

Abstract
A limited understanding of the pathology underlying chronic wounds has hindered the development of effective diagnostic markers and pharmaceutical interventions. This study aimed to elucidate the molecular composition of various common chronic ulcer types to facilitate drug discovery strategies. We conducted a comprehensive analysis of leg ulcers (LUs), encompassing venous and arterial ulcers, foot ulcers (FUs), pressure ulcers (PUs), and compared them with surgical wound healing complications (WHCs). To explore the pathophysiological mechanisms and identify similarities or differences within wounds, we dissected wounds into distinct subregions, including the wound bed, border, and peri‐wound areas, and compared them against intact skin. By correlating histopathology, RNA sequencing (RNA‐Seq), and immunohistochemistry (IHC), we identified unique genes, pathways, and cell type abundance patterns in each wound type and subregion. These correlations aim to aid clinicians in selecting targeted treatment options and informing the design of future preclinical and clinical studies in wound healing. Notably, specific genes, such as PITX1 and UPP1, exhibited exclusive upregulation in LUs and FUs, potentially offering significant benefits to specialists in limb preservation and clinical treatment decisions. In contrast, comparisons between different wound subregions, regardless of wound type, revealed distinct expression profiles. The pleiotropic chemokine‐like ligand GPR15L (C10orf99) and transmembrane serine proteases TMPRSS11A/D were significantly upregulated in wound border subregions. Interestingly, WHCs exhibited a nearly identical transcriptome to PUs, indicating clinical relevance. Histological examination revealed blood vessel occlusions with impaired angiogenesis in chronic wounds, alongside elevated expression of genes and immunoreactive markers related to blood vessel and lymphatic epithelial cells in wound bed subregions. Additionally, inflammatory and epithelial markers indicated heightened inflammatory responses in wound bed and border subregions and reduced wound bed epithelialization. In summary, chronic wounds from diverse anatomical sites share common aspects of wound pathophysiology but also exhibit distinct molecular differences. These unique molecular characteristics present promising opportunities for drug discovery and treatment, particularly for patients suffering from chronic wounds. The identified diagnostic markers hold the potential to enhance preclinical and clinical trials in the field of wound healing.
Keywords: chronic wounds, histopathology, quantitative immunohistochemistry, RNA sequencing, wound subregions
1. INTRODUCTION
Acute wound healing is a well‐documented, highly regulated process involving multiple genes, pathways, and cell types that interact with one another in a timely and meticulously orchestrated manner. 1 Dysregulation of any aspect of this process can result in impaired healing. A chronic wound develops if these processes fail to restore tissue functionality and integrity within 1–3 months of the acute injury. 2 The number of individuals living with chronic wounds is currently growing due to the increased incidence and prevalence of multifactorial diseases and the aging population. Thus, wound care and treatment have become a multi‐billion dollar economic burden. 3
Chronic wounds are typically classified by their underlying pathophysiology. Examples of this classification system include diabetic ulcers, vascular ulcers (venous and arterial), and PUs. 4 Distinct from these chronic wounds are WHCs that result from the rupture of surgical incisions for reasons that remain to be resolved. 5 , 6 In this regard, Regranex (becaplermin) represents the only growth factor therapy approved by American and European authorities for the treatment of foot ulceration. 7 Although a variety of therapeutic approaches are available, there are currently no safe and effective pharmaceutical interventions that can be used for the specific treatment of skin ulcers of any type. 8 Many of these methods focus on the prevention and treatment of etiologic and systemic factors such as wound debridement, the relief of pressure, negative pressure therapy, treatment of the underlying disease (e.g., diabetes), and the application of local dressings to encourage wound healing. 9 Efforts to elucidate the molecular processes associated with chronic wound pathology are of paramount importance for the development of optimized local and specific treatment options.
Macroscopically, chronic wounds are variable but commonly consist of a core, namely the wound bed surrounded by the wound border. 10 The wound border is a region with pathological alterations, separated from surrounding intact skin by a transition zone known as the peri‐wound region. 11 A baseline assessment of a chronic wound facilitates diagnosis and treatment evaluation, including short‐ and long‐term management goals. 4 , 9 , 12 Thus, wound subregion‐specific diagnostic molecular markers would permit clinicians to better assess therapeutic options.
Typically, chronic wounds exhibit a prolonged proinflammatory phase characterized by extensive neutrophil and macrophage infiltration. 13 , 14 This process can be monitored by the detection of myeloperoxidase (MPO), an oxidative enzyme expressed by proinflammatory neutrophils. 13 Chronic wounds also exhibit diminished functional revascularization due to impaired vasculogenesis and angiogenesis, as reflected by the levels of the pan‐endothelial marker, CD31 (PECAM‐1). 15 , 16 The proinflammatory cytokine Interleukin‐6 (IL‐6) and ACTA2 (Actin Alpha 2, Alpha‐Smooth Muscle Actin), expressed in myofibroblasts and pericytes or vascular smooth muscle cells, both play significant roles in the wound healing process. 17 , 18 , 19 Another universal characteristic of chronic wounds is their inability to re‐epithelialize to cover the wound bed. 20 Keratin 5 (KRT5) is a keratinocyte marker implicated in the proliferation and migration potential of cells in the basal layer of stratified epithelia, 21 and a marker for re‐epithelialization and closure during wound healing. 22 Likewise, quantification of nuclear Ki67 can be used to monitor total cell proliferation. 23 Increased expression of Ki67 in cells within the epidermis at the wound border and likewise in the stratifying epidermis within the bed of acute wounds is a hallmark of re‐epithelialization associated with the normal healing process. 24 Immunohistochemistry (IHC)‐based data, which is widely used to monitor wound pathology, provides valuable information about the presence of cells expressing certain markers within wounds. The method often yields only qualitative or semi‐quantitative information without providing information about the underlying disease‐relevant molecular pathological conditions. Thus, we aimed to understand the molecular disposition of chronic wounds by utilizing an in‐depth molecular characterization using a combination of several methods.
Our study presents an integrated assessment of wound histopathology,immunohistochemistry (IHC) including quantitative IHC (qIHC), and genome‐wide RNA sequencing (RNA‐Seq) to evaluate the molecular events within common human ulcer types, WHC's, and specific subregions. A chronic ulcer was defined as a wound that was present for at least 4 weeks at the initial presentation at the department of plastic surgery and was assessed by patient history. We identified common pathways, cell types, and gene expression patterns characteristic of chronic wounds. Moreover, we identified genes unique to specific ulcer types and subregions which may have the potential to serve as diagnostic signatures. The results of this study will contribute to the reproducible diagnosis of wounds with the potential to assist in personalized treatments.
2. MATERIALS AND METHODS
2.1. Resection of chronic wounds, WHCs and intact skin
Human tissue resections were obtained in accordance with the local ethics approval protocol. Informed consent was obtained from each donor (Ethikkommission Nordwest‐ und Zentralschweiz, EKNZ, Ref. Nr. 126/13). All patients were referred to the Plastic Surgery Department, University Hospital, Basel, Switzerland, for surgical wound care after failure of conservative therapies. In cases of deep‐pressure injuries, the standard of care was surgical therapy. The presence of signs of infection was a contraindication for surgery unless there was necrotic tissue. Standard in hospital care of ulcers were either moist dressings, hydrocolloid dressings or negative pressure wound therapy. The tissue resections were obtained from surgical debridement material. This was accomplished through en bloc debridement of the wounds so that the study could be performed on the whole tissue (Figure 1A). Resections from chronic wounds, WHCs and intact skin obtained from the University Hospital Basel, Switzerland, were preserved in thermo packs and transferred to the Novartis facility. One intact skin resection (01005) was provided by Dr. de Roche, Merian Iselin Clinic Basel, Switzerland. Tissue resections were imaged, and their dimensions were captured upon arrival.
FIGURE 1.
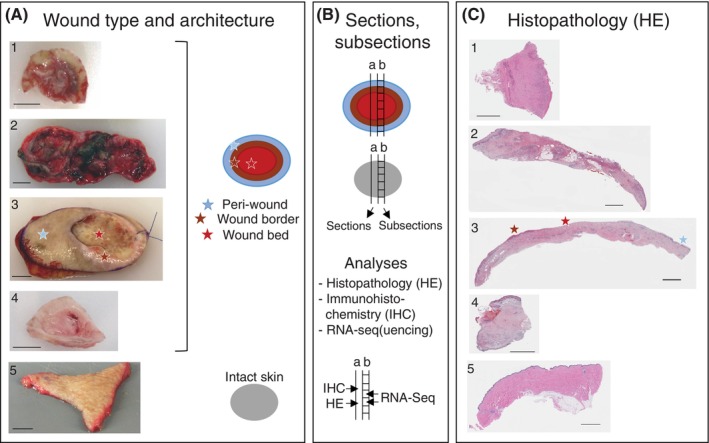
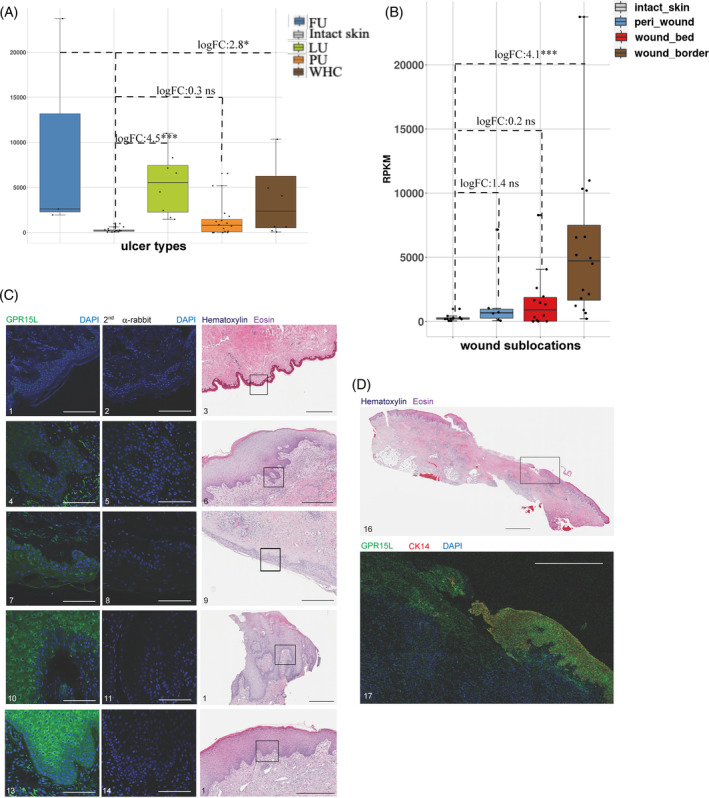
Clinical and molecular analyses of chronic wounds and wound healing complications (WHCs). (A) Classification of chronic wounds was based on ulcer type and anatomical location of the donor. WHC resections were derived from complications of surgical wounds. Intact skin samples were taken from plastic surgeries on patients without chronic wounds and served as controls. Wound resections were collected by the treating physician from patients undergoing debridement surgery. The photographs shown include examples of 1, foot ulcer (FU); 2, leg ulcer (LU); 3, pressure ulcer (PU); 4, WHC; 5, intact skin. The schematic on the right indicates the colour code used to differentiate wound subregions and intact skin specimen evaluated in this study. Light‐blue, brown, and red indicate peri‐wound, wound border and wound bed subregions, respectively, of ulcers and WHCs; grey indicates intact skin. Peri‐wound denotes the transition zone between the wound border and surrounding intact skin, wound border the region adjacent to the wound bed, and wound bed the center of the wound that is devoid of the epidermis. Star symbols in the respective colours denote the different wound subregions which are based on visual inspection of the wound specimen, as exemplified for the PU sample. Scale bars correspond to 1 cm (about 0.39 in) length. (B) Tissue sectioning and sub‐sectioning, schematic representation. Full trans‐sectional tissue sections were formalin‐fixed and embedded in paraffin prior to (a) Haematoxylin and eosin (HE) and IHC analysis (including qIHC). Adjacent, non‐fixed sections were divided into subsections according to wound architecture for (b) gene expression analyses by RNA‐Seq in individual wound subregions; similarly sized intact skin subsections served as controls. (C) Histopathology. HE‐stained sections from wound and intact skin specimen, numbering and star symbols as in A. Scale bars correspond to 2.5 mm (about 0.1 in) length.
All tissues were dissected in the laboratory using the following protocol. The first section (i.e., the margin) was removed and discarded. The second section was transferred to a 10% neutral‐buffered formalin solution (Sigma‐Aldrich) and was fixed at room temperature in the dark for 24–72 h before embedding in paraffin. The third section was dissected into a maximum of four subsections. Each subsection was individually flash‐frozen and stored in liquid nitrogen for subsequent RNA isolation. The subsections were generated by a pathologist who divided the tissue samples into sections that contained the wound bed, wound border, or peri‐wound regions. A fourth section was embedded in optimal cutting temperature (OCT) reagent, frozen in liquid nitrogen, and stored at −80°C. Depending on the size and nature of the materials obtained, this procedure was repeated several times to generate multiple sections and subsections from each tissue sample. Schema of the wound subregions (bed, border and peri‐wound, as well as intact skin) are shown in Figure 1B.
The age of the chronic wound of each patient was determined by patient history at the time of first presentation at the department of plastic surgery and defined as the duration between original wound diagnosis and wound resection. Wound resections from 22 patients and 11 intact skin resections obtained from 11 donors who were undergoing plastic surgery were subdivided into 156 and 68 individual subsections, respectively, for mRNA expression analysis (Figure 1B and Supplementary Table 1). The quality of the total RNA isolated from each of the 224 individual tissue samples was verified using an Agilent Bioanalyzer. RNA‐Seq and data analysis were performed as described below. Histopathology and IHC analyses were performed on formalin‐fixed, paraffin‐embedded (FFPE) tissue sections adjacent to those analysed by RNA‐Seq to facilitate correlations linking gene expression, detection of specific immunoreactive proteins, and histopathology assessments (Figure 1B,C). The same procedures were applied to samples of intact skin tissue. A list of donor samples is provided in Supplementary Table 1 (donors 50 042 [burn wound, wound bed] and 50 044 [WHC, used for qIHC only] are omitted).
