

Abstract
Multiple clinical trials targeting the gut microbiome are being conducted to optimize treatment outcomes for immune checkpoint blockade (ICB). To improve the success of these interventions, understanding gut microbiome changes during ICB is urgently needed. Here through longitudinal microbiome profiling of 175 patients treated with ICB for advanced melanoma, we show that several microbial species-level genome bins (SGBs) and pathways exhibit distinct patterns from baseline in patients achieving progression-free survival (PFS) of 12 months or longer (PFS ≥12) versus patients with PFS shorter than 12 months (PFS <12). Out of 99 SGBs that could discriminate between these two groups, 20 were differentially abundant only at baseline, while 42 were differentially abundant only after treatment initiation. We identify five and four SGBs that had consistently higher abundances in patients with PFS ≥12 and <12 months, respectively. Constructing a log ratio of these SGBs, we find an association with overall survival. Finally, we find different microbial dynamics in different clinical contexts including the type of ICB regimen, development of immune-related adverse events and concomitant medication use. Insights into the longitudinal dynamics of the gut microbiome in association with host factors and treatment regimens will be critical for guiding rational microbiome-targeted therapies aimed at enhancing ICB efficacy.
Subject terms: Melanoma, Microbiome, Statistical methods
Understanding the dynamics of the gut microbiota over the course of cancer treatment regimens and their associated adverse events can help identify microbial features that associate with response to immune checkpoint blockade.
Main
Immune checkpoint blockade (ICB) has revolutionized the field of oncology by prolonging the survival of patients with different tumor types at advanced stages1. However, only a subset of patients responds to ICB, and the treatment can induce a variety of immune-related adverse events (irAEs), including colitis2,3. Cross-sectional studies have assessed the gut microbiome before ICB initiation4–12, but the field is hampered by a lack of consensus as different studies often report different microbial biomarkers of response4—a heterogeneity that is probably the result of many methodological, biological and/or clinical confounders but that also arises from the high intra- and inter-individual variation of the gut microbiome13–15. Despite the lack of a thorough understanding of underlying mechanisms, multiple microbiome-directed clinical trials are ongoing in the oncoimmunology field, including fecal microbiota transplantation (FMT) trials16. To better interpret the findings from these trials and to increase our understanding of gut microbiome dynamics more generally and in the context of ICB, there is an urgent need for longitudinal microbiome studies along the course of ICB treatment.
In this Article, we therefore describe the profiling of the gut microbiome (via shotgun metagenomics followed by MetaPhlAn417 and microbial metabolic (MetaCyc)18 analyses) at four time points during the first 12 weeks of treatment in a multicenter cohort comprising 175 patients treated with ICB for advanced melanoma (Extended Data Fig. 1). First, because patients received an immunotherapy infusion at each study visit (thus, the effect of ICB on the gut microbiome may increase as the treatment progresses), we hypothesize that many microbial abundances may increase or decrease over the treatment period. Second, because baseline abundances of several microbial taxa have already been shown to differ between ICB response and nonresponse, we further hypothesize that patients responding and not responding to the treatment exhibit different patterns of microbial increase/decrease. To model this, we used a Bayesian regression model with higher-order interactions, allowing patients with progression-free survival (PFS) ≥12 months and patients with PFS <12 months to exhibit different longitudinal (linear) trajectories for each microbial feature. While we focus on the overall comparison between patients with PFS ≥12 and PFS <12 months averaging over the effect of multiple confounders, our methodology also allowed us to analyze microbial dynamics between patients with PFS ≥12 and PFS <12 months in three relevant clinical scenarios, namely therapy regimen (mono versus combination ICB), the development of ICB-induced colitis and concomitant proton-pump inhibitor (PPI) use. The latter two have well-studied effects on the gut microbiome19,20.
Extended Data Fig. 1. Study description with sample numbers across study visits.

Samples were collected within 5 sub-cohorts: two prospectively recruited within parallel observational studies (The PRIMM cohorts), and three retrospectively pooled cohorts. Fecal samples were collected at 4 timepoints: at baseline (T0) and at every treatment cycle (T1 to T3) over a period of 12 weeks. The time between two samples was 3 or 4 weeks, depending on the treatment regimen, with Ipilimumab/Nivolumab combination therapy and Pembrolizumab monotherapy administered 3-weekly and Nivolumab monotherapy administered 4-weekly. Treatment continued after the 12 weeks until the patient responded or until the treatment had to pause/stop due to irAEs. Not all subjects provided fecal samples at all study visits. Therefore, gut microbial dynamics were modeled at the level of the population including a random effect for the patient identifier (see Methods). Sample numbers represent patients with complete metadata (that is, no missingness) for all considered covariates/confounders. For the survival analysis, because we adjusted for a smaller number of covariates/confounders, there were n = 147 at baseline (PRIMM-UK = 41; PRIMM-NL = 53; Barcelona = 12; Leeds = 17; Manchester = 24) rather than n = 136 as indicated here. Tumor staging by CT or PET-scans was performed at study entry and at regular intervals during treatment. Tumor response was classified using the Response Evaluation Criteria in Solid Tumors (RECIST) v.1. Endpoints were defined as Progression-free survival at 12 months (PFS12) and overall survival (OS). Immune-related adverse events (irAEs) were assessed using the Common Terminology Criteria for Adverse Events (CTCAE) v.5 (see Table 1). ICB, Immune checkpoint blockade; PRIMM, Predicting Response to Immunotherapy for Melanoma With Gut Microbiome and Metabolomics; NL, Netherland; UK, United Kingdom. The figure was generated in BioRender.com.
Results
Cohort characteristics
Cohort characteristics are summarized in Table 1. We recruited 175 patients from five distinct cohorts across the Netherlands, the United Kingdom and Spain who were treated with ICB for unresectable stage 3 and stage 4 cutaneous melanoma, as previously described4–6,9–12. A total of 117 (67%) patients received single agent treatment with an anti-programmed cell death (PD)-1 antibody (nivolumab or pembrolizumab), while 58 (33%) patients received combination therapy with anti-PD-1 and anti-cytotoxic T-lymphocyte-associated antigen (CTLA)-4 antibody (ipilimumab). We used the Response Evaluation Criteria in Solid Tumors (RECIST v.1.1) to determine tumor response (Methods). To capture patients who are alive or progression-free at late time points, we defined clinical endpoints as PFS at 12 months (PFS12) and overall survival (OS). PFS was defined as the time from the initial immunotherapy to disease progression or death, comparing patients achieving a PFS of 12 months or longer and patients with a PFS of less than 12 months. PFS12 was reached by 83 (47%) participants, and the overall median OS was 34.1 months (minimum of 0.39 months, maximum of 93.4 months; censoring date, 28 March 2023). OS was defined for a subset of patients (n = 147 patients) as the time in months from initiation of treatment to occurrence of death from any cause. Patients were followed over a maximum period of 7.3 years (median of 4.3 years) after providing the first fecal sample. Fecal samples were collected at baseline and three subsequent treatment visits over a period of 12 weeks (Methods and Extended Data Fig. 1).
Table 1.
Cohort characteristics at study entry
| PRIMM–UK (n = 54) | PRIMM–NL (n = 74) | Manchester (n = 17) | Leeds (n = 19) | Barcelona (n = 11) | All cohorts (n = 175) | P value | |
|---|---|---|---|---|---|---|---|
| Age (years), median (range) | 64 (19–94) | 60 (21–85) | 66 (38–87) | 57 (35–88) | 64 (37–88) | 63 (19–94) | 0.127 |
| Sex (female), n (%) | 19 (35) | 37 (50) | 7 (41) | 7 (37) | 5 (45) | 75 (43) | 0.530 |
| BMI (kg m−2), mean (range) | 28.6 (18.83–47.66) | 27.02 (15.43–40.74) | 26.92 (18.99–40.40) | 28.46 (20.90–38.57) | 26.27 (20.96–36.08) | 27.63 (15.43–47.66) | 0.075 |
| Metastatic stage, n (%) | 0.006 | ||||||
| Stage 3 unresectable | 5 (9) | 2 (3) | 0 (0) | 1 (5) | 0 (0) | 8 (5) | |
| Stage 4 M1a | 12 (22) | 7 (9) | 5 (29) | 3 (16) | 4 (36) | 31 (18) | |
| Stage 4 M1b | 12 (22) | 14 (19) | 2 (12) | 5 (26) | 5 (45) | 38 (22) | |
| Stage 4 M1c | 20 (37) | 23 (31) | 8 (47) | 7 (37) | 2 (18) | 60 (34) | |
| Stage 4 M1d | 5 (9) | 28 (38) | 2 (12) | 3 (16) | 0 (0) | 38 (22) | |
| BRAF mutant, n (%) | 18 (33) | 42 (57) | 2 (12) | 9 (47) | 3 (27) | 74 (42) | 0.004 |
| ECOG performance status ≥1, n (%) | 36 (67) | 17 (23) | 8 (47) | 2 (11) | 1 (9) | 64 (37) | 1.729 × 10−6* |
| Outcomes following ICB | |||||||
| PFS ≥12 months, n (%) | 27 (50) | 32 (43) | 8 (47) | 11 (58) | 5 (45) | 83 (47) | 0.824 |
| irAEs, n (%) | 38 (70) | 44 (59) | 9 (53) | 9 (47) | 7 (64) | 107 (61) | 0.399 |
| Colitis, n (%) | 13 (24) | 10 (14) | 3 (18) | 4 (21) | 3 (27) | 33 (19) | 0.570 |
| Treatment details | |||||||
| ICB combination therapy (anti-CTLA-4/anti-PD-1), n (%) | 29 (54) | 15 (20) | 2 (12) | 11 (58) | 1 (9) | 58 (33) | 1.60 × 10−5* |
| Previous BRAF or MEK inhibition, n (%) | 10 (19) | 28 (38) | 2 (12) | 0 (0) | 1 (9) | 41 (23) | 0.001* |
| PPI use at baseline, n (%) | 13 (24) | 24 (32) | 4 (24) | 6 (32) | 1 (9) | 48 (27) | 0.495 |
| Antibiotics use at baseline, n (%) | 9 (17) | 11 (15) | 2 (12) | 3 (16) | 0 (0) | 25 (14) | 0.694 |
Baseline characteristics are presented as mean and s.d. or median (range) for continuous variables and as counts and percentages for categorical variables. χ2 tests for categorical variables and two-sided Wilcoxon tests for continuous data were performed to calculate differences between cohorts. P values written in bold indicate nominally significant differences between cohorts (P < 0.05). *Statistical significance under a false discovery rate of 5%. UK, United Kingdom; NL, the Netherlands.
