealing with lymphocyte subsets. Sixth, our analytic methods provide data on total DNA methylation—this includes hydroxymethylation, a stable epigenetic mark that can contribute to regulation of gene expression independently from standard methylation (45). Profiling hydroxymethylation separately may better inform understanding of CD70 regulation in lupus. Nonetheless, there is substantial interplay between disease status, race, and age in CD70 regulation in SLE.
We provide evidence from this large-scale immunoepidemiology study of differential gene-specific DNA methylation in persons with SLE compared to controls from the general population. We further report associations particular to SLE between methylation of this immune-relevant gene and both age and race, possibly a consequence of “weathering” or accelerated aging. Additional studies of aging effects on methylation of genes involved in autoimmunity may improve understanding of the variability in susceptibility to development and progression of disease, including patterns of morbidity and mortality, and potential intervention strategies.
Supplementary Material
ACKNOWLEDGMENTS
The authors wish to thank the MILES Cohort participants and staff; Tracy Fuller for data management; Aubrie Andrews for research clinic management; Donna Ray, PhD for conducting pyrosequencing assays; Wenqian Li, MS for assistance with the statistical plan for pyrosequencing data; Professor Emeritus Bruce Richardson, MD PhD, for mentorship and early guidance planning the MILES Biorepository to accommodate epigenetics; The University of Michigan Epigenomics Core and Advanced Genomics Core for completing the EPIC array analysis.
Funding
The Michigan Lupus Epidemiology & Surveillance (MILES) Program is supported by the Centers for Disease Control and Prevention (CDC) of the U.S. Department of Health and Human Services (HHS) under grant number U01DP006489 as part of a financial assistance award totaling $2,600,000 with 100 percent funded by CDC/HHS, and grants U58/CCU522826, U58/DP001441, U01/DP003250 and U01/DP006265.
This study was also supported by the National Institutes of Health (NIH) through the National Institute of Environmental Health Sciences (NIEHS), the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS), and the National Center for Advancing Translational Sciences (NCATS) (grants K01ES019909, P30ES017885, T32AR007080, UL1TR002240).
Footnotes
Competing Interests
The authors declare that they have no competing interests.
Ethical Approval
Ethics approval was obtained from the Institutional Review Boards of the University of Michigan and Michigan Department of Health and Human Services. Study participants provided written, informed consent.
DISCLAIMER
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the CDC.
Availability of data and materials
Data supporting this study are not publicly available as these data are considered sensitive personal and health data, and pertain to a relatively uncommon disease in a defined geographic region.
REFERENCES
- 1.Somers EC, Richardson BC. Environmental exposures, epigenetic changes and the risk of lupus. Lupus 2014;23:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Richardson B, Somers EC. Environmental and drug-induced lupus. In: Lahita R, ed. Lahita’s Systemic Lupus Erythematosus. 6th ed. London: Academic Press; 2021:381–388. [Google Scholar]
- 3.Marder W, Vinet É, Somers EC. Rheumatic autoimmune diseases in women and midlife health. Women’s Midlife Heal 2015;1:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jonsson H, Nived O, Sturfelt G, Silman A. Estimating the incidence of systemic lupus erythematosus in a defined population using multiple sources of retrieval. Br J Rheumatol 1990;29:185–8. [DOI] [PubMed] [Google Scholar]
- 5.Somers EC, Thomas SL, Smeeth L, Schoonen WM, Hall AJ. Incidence of systemic lupus erythematosus in the United Kingdom, 1990–1999. Arthritis Rheum 2007;57:612–8. [DOI] [PubMed] [Google Scholar]
- 6.Somers EC, Marder W, Cagnoli P, Lewis EE, DeGuire P, Gordon C, et al. Population-based incidence and prevalence of systemic lupus erythematosus: The Michigan lupus epidemiology and surveillance program. Arthritis Rheumatol 2014;66:369–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Johnson AE, Gordon C, Palmer RG, Bacon PA. The prevalence and incidence of systemic lupus erythematosus in Birmingham, England. Relationship to ethnicity and country of birth. Arthritis Rheum 1995;38:551–8. [DOI] [PubMed] [Google Scholar]