2.2. Histology and quantitative immunohistochemistry
Formalin‐fixed tissue sections were transferred into cassettes where they were dehydrated and embedded in paraffin overnight using a VIP‐Tissue‐Tek VIP (Finetek Europe B.V., The Netherlands). The FFPE skin blocks were sectioned at 3 μm thickness and affixed to SuperFrost Plus slides and allowed to dry overnight at room temperature. Tissue sections were stained with haematoxylin and eosin (HE) using Ehrlich's method in a multi‐stainer (Leica ST5020) followed by an automated glass cover slipper (Leica CV5030). The HE‐stained tissue sections were evaluated by experienced pathologists.
Routine IHC was performed to detect cytokeratin 14, TMPRSS11D, S100A8/S100A9, and GRP15L using FFPE tissue sectioned at 5 μm thickness and processed as described above. Tissue slices were de‐paraffinized by two 10 min incubations in 100% xylene at room temperature followed by sequential 3 min incubations in 100%, 95%, 70%, and 50% ethanol followed by a final water rinse. Antigen retrieval was conducted using a KOS Microwave Multifunctional Tissue Processor (Milestone) and 10× Heat Mediated Antigen Retrieval Solution pH 6.0 (Abcam Universal HIER antigen retrieval reagent) diluted in double‐distilled water. Slides immersed in retrieval buffer were cooled on ice for 30 min, washed three times (5 min per wash) in phosphate‐buffered saline (PBS) with 0.025% Triton‐X100 (wash buffer), permeabilized for 15 min in PBS with 0.1% Triton‐X100 at room temperature, and blocked for 1 h in blocking buffer (Abcam) at room temperature. After another two consecutive washing steps for 5 min each in wash buffer, primary antibodies in universal antibody diluent (Abcam) were applied followed by overnight incubation at 4°C. The antibodies used in this study included mouse anti‐human cytokeratin 14 (Abcam, ab7800) at 1:200, rabbit anti‐human TMPRSS11D (Abcam, ab127031) at 1:250, mouse anti‐human S100A8/S100A9 (Abcam, ab22506) at 1 μg/mL, and rabbit anti‐human GPR15L (Abcam, ab151109) at 1:200. After overnight incubation, the slides were washed three times at 5 min each in wash buffer followed incubation with secondary antibodies diluted in universal antibody diluent (Abcam) for 1 h at room temperature in the dark. Secondary antibodies included goat anti‐rabbit Alexa 488 (Invitrogen), goat anti‐mouse Alexa 555 (Invitrogen), and goat anti‐rabbit Alexa 647 (Invitrogen). Slices were washed twice in wash buffer as described above. Cell nuclei were stained with DAPI (Invitrogen) diluted in wash buffer for 5 min at room temperature in the dark. Slides were then washed twice and covered with ProLong Gold antifade reagent (Invitrogen). ACTA2 staining was performed using the automated Ventana instruments Ventana Discovery® XT (Roche Diagnostics Schweiz AG, Rotkreuz, Switzerland). All chemicals were provided by Roche Diagnostics. The FFPE sections were deparaffinized and rehydrated under solvent‐free conditions (Discovery Wash solution, reference 07311079001, Roche Diagnostics) followed by an antigen retrieval step (Discovery CC1). Subsequently, slides were blocked using casein solution in PBS for 32 min and avidin/biotin for 4 min for each reagent. Primary antibody directed against ACTA2 was used: M0851, Dako, 1:1000 dilution. The primary antibody was applied on tissue sections and incubated at 37°C for 32 min. Subsequently, a biotin‐conjugated anti‐mouse antibody was applied for 16 min, followed by a DAB detection kit (reference 05266360001, Roche Diagnostics) following the Roche Diagnostics manufacturer recommendations. Slides were counterstained with haematoxylin and bluing reagent and mounted using Eukitt mounting medium. The stained slides were scanned using an NanoZoomer 2.0 HT, scanning software NDP‐Scan Version 3.4, Hamamatsu Photonics France, Swiss Office, Solothurn, Switzerland).
Quantitative (q)IHC analysis based on the marker protocol designed at Novartis was performed at TPL Path Labs, Freiburg im Breisgau, Germany. Staining procedures were performed using the automated Ventana Discovery XT or Discovery ULTRA instruments (Roche Diagnostics Schweiz AG, Rotkreuz, Switzerland). All chemicals were provided by Roche Diagnostics. For qIHC analysis, FFPE sections were deparaffinized and rehydrated in solvent‐free conditions (Discovery Wash solution, reference 07311079001, Roche Diagnostics) followed by antigen retrieval (Discovery CC1 for CD31, MPO, and Ki67 and RiboCC for KRT5). Non‐specific binding was blocked by a 32 min incubation in a casein solution in PBS. Primary antibodies included anti‐CD31 (Roche Diagnostics, 05463475001, ready to use dilution), anti‐MPO (Roche Diagnostics, 05267692001, ready to use dilution), anti‐KRT5 (Covance, PRB‐160P at a 1:18000 dilution) anti‐Ki67 (ThermoFisher, RM‐9106 at a 1:800 dilution). The primary antibodies were applied to tissue sections and incubated at room temperature for 16 min (anti‐CD31), 8 min (anti‐MPO), or 3 h (anti‐KRT5 and anti‐Ki67). Subsequently, a horseradish peroxidase (HRP)‐conjugated multimer (OmniMap to detect anti‐MPO and UltraMap to detect anti‐CD31, anti‐KRT5, and anti‐Ki67) was applied for 16 min. Signals were developed using the purple detection kit (reference 07053983001, Roche Diagnostics) following the manufacturer's recommendations. Slides were counterstained with haematoxylin and bluing reagent and mounted with Eukitt mounting medium.
For In Situ Hybridization (ISH), the FFPE blocks of human skin samples, wounded or healthy, were sectioned at 3 μm thickness and collected on SuperFrost Plus slides. The gene IL‐6 probe (probe 310 379) was investigated on mRNA level using RNAscope ISH technology commercialized by Advanced Cell Diagnostics Ltd, using the procedure mRNA Universal. The house‐keeping gene Ubiquitin C probe (probe 312 029) has been used to assess sample RNA quality. FFPE tissue sections were placed in a Discovery Ultra instrument (Roche Diagnostics Schweiz AG, Rotkreuz, Switzerland) and processed using the mRNA Universal procedure with predefined parameters for deparaffinization, demasking, hybridization and amplification steps (with an amplification 5‐step set at 2 h). Slides were counterstained with Haematoxylin II and Bluing reagent for 8 min each, dried at 60°C, dipped briefly in pure xylene and mounted using EcoMount medium. The stained slides were scanned using an NanoZoomer 2.0 HT, scanning software NDP‐Scan Version 3.4, Hamamatsu Photonics France, Swiss Office, Solothurn, Switzerland).
2.3. Slide digitalization and image analysis
The stained slides were scanned for image analysis with an Aperio slide scanner (Aperio Scanscope AT Turbo, Leica Biosystems, Muttenz, Switzerland). Automated quantitative assessment of positively stained and total tissue areas was performed on whole slide scan data using HALO image analysis software (Version 2.2, Indica Labs, Corrales, New Mexico, USA). Tissue regions of interest (ROIs) were manually outlined on the HE‐stained slide scans by the reviewing pathologist. These outlines were transferred from the HE‐stained slide images to the corresponding IHC‐stained slides. As a first step towards evaluating correlations, algorithms were configured individually for each antibody used in the IHC protocol. For anti‐CD31 and anti‐KRT5, areas detected as positively stained were normalized to the areas identified as ROIs by HE staining. Another algorithm was used to classify nuclei based on the expression of Ki67. For quantitative evaluation of MPO, staining in the cytoplasm was used to evaluate staining in the nuclei, and nuclei were classified as positive or negative depending on cytoplasmic MPO expression levels. The number of MPO‐positive nuclei was determined and normalized to the area of the respective ROI. Ki67 staining in epithelial areas was assessed from the length of the epithelium as derived by manual annotation and used as a reference for the normalization of nuclear counts.
2.4. RNA isolation and quantification
Frozen tissue samples were pulverized in Covaris TT1XT using Covaris cryoPREP Impactor and transferred to Lyse Matrix D tubes containing 1.4 mm (about 0.06 in) ceramic spheres (MP Biomedicals). The samples were homogenized after the addition of RLT plus buffer (RNeasy Plus Mini Kit, Qiagen) with beta‐mercaptoethanol (1:100 dilution; Sigma) for 2 × 20 sec at 6800 rpm with a 30 sec break between cycles. RNA was isolated from the resulting homogenate using the RNeasy Plus Mini Kit (Qiagen). RNA concentrations were measured on a Nanodrop instrument (ThermoFisher). RNA samples were desalted and concentrated by precipitation with a 1/10 volume of 5 M NaCl and 7/10 volume of 2‐propanol (Fluka) followed by centrifugation at 20000 × g for 30 min at 4°C. RNA‐containing pellets were washed once with 70% EtOH (Sigma), centrifuged at 20000 × g for an additional 15 min at 4°C, air‐dried, and resuspended in double‐distilled water. RNA quality was determined using an Agilent Bioanalyzer RNA 6000 Nano (minimum RIN value of 7).
2.5. RNA sequencing and analysis
Total RNA extracted from human chronic skin wounds, surgical wound healing complications (WHCs), and intact skin was profiled through RNA sequencing. RNA libraries were prepared using the Illumina TruSeq Stranded mRNA protocol and subsequently sequenced using the Illumina HiSeq2500 platform, paired end with a read length of 2 × 76 base pairs (bp). The obtained output was processed using the manufacturer's software to generate FASTQ sequence files, with read quality being assessed using FastQC (version 0.10). The sequencing reads exhibited excellent quality, with a mean Phred score >30 observed at all base positions.
For alignment, the raw reads were mapped to the human genome (Homo sapiens, build GRCh38.p10, Ensembl 90, August 2017), and the results were annotated using Ensembl v90 to quantify gene expression. Quality control procedures were performed using Picard tools (http://picard.sourceforge.net). The exon quantification pipeline (EQP), 25 based on Bowtie 2.0, was employed to generate count‐based estimates of gene expression by aligning reads to the human genome. These values, representing gene expression, were presented as counts.
Subsequent RNA‐Seq data analysis was conducted using R/Bioconductor. Limma, an R package traditionally used for microarray data, was extended to facilitate differential expression for RNA‐Seq data. When combined with the voom function, limma applies statistical methods for data import, pre‐processing, including data transformation, quality assessment, normalization, linear modelling, and differential expression analyses tailored to RNA‐Seq data. 26 , 27
The obtained counts were subjected to transformation using voom to scale and normalize the results. Normalization was performed utilizing the trimmed mean of M‐values method (TMM) from the calcNormFactors function within the EdgeR package, which facilitated library‐scaled normalization. To facilitate data exploration, the data was log‐transformed. Precision weights, signifying gene‐wise variation among samples, were calculated and used as input for limma. The limma model was employed to fit the linear model to the voom‐transformed matrix. Genes with an expression value less than 1 read/kilobase of transcript per million mapped reads (RPKM), not expressed in at least 75% of the samples, and in at least one group (wound subregion or ulcer type) being filtered out from the analysis.
The limma model was employed to describe the log‐expression of each gene, utilizing both the design matrix and the contrast matrix for conducting pairwise comparisons among different ulcer types or subregions The design matrix was described using the formula ‘model.matrix (~‐1 + Group, pData)’. The ‘Group’ coefficients represented the average for each wound subregion (or ulcer type). The study design outlined in Supplementary Table 1 was not designed to model individual effects. The design shows the collection of different wound subregions from certain individuals, whereas others only contributed to a single wound subregion (e.g., patient 50 033).
Differential gene expression was assessed by comparing each wound subregion to intact skin, resulting in the computation of normalized log2 count values for individual genes. Differentially expressed genes were identified using empirical Bayes (eBayes) moderation, 26 which calculated precision weights for each observation. Genes with a Log2(fold change) greater than +1 were defined as upregulated, whilst those with a Log2(fold change) less than −1 were downregulated. The resulting p‐values were adjusted using the Benjamini‐Hochberg method and statistical significance set at ≤0.01.
2.6. Gene set enrichment analysis (GSEA)
Gene set enrichment analysis (GSEA) was performed using a two‐sample, one‐sided Kolmogorov–Smirnov (KS) test to identify up‐ and downregulated gene sets utilizing differential expression values (i.e., fold changes and p‐values). Gene Ontology (GO), Interpro, NCBI Biosystems, MSIGDB, and Human Phenotype Ontology were used to examine the RNA‐Seq results within the context of existing biological knowledge. The resulting p‐values were adjusted using the Benjamini‐Hochberg method to control the false discovery rate (FDR). Only enrichments scores with a −log10 adjusted p‐value >2 were retained.
2.7. Inter‐ and intra‐patient variability
Inter‐ and intra‐patient variability with respect to gene expression was calculated using the coefficient of variation (CV), which is the ratio of the standard deviation (SD) to the mean.
2.8. Cell type abundance determined by gene expression profiling
Cell type abundance was determined using R xCell package—a high‐performing gene signature‐based method used to infer several immune and stromal cell types. 28 Various cell types involved in wound healing were considered and are listed in Supplementary Table 7 with scaled median values across subregions, generated using xCell.
2.9. Logistic regression to classify wound subregions
The full list of samples (n = 244) was randomly split and stratified into a train and test set with a ratio of 70:30, using the ‘createDataPartition’ method within the CARET package, focusing on wound subregion and intact skin alone. Each sample was derived from a different physical wound or intact skin and is thus treated as independent for the purpose of this analysis. The supervised machine learning algorithm, Logistic Regression with Lasso shrinkage was performed to determine transcripts of genes which may aid in classifying the various wound subregions and intact skin. The model design is depicted using the formula, Group~., where Group refers to the wound subregions and intact skin. Additional covariates were excluded due to the experimental design and small patient sample size.
The multicategory receiver operating characteristic (ROC) was used to test the model's ability to classify/distinguish the wound subregions (bed, border, peri‐wound) and intact skin. The multinomial log‐linear function of the nnet package 29 was used on the training data and the Hypervolume under the Manifold (HUM) value was used to test the classification.
A binary classification (one subregion versus all others) was also performed to identify genes that might serve as unique markers of a given subregion of interest. Subregions were classified using R caret package which utilizes glmnet. 30 A leave‐one‐out cross‐validation was performed on the training set and the optimal hyperparameters were alpha = 0 and lambda = 0.2 for elastic net regression and feature selection. Feature selection was also performed using R randomForest package 31 to compare against the features selected using Elastic Net. The final feature set was the intersection of the features from ElasticNet and randomForest. glmnet 30 describes the usual problem formulation as shown above. The tuning parameter 𝜆 is used to control the overall strength of the regularization or penalty term. α denotes the elasticNet penalty term which varies from pure lasso regression (α = 1) to pure ridge regression (α = 0).
