

Abstract
Transaldolase deficiency predisposes to chronic liver disease progressing from cirrhosis to hepatocellular carcinoma (HCC). Transition from cirrhosis to hepatocarcinogenesis depends on mitochondrial oxidative stress, as controlled by cytosolic aldose metabolism through the pentose phosphate pathway (PPP). Progression to HCC is critically dependent on NADPH depletion and polyol buildup by aldose reductase (AR), while this enzyme protects from carbon trapping in the PPP and growth restriction in TAL deficiency. Although AR inactivation blocked susceptibility to hepatocarcinogenesis, it enhanced growth restriction, carbon trapping in the non-oxidative branch of the PPP and failed to reverse the depletion of glucose 6-phosphate (G6P) and liver cirrhosis. Here, we show that inactivation of the TAL-AR axis results in metabolic stress characterized by reduced mitophagy, enhanced overall autophagy, activation of the mechanistic target of rapamycin (mTOR), diminished glycosylation and secretion of paraoxonase 1 (PON1), production of antiphospholipid autoantibodies (aPL), loss of CD161+ NK cells, and expansion of CD38+ Ito cells, which are responsive to treatment with rapamycin in vivo. The present study thus identifies glycosylation and secretion of PON1 and aPL production as mTOR-dependent regulatory checkpoints of autoimmunity underlying liver cirrhosis in TAL deficiency.
Keywords: transaldolase, aldose reductase, pentose phosphate pathway, tricarboxylic acid cycle, mitochondrial oxidative stress, cirrhosis, hepatocarcinogenesis, mitophagy, autophagy, paraoxonase 1, glucose 6-phosphate, UDP-GlcNAc, PON1, Ito cell, rapamycin, mechanistic target of rapamycin
Graphical Abstract
INTRODUCTION
Liver cirrhosis is a significant cause of morbidity and mortality in the United States (1) and globally (2). Mortality of patients with cirrhosis often results from the progression to hepatocellular carcinoma (HCC), fifth most common cancer and the third leading cause of cancer-related mortality worldwide (3). As much as 80% of HCC cases develop in cirrhotic livers (4). Both the development of cirrhosis and the progression to HCC have been linked to mitochondrial oxidative stress (5). In turn, protection from oxidative stress depends on the maintenance of a reducing environment, such as de novo synthesis of intracellular antioxidants, such as reduced glutathione (GSH) and NADPH (6). GSH may also be regenerated from its oxidized form, GSSG, at the expense of NADPH, which primarily originates from the pentose phosphate pathway (PPP) in all mammalian cells (6).
The PPP has been formulated to operate in two functionally connected branches, oxidative and non-oxidative. Glucose 6-phosphate dehydrogenase (G6PD) is the rate-limiting NADPH-producing enzyme of the oxidative branch (6). Complete deficiency of G6PD is not compatible with cell survival (7). Diminished activity of G6PD is caused by mutation in genomic DNA that represent the most common genetic defect in humans (8), which have been associated with the development of HCC (9). In contrast, the complete deficiency of transaldolase (TAL), a rate limiting enzyme of the non-oxidative branch of the PPP, does not abrogate cell survival (10). Despite complete TAL deficiency, both humans (11) and mice develop to adulthood, although males exhibit infertility due to structural and functional damage of sperm mitochondria (12). Importantly, the deficiency of TAL also predisposes to HCC both in mice (13) and humans (14, 15). 50% of mice with deficiency of transaldolase (TALKO) die of HCC at mean age of 74 weeks (13), matching the average age of 65 years upon HCC diagnosis in humans (16).
Progression from cirrhosis to HCC has been recently linked to the activation of aldose reductase (AR) in TALKO mice (17). A critical role of AR in hepatocarcinogenesis of TALKO mice has been attributed to the conversion of the accumulated PPP sugars to polyols at the expense of NADPH. Polyol levels are markedly accumulated in the urine of TALKO mice (18) and patients with TAL deficiency (11), including those with HCC (14, 15). The accumulation of polyols in the urine is consistent with their osmotic toxicity to hepatocytes (19) and other types of cells (20). The overexpression of AR has also been linked to drug resistance of HCC in general (21). High-resolution mass spectrometry, stable isotope tracing, RNA sequencing, and confirmatory functional studies unveiled carbon sequestration in the non-oxidative branch of the PPP that caused growth restriction and NADPH depletion and restricted substrates for the tricarboxylic acid (TCA) cycle, amino acid, amino sugar, and phospholipid biosynthesis in TAL deficiency. Amongst NADPH-consuming enzymes, AR was strikingly and consistently upregulated at the RNA and protein levels in TALKO mice and TALKO hepatomas (13). The inactivation of AR reversed NADPH depletion and polyol buildup in the absence of TAL and blocked mitochondrial oxidative stress and liver disease progression from cirrhosis to HCC. Mice double-deficient for TAL and AR (DKO) mice were also resistant to APAP-induced liver necrosis. Although AR inactivation blocked mitochondrial oxidative stress and hepatocarcinogenesis, it exacerbated carbon sequestration in the PPP, caused further growth restriction, and failed to block liver cirrhosis.
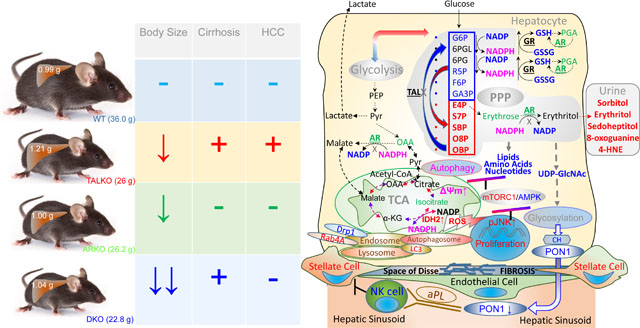
Genetic and environmental factors have been identified as triggers of cirrhosis, which generally involves oxidative stress-mediated hepatocyte injury and the expansion of pro-fibrotic hepatic stellate cells (HSC) or Ito cells (22, 23). Proliferation of HSC and cirrhosis depend on the activation of the mechanistic target of rapamycin (mTOR) in mice (24–26) and humans (27). Liver cirrhosis has also been associated with the production of antiphospholipid autoantibodies (aPL) (28–30). Along these lines, patients with aPL are predisposed to cirrhosis (31, 32). Although the generation of aPL is incompletely understood, oxidization of phospholipid antigens has been widely implicated (33, 34). Production of aPL has been associated with mTOR activation in the liver (35). Notably, paraoxonase 1 (PON1) (36, 37), which is synthesized and secreted by the liver (38), protects both hepatocytes (39, 40) and circulating blood antigens from oxidative damage (41). Interestingly, liver cirrhosis has been independently linked to diminished serum levels of paraoxonase 1 (PON1) (42–44). While PON1 levels are diminished in patients with liver cirrhosis (45), its mechanistic bases and pathological consequences have not been delineated. Of note, RNAseq studies identified profoundly diminished expression of PON1 in TALKO livers, which was not reversed by AR inactivation in DKO mice (17). Here, we show that Ito cells expanded in the liver and PON1 protein levels and enzymatic activities are diminished in the livers and sera of TALKO and DKO mice. Moreover, aPL production against apolipoprotein H (Apo-H, also called β2-glycoprotein 1) and cardiolipin, is increased in TALKO and DKO mice. Diminished secretion of PON1 was attributed to the depletion of substrates, F6P, UTP, UDP-glucose and UDP-GlcNAc, which are required for glycosylation and secretion by the liver into the bloodstream (46). Importantly, shift to the lower molecular weight PON1 isoform in TALKO and DKO livers is effectively mimicked by in vitro treatment of WT liver lysates with peptide-N-glycosidase F, also known as PNGase F, which releases N-linked oligosaccharides from glyco-proteins by hydrolyzing the amide group of the asparagine (N) side chains (47). High-energy collisional dissociation tandem mass spectroscopy (HCD-MS2) of immunoprecipitated PON1 identified glycosylation of asparagine residue 253 with Man8GlcNAc2. These findings suggest that deficient recycling of 6-carbon substrates by the PPP elicits mTOR activation and limits glycosylation and PON1 secretion into the bloodstream of TALKO and DKO mice. PON1 deficiency elicited autoimmunity characterized by the generation of aPL and expansion of pro-fibrotic HSC in the liver. Treatment with rapamycin enhanced serum PON1 activity and blocked aPL production and the expansion of HSC in TALKO mice. This study identifies glycosylation-dependent PON1 secretion as a trigger of aPL-mediated autoimmunity that underlies liver cirrhosis in TAL deficiency.
RESULTS
Production of antiphospholipid antibodies (aPL), depletion of NK cells, and accumulation of Ito cells underlie autoimmunity-mediated cirrhosis in TALKO mice
Life expectancy of C57Bl/6 wild-type (WT) mice ranges between 140–160 weeks (48). TALKO mice die due to HCC at a mean age of 74 weeks (13).Premature death of TALKO mice approximates the average age of HCC diagnosis at 65 years in patients (16). The development of HCC has been attributed to the overexpression of aldose reductase (AR) in livers and hepatomas of TALKO mice, since AR deletion effectively blocked hepatocarcinogenesis in double-knockout (DKO) mice lacking both TAL and AR (17). However, hepatomegaly and cirrhosis persisted in DKO mice (17).Moreover, TALKO and ARKO mice showed restricted growth and body size, with DKO mice being even smaller than each of the single knockouts. The overall limitation of body size by inactivation of the TAL-AR axis was underlain by carbon sequestration in the non-oxidative branch of the PPP and restriction of substrates for the biosynthesis of amino acids (glutamine, alanine, valine, leucine/isoleucine, aspartate, and serine) which are significant metabolic predictors of body size (49).
Cirrhosis persisted in DKO mice, as evidenced by fibrosis in Gömöri-trichrome-stained liver tissues (Figure 1A). During in situ perfusion via the portal vein (13, 50, 51), formation of a striking vascular reticulum was observed in livers of TALKO and DKO mice (Figure 1B). Pro-fibrotic fat-storing Ito or hepatic stellate cells (HSC) (52, 53) were expanded in livers of TALKO (4.75-fold; p=0.007) and DKO mice relative to WT controls (6-fold; p=0.042). HSC undergo trans-differentiation into myofibroblasts, and promote vascular resistance, portal hypertension (52) and cirrhosis (52). Ito cells express CD38 (54) that is predictive of cirrhosis severity (55). Following in situ liver perfusion and sedimentation of hepatocytes at 50 x g, CD38+ cells were expanded > 2-fold in hepatocyte supernatants of TALKO mice (Figure 1C). Moreover, surface expression of CD38 was also increased 3.6-fold in TALKO mice, as measured by mean fluorescence intensity (MFI, p=0.04). CD38 is an ectoenzyme that hydrolyzes NAD, therefore, its overexpression may underlie the depletion of NAD by 40% in TALKO mice and by 53% in DKO mice (Figure 1C). These findings are compatible with elevated NAD levels in the liver of mice lacking CD38 (56). Fibrosis is subject to regulation by NK cells that can directly kill Ito cells (57, 58). Relative to WT controls, intrahepatic CD161+ NK cells were depleted in TALKO and DKO mice (Figure 1D). NK T cells were not depleted significantly (Figure 1D).
Figure 1. Cirrhosis in TAL deficiency is resistant to inactivation of AR.
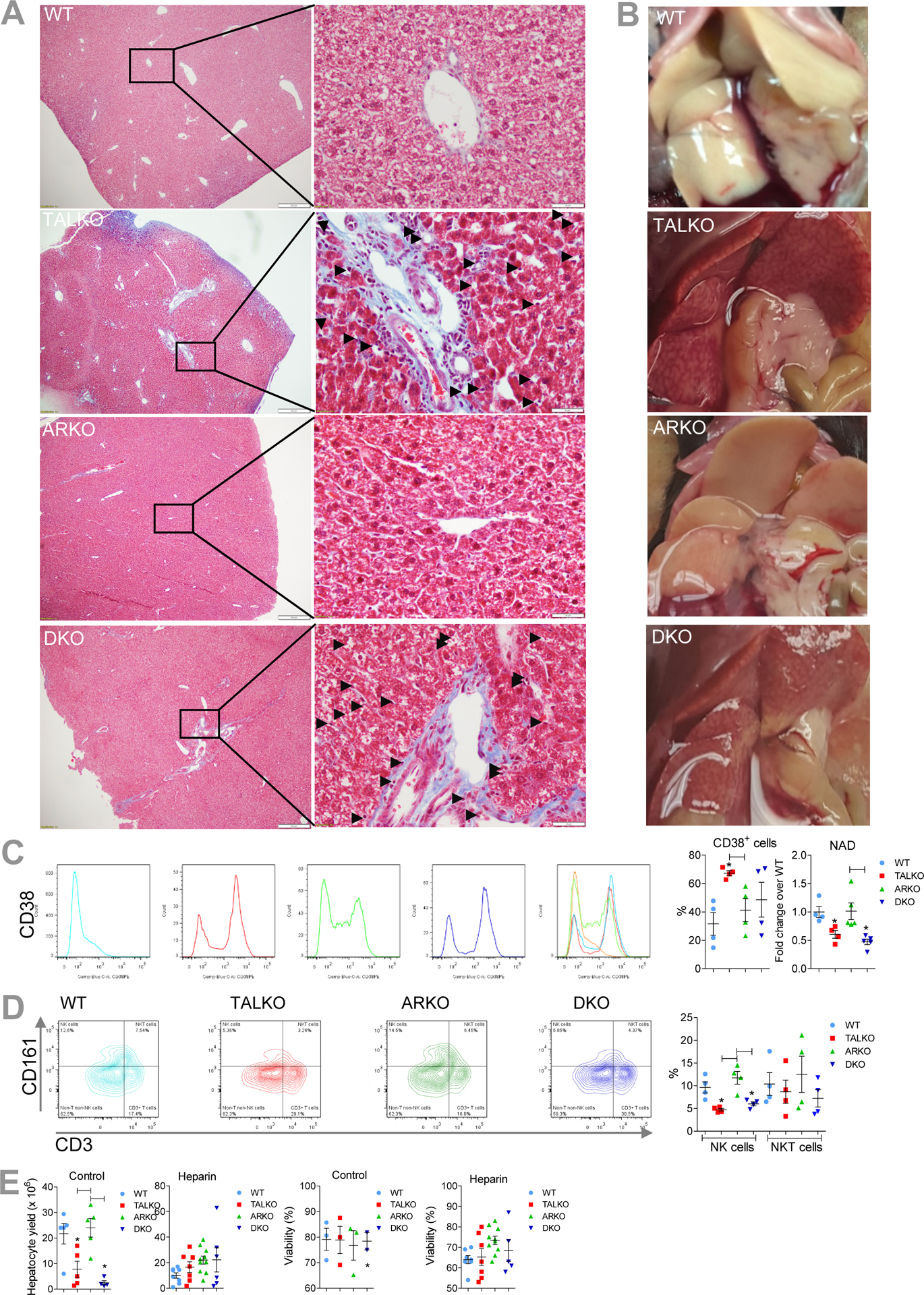
A, Detection of fibrosis in Gömöri-trichrome-stained liver tissues from TALKO and DKO mice. Pro-fibrotic Ito cells or fat-storing hepatic stellate cells are indicated with arrows in areas of higher magnification. B, Formation of vascular reticulum in livers of TALKO and DKO mice upon in situ perfusion via the inferior vena cava. C, Assessment of CD38+ in WT, TALKO, ARKO, and DKO mice by flow cytometry of non-hepatocyte fraction isolated after in situ liver perfusion. NAD levels were assessed in liver extracts in WT, TALKO, ARKO, and DKO mice by LC- MS/MS. D, Assessment of NK and NKT cells in WT, TALKO, ARKO, and DKO mice by flow cytometry of non-hepatocyte fraction isolated after in situ liver perfusion. E, Effect of heparin administration on hepatocyte yield and viability after in situ liver perfusion. Mice were injected with or without 5 U heparin per g of body weight in 100 μl of PBS prior to perfusion (61).