Taxonomic profiling was performed at the level of species-level genome bins (SGBs) using MetaPhlAn4, which represent both existing and yet-to-be-characterized microbial species17. We first analyzed which SGBs’ and MetaCyc pathways’ relative abundances were differentially abundant between patients with PFS ≥12 and PFS <12 months averaging over the effect of confounders such as therapy regimen, development of ICB-induced colitis and other irAEs, concomitant use of PPIs, previous use of antibiotics, previous v-raf murine sarcoma viral oncogene homolog B1 (BRAF) or mitogen-activated protein kinase (MEK)-targeted therapy, and cancer center (Methods). Each regression parameter in our Bayesian model was represented by a marginal posterior probability distribution. We computed post hoc contrasts (see Supplementary Table 1 for the number of patient samples per contrast and study visit) for which we concluded that a microbial SGB or pathway is differentially abundant between cases and controls if 90% of its posterior distribution does not cover zero (that is, 90% Bayesian confidence level (BCL); other BCLs are reported in Supplementary Tables 2–12). At 90% BCL, we observed 62 (14.3%) and 41 (9.4%) SGBs that exhibited increasing or decreasing slopes in patients with PFS ≥12 and PFS <12 months, respectively (Supplementary Table 2), and 99 (22.8%) SGBs that were able to discriminate between patients with PFS ≥12 and PFS <12 months in at least one study visit (90% BCL; range: 342 (50% BCL)–3 (100% BCL); Supplementary Table 3). Of these 99 SGBs, 20 were differentially abundant only at baseline, 42 were differentially abundant only after the start of ICB and 5 and 4 remained at consistently higher abundances in patients with PFS ≥12 and PFS <12 months, respectively, at baseline and all subsequent study visits (Fig. 1a and Supplementary Table 4). To aid in the interpretation, Fig. 1a,b displays the longitudinal trajectories (that is, slopes) of two example SGBs and one MetaCyc pathway for patients with PFS ≥12 and PFS <12 months, respectively. A clear example is Sellimonas intestinalis (SGB4617), which is not differentially abundant between patients with PFS ≥12 and PFS <12 months at baseline (as illustrated by a gray cell at T0 in Fig. 1a). Beyond baseline, however, the expected abundance (represented in centered log ratio coordinates) increases sharply (as illustrated by a vivid red cell in Fig. 1a) in patients with PFS <12 months while decreasing slightly (as illustrated by a light blue cell in Fig. 1a) in patients with PFS ≥12 months. Thus, the average difference between these two patient groups increases (in absolute terms) across the study visits (as illustrated by the increasingly darker brown shades from T1 towards T3 in Fig. 1a). The five SGBs that had consistently higher abundances in patients with PFS ≥12 months were Agathobaculum butyriciproducens (SGB14993 group), Intestinibacter bartlettii (SGB6140), Dorea sp. AF24 7LB (SGB4571), Lactobacillus gasseri (SGB7038 group) and Lacrimispora celerecrescens (SGB4868), whereof the latter two also exhibited increasing abundances (that is, positive slopes) over the study period (Fig. 1a and Supplementary Table 4). Three of these species have recently been associated with response in two new studies utilizing MetaPhlAn4, one meta-analysis21 and one phase 1 FMT trial of ICB-naive patients22. These five species represent fiber degrading taxa capable of short chain fatty acid (SCFA) synthesis that has been linked to plant-based diets12,23,24. Consequently, we also observed higher abundances of metabolic pathways (PWY-6396: superpathway of 2,3-butanediol biosynthesis; PWY-P124: Bifidobacterium shunt; PWY-6435: 4-hydroxybenzoate biosynthesis V; and PWY-5088: l-glutamate degradation VIII (to propanoate)) involved in the synthesis of SCFAs or their precursors in patients with PFS ≥12 months across multiple study visits (Fig. 1c and Supplementary Table 5), supporting a potential benefit of microbially produced SCFAs and an adjuvant role of fiber for ICB7,25. Patients with PFS <12 months, on the other hand, were enriched across all four study visits with Ruthenibacterium lactatiformans (SGB15271), Prevotella copri clade A (SGB1626), Ruminococcaceae unclassified (SGB15265 group) and an unidentified SGB from the phylum Bacteroidetes (SGB1957; Fig. 1a and Supplementary Table 4). In previous baseline studies, P. copri has been associated with ICB response26,27. However, in the recent meta-analysis by Thomas et al.21, this particular SGB (SGB1626) was only associated with response in 5/12 cohorts and with 3/9 different statistical methods. We found that patients with PFS <12 months exhibited higher abundances of several pathways involved in menaquinol (vitamin K) synthesis at baseline and during early treatment (Fig. 1c and Supplementary Table 5). Menaquinol synthesis pathways are enriched in various chronic inflammatory and cardiovascular diseases28–31. Fecal menaquinone levels have been correlated with the abundance of Prevotella and Bacteroides species and are susceptible to microbiome-targeted diets32, suggesting that menaquinol could represent an early marker of nonresponse that is amenable to dietary intervention. In contrast, patients achieving PFS12 exhibited higher abundances of a polyamine synthesis pathway (POLYAMINSYN3-PWY: superpathway of polyamine biosynthesis II) across the three study visits after baseline, but not at baseline. Polyamines are autophagy inducers33 that are implicated in immune regulation and have been shown to improve anti-cancer immunity in mice, synergizing with anti-PD ligand 1 immunotherapy22,34. Polyamines, including spermidine, are naturally occurring in foods and can be synthesized by the gut microbiome, suggesting a potential beneficial role for spermidine-enriched diets35.
Fig. 1. High-level view of gut microbiome dynamics in patients with PFS ≥12 and PFS <12 months.

a, For each microbial SGB listed, slopes are shown (that is, whether it is increasing or decreasing over study visits) in patients with PFS ≥12 (n = 83) and PFS <12 months (n = 92), respectively. For increased readability, SGBs differentially abundant in only one study visit have been removed (see Extended Data Fig. 2 for all SGBs). Red and blue colors indicate whether the focal SGB is increasing or decreasing in its abundance over study visits, respectively, with the strength of the colors corresponding to the steepness of the slope, with darker shades indicating steeper increases/decreases. It then shows, in the teal–brown heatmap, the average difference between the two slopes (that is, between patients with PFS ≥12 and PFS <12 months) across the different study visits. Non-gray cells in the heatmap correspond to the focal SGB’s log fold change in abundance between patients with PFS ≥12 and PFS <12 months, respectively. Teal cells correspond to study visits for which the abundance of the focal SGB is higher in patients with PFS ≥12 than with PFS <12 months, and vice versa for brown cells (at 90% BCL). Gray cells denote differences between patients with PFS ≥12 and PFS <12 months whose 90% credible interval cover zero. b, Three example features and how they increase and/or decrease in their expected abundance (represented in centered log ratio coordinates) over the study visits in patients with PFS ≥12 months (yellow slope) and in patients with PFS <12 months (purple slope). For each microbial SGB or pathway, the inset figure then displays the average difference between the two slopes at each study visit, including its 90% credible interval. These averages are the same as depicted in the teal–brown heatmap in a, and significance is deemed by evaluating whether or not the 90% credible interval covers zero. c, Microbial pathways are shown, similar to the format in a. The number (n) represents the number of patient samples at each visit for patients with PFS ≥12 and PFS <12 months.
To assess whether the five and four SGBs that had consistently higher abundances in patients with PFS ≥12 and PFS <12 months, respectively, could serve as a predictive marker for PFS12, we constructed a balance (a type of log ratio) between these SGBs and tested whether it could predict PFS12 in each study visit (Fig. 2a). We found that this balance could discriminate between patients with PFS ≥12 and PFS <12 months in all but the last study visit (two-sided Wilcoxon test: PT0 = 0.00085, PT1 = 0.0007, PT2 = 0.0005 and PT3 = 0.1; Fig. 2b) with a moderate predictive ability across visits (area under the curve (AUC) from 100 times repeated five-fold cross-validation, measured as mean AUC ± standard deviation (s.d.): AUCT0 0.659 ± 0.092, AUCT1 0.666 ± 0.091, AUCT2 0.739 ± 0.118 and AUCT3 0.655 ± 0.129; Fig. 2c). When we expanded this balance to include SGBs that were differentially abundant in all but the last study visit, its predictive ability increased across all study visits (AUCT0 0.771 ± 0.088, AUCT1 0.706 ± 0.094, AUCT2 0.783 ± 0.118 and AUCT3 0.765 ± 0.138; Extended Data Fig. 3a,b). Stratifying patients on the basis of whether they harbored higher or lower than median values of these two balances showed that patients above the median exhibited longer OS compared to patients below the median (first balance: OSHigh of 35.4 versus OSLow 28.4 months; hazard ratio (HR) of 1.669, P = 0.035; Fig. 2d; second balance: OSHigh of 37.0 versus OSLow of 26.9 months; HR of 1.792, P = 0.014; Extended Data Fig. 3c). Results did not quantitatively change when we substituted OS with continuous PFS (first balance: HR of 1.685, P = 0.022; second balance: HR = 2.25, P = 0.0004) and/or when we treated each balance as a continuous score (first balance: HROS = 0.828, POS = 0.001; second balance: HROS of 0.752, POS = 0.0002; Extended Data Fig. 4; first balance: HRPFS of 0.829, PPFS = 0.0005; second balance: HRPFS of 0.727, PPFS = 8.93 × 10−6).
Fig. 2. A longitudinal balance of microbial taxa (SGBs) predicts OS at baseline.

a, Schematic illustration of a balance between the five SGBs that were consistently higher in patients with PFS ≥12 months (A. butyriciproducens SGB14993 group, I. bartlettii SGB6140, Dorea sp. AF24 7LB SGB4571, L. gasseri SGB7038 group and L. celerecrescens SGB4868) and the four SGBs that were found to be consistently higher in patients with PFS <12 months (R. lactatiformans SGB15271, R. unclassified SGB15265 group, P. copri clade A SGB1626 and an unidentified SGB from the phylum Bacteroidetes SGB1957). In patients with PFS ≥12 and PFS <12 months, the balance is tilted to the left and right side, respectively. b, The balance’s ability to discriminate between patients with PFS ≥12 (n = 83, n0 = 62, n1 = 77, n2 = 38 and n3 = 30) and PFS <12 months (n = 92, n0 = 74, n1 = 69, n2 = 34 and n3 = 24) months across study visits (two-sided Wilcoxon test: PT0 = 0.00085, PT1 = 0.0007, PT2 = 0.0005 and PT3 = 0.1). Boxplots represent minima, Q1, Q2, Q3 and maxima. c, The balance’s predictive ability expressed as the AUC computed from 100 times repeated five-fold cross-validation. Each line shows, for each study visit, the average across the 100 times repeated five-fold cross-validations with the shaded area representing the 95% CI (mean AUC ± s.d.: AUCT0 0.659 ± 0.092, AUCT1 0.666 ± 0.091, AUCT2 = 0.739 ± 0.118 and AUCT3 0.655 ± 0.129). The dashed diagonal line represents random chance. d, Kaplan–Meier curves and multivariable Cox regression of OS in months for 146 patients at baseline according to high (above median; teal) and low (below median; orange) values of the balance after adjusting for age, sex, BMI, previous therapy, PPI and antibiotics use.
Extended Data Fig. 3. Extended longitudinal balance.
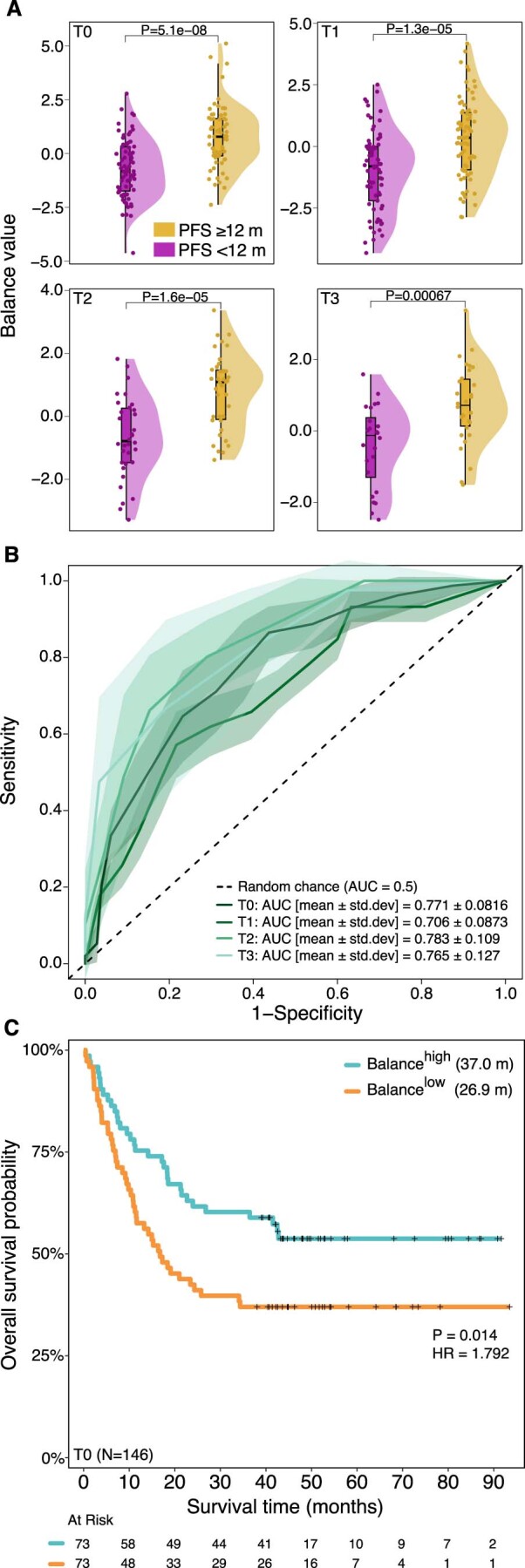
The extended balance has 12 SGBs in the numerator (A. butyriciproducens [SGB14993 group], I. bartlettii [SGB6140], D. sp AF24 7LB [SGB4571], L. gasseri [SGB7038 group], L. celerecrescens [SGB4868], R. sp NSJ 71 [SGB4290], GGB9640 [SGB15115], E. rectale [SGB4933 group], E. entriosum [SGB5045], E. sp AM28 29 [SGB6796 group], Clostridium sp AF15 49 [SGB5111], and A. bouchesdurhonensis [SGB17152]) and 9 SGBs in the denominator (R. lactatiformans [SGB15271], R. unclassified [SGB15265 group], P. copri clade A [SGB1626], GGB1420 [SGB1957], Gemmiger [SGB15299], B. obeum [SGB4809], Clostridiales unclassified [SGB15145], P. vulgatus [SGB1814], and B. clarus [SGB1832]). Panel (A) balance’s ability to discriminate between patients with PFS ≥ 12 (n = 83; n0 = 62; n1 = 77; n2 = 38; n3 = 30) and PFS < 12 (n = 92; n0 = 74; n1 = 69; n2 = 34; n3 = 24) months across study visits. Boxplots represent minima, Q1, Q2, Q3, and maxima. Panel (B) the balance’s predictive ability expressed as the Area Under the Curve (AUC) computed from 100 times repeated five-fold cross-validation (CV). Each line shows, for each study visit, the average across the 100 repeated five-fold CVs with the shaded area representing the 95% confidence interval. Panel (C) Kaplan–Meier curves and multivariable Cox regression analysis of overall survival in 146 patients at baseline (one patient was removed due to missingness in the included predictor variables) according to high (above median) and low (below median) values of the balance after adjusting for age, sex, BMI, previous therapy, PPI and antibiotic use.