2.10. Statistics, algorithms, and visualization
Statistics, algorithms, and visualization were performed using R version 4.0.2 (R Development Core Team, 2005).
3. RESULTS
3.1. Protocol for comparative clinical, histopathological and molecular analysis of wounds
We designed a protocol for the parallel assessment of cutaneous wounds using clinical information (Supplementary Table 1), histopathology (HE staining), IHC/qIHC, and transcriptional profiling (RNA‐Seq). Chronic wound and WHC resections of 22 patients (5LU, 2 FU, 9 PU, 6 WHC; mean age 62 ± 18 years, 50% females and 50% males) obtained by standard‐of‐care debridement surgeries were evaluated. Nineteen of these patients donated wound tissue during a single visit (either visit 1 or 2), and 3 patients donated wound tissues on 2 consecutive visits (Supplementary Table 1). Intact skin from 11 healthy donors served as controls (mean age 55 ± 8 years, 91% females and 9% males; calculations based on 10 healthy donors of which age was known). Wound and intact skin specimens were processed immediately following surgery (Figure 1A). To correlate histopathology and immunoreactive protein levels against mRNA expression from the same wound specimen, adjacent sections were prepared by cutting through the entire wound or intact skin tissues. Immunostaining (IHC) and histopathology (HE) were performed on entire FFPE sections, whereas RNA‐Seq was performed from adjacent tissue sections that were further separated into subsections derived from peri‐wound, wound border, and wound bed subregions (Figure 1B). A similar procedure was performed for intact skin resections without sub‐sectioning. Since the manifestation of chronic wounds is generally heterogenous, 4 with varying wound border, bed, and peri‐wound ratios, wound subregions were harvested as present on a per patient basis (Supplementary Table 1). Histopathology assessment of tissue sections was performed by HE staining (Figure 1C). Integrative analysis was performed using ulcer‐specific information, histopathology, IHC/qIHC and mRNA expression.
3.2. Histopathologic characterization of wounds
The histopathological evaluation of all ulcers and WHCs revealed several consistent morphological features, including squamous cell hyperplasia, hyperkeratosis, necrotic tissue, inflammatory cell infiltrates, connective tissue formation, and aberrant angiogenesis (Supplementary Table 2). Wound beds lacked an epithelial layer and exhibited exophytic growth of poorly organized connective tissue that was associated with both intact and degenerating inflammatory cells and necrotic excrescence, for example, PU (Supplementary Figure 1). We noted hyperactive endothelial cell proliferation consistent with an increase in blood vessel formation. However, the vessels were mostly nonfunctional as they did not have adequate lumina containing red blood cells. Compared to intact skin, chronic ulcers and WHCs exhibited prominent necrosis and angiogenesis, while the connective tissue consisted of both mature and immature collagen fibres. Moreover, the inflammatory cell infiltrates included similar cell populations throughout all chronic wounds and WHCs (Supplementary Table 2).
Comparisons between the different ulcer types revealed distinctive areas of newly formed connective tissue intermingled with the pre‐existing substrate. While angiogenesis was more pronounced in LUs, gradients of collagen maturation initiating from regions below the necrotic tissue were more prominent in PUs and WHCs. The connective tissue in WHCs was hypocellular and consisted of fibrocytes with a few fibroblasts and inflammatory cells (Supplementary Table 2).
Collectively, our analysis revealed that chronic ulcers and WHCs exhibited abnormal endothelial cell morphology and nonfunctional blood vessels. Notably, PUs and WHCs exhibit similar tissue histology.
3.3. Evaluation of inflammation, vascularization, and re‐epithelialization in wounds by quantitative IHC
Next, we determined how the histopathological findings correlated to cellular manifestations in wounds by qIHC. We employed a set of antibodies specific for markers of biological processes and assessed the relative localization and abundance in a quantitative manner to compare the presence of these markers in the dermal and epidermal compartments of wound subregions. The wound subregions of aggregated wound types (LU, FU PU and WHC) were compared against the dermal and epidermal compartments of intact skin. Immunoreactive CD31 (Figure 2A), Ki67 (Figure 2B), KRT5 (Figure 2C) and MPO (Figure 2D) were used to evaluate vascularization, proliferation, re‐epithelialization and inflammation, respectively (Supplementary Figure 2).
FIGURE 2.
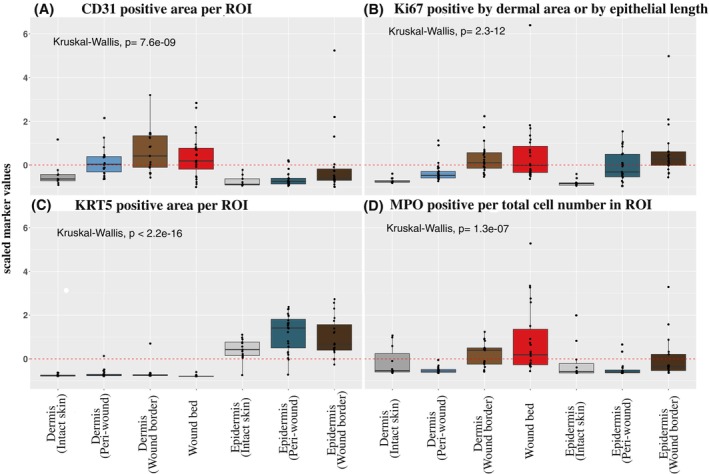
Quantification of vasculature, epidermal proliferation, re‐epithelialization and immune cell markers. A quantitative evaluation of immunoreactive markers (qIHC) was performed on tissue sections from chronic ulcer, WHC and intact skin samples. Shown are scaled immunoreactive protein expression values for (A) CD31, (B) Ki67, (C) KRT5 and (D) MPO quantified in the dermal and epidermal compartments of peri‐wound, wound border and bed subregions. Intact skin dermal and epidermal compartments were monitored as controls. A subset of immune cells in (A) revealed CD31 signals that were filtered out by image analysis to permit CD31 quantification in the vasculature. Values for CD31 and KRT5 expression levels were normalized to the ROIs and presented as positively stained areas. Values for MPO expression were normalized to total cell numbers. Ki67 expression levels were normalized to the ROI areas in the dermis or epithelial length for epidermal regions. For qIHC analyses, samples from the following donors have been used (donor identifiers in parentheses for each wound type/intact skin): 2 FUs (50 002, 50 038); 4 LUs (50 037, 50 040, 50 046, 50 052); 8 PUs (50 007, 50 013, 50 021, 50 025, 50 036, 50 041, 50 048, 50 051); 5 WHCs (50 030, 50 032, 50 044, 50 049, 50 050); 5 intact skin (00048, 00051, 00096, 00099, 00108). The aggregated data of all wound types (LU, FU, PU, WHC) is compared against intact skin (dermal and epidermal compartments for intact skin and wound subregions). A global comparison of the means was performed using Kruskal–Wallis test used to determine significance.
The endothelial cell marker CD31 showed an increased level of abundance in the dermal compartments of the wound border, bed and peri‐wound subregions, compared to intact skin. This is in line with the increased endothelial cell proliferation and formation of non‐functional blood vessels observed by histology (Supplementary Table 2). Low protein abundance levels of CD31 were identified in the epidermis of intact skin, peri‐wound and wound border regions, reflecting the absence of vasculature (Figure 2A). In contrast to intact skin, elevated levels of both Ki67 (Figure 2B) and KRT5 (Figure 2C) were observed in the epidermal layers of the peri‐wound and wound border regions, reflecting increased cell proliferation. Proliferation within the wound border was suprabasal, that is, not restricted to the basal layer as typically seen in healthy epidermis. 32 Ki67 was significantly upregulated in the wound bed and in the wound border dermis, and to a lesser degree in the dermal layer of the peri‐wound regions. KRT5 staining was not detected in any of the dermal layers. The dermis of both wound border and bed exhibited elevated levels of MPO, reflecting neutrophil infiltration (Figure 2D), in contrast to the dermal compartments of peri‐wound subregions and intact skin. Low levels of MPO were observed in the epidermal compartments of peri‐wound and wound border subregions and in intact skin which reflected the relative paucity of neutrophils in these subregions.
In summary, qIHC data extended histology findings by providing quantitative information. Chronic wounds and WHCs exhibited increased epidermal proliferation, dermal formation of defective blood vessels and dermal neutrophil infiltration in the respective subregions.
3.4. Multidimensional scaling to visualize variations between wound subregions
We conducted gene expression analyses on all 148 wound subsections derived from wound subregions of 22 patients and on 47 intact skin samples derived from 11 donors, as described in Supplementary Table 1 (Four wound bed subsections obtained from donor 50 042 burn wounds were also included). Before performing downstream transcriptome analyses, we carried out multidimensional scaling (MDS) to determine if any molecular variations exist between the different subregions of chronic wounds and WHCs. This analysis revealed that samples derived from the individual wound subregions and intact skin clustered in distinct groups regardless of patient, wound type, or anatomical location (Figure 3). We therefore hypothesized that the distinctions between subregions were due to the presence of different cell types, cell type abundance and subregion‐specific gene expression patterns. We proceeded to identify differentially expressed genes to determine those associated with wound type (irrespective of subregion) and/or wound subregion (wound type‐agnostic), or patient‐specific heterogeneity, as described in the sections below. The full list of differentially expressed genes and log2FC and p‐values (FDR) are shown in Supplementary Table 3.
FIGURE 3.
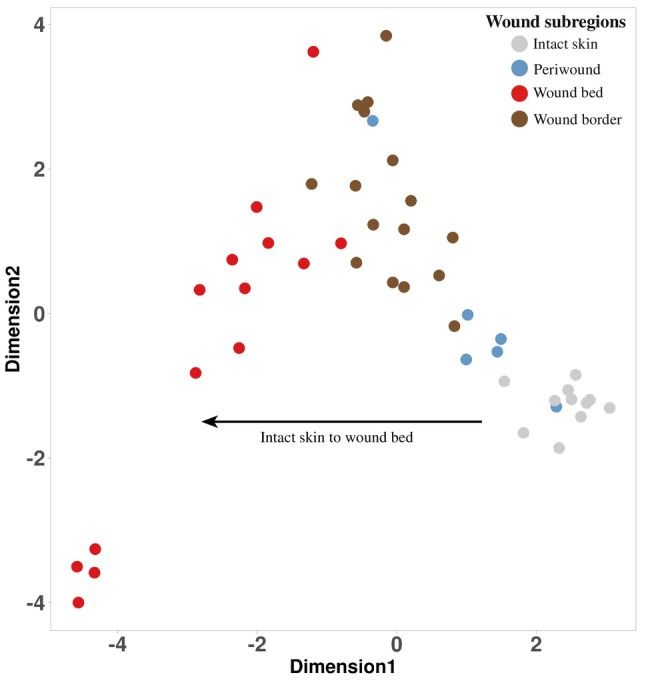
Multidimensional scaling plot to visualize wound subregions. A multidimensional scaling (MDS) plot presenting the first two dimensions displayed the data associated with wound subregions (aggregated across wound type) and intact skin. The MDS plot was generated based on averaging the gene expression values per patient, subregion to highlight a specific subregion trajectory. Each point represented the average gene expression for each sample (see legend at top right).
3.5. Comparative differential gene expression analysis revealed unique patterns associated with specific wound subregions
To visualize gene expression changes that occur in wounds, a comparative gene expression analysis was performed in aggregated wound bed and border samples and compared against intact skin (Figure 4A). A total of 1870 genes comprising those involved in extracellular matrix (ECM) regulation were increased, and 2159 genes including many keratins showed decreased expression in wounds. Marked in Figure 4A are genes that were most prominently up‐ and downregulated, or are known to be involved in inflammation, vascularization, and re‐epithelialization described in this study.
FIGURE 4.
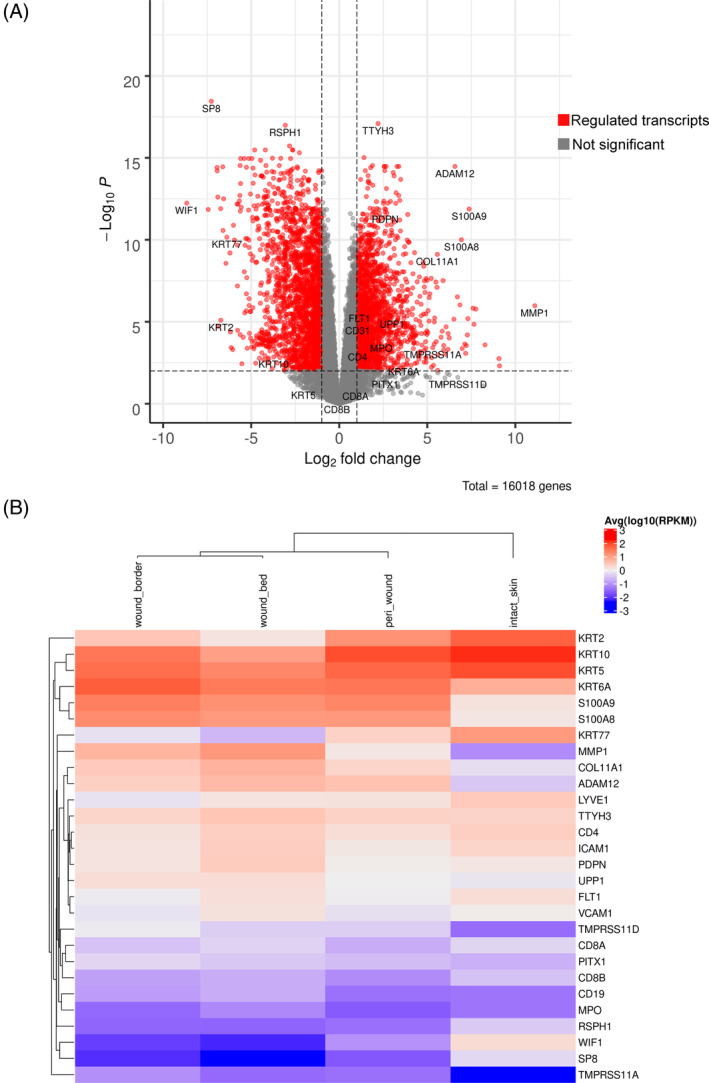
Global analysis of the transcriptomes of wound tissue versus intact skin. (A) Gene expression in wounds (aggregated wound bed, wound border and peri‐wound samples) compared with intact skin. The x‐axis specifies log2 fold changes (FC), and the y‐axis specifies the −log10 of the Benjamini–Hochberg‐adjusted p‐values (false discovery rate). The black vertical and horizontal lines reflect the filtering criteria, including a fold change (FC) > +2.0 (log2[FC] >1) to indicate upregulated transcripts (n = 1870) and log2[FC] > −1 to indicate downregulated expression (n = 2159). The genes highlighted are those described in the study and include those that were identified as most prominently up‐ and downregulated. (B) Heatmap clustering of highlighted genes shown in A. Shown is the average and scaled level of highlighted differentially expressed genes associated with a Benjamini–Hochberg‐adjusted p‐value of <0.01. Gene expression values are coloured from blue (low) to white (moderate) to red (high) based on the quantiles shown in the legend (at right) with individual genes shown in each row. Values in each column (log10[RPKM]) correspond to specific wound subregions (bed, border and peri‐wound) and intact skin. The column dendrogram was calculated based on a Pearson distance metric.