Liver cirrhosis is associated with increased intrahepatic vascular resistance (IHVR), which has been associated with the accumulation of HSC that exhibit contractility and promote fibrosis (59). Of note, heparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats (60). Following liver perfusion, hepatocyte yields were reduced in TALKO and DKO mice (Figure 1E). Heparin injection of mice prior to liver perfusion in situ (61) effectively blocked vascular reticulum formation and normalized hepatocyte yields in TALKO and DKO mice (Figure 1E).
The TAL-AR axis regulates expression of genes involved in cirrhosis, mitochondrial metabolism and pro-inflammatory mTOR pathway activation in the liver
Transcriptional control of liver disease by the TAL-AR axis was delineated by RNAseq analysis of livers from pre-disease mice. TAL deficiency influenced 300 genes at false discovery rate (FDR) p value < 0.05 (17). The metabolome-linked RNAseq changes were systematically confirmed at the protein level. As earlier documented, the expression of genes involved in mitochondrial oxidative stress were moderated (NDUFS3, Bak, NQO1), while those connected to carbon trapping in the PPP (GPI1, ME1, IDH2) and mitochondrial mass (VDAC) persisted in DKO mice (17).
With respect to carcinogenesis, transcription of H19, a long non-coding RNA, was increased 239-fold in TALKO livers but reduced 5.5-fold by AR inactivation (Figure 2A). H19 stimulates carcinogenesis through interacting with IGF2 (62), both of which are clustered in synthenic chromosomal loci in men and mice (Figure 2B). Notably, the TALDO1 locus encoding TAL (63) and zinc finger protein 143 (ZNF143), a transcription factor that controls the expression of TAL (64), are also located in the very same synthenic loci in men and mice (Figure 2B). In addition to TAL, ZNF143 also controls the expression of AR (65), which is located on human chromosome 7 (66) and mouse chromosome 6, respectively (67). Interestingly, ZNF143 expression was low in TALKO livers, increased in ARKO livers, and normalized in DKO livers (Figure 2C). While IGF2 expression on the mRNA level was not affected, IGF2 receptor mRNA levels were increased 1.41-fold in TALKO livers (raw p=0.0035) and 1.47-fold in DKO livers over WT controls (raw p=0.0014). Nevertheless, IRF2 protein levels were increased in both TALKO (1.7-fold, p=0.0557) and DKO livers (1.9-fold, p=0.0011; Figure 1C). While the separate deletions of TAL and AR exerted opposite effects on the expression of ZNF143, the inactivation of TAL-AR axis synergistically enhanced IGF2 expression, (Figure 1C). Igf2−/− mice exhibit growth retardation and hepatic steatosis (68). Along this line, increased expression of IGF2 promotes the development of cirrhosis in the setting of NAFLD (69).
Figure 2. The TALAR axis regulates expression of genes involved cirrhosis, mitochondrial metabolism and mTOR activation in the livers.
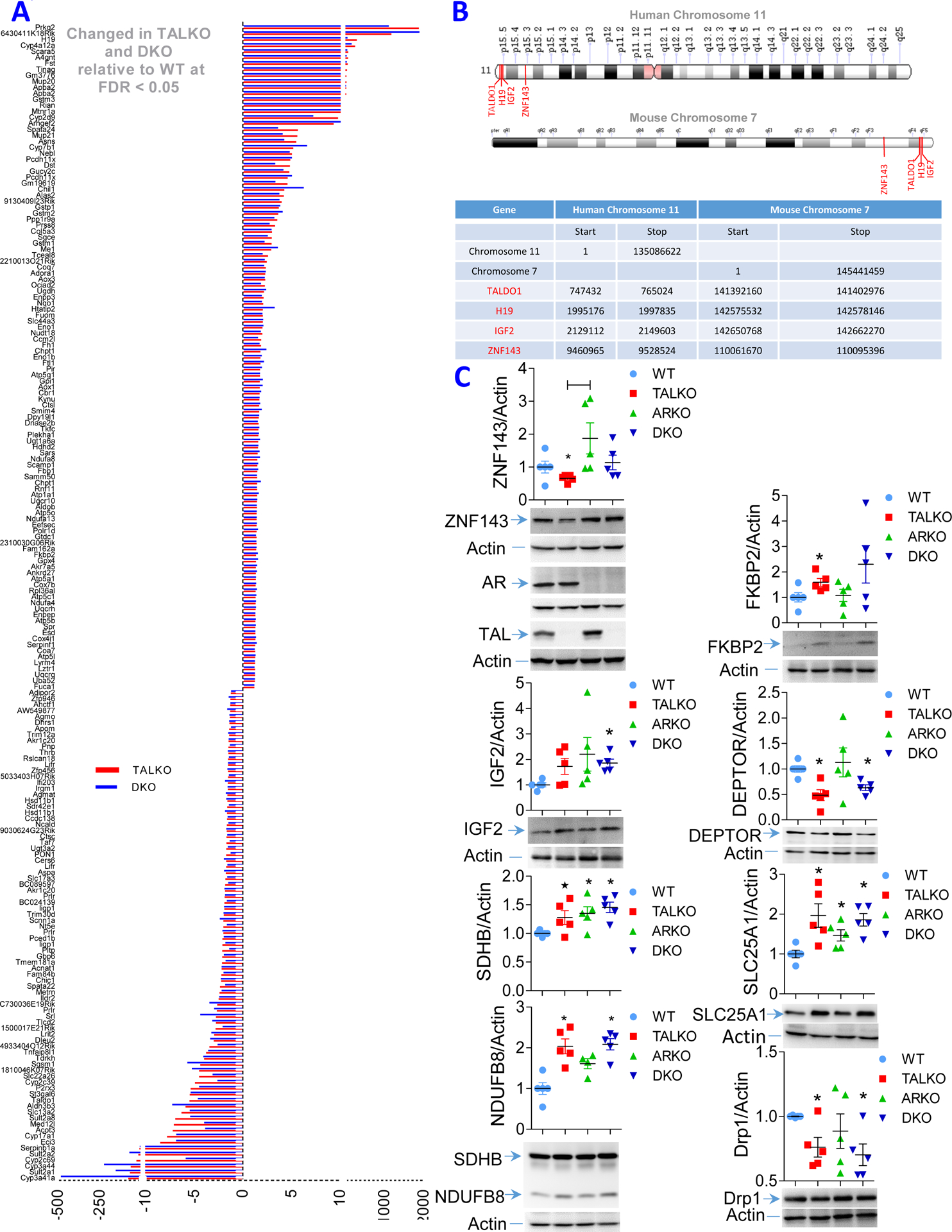
Five age-matched mice were used for each of four genotypes: WT, TALKO, ARKO, and DKO.. A, Concordant changes in expression of 193 genes by RNAseq analysis in TALKO and DKO livers relative to WT controls at false discovery rate (FDR) p value < 0.05. B, Chromosomal proximity of H19, Igf2, TALDO1, and ZNF143 in synthenic genomic loci in humans and mice. Upper panel, schematic mapping of H19, Igf2, TALDO1, and ZNF143 along human chromosome 11 and mouse chromosome 7. Lower panel, nucleotide positions of H19, Igf2, TALDO1, and ZNF143 along human chromosome 11 and mouse chromosome 7. C, Western blot detection of ZNF143, IGF2, FKBP2, Deptor, NDUFB8, SDHB, SLC25A1, and Drp1. Representative blots and bar charts of cumulative analysis of five mice per genotype are shown for each gene. *, two-tailed t-test p < 0.05.
H19 stimulates hepatocarcinogenesis by promoting the expression of IGF2 (70, 71) through interacting with CTCF during gene transcription (72). In turn, CTCF depends on ZNF143 in coordinating chromatin remodeling (73–75). Of note, the IGF2 receptor binds hexose- 6-phosphate that affects its ability to transport IGF2 across the lysosomal membrane (76). Thus, the accumulation of IGF2 may be related to coordinate loss of hexose-6-phosphate due to C4, C5, C7, and C8 carbon trapping in the non-oxidative branch of the PPP (17). Given the coordinate regulation via the TAL-AR axis, this study suggests that metabolic control of carcinogenesis by ZNF143 and IGF2 are functionally connected through aldose metabolism.
The mechanistic target of rapamycin (mTOR) has emerged as a major sensor of metabolic stress and mediator of cell proliferation (77) and fibrosis in the liver (78). Among the 300 genes significantly altered by RNAseq at FDR p<0.05, expression of FKBP2 was upregulated in TALKO mice and further accentuated in DKO mice (Figure 2A). Both the extent and direction of changes in FKBP2 expression were confirmed on the protein level in TALKO and DKO mice (Figure 2C). FKBP2 is a 13-kD protein that serves as a cellular receptor for rapamycin (79). We also noted a reduction of Deptor, an inhibitor of mTOR complex 1 (mTORC1) (80), by RNAseq (raw p=0.038), which was corroborated on the protein level (Figure 2C). Similar to FKBP2, Deptor remained depleted in DKO mice (Figure 2C).
TAL deficiency restricts mitophagy and elicits the accumulation of mitochondria in hepatocytes
TALKO hepatocytes exhibit mitochondrial dysfunction, characterized by the elevation of the mitochondrial transmembrane potential (ΔΨm) or mitochondrial hyperpolarization (MHP) and increased ROI production, which were effectively reversed by the inactivation of AR (17). However, mitochondrial mass and ATP production remained enhanced in DKO hepatocytes (17). Similar to the outer mitochondrial membrane protein VDAC1 (17), inner mitochondria membrane proteins, such as NDUFB8 of ETC complex I and SDHB of ETC complex II, were accumulated both in TALKO and DKO mice (Figure 2C). Expression of SLC25A1, the mitochondrial antiporter that exports citrate from the mitochondria into the cytosol was upregulated in TALKO, ARKO, and DKO livers overt WT controls (Figure 2C). In contrast, Drp1, which initiates mitochondrial fission, was depleted in TALKO and DKO mice (Figure 2C), suggesting that diminished turnover by mitophagy may underlie the accumulation of mitochondria in TAL deficiency.
As evidenced by confocal microcopy, the numbers of small mitochondria were decreased and mitochondria were overall enlarged and elongated in TALKO, ARKO, and DKO mice (Figure 3A). Autophagy allows for recycling of organelles, such as mitochondria, which is also called mitophagy. The recycling of organelles generates nutrients during state of starvation and protects membranes and DNA integrity from oxidative stress (81). Mitochondria also supply membrane for autophagosome formation during metabolic stress (82). As shown in Figure 3B, budding of autophagosomes from mitochondria was reduced in TALKO mice relative to WT controls. Autophagosome formation was further reduced in DKO relative to WT and TALKO mice (Figure 3B). Moreover, we evaluated the involvement of LC3, an essential component of autophagosome formation. The transition of LC3-I to LC3-II, LC3-II/LC3-I ratio, was reduced by 50% on isolated mitochondria of TALKO mice, which was moderated in DKO mice (Figure 3C). By contrast, autophagy protein p62/SQTM1 was accumulated on DKO and to a lesser extent on TALKO mitochondria (Figure 3C). In accordance with the depletion of Drp1 (Figure 2C), reduced autophagosome formation (Figure 3B), reduced LC3 processing and reduced p62 consumption (Figure 3C), the accumulation of mitochondria mass was also substantiated by greater MTG fluorescence in hepatocytes of TALKO and DKO mice (Figure S1).
Figure 3. Regulation of mitochondrial homeostasis by the TAL-AR axis.