- 8.Geronimus AT, Hicken MT, Pearson JA, Seashols SJ, Brown KL, Cruz TD. Do US Black Women Experience Stress-Related Accelerated Biological Aging?: A Novel Theory and First Population-Based Test of Black-White Differences in Telomere Length. Hum Nat 2010;21:19–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Simons RL, Lei M-K, Klopack E, Beach SRH, Gibbons FX, Philibert RA. The effects of social adversity, discrimination, and health risk behaviors on the accelerated aging of African Americans: Further support for the weathering hypothesis. Soc Sci Med 2021;282:113169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gorelik GJ, Yarlagadda S, Patel DR, Richardson BC. Protein kinase Cδ oxidation contributes to ERK inactivation in lupus T cells. Arthritis Rheum 2012;64:2964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorelik G, Fang JY, Wu A, Sawalha AH, Richardson B. Impaired T Cell Protein Kinase Cδ Activation Decreases ERK Pathway Signaling in Idiopathic and Hydralazine-Induced Lupus. J Immunol 2007;179:5553–5563. [DOI] [PubMed] [Google Scholar]
- 12.Quddus J, Johnson KJ, Gavalchin J, Amento EP, Chrisp CE, Yung RL, et al. Treating activated CD4+ T cells with either of two distinct DNA methyltransferase inhibitors, 5-azacytidine or procainamide, is sufficient to cause a lupus-like disease in syngeneic mice. J Clin Invest 1993;92:38–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lu Q, Wu A, Richardson BC. Demethylation of the same promoter sequence increases CD70 expression in lupus T cells and T cells treated with lupus-inducing drugs. J Immunol 2005;174:6212–9. [DOI] [PubMed] [Google Scholar]
- 14.Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, et al. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum 2004;50:1850–60. [DOI] [PubMed] [Google Scholar]
- 15.Wang D, Du J, Song Y, Wang B, Song R, Hao Y, et al. CD70 contributes to age-associated T cell defects and overwhelming inflammatory responses. Aging (Albany NY) 2020;12:12032–12050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lim SS, Drenkard C, McCune WJ, Helmick CG, Gordon C, DeGuire P, et al. Population-based lupus registries: Advancing our epidemiologic understanding. Arthritis Care Res 2009;61:1462–1466. [DOI] [PubMed] [Google Scholar]
- 17.Somers EC, Lee J, Hassett AL, Zick SM, Harlow SD, Helmick CG, et al. Prescription Opioid Use in Patients With and Without Systemic Lupus Erythematosus - Michigan Lupus Epidemiology and Surveillance Program, 2014–2015. MMWR Morb Mortal Wkly Rep 2019;68:819–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Minhas D, Marder W, Harlow S, Hassett AL, Zick SM, Gordon C, et al. Access and cost-related non-adherence to prescription medications among lupus cases and controls: the Michigan Lupus Epidemiology & Surveillance (MILES) Program. Arthritis Care Res (Hoboken) 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Centers for Disease Control and Prevention (CDC), National Center for Health Statistics (NCHS). National Health and Nutrition Examination Survey. Hyattsville, MD US Dep Heal Hum Serv. Available at: http://www.cdc.gov/nchs/nhanes.htm.
- 20.Karlson EW, Daltroy LH, Rivest C, Ramsey-Goldman R, Wright EA, Partridge AJ, et al. Validation of a Systemic Lupus Activity Questionnaire (SLAQ) for population studies. Lupus 2003;12:280–6. [DOI] [PubMed] [Google Scholar]
- 21.Costenbader KH, Khamashta M, Ruiz-Garcia S, Perez-Rodriguez MT, Petri M, Elliott J, et al. Development and initial validation of a self-assessed lupus organ damage instrument. Arthritis Care Res (Hoboken) 2010;62:559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tost J, Gut IG. Analysis of gene-specific DNA methylation patterns by pyrosequencing technology. Methods Mol Biol 2007;373:89–102. [DOI] [PubMed] [Google Scholar]
- 23.Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics 2016;8:389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fortin JP, Triche TJ, Hansen KD. Preprocessing, normalization and integration of the Illumina HumanMethylationEPIC array with minfi. Bioinformatics 2017;33:558–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu Z, Langie SA, Boever P De, Taylor JA, Niu L. RELIC: a novel dye-bias correction method for Illumina Methylation BeadChip. BMC Genomics 2017;18:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013;29:189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform 2009;42:377–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sawalha AH, Jeffries M. Defective DNA methylation and CD70 overexpression in CD4+ T cells in MRL/lpr lupus-prone mice. Eur J Immunol 2007;37:1407–1413. [DOI] [PubMed] [Google Scholar]