Next, we determined the relative expression levels of the differentially expressed genes highlighted in Figure 4A in the peri‐wound, wound border and bed subregions (Figure 4B). While several genes showed a similar expression pattern in all subregions compared to intact skin, other genes revealed subregion specificity. For example, a disintegrin and metalloproteinase domain 12 (ADAM12) was one of the most highly upregulated genes in all subregions of wounds compared with intact skin (Figure 4A,B). Previous studies have demonstrated that the upregulation of ADAM12 inhibits the migration and/or proliferation of keratinocytes through its role as a mediator of wound healing. 33 The likely contribution to the pathogenesis of chronic wounds had prompted its exploration as a candidate target for the treatment of chronic wounds.
Conversely, the transcription factor specificity protein 8 (SP8), 34 the WNT inhibitory factor 1 (WIF1) 35 and the radial spoke head component 1 (RSPH1) 36 showed lower expression in wound bed, border and peri‐wound subregions compared to intact skin. KRT2, KRT10 and KRT77 revealed low expression in bed and border subregions compared to intact skin, with intermediate expression in the peri‐wound region. Wound bed and border exhibited increased MMP1 expression compared with intact skin and peri‐wound samples. MMP1, an interstitial collagenase, contributes to extracellular matrix remodelling, chemokine regulation and keratinocyte migration. In excess, MMP1 can lead to re‐epithelization delay. 20 , 37 , 38 Metalloproteases play pivotal roles in chronic and acute wounds, whereas excess protease activity blocks healing. 39 In summary, several genes involved in ECM remodelling and keratinocyte biology were dysregulated in wound bed, border and peri‐wound subregions.
To identify genes enriched in pathways and processes dysregulated in chronic wounds and WHCs, GSEA was performed on the wound subregions compared with intact skin differential expression (DE) results. Enrichment (Kolmogorov–Smirnov (KS) antilog adjusted p‐value) in immune and inflammatory response, response to infection, modulation of the extracellular matrix 40 and angiogenesis was observed in all wound tissues (Figure 5A,B; Supplementary Table 4).
FIGURE 5.
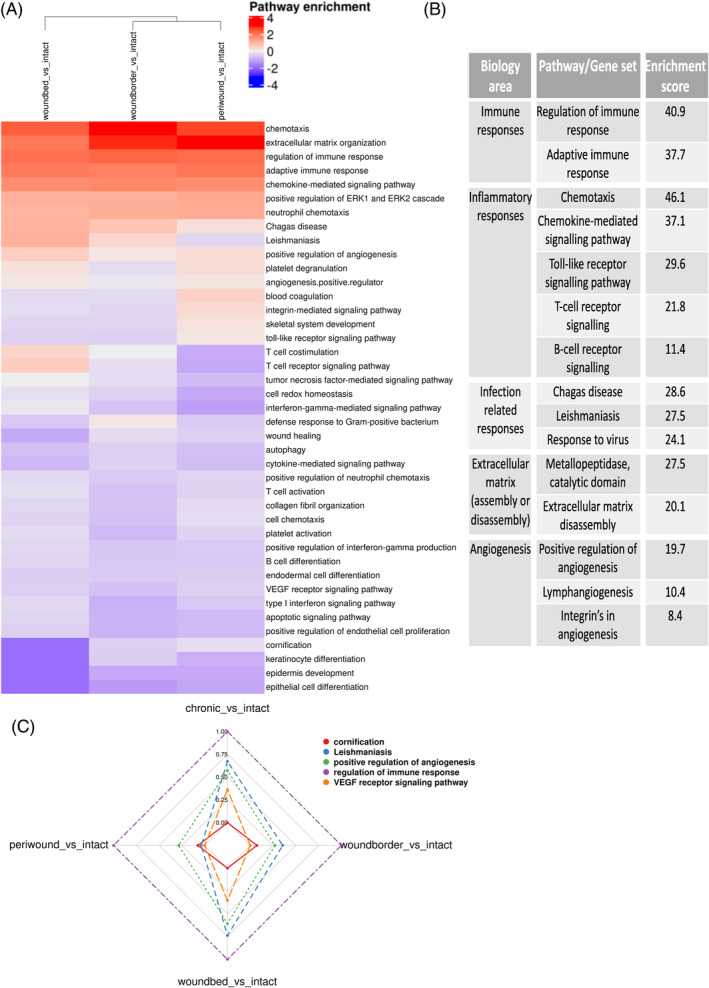
Molecular pathways and processes dysregulated in human chronic wounds and WHCs. (A) Heatmap clustering of pairwise comparisons of up‐ and downregulated pathways enriched in each wound subregion versus intact skin. Gene set enrichment analysis (GSEA) was performed using Biosystem, Interpro, and Gene Ontology sets, and only upregulated genes compared against intact skin were included for further analysis. The rows correspond to enriched pathways, and columns correspond to comparisons of wound subregions with intact skin. The column dendrogram is calculated using the Euclidean distance metric generated from the enrichment results for each comparison. The scaled, log10 converted enrichment results are colour‐coded (blue: downregulated enriched, white: not regulated, red: upregulated enriched; see the legend, top right). (B) Summary of some of the dysregulated pathways with enrichment results, grouped by biological area. (C) Radar plot displaying a selection of biological processes/pathways enriched across the different wound regions compared to intact skin samples. The selected pathways (color‐coded, top right) include cornification, Leishmaniasis, positive regulation of angiogenesis, regulation of immune response and VEGF receptor signalling pathway, representing the various processes dysregulated in chronic wounds and WHCs.
To visualize multiple values (gene sets/pathways) over common variables (wound subregions), the radar plot (Figure 5C) was used to further highlight the pathways cornification, Leishmaniasis, positive regulation of angiogenesis, regulation of immune response and VEGF receptor signalling dysregulated in chronic wounds and WHCs. The analysis revealed a general downregulation of VEGF receptor signaling across all wound subregions and a notable enrichment in the regulation of immune response within these subregions. Although there is a general decrease in VEGF receptor signaling, the radar plot shows a specific increase within chronic wounds, especially in the wound bed.
Conversely, pathways that were decreased in wounds compared to intact skin were identified. Most notably, those associated with impaired or absent re‐epithelialization, including those featuring keratins (e.g., keratin‐2 (KRT2) and keratin‐15 (KRT15)), amongst others shown in blue were identified (Figure 5A, Supplementary Figure 3).
Thus, several pathways and processes were increased or decreased in chronic wounds and WHCs, revealing the pathobiology present in wounds.
3.6. Dysregulation of genes associated with inflammation, vascularization, and re‐epithelialization pathways in chronic wounds and WHCs
We focused on molecular pathways known to be active during the various stages of the normal healing process. 9 , 11 This required first evaluating the pathways involved in the inflammatory response. Our findings revealed an upregulation of genes encoding the neutrophil chemoattractant MPO, the chemokine (C‐X‐C motif) ligand‐1 (CXCL1), T‐cell antigens CD4, CD8A, and the B‐cell marker CD19 (Figure 6A). These observations were all consistent with the observed increase in inflammatory cell infiltrates that are characteristic of chronic wounds. 13 , 14 These genes were most prominently elevated in the wound bed and border subregions as compared to intact skin. Of these genes, only CD4 and MPO were upregulated in the peri‐wound subregion, allowing a molecular classification of this subregion. Interestingly, our results revealed upregulation of proinflammatory mediators S100A8 and S100A9 41 , 42 particularly in the wound border and to a lesser extent in the wound bed and peri‐wound subregions of wound lesions (Figure 6A).
FIGURE 6.
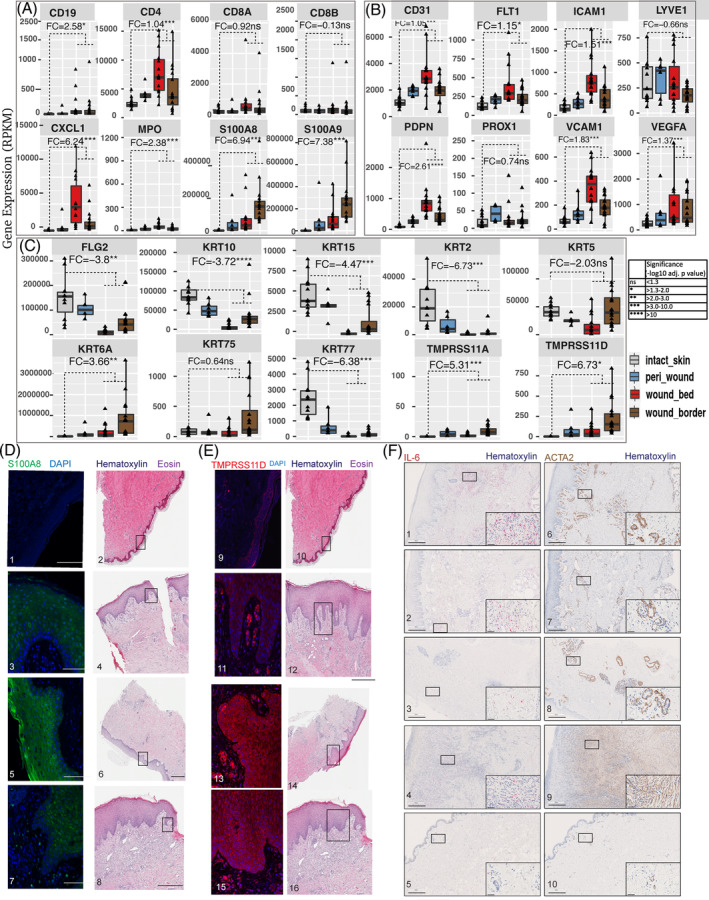
Differential expression of genes associated with the wound phenotype. Bar chart visualization of differently expressed genes in subregions of wounds. The genes were categorized into three groups, including (A) inflammation, (B) vascularization and (C) re‐epithelization. Each graph represents the expression (in RPKM) of the gene shown at the top of each plot. The bars are colour‐coded by wound subregion (legend on the right). The fold changes (FCs) displayed within each graph were generated by a comparison between expression levels in the specific wound subregion compared to intact skin. Relevant p‐values are represented by asterisks as per the legend on the bottom right. Detection of immunoreactive (D) S100A8/9 and (E) TMPRSS11D in intact skin and LUs, PUs and WHCs. HE‐stained tissues representing the various ulcer types are shown on the right side of each panel; DAPI was used to stain cell nuclei. Tissue samples were 1,2,9,10, intact skin (00082, visit 1); 3,4,11,12, LU (50 037, visit 1); 5,6,13,14, PU (50 051, visit 1); 7,8,15,16, WHC (50 050, visit 2). Black scale bars, 500 μm; white scale bars, 100 μm. (F) Analysis of IL‐6 mRNA using an RNAscope probe (1–5, red) and ACTA2 protein by IHC (6–9, brown) expression in wounds (1,6, LU [50 037, visit 1]; 2,7, WHC [50 050, visit 2]; 3,8, PU [50 051, visit 1]; 4,9, FU [50 038, visit 1]) and intact skin (5,10, brown; [00108, visit 1]). Haematoxylin was used to counterstain the sections. Magnified insets in each panel at the bottom right denote representative areas depicted in each panel. Scale bars are indicated in each panel and magnified inset, respectively; scale bars in panels, 500 μm; scale bars in insets, 50 μm.
Our findings also highlighted a differential regulation of genes encoding proteins that contribute to angiogenesis and formation of lymphatic vessels. We detected the upregulation of CD31, especially within the wound bed, in association with rounded endothelial cells, suggesting impaired vascularization (Figure 6B). Similarly, the vascular endothelial growth factor receptor family member, fms related receptor tyrosine kinase 1 (FLT1), intercellular adhesion molecule 1 (ICAM1), vascular cell adhesion molecule 1 (VCAM1), vascular endothelial growth factor A (VEGFA), and podoplanin (PDPN) were all upregulated in wounds compared with intact skin, with relative expression levels increasing from peri‐wound to wound border and bed regions (Figure 6B). The expression patterns displayed by prospero homeobox 1 (PROX1), which regulates the development of lymphatic endothelial cells, 43 and lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1) 44 were similar in chronic wounds, WHCs and intact skin. Interestingly, PROX1 was significantly upregulated in peri‐wounds compared with intact skin (log2FC 1.6, 2.2 log10 adjusted p‐value), which was not observed in wound border or wound bed subregions. Collectively, the expression of blood and lymphatic vessel endothelial cell markers reflected abnormal vascular physiology associated with wounds.
Expression of specific keratinocyte‐specific markers was performed to investigate the re‐epithelialization process. Keratins 6, 16, and 17 (KRT6, KRT16, and KRT17) are critical early barrier alarmins and first responders to pathogens and skin injury. 45 Our findings revealed that KRT6A was among the most prominent of the differentially expressed genes in a comparison between chronic wounds of all types, WHCs and intact skin, and specifically differentiates between the wound border and wound bed (Figure 6C).