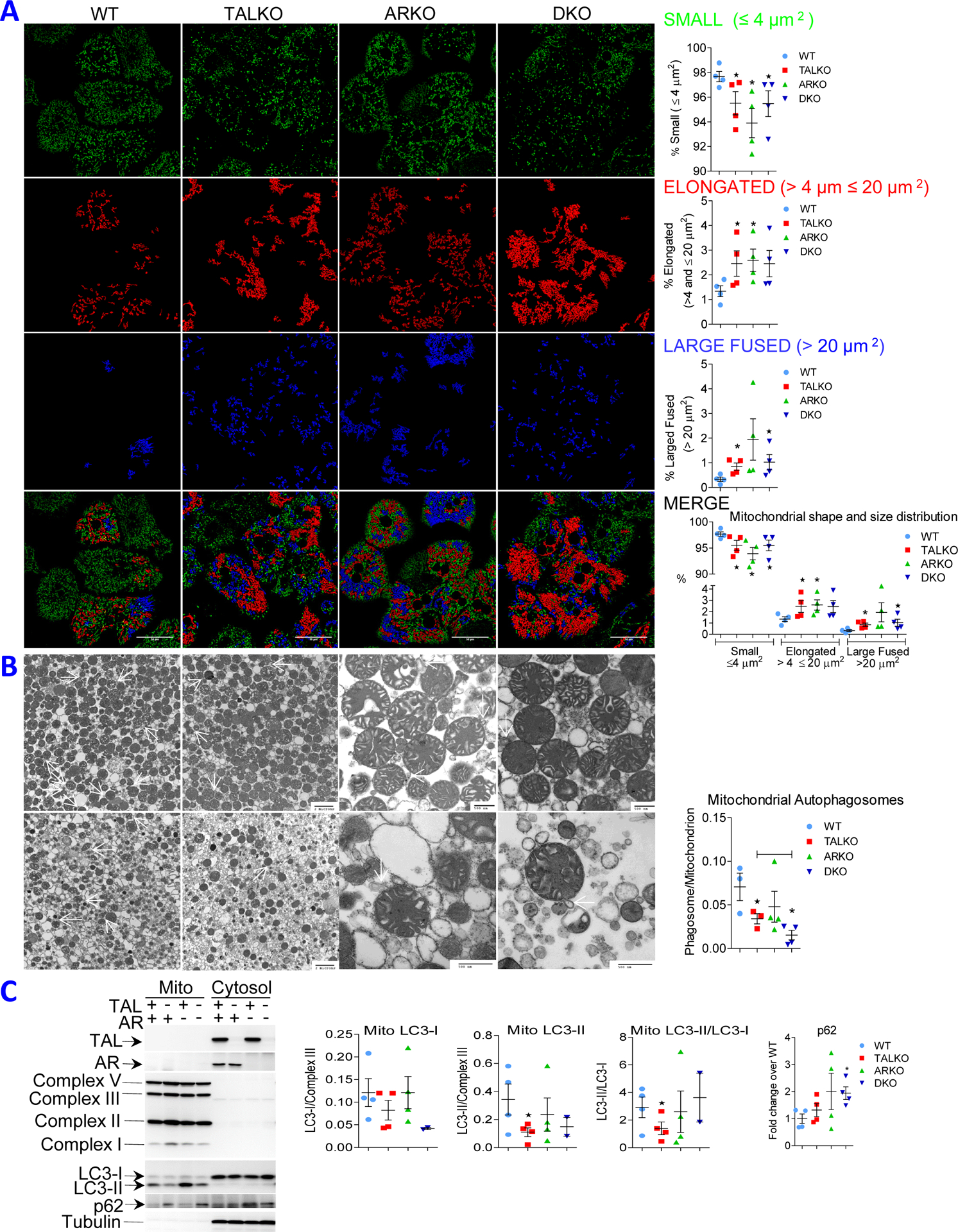
A, Immunofluorescence microscopy of mitochondria. Left panel, representative images colored by size and shape. Right panel, cumulative analyses represent mean ± SEM of 5 mice per genotype. *, p < 0.05 relative to WT. B, Electron microscopy of liver mitochondria. Left panel, formation of autophagosomes from mitochondrial membranes, also termed phagophores (autophagosomes/mitochondrion), are indicated by white arrows. Right panel, cumulative analysis of mitochondrial autophagosome formation. C, Western blot analysis of LC3-I and LC3-II isoforms and mTOR in purified mitochondrial and cytosol fractions of liver from WT, TALKO, ARKO, and DKO mice. Left panel, representative blots. Right panel, cumulative analysis of LC3-I, LC3-II, LC3-II/LC3-I, and p-mTORS2448 in 4–5 mice per genotype. *, p < 0.05 relative to WT.
To delineate the mechanism underlying diminished mitochondrial autophagosome formation, we evaluated the phosphorylation of Drp1 since its protein levels were reduced in TALKO and DKO mice (Figure 2C). Phosphorylation at S616 activates Drp1 to promote the mitochondrial fission, while phosphorylation of Drp1 at S637 has opposite effects (83). Importantly, mitochondrial pDrp1S616 levels were reduced on isolated mitochondria of TALKO and DKO mice (Figure S2A). With respect to a potential mechanism underlying Drp1 depletion, Rab4A protein levels were increased on mitochondria of TALKO and DKO mice (Figure S2B). Rab4A may enhance recycling and promotes lysosomal degradation of Drp1 under metabolic stress (84, 85). Along this line, mass spectroscopy identified GDP dissociation inhibitor 2 (GDI2) and β-catenin among proteins pulled down by GST-Rab4A, but not by GST-coupled GSH agarose beads, which were consistent with co-immunoprecipitation of Rab4A with GDI2 (86) and β-catenin (87). Similar to Rab4A, GDI2 (Figure S2C) and β-catenin were accumulated on mitochondria of TALKO livers (Figure S2D). Autophagy effector protein SQSTM1/p62 was moderately enriched on TALKO mitochondria and significantly accumulated on DKO mitochondria, suggesting its diminished consumption for mitochondrial autophagosome formation in DKO mice (Figure S2E). Thus, the overall depletion of Drp1 and loss of pDrp1S616 on isolated mitochondria occurred with diminished mitophagy and build-up mitochondrial mass in TALKO and DKO mice (Figures 3, S1 and S2).
Autoimmunity is characterized by aPL production underlain by diminished glycosylation, secretion, and enzymatic activity of paraoxonase1 (PON1) in TAL deficiency
Persistence of cirrhosis was associated with aPL production in TALKO and DKO mice (Figure 1). Although the generation of aPL is incompletely understood, oxidization of phospholipid antigens has been widely implicated as a trigger of auto-antigenicity (33, 34). Notably, paraoxonase 1 (PON 1) (36) (37), which is synthesized and secreted by the liver (38), protects both hepatocytes (39, 40, 45) and circulating blood antigens from oxidative damage (41). PON1 has been implicated in suppressing mTOR activity and enhancing autophagy (88, 89). PON1 was among the 300 genes affected by TAL deficiency at FDR p<0.05 (17). PON1 mRNA levels were significantly reduced both in TALKO and DKO livers (Figure 2A). Follow-up western blot analyses revealed complex changes in PON1 expression (Figure 4A). Total PON1 protein levels were increased in TALKO and ARKO mice and further increased in DKO mice (Figure 4A). However, both TALKO and DKO livers predominantly expressed a lower molecular weight isoform (Figure 4A). PON1 serum levels were strikingly diminished in TALKO mice (−79.8%; p=0.0014) and, to a lesser extent, in DKO mice (−63.8%; p=0.0158; Figure 4B). Along these lines, paraoxonase enzymatic activities were reduced in sera of TALKO and DKO mice (Figure 4B). These findings are consistent with earlier observations that lower molecular weight PON1 isoforms have less enzymatic activity (90).
Figure 4. Diminished glycosylation of PON1 in TAL deficiency.
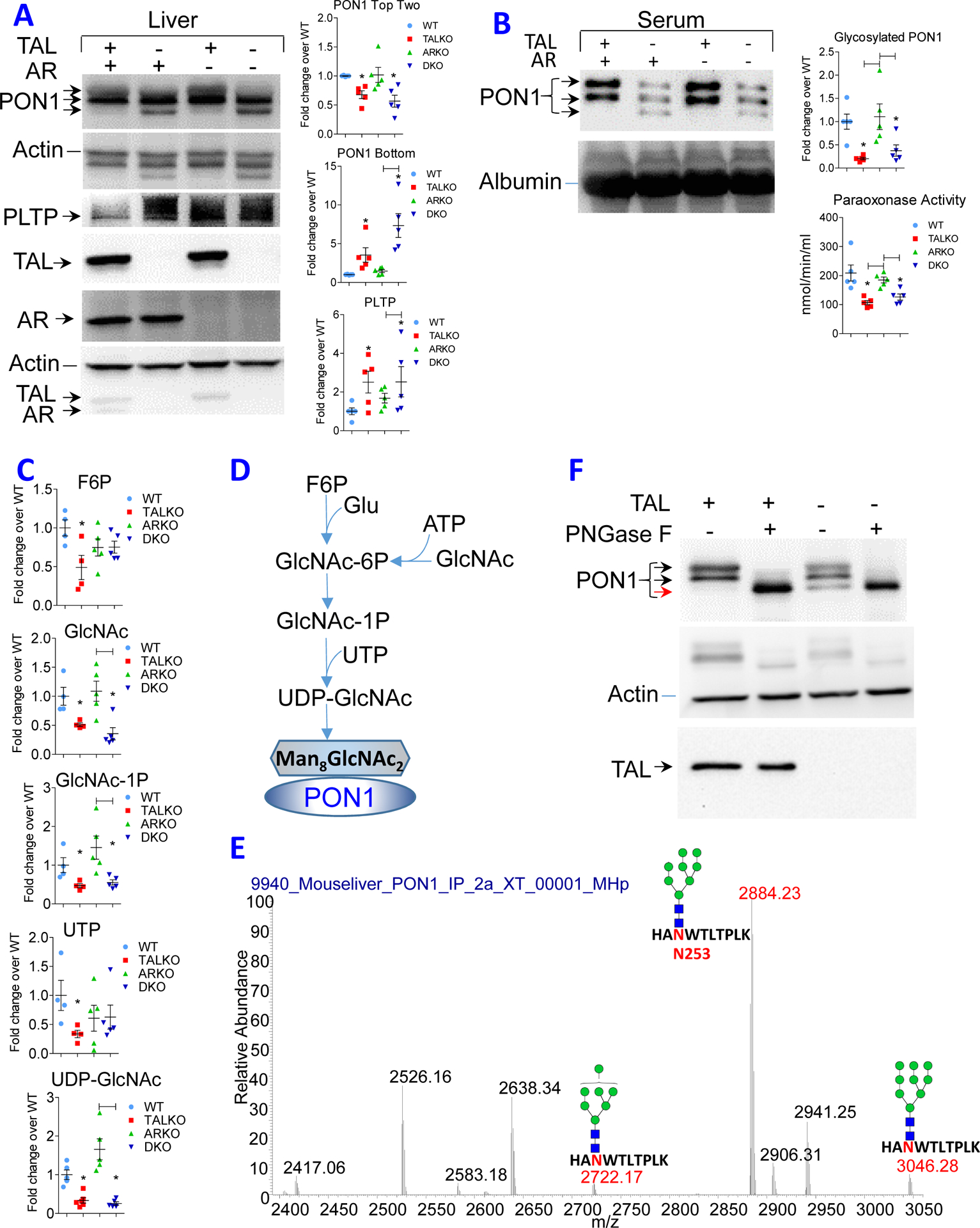
A, Western blot analysis of PON1 and PLTP expression in liver of WT, TALKO, ARKO, and DKO mice using five animals per genotype. Left panel, representative western blots. Each lysate was validated by expression of TAL and AR, using β-actin as loading control. Right panel, Bar charts show expression of top two isoforms and lowest molecular weight isoform of PON1 and PLTP relative to β-actin. *, p < 0.05 relative to WT. B, Western blot analysis of PON1 expression and paraoxonase activity in the serum of WT, TALKO, ARKO, and DKO mice. C, Detection of glycosylation substrates in the liver of WT, TALKO, ARKO, and DKO mice. *, p < 0.05 relative to WT; differences at p < 0.05 between other mouse strains are indicated by brackets. D, Schematic diagram of UDP- GlcNAc biosynthesis required for glycosylation of PON1. E, Mapping of N-glycosylation at position N253 in PON1 peptide “HANWTLTPLK” in WT liver extracts. High-energy collisional dissociation tandem mass spectroscopy (HCD-MS2) predominantly detected Man8GlcNAc2 with lesser amounts of Man9 and Man7 N-glycans. F, Treatment of hepatocyte lysates in vitro with peptide-N-glycosidase F, PNGase F. Western blot represents five experiments. Black arrows indicate glycosylated isoforms, while red arrow indicates non-glycosylated isoform.
Phospholipid transfer protein (PLTP) regulates the secretion of PON1 (91). By RNAseq, PLTP transcription was diminished (Figure 2A) while protein levels were increased in TALKO and DKO livers (Figure 4A). Interestingly, PLTP regulates PON1 secretion (91) in a glycosylation-dependent manner (46). Key substrates of glycosylation, F6P, UTP, UDP-glucose (92, 93) and UDP-GlcNAc(94), were deficient in TALKO livers (Figure 4C). While UTP depletion was moderated, but UDP-GlcNAc depletion persisted in the absence of AR (Figure 4C). UDP-GlcNAc represents the final substrate in the biosynthetic chain required for glycosylation (Figure 4D) (94). We immunoprecipitated PON1 (Figure 4E) and analyzed the glycosylation state of its trypsin-digested peptides by LC-MS/MS. Among four potential sites of N-glycosylation, N253 in peptide “HANWTLTPLK” was N-glycosylated in WT liver extracts. High-energy collisional dissociation tandem mass spectroscopy (HCD-MS2) predominantly detected Man8GlcNAc2 with lesser amounts of Man9 and Man7 N-glycans (Figure 4E). Importantly, shift to the lower molecular weight PON1 isoform in TALKO and DKO livers was effectively mimicked by in vitro treatment of WT liver lysates with peptide-N-glycosidase F, also known as PNGase F (Figure 4F), which releases N-linked oligosaccharides from glyco-proteins by hydrolyzing the amide group of the asparagine (N) side chains (47). These findings suggest that deficient glycosylation blocks the secretion of PON1 into the bloodstream of TALKO and DKO mice.
mTOR activation is a driver of autoimmunity in TALKO mice: PON1 deficiency, aPL production and expansion of Ito cells are responsive to treatment with rapamycin in vivo
Given the diminished formation of autophagosomes by mitochondria, we evaluated overall autophagy as a physiological response to metabolic stress in the liver of TALKO mice. In contrast to isolated mitochondria, LC3-I levels were reduced by 38% (Figure 5A), suggesting that restrained mitophagy occurred with overall increased autophagy in livers of TALKO mice. In support of increased autophagy, cytosolic p62 levels were also diminished in TALKO mice (Figure 5A). Overall enhanced autophagy was corroborated by accumulation of lysosomes in TALKO but not in DKO hepatocytes (Figure S1). Each of these checkpoints of enhanced autophagy, reduced LC3-I and p62 (Figure 5A) and accumulation of lysosomes, was reversed in DKO hepatocytes (Figure S1).