Extended Data Fig. 4. Treating balances as continuous independent variables.

Panel A-C shows a multivariable Cox regression analysis of overall survival (OS) in months for 146 patients at baseline (one patient was removed due to missingness in the included predictor variables) treating (a) the first balance (Fig. 2a), (b) the second balance (that is, the extended longitudinal balance), and (c) the third balance (that is, the ‘baseline only’ balance) as a continuous independent variable. While the histograms show the distribution of each balance (right y-axes), each regression line represent the hazard ratio as a smooth function of each balance (left y-axes). All models are adjusted for age, sex, BMI, PPI and antibiotics use, and previous therapy.
We next tested the generalizability of the balance described in Fig. 2a by computing it for patients from six independent melanoma cohorts5,7–10,12. Despite small sample sizes and large heterogeneity in terms of DNA isolation protocols and sequencing platforms (Supplementary Table 6), this analysis showed that the balance achieves a comparable AUC in several of the independent cohorts (Extended Data Fig. 5a,b). However, only in the cohort with a reasonably large number of patients (N = 112) did we find that the balance could discriminate between patients with PFS ≥12 and PFS <12 months (two-sided Wilcoxon test, P = 0.04; Extended Data Fig. 5c). While limited in sample size (N = 27), the balance also predicted OS in one of the independent cohorts (P = 0.024; Extended Data Fig. 5d).
Extended Data Fig. 5. Generalizability of the longitudinal balance (Fig. 2a) across six independent melanoma cohorts.

Panel (A) shows the AUC for each independent baseline cohort, including the current study (in red). Panel (B) shows the AUC for McCullochJA_2022’s post-ICB cohort. Panel (C) shows the average difference in the balance score between patients with PFS < 12 months versus PFS ≥ 12 months from the SpencerCN_2021 cohort. Finally, panel (D) shows Kaplan-Meier curves and multivariable Cox regression analysis of overall survival (OS) in months from 27 patients from McCullochJA_2022’s baseline cohort according to high (≥75 percentile) and low (<75 percentile) values of the balance after adjusting for age, sex, BMI and PPI-use.
Our analysis also revealed several SGBs only associated with PFS12 at baseline but not thereafter, many of which have not been previously reported in association with ICB, potentially owing to a lower resolution in taxonomic profiling (Extended Data Fig. 2 and Supplementary Table 4). For example, patients with PFS ≥12 months were enriched with Romboutsia timonensis (SGB6148), Limosilactobacillus fermentum (SGB7106) and Blautia schinkii (SGB4825), while patients with PFS <12 months had higher abundances of Eubacterium siraeum (SGB4198 group), Oscillibacter sp. ER4 (SGB15254) and Dysosmobacter sp. NSJ 60 (SGB15124) at baseline but not at subsequent study visits (Extended Data Fig. 2 and Supplementary Table 4). Stratifying patients on the basis of the median value of a balance between the 9 and 11 SGBs that, only at baseline, had higher abundances in patients with PFS ≥12 and PFS <12 months, respectively, we could predict OS at baseline (OSHigh of 35.5 months versus OSLow of 28.4 months; HR of 1.639, P = 0.034; Extended Data Fig. 6).
Extended Data Fig. 2. Extension of Fig. 1.

This includes all SGBs, that is, also those that were differentially abundant in only one study visit.
Extended Data Fig. 6. Baseline only balance.
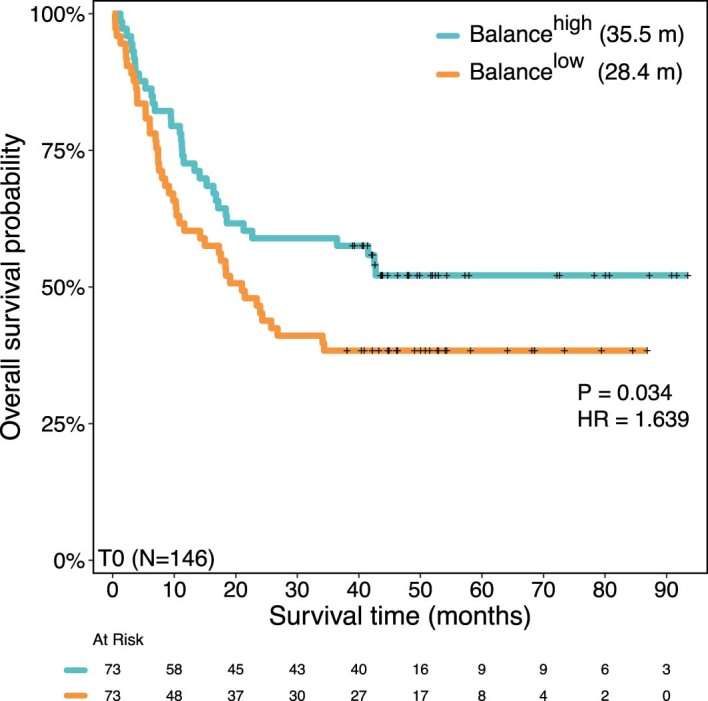
A ‘baseline only’ balance containing the 9 species associated with patients with PFS ≥ 12 months at baseline only in the numerator, and the 11 SGBs associated with patients with PFS < 12 months at baseline only in the denominator (see Fig. 1a). Kaplan-Meier curves and multivariable Cox regression analysis of overall survival in 146 patients at baseline (one patient was removed due to missingness in the included predictor variables) according to high (above median) and low (below median) values of the balance after adjusting for age, sex, BMI, previous therapy, PPI and antibiotic use.
Microbial associations that emerge after ICB initiation
While microbial taxa that are able to differentiate between patients with PFS ≥12 and PFS <12 months at baseline may serve as important predictive and/or prognostic biomarkers, studying microbial taxa longitudinally could derive novel mechanistic insights in addition to becoming a new way to monitor ICB efficacy and irAEs. Therefore, we next identified SGBs that were only discriminative of patients with PFS ≥12 and PFS <12 months after ICB initiation. We found higher abundances of several SCFA producers from the Lachnospiraceae family, which included Coprococcus comes (SGB4577 group), Coprococcus catus (SGB4670), Gemmiger (SGB15295 group) and Anaerobutyricum hallii (SGB4532) in patients with PFS ≥12 months after ICB initiation (Fig. 1a and Supplementary Table 4). These species have previously been associated with increased response and OS in patients treated with immunotherapy8,12,36,37, but also with general health and a lower risk for metabolic and chronic inflammatory diseases28. Patients with PFS <12 months, on the other hand, showed an increase in Clostridium spiroforme (SGB6747), several other Lachnospiraceae (Blautia hydrogenotrophica (SGB4677), Blautia wexlerae (SGB4837 group), Ruminococcus torques (SGB4608), Sellimonas intestinalis (SGB4617) and Eisenbergiella tayi (SGB4988) and Erysipelotrichaceae (Turicibacter sanguinis (SGB6847) and Faecalibacillus faecis (SGB6750)) species only after the start of ICB (Fig. 1a and Supplementary Table 4). Recent studies have reported that Eisenbergiella sp., B. wexlerae, C. spiroforme and Erysipelotrichaceae were associated with resistance to ICB4,36 and enriched in patients with more aggressive tumors38.
Abundance patterns in patients with PFS ≥12 and <12 months during ICB
Next, we took a closer look at specific abundance patterns in patients with PFS ≥12 and PFS <12 months over the study period. Here, we assess whether microbial abundances reversed, converged or diverged from baseline in patients with PFS ≥12 and PFS <12 months over the study period. We found that 22.8% (90% BCL; range: 74.7% (50% BCL)–0.7% (100% BCL)) of the SGBs increased or decreased after treatment initiation (Supplementary Table 3). Focusing on the aforementioned 99 SGBs that could discriminate between patients with PFS ≥12 and PFS <12 months, we identified 22 SGBs for which patients with PFS ≥12 and PFS <12 months exhibited intersecting slopes (Fig. 3b,c and Extended Data Fig. 7, dynamics 3ab). In these cases, patients with PFS ≥12 and PFS <12 months had different initial abundances at baseline, with slopes crossing after the start of the treatment generating reverse abundance patterns at baseline compared to the last study visit. We found, for example, several SGBs that have been associated with various chronic and immune-mediated diseases, such as Streptococcus thermophilus (SGB8002) and T. sanguinis (SGB6847), which are dominant in the oral cavity, and B. schinkii (SGB4825), to exhibit opposite abundance patterns in patients with PFS ≥12 and PFS <12 months at baseline compared to the last study visit with patients with PFS <12 months and patients with PFS ≥12 months exhibiting positive and negative slopes, respectively. Other SGBs showed similar baseline abundances in patients with PFS ≥12 and PFS <12 months to only diverge after the start of ICB (Fig. 3a and Extended Data Fig. 7, dynamics 1ab). For example, we found increasingly separating abundances of Christensenellaceae bacterium NSJ 53 (SGB82545), E. tayi (SGB4988; Fig. 3a), Mediterraneibacter massiliensis (SGB4595), S. intestinalis (SGB4617) and Hydrogeniiclostridium mannosilyticum (SGB14890) that increased in patients with PFS <12 months and decreased in patients with PFS ≥12 months after the initiation of ICB (Extended Data Fig. 7, dynamics 2a and Supplementary Table 2).
Fig. 3. Different taxon dynamics in patients with PFS ≥12 and PFS <12 months.

a–d, Four different dynamics exemplified by different microbial SGBs with different dynamics in patients with PFS ≥12 (n0 = 62, n1 = 77, n2 = 38 and n3 = 30) and PFS <12 (n0 = 74, n1 = 69, n2 = 34 and n3 = 24) months, where the slopes of patients with PFS ≥12 months (yellow slopes) and patients with PFS <12 months (purple slopes) diverge from similar baseline abundances (a, dynamics 2a, Extended Data Fig. 7a), where the slopes of patients with PFS ≥12 and PFS <12 months are crossing (b, generating opposite abundance patterns when comparing baseline to the last study visit, dynamics 3b, Extended Data Fig. 7b), where the slope of the patients with PFS <12 months is relatively unchanged across the study visits compared to the slope of the patients with patients with PFS ≥12 months (c, dynamics 1c, Extended Data Fig. 7c); where the slope of the patients with PFS ≥12 months is relatively unchanged across the study visits compared to the slope of the patients with PFS <12 months (d, dynamics 2c, Extended Data Fig. 7c). The y axis shows the expected abundance (represented in centered log ratio coordinates) for each study visit (x axis). The corresponding inset figures show the average difference between patients with PFS ≥12 and PFS <12 months at each study visit, including its 90% credible interval. The number (n) represents the number of patient samples at each visit for patients with PFS ≥12 and PFS <12 months.
Extended Data Fig. 7. Schematic illustration showing the different types of microbial dynamics we observe between patients with PFS ≥ 12 months and PFS < 12 months.

Panels A-E are schematic illustrations (that is, cartoons) showing the breakdown of the different types of taxon dynamics we observe in the overall comparison between patients with PFS ≥ 12 months and patients with PFS < 12 months. Yellow and purple slopes correspond to patients with PFS ≥ 12 and PFS < 12 months, respectively. Panel (A) shows dynamics where patients with PFS ≥ 12 and PFS < 12 months are differentially abundant only after T0 (that is, dynamics 1a and 2a). Dynamics 3a is a particular case of dynamics 1a and 2a where the slopes for patients with PFS ≥ 12 and PFS < 12 months intersect. Panel (B) shows dynamics where patients with PFS ≥ 12 and PFS < 12 months are differentially abundant at early but not at late visits (that is, dynamics 1b and 2b). Dynamics 3b is a special case of dynamics 1b and 2b where the patients with PFS ≥ 12 and PFS < 12 months slopes intersect. Panel (C) shows dynamics where the slope of one of the groups is zero (or close to zero) while the other group is either increasing or decreasing, respectively (that is, dynamics 1c and 2c). In panels (D) and (E), included the inset figures, patients with PFS ≥ 12 and PFS < 12 months exhibit parallel lines (that is, no statistical interactions); Panel 1d and 2d shows dynamics where both patients with PFS ≥ 12 and PFS < 12 months are either increasing or decreasing, respectively, while in panels 1f and 2f, the slopes of patients with PFS ≥ 12 and PFS < 12 months are zero (or close to zero). The number in each plot corresponds to the number of microbial SGBs that follow each type of different dynamics.