Increased mRNA expression of S100A8 and S100A9 mentioned above were further evaluated by IHC analysis. IHC showed elevated levels of immunoreactive S100A8 and S100A9 primarily in the epidermis of the wound border, whereas no immunoreactive S100A8 or S100A9 was detected in the dermis (Figure 6D). We also detected the upregulation of transmembrane serine proteases 11A (TMPRSS11A) and 11D (TMPRSS11D) mRNAs in peri‐wound, wound bed, and, most prominently, wound border subregions compared with intact skin (Figure 6C). TMPRSS11A and TMPRSS11D are both enzymes that have been implicated in epidermal barrier function and maintaining homeostasis. 46 , 47 These findings also correlated with elevated levels of immunoreactive TMPRSS11D protein in the wound border subregions, specifically within the dermal and epidermal layers of PUs and WHCs (Figure 6E).
Keratins 2, 10, 15, and 77 (KRT2, KRT10, KRT15, KRT77) and Filaggrin 2 (FLG2) were significantly downregulated in all subregions of wounds when compared with intact skin. As expected, the basal epidermal marker KRT5 was reduced in the wound bed. 21 Overall, our findings revealed the DE of several genes (e.g., CD31, FLT1, KRT2) implicated in inflammation, vascularization, and re‐epithelialization in distinct patterns in wounds compared to intact skin.
To characterize inflammatory and endothelial pathology in chronic wounds and WHCs, we studied the expression of the proinflammatory cytokine IL‐6 48 using ISH and ACTA2 by IHC. 18 IL‐6 revealed increased expression in the dermis of all wound types compared to intact skin (Figure 6F). LU and WHC samples displayed higher IL‐6 levels compared with PU and FU samples, and all wound types revealed occasional IL‐6‐positive cells in the epidermis. Similarly, all wound types exhibited higher levels of ACTA2 expression in the dermal compartment compared to intact skin. LU, WHC and PU samples showed excessive dilated vessels with many cells expressing high levels of ACTA2, whereas the FU sample was highly fibrotic and showed high expression levels of ACTA2 in myofibroblasts and in some dilated vessels (pericytes and smooth muscle). 49 The intact skin sample revealed low expression levels of IL‐6 and ACTA2. ACTA2 expression was detected in skeletal muscle (e.g., erector of the hair follicle), pericytes and smooth muscle of blood vessels, and only few myofibroblasts were present in the intact dermis. Thus, all four wound types exhibited a strong proinflammatory milieu with a high level of neovascularization.
3.7. Assessment of intra‐ and inter‐donor variability
Intra‐ and inter‐donor variability was assessed by calculating coefficients of variation (CV) for each differentially expressed transcript using the RPKM values (Supplementary Tables 5 and 6, respectively). Evaluation of the calculated CVs revealed consistent gene expression levels in intact skin samples obtained from the same donor, demonstrating reproducibility. In chronic wound and WHC samples, we detected low intra‐ and inter‐donor variability in the wound bed, permitting us to perform a uniform and reproducible assessment of this subregion. Higher levels of variability were detected in wound border samples when comparing multiple subregions obtained from the same donor. Gene expression profiling could allow the identification of distinct molecular markers for wound subregion prediction and the guiding of wound debridement's, upon further evaluation of individual markervariability. Nevertheless, it is crucial to identify the variability of potential markers, since they may show different degrees of variation in individual patients.
3.8. Cell type abundance implied by gene expression profiling correlated with wound histology and immunohistochemistry
The identification and quantification of different cell types present in chronic wounds will contribute insights into their progression towards healing. The xCell R package 28 was used to monitor the distribution of cell types within each subregion (Figure 7A; Supplementary Table 7). Our results revealed a comparative reduction in epithelial cells/keratinocytes in the wound bed compared with wound border and peri‐wound subregions as well as intact skin, in line with the histological characterization. Changes in the abundance of several cell types involved in immune and inflammatory conditions reflected the ongoing adaptive and innate immune cell responses. 50 Monocytes, macrophages (M1, M2) and neutrophils were primarily enriched in wound bed and to a lesser extent in wound border subregions. A relative increase of interstitial dendritic cells (iDC) and fibroblasts in the wound bed and to a lesser extent in the wound border and peri‐wound subregions compared to intact skin was observed. In contrast, conventional/classical dendritic cells (cDC) showed a minor decrease in the wound bed and border subregions compared with peri‐wounds and intact skin. Increased fibroblast presence is in line with the notion that fibroblasts play important roles in several phases of the normal wound healing process, however, interestingly we observe the highest levels in the wound bed compared with other subregions of the wound. Importantly, endothelial cell content was increased in the wound bed and less in border and peri‐wound subregions compared to intact skin, in parallel with elevated levels of VEGFA, that induce endothelial sprouting 51 (Figure 6B). This increase correlated with increased CD31 presence in chronic wounds and WHCs (Figure 2) and histopathology (Supplementary Table 2). Adipocytes did not substantially change in wound subregions compared to intact skin. Gene expression analysis therefore allowed us to deconvolute the occurrence of different cell types in chronic wounds and WHCs, supporting the assessment of wound pathology.
FIGURE 7.
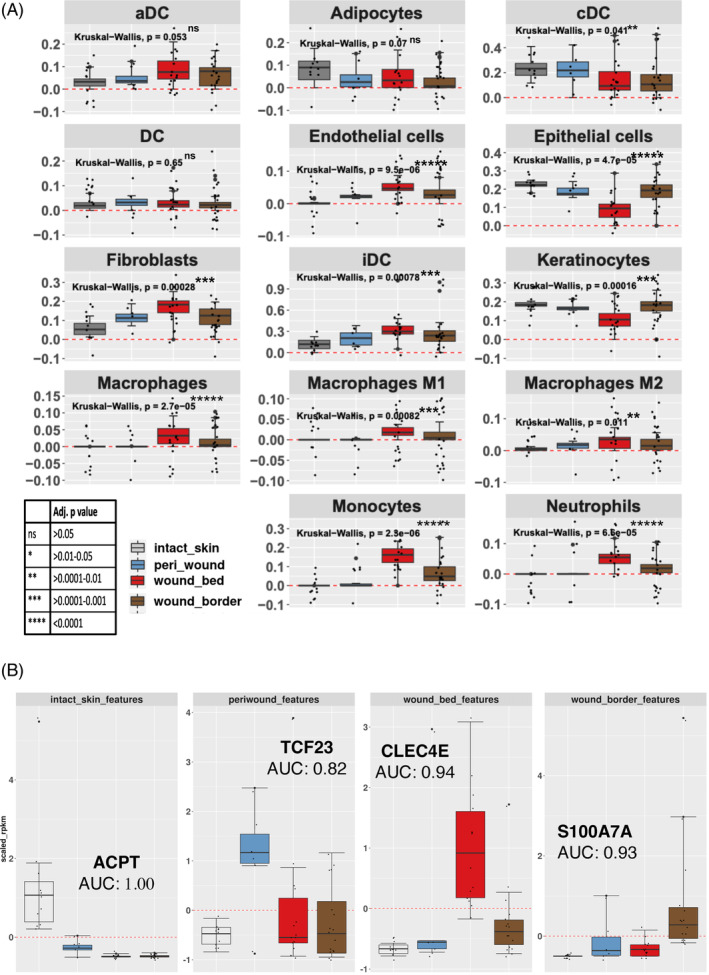
Logistic regression of cell type abundance to classify wound subregions. (A) Cell types involved in wound healing and shown in Supplementary Table 7 were inferred utilizing the xCell R library. Each graph denotes the expression of the cohort of genes that represent the cell type shown in the header. Each bar represents the scaled average expression (RPKM) for each subregion (see legend, bottom left). Each dot represents the average expression of a given gene (RPKM) that contributes to the identification of the cell type. (B) Elastic Net logistic regression analysis was used to identify candidate classification markers for specifically discriminating wound subregions and assessing using the AUC values as shown. The genes shown were selected from the subset identified by this method (using the shrinkage term within ElasticNet methods), as examples to explore their discriminatory power. Each bar represents the scaled median expression value for specific gene expression markers for each subregion identified (legend in A). Identified candidate marker genes differentiated between the various subregions. ACPT is a marker gene that was decreased in all three wound subregions compared to intact skin (AUC = 1.00). DC, dendritic cells; aDC, activated dendritic cells; cDC, conventional/classical dendritic cells; iDC, interstitial dendritic cells.
3.9. Candidate gene markers to classify wound subregions
We aimed to identify marker genes characteristic of a wound subregion. To do so, the machine learning algorithm, HUM 52 was used to evaluate the discriminative power of multiple classes, the wound bed, wound border, and peri‐wound subregions and intact skin using the RNA‐Seq gene expression profiles. The results presented a multi‐class area under the curve (AUC) value of 0.9 (standard error 0.12; 95% CI, 0.6–1.0; n = 244 samples) which suggested that the expression of specific transcripts could be used to discriminate between the various subregions. Further analysis using Elastic Net regression was performed along with ‘leave one sample out’ for cross‐validation(LOOCV). 53 , 54 , 55 This facilitated the identification of genes that we ascribed to define wound subregion identity. An evaluation of the wound bed versus other subregions resulted in an accuracy of 0.75 (AUC of 0.8). Using Caret for feature selection resulted in 15 999 predictors (accuracy of 0.9, standard deviation [SD] of 0.3). Interestingly, as few as 2 genes resulted in an accuracy of 0.7 (SD of 0.46). Intersecting these results with random forest, reduced the list to a total of 25 genes. Wound border versus other subregions resulted in 41 predictors with an accuracy of 0.6 (AUC of 0.73), which were reduced to 33 genes through determining overlaps with random forest feature selection results. Peri‐wound region versus other subregions resulted in 29 predictors (Accuracy, 0.74 SD:0.44) further reduced to 20 genes by random forest, feature selection. Whereas, intact skin versus all wound subregions resulted in an accuracy of 1 (AUC = 1), where feature selection analysis identified 15 predictors (marker genes) that were either decreased in all three wound subregions compared to intact skin, such as acid phosphatase 4 (ACPT), 56 or specifically in one subregion (Figure 7B). The latter genes included C‐type lectin domain family four, member E (CLEC4E, Mincle), 57 which was upregulated in wound bed, S100 calcium‐binding protein A7A (S100A7A), 58 which was upregulated in wound border, and the basic helix–loop–helix transcription factor 23 (TCF23), 59 which was upregulated in peri‐wound subregions (Figure 7B). These genes were selected to explore the discriminatory power, thus emphasizing their individual effects in classifying the various subregions. The full list of marker genes identified through feature selection is shown in Supplementary Table 8. We concluded that further analyses to refine the list of markers to smaller subsets and determining the relative expression of the marker genes in wound biopsies relative to intact skin, for example, by qRT‐PCR, are the first steps towards enabling molecular prediction to determine wound subregions, thus supporting proper wound diagnosis.
3.10. Differential gene expression patterns revealed differences between the various ulcer types
Transcriptome comparisons among the three aggregated wound subregions bed, border and peri‐wound were performed to determine differences between the various ulcer types and intact skin. This was achieved through pairwise comparisons between PU, LU, FU, WHC and intact skin (Figure 8A). Hypergeometric enrichment scores for pathways enriched in LUs (LU vs. PU enrichment score) included glycolysis (10.1), hypoxia‐inducible factor (HIF)1A transcription factor associated network signalling (9.3), inflammatory response (10.5), keratinocyte differentiation (7.5), epidermal development (7.1), positive regulation of angiogenesis (4.3), epidermal thickening (2.63), gangrene (2.4), and parakeratosis (2.2) (Supplementary Table 9). Biological terms characteristic of PUs (PU vs. LU) included interferon signalling (3.1), Staphylococcus infection (2.6), complement cascade (2), and glomerulonephritis (4.4). Other pathways with lower enrichment scores included those related to the complement system and microbial defence (Supplementary Table 10).
FIGURE 8.
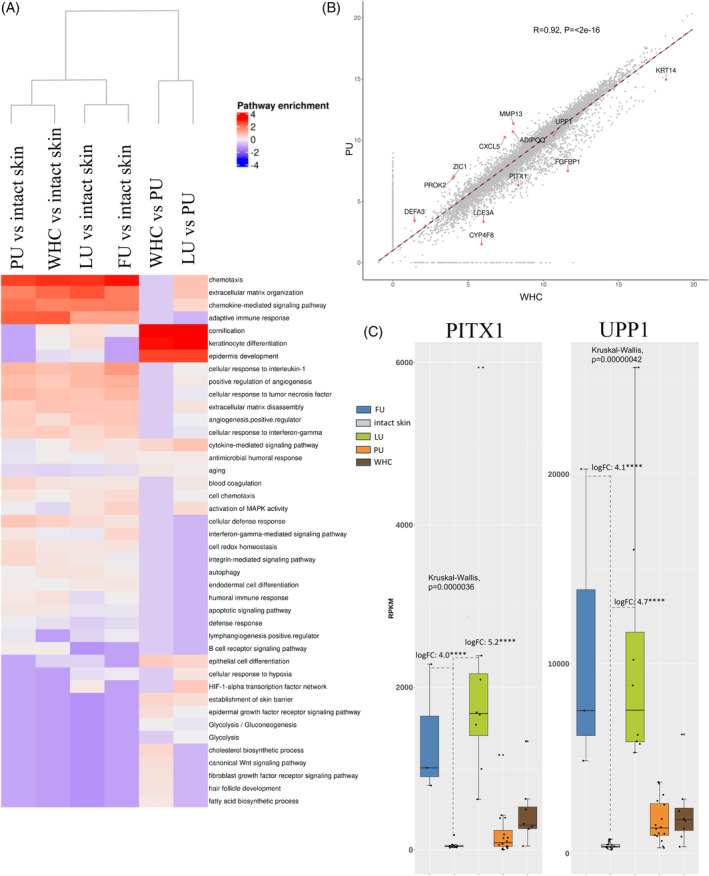
Molecular comparisons across different wound types. (A) Heatmap clustering of enriched regulated pathways detected for each wound type versus intact skin. The following comparisons are shown, PU, LU, FU and WHC versus intact skin, each, WHC versus PU and LU versus PU. Biosystem, Interpro, and Gene Ontology sets were used for gene set enrichment analysis (GSEA). Upregulated genes identified in pairwise comparisons were included in this analysis. The rows correspond to enriched pathways and columns correspond to comparisons between ulcer types. The column dendrogram was generated using the Euclidean distance metric based on the enrichment results for each comparison. The scaled, log10 transformed, enrichment results are colour‐coded (blue, downregulated enriched; white, not regulated; red, upregulated enriched; see legend, top right). The heatmap highlights the difference in enrichment results based on comparisons between ulcer types. (B) Scatter plot illustrating the correlations linking PUs and WHCs using global gene expression values (i.e., scaled RPKMs) with a Pearson correlation of 0.92 (p‐value, <2e−16) indicating a high degree of similarity. Genes highlighted here are those representing the most prominent differences between the two ulcer types. (C) Increased expression of PITX1 and UPP1 in LUs and FUs. Bar graphs illustrating the average expression (RPKM) of PITX1 and UPP1, across the various ulcer types. The bars are colour‐coded by ulcer type (legend top). Each dot represents the average gene expression for one donor. Error bars represent the standard deviation of gene expression across all donors. Expression levels of both PITX1 and UPP1 were elevated in LUs and FUs compared to other wound types and intact skin.