Figure 5. Rapamycin restrains autophagy and oxidative stress and reduces aPL production and Ito cell expansion in TALKO mice.
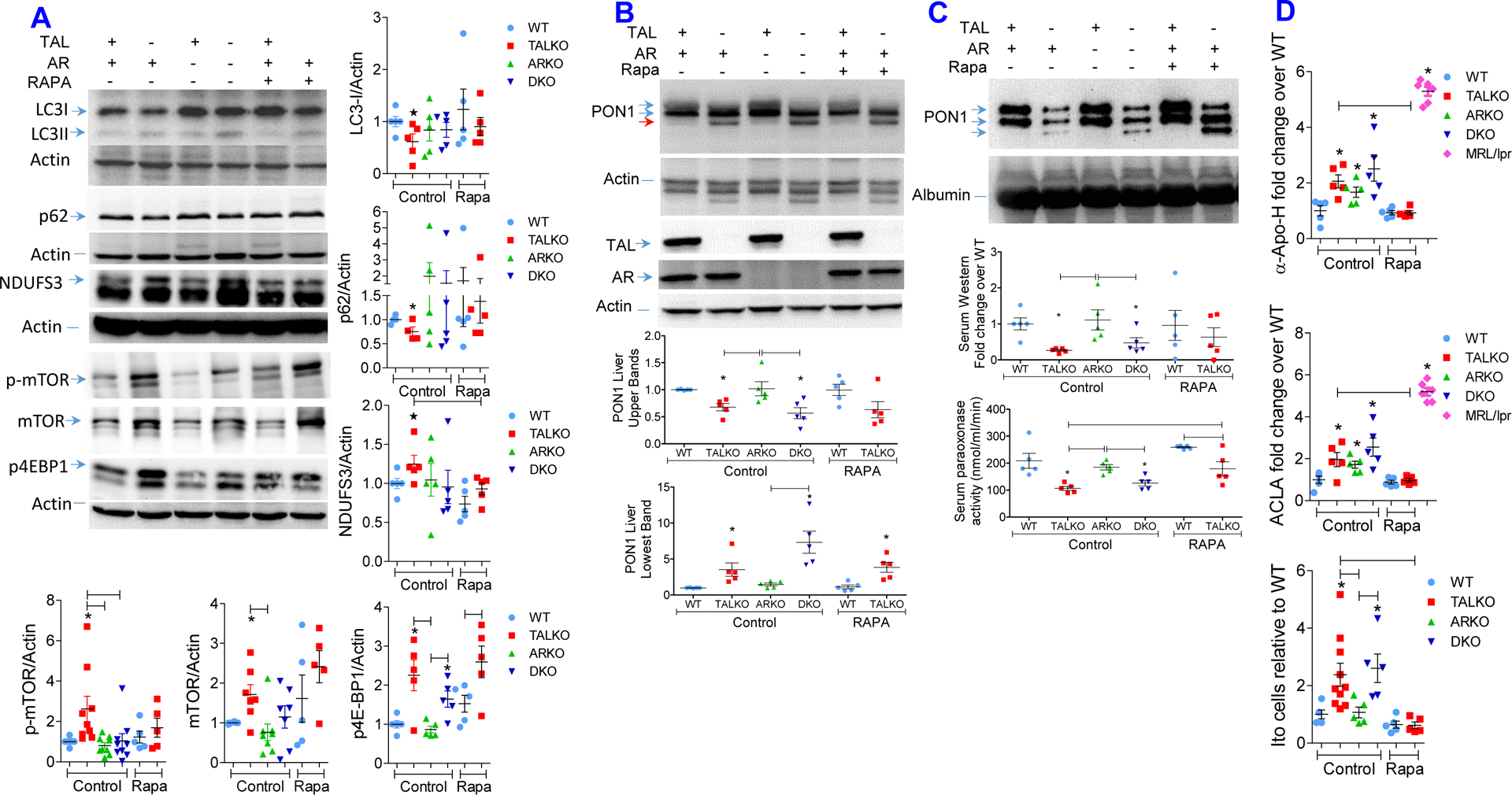
WT and TALKO mice were with rapamycin (3 mg/kg sc 3 times weekly) from 35 weeks of age for 10 weeks. A, Western blot analysis of LC3, p62 and NDUFS3 expression, and mTORC1 pathway activation in livers of WT, TALKO, ARKO and DKO mice. Representative western blots (left panels) and cumulative analysis of LC3-I, p62, NDUFS3, phosphorylated (p-mTOR) and total mTOR, and p4E-BP1 protein levels were been determined relative to β-actin loading controls (right panels) in age-matched WT, TALKO, ARKO, and DKO mice and compared to WT and TALKO mice treated with rapamycin. *, p < 0.05 relative to WT based on two-tailed t-test. B, Effect of in vivo rapamycin treatment on PON1 protein levels in the liver. C, Effect of in vivo rapamycin treatment on PON1 protein levels in the serum. D, Effect of in vivo rapamycin treatment on serum aPL, ACLA and anti-Apo-H, antibody levels and Ito cell counts in the liver. Analyses were performed in age-matched untreated control WT, TALKO, ARKO and DKO female mice and rapamycin-treated WT and TALKO female mice. *, p < 0.05 relative to WT based on two-tailed t-test.
Interestingly, enhanced autophagy occurred with the activation of the mechanistic target of rapamycin (mTOR) that has been identified as a driver of aPL-mediated inflammation (35) as well as fibrosis in the liver (78). RNAseq and follow-up western blot analysis revealed increased expression of FKBP2 and depletion of Deptor (Figure 2A and 2C). In accordance with the depletion of DEPTOR, an mTOR inhibitor (80), mTOR activation was corroborated by its greater phosphorylation in TALKO livers (Figure 5A). Moreover, protein levels of its phosphorylated signature substrate 4E-BP1 (p4E-BP1) were increased in livers of TALKO (2.39±0.54-fold, p=0.0148) and DKO mice (1.65±0.28-fold, p=0.0214; Figure 5A).
Proliferation of stellate cells and cirrhosis depend on mTORC1 in mice (24–26) and human subjects (27).Since mTORC1 was activated in TALKO mice (Figure 5A), we treated these mice with rapamycin (3 mg/kg sc 3 times weekly) from 35 weeks of age for 10 weeks.
While its glycosylation was unaffected (Figure 5B, deficient PON1 secretion and paraoxonase activity were normalized in the sera of TALKO mice upon treatment with rapamycin in vivo (Figure 5C).
Liver cirrhosis and vascular damage have long been associated with the production of antiphospholipid autoantibodies (aPL)(28–30). In turn, patients with aPL are predisposed to liver cirrhosis (31, 32). Moreover, aPL may trigger the loss of NK cells (95) that favors the expansion of Ito cells in TAL deficiency (Figure 1E). Indeed, aPL production against Apo-H and cardiolipin was increased in TALKO and DKO mice. Antigenicity of aPL has been associated with NDUFS3-dependent oxidative stress, which is responsive to rapamycin treatment (35). Indeed, rapamycin reduced the expression of NDUFS3 in TALKO mice (Figure 5A). In accordance with earlier findings (35), rapamycin abrogated aPL production in TALKO mice (Figure 5D). Blockade of mTORC1 (Figure 5A) markedly reduced the numbers of Ito cells upon sacrifice at 45 weeks of age (Figure 5D).
DISCUSSION
The present study provides evidence that the TAL-AR axis has profound implications for autoimmunity in the liver, which underlies cirrhosis, a precursor of carcinogenesis in the liver. With respect to carcinogenesis, TAL and AR exerted opposite effects on expression of ZNF143 which has been implicated in colon (96), breast (97), and HCC (98, 99). ZNF143 plays a central role in CTCF-dependent chromatin remodeling (73, 74). In turn, CTCF interacts with H19 (72) that stimulates hepatocarcinogenesis by promoting the expression of IGF2 (70, 71). TALDO1, H19, IGF2, and ZNF143 are clustered within synthenic chromosomal loci in men and mice. Interestingly, the IGF2 receptor binds hexose −6-phosphate that affects its ability to transport IGF2 across the lysosomal membrane (76). Thus, the depletion of IGF2 may be related to coordinate loss of hexose-6-phosphate due to C4-C8 carbon trapping in the non-oxidative branch of the PPP (17). Given its coordinate regulation via the TAL-AR axis, this study suggests that transcriptional control of carcinogenesis by ZNF143 and H19 are genetically and functionally connected through aldose metabolism (Graphical Abstract).
The elimination of HCC in TALKO mice by deleting AR provided a causal insight into carcinogenesis and a mechanistic rationale for targeting AR for treatment of HCC (17). The ability of AR blockade to inhibit cancer cell proliferation occurred with the reversal of mitochondrial oxidative stress and the accumulation of polyols. However, the inactivation of AR increased carbon trapping in the non-oxidative branch of the PPP and failed the reverse autoimmunity and the depletion of G6P and UDP-GlcNAc. Carbon trapping in the PPP limited the biosynthesis of amino acids and the availability of substrates for the mitochondrial tricarboxylic acid cycle (17) and elicited a compensatory accumulation of mitochondria via enhanced expression of Rab4A and depletion of Drp1 (84, 85). In accordance with earlier findings, overexpression of Rab4A also occurred with enhanced overall autophagy and mTOR activation (84, 100). These persistent metabolic changes triggered autoimmunity characterized by mTOR activation, the accumulation of Ito cells in the liver and deficient glycosylation and depletion of PON1 and aPL production in the sera of TALKO mice, which were all responsive to treatment with rapamycin. The present study thus identifies glycosylation and secretion of PON1 and aPL production as mTOR-dependent regulatory checkpoints of autoimmune liver disease in TAL deficiency.
Our study shows that TAL deficiency restrains the secretion of the antioxidant protein PON1 by the liver into the bloodstream (101, 102). PON1 is secreted by the liver into the serum where it functions as an antioxidant (101, 102). Secretion of PON1 by the liver is dependent on glycosylation (46), and it is mediated by PLTP (91). Expression of PLTP was diminished on the RNA level, while PLTP protein levels were increased in TALKO and DKO livers, suggesting its compensatory accumulation due to diminished PON1 glycosylation. Essential precursor substrates of glycosylation, G6P/F6P, UTP, UDP-glucose (103, 104) and the final substrate of the biosynthetic chain required for glycosylation, UDP-GlcNAc (94) were depleted in TALKO and DKO livers. HCD-MS2 predominantly detected Man8GlcNAc2 at position N253 of PON1, which appeared to be lacking in TALKO livers. Importantly, shift to the low molecular weight PON1 isoform in TALKO and DKO livers was effectively mimicked by in vitro treatment of WT liver lysates with PNGase F that released N-linked oligosaccharides from glyco-proteins by hydrolyzing the amide group of the asparagine (N) side chains (47). Absence of Man8GlcNAc2 modification has been previously implicated in deficient secretion of HCG by human choriocarcinoma cells (105).These findings indicate that deficient glycosylation may block PON1 secretion into the bloodstream of TALKO and DKO mice.
Liver cirrhosis has been associated with the production of aPL (28–30). Alternatively, aPL confer predisposition to cirrhosis (31, 32). Anticoagulation with heparin may limit thrombotic events and improve the clinical outcomes in patients with cirrhosis (106, 107). While the pathogenesis of aPL is incompletely understood, oxidization of phospholipid antigens has been documented as the initial trigger eliciting the autoantigenicity of the primary target of aPL, Apo-H (ß2GPI) (33, 34). Of note, PON1 protects blood antigens, such as Apo-H (ß2GPI), from oxidation (36, 37). Therefore, the diminished glycosylation and diminished secretion of PON1 by the liver of TAL-deficient mice, which lead to the depletion of PON1 in the serum, may spread oxidative stress through the bloodstream. Along these lines, the enhanced oxidation promotes the antigenicity of Apo-H and triggers aPL-mediated autoimmunity directed against Apo-H (ß2GPI) as well as cardiolipin through epitope spreading (33, 108). Thus, PON1 deficiency and aPL production are newly implicated in autoimmunity-mediated cirrhosis in TAL deficiency.
In accordance with a role of mTOR activation in cirrhosis of mice (24–26) and humans (24–26), rapamycin enhanced the secretion of PON1 into the bloodstream and abrogated the production of Apo-H and cardiolipin antibodies as well as the expansion of Ito cells in TALKO mice in vivo. Although the role of glycosylation in secretion of PON1 requires further investigation, these results identify TAL-mTOR-dependent PON1 secretion, as a mechanistic target for the therapeutic action of rapamycin in autoimmunity and cirrhosis of the liver. These findings mimic aPL-mediated liver disease in patients (109, 110) and mice with SLE (35, 111). Among 24 serum proteins, loss of PON1 was prominently associated with microvascular invasion (MVI) and gross vascular invasion (GVI) of tumor cells in 90 patients with HCC (112). Thus, loss of PON1 may also facilitate the development of HCC in TAL deficiency.
Of note, sirolimus affords protection against the recurrence of HCC in human subjects following liver transplantation (113–117). To the extent that cirrhosis is a precursor to HCC, life-long rapamycin treatment may also prevent hepatocarcinogenesis in TAL deficiency. However, genetic inactivation of mTORC1 predisposes to carcinogen-induced HCC in mice (118). Therefore, future studies are needed to address the impact of life-long treatment with rapamycin and genetic inactivation of mTORC1 on autoimmunity, cirrhosis, and hepatocarcinogenesis in TAL deficiency.
Materials and Methods
Mice.
Mice with heterozygous deletion of transaldolase (TALDO1+/−) were created and fully backcrossed for >10 generations onto the C57BL/6 strain, as earlier described (12). TALDO1+/− and Akr1b3-deficient aldose reductase (AR) knockout (ARKO) strains (119) were crossed to generate double-knockout (DKO) mice lacking both TAL and AR. Both TAL-deficient and DKO mice were breed as heterozygotes for the TALDO1 locus, as homozygous TAL deficiency causes male infertility due to mitochondrial dysfunction in sperm cells (12). Wild-type (WT or TAL+/+AR+/+), TAL-deficient (TALKO or TAL−/−AR+/+), AR-deficient (ARKO or TAL+/+AR−/−), and double-deficient (DKO, or TAL−/−AR−/−) mice were matched for age and gender in each experiments. Sensitivity to acetyl-p-aminophenol (APAP) was tested as earlier described (13). Briefly, APAP was dissolved in phosphate buffered saline (PBS) at 70°C, allowed to cool to 37°C and then injected intraperitoneally at 800 mg/kg. APAP-treated mice were continuously monitored up to 24 hours and euthanized at the earliest sign of distress. N-acetylcysteine (NAC) was provided to mice at 10 g/l in drinking water throughout their life, including treatment of their parents, as earlier described (12, 13). Animal experimentation has been approved by the Committee on the Human Use of Animals in accordance with NIH Guide for the Care and Use of Laboratory Animals.