Interestingly, we found 16 SGBs that remained relatively unchanged in patients with PFS <12 months over the study period but showed larger changes in patients with PFS ≥12 months (Extended Data Fig. 7c, dynamics 1c). For example, only patients with PFS ≥12 months exhibited increasing abundances of Lachnospiraceae bacterium OF09 6 (SGB4966) and Eubacterium siraeum (SGB4198; Fig. 3c) and decreasing abundances of F. faecis (SGB6750) and Fusicatenibacter saccharivorans (SGB4874). Recent immunotherapy studies in renal cell carcinoma reported that E. siraeum was associated with improved survival and overall response rate8,12,36,37, whereas F. saccharivorans and Erysipelotrichaceae members such as F. faecis were associated with resistance to ICB39. Lastly, we found 14 SGBs, including Bilophila wadsworthia (SGB15452; Fig. 3d) and several Clostridium SGBs, which remained relatively unchanged in patients with PFS ≥12 months across all study visits while exhibiting larger changes in patients with PFS <12 months (Extended Data Fig. 7c, dynamics 2c). While these findings support previous studies showing that the gut microbiome can discriminate between response and nonresponse at baseline, they also suggest that ICB may induce different changes in the gut microbiome of patients with PFS ≥12 and PFS <12 months, respectively. Thus, therapeutic targets that are based on baseline data only risk producing opposite or even unexpected effects.
The clinical context influences abundance patterns
Anti-PD-1 monotherapy versus anti-CTLA-4/anti-PD-1 combination therapy
We next analyzed microbial dynamics for different clinical scenarios and identified common and diverging signals of monotherapy (anti-PD-1) and combination therapy (anti-PD-1 and anti-CTLA-4). To avoid confounding by colitis and PPI use, which individually has considerable effects on the gut microbiome19,20, we compared patients with PFS ≥12 months versus patients with PFS <12 months on monotherapy (Extended Data Fig. 8 and Supplementary Table 7) or combination therapy (Extended Data Fig. 9 and Supplementary Table 8) who did not develop colitis and did not use PPIs, while also averaging over the effects of irAEs that were not colitis, previous antibiotics use, previous therapy and cancer center (Methods and Supplementary Information). We found 28 associations in common between monotherapy (27% of all associations at 90% BCL) and combination therapy (30% of all associations at 90% BCL), whereof 10 and 12 differentially abundant SGBs were shared between patients with PFS ≥12 and PFS <12 months, respectively. Interestingly, the remaining six SGBs (of the 28 shared) exhibited opposite patterns in patients with PFS ≥12 versus patients with PFS <12 months on monotherapy compared to combination therapy (Extended Data Figs. 8 and 9). These included Coprococcus eutactus (SGB5121), Butyricicoccus sp. AM29 23AC (SGB14991) and Parabacteroides merdae (SGB1949), which had opposite slopes in patients with PFS ≥12 versus patients with PFS <12 months on monotherapy compared to combination therapy (Fig. 4). Patients with PFS <12 months treated with monotherapy showed increasing abundances of several Bacteroides species (except for B. intestinalis) across most or all study visits, which were not observed for combination therapy (Extended Data Figs. 8 and 9 and Supplementary Tables 7 and 8). These results confirm previous observations of biphasic effects for the Bacteroides genus dependent on the specific treatment agent(s) used9,40,41. SGBs that exhibited higher abundances in patients with PFS ≥12 months compared to patients with PFS <12 months, regardless of therapy regimen, included Lacticaseibacillus rhamnosus (SGB7144), an unknown Firmicutes (SGB47850), three members from Lachnospiraceae (Dorea sp. AF24 7LB (SGB4571), Dorea formicigenerans (SGB4575), as reported previously12, and C. comes (SGB4577 group)) and four unidentified species from the family Ruminococcaceae (Ruminococcaceae bacterium (SGB15356), GGB9705 (SGB15224), GGB9712 (SGB15244) and GGB9677 (SGB15180); Extended Data Figs. 8 and 9 and Supplementary Tables 7 and 8).
Extended Data Fig. 8. Patients with PFS ≥ 12 and PFS < 12 months on monotherapy.

Panel (A) shows, for each microbial SGB listed, its slopes in patients with PFS ≥ 12 months and PFS < 12 months on monotherapy, respectively. Red and blue colors indicate whether the focal SGB is increasing or decreasing in its abundance over study visits, respectively. It then shows the average difference between patients with PFS ≥ 12 and PFS < 12 months across the different study visits. Non-gray cells in the heatmap correspond to the focal SGB’s log-fold change in abundance between patients with PFS ≥ 12 and PFS < 12 months, respectively. Teal cells correspond to study visits for which the abundance of the focal SGB is higher in in patients with PFS ≥ 12 than with PFS < 12 months on monotherapy, and vice versa for brown cells (at 90% BCL). Gray cells denote differences between patients with PFS ≥ 12 and PFS < 12 months on monotherapy whose 90% CI overlapped with zero.
Extended Data Fig. 9. Patients with PFS ≥ 12 and PFS < 12 months on combination therapy.

Panel (A) shows, for each microbial SGB listed, its slopes in patients with PFS ≥ 12 months and PFS < 12 months on combination therapy, respectively. Red and blue colors indicate whether the focal SGB is increasing or decreasing in its abundance over study visits, respectively. It then shows the average difference between patients with PFS ≥ 12 and PFS < 12 months across the different study visits. Non-gray cells in the heatmap correspond to the focal SGB’s log-fold change in abundance between patients with PFS ≥ 12 and PFS < 12 months, respectively. Teal cells correspond to study visits for which the abundance of the focal SGB is higher in in patients with PFS ≥ 12 than with PFS < 12 months on combination therapy, and vice versa for brown cells (at 90% BCL). Gray cells denote differences between patients with PFS ≥ 12 and PFS < 12 months on combination therapy whose 90% CI overlapped with zero.
Fig. 4. Divergent signals in monotherapy versus combination therapy.

a–c, Three examples out of the six SGBs that exhibited divergent patterns in monotherapy (PFS ≥12: n0 = 41, n1 = 49, n2 = 29 and n3 = 25; PFS <12: n0 = 49, n1 = 48, n2 = 25 and n3 = 18) compared to combination therapy (PFS ≥12: n0 = 21, n1 = 28, n2 = 9 and n3 = 5; PFS <12: n0 = 25, n1 = 21, n2 = 9 and n3 = 6): C. eutactus (SGB5121) (a), Butyricicoccus sp. AM29 23AC (SGB14991) (b) and P. merdae (SGB1949) (c). The y axis shows the expected abundance (represented in centered log ratio coordinates) for each study visit (x axis). Left: anti-PD-1 monotherapy. Right: anti-PD-1/anti-CTLA-4 combination therapy. The corresponding inset figures show the average difference between patients with PFS ≥12 and PFS <12 months at each study visit, including its 90% credible interval. The number (n) represents the number of patient samples at each visit for patients with PFS ≥12 and PFS <12 months.
ICB-induced colitis
We then aimed to identify SGBs associated with development or no development of colitis, averaging over the effects of all other predictors in our model, including PFS12 and therapy regimen (Extended Data Fig. 10 and Supplementary Table 9). We were particularly interested in colitis given the role of the gut microbiome in maintaining colonic immune homeostasis. Colitis was defined using the Common Terminology Criteria for Adverse Events (CTCAE) version 5, excluding intestinal symptoms of non-immune etiology. We found that butyrate producers, such as Roseburia inulinivorans (SGB4940) and Roseburia hominis (SGB4936), A. butyriciproducens (SGB14993 group), Eubacterium rectale (SGB4933 group), Bacteroides thetaiotaomicron (SGB1861) and two Faecalibacterium prausnitzii subspecies (SGB15342 and SGB15317) had higher abundances in patients who did not develop colitis after the start of ICB (Extended Data Fig. 10 and Supplementary Table 9). While all of these SGBs, apart from R. inulinivorans (SGB4940), exhibited negative slopes in both patients affected and patients unaffected by colitis, the decrease was larger in patients who developed colitis. It has been suggested that butyrate may be protective against ICB-induced colitis42; thus a further reduction in the abundance of butyrate producing bacteria during ICB may predispose patients with already lower baseline abundances to colitis. We found that the patient group who did not develop ICB-induced colitis exhibited a higher abundance of F. saccharivorans (SGB4874), which has been shown to induce anti-inflammatory effects in ulcerative colitis43 but has also been associated with resistance to ICB39. While Akkermansia muciniphila has been associated with response in several baseline studies4,11, we found that A. muciniphila (SGB9226) had higher baseline abundances in patients who developed colitis but decreased sharply in abundance thereafter (Extended Data Fig. 10 and Supplementary Table 9). In comparison, the group who did not develop colitis exhibited lower but somewhat increasing abundances of the same SGB (Extended Data Fig. 10 and Supplementary Table 9). While in our cohorts only this particular SGB was identified, there are four different A. muciniphila SGBs in the new MetaPhlAn4 database. Finally, monitoring microbial taxa that are associated with colitis is an important first step toward developing strategies to ameliorate its effects. As a proof of concept, we tested whether a balance between the SGBs associated with development of colitis and the SGBs associated with no development of colitis at baseline (that is, at T0 in Extended Data Fig. 10) could predict colitis development at baseline. We found that this balance could discriminate between the two groups (two-sided Wilcoxon test, PT0 = 0.00055) with an acceptable predictive ability (AUC mean ± s.d. of 0.723 ± 0.121; Fig. 5).
Extended Data Fig. 10. Patients who developed and not developed colitis.

The figure shows, for each microbial SGB listed, its slopes in patients who developed and not developed colitis, respectively, regardless of response to therapy. Red and blue colors indicate whether the focal SGB is increasing or decreasing in its abundance over study visits, respectively. It then shows the average difference between patients with and without colitis across the different study visits. Non-gray cells in the heatmap correspond to the focal SGB’s log-fold change in abundance between patients with and without colitis, respectively. Teal cells correspond to study visits for which the abundance of the focal SGB is higher in in patients who developed colitis compared to those resistant to colitis, and vice versa for brown cells (at 90% BCL). Gray cells denote differences between patients with and without colitis whose 90% CI overlapped with zero.
Fig. 5. A balance predictive of ICB-induced colitis at baseline.

A balance between the 10 SGBs associated with the presence of colitis (red; n = 24 patients) and the 12 SGBs associated with the absence of colitis (blue; n = 112 patients) at baseline is predictive of colitis development at baseline. Left: the balance’s discriminatory ability (two-sided Wilcoxon test, PT0 = 0.00055). Boxplots represent minima, Q1, Q2, Q3 and maxima. Right: the same balance’s predictive ability expressed as the averaged AUC computed from a 100 times repeated five-fold cross-validation (AUC mean ± s.d. of 0.723 ± 0.121). The dashed diagonal line represents random chance.
In our dataset, we found a relationship between PFS12 and irAEs that were not colitis (Fisher’s exact test: P = 0.002; Supplementary Fig. 1). Compared to patients who achieved a PFS ≥12 months and developed colitis, we found that patients who achieved PFS ≥12 months but did not develop colitis exhibited higher abundances of four SGBs across the entire study period (Blautia sp. AF19 10LB (SGB4810), Lachnospiraceae bacterium (SGB4706), Gordonibacter pamelaeae (SGB14807) and Clostridium sp. AF20 17LB (SGB4714); Supplementary Fig. 2 and Supplementary Table 10). On the other hand, we found seven SGBs that exhibited higher abundances, throughout the study period, in patients with PFS ≥12 months who developed colitis, including several unclassified Clostridia species (Supplementary Fig. 2 and Supplementary Table 10). Interestingly, while patients with PFS ≥12 months without colitis showed enrichment in several F. prausnitzii SGBs (SGB15317, SGB15318 group, and SGB15342), A. butyriciproducens (SGB14993 group) and R. hominis (SGB4936), the abundance of A. muciniphila (SGB9226) was higher (but decreasing) in patients with PFS ≥12 months who developed colitis (Supplementary Fig. 2 and Supplementary Table 10). F. prausnitzii has previously been associated with the absence of colitis37; hence our findings further support approaches targeting different subspecies of F. prausnitzii to counteract colitis while maintaining ICB efficacy.
While we found a difference in the proportion of patients who develop colitis on monotherpay (0.128) compared to combination therapy (0.310) (two-sided test of equal proportions: Δ = 0.182; 95% CI: 0.036, 0.329; χ2 = 7.259; P = 0.007), we did not find a difference in the proportion of patients with PFS ≥12 months who developed colitis on monotherapy (0.051) compared to combination therapy (0.138) (two-sided test of equal proportions: Δ = 0.087; 95% CI: −0.024, 0.197; χ2 = 2.866; P = 0.09). When we compared colitis development under combination versus monotherapy, we observed higher and increasing abundances of SGBs belonging to the Streptococcus, Veillonella, Bacteroides and Eggerthella genera, an overall signature resembling the gut microbiome of patients with inflammatory bowel disease20 (Supplementary Fig. 3 and Supplementary Table 11).