GSEA Pathway enrichment analysis highlighted critical inflammatory responses (e.g., chemotaxis, chemokines, adaptive immune responses) in all ulcer types compared with intact skin (Figure 8A; Supplementary Table 11 for upregulated pathways; Supplementary Table 12 for downregulated pathways). The full list of differentially expressed genes used as input for enrichment analysis is included in Supplementary Table 13. Although some differences were noted among the different ulcer types, our findings revealed an overall high degree of similarity (LU vs. WHC Pearson correlation: 0.86, LU vs. PU Pearson correlation: 0.86).
Importantly, we found that the transcriptome profile of WHCs was analogous to PUs, with a Pearson correlation of 0.92 (p‐value, <2e−16; Figure 8B). Of note, the similarities in gene expression patterns were consistent with the observed histological similarities reported for PUs and WHCs (Supplementary Table 2). Moreover, our analysis revealed two genes, paired‐like homeodomain 1 (PITX1) and uridine phosphorylase 1 (UPP1), that were specifically upregulated in LUs and FUs compared to the other wound types and intact skin (Figure 8C).
To assess the relevance of our identified chronic wound gene signature to wound healing, we compared the expression of genes in chronic wounds versus intact skin of our study to genes expressed in non‐healing versus healing venous leg ulcers (VLUs) from a previous study. 60
We found that 14 out of the top 15 upregulated genes in non‐healing ulcer edges compared to healing ulcer edges were also present in our chronic wound dataset. Notably, 11 of these genes (ALOX5AP, BCAT1, COL10A1, COL11A1, CYP1B1, RUNX2, SELE, SERPINA1, SFRP4, SULF1, TLR1) showed significant upregulation in the wound bed/border regions of our study, while one gene (HSD17B6) exhibited a slight increase. However, ITGBL1 remained unchanged, and CLU showed decreased expression. These findings strongly suggest non‐healing characteristics in chronic wounds. 60
Furthermore, among the top 15 upregulated genes in non‐healing ulcer beds compared to healing ulcers, 11 were identified in our chronic wound study. In our dataset, 5 genes (CYP1B1, FAM26F, PDGFRA, PI15, THBS1) exhibited significant upregulation, one gene (FKBP5) showed downregulation, and five genes (DPT, FBLN1, GLCCI1, PLTP, SAP30) did not show significant regulation. This indicates a good correlation between our study and the published signature, emphasizing non‐healing characteristics. 60
Additionally, we identified 9 out of 15 top downregulated genes in the non‐healing ulcer beds, with 5 genes (ADAMTS14, HBEGF, MPEG1, PCDH17, PPIF) showing significantly increased expression in the wound bed/border regions in our dataset, while 4 genes (NAPA, RBBP6, SASH1, TPSB2) remained unchanged. These results suggest a potential for healing within chronic wounds, aligning with our histological findings (Supplementary Table 2).
Furthermore, a comparison of our chronic wound versus intact skin data against a published signature of hard‐to‐heal chronic VLUs 61 identified 15 overlapping genes. Among them, the expression of 2 genes (CXCR2 [IL8RB]) and S100A7 [psoriasin]) was significantly increased, while 3 genes (CLDN5, IL22RA1, VEGF‐D) showed decreased expression in our dataset. Ten genes did not exhibit significant expression changes (AMFR, ARHGDIA [RhoGDI‐G], CTNNB1, KAI1, EMCN, IL17BR, WASL [N‐WASP], PAWR [PAR4], PTPKR, VEGF‐C) (Supplementary Figure 4B). Additionally, a subset of 8 genes (CLDN5, CXCR2, EMCN, IL17RB, IL22RA1, PTPKR, S100A7, VEGF‐C) from a 14‐gene signature (WD14) derived from the 25‐gene signature was identified in our study. This subset included 4 out of 5 genes with significant expression changes (CLDN5, CXCR2 [IL8RB], IL22RA1, S100A7). Although a direct comparison of all genes was challenging due to the inclusion of acute wound data in the published dataset, the observed imperfect correlation with wound healing potential suggests that the chronic wounds analysed in our study did not exhibit a strong healing capacity, as expected.
3.11. The gene C10orf99 encoding GPR15L was upregulated in LUs, FUs, PUs and WHCs
Our findings revealed the upregulation of C10orf99 in LUs, FUs, PUs and WHCs (Figure 9A). C10orf99, which encodes the protein G protein‐coupled receptor 15 ligand (GPR15L), is a recently discovered CC chemokine‐like ligand of the G protein‐coupled receptor, GPR15. GPR15L plays a key role in recruiting lymphocytes to mucosal tissues 62 and has been described as an antimicrobial peptide (AP‐57). 63 , 64 Levels of C10orf99 mRNA were higher within the wound borders than in the wound bed and intact skin tissue (Figure 9B). This finding is consistent with the observation that pathogen‐challenged epithelial cells or epithelial cell precursors secrete this peptide and thus promote keratinocyte proliferation. 63 , 64 Increased C10orf99 mRNA expression in FUs, LUs, WHCs, and, to a lesser extent, PUs correlated with increased levels of immunoreactive GPR15L in the epidermal regions of these wounds (Figure 9C). Furthermore, we detected increased expression of GPR15L in the thickened epidermal compartment at the wound border, at sites that overlapped with those expressing cytokeratin 14 (CK14), which is a marker for mitotically active epithelial cells 65 (Figure 9D). In summary, our studies revealed increased expression of C10orf99 at both mRNA and protein levels mainly in the border subregions of chronic wounds and WHCs, implying that this chemokine‐like ligand might contribute to wound pathology.
FIGURE 9.
Upregulated expression of C10orf99 in chronic wounds and WHC. Expression of C10orf99 (encoding GPR15L) was significantly increased in LUs, FUs, PUs and WHCs. (A) Levels of C10orf99 mRNA were aggregated from both wound border and bed subregions. Each bar represented the median level of C10orf99 expression for each ulcer type as shown in the legend, top right. Each dot represented the average C10orf99 expression for each subject. (B) Average C10orf99 levels of all ulcer types as shown in A were higher in the wound border compared to the wound bed. The graph highlighted the median expression levels of C10orf99 in each subregion, colour‐coded as shown in the legend on the top left. (C) Increased levels of GPR15L detected in chronic wounds and WHCs compared to intact skin. HE‐stained tissues representing each wound type and intact skin are shown on the right. Signal specificity was confirmed by the omission of the anti‐GPR15L antibody for each tissue type (middle panel). Tissue samples 1,2,3, intact skin (00099, visit 1); 4,5,6, LU (50 037, visit 1); 7,8,9, PU (50 051, visit 1); 10,11,12, FU (50 002, visit 1); 13,14,15, WHC (50 050, visit 2). Black scale bars, 500 μm; white scale bars, 100 μm. (D) Anti‐GPR15L staining of a full LU section revealed increased levels of immunoreactive protein in the wound border compared to the wound bed. Top, HE; bottom, IHC). Tissue samples 16,17, LU (50 037, visit 1). Black scale bar, 2500 μm; white scale bar, 1000 μm.
4. DISCUSSION
In this study, the molecular basis of chronic cutaneous wounds and WHCs was investigated. We performed an in‐depth comparative analysis of the common ulcer types, LU, FU and PU, and compared their attributes to those identified in WHCs and intact human skin. This comparative analysis was performed in defined wound subregions, bed, border, and peri‐wound. Histology, IHC, qIHC, and de novo mRNA expression analyses were applied to reveal novel insights involving a broad range of genes, molecular pathways, and biological processes. Our analysis of DE genes was used to define unique wound signatures that reflect the underlying pathology. We found that LUs, FUs and PUs showed common molecular pathway dysregulation, while each ulcer type also displayed unique pathological features. Interestingly, PUs and WHCs showed almost identical molecular dysregulation. These correlations may assist clinicians in choosing treatment options and support researchers in designing more precise preclinical and clinical studies in wound healing, tailored to specific wound types and disease states.
We employed bulk RNA‐Sseq to ascertain the mechanisms and pathways that differ across chronic wounds, WHCs and intact skin. As bulk RNA‐Seq is well‐suited to identify average global gene expression across multiple cell types in a tissue, it is not traditionally used to study cell types. 66 Conversely, single‐cell RNA sequencing (scRNA‐Seq), which is designed to measure the expression level of transcripts within individual cells and study distinct cell type composition in a tissue, is still confronted with a few limitations. 67 To overcome these challenges and bridge the full potential of bulk and single‐cell RNA‐Seq, methods such as gene deconvolution have been established to extrapolate the cellular composition using bulk RNA‐Seq data. 67 Using gene expression deconvolution, we could infer the presence of distinct cell types in wounds. As a result, we observed distinct gene markers indicative of wound subregion identity and wound type. Thus, our work has the potential to support wound diagnosis and treatment.
Excessive inflammation was common in all ulcer types. The inflammatory markers MPO, CD19, CD4, and CD8A were all upregulated in the wound bed, and, to a lesser extent, in the wound border compared to intact skin. Most notably, the chemokine CXCL1, a chemoattractant for several types of immune cells including neutrophils, 68 was persistently elevated in the bed subregion of chronic wounds and WHCs. This finding suggested a role for neutrophils in supporting the persistent wound pathology. Neutrophils have been described to play a role in several inflammatory diseases, including diabetes and nonalcoholic fatty liver disease, by releasing proteases, forming extracellular traps and activating other immune cells. 69 Our study suggested a similar role in chronic cutaneous wounds and WHCs.
The proinflammatory cytokine IL‐6 is primarily expressed in myeloid lineage cells and antigen‐presenting cells (APCs) in intact skin and wounds, but it is also expressed in mesenchymal stromal cells in wounds. 70 , 71 Pleiotropic IL‐6 plays a crucial role in acute inflammatory response during the wound healing response. 72 Its expression occurs in a biphasic manner, returning to baseline levels after the various phases of healing. Wounds, however, can become chronic if excessive proinflammatory cytokines, including IL‐6, persist during the healing process. 17 , 72 Our data showed that IL‐6 mRNA was elevated in all wound types analysed compared to intact skin (Figure 6F), thus demonstrating an exacerbated proinflammatory state that correlated with wound chronicity.
Impaired perfusion is another hallmark of chronic wounds that results from defective endothelial cell function and progenitor cell recruitment to the site of an injury. 15 Our findings included the significant upregulation of PDPN mRNA in chronic wounds and WHCs, particularly in the wound bed. Increased levels of PDPN may reflect elevations in lymphatic endothelial cell content, which would be consistent with increased levels of pan‐endothelial CD31 detected in these chronic wounds and WHCs by IHC. However, it is unlikely that increases in endothelial cells contribute to improved vessel functionality in chronic wounds and WHCs. The high fraction of occluded blood vessels detected by histopathological analysis strongly suggested that the function of these vessels was compromised. Blood vessel occlusion would result in a decrease in available blood supply, which may contribute to wound persistence.
Pericytes and dermal myofibroblasts express ACTA2. 18 Pericytes regulate the vessel tone and permeability of mature vessels, modulate angiogenesis and proliferation in the wound bed, and promote endothelial migration and cell survival, thus facilitating wound healing. 19 , 73 Pericyte contraction can exacerbate the occlusion of blood vessel lumen, reminiscent of our findings described in this study. In fact, the modulation of ACTA2 expression could occur via IL‐6, linking inflammation to endothelial cell proliferation and blood vessel formation. 74 Further study will be needed to determine whether increases in lymphatic endothelial cells are correlated with modulated lymphatic system function.
Keratinocytes represent a large cellular component of the skin and play critical roles in the initiation, maintenance, and completion of the wound healing process. 75 , 76 The results of our study revealed the downregulation of numerous keratins together with the upregulation of genes that may contribute to keratinocyte hyperproliferation and thus persistence of the non‐healing state. 77 KRT2, 10, 15 and 77 were downregulated within the wound bed and border compared with intact skin and peri‐wounds, which may explain the observed inability of keratinocytes to migrate and cover the wound bed. 78 , 79 , 80 Previous studies revealed increased expression of KRT15 in response to treatment with a BET bromodomain inhibitor, which is a regulator of keratinocyte plasticity that enhances skin wound healing in vivo. 81 , 82 Moreover, we found elevated expression of KRT6 and, to a lesser degree, KRT75 in the border subregions of chronic wounds and WHCs. KRT6 was reported to contribute to the hyperproliferation observed in these wounds, potentially due to a role in innate immune system function, and KRT75 was associated with aging keratinocytes and loss of stemness. 45 , 83 Our findings substantiated the decreased keratinocyte stem cell activity in wound borders. We also detected a prominent upregulation of the serine proteases TMPRSS11A and TMPRSS11D in wound border subregions. These mediators have been implicated both in the recruitment of neutrophils into the epidermis and the presence of p21‐induced cellular senescence and thus may have a negative impact on wound re‐epithelialization. 84 , 85 , 86
Pairwise comparisons between ulcer types resulted in enrichment of certain pathways in LUs, including those reflecting elevated levels of inflammatory activity, hypoxia, and reperfusion. These responses may induce an adaptive response that results in an increased rate of glycolysis in order to conserve energy or promote cell death. 12 , 87 , 88 , 89 By contrast, a similar evaluation of PUs revealed enrichment of pathways associated with the complement system and antimicrobial defence, which correlated with the presence of bacteria in some chronic wound tissue samples. 90 , 91 We identified two genes, UPP1 and PITX1, that were specifically upregulated in FUs and LUs. PITX1 encodes a transcription factor that has been implicated in reprograming keratinocytes towards wound resolution 76 and the transcription regulation of genes associated with the normal development and structure of the lower limbs. 92 Additional studies have identified a role for PITX1 in the reprogramming of keratinocytes to increase their migration potential and ensure wound closure. 76 UPP1, a gene described to play a role in DNA synthesis, has previously been shown to be increased in the wound border subregion compared to non‐ulcerated skin in venous stasis ulcers. 93 Our data suggests that UPP1 and PITX1 may contribute to wound pathology in FUs and LUs, two ulcer types with high clinical similarity.