Liver histology.
At the time of sacrifice, liver was removed from each mouse, cut into ~ 5 mm diameter blocks, and transferred into 10% formalin. Samples were paraffin-embedded, sectioned, and visualized with hematoxylin-eosin or Gömöri-trichrome staining. Slides were scored independently by expert pathologists blinded to genetic background or treatment groups. Images were taken with an Olympus CX43RF microscopy equipped with a DP-22 CCD color camera using CellSense Software.
Isolation of mitochondria.
Liver mitochondria were isolated by differential centrifugation as previously described (120). Livers were removed, cut into small pieces which were mechanically disrupted in a 15-ml Dounce homogenizer on ice in liver mitochondria isolation medium (LMIM) containing 250mM sucrose, 10mM Tris, 1mM EGTA, pH 7.4. Homogenate was centrifuged at 1,000 × g for 3 minutes at 4°C. The supernatant was transferred to new tubes and then centrifuged at 10,000 × g for 10 minutes at 4°C. The pellet was resuspended in LMIM and centrifuged again at 10,000 × g for 10 minutes at 4°C. The mitochondrial pellet was then resuspended in a Ca2+ chelating buffer containing 195 mM mannitol, 25 mM sucrose, 40 mM HEPES pH 7.2, 1 mM EGTA, 10 mM NaCl, and 5 mM succinate at room temperature. This mitochondrial solution was homogenized in a 2-ml Dounce homogenizer and then stirred at room temperature for 10 minutes and then on ice for 5 minutes. Mitochondria were centrifuged at 10,000 × g for 10 minutes at 4°C and resuspended in liver swelling buffer (LSB), which contained 195 mM mannitol, 25 mM sucrose, 40 mM HEPES pH 7.2 at 4°C. Mitochondria were centrifuged at 10,000 × g for 10 minutes at 4°C twice and then resuspended in 1 mL of LSB and protein content was measured by Bradford assay (121). Mitochondria were kept on ice for 1 hour before using in downstream analyses. NaCl, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), sucrose, and ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) were obtained from Sigma-Aldrich (St. Louis, MO). Tris-HCl was obtained from USB (Cleveland, OH). Succinic acid was obtained from Acros Organics (Geel, Belgium).
Liver perfusion.
Mice were anesthetized with pentobarbital sodium. For imaging of vasculature in fibrotic livers, mice were perfused via the inferior vena cava with Hank’s balanced salt solution (HBSS) without Mg2+ and Ca2+ containing 0.5mM EGTA, pH 7.4 at 37°C. Once the vena cava was cannulated and snip was made in the portal vein to ensure complete liver perfusion. 60mL of perfusion buffer was used for each liver. For hepatocyte isolation we used the 2-step perfusion method (13, 50, 51). Mice were injected with or without 5 U of heparin (Millipore-Sigma Cat. No. H3149) per g of body weight in 100 μl of PBS prior to perfusion (61). For analysis of metabolic activity within intact hepatocytes, livers were perfused through the portal vein in situ, as described below (51). For ultrasound assessment of hepatic tissue density and blood flow, mice were anesthetized with 2% isoflurane in 1 L of oxygen per minute during imaging procedure. Liver density was measured with a 40 MHz transducer using the Vevo 3100 (Visual Sonics, Toronto, Canada).
Confocal immunofluorescence microscopy.
Primary hepatocytes were cultured overnight on 4-well #1.5 glass-bottom slides (Lab-Tek II, cat. No.: 155382) that had been pre-coated with type I collagen (Corning, cat. no.: 354236). Slides were washed and incubated in phenol red free Dulbecco’s Modified Eagle Medium (DMEM) containing 10% fetal bovine serum, 2 mM L-glutamine, 1% penicillin/streptomycin and 0.5 μM MTG (Invitrogen, cat. No.: M7514) at 37°C in 5% CO2 air. After 60 min incubation, slides were washed twice with DMEM and incubated with 0.5 μM LysoTracker Red (LTR, Invitrogen, cat. no.: L7528) for 20 min, followed by one wash and resuspension in medium with 0.5 μM LTR. Confocal images (z-series) were captured with Zeiss LSM 780 inverted laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany) using a 40×/1.3 Plan-NeoFluar oil-immersion objective at 0.45 μm z-step intervals with lateral pixel dimensions of 0.22 μm. Images were acquired with transmission photomultiplier tube detector. Signal intensity gain was calibrated on cells that have been unstained. Sequential scanning was used to record MTG (excitation: 490 nm, emission: 516 nm), LTR (excitation: 577 nm, emission: 590 nm), and Hoechst 33342 (excitation: 350 nm, emission: 461 nm); the RGB images were converted to 8-bit grayscales and pseudo-colored in green, red, and blue, respectively. Captured z-series were imported and analyzed using Image J (http://rsbweb.nih.gov/ij/). Mean intensity values were taken in each channels for each pixel, as earlier described (122, 123). Manders’ overlap coefficient was obtained using JACOP plugin (available at http://rsb.info.nih.gov/ij/plugins/track/jacop.html). For each genotype, data were generated from analyses of ≥ 4 mice and > 20 cells. Colocalized signal between MTG and LTR was quantified and divided by the mean value of the corresponding channel to determine colocalization ratio (124). Colocalization between MTG and LTR was assessed using an in-house developed macro. Briefly, the FIJI macro converted the red and green channel to binary images followed by calculating the total number of overlapping pixels that could not be spatially resolved between the two channels in each z-slice for the entire z-series. Mitochondrial morphology was quantified as earlier described (125). For analysis of mitochondrial shape distribution, a FIJI-macro was developed that categorized the shape and morphology of mitochondria based on size (μm2) and roundness (0 to 1). Mitochondria larger than 20 μm2 with roundness > 0.6 were classified as large fused mitochondria. Mitochondria with size between 4 μm2 and 20 μm2 with roundness < 0.6 were classified as elongated mitochondria. Mitochondria with size smaller than 4 μm2 were classified as small segregated. Sub-resolution beads were imaged with the same acquisition parameters and used to collect z-series to validate the FIJI-macro (126).
Electron microscopy.
Freshly isolated mitochondria were fixed overnight in PBS with 2.5% glutaraldehyde, post-fixed in 1% OsO4, dehydrated in graded ethanol series, infiltrated with propylene oxide, and embedded in Araldite 502 epoxy resin (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections were stained with uranyl acetate and Reynold’s lead citrate prior to examination with a Tecnai BioTWIN 12 transmission electron microscope (FEI, Hillsboro, OR).
Isolation of hepatocytes for metabolic studies.
For analysis of metabolic activity within intact hepatocytes, livers were perfused through the portal vein in situ (51). Briefly, mice were sedated and anesthetized with pentobarbital via intraperitoneal injection according to IACUC approved protocols. Hepatocytes were isolated via liver perfusions, as previously described (13, 50, 51). Mice were injected with 5 U of heparin per g of body weight in 100 μl of PBS prior to perfusion (61). The abdomen and peritoneum were dissected and the intestines were reflected to allow access to the inferior vena cava and portal vein. Mice were first perfused with 50 mL of Buffer A (HBSS pH 7.4 without calcium or magnesium (Catalog No. 21–022-CM, Corning) plus 0.5 mM EGTA (Catalog No. E4378, Sigma-Aldrich) followed by 50 mL of Buffer B (HBSS pH 7.4 without calcium or magnesium with 1 mM CaCl2 (Catalog No. C7902, Sigma-Aldrich), 15mM HEPES (Catalog No. H4034, Sigma-Aldrich), and 100 U/mL Type 4 collagenase (Catalog No. LS004212, Worthington Biochemical, Lakewood, NJ). Both perfusion buffers were pre-warmed at 37°C. The portal vein was cannulated with a 25-gauge needle and buffer A flow was initiated at a rate of 5 mL/minute. When the liver began to blanch, the inferior vena cava was severed and buffer A was perfused at 10 mL/minute which was followed by perfusion of buffer B at 10mL/minute. The liver was then excised and placed in a Petri dish containing 5mL of buffer B. The capsule was then cut open to allow the release of hepatocytes into solution. Hepatocytes were filtered through a 70 μm filter and washed with isolation medium (high glucose Dulbecco’s Modified Eagle Medium (DMEM), L-glutamine free, Na-Pyruvate free (Corning Catalog No. 150–17-CV, with 1% antibiotics (30–004-CI, Corning). Hepatocytes were centrifuged at 50 × g for 3 minutes at 4°C. The pellet was washed and pelleted twice more in isolation medium. Prior to seeding of hepatocytes, XF 96-well culture plates (Catalog No. 101085–004, Seahorse Bioscience, North Billerica, MA) were pre-coated in PBS with 100 μg/ml collagen (50 μl/well) overnight followed by washes in adherence medium(13). Cells were plated at 10,000 cells/well in adherence medium (high glucose DMEM, L-glutamine free, Na-Pyruvate free with 1% antibiotics, and 10% fetal bovine serum (Catalog No. 26140079, ThermoFisher) in a collagen-precoated XF 96-well culture plate. After 4 hours, the adherence medium was removed and replaced with culture medium (high glucose DMEM, L-glutamine free, Na-Pyruvate free with and 1% antibiotics). Cells were cultured overnight on collagen-coated 96-well Seahorse metabolic plates in 5% CO2 at 37°C for 24 h prior to analysis. Next, the culture medium was removed, and the cells were washed with either liver glycolysis medium (Seahorse XF Base Medium (Catalog No. 102353–100, Seahorse Bioscience), 2mM glutamine,) or mitochondrial stress test medium (Seahorse XF Base Medium, 2mM glutamine, 1mM pyruvate, 25 mM glucose). Cells were then equilibrated with 175 μl of either glycolysis or mitochondrial stress test medium. Hepatocytes were incubated at 37°C in ambient atmosphere for 60 minutes before using a Seahorse XFe96 Analyzer (North Billerica, MA). For the glycolysis assay, we injected 10 mM glucose, 1 μM oligomycin, and 50 mM 2-deoxyglucose. For the mitochondrial stress test, we injected 1 μM oligomycin, 2 μM carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP), and 500 nM rotenone/antimycin A.
Flow cytometry of mitochondria and liver cells.
Following in situ liver perfusions, flow cytometry was performed on isolated hepatocytes sedimented at 50 × g, as earlier described (17). Cell supernatants were analyzed for presence of NK cells and stellate cells, using fluorochrome-conjugated antibodies to CD161, CD3, and CD38, respectively (BD Biosciences, San Jose, CA).
Metabolome analysis by LC-MS/MS.
Liver slices weighing approximately 100 mg were resuspended in 400 μl of 80% methanol (−80°C) and homogenized using the Power Gen 500 (Fisher Scientific, Waltham, MA). After freezing at −80°C and thawing once, the sample was centrifuged at 13,000 × g for 30 min at 4°C, and 400μl of supernatant was saved. A 2nd 400 μl of 80 % methanol (−80°C) was added to the pellet, the sample was vortexed, centrifuged at 13,000 × g for 30 min at 4°C, and the 2nd 400 μl of supernatant was saved. The two 400-μl supernatants were combined, dried in a SpeedVac (Savant AS160, Farmingdale, NY), and stored −80 °C until analysis. For nucleotide analysis, liver pieces weighing approximately 100 mg were resuspended in 400μl of 0.5M KOH with 10 μl of octanol to prevent foaming. After freezing at −80°C and thawing 3 times, the sample was centrifuged at 13,000 × g for 30 min at 4°C, and then samples were neutralized with 80 μl of KHCO3. The sample was centrifuged again at 13,000 × g for 30 min at 4°C and filtered through 0.45μm PVDF filters. 1.47 ml of 100% methanol (−80°C) was added, the sample was vortexed, centrifuged at 13,000 × g for 5 min at 4°C, and the supernatant was dried in a SpeedVac. Each sample was resuspended in 20 μl of LC/MS grade water, and 10 μl per sample was injected into a 5500 QTRAP, a hybrid triple quadrupole/linear ion trap mass spectrometer, using a quantitative polar metabolomics profiling platform with selected reaction monitoring (SRM) that covers all major metabolic pathways. The platform uses hydrophilic interaction liquid chromatography with positive/negative ion switching to analyze up to 308 metabolites (289 Q1/Q3 transitions) from a single 15-min targeted liquid chromatography–tandem mass spectrometry (LC–MS/MS) acquisition with a 3-ms dwell time and a 1.55-s duty cycle time (127).
Metabolite and pathway analysis.
Quantitative enrichment analysis of ~500 detected metabolites was utilized for pathway analysis employing the web-based MetaboAnalyst 5.0 software (17, 128). Mice were matched for age and gender and were injected in the same run. The signal stability was assured by normalizing the controls between runs to the sum of all signals between separate runs using Metaboanalyst. The enrichment analysis was based on global analysis of covariance (Ancova). A Google-map style interactive visualization system was utilized for data exploration and creation of a 3-level graphical output: metabolome view, pathway view, and compound view. The ‘metabolome view’ shows all metabolic pathways arranged according to the scores from enrichment analysis (y axis: −log p) and from topology analysis (x axis: impact: number of detected metabolites with significant p value)27. The pathway topology analysis used two well-established node centrality measures to estimate node importance: degree centrality and betweenness centrality. Degree centrality depends on the number of links connected to a given node. For directed pathway graphs, there are two types of degrees: in-degree for links came from other nodes, and out-degree for links initiated from the current node. Here, we only considered the out-degree for node importance measure. Upstream nodes are considered to have regulatory roles for the downstream nodes, and not vice versa. The betweenness centrality measures the number of shortest paths going through the node. Since metabolic networks are directed, we used relative-betweenness centrality for a metabolite importance measure based on metabolite topology weighed by relative-betweenness centrality (128). The degree centrality measures focus more on local connectivity, while the betweenness centrality measures focus more on global network topology. The node importance values calculated from centrality measures were further normalized by the sum of the importance of the pathway. Therefore, the total/maximum importance of each pathway reflects the importance measure of each metabolite node that is actually the percentage relative to the total pathway importance, and the pathway impact value is the cumulative percentage from the matched metabolite nodes. The altered compounds have been grouped and presented together for each pathway.