PPI use
Finally, we investigated the effect of PPI use on patients with PFS ≥12 and PFS <12 months (Supplementary Fig. 4 and Supplementary Table 12). To avoid confounding by combination therapy and colitis, we focused on the group of patients who were treated with monotherapy and did not develop ICB-induced colitis. Here we found that PPI users on monotherapy shared 33 associations with nonusers on monotherapy (at 90% BCL; Supplementary Table 13). For a few SGBs, patients with PFS ≥12 months exhibited different slopes for users and nonusers. For example, S. thermophilus (SGB8002) exhibited increasing abundances in nonusers with PFS ≥12 months and decreasing abundances in users with PFS ≥12 months. Similarly, C. bacterium NSJ 53 (SGB82545) and B. caccae (SGB1877) exhibited increasing and decreasing abundances in nonusers and users, respectively, with PFS <12 months. While the Christensenellaceae family has been associated with health, B. caccae, B. stercoris and P. vulgatus have been linked to diseases such as inflammatory bowel disease and colorectal cancer44,45.
Discussion
In this study, we longitudinally profiled the gut microbiome in a multicenter cohort of 175 patients with advanced melanoma undergoing ICB. Through Bayesian regression models with higher-order interactions, we characterized microbiome changes in patients with PFS ≥12 or PFS <12 months during ICB, including in different clinical contexts such as therapy regimen, development of colitis and PPI use.
Previous studies conducted at baseline have led to an accumulating interest in SCFA producers as targets for increasing ICB efficacy, whereas species predictive of resistance to ICB have been associated with chronic immune-mediated or metabolic diseases46. However, longitudinal studies of the gut microbiome dynamics during treatment with ICB have been lacking. We show that, during ICB, a number of SGBs have contrasting dynamics to what would be expected from baseline and that the same SGB can exhibit different trajectories depending on the clinical context. While the abundance of SCFA producers remained at a higher abundance or even increased in patients with PFS ≥12 months during treatment, the abundance of SGBs considered ‘immunogenic’ exhibited larger changes from baseline, with different dynamics in different clinical contexts. Patients with PFS <12 months showed higher or increasing abundances of taxa that have been associated with inflammatory diseases, such as B. clarus, S. intestinalis and E. tayi. However, when considering different clinical contexts, we also found several taxa regarded as ‘proinflammatory’ (for example, P. merdae, Desulfovibrio piger and Streptococcus oralis) to be enriched in patients with PFS ≥12 months (Supplementary Fig. 5).
Comparing our results to two recent studies employing MetaPhlAn4, we found many common SGBs associated with ICB response at baseline, including several SGBs that also were differentially abundant during therapy and in different clinical contexts in our study. For example, Thomas et al.21 found that Eubacterium sp. AM28 29 (SGB6796 group) was associated with response in nine melanoma cohorts, four of which were not included in our study5,9,11,36. The same SGB was enriched in responders 1 month after FMT22. In our study, it was associated with PFS ≥12 months at baseline through to the second study visit, and at baseline in patients with PFS ≥12 months on combination therapy. Another SGB, L. celerecrescens (SGB4868), which is part of the balance described in Fig. 2a, was associated with response in six melanoma cohorts, five of which were not included in our study9,11,12,36,47 analyzed by ref. 21 and also enriched in all responders one month after FMT22. The replicability of our results, both the main balance and specific SGBs, shows the robustness of our longitudinal analysis (Supplementary Tables 14 and 15).
Our findings provide an important roadmap for designing and interpreting microbiome-based intervention studies. Owing to the distinct longitudinal dynamics observed in this study, therapeutic targets developed only from baseline findings may produce opposite or unexpected results. This can further vary depending on the clinical context. While we confirm a higher or increasing abundance of several species that are currently being studied as consortia therapies, including A. butyriciproducens, A. hallii, C. catus, E. rectale, Bifidobacterium adolescentis, F. prausnitzii48 and B. thetaiotaomicron49, other members of these consortia showed an increase in patients with PFS <12 months in our study, such as R. torques, Parabacteroides distasonis48 and B. clarus49, or had opposite trajectories depending on the therapy regimen (for example, increase of several Bacteroides SGBs and P. merdae in patients with PFS <12 months on anti-PD-1 monotherapy)48.
Our results could also be used to disentangle the effect of FMT from the longitudinal effect of ICB and important confounders on the gut microbiome. Recent phase 1 clinical trials suggest that FMT from responders50,51 or healthy donors22 combined with anti-PD-1 can induce response in a subset of ICB-refractory (OR 20–30% in refs. 50,51) and ICB-naive patients (OR 65% in ref. 22). Without performing FMTs, we observe similar taxa changes in patients with PFS ≥12 months (Supplementary Tables 14 and 15), suggesting that FMT synergizes with ICB to improve responses. Inter-individual variability in the response and engraftment of strains is widely described after FMT for different clinical contexts52,53 in which various treatment and host factors play a role16,22,54,55. We observed different dynamics of the shared SGBs depending on the clinical context (Supplementary Tables 14 and 15), findings that could be used to help optimize donor-recipient stratification in future trials.
To conclude, this study underlines the dynamic nature of the gut microbiome and indicates that longitudinal profiling at finer taxonomic resolution in association with host factors is critical for guiding microbiome-targeted interventions aimed at improving treatment outcomes. Limitations of this study include (but are not restricted to) simplifying microbial dynamics to linear trajectories, comparability with previous studies using different taxonomic databases and a smaller number of patient samples for some of the post hoc comparisons, which limits the generalizability of some of our results. To further validate our findings and move the clinical gut microbiome field forward from a biomarker perspective to actionable treatments, continued efforts should go into longitudinal profiling of ICB patients at larger scales, linking the gut microbiome, metabolome and immunome to treatment outcome.
Methods
Study design and cohort description
The prospective PRIMM cohorts
We prospectively recruited 128 patients with advanced melanoma who were treated with ICB between August 2015 and January 2020 in the UK studies Predicting Response to Immunotherapy for Melanona with Gut Microbiome and Metabolomics (PRIMM–UK, n = 54) and the Netherlands studies (PRIMM–NL, n = 74, made up of eligible patients from the COLIPI, POINTING and OncoLifeS studies). PRIMM–UK (NCT03643289) is sponsored by East and North Hertfordshire NHS Trust with ethical approval from King’s College London. OncoLifeS (METc number 2010/109), COLIPI (METc number 2012/085, NCT02600143) and POINTING (METc number 2018/350, NCT04193956) have all been approved by the Medical Ethical Committee (in Dutch: Medisch Ethische Toetsingsingscommissie or METc) of the University Medical Center Groningen in the Netherlands. OncoLifeS information is available on the Netherlands Trial Register56. Fecal samples were collected from these patients before initiation of ICB and longitudinally at up to four treatment (study) visits: at baseline and before each subsequent treatment cycle over a period of 12 weeks (Supplementary Fig. 1). The time between two samples was 3 or 4 weeks, depending on the treatment regimen, with ipilimumab/nivolumab combination therapy and pembrolizumab monotherapy administered three times weekly and nivolumab monotherapy administered four times weekly. Written informed consent was obtained from all patients.
Other enrolled cohorts
Patients within the PRIMM cohorts were recruited in parallel, using aligned protocols4. Additional patients, treated between March 2015 and November 2019, were enrolled from cohorts outside the setting of the PRIMM study: Leeds (n = 19), Barcelona (n = 11) and Manchester (n = 17). Fecal samples were collected at time points similar to those used in our included prospective studies. Patient samples within the Manchester cohort were collected with written full-informed patient consent under Manchester Cancer Research Centre Biobank ethics application 07/H1003/161+5 (updated in 18/NW/0092) and approval for the work under Manchester Cancer Research Centre Biobank Access Committee application 13_RIMA_01. Barcelona cohort samples were subjected to the ethical committee of Hospital Clínic of Barcelona approval (registry HCB/2015/1032). Data and samples from Leeds were collected in a study named ‘Developing a blood test of immunity in illness: a study examining the peripheral blood transcriptome in patients with cancer, autoimmune disease, immunodeficiency or iatrogenic immune suppression’ (research ethics committee reference 15/NW/0933). Informed written consent was obtained for collection of samples and data, sharing anonymized data and working with collaborators whether academic or commercial.
Inclusion criteria
Patients who fulfilled the following criteria were eligible for the analysis: (1) histologically or cytologically confirmed non-resectable advanced (stage 3 or 4) cutaneous melanoma, (2) treatment with ICB (nivolumab or pembrolizumab) or a combination of ipilimumab and nivolumab at the recommended dose as a first-line immune checkpoint inhibitor, (3) 18 years of age or older and (4) availability of baseline characteristics presented in Table 1.
Assessment of treatment outcomes
Response to ICB was classified according to RECIST v1.1 criteria. Based on radiographic response, patients were classified as responders (complete response, partial response and stable disease), or nonresponders (progressive disease).
Clinical endpoints were defined as PFS12 and OS. PFS was defined as the time from the initial immunotherapy to disease progression. OS was defined as time in months from initiation of treatment to occurrence of death from any cause. IrAEs, including colitis, were assessed using the CTCAE version 5 (19). Side effects that were clearly of non-immune etiology were excluded.
Sample and data collection
Patients received oral and written instructions regarding the stool collection procedure. Patients within PRIMM–UK and PRIMM–NL were requested to collect approximately 3–5 ml plain feces using a collection kit that could be used at home and then store the sample in their freezer directly after collection. PRIMM–NL samples were transported to the hospital in a frozen insulated cooling bag to prevent thawing. Due to the geographic disbursal of PRIMM–UK patients, samples were collected and placed in Thermo Fisher Scientific kits and sent by special post to the laboratory at King’s College London. After arrival in the hospital, the samples were directly stored at −80 °C. Plain stool samples from the Manchester cohort were either collected on site at the hospital and stored directly at −80 °C within 4–6 h of collection or collected in sample containers and sent by special post to the laboratories of CRUK Manchester Institute and stored directly at −80 °C upon arrival. Patients within the Barcelona cohort used the OMNIgene GUT collection kit (DNA Genotek). Fecal DNA was extracted from 1 to 14 days after sample collection using the PowerFecal DNA Isolation Kit (previously MoBio, currently Qiagen) and kept frozen until needed. Patients from Leeds also collected stool at home using the OMNIgene GUT collection kit (DNA Genotek), and samples were returned to the research nurse.
Baseline demographics, including sex, age, body mass index (BMI), Eastern Cooperative Oncology Group (ECOG) performance status and medication use, were collected, along with tumor staging and previous anti-cancer therapy data. Demographic data were collected as part of a screening visit up to 14 days before ICB treatment began. All baseline antibiotics or PPI use within 3 months of commencing ICI treatment was documented. Tumor staging took place up to 1 month before the start of treatment.
Radiological evaluation, consisting of a computed tomography (CT) scan of the thorax, abdomen and pelvis and magnetic resonance imaging of the brain, was performed at baseline (that is, before the first dose of immunotherapy). A small number of patients had positron emission tomography scans with a CT component. Follow-up radiological evaluation was performed every 10–14 weeks as long as the patient received systemic therapy. Additional CT and/or magnetic resonance imaging scans were performed when there was suspicion of progression. If the first radiological evaluation after the start of therapy was inconclusive, then a confirmatory scan was performed 4–12 weeks later.
Metagenomics processing
DNA extraction and sequencing
DNA was isolated for all cohorts at King’s College London using Thermo Fisher Scientific’s MagMax Core protocols for nucleic acid purification and mechanical lysis. Samples with a high-quality DNA profile (>15 ng µl−1 of DNA) were further processed. Sequencing libraries were prepared using the Illumina Nextera DNA Flex Library Prep Kit according to the manufacturer’s protocols. Libraries were multiplexed using dual indexing and sequenced for 300 bp paired-end reads using the Illumina NovaSeq6000 platform according to the manufacturer’s protocols. We obtained a total of 1,283 Gb with an average of 53,919,210 reads per sample before quality control and pre-processing.
Metagenome quality control and pre-processing
Shotgun metagenomic sequencing was performed at the NGS Core Facility at University of Trento. The quality of all sequenced metagenomes was controlled using the pre-processing pipeline implemented in ref. 57. Of all the samples collected across the five observational cohorts, we considered those that passed all the quality control steps of the metagenomic sequencing pipeline and had more than 1 Gb of sequencing data. This resulted in a total of 447 samples from 195 patients that were then subjected to strict quality control and were processed into taxonomic and predicted pathway abundances.
Microbiome taxonomic and functional potential profiling
Taxonomic and functional metagenomic profiling was performed using MetaPhlAn417 with the vJan21 SGB database release and HuMAnN358 with default parameters. Before prevalence filtering (see below), we identified a total of 2,223 microbial SGBs and 518 microbial pathways.
Selection of independent variables for the longitudinal model
We were interested in modeling study visit varying intercept and slopes for patients with PFS ≥12 and PFS <12 months, respectively, in three main clinical contexts: (1) the type of immunotherapy patients received (that is, mono versus combination therapy), (2) if patients had developed any grade of ICB-induced colitis (no versus yes) and (3) if patients received concomitant PPIs (no versus yes). Beyond these three independent variables, we also controlled for previous antibiotics use, previous BRAF or MEK-targeted therapy, time since first injection (in days), cancer center, other forms of irAEs (that is, not colitis), age, sex and BMI. We also included a patient identifier to account for the repeated measurements. In the end, the included variables represented a balance between (1) minimizing collinearity between independent variables, (2) loss of patient samples due to missingness in independent variables and (3) the number of included independent variables versus the number of modeled samples. These selection criteria resulted in 408 samples from 175 patients.