C10orf99 was consistently elevated in the wound border subregions of LUs, FUs, PUs and WHCs, suggesting a global role in wound pathology. The protein product of this gene, the chemoattractant ligand GPR15L, is expressed in human epithelial cells that are in close contact with microbes in the environment, including the skin, where it serves as a regulator of effector T‐cell responses and cutaneous inflammation. 63 , 94 GPR15L is a secreted peptide with an antimicrobial function 64 and has been implicated in the pathogenesis of psoriasis due to its capacity to promote keratinocyte proliferation. 95 , 96 Our findings revealed increased levels of both GPR15L and its receptor that coincided with sustained wound inflammation. These observations correlated with the elevated immune response, chemotaxis, and chemokine‐mediated signalling pathways in our pathway enrichment analysis. Additional studies are necessary to determine whether GPR15L represents a novel wound biomarker and/or therapeutic target.
A main outcome of our study was that the transcriptomes and histopathological profiles of both PUs and WHCs are remarkably alike. PUs result from continuous pressure applied to the skin and are often accompanied by critical comorbidities. By contrast, WHCs are the result of post‐surgical wound dehiscence or complications of fractures. The gene expression profiles and histopathological similarities may be caused by conditions that involve strain, that is, tension or long‐term pressure, on the skin. These data emphasized the need to reduce tension on the skin following surgery. Moreover, we observed that the transcriptome profiles of wound bed samples were similar across the four wound types as shown by the low inter‐patient variability. The MDS plot displays the distinct profiles of the various wound subregions despite the differences in anatomical location and disease type. These findings expand on a previous study that assessed the expression profiles of non‐healing and non‐ulcerated edges of venous ulcers derived from 3 patients, as assessed by microarray analysis, with the aim to identify distinct molecular markers to guide surgical debridement. 93 Few studies have compared different ulcer types derived from various anatomical locations. We observed a high degree of similarity between the wound types analysed despite the underlying causes. This illustrates that chronic, non‐healing wounds most likely follow the same trajectory as described in a previous review focusing on inflammation in chronic wounds of venous, arterial, pressure and diabetic ulcers. 13
We hypothesized that changes in gene expression patterns in chronic wounds and WHCs are the result of an imbalance in cell recruitment and activation which can impair the progression of wound healing. For example, we detected a decrease in adipocytes in wound bed. Concordantly, adipose tissue‐derived stem cells have shown promise in the treatment of chronic wounds. 97 Neutrophil infiltration into the wound bed and absence of epidermal basal cells in wound bed and border subregions were revealed by gene expression deconvolution and histological analysis.
Machine learning classification methods enabled us to identify several genes with relative expression patterns that were characteristic of specific subregions of a wound. We identified wound bed‐specific elevated levels of CLEC4E, thus serving as a marker gene candidate for the wound bed. This C‐type lectin receptor has been recognized as a driver of allergic skin inflammation. 57 We further postulated that elevated levels of S100A7A identify wound border subregions. S100A7A encodes an antimicrobial peptide that has been identified as a potential drug target in patients diagnosed with acne vulgaris. 58 Finally, our findings revealed that TCF23 was expressed primarily in the peri‐wounds subregion. TCF23 encodes a progesterone‐induced transcription factor required for decidualization of human endometrial stromal cells. 59 We noted that the association between TCF23 and peri‐wound regions was comparatively low because of the high variability observed within this subregion. These molecular markers have the potential to facilitate reproducible subregion classification for wound assessment and wound care.
In summary, this study provides a comprehensive molecular map of common human ulcer types and WHCs based on the architecture and gene expression patterns within wounds. We have identified common pathways, cell types, and gene expression patterns associated with chronic wounds and WHCs. Notably, we have highlighted candidate markers such as PITX1, UPP1, C10orf99, and TMPRSS11A/D for assessing chronic wounds and WHCs. These molecular insights offer the potential to identify drug targets and wound biomarkers. The study underscores both the differences and similarities between ulcer types, with the potential to guide treatment decisions. Common pathway dysregulation across all wound types may suggest treatment options that transcend specific wound types, whilst wound type‐specific pathway alterations can assist clinicians in tailoring treatments. This research may aid in identifying wound healing biomarkers, inform the design of future preclinical and clinical studies, and guide debridement strategies for wound subregions. Expanding the study to larger cohorts of wound specimens in the future will enhance its statistical power and clinical relevance.
Overall, this study holds promise for improving the accurate classification, diagnosis, and personalized treatment of wound subregions and ulcer types, contributing to better patient care.
4.1. Limitations
This study has several limitations that may introduce bias. Ethical challenges in obtaining chronic wound biopsies directly from patients in situ necessitated the analysis of discarded wound samples from debridement surgeries. Additionally, the collection of LU samples was limited to one site, and the study analysed a restricted number of wound samples per type due to donor availability constraints. Despite these limitations, our findings hold significance, and valid conclusions can be drawn.
It is essential to note that our study was not designed as a longitudinal study, and therefore, we cannot establish healing trajectories. Furthermore, when comparing our chronic wound data to published gene signatures, it should be acknowledged that the focus in the published studies 60 , 61 was on VLUs, whereas our study encompassed LUs, FUs, PUs, and WHCs.
FUNDING INFORMATION
This study was funded by Novartis Biomedical Research, Novartis Pharma AG, Basel, Switzerland.
CONFLICT OF INTEREST STATEMENT
All authors contributing to this manuscript declare that they have nothing to disclose and do not have a conflict of interest. All authors except D.W., M.V.S., D.S., B.W. and R.W., were or are employees of Novartis Pharma AG.
Supporting information
Supplementary Figure 1. Abnormal connective tissue and angiogenesis in chronic wounds and WHCs. Histopathological evaluation (HE) of a representative chronic wound resection (PU, patient 50 025) is illustrated. (a) HE staining of a transversal section through a PU. Indicated by the long arrow is separation between wound border (right) and wound bed (left). A necrotic region is marked by arrow 1, a region with acanthosis by arrow 2. Granulation tissue is present in the dermal compartment. Insets B and C correspond to magnifications shown in B and C, respectively. (b) Magnified inset B from wound bed region. Evident are severe necrosis (ulceration), marked accumulation of inflammatory cells (neutrophils [arrow 3], macrophages, lymphocytes), granulation tissue formation, haemorrhages (arrow 4), blood vessels (angiogenesis) with activated endothelial cells (arrows 5), activated fibroblasts (arrows 6), and vascular fibrinoid necrosis and vasculitis (arrow 7). (c) Magnified inset C from wound border region. Manifestations of acanthosis (arrow 8) marked mixed inflammatory neutrophil, macrophage and lymphocyte infiltrates (arrow 9), haemorrhages (arrows 10), and granulation tissue formation (arrow 11) are visible.
Supplementary Figure 2. Quantification of protein markers in epidermal and dermal layers of peri‐wound regions or intact skin by qIHC analysis. An example is shown for Ki67 (intact skin), CD31 (WHC), KRT5 (WHC) and MPO (PU), each. CD31 and KRT5 expression was normalized to the regions of interest (ROIs) and represented as staining per area. MPO expression was normalized to total cell numbers. Ki67 expression was normalized to dermal ROIs or epidermal epithelial length. Left column, respective marker staining; right column, overlay image of marker‐positive nuclei or area. Bar lengths correspond to 50 μm for Ki67, 200 μm for CD31, 500 μm for KRT5, and 50 μm for MPO.
Supplementary Figure 3. Heatmap visualizing the most enriched pathways observed from downregulated genes attained through wound subregion versus intact skin comparisons.
Supplementary Figure 4. Comparison of chronic wounds versus intact skin gene expression data from our study to non‐healing versus healing and of hard‐to‐heal chronic VLUs gene signatures. (A) All top 15 upregulated and downregulated genes in the edge and bed of non‐healing VLUs listed in a published non‐healing versus healing signature dataset 60 (Tables 1–4) that were detected in our chronic wounds versus intact skin dataset are shown. (B) All 15 genes from the 25 gene signature 61 that were identified in our chronic wounds versus intact skin dataset are shown. Not identified: ARP2, BMP15, CAR1, CREB1l, IL17C, PEDF, RON, TEM4, TEM7R, WAVE2. *logFC ≤±2, **logFC ≤±3, ***logFC <±10 and adj p value <0.05; ns, non‐significant. The fold changes and significance values presented were derived from comparisons between chronic wounds (wound bed and border) and intact skin.
Supplementary Table 1. Patient source and characteristics of wound and intact skin samples analysed in this study. Columns, from left to right. Wound type: LU (leg ulcer), FU (foot ulcer), PU (pressure ulcer) and WHC (wound healing complications) resections, Intact (intact skin) samples served as controls. Patient: Patient identifier. Gender, F (female) and M (male). Race: Cauc. (Caucasian), Asian, NS (Not specified). Age: Age (years) of patient/donor at the time of resection. Disease: The following diseases were reported as ongoing at the time of resection; CV (cardiovascular disease), HT (hypertension), PD (pulmonary disease), RD (renal disease), SA (seasonal allergy), ND (neurological disease), SD (skin disease, other), DB (diabetes), GI (gastrointestinal disease), CA (cancer), OD (other disease), DA (drug allergy). Anatomical location: Body location from which wound and intact skin resections were performed. Vis: Visits V1 or V2, first or second patient visit during which resections were performed. Ulcer size: Dimension of ulcers, in cm; NA, not indicated. Ulcer stage: Stage 2, 3 or 4 according to scale; NA, not indicated. Wound age: Defined as the duration between original wound diagnosis (determined by patient history at the time of first presentation at the department of plastic surgery) and wound resection. Wound subregion: Numbers indicate quantities of sub‐sections derived from each Bed (wound bed), Border (wound border) and Peri (peri‐wound) section, respectively, that were used for RNA‐Seq. Intact: Numbers indicate quantities of intact skin specimen per donor resection used for RNA‐Seq. Stippled boxes denote not applicable.
Supplementary Table 2. Histopathological assessment of chronic wound, WHC and intact skin specimen. HE stained sections of human LU, FU, PU, WHC and intact skin specimen were examined by a pathology expert. Left column, skin specimen examined; middle column, wound subregions analysed; right column, histopathological findings. NA, not applicable.
Supplementary Table 3. Comparative gene expression analysis results of aggregated wound bed, border and peri‐wound samples compared to intact skin. Table of genes, fold changes and Benjamini‐Hochberg adjusted p‐values.
Supplementary Table 4. GSEA (pathway enrichment) results generated from the gene expression results from pairwise comparisons between wound subregions and intact skin. The full list of pairwise, wound subregion comparisons; differentially expressed genes with relevant statistics are reported.
Supplementary Table 5. Intra‐patient CV values: Generated to determine wound subregion and intact skin variability, to determine the reproducibility of samples derived from each patient. The table includes the patient identifier, wound subregion/intact skin, wound subregion mean RPKM, SD, variance and covariance values.
Supplementary Table 6. Inter‐patient CV values: Generated to determine wound subregion and intact skin agnostic of patient. The table includes mean RPKM, SD, variance, and covariance values for each wound subregion.
Supplementary Table 7. Scaled median values of gene‐signatures of cell types representing different areas of wound healing biology, including inflammation, epithelial or endothelial biology across subregions, generated using xCell.
Supplementary Table 8. Caret and randomForest R packages feature selection, intersection results. Gene lists shown were attained by comparing each subregion or intact skin against the others. Genes highlighted in blue are shown in Figure 7b as an exploration of the discriminatory power of the selected gene/transcript as examples of individual effect.
Supplementary Table 9. Hypergeometric testing for pathway enrichment results generated from comparing PU vs. LU gene expression upregulated values. The table includes gene set/pathway identifier, enrichment score, geneset/pathway name and the list of PU vs. LU DE genes associated with the named pathway/geneset.
Supplementary Table 10. Hypergeometric testing for pathway enrichment results generated from comparing PU vs. LU gene expression down‐regulated values. The table includes gene set/pathway identifier, enrichment score, gene set/pathway name and the list of PU vs. LU DE genes associated with the named pathway/gene set.
Supplementary Table 11. GSEA (pathway enrichment) Upregulated results generated from the gene expression results from pairwise comparisons between ulcer type and intact skin. The full list of pairwise, ulcer type comparisons‐differentially expressed genes with relevant statistics are reported.
Supplementary Table 12. GSEA Down‐regulated results generated from the gene expression results from pairwise comparisons between ulcer type and intact skin. The full list of pairwise, wound ulcer type comparisons‐differentially expressed genes with relevant statistics are reported.
Supplementary Table 13. Comparative gene expression analysis results generated from pairwise comparisons of the different ulcer types. The ulcer types comprise of aggregated wound bed, border and peri‐wound samples compared with intact skin. The results include the contrast, gene name, fold changes and adjusted p‐values.
ACKNOWLEDGEMENTS
We would like to thank Florian Nigsch, Michael Rebhan, Jan Tchorz, Anne Granger, Ieuan Clay, Yann Abraham, Diethilde Theil and Armelle Grevot for their scientific contributions and discussions. We thank Karine Bigot and Johann Müller for their technical assistance.
Richards SM, Gubser Keller C, Kreutzer R, et al. Molecular characterization of chronic cutaneous wounds reveals subregion‐ and wound type‐specific differential gene expression. Int Wound J. 2024;21(4):e14447. doi: 10.1111/iwj.14447
Shola Michelle Richards is the first author.
DATA AVAILABILITY STATEMENT
All processed or analysed data are available as supplementary files. Raw, anonymised data may be made available upon reasonable request due to ethical reasons.