Metabolite concentrations were evaluated for their ability to discriminate between wild-type (WT), TALKO, ARKO, and DKO by partial least squares-discriminant analysis (PLS-DA) using MetaboAnalyst (129). PLS-DA is a supervised method that uses a multi-variate regression technique to extract via linear combination of metabolites (X) the information that can predict the subject group membership (Y). The classification and cross validation were performed using the wrapper function offered by the caret package in MetaboAnalyst software27. In order to assess that the class discrimination is statistically significant, a permutation test was performed. In each permutation, a PLS-DA model was built between the data (X) and the permuted class labels (Y) using the optimal number of components determined by cross validation for the model based on the original class assignment. The ratio of the between sum of the squares and the within sum of squares (B/W-ratio) for the class assignment prediction of each model was calculated. PLS-DA models were validated by permutation test p value <0.05. Contribution of individual metabolites to PLS-DA was assessed by variable importance in projection (VIP) and coefficient scores. Individual compounds were also compared between WT, TALKO, ARKO, and DKO by paired or unpaired t-test with Welch’s correction using Prism software (GraphPad, San Diego, CA).
RNA sequencing.
RNA was extracted from livers using the Qiagen miRNeasy kit (Qiagen, Hilden, Germany). RNA quality and quantity were determined using the RNA 6000 Nano Kit on the Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA). Sequencing library preparation was done with the Illumina TruSeq Stranded Total RNA with RiboZero Gold kit (Illumina, San Diego, CA). Sequencing libraries were quantified using the KAPA Library Quantification Complete Kit Universal (KAPA Biosystems, Wilmington, MA). The pooled library (1.4pM) was loaded onto the NextSeq 500 instrument, using the NextSeq 500/550 High Output v2 Kit for 75 cycles (Illumina, San Diego, CA). RNA-Seq data have been securely transferred, stored, and analyzed in the Illumina BaseSpace Sequence Hub. RNA Express software was used to assign aligned reads to genes, and perform differential gene expression analysis. Cufflinks software was used to profile gene expression and to detect transcript isoforms. Leveraging KEGG, Ingenuity, and Panther GeneOntology databases, integrated analysis of individually matched metabolome and RNAseq results was carried out with MetaboAnalyst. Changes between genotypes at false discovery rate (FDR) p value < 0.05 were further evaluated by western blot.
Western blot analyses.
Liver protein lysates were prepared by sonication in 300 μl of lysis buffer (20mM Tris-HCl, 150mM NaCl, 1mM Na2EDTA, 1mM EGTA, 1% Triton, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM Na3VO4, 1μg/mL leupeptin, 1mM phenylmethanesulfonyl fluoride. Mitochondria were directly dissolved in lysis buffer (20mM Tris-HCl, 150mM NaCl, 1mM Na2EDTA, 1mM EGTA, 1% Triton, 2.5mM sodium pyrophosphate, 1mM β-glycerophosphate, 1mM Na3VO4, 1μg/mL leupeptin; catalog no. 9803, Cell Signaling Technology, Danvers, MA) with 1mM phenylmethanesulfonyl fluoride (PMSF) (catalog no. P7626, Sigma-Aldrich). 40 μg of protein lysates, unless otherwise indicated, were resuspended in Laemmli buffer (20% glycerol, 125mM Tris-HCl, 4% sodium dodecyl sulfate, 10% β-mercaptoethanol, 0.075% bromophenol blue) (130), separated in a 12% SDS-polyacrylamide gel electrophoreses (SDS-PAGE), and electroblotted to nitrocellulose. TAL was detected with rabbit antibody 170 (131). Aldose reductase (AR) was detected with H9 mouse hybridoma antibody developed in-house. Rabbit polyclonal Drp1, AMPK, and phospho-AMPK antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal antibodies to 4E-BP1 and phospho-4E-BP1 were purchased from Cell Signaling (Danvers, MA). Rabbit monoclonal Rab4A, NDUFS3, OXPHOS cocktail including ATP5A, UQCRC2, MTCO1, SDHB, and NDUFB8 antibodies, rabbit polyclonal GPI1, ME1, PON1, Albumin, and PLTP antibodies were obtained from Abcam (Cambridge, UK). SQSTM1/p62 antibody was obtained from Abnova (Taipei, Taiwan). β-catenin antibody was obtained from BD (Franklin Lakes, NJ). Rabbit polyclonal Rab4A (Catalog No. sc-312) and Drp1 antibodies (Catalog No. sc-32898), mouse monoclonal p70S6 kinase (p70S6K) (Catalog No. sc-8418) and phospho-p70S6K antibodies (Catalog No. sc-8416) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit monoclonal p70S6K (Catalog No. 2708), Akt (Catalog No. 4685), rabbit polyclonal phospho-p70S6K (Catalog No. 9205), and phospho-Akt antibodies (Catalog No. 4058) were purchased from Cell Signaling Technology. Rabbit monoclonal Rab4A (Catalog No. ab108974), NDUFS3 (Catalog No. ab14711), SDHA (Catalog No. 14715), mitochondrial cytochrome c oxidase subunit 1 (MTCO1) of complex IV (Catalog No. 14705), and complex I immunocapture antibody (Catalog No. ab109798) were obtained from Abcam (Cambridge, UK). Apolipoprotein-H antibody (Catalog No. AF6575) was purchased from R&D Systems (Minneapolis, MN). β-actin antibody (Catalog No. Mab1501R) was purchased from Millipore (Billerica, MA).
Paraoxonase assay.
Paraoxonase activity was assayed based on hydrolysis of a fluorogenic organophosphate substrate using the EnzChek Paraoxonase Assay kit from ThermoFisher Scientific (Waltham, MA).
Analysis of antiphospholipid antibody (aPL) production.
Flat-bottom 96-well polystyrene plates were coated with cardiolipin (100 ng/well, Sigma cat.no. c1649) or Apoliprotein H (Apo- H, 100 pg/well, R&D Systems Cat.No. 6575-AH-050), also called β2glycoprotein I (β2GPI) in 0.01 M NaHCO3 (pH 9.55) overnight (132). Antibodies were incubated with antigen in PBS with 0.1% Tween-20 (Tween-20/PBS) at a 100-fold dilution for 1 hour. Then, plates were washed 6 times with 0.1% Tween-20/PBS, and incubated with 2000-fold diluted, HRP-conjugated secondary goat antibody directed against mouse IgG (heavy and light chain) from Jackson Immuno Research Laboratories s (Cat.no. 115–035-146). After washing 6 times with 0.1% Tween-20/PBS, plates were developed with 3,3′,5,5′-Tetramethylbenzidine (TMD, Alpha Diagnostic International). Results were read on a Biotek Synergy II plate reader equipped with Gen5 software at absorbances of 450 nm and 630 nm, with the 630 nm subtracted from the 450 nm measurement for background reduction. Data show fold-changes of OD at 630 nm relative to wells developed with secondary anti-mouse antibody alone.
Statistical analysis.
Statistical analyses were performed using Statview 5.0.1 (SAS Institute, Cary, NC) and GraphPad Prism 5.0 Software (San Diego, CA). Data were expressed as the mean ± standard error of the mean (SEM) of individual experiments. Pairwise repeated measures analysis of variance (ANOVA), two-way ANOVA, and Student’s t-test were used for analysis. Changes were considered significant at p value < 0.05. Corrections for multiple comparisons were made as indicated in each experiment.
Supplementary Material
Figure S1. Confocal microscopy of mitochondria and lysosomes in hepatocytes isolated from WT, TALKO, ARKO and DKO mice following in situ perfusion. Mitochondria were detected by MTG, lysosomes were detected with LTR, and nuclei were detected by Hoechst 33342 fluorescence. Representative images (left panels) and cumulative analysis are shown for each genotype (right panels). Five or more cells were analyzed in five of more mice per genotype. Fluorescence was quantified on the basis of pixel counts per cell. *, p < 0.05 relative to WT based on two-tailed t-test.
Figure S2. Western blot analysis of mitochondria isolated from livers of WT, TALKO, ARKO and DKO mice. Representative western blots and cumulative analysis of pDrp1S616, Drp1, Rab4A, GDI2, β-catenin, and SQSTM1/p62 protein levels in comparison to β-actin loading controls. *, p < 0.05 relative to WT based on two-tailed t-test.
HIGHLIGHTS.
Carbon trapping in the pentose phosphate pathway deprives substrates from the tricarboxylic acid cycle and causes the accumulation of mitochondria and activation of the mechanistic target of rapamycin (mTOR) in transaldolase deficiency.
UDP-GlcNAc depletion restrains the glycosylation and secretion of paraoxonase 1 (PON1) underlying the production of antiphospholipid autoantibodies (aPL), the loss of CD161+ NK cells, and the expansion of CD38+ hepatic stellate cells (HSC) that characterize cirrhosis in transaldolase deficiency.
Rapamycin treatment in vivo restores PON1 secretion and blocks aPL production and HSC accumulation in transaldolase deficiency.
Acknowledgments
This work was supported in part by grants RO1 DK078922 (A.P.), R01 AI072648 (A.P.), P30CA006516 (J.M.A.) and P01CA120964 (J.M.A.) from the National Institutes of Health, and the Central New York Community Foundation (AP).
Reference List
- 1.Scaglione S, Kliethermes S, Cao G, Shoham D, Durazo R, Luke A, et al. The Epidemiology of Cirrhosis in the United States: A Population-based Study. Journal of Clinical Gastroenterology 2015;49(8):690–696. [DOI] [PubMed] [Google Scholar]
- 2.Sepanlou SG, Safiri S, Bisignano C, Ikuta KS, Merat S, Saberifiroozi M, et al. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol 2020. Mar 1;5(3):245–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021. May 1;71(3):209–249. [DOI] [PubMed] [Google Scholar]
- 4.Llovet JL, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW, et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer 2022;3:386–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Longo M, Paolini E, Meroni M, Dongiovanni P. Remodeling of Mitochondrial Plasticity: The Key Switch from NAFLD/NASH to HCC. Int J Mol Sci 2021;22(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perl A, Hanczko R, Telarico T, Oaks Z, Landas S. Oxidative stress, inflammation and carcinogenesis are controlled through the pentose phosphate pathway by transaldolase. Trends Mol Med 2011;7:395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F. Targeted disruption of the housekeeping gene encoding glucose 6- phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J 1995;14:5209–5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vulliamy T, Mason P, Luzzatto L. The molecular basis of glucose-6-phosphate dehydrogenase deficiency. [Review] [41 refs]. Trends Genet 1992. Apr;8 (4):138–143. [DOI] [PubMed] [Google Scholar]
- 9.Li M, He X, Guo W, Yu H, Zhang S, Wang N, et al. Aldolase B suppresses hepatocellular carcinogenesis by inhibiting G6PD and pentose phosphate pathways. Nat Cancer 2020;1(7):735–747. [DOI] [PubMed] [Google Scholar]
- 10.Qian Y, Banerjee S, Grossman CE, Amidon W, Nagy G, Barcza M, et al. Transaldolase deficiency influences the pentose phosphate pathway, mitochondrial homoeostasis and apoptosis signal processing. Biochem J 2008. Oct 1;415(1):123–134. [DOI] [PubMed] [Google Scholar]
- 11.Williams M, Valayannopoulos V, Altassan R, Chung WK, Heijboer AC, Keng WT, et al. Clinical, biochemical, and molecular overview of transaldolase deficiency and evaluation of the endocrine function: Update of 34 patients. J Inher Metab Dis 2019. Jan 1;42(1):147–158. [DOI] [PubMed] [Google Scholar]
- 12.Perl A, Qian Y, Chohan KR, Shirley CR, Amidon W, Banerjee S, et al. Transaldolase is essential for maintenance of the mitochondrial transmembrane potential and fertility of spermatozoa. Proc Natl Acad Sci USA 2006;103:14813–14818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hanczko R, Fernandez D, Doherty E, Qian Y, Vas Gy, Niland B, et al. Prevention of hepatocarcinogenesis and acetaminophen-induced liver failure in transaldolase-deficient mice 119 by N-acetylcysteine. J Clin Invest 2009; 119:1546–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.LeDuc C, Crouch E, Wilson A, Lefkowitch J, Wamelink M, Jakobs C, et al. Novel Association of Early Onset Hepatocellular Carcinoma with Transaldolase Deficiency. JIMD Rep 2014;12:121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grammatikopoulos T, Hadzic N, Foskett P, Strautnieks S, Samyn M, Vara R, et al. Liver Disease and Risk of Hepatocellular Carcinoma in Children With Mutations in TALDO1. Hepatol Commun 2021. Oct 22;n/a(n/a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.El-Serag HB. Hepatocellular carcinoma: Recent trends in the United States. Gastroenterology 127(5):S27–S34. [DOI] [PubMed] [Google Scholar]
- 17.Oaks Z, Patel A, Huang N, Choudhary G, Winans T, Faludi T, et al. Cytosolic aldose metabolism contributes to progression from cirrhosis to hepatocarcinogenesis. Nat Metab 2023;5:41–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gy Vas, Conkrite K, Amidon W, Qian Y, Banki K, Perl A. Study of transaldolase deficiency in urine samples by capillary LC-MS/MS. J Mass Spec 2005;41:463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilmarth PA, Short KK, Fiehn O, Lutsenko S, David LL, Burkhead JL. A systems approach implicates nuclear receptor targeting in the Atp7b −/− mouse model of Wilson’s disease. Metallomics 2012;4(7):660–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nordgaard C, Vind AC, Stonadge A, Kjobsted R, Snieckute G, Antas P, et al. ZAK-β activated by cellular compression and mediates contraction-induced MAP kinase signaling in skeletal muscle. EMBO J 2022. Jul 28;n/a(n/a):e111650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KW, Ko BC, Jiang Z, Cao D, Chung SS. Overexpression of aldose reductase in liver cancers may contribute to drug resistance. Anticancer Drugs 2001. Feb;12:129–132. [DOI] [PubMed] [Google Scholar]