- 29.Oelke K, Lu Q, Richardson D, Wu A, Deng C, Hanash S, et al. Overexpression of CD70 and overstimulation of IgG synthesis by lupus T cells and T cells treated with DNA methylation inhibitors. Arthritis Rheum 2004;50:1850–1860. [DOI] [PubMed] [Google Scholar]
- 30.Lee W-W, Yang Z-Z, Li G, Weyand CM, Goronzy JJ. Unchecked CD70 expression on T cells lowers threshold for T cell activation in rheumatoid arthritis. J Immunol 2007;179:2609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Keshavarz-Fathi M, Sanati G, Sadr M, Mohebbi B, Ziaee V, Rezaei N. DNA Methylation of CD70 Promoter in Juvenile Systemic Lupus Erythematosus. Fetal Pediatr Pathol 2022;41:58–67. [DOI] [PubMed] [Google Scholar]
- 32.Jjingo D, Conley AB, Yi SV, Lunyak VV, Jordan IK. On the presence and role of human gene-body DNA methylation. Oncotarget 2012;3:462–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McMichael AJ. Standardized mortality ratios and the “healthy worker effect”: Scratching beneath the surface. J Occup Med 1976;18:165–8. [DOI] [PubMed] [Google Scholar]
- 34.Teschendorff AE, West J, Beck S. Age-associated epigenetic drift: implications, and a case of epigenetic thrift? Hum Mol Genet 2013;22:R7–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jung M, Pfeifer GP. Aging and DNA methylation. BMC Biol 2015;13:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Chen BH, Assimes TL, Ferrucci L, Horvath S, Levine ME. The role of epigenetic aging in education and racial/ethnic mortality disparities among older U.S. Women. Psychoneuroendocrinology 2019;104:18–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Izmirly PM, Ferucci ED, Somers EC, Wang L, Lim SS, Drenkard C, et al. Incidence rates of systemic lupus erythematosus in the USA: estimates from a meta-analysis of the Centers for Disease Control and Prevention national lupus registries. Lupus Sci Med 2021;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Izmirly PM, Parton H, Wang L, McCune WJ, Lim SS, Drenkard C, et al. Prevalence of Systemic Lupus Erythematosus in the United States: Estimates From a Meta-Analysis of the Centers for Disease Control and Prevention National Lupus Registries. Arthritis Rheumatol (Hoboken, NJ) 2021;73:991–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martz CD, Allen AM, Fuller-Rowell TE, Spears EC, Lim SS, Drenkard C, et al. Vicarious Racism Stress and Disease Activity: the Black Women’s Experiences Living with Lupus (BeWELL) Study. J Racial Ethn Health Disparities 2019;6:1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunter EA, Spears EC, Martz CD, Chung K, Fuller-Rowell TE, Lim SS, et al. Racism-related stress and psychological distress: Black Women’s Experiences Living with Lupus study. J Health Psychol 2020:1359105320913085. [DOI] [PubMed] [Google Scholar]
- 41.Sullivan J, Mirbahai L, Lord JM. Major trauma and acceleration of the ageing process. Ageing Res Rev 2018;48:32–39. [DOI] [PubMed] [Google Scholar]
- 42.Franceschi C Inflammaging as a major characteristic of old people: can it be prevented or cured? Nutr Rev 2007;65:S173–6. [DOI] [PubMed] [Google Scholar]
- 43.Ligthart S, Marzi C, Aslibekyan S, Mendelson MM, Conneely KN, Tanaka T, et al. DNA methylation signatures of chronic low-grade inflammation are associated with complex diseases. Genome Biol 2016;17:255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bombardier C, Gladman DD, Urowitz MB, Caron D, Chang CH. Derivation of the SLEDAI. A disease activity index for lupus patients. The Committee on Prognosis Studies in SLE. Arthritis Rheum 1992;35:630–40. [DOI] [PubMed] [Google Scholar]
- 45.Tsagaratou A, Äijö T, Lio C-WJ, Yue X, Huang Y, Jacobsen SE, et al. Dissecting the dynamic changes of 5-hydroxymethylcytosine in T-cell development and differentiation. Proc Natl Acad Sci U S A 2014;111:E3306–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Heijmans BT, Mill J. Commentary: The seven plagues of epigenetic epidemiology. Int J Epidemiol 2012;41:74–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data supporting this study are not publicly available as these data are considered sensitive personal and health data, and pertain to a relatively uncommon disease in a defined geographic region.