Prevalence filtering of microbiome taxonomic and functional profiles
We retained microbial features that were present in at least 20% of the baseline samples, which also had a prevalence of least 10% among the longitudinal samples. Applying this stringent filtering criterion, we retained in the 408 samples; 434 and 395 microbial SGBs and pathways, respectively. This was done using phyloseq (v.1.42.0) and tidyverse (v.2.0.0) R packages.
Independent melanoma cohorts for validation
To validate the balance described in Fig. 2a, we downloaded the raw sequences from six publicly available melanoma cohorts (three using radiographic response based on RECIST1.1 criteria, one using PFS12 and two using both RECIST and PFS12) that characterized gut microbiome composition at baseline (Supplementary Table 6). We kept the response definition used in the original publication. One of the cohorts5 also characterized gut microbiome composition within 4 months after the start of ICB. We treated these pre- and post-ICB samples from ref. 5 as two cohorts. We re-processed the raw sequences using MetaPhlAn4 (using the same database and settings as described above) and computed the balance (Fig. 2a) for all samples in each independent cohort. Not all SGBs were present in all independent cohorts. For example, we did not find taxon GGB1420 SGB1957 (SGB1957) in any of the independent cohorts after 10% prevalence filtering (see Supplementary Table 16 to see which SGBs were missing in each independent cohort). To test whether the balance could predict response anew in each independent cohort, we fit a simple logistic model [glm(response_definition ~ balance_score, family = ’binomial’)] to all samples in each cohort and computed the AUC (on the training data, as we fit all samples). We could not include any other independent variables in the models because most cohorts did not report information such as age, sex, BMI or other clinical variables.
Statistical analysis
Compositional data analysis
Metagenomic sequencing produces compositional data, which means that information can only be obtained in the form of relative abundances that are independent of the total microbial load in a given sample. As a result, an increase in one microbial feature (for example, a taxon or metabolic pathway) relative abundance necessarily requires an equivalent decrease in the relative abundance of the remaining community of features present in the same sample. If this statistical property is not accounted for, the likelihood of introducing false positives in differential abundance analysis59,60 and negative correlation biases in correlation-based analysis61,62 increases heavily. While standard statistical methodology assumes that the analyzed data are represented by variables free to vary from −∞ to ∞ within Euclidean geometry, compositional data occupy the simplex that is a restricted space where variables are strictly positive and vary from 0 to 1 or 0 to 100, if data are represented as proportions or percentages (such as relative abundances), respectively. A log ratio transformation maps the simplex to Euclidean real space (that is, the Aitchison geometry) where standard statistical methodology can be applied. There are several available log ratio transformations, each using a different reference frame (that is, the denominator). For example, the additive and centered log ratio (alr and clr, respectively) transformation is defined as
| 1 |
| 2 |
where denotes a sample (that is a composition) containing D ‘counted’ microbial features. In the alr transformation (equation (1)), the choice of the denominator or the reference frame is arbitrary and could represent any specified feature. In the clr transformation (equation (2)), however, the denominator is defined by the geometric mean g(x) of the focal sample, or put simply, the ‘average’ feature in the focal sample.
Differential ranking
There already exist several developed methods to find changes in compositional data between cases and controls that avoid the biases caused by the compositional nature of metagenomic sequencing data (for example, ALDEx263, ANCOM64 and Gneiss65). What these methods typically have in common is that they internally use some log ratio transformation, which is conserved regardless of whether the data are relative or absolute. A recent approach called differential ranking is robust to the choice of the alr reference feature, and ranks produced from relative abundances are identical to the ranks of absolute abundances66,67. More specifically, the term ‘differential’ refers to the logarithm of the fold change in abundance of a microbial feature between cases and controls. Differential rankings can therefore be used to detect differentially abundant features knowing that the results are not affected by the compositional nature of the data. It is important to note, however, that high-ranking (positive) features have not necessarily increased in absolute terms between the cases compared to the controls but can still have decreased, although to a lesser extent than the lower-ranking features.
A logistic normal model to estimate differential rankings from proportions
Almost all of the methods developed for compositional sequencing data are intended for counts (for example, 16S rRNA gene amplicons). However, if the processed sequencing data are expressed as proportions with unknown sample totals, then these methods may require changes before being applied. The R package fido68 (1.0.4) implements a Bayesian multinomial logistic normal regression model called Pibble that can be adapted to model proportions (that is, only fitting the logistic normal model). Furthermore, the coefficients estimated by Pibble can be ranked and interpreted as differential rankings with statistical significance achieved through Bayesian inference68,69. Pibble is constructed to model any observed sequencing counts using a multinomial distribution, with the underlying microbial feature composition as random variables modeled by a logistic normal distribution. More specifically, the observed relative abundances are considered to be drawn from a multinomial distribution parameterized by a set of proportions (πj), which have an analogous representation in the alr space, with the transformed variables drawn from a multivariate normal distribution that exists in Euclidean real space68,69. While both the multinomial Dirichlet model and the multinomial logistic normal model can handle over-dispersed count data70,71, the logistic normal model also allows for both positive and negative covariation between microbial features69. In short, the Pibble model is defined as follows:
| 3 |
| 4 |
| 5 |
| 6 |
| 7 |
with Y representing a D × N count matrix with the jth column representing a sample (that is composition) containing the D ‘counted’ (microbial) features (equation (3)). Equation (4) represents a transformation between the multinomial parameters (πj sum to 1) that exist on the simplex, and the transformed parameters ηj that exist in Euclidean real space. As is common for multinomial regression, Pibble uses the inverse alr transformation (also called the softmax transform in the machine learning literature) to produce a relative abundance matrix (that is, proportions varying between 0 and 1). This also implies that . The Q modeled covariates are included in the Q × N matrix denoted X. Importantly, equation (5) simply represents a multivariate linear model with X containing the Q modeled covariates, Λ a matrix containing the corresponding estimated regression coefficients that can be ranked to produce the differential rankings and a D × D matrix containing the residual covariance between all log ratios. The matrix containing the estimated regression coefficients (that is, Λ) is modeled as a matrix normal distribution, which is simply a generalization of the multivariate normal distribution capable of describing the covariation between the rows (that is, features ) and between the columns (that is, samples, Γ) of Λ (equation (6)). Finally, is modeled as a inverse Wishart distribution (W−1), which is a common distribution over covariance matrices (equation (7))68,69.
Owing to the large flexibility of the Pibble model, it is possible to directly model sequencing data expressed as proportions (that is, relative abundances) using the logistic normal model (that is, starting from equation (4)). The only drawback of this is that variation in the counts cannot be modeled, but this information is naturally lost once data are normalized (and if the information on sample totals is not kept). Importantly, once the model is fit, the results can be viewed as if any log ratio transform had been used (instead of the alr in equation (4)), including the clr. Lastly, because equation (5) simply represents a multivariate linear model, interactions between predictor variables can also be modeled. Pibble uses the collapse–uncollapse sampler, which was developed particularly for this class of models68,69. We used the same priors as suggested by refs. 68,69.
A linear model with higher-order interactions
We hypothesized that microbial abundances may change over the course of the treatment period because patients received an immunotherapy injection at each treatment visit, thus probably increasing the cumulative effect of the therapy on the gut microbiome across the study period. We further hypothesized that patients with PFS ≥12 and PFS <12 months may exhibit different patterns of change. To model this, we included higher-order interactions, thereby assuming that microbial abundances change linearly across study visits. In equation (5), we modeled the relationship between X (study visits/cumulative number of treatment injections) and Y (the log ratio value for any given microbial feature) to be contingent not only on Z whether patients achieved PFS ≥12 months, but also on the moderator variable W, which in our case represents one of three treatment characteristics of interest. Therefore, the three three-way interactions we modeled all included the same X and Z variables but with a different treatment characteristic of interest (that is, the moderator variable W1–3; see equation (8)). The different treatment characteristics for W that we modeled were: W1, the type of immunotherapy patients received (that is mono versus combination therapy); W2, if patients had developed any grade of ICB-induced colitis (no versus yes); and W3, if patients received concomitant PPIs (no versus yes). Beyond the different treatment characteristics, we also controlled for whether patients have had previous chemotherapy, time since first injection (in days), the cancer center patients were treated at, whether they experienced other forms of irAEs (that is, not colitis), age, sex and BMI. We also included a patient identifier to account for the repeated measurements. Lastly, before model fitting, all continuous variables (that is, age and BMI) were mean centered, and all ‘peripheral’ categorical variables (that is, previous therapy, center, other forms of irAEs, patient identification and sex) were coded using weighted sum contrasts (as opposed to treatment contrasts). The latter effectively mean-centers categorical variables with the result being that the intercept represents the average of all independent variables not included in the three-way interactions. To note is that all 175 patients in the main analysis have information on all of these metadata (that is, there is nothing missing/not available).
Without including any of the ‘peripheral’ independent variables, which we adjusted for (that is, center, time to/since first injection, other forms of irAEs, patient identification, age, sex and BMI), we can write our linear regression model as
| 8 |
where Z and W1–3 are binary variables dummy coded to be either 0 or 1, always with 0 as the reference category. Thus, the β2 coefficient for Z (PFS12: 0 is PFS <12, 1 is PFS ≥12) represents the value when all treatment characteristics of interest (W1–3) are at their reference level (that is, monotherapy (W1), no colitis (W2) and no PPIs (W3)) and when the independent variable X has a value of zero (that is, baseline). We can further rewrite equation (8) to illustrate that the relationship between X and Y is conditional on Z and W1–3 as follows:
| 9 |
where the first and second parentheses represent the intercepts and the slopes graphing Y against X.
Post hoc contrasts to compute the comparisons of interest
To create the relevant comparisons between cases and controls, we constructed so-called post hoc contrasts (linear combinations of coefficients) directly from the fitted model. To compute these, we first constructed reference grids (Supplementary Information), which contain all relevant combinations of the categorical independent variables that we wanted to average over. Based on these reference grids, we computed marginal means of cases and controls, which we then could statistically compare. Because we already mean centered all ‘peripheral’ independent (continuous and categorical) variables, we only consider the coefficients associated with the treatment characteristic of interest (W1–3), which is shown in equation (8). The post hoc contrasts we computed were (1) PFS ≥12 versus PFS <12 months, (2) colitis versus no colitis, (3) PFS ≥12 months with and without colitis, (4) patients on combination versus monotherapy with colitis, (5) PFS ≥12 versus PFS <12 months on monotherapy without colitis and no PPIs, (6) PFS ≥12 versus PFS <12 months on combination therapy without colitis and no PPIs and (7) PFS ≥12 versus PFS <12 months on PPIs, monotherapy and without colitis. In Supplementary Information, we show the mathematical procedure to compute these post hoc contrasts for (1), (2) and (3), but the same logic applies when computing to the remaining contrasts.
Balance analysis
A balance is a type of log ratio defined as the ratio between the geometric means of two subsets of features72,73. Following the definition in ref. 73, mathematically a balance is defined as follows. Let X = (X1,X2,X3,…,Xk) be a sample with k features. Given two non-overlapping subsets of features in X denoted by X+ and X−, indexed by I+ and I−, and comprising k+ and k−, the balance between X+ and X− is defined as the log ratio of the geometric mean of the two subsets of features as follows:
| 10 |
Expanding the logarithm, we can simplify the above equation as
| 11 |
Note that we have removed the normalization constant from the original definition as it is later shown by ref. 74 to be unnecessary. Using the relative abundances of the focal SGBs, we computed different balances using the above mathematical formula implemented in custom-written R scripts. Because each balance consists of a selection of SGBs or ‘top hits’ from the longitudinal model that already adjusts for a large number of confounders, the effect of different confounders (for example, cancer center) on each balance score, has already been averaged out.
Survival analysis
To test whether a focal balance was associated with OS at baseline, we used the multivariable Cox proportional hazard regression model as implemented in the coxph() function in the R package survival (3.5-5). We either considered the start of therapy to (1) death from any cause (OS) or (2) progression or death from any cause (PFS) as the time-to-event data. If patients were event-free (that is, alive and/or progression-free) at the last follow-up (28 March 2023), they were right censored. We used these models to estimate HRs including their 95% confidence intervals (CIs) for OS, adjusting for sex, age, BMI, PPI, antibiotics use, previous chemotherapy, colitis and other irAEs. We had n = 146 patients for which these data were complete (that is, no missings). The proportional hazard assumption was checked testing the trend of the Schoenfeld residuals with the cox.zph() function in the survival package (3.5-5). We did not observe any violations in this assumption. Finally, survival curves were estimated using the Kaplan–Meier method as implemented in the survfit2() function in the R package ggsurvfit (0.3.0).
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Online content
Any methods, additional references, Nature Portfolio reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at 10.1038/s41591-024-02803-3.