- 22.Peiseler M, Schwabe R, Hampe J, Kubes P, Heikenwalder M, Tacke F. Immune mechanisms linking metabolic injury to inflammation and fibrosis in fatty liver disease; novel insights into cellular communication circuits. J Hepatol 2022. Oct 1;77(4):1136–1160. [DOI] [PubMed] [Google Scholar]
- 23.Engelmann C, Clária J, Szabo G, Bosch J, Bernardi M. Pathophysiology of decompensated cirrhosis: Portal hypertension, circulatory dysfunction, inflammation, metabolism and mitochondrial dysfunction. J Hepatol 2021. Jul 1;75:S49–S66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Neef M, Ledermann M, Saegesser H, Schneider V, Reichen J. Low-dose oral rapamycin treatment reduces fibrogenesis, improves liver function, and prolongs survival in rats with established liver cirrhosis. J Hepatol 2006;45(6):786–796. [DOI] [PubMed] [Google Scholar]
- 25.Bridle KR, Popa C, Morgan ML, Sobbe AL, Clouston AD, Fletcher LM, et al. Rapamycin inhibits hepatic fibrosis in rats by attenuating multiple profibrogenic pathways. Liver Transplant 2009. Oct 1;15(10):1315–1324. [DOI] [PubMed] [Google Scholar]
- 26.Zhu J, Wu J, Frizell E, Liu SL, Bashey R, Rubin R, et al. Rapamycin inhibits hepatic stellate cell proliferation in vitro and limits fibrogenesis in an in vivo model of liver fibrosis. Gastroenterology 1999;117(5):1198–1204. [DOI] [PubMed] [Google Scholar]
- 27.Czaja AJ. Current and prospective pharmacotherapy for autoimmune hepatitis. Exp Opin Pharmacother 2014. Aug 1;15(12):1715–1736. [DOI] [PubMed] [Google Scholar]
- 28.Uthman I, Khamashta M. The abdominal manifestations of the antiphospholipid syndrome. Rheumatology 2007;46(11):1641–1647. [DOI] [PubMed] [Google Scholar]
- 29.Chedid A, Chadalawada KR, Morgan TR, Moritz TE, Mendenhall CL, Hammond JB, et al. Phospholipid antibodies in alcoholic liver disease. Hepatology 1994;20(6):1465–1471. [DOI] [PubMed] [Google Scholar]
- 30.Quintarelli C, Ferro D, Valesini G, Basili S, Tassone G, Violi F. Prevalence of lupus anticoagulant in patients with cirrhosis: relationship with beta-2-glycoprotein I plasma levels. J Hepatol 1994;21(6):1086–1091. [DOI] [PubMed] [Google Scholar]
- 31.Asherson RA, Cervera R, Piette JC, Font J, Lie JT, Burcoglu A, et al. Catastrophic Antiphospholipid Syndrome: Clinical and Laboratory Features of 50 Patients. Medicine (Baltimore) 1998;77(3):195–207. [DOI] [PubMed] [Google Scholar]
- 32.Ambrosino P, Lupoli R, Spadarella G, Tarantino P, Di Minno A, Tarantino L, et al. Autoimmune liver diseases and antiphospholipid antibodies positivity: A meta-analysis of literature studies. J.Gastrointestin. Liver Dis 2015;24(1):25–34. [DOI] [PubMed] [Google Scholar]
- 33.McNeil HP, Simpson RJ, Chesterman CN, Krilis SA. Anti-phospholipid antibodies are directed against a complex antigen that includes a lipid-binding inhibitor of coagulation: beta 2-glycoprotein I (apolipoprotein H). Proc Natl Acad Sci USA 1990. Jun 1;87(11):4120–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ioannou Y, Zhang JY, Qi M, Gao L, Qi JC, Yu DM, et al. Novel assays of thrombogenic pathogenicity in the antiphospholipid syndrome based on the detection of molecular oxidative modification of the major autoantigen β2-glycoprotein I. Arthritis Rheum 2011. Sep 1;63(9):2774–2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oaks Z, Winans T, Caza T, Fernandez D, Liu Y, Landas SK, et al. Mitochondrial dysfunction in the liver and antiphospholipid antibody production precede disease onset and respond to rapamycin in lupus-prone mice. Arthritis Rheumatol 2016. Jun 1;68:2728–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert M, Boullier A, Hachulla E, Fruchart JC, Teissier E, Hatron PY, et al. Paraoxonase activity is dramatically decreased in patients positive for anticardiolipin antibodies. Lupus 2000;9(4):299–300. [DOI] [PubMed] [Google Scholar]
- 37.Delgado Alves J, Ames PRJ, Donohue S, Stanyer L, Noorouz-Zadeh J, Ravirajan C, et al. Antibodies to high-density lipoprotein and β2-glycoprotein I are inversely correlated with paraoxonase activity in systemic lupus erythematosus and primary antiphospholipid syndrome. Arthritis Rheum 2002. Oct 1;46(10):2686–2694. [DOI] [PubMed] [Google Scholar]
- 38.She ZG, Chen HZ, Yan Y, Li H, Liu DP. The Human Paraoxonase Gene Cluster As a Target in the Treatment of Atherosclerosis. Antioxid Redox Signal 2012. Mar 15;16(6):597–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Desai S, Baker SS, Liu W, Moya DA, Browne RW, Mastrandrea L, et al. Paraoxonase 1 and oxidative stress in paediatric non-alcoholic steatohepatitis. Liver Int 2014. Jan;34(1):110–117. [DOI] [PubMed] [Google Scholar]
- 40.Abdel-Wahhab KG, Fawzi H, Mannaa FA. Paraoxonase-1 (PON1) inhibition by tienilic acid produces hepatic injury: Antioxidant protection by fennel extract and whey protein concentrate. Pathophysiology 2016. Mar;23(1):19–25. [DOI] [PubMed] [Google Scholar]
- 41.Shih DM, Gu L, Xia YR, Navab M, Li WF, Hama S, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature 1998. Jul 16;394(6690):284–287. [DOI] [PubMed] [Google Scholar]
- 42.Ferre N, Camps J, Cabre M, Paul A, Joven J. Hepatic paraoxonase activity alterations and free radical production in rats with experimental cirrhosis. Metabolism 2001. Sep 1;50(9):997–1000. [DOI] [PubMed] [Google Scholar]
- 43.Fedelesova M, Kupcova V, Luha J, Turecky L. Paraoxonase activity in sera of patients with non- alcoholic fatty liver disease. Bratisl Med J 2017;118(12):719–720. [DOI] [PubMed] [Google Scholar]
- 44.Camps J, Marsillach J, Joven J. Measurement of serum paraoxonase-1 activity as a potential biomarker for chronic liver impairment. Clin Chim Acta 2007;386(1):114–115. [DOI] [PubMed] [Google Scholar]
- 45.Ferre N, Marsillach J, Camps J, Mackness B, Mackness M, Riu F, et al. Paraoxonase-1 is associated with oxidative stress, fibrosis and FAS expression in chronic liver diseases. J Hepatol 2006. Jul;45(1):51–59. [DOI] [PubMed] [Google Scholar]
- 46.Marsillach J, Aragones G, Mackness B, Mackness M, Rull A, Beltran-Debon R, et al. Decreased paraoxonase-1 activity is associated with alterations of high-density lipoprotein particles in chronic liver impairment. Lipids Health Dis 2010. May 14;9:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Powell LD, Varki AP, Freeze HH. Release of Saccharides from Glycoconjugates. Current Protocols in Immunology. John Wiley & Sons, Inc., 2001. [DOI] [PubMed] [Google Scholar]
- 48.Schriner SE, Linford NJ, Martin GM, Treuting P, Ogburn CE, Emond M, et al. Medecine: Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 2005;308(5730):1909–1911. [DOI] [PubMed] [Google Scholar]
- 49.Sohn MJ AUID Chae WA, Ko JS Cho JY AUID, Kim JE Choi JY, et al. Metabolomic Signatures for the Effects of Weight Loss Interventions on Severe Obesity in Children and Adolescents. LID - 10.3390/metabo12010027 [doi] LID - 27.(2218–1989 (Print)). [DOI] [PMC free article] [PubMed]
- 50.Berry MN, Phillips JW. The isolated hepatocyte preparation: 30 years on. Biochem Soc Trans 2000. Feb;28(2):131–135. [DOI] [PubMed] [Google Scholar]
- 51.Severgnini M, Sherman J, Sehgal A, Jayaprakash NK, Aubin J, Wang G, et al. A rapid two-step method for isolation of functional primary mouse hepatocytes: cell characterization and asialoglycoprotein receptor based assay development. Cytotechnology 2012. Mar 22;64(2):187–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reynaert H, Thompson MG, Thomas T, Geerts A. Hepatic stellate cells: role in microcirculation and pathophysiology of portal hypertension. Gut 2002. Apr 17;50(4):571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Suematsu M, Aiso S. Professor Toshio Ito: a clairvoyant in pericyte biology. The Keio Journal of Medicine 2001;50(2):66–71. [DOI] [PubMed] [Google Scholar]
- 54.March S, Graupera M, Rosa Sarrias Ma, Lozano F, Pizcueta P, Bosch J, et al. Identification and Functional Characterization of the Hepatic Stellate Cell CD38 Cell Surface Molecule. Am J Pathol 2007. Jan 29;170(1):176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abdeen SM, Olusi SO, Askar HA, Thalib L, Al-Azemi A, George S. The predictive value of CD38 positive hepatic stellate cell count for assessing disease activity and fibrosis in patients with chronic hepatitis. Acta Histochem 2009. Nov;111(6):520–530. [DOI] [PubMed] [Google Scholar]
- 56.Bengtsson AA, Trygg J, Wuttge DM, Sturfelt G, Theander E, Donten M, et al. Metabolic Profiling of Systemic Lupus Erythematosus and Comparison with Primary Sjogren’s Syndrome and Systemic Sclerosis. PLoS ONE 2016. Jul 21;11(7):e0159384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fasbender F, Widera A, Hengstler JG, Watzl C. Natural Killer Cells and Liver Fibrosis. Frontiers in Immunology 2016. Jan 29;7:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao B, Radaeva S. Natural killer and natural killer T cells in liver fibrosis. Biochim Biophys Acta 2013. Jul 26;1832(7):1061–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vilaseca M, Garcia-Caldero H, Lafoz E, Garcia–Irigoyen O, Avila MA, Reverter JC, et al. The anticoagulant rivaroxaban lowers portal hypertension in cirrhotic rats mainly by deactivating hepatic stellate cells. Hepatology 2017;65(6):2031–2044. [DOI] [PubMed] [Google Scholar]
- 60.Cerini F, Vilaseca M, Lafoz E, Garcia-Irigoyen O, Garcia-Caldero H, Tripathi DM, et al. Enoxaparin reduces hepatic vascular resistance and portal pressure in cirrhotic rats. J Hepatol 2016;64(4):834–842. [DOI] [PubMed] [Google Scholar]
- 61.Wan MX, Zhang XW, Torkvist L, Thorlacius H. Low molecular weight heparin inhibits tumor necrosis factor α-induced leukocyte rolling. Inflamm Res 2001;50(12):581–584. [DOI] [PubMed] [Google Scholar]
- 62.Raveh E, Matouk IJ, Gilon M, Hochberg A. The H19 Long non-coding RNA in cancer initiation, progression and metastasis - a proposed unifying theory. Mol Cancer 2015. Nov 4;14:184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Banki K, Eddy RL, Shows TB, Halladay DL, Bullrich F, Croce CM, et al. The human transaldolase gene (TALDO1) is located on chromosome 11 at p15.4-p15.5. Genomics 1997;45:233–238. [DOI] [PubMed] [Google Scholar]
- 64.Grossman CE, Qian Y, Banki K, Perl A. ZNF143 Mediates Basal and Tissue-specific Expression of Human Transaldolase. J Biol Chem 2004;279:12190–12205. [DOI] [PubMed] [Google Scholar]
- 65.Barski OA, Papusha VZ, Kunkel GR, Gabbay KH. Regulation of aldehyde reductase expression by STAF and CHOP. Genomics 2004. Jan; 83(1):119–129. [DOI] [PubMed] [Google Scholar]
- 66.Wolford JK, Yeatts KA, Eagle ARR, Nelson RG, Knowler WC, Hanson RL. Variants in the gene encoding aldose reductase (AKR1B1) and diabetic nephropathy in American Indians. Diabet Med 2006. Apr 1;23(4):367–376. [DOI] [PubMed] [Google Scholar]
- 67.Ho HTB, Jenkins NA, Copeland NG, Gilbert DJ, Winkles JA, Louie HWY, et al. Comparisons of genomic structures and chromosomal locations of the mouse aldose reductase and aldose reductase-like genes. Eur J Biochem 1999. Feb 1;259(3):726–730. [DOI] [PubMed] [Google Scholar]
- 68.Lopez MF, Zheng L, Miao J, Gali R, Gorski G, Hirschhorn JN. Disruption of the Igf2 gene alters hepatic lipid homeostasis and gene expression in the newborn mouse. Am J Physiol Endocrin Metab 2018. Jul 17;315(5):E735–E744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cianfarani S, Inzaghi E, Alisi A, Germani D, Puglianiello A, Nobili V. Insulin-Like Growth Factor-I and -II Levels Are Associated with the Progression of Nonalcoholic Fatty Liver Disease in Obese Children. J Pediatr 2014. Jul 1;165(1):92–98. [DOI] [PubMed] [Google Scholar]
- 70.Ramani K, Mavila N, Ko KS, Mato JM, Lu SC. Prohibitin 1 Regulates the H19-Igf2 Axis and Proliferation in Hepatocytes. Journal of Biological Chemistry 2016. Nov 11;291(46):24148–24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Czarny MJ, Babcock K, Baus RM, Manoharan H, Pitot HC. Hepatocellular carcinomas of the albumin SV40 T-antigen transgenic rat display fetal-like re-expression of Igf2 and deregulation of H19. Mol Carcinog 2007. Sep 1;46(9):747–757. [DOI] [PubMed] [Google Scholar]