REFERENCES
- 1. Eming SA, Martin P, Tomic‐Canic M. Wound repair and regeneration: mechanisms, signaling, and translation. Sci Transl Med. 2014;6:265sr6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Nunan R, Harding KG, Martin P. Clinical challenges of chronic wounds: searching for an optimal animal model to recapitulate their complexity. DMM Dis Model Mech. 2014;7:1205‐1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Järbrink K, Ni G, Sönnergren H, et al. The humanistic and economic burden of chronic wounds: a protocol for a systematic review. Syst Rev. 2017;6:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care. 2015;4:560‐582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sørensen LT, Hemmingsen U, Kallehave F, et al. Risk factors for tissue and wound complications in gastrointestinal surgery. Ann Surg. 2005;241:654‐658. doi: 10.1097/01.sla.0000157131.84130.12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kromuszczyńska J, Kołodziej Ł, Jurewicz A. Wound healing complications in patients with and without systemic diseases following hallux valgus surgery. PLoS One. 2018;13:e0197981. doi: 10.1371/journal.pone.0197981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Papanas N, Maltezos E. Becaplermin gel in the treatment of diabetic neuropathic foot ulcers. Clin Interv Aging. 2008;3:233‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zaulyanov L, Kirsner RS. A review of a bi‐layered living cell treatment (Apligraf) in the treatment of venous leg ulcers and diabetic foot ulcers. Clin Interv Aging. 2007;2:93‐98. doi: 10.2147/ciia.2007.2.1.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han G, Ceilley R. Chronic wound healing: a review of current management and treatments. Adv Ther. 2017;34:599‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Harries RL, Bosanquet DC, Harding KG. Wound bed preparation: TIME for an update. Int Wound J. 2016;13:8‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Demidova‐Rice TN, Hamblin MR, Herman IM. Acute and impaired wound healing: pathophysiology and current methods for drug delivery, part 1: normal and chronic wounds: biology, causes, and approaches to care. Adv Skin Wound Care. 2012;25:304‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grey JE, Enoch S, Harding KG. Wound assessment. BMJ. 2006;332:285‐288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhao R, Liang H, Clarke E, Jackson C, Xue M. Inflammation in chronic wounds. Int J Mol Sci. 2016;17:1‐14 (Multidisciplinary Digital Publishing Institute (MDPI). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage phenotypes regulate scar formation and chronic wound healing. Int J Mol Sci. 2017;18:1‐10. doi: 10.3390/ijms18071545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Demidova‐Rice TN, Durham JT, Herman IM. Wound healing angiogenesis: innovations and challenges in acute and chronic wound healing. Adv Wound Care. 2012;1:17‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Güç E, Briquez PS, Foretay D, et al. Local induction of lymphangiogenesis with engineered fibrin‐binding VEGF‐C promotes wound healing by increasing immune cell trafficking and matrix remodeling. Biomaterials. 2017;131:160‐175. [DOI] [PubMed] [Google Scholar]
- 17. Li Y, Zhao J, Yin Y, Li K, Zhang C, Zheng Y. The role of IL‐6 in fibrotic diseases: molecular and cellular mechanisms. Int J Biol Sci. 2022;18:5405‐5414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McAndrews KM, Miyake T, Ehsanipour EA, et al. Dermal αSMA + myofibroblasts orchestrate skin wound repair via β1 integrin and independent of type I collagen production. EMBO J. 2022;41:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thomas HM, Cowin AJ, Mills SJ. The importance of pericytes in healing: wounds and other pathologies. Int J Mol Sci. 2017;18:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rousselle P, Braye F, Dayan G. Re‐epithelialization of adult skin wounds: cellular mechanisms and therapeutic strategies. Adv Drug Deliv Rev. 2019;146:344‐365. (Elsevier B.V.). [DOI] [PubMed] [Google Scholar]
- 21. Alam H, Sehgal L, Kundu ST, Dalal SN, Vaidya MM. Novel function of keratins 5 and 14 in proliferation and differentiation of stratified epithelial cells. Mol Biol Cell. 2011;22:4068‐4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bruen KJ, Campbell CA, Schooler WG, et al. Real‐time monitoring of keratin 5 expression during burn re‐epithelialization. J Surg Res. 2004;120:12‐20. [DOI] [PubMed] [Google Scholar]
- 23. Jalava P, Kuopio T, Juntti‐Patinen L, Kotkansalo T, Kronqvist P, Collan Y. Ki67 immunohistochemistry: a valuable marker in prognostication but with a risk of misclassification: proliferation subgroups formed based on Ki67 immunoreactivity and standardized mitotic index. Histopathology. 2006;48:674‐682. [DOI] [PubMed] [Google Scholar]
- 24. Patel GK, Wilson CH, Harding KG, Finlay AY, Bowden PE. Numerous keratinocyte subtypes involved in wound re‐epithelialization. J Invest Dermatol. 2006;126:497‐502. doi: 10.1038/sj.jid.5700101 [DOI] [PubMed] [Google Scholar]
- 25. Schuierer S, Roma G. The exon quantification pipeline (EQP): a comprehensive approach to the quantification of gene, exon and junction expression from RNA‐seq data. Nucleic Acids Res. 2016;44:e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Law CW, Chen Y, Shi W, Smyth GK. Voom: precision weights unlock linear model analysis tools for RNA‐seq read counts. Genome Biol. 2014;15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu R, Holik AZ, Su S, et al. Why weight? Modelling sample and observational level variability improves power in RNA‐seq analyses. Nucleic Acids Res. 2015;43:e97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 2017;18:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Modern Applied Statistics with S | W.N. Venables | Springer. https://www.springer.com/de/book/9780387954578
- 30. Friedman J, Hastie T, Tibshirani R. Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33:1‐22. [PMC free article] [PubMed] [Google Scholar]
- 31. Breiman L, Cutler A. Breiman and Cutler's random forests for classification and regression. Package ‘randomForest’ 29. 2012. doi: 10.5244/C.22.54 [DOI]
- 32. Fuchs E, Horsley V. More than one way to skin. Genes Dev. 2008;22:976‐985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Harsha A, Stojadinovic O, Brem H, et al. ADAM12: a potential target for the treatment of chronic wounds. J Mol Med (Berl). 2008;86:961‐969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kennedy MW, Chalamalasetty RB, Thomas S, Garriock RJ, Jailwala P, Yamaguchi TP. Sp5 and Sp8 recruit β‐catenin and Tcf1‐Lef1 to select enhancers to activate Wnt target gene transcription. Proc Natl Acad Sci U S A. 2016;113:3545‐3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Poggi L, Casarosa S, Carl M. An eye on the Wnt inhibitory factor Wif1. Front Cell Dev Biol. 2018;6:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Onoufriadis A, Shoemark A, Schmidts M, et al. Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects. Hum Mol Genet. 2014;23:3362‐3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Toriseva M, Kahari VM. Proteinases in cutaneous wound healing. Cell Mol Life Sci. 2009;66:203‐224. doi: 10.1007/s00018-008-8388-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Whitney JD. Overview: acute and chronic wounds. Nurs Clin North Am. 2005;40:191‐205. [DOI] [PubMed] [Google Scholar]
- 39. Caley MP, Martins VLCC, O'toole EA. Metalloproteinases and wound healing. Adv Wound Care. 2015;4:225‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abdoli A, Maspi N, Ghaffarifar F. Wound healing in cutaneous leishmaniasis: a double edged sword of IL‐10 and TGF‐β. Comp Immunol Microbiol Infect Dis. 2017;51:15‐26. [DOI] [PubMed] [Google Scholar]
- 41. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. 2018;9:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pruenster M, Vogl T, Roth J, Sperandio M. S100A8/A9: from basic science to clinical application. Pharmacol Ther. 2016;167:120‐131. [DOI] [PubMed] [Google Scholar]
- 43. Johnson NC, Dillard ME, Baluk P, et al. Lymphatic endothelial cell identity is reversible and its maintenance requires Prox1 activity. Genes Dev. 2008;22:3282‐3291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gordon EJ, Gale NW, Harvey NL. Expression of the hyaluronan receptor LYVE‐1 is not restricted to the lymphatic vasculature; LYVE‐1 is also expressed on embryonic blood vessels. Dev Dyn. 2008;237:1901‐1909. [DOI] [PubMed] [Google Scholar]
- 45. Zhang X, Yin M, Zhang LJ. Keratin 6, 16 and 17‐critical barrier alarmin molecules in skin wounds and psoriasis. Cell. 2019;8:1‐14. doi: 10.3390/cells8080807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Callies LLK, Tadeo D, Simper J, Bugge TH, Szabo R. Iterative, multiplexed CRISPR‐mediated gene editing for functional analysis of complex protease gene clusters. J Biol Chem. 2019;294:15987‐15996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang Z, Hu Y, Yan R, et al. The transmembrane serine protease HAT‐like 4 is important for epidermal barrier function to prevent body fluid loss. Sci Rep. 2017;7:45262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin Z‐Q, Kondo T, Ishida Y, Takayasu T, Mukaida N. Essential involvement of IL‐6 in the skin wound‐healing process as evidenced by delayed wound healing in IL‐6‐deficient mice. J Leukoc Biol. 2003;73:713‐721. [DOI] [PubMed] [Google Scholar]
- 49. Rockey DC, Weymouth N, Shi Z. Smooth muscle α actin (Acta2) and myofibroblast function during hepatic wound healing. PLoS One. 2013;8:e77166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Rosales C, Lowell CA, Schnoor M, Uribe‐Querol E. Neutrophils: their role in innate and adaptive immunity 2017. J Immunol Res. 2017;2017:1‐2. doi: 10.1155/2017/9748345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound healing: a cellular perspective. Physiol Rev. 2019;99:665‐706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Novoselova N, Della Beffa C, Wang J, Li J, Pessler F, Klawonn F. HUM calculator and HUM package for R: easy‐to‐use software tools for multicategory receiver operating characteristic analysis. Bioinformatics. 2014;30:1635‐1636. [DOI] [PubMed] [Google Scholar]
- 53. Vehtari A, Gelman A, Gabry J. Practical Bayesian model evaluation using leave‐one‐out cross‐validation and WAIC. Stat Comput. 2015;27:1413‐1432. [Google Scholar]
- 54. Vehtari A, Gelman A, Gabry J. Practical Bayesian model evaluation using leave‐One‐out cross‐validation and WAIC *. 2016.
- 55. Cheng H, Garrick DJ, Fernando RL. Efficient strategies for leave‐one‐out cross validation for genomic best linear unbiased prediction. J Anim Sci Biotechnol. 2017;8:1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mu Y, Huang X, Liu R, et al. ACPT gene is inactivated in mammalian lineages that lack enamel or teeth. PeerJ. 2021;9:e10219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Kostarnoy AV, Gancheva PG, Lepenies B, et al. Receptor Mincle promotes skin allergies and is capable of recognizing cholesterol sulfate. Proc Natl Acad Sci U S A. 2017;114:E2758‐E2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Al‐Sudany NK, Mohammed NH, Alrifai SB. Downregulation of S100a7a antimicrobial peptide in acne vulgaris patients after isotretinoin therapy. Dermatol Ther. 2019;32:e13136. [DOI] [PubMed] [Google Scholar]
- 59. Kommagani R, Szwarc MM, Kovanci E, et al. A murine uterine transcriptome, responsive to steroid receptor coactivator‐2, reveals transcription factor 23 as essential for decidualization of human endometrial stromal cells. Biol Reprod. 2014;90:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Charles CA, Tomic‐Canic M, Vincek V, et al. A gene signature of nonhealing venous ulcers: potential diagnostic markers. J Am Acad Dermatol. 2008;59:758‐771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bosanquet DC, Sanders AJ, Ruge F, et al. Development and validation of a gene expression test to identify hard‐to‐heal chronic venous leg ulcers. Br J Surg. 2019;106:1035‐1042. [DOI] [PubMed] [Google Scholar]
- 62. Lahl K, Sweere J, Pan J, Butcher E. Orphan chemoattractant receptor GPR15 mediates dendritic epidermal T‐cell recruitment to the skin. Eur J Immunol. 2014;44:2577‐2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ocón B, Pan J, Dinh TT, et al. A mucosal and cutaneous chemokine ligand for the lymphocyte chemoattractant receptor GPR15. Front Immunol. 2017;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Yang M, Tang M, Ma X, et al. AP‐57/C10orf99 is a new type of mutifunctional antimicrobial peptide. Biochem Biophys Res Commun. 2015;457:347‐352. [DOI] [PubMed] [Google Scholar]
- 65. Coulombe PA, Kopan R, Fuchs E. Expression of keratin K14 in the epidermis and hair‐follicle: insights into complex programs of differentiation. J Cell Biol. 1989;109:2295‐2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li X, Wang CY. From bulk, single‐cell to spatial RNA sequencing. Int J Oral Sci. 2021;13:1‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kuksin M, Morel D, Aglave M, et al. Applications of single‐cell and bulk RNA sequencing in onco‐immunology. Eur J Cancer. 2021;149:193‐210. [DOI] [PubMed] [Google Scholar]
- 68. Ridiandries A, Tan JTM, Bursill CA. The role of chemokines in wound healing. Int J Mol Sci. 2018;19:1‐20. doi: 10.3390/ijms19103217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Herrero‐Cervera A, Soehnlein O, Kenne E. Neutrophils in chronic inflammatory diseases. Cell Mol Immunol. 2022;19:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Tie R, Li H, Cai S, et al. Interleukin‐6 signaling regulates hematopoietic stem cell emergence. Exp Mol Med. 2019;51:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Liu C, Xu Y, Lu Y, et al. Mesenchymal stromal cells pretreated with proinflammatory cytokines enhance skin wound healing via IL‐6‐dependent M2 polarization. Stem Cell Res Ther. 2022;13:1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Xiao T, Yan Z, Xiao S, Xia Y. Proinflammatory cytokines regulate epidermal stem cells in wound epithelialization. Stem Cell Res Ther. 2020;11:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Bodnar RJ, Satish L, Yates CC, Wells A. Pericytes: a newly recognized player in wound healing. Wound Repair Regen. 2016;24:204‐214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Gallucci RM, Lee EG, Tomasek JJ. Il‐6 modulates alpha‐smooth muscle Actin expression in dermal fibroblasts from IL‐6‐deficient mice. J Invest Dermatol. 2006;126:561‐568. [DOI] [PubMed] [Google Scholar]
- 75. Pastar I, Stojadinovic O, Tomic‐Canic M. Role of keratinocytes in healing of chronic wounds. Surg Technol Int. 2008;17:105‐112. [PubMed] [Google Scholar]
- 76. Iglesias‐Bartolome R, Uchiyama A, Molinolo AA, et al. Transcriptional signature primes human oral mucosa for rapid wound healing. Sci Transl Med. 2018;10:1‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wang F, Chen S, Liu HB, Parent CA, Coulombe PA. Keratin 6 regulates collective keratinocyte migration