Supplementary information
Supplementary Figs. 1–5 and methods.
Supplementary Table 1. Number of patient samples in each post hoc contrast.Supplementary Table 2. For each BCL, the (nonzero) slope for each taxon in patients with PFS ≥12 and PFS <12 months, respectively, including their phylum and family belongings.Supplementary Table 3. Number of differentially abundant taxa at different thresholds.Supplementary Table 4. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months across visits, including their phylum and family belongings.Supplementary Table 5. For each BCL, the differentially abundant pathways between patients with PFS ≥12 and PFS <12 months across visits, including their phylum and family belongings.Supplementary Table 6. Independent cohorts used for validation.Supplementary Table 7. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on monotherapy across visits, including their phylum and family belongings.Supplementary Table 8. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on combination therapy across visits, including their phylum and family belongings.Supplementary Table 9. For each BCL, the differentially abundant taxa between patients who developed colitis and patients who did not develop colitis across visits, including their phylum and family belongings.Supplementary Table 10. For each BCL, the differentially abundant taxa between patients with PFS ≥12 months who developed colitis and patients with PFS ≥12 months who did not develop colitis across visits, including their phylum and family belongings.Supplementary Table 11. For each BCL, the differentially abundant taxa between patients who developed colitis on monotherapy versus patients who developed colitis on combination therapy across visits, including their phylum and family belongings.Supplementary Table 12. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on PPI across visits, including their phylum and family belongings.Supplementary Table 13. For BCL of 0.9, the 33 differentially abundant SGBs shared between PPI users (Table 7) and nonusers (Table 12) across visits, including their phylum and family belongings.Supplementary Table 14. SGBs that exhibited similar directional effects in the meta-analysis by Thomas et al. (2023) and in our analysis.Supplementary Table 15. SGBs that exhibited similar directional effects in Routy et al. (2023) and in our analysis.Supplementary Table 16. SGBs in the balance described in Fig. 2a. SGBs that were missing in each independent cohort after 10% prevalence filtering.
Acknowledgements
This work was supported by the Seerave Foundation. The work was also supported by the European Research Council (ERC-STG project MetaPG to N.S.), MIUR ‘Futuro in Ricerca’ (grant RBFR13EWWI_001 to N.S.), the European Horizon 2020 program (ONCOBIOME-825410 project and MASTER-818368 project to N.S.), the National Cancer Institute of the National Institutes of Health (grant 1U01CA230551 to N.S. and L.W.), the Premio Internazionale Lombardia e Ricerca 2019 (to N.S.), the Spanish Fondo de Investigaciones Sanitarias (grants PI15/00716 and PI15/00956) of the Instituto de Salud Carlos III, Spain, co-financed by European Development Regional Fund ‘A way to achieve Europe’ to E.R.D.F., the Wellcome Trust (grant 100282/Z/12/Z to N.C.-L.), and the Dutch Cancer Society (grant 10034 POINTING to E.G.E.d.V.). N.C.-L. is the recipient of a PhD fellowship (FPU17/05453) from the Ministerio de Educación, Cultura y Deportes, Spain. The Leeds group was supported by the Medical Research Council (grant MR/MO19012/1). S.V. was supported by a Harry J. Lloyd Charitable Trust Career Development Award. We acknowledge the Seerave Foundation, which funded this work and many other projects investigating the gut microbiome in human health and disease. We thank the team of the NGS Core Facility at University of Trento for support in sample preparation and for metagenomic sequencing and the high-performance computing team at the University of Trento. We also thank Oncobiome and the Institut Gustave Roussy for their involvement and support of our initiative. The collection of samples from the Hospital Clínic in Barcelona was funded by grant PI15/00716 from Fondo de Investigaciones Sanitarias, Instituto de Salud Carlos III, Spain. Funders did not influence the study design or data analysis. We also thank H. Donker and D. Neijzen for providing feedback on the statistical models. Lastly, we would like to thank all the patients who selflessly took the time to collect samples for this project.
Extended data
Author contributions
K.A.L., L.A.B., N.C.-L., P.S.-B. and M. Harland collected the clinical data. N.D., K.B., M.P. and S.V. provided organizational and scientific assistance relating to sample collection and processing of the samples. A.M.T., P.M., F. Armanini, F. Asnicar, A.B.-M., F.P., M.Z. and A.V, supervised by N.S., were responsible for the bioinformatics pipeline. J.R.B. performed all statistical analyses, and J.R.B. and L.A.B. wrote the paper supervised by G.A.P.H. and R.K.W. E.G.E.d.V., R.S.N.F., G.A.P.H. and R.K.W. reviewed all versions of the paper. All authors reviewed the manuscript before submission. R.B., P.L., R.M., J.N.-B., H.M.S., M. Harries, E.G.E.d.V., P.N., R.S.N.F., S.P., V.B., T.D.S., G.A.P.H. and R.K.W. provided supervision over the studies and collection protocols within which samples were collected. All authors approved the final manuscript.
Peer review
Peer review information
Nature Medicine thanks Diwakar Davar, Jonathan Peled, and Saman Maleki Vareki for their contribution to the peer review of this work. Primary Handling Editor: Alison Farrell, in collaboration with the Nature Medicine team.
Data availability
The longitudinally profiled metagenomes have been deposited in the European Nucleotide Archive under accession number PRJEB70966. Baseline samples are already deposited under accession number PRJEB43119. All MetaPhlAn4 and HUMAnN3 profiles will also be available within the latest version of curatedMetagenomicData (https://bioconductor.org/packages/curatedMetagenomicData). All relevant patient data used in this study can be requested by emailing the first author (bjork.johannes@gmail.com). The six previously published studies used for validation are available under accession numbers: PRJNA770295, PRJNA541981, PRJNA762360, PRJNA399742, PRJNA397906, PRJEB22893 and PRJEB22894 (see Supplementary Table 6).
Code availability
All code is available in the first author’s GitHub page (https://github.com/johannesbjork/Longitudinal-gut-microbiome-changes-in-ICB-treated-advanced-melanoma) and mirrored at WeersmaLab (https://github.com/WeersmaLabIBD).
Competing interests
R.K.W. acted as a consultant for Takeda; received unrestricted research grants from Takeda, Johnson & Johnson, Tramedico and Ferring; and received speaker fees from MSD, AbbVie and Janssen Pharmaceuticals. E.G.E.d.V. reports an advisory role at Daiichi Sankyo, NSABP and Sanofi (paid to University Medical Center Groningen) and research funding from Amgen, AstraZeneca, Bayer, Chugai Pharma, CytomX Therapeutics, G1 Therapeutics, Genentech, Nordic Nanovector, Radius Health, Regeneron, Roche, Servier and Synthon (paid to University Medical Center Groningen). S.P. received speaker fees from Almirall, BMS, ISDIN, La Roche Posay, Leo Pharma, Regeneron, Roche and Sanofi; acted as advisory board member of Almirall, ISDIN, La Roche Posay, Pfizer, Roche, Regeneron, Sanofi and Sun Pharma; and received research funding from Abbie, AMGEN, ISDIN, La Roche Posay, Leo Pharma and Novartis. R.B. has received honoraria from, and sits on advisory boards of, Novartis, BMS and MSD. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Johannes R. Björk, Laura A. Bolte.
Contributor Information
Johannes R. Björk, Email: bjork.johannes@gmail.com
Rinse K. Weersma, Email: r.k.weersma@umcg.nl
Extended data
is available for this paper at 10.1038/s41591-024-02803-3.
Supplementary information
The online version contains supplementary material available at 10.1038/s41591-024-02803-3.
References
- 1.Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359:1350–1355. doi: 10.1126/science.aar4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hammers HJ, et al. Safety and efficacy of nivolumab in combination with ipilimumab in metastatic renal cell carcinoma: the CheckMate 016 study. J. Clin. Oncol. 2017;35:3851–3858. doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sznol M, et al. Pooled analysis safety profile of nivolumab and ipilimumab combination therapy in patients with advanced melanoma. J. Clin. Oncol. 2017;35:3815–3822. doi: 10.1200/JCO.2016.72.1167. [DOI] [PubMed] [Google Scholar]
- 4.Lee KA, et al. Cross-cohort gut microbiome associations with immune checkpoint inhibitor response in advanced melanoma. Nat. Med. 2022;28:535–544. doi: 10.1038/s41591-022-01695-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McCulloch JA, et al. Intestinal microbiota signatures of clinical response and immune-related adverse events in melanoma patients treated with anti-PD-1. Nat. Med. 2022;28:545–556. doi: 10.1038/s41591-022-01698-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Simpson RC, et al. Diet-driven microbial ecology underpins associations between cancer immunotherapy outcomes and the gut microbiome. Nat. Med. 2022;28:2344–2352. doi: 10.1038/s41591-022-01965-2. [DOI] [PubMed] [Google Scholar]
- 7.Spencer CN, et al. Dietary fiber and probiotics influence the gut microbiome and melanoma immunotherapy response. Science. 2021;374:1632–1640. doi: 10.1126/science.aaz7015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Peters BA, et al. Relating the gut metagenome and metatranscriptome to immunotherapy responses in melanoma patients. Genome Med. 2019;11:61. doi: 10.1186/s13073-019-0672-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gopalakrishnan V, et al. Gut microbiome modulates response to anti–PD-1 immunotherapy in melanoma patients. Science. 2018;359:97–103. doi: 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matson V, et al. The commensal microbiome is associated with anti-PD-1 efficacy in metastatic melanoma patients. Science. 2018;359:104–108. doi: 10.1126/science.aao3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Routy B, et al. Gut microbiome influences efficacy of PD-1-based immunotherapy against epithelial tumors. Science. 2018;359:91–97. doi: 10.1126/science.aan3706. [DOI] [PubMed] [Google Scholar]
- 12.Frankel AE, et al. Metagenomic shotgun sequencing and unbiased metabolomic profiling identify specific human gut microbiota and metabolites associated with immune checkpoint therapy efficacy in melanoma patients. Neoplasia. 2017;19:848–855. doi: 10.1016/j.neo.2017.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olsson LM, et al. Dynamics of the normal gut microbiota: a longitudinal one-year population study in Sweden. Cell Host Microbe. 2022;30:726–739.e3. doi: 10.1016/j.chom.2022.03.002. [DOI] [PubMed] [Google Scholar]
- 14.Chen L, et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell. 2021;184:2302–2315.e12. doi: 10.1016/j.cell.2021.03.024. [DOI] [PubMed] [Google Scholar]
- 15.Franzosa EA, et al. Identifying personal microbiomes using metagenomic codes. Proc. Natl Acad. Sci. USA. 2015;112:E2930–E2938. doi: 10.1073/pnas.1423854112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lavelle A, Sokol H. Understanding and predicting the efficacy of FMT. Nat. Med. 2022;28:1759–1760. doi: 10.1038/s41591-022-01991-0. [DOI] [PubMed] [Google Scholar]
- 17.Blanco-Míguez A, et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2023 doi: 10.1038/s41587-023-01688-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caspi R, et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 2014;42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Halfvarson J, et al. Dynamics of the human gut microbiome in inflammatory bowel disease. Nat. Microbiol. 2017;2:17004. doi: 10.1038/nmicrobiol.2017.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imhann F, et al. Proton pump inhibitors affect the gut microbiome. Gut. 2016;65:740–748. doi: 10.1136/gutjnl-2015-310376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas AM, et al. Gut OncoMicrobiome Signatures (GOMS) as next-generation biomarkers for cancer immunotherapy. Nat. Rev. Clin. Oncol. 2023;20:583–603. doi: 10.1038/s41571-023-00785-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Routy B, et al. Fecal microbiota transplantation plus anti-PD-1 immunotherapy in advanced melanoma: a phase I trial. Nat. Med. 2023;29:2121–2132. doi: 10.1038/s41591-023-02453-x. [DOI] [PubMed] [Google Scholar]
- 23.Asnicar F, et al. Microbiome connections with host metabolism and habitual diet from 1,098 deeply phenotyped individuals. Nat. Med. 2021;27:321–332. doi: 10.1038/s41591-020-01183-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nomura M, et al. Association of short-chain fatty acids in the gut microbiome with clinical response to treatment with nivolumab or pembrolizumab in patients with solid cancer tumors. JAMA Netw. Open. 2020;3:e202895. doi: 10.1001/jamanetworkopen.2020.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietrzak B, et al. A clinical outcome of the Anti-PD-1 therapy of melanoma in polish patients is mediated by population-specific gut microbiome composition. Cancers. 2022;14:5369. doi: 10.3390/cancers14215369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jin Y, et al. The diversity of gut microbiome is associated with favorable responses to anti-programmed death 1 immunotherapy in Chinese patients with NSCLC. J. Thorac. Oncol. 2019;14:1378–1389. doi: 10.1016/j.jtho.2019.04.007. [DOI] [PubMed] [Google Scholar]
- 28.Gacesa R, et al. Environmental factors shaping the gut microbiome in a Dutch population. Nature. 2022;604:732–739. doi: 10.1038/s41586-022-04567-7. [DOI] [PubMed] [Google Scholar]
- 29.Vich Vila A, et al. Gut microbiota composition and functional changes in inflammatory bowel disease and irritable bowel syndrome. Sci. Transl. Med. 2018;10:eaap8914. doi: 10.1126/scitranslmed.aap8914. [DOI] [PubMed] [Google Scholar]
- 30.Dash NR, Al Bataineh MT. Metagenomic analysis of the gut microbiome reveals enrichment of menaquinones (vitamin K2) pathway in diabetes mellitus. Diabetes Metab. J. 2021;45:77–85. doi: 10.4093/dmj.2019.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurilshikov, A. et al. Gut microbial associations to plasma metabolites linked to cardiovascular phenotypes and risk. Circ. Res.10.1161/circresaha.118.314642 (2019). [DOI] [PubMed]
- 32.Karl JP, et al. Fecal menaquinone profiles of overweight adults are associated with gut microbiota composition during a gut microbiota-targeted dietary intervention. Am. J. Clin. Nutr. 2015;102:84–93. doi: 10.3945/ajcn.115.109496. [DOI] [PubMed] [Google Scholar]
- 33.Madeo F, Eisenberg T, Pietrocola F, Kroemer G. Spermidine in health and disease. Science. 2018;359:eaan2788. doi: 10.1126/science.aan2788. [DOI] [PubMed] [Google Scholar]
- 34.Lévesque S, et al. A synergistic triad of chemotherapy, immune checkpoint inhibitors, and caloric restriction mimetics eradicates tumors in mice. Oncoimmunology. 2019;8:e1657375. doi: 10.1080/2162402X.2019.1657375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Montégut L, de Cabo R, Zitvogel L, Kroemer G. Science-driven nutritional interventions for the prevention and treatment of cancer. Cancer Discov. 2022;12:2258–2279. doi: 10.1158/2159-8290.CD-22-0504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Derosa L, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur. Urol. 2020;78:195–206. doi: 10.1016/j.eururo.2020.04.044. [DOI] [PubMed] [Google Scholar]
- 37.Chaput N, et al. Baseline gut microbiota predicts clinical response and colitis in metastatic melanoma patients treated with ipilimumab. Ann. Oncol. 2017;28:1368–1379. doi: 10.1093/annonc/mdx108. [DOI] [PubMed] [Google Scholar]
- 38.Terrisse S, et al. Intestinal microbiota influences clinical outcome and side effects of early breast cancer treatment. Cell Death Differ. 2021;28:2778–2796. doi: 10.1038/s41418-021-00784-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salgia NJ, et al. Stool microbiome profiling of patients with metastatic renal cell carcinoma receiving anti–PD-1 immune checkpoint inhibitors. Eur. Urol. 2020;78:498–502. doi: 10.1016/j.eururo.2020.07.011. [DOI] [PubMed] [Google Scholar]
- 40.Roy S, Trinchieri G. Microbiota: a key orchestrator of cancer therapy. Nat. Rev. Cancer. 2017;17:271–285. doi: 10.1038/nrc.2017.13. [DOI] [PubMed] [Google Scholar]
- 41.Vétizou M, et al. Anticancer immunotherapy by CTLA-4 blockade relies on the gut microbiota. Science. 2015;350:1079–1084. doi: 10.1126/science.aad1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang AE, et al. Targeting the gut microbiome to mitigate immunotherapy-induced colitis in cancer. Trends Cancer Res. 2021;7:583–593. doi: 10.1016/j.trecan.2021.02.005. [DOI] [PubMed] [Google Scholar]
- 43.Takeshita K, et al. A single species of clostridium subcluster XIVa decreased in ulcerative colitis patients. Inflamm. Bowel Dis. 2016;22:2802–2810. doi: 10.1097/MIB.0000000000000972. [DOI] [PubMed] [Google Scholar]
- 44.Read E, Curtis MA, Neves JF. The role of oral bacteria in inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2021;18:731–742. doi: 10.1038/s41575-021-00488-4. [DOI] [PubMed] [Google Scholar]
- 45.Thomas AM, et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019;25:667–678. doi: 10.1038/s41591-019-0405-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Derosa L, et al. Microbiota-centered interventions: the next breakthrough in immuno-oncology? Cancer Discov. 2021;11:2396–2412. doi: 10.1158/2159-8290.CD-21-0236. [DOI] [PubMed] [Google Scholar]
- 47.Derosa L, et al. Intestinal Akkermansia muciniphila predicts clinical response to PD-1 blockade in patients with advanced non-small-cell lung cancer. Nat. Med. 2022;28:315–324. doi: 10.1038/s41591-021-01655-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Spreafico A, et al. First-in-class Microbial Ecosystem Therapeutic 4 (MET4) in combination with immune checkpoint inhibitors in patients with advanced solid tumors (MET4-IO trial) Ann. Oncol. 2023;34:520–530. doi: 10.1016/j.annonc.2023.02.011. [DOI] [PubMed] [Google Scholar]
- 49.Tanoue T, et al. A defined commensal consortium elicits CD8 T cells and anti-cancer immunity. Nature. 2019;565:600–605. doi: 10.1038/s41586-019-0878-z. [DOI] [PubMed] [Google Scholar]
- 50.Baruch EN, et al. Fecal microbiota transplant promotes response in immunotherapy-refractory melanoma patients. Science. 2021;371:602–609. doi: 10.1126/science.abb5920. [DOI] [PubMed] [Google Scholar]
- 51.Davar D, et al. Fecal microbiota transplant overcomes resistance to anti–PD-1 therapy in melanoma patients. Science. 2021;371:595–602. doi: 10.1126/science.abf3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ianiro G, et al. Variability of strain engraftment and predictability of microbiome composition after fecal microbiota transplantation across different diseases. Nat. Med. 2022;28:1913–1923. doi: 10.1038/s41591-022-01964-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schmidt TSB, et al. Drivers and determinants of strain dynamics following fecal microbiota transplantation. Nat. Med. 2022;28:1902–1912. doi: 10.1038/s41591-022-01913-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Halsey TM, et al. Microbiome alteration via fecal microbiota transplantation is effective for refractory immune checkpoint inhibitor-induced colitis. Sci. Transl. Med. 2023;15:eabq4006. doi: 10.1126/scitranslmed.abq4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Baruch EN, Gaglani T, Wargo JA. Fecal microbiota transplantation as a mean of overcoming immunotherapy resistant cancers – hype or hope? Ther. Adv. Med. Oncol. 2021;13:17588359211045853. doi: 10.1177/17588359211045853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.OncoLifeS Oncological Life Study: living well as a cancer survivor (University Medical Center Groningen, 2022)
- 57.SegataLab. GitHubhttps://github.com/SegataLab/preprocessing (2023).
- 58.Beghini F, et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife. 2021;10:e65088. doi: 10.7554/eLife.65088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hawinkel S, Mattiello F, Bijnens L, Thas O. A broken promise: microbiome differential abundance methods do not control the false discovery rate. Brief. Bioinform. 2019;20:210–221. doi: 10.1093/bib/bbx104. [DOI] [PubMed] [Google Scholar]
- 60.Gloor GB, Macklaim JM, Pawlowsky-Glahn V, Egozcue JJ. Microbiome datasets are compositional: and this is not optional. Front. Microbiol. 2017;8:2224. doi: 10.3389/fmicb.2017.02224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jian C, Luukkonen P, Yki-Järvinen H, Salonen A, Korpela K. Quantitative PCR provides a simple and accessible method for quantitative microbiota profiling. PLoS ONE. 2020;15:e0227285. doi: 10.1371/journal.pone.0227285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gloor GB, Wu JR, Pawlowsky-Glahn V, Egozcue JJ. It’s all relative: analyzing microbiome data as compositions. Ann. Epidemiol. 2016;26:322–329. doi: 10.1016/j.annepidem.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 63.Fernandes AD, et al. Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome. 2014;2:15. doi: 10.1186/2049-2618-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mandal S, et al. Analysis of composition of microbiomes: a novel method for studying microbial composition. Microb. Ecol. Health Dis. 2015;26:27663. doi: 10.3402/mehd.v26.27663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Morton JT, et al. Balance trees reveal microbial niche differentiation. mSystems. 2017;2:e00162–16. doi: 10.1128/mSystems.00162-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lin H, Peddada SD. Analysis of microbial compositions: a review of normalization and differential abundance analysis. NPJ Biofilms Microbiomes. 2020;6:60. doi: 10.1038/s41522-020-00160-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Morton JT, et al. Establishing microbial composition measurement standards with reference frames. Nat. Commun. 2019;10:2719. doi: 10.1038/s41467-019-10656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Silverman, J. D., Roche, K., Holmes, Z. C., David, L. A., & Mukherjee, S. R package fido (v.1.0.4). Githubhttps://jsilve24.github.io/fido/ (2022).
- 69.Silverman, J. D., Roche, K., Holmes, Z. C., David, L. A. & Mukherjee, S. Bayesian multinomial logistic normal models through marginally latent matrix-T processes. J. Mach. Learn. Res. 23, 1–42 (2022).
- 70.Chen, J. & Li, H. Variable selection for sparse dirichlet-multinomial regression with an application to microbiome data analysis. Ann. Appl. Stat. 10.1214/12-AOAS592 (2013). [DOI] [PMC free article] [PubMed]
- 71.Xia F, Chen J, Fung WK, Li H. A logistic normal multinomial regression model for microbiome compositional data analysis. Biometrics. 2013;69:1053–1063. doi: 10.1111/biom.12079. [DOI] [PubMed] [Google Scholar]
- 72.Calle ML, Pujolassos M, Susin A. coda4microbiome: compositional data analysis for microbiome cross-sectional and longitudinal studies. BMC Bioinform. 2023;24:82. doi: 10.1186/s12859-023-05205-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rivera-Pinto J, et al. Balances: a new perspective for microbiome analysis. mSystems. 2018;3:e00053–18. doi: 10.1128/mSystems.00053-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gordon-Rodriguez E, Quinn TP, Cunningham JP. Learning sparse log-ratios for high-throughput sequencing data. Bioinformatics. 2021;38:157–163. doi: 10.1093/bioinformatics/btab645. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figs. 1–5 and methods.
Supplementary Table 1. Number of patient samples in each post hoc contrast.Supplementary Table 2. For each BCL, the (nonzero) slope for each taxon in patients with PFS ≥12 and PFS <12 months, respectively, including their phylum and family belongings.Supplementary Table 3. Number of differentially abundant taxa at different thresholds.Supplementary Table 4. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months across visits, including their phylum and family belongings.Supplementary Table 5. For each BCL, the differentially abundant pathways between patients with PFS ≥12 and PFS <12 months across visits, including their phylum and family belongings.Supplementary Table 6. Independent cohorts used for validation.Supplementary Table 7. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on monotherapy across visits, including their phylum and family belongings.Supplementary Table 8. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on combination therapy across visits, including their phylum and family belongings.Supplementary Table 9. For each BCL, the differentially abundant taxa between patients who developed colitis and patients who did not develop colitis across visits, including their phylum and family belongings.Supplementary Table 10. For each BCL, the differentially abundant taxa between patients with PFS ≥12 months who developed colitis and patients with PFS ≥12 months who did not develop colitis across visits, including their phylum and family belongings.Supplementary Table 11. For each BCL, the differentially abundant taxa between patients who developed colitis on monotherapy versus patients who developed colitis on combination therapy across visits, including their phylum and family belongings.Supplementary Table 12. For each BCL, the differentially abundant taxa between patients with PFS ≥12 and PFS <12 months on PPI across visits, including their phylum and family belongings.Supplementary Table 13. For BCL of 0.9, the 33 differentially abundant SGBs shared between PPI users (Table 7) and nonusers (Table 12) across visits, including their phylum and family belongings.Supplementary Table 14. SGBs that exhibited similar directional effects in the meta-analysis by Thomas et al. (2023) and in our analysis.Supplementary Table 15. SGBs that exhibited similar directional effects in Routy et al. (2023) and in our analysis.Supplementary Table 16. SGBs in the balance described in Fig. 2a. SGBs that were missing in each independent cohort after 10% prevalence filtering.
Data Availability Statement
The longitudinally profiled metagenomes have been deposited in the European Nucleotide Archive under accession number PRJEB70966. Baseline samples are already deposited under accession number PRJEB43119. All MetaPhlAn4 and HUMAnN3 profiles will also be available within the latest version of curatedMetagenomicData (https://bioconductor.org/packages/curatedMetagenomicData). All relevant patient data used in this study can be requested by emailing the first author (bjork.johannes@gmail.com). The six previously published studies used for validation are available under accession numbers: PRJNA770295, PRJNA541981, PRJNA762360, PRJNA399742, PRJNA397906, PRJEB22893 and PRJEB22894 (see Supplementary Table 6).
All code is available in the first author’s GitHub page (https://github.com/johannesbjork/Longitudinal-gut-microbiome-changes-in-ICB-treated-advanced-melanoma) and mirrored at WeersmaLab (https://github.com/WeersmaLabIBD).