- 72.Holwerda S, De Laat W. Chromatin loops, gene positioning, and gene expression. Front Genet 2012;3:217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailey SD, Zhang X, Desai K, Aid M, Corradin O, Cowper-Sallari R, et al. ZNF143 provides sequence specificity to secure chromatin interactions at gene promoters. Nature Comm 2015. Feb 3;6:6186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhou Q, Yu M, Tirado-Magallanes R, Li B, Kong L, Guo M, et al. ZNF143 mediates CTCF-bound promoter-enhancer loops required for murine hematopoietic stem and progenitor cell function. Nat Commun 2021;12(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ye B, Yang G, Li Y, Zhang C, Wang Q, Yu G. ZNF143 in Chromatin Looping and Gene Regulation. Front Genet 2020;11:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bochel AJ, Williams C, McCoy AJ, Hoppe HJ, Winter AJ, Nicholls RD, et al. Structure of the Human Cation-Independent Mannose 6-Phosphate/IGF2 Receptor Domains 7–11 Uncovers the Mannose 6-Phosphate Binding Site of Domain 9. Structure 2020;28(12):1300–1312. [DOI] [PubMed] [Google Scholar]
- 77.Perl A mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann NY Acad Sci 2015. Apr 1;1346(1):33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Patsenker E, Schneider V, Ledermann M, Saegesser H, Dorn C, Hellerbrand C, et al. Potent antifibrotic activity of mTOR inhibitors sirolimus and everolimus but not of cyclosporine A and tacrolimus in experimental liver fibrosis. J Hepatol 2011. Aug 1;55(2):388–398. [DOI] [PubMed] [Google Scholar]
- 79.Jin YJ, Albers MW, Lane WS, Bierer BE, Schreiber SL, Burakoff SJ. Molecular cloning of a membrane-associated human FK506- and rapamycin-binding protein, FKBP-13. Proc Natl Acad Sci USA 1991. Aug 1;88(15):6677–6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, et al. DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival. Cell 2009. May 29;137(5):873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Picca A, Calvani R, Coelho-Junior HJ, Marzetti E. Cell Death and Inflammation: The Role of Mitochondria in Health and Disease. Cells 2021;10(3):537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, et al. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010;141(4):656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Archer SL. Mitochondrial Dynamics - Mitochondrial Fission and Fusion in Human Diseases. N Engl J Med 2013. Dec 5;369(23):2236–2251. [DOI] [PubMed] [Google Scholar]
- 84.Caza TN, Fernandez D, Talaber G, Oaks Z, Haas M, Madaio MP, et al. HRES-1/RAB4-Mediated Depletion of DRP1 Impairs Mitochondrial Homeostasis and Represents a Target for Treatment in SLE. Ann Rheum Dis 2014;73:1887–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Talaber G, Miklossy G, Oaks Z, Liu Y, Tooze SA, Chudakov DM, et al. HRES-1/Rab4 promotes the formation of LC3+ autophagosomes and the accumulation of mitochondria during autophagy. PLoS ONE 2014;9(1):e84392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shisheva A, Czech MP. Association of Cytosolic Rab4 with GDI Isoforms in Insulin-Sensitive 3T3-L1 Adipocytes. Biochemistry 1997. Jun 1;36(22):6564–6570. [DOI] [PubMed] [Google Scholar]
- 87.Mruk DD, Lau ASN, Sarkar O, Xia W. Rab4A GTPase-Catenin Interactions Are Involved in Cell Junction Dynamics in the Testis. J Androl 2007. Sep 10;28(5):742–754. [DOI] [PubMed] [Google Scholar]
- 88.Liu Q, Xiao JJ, Wang S, Li Y, Yang LJ, Lu QY, et al. Paraoxonase 1 Ameliorates Renal Lipotoxicity by Activating Lipophagy and Inhibiting Pyroptosis. Am J Pathol 2022;192(11):1531–1545. [DOI] [PubMed] [Google Scholar]
- 89.Witucki L, Jakubowski H. Depletion of Paraoxonase 1 (Pon1) Dysregulates mTOR, Autophagy, and Accelerates Amyloid Beta Accumulation in Mice. Cells 2023;12(5):746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Furlong CE, Richter RJ, Chapline C, Crabb JW. Purification of rabbit and human serum paraoxonase. Biochemistry 1991;30(42):10133–10140. [DOI] [PubMed] [Google Scholar]
- 91.Dullaart RPF, Gruppen EG, linga-Thie GM. Paraoxonase-1 activity is positively related to phospholipid transfer protein activity in type 2 diabetes mellitus: Role of large HDL particles. Clin Biochem 2016. Apr;49(6):508–510. [DOI] [PubMed] [Google Scholar]
- 92.Sousa M, Parodi AJ. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J 1995. Sep 1;14(17):4196–4203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Vanstapel F, Blanckaert N. Carrier-mediated translocation of uridine diphosphate glucose into the lumen of endoplasmic reticulum-derived vesicles from rat liver. J Clin Invest 1988. Sep;82(3):1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Roseman S. Reflections on Glycobiology. J Biol Chem 2001. Nov 9;276(45):41527–41542. [DOI] [PubMed] [Google Scholar]
- 95.Ames PRJ, Tommasino C, Fossati G, Matsuura E, Margarita A, Saulino A, et al. Lymphocyte subpopulations and intima media thickness in primary antiphospholipd syndrome. 2005;14(10):809–813. [DOI] [PubMed] [Google Scholar]
- 96.Verma V, Paek AR, Choi BK, Hong EK, You HJ. Loss of zinc-finger protein 143 contributes to tumour progression by interleukin-8-CXCR axis in colon cancer. J Cell Mol Med 2019. Apr 1;0(0):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Paek RA, Mun YJ, Jo JM, Choi H, Lee JY, Cheong H, et al. The Role of ZNF143 in Breast Cancer Cell Survival Through the NAD(P)H Quinone Dehydrogenase 1-p53-Beclin1 Axis Under Metabolic Stress. Cells 2019;8(4):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang L, Huo Q, Ge C, Zhao F, Zhou Q, Chen X, et al. ZNF143-Mediated H3K9 Trimethylation Upregulates CDC6 by Activating MDIG in Hepatocellular Carcinoma. Cancer Res 2020. Jun 15;80:2599–2611. [DOI] [PubMed] [Google Scholar]
- 99.Wang Z, Chen X, Zhou L, Zhao X, Ge C, Zhao F, et al. FBXO9 Mediates the Cancer-Promoting Effects of ZNF143 by Degrading FBXW7 and Facilitates Drug Resistance in Hepatocellular Carcinoma. Front Oncol 2022;12:930220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Godavarthy A, Kelly R, Jimah J, Beckford M, Caza T, Fernandez D, et al. Lupus-associated endogenous retroviral LTR polymorphism and epigenetic imprinting promote HRES-1/Rab4 expression and mTOR activation. JCI Insight 2020. Jan 16;5(1):e134010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Shih DM, Xia YR, Wang XP, Miller E, Castellani LW, Subbanagounder G, et al. Combined Serum Paraoxonase Knockout/Apolipoprotein E Knockout Mice Exhibit Increased Lipoprotein Oxidation and Atherosclerosis. J Biol Chem 2000. Jun 9;275(23):17527–17535. [DOI] [PubMed] [Google Scholar]
- 102.Watson AD, Berliner JA, Hama SY, La Du BN, Faull KF, Fogelman AM, et al. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest 1995. Dec;96(6):2882–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Oikari S, Makkonen K, Deen AJ, Tyni I, Karna R, Tammi RH, et al. Hexosamine biosynthesis in keratinocytes: roles of GFAT and GNPDA enzymes in the maintenance of UDP-GlcNAc content and hyaluronan synthesis. Glycobiology 2016. Jul 1;26(7):710–722. [DOI] [PubMed] [Google Scholar]
- 104.Miszkiel A, Wojciechowski M. Long range molecular dynamics study of interactions of the eukaryotic glucosamine-6-phosphate synthase with fructose-6-phosphate and UDP-GlcNAc. J Mol Graph Model 2017;78:14–25. [DOI] [PubMed] [Google Scholar]
- 105.Peters BP, Krzesicki RF, Hartle RJ, Perini F, Ruddon RW. A kinetic comparison of the processing and secretion of the alpha beta dimer and the uncombined alpha and beta subunits of chorionic gonadotropin synthesized by human choriocarcinoma cells. J Biol Chem 1984. Dec 25;259(24):15123–15130. [PubMed] [Google Scholar]
- 106.Leonardi F, Maria ND, Villa E. Anticoagulation in cirrhosis: a new paradigm? Clin Mol Hepatol 2017. Mar 14;23(1):13–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Villa E, Camm+á C, Marietta M, Luongo M, Critelli R, Colopi S, et al. Enoxaparin Prevents Portal Vein Thrombosis and Liver Decompensation in Patients With Advanced Cirrhosis. Gastroenterology 2012. Nov 1;143(5):1253–1260. [DOI] [PubMed] [Google Scholar]
- 108.Sun X, Seidman JS, Zhao P, Troutman TD, Spann NJ, Que X, et al. Neutralization of Oxidized Phospholipids Ameliorates Non-alcoholic Steatohepatitis. Cell Metab 2020;31(1):189–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu Y, Yu J, Oaks Z, Marchena-Mendez I, Francis L, Bonilla E, et al. Liver injury correlates with biomarkers of autoimmunity and disease activity and represents an organ system involvement in patients with systemic lupus erythematosus. Clin Immunol 2015;160:319–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lai Z, Kelly R, Winans T, Marchena I, Shadakshari A, Yu J, et al. Sirolimus in patients with clinically active systemic lupus erythematosus resistant to, or intolerant of, conventional medications: a single-arm, open-label, phase 1/2 trial. Lancet 2018;391:1186–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Perl A. Mechanistic Target of Rapamycin Pathway Activation in Rheumatic Diseases. Nat Rev Rheumatol 2016;12:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Huang C, Wang Y, Liu S, Ding G, Liu W, Zhou J, et al. Quantitative Proteomic Analysis Identified Paraoxonase 1 as a Novel Serum Biomarker for Microvascular Invasion in Hepatocellular Carcinoma. J Proteome Res 2013. Apr 5;12(4):1838–1846. [DOI] [PubMed] [Google Scholar]
- 113.Lee KW, Kim SH, Yoon KC, Lee JM, Cho JH, Hong SK, et al. Sirolimus Prolongs Survival after Living Donor Liver Transplantation for Hepatocellular Carcinoma Beyond Milan Criteria: A Prospective, Randomised, Open-Label, Multicentre Phase 2 Trial. J Clin Med 2020. Oct 12;9(10):3264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Schnitzbauer AA, Filmann N, Adam R, Bachellier P, Bechstein WO, Becker T, et al. mTOR Inhibition Is Most Beneficial After Liver Transplantation for Hepatocellular Carcinoma in Patients With Active Tumors. 2020;272(5):855–862. [DOI] [PubMed] [Google Scholar]
- 115.Tejedor-Tejada J, Alonso-Martin C, Almohalla-Alvarez C, Valenzuela EF, Munoz RN, Delgado LS, et al. Immunosuppressive Treatment With mTOR Inhibitors for Malignancies After Liver Transplantation: Long-Term Survival Retrospective Analysis. Transplant Proc. 2020;52(5):1507–1510. [DOI] [PubMed] [Google Scholar]
- 116.Invernizzi F, Iavarone M, Zavaglia C, Mazza S, Maggi U, Cesarini L, et al. Experience with early sorafenib treatment with mTOR inhibitors in hepatocellular carcinoma recurring after liver transplantation. 2020;568–574. [DOI] [PubMed] [Google Scholar]
- 117.Zhang ZH, Li LX, Li P, Lv SC, Pan B, He Q. Sirolimus in Liver Transplant Recipients with Hepatocellular Carcinoma: An Updated Meta-Analysis. 2019;32(7):632–641. [DOI] [PubMed] [Google Scholar]
- 118.Umemura A, Park EJ, Taniguchi K, Lee JH, Shalapour S, Valasek MA, et al. Liver damage, inflammation, and enhanced tumorigenesis after persistent mTORC1 inhibition. 2014;20(1):133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Ho HT, Chung SK, Law JW, Ko BC, Tam SC, Brooks HL, et al. Aldose reductase-deficient mice develop nephrogenic diabetes insipidus. Mol Cell Biol 2000;20(16):5840–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brookes PS, Shiva S, Patel RP, Darley-Usmar VM. Measurement of mitochondrial respiratory thresholds and the control of respiration by nitric oxide. Methods in Enzymology Nitric Oxide, Part D: Oxide Detection, Mitochondria and Cell Functions, and Peroxynitrite Reactions. Volume 359 ed. Academic Press, 2002. 305–319. [DOI] [PubMed] [Google Scholar]
- 121.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 122.BOLTE S, Cordeliêres FP. A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 2006. Dec 1;224(3):213–232. [DOI] [PubMed] [Google Scholar]
- 123.Venco P, Bonora M, Giorgi C, Papaleo E, Iuso A, Prokisch H, et al. Mutations of C19orf12, coding for a transmembrane glycine zipper containing mitochondrial protein, cause mis-localization of the protein, inability to respond to oxidative stress and increased mitochondrial Ca(2+). Front Genet 2015. May 19;6:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mancuso MR, Davis R, Norberg SM, O’Brien S, Sennino B, Nakahara T, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest 2006;116(10):2610–2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 2011. Jun 21;108(25):10190–10195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lin YH, Chang HM, Chang FP, Shen CR, Liu CL, Mao WY, et al. Protoporphyrin IX accumulation disrupts mitochondrial dynamics and function in ABCG2-deficient hepatocytes. FEBS Lett 2013. Oct 1;587(19):3202–3209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Confocal microscopy of mitochondria and lysosomes in hepatocytes isolated from WT, TALKO, ARKO and DKO mice following in situ perfusion. Mitochondria were detected by MTG, lysosomes were detected with LTR, and nuclei were detected by Hoechst 33342 fluorescence. Representative images (left panels) and cumulative analysis are shown for each genotype (right panels). Five or more cells were analyzed in five of more mice per genotype. Fluorescence was quantified on the basis of pixel counts per cell. *, p < 0.05 relative to WT based on two-tailed t-test.
Figure S2. Western blot analysis of mitochondria isolated from livers of WT, TALKO, ARKO and DKO mice. Representative western blots and cumulative analysis of pDrp1S616, Drp1, Rab4A, GDI2, β-catenin, and SQSTM1/p62 protein levels in comparison to β-actin loading controls. *, p < 0.05 relative to WT based on two-tailed t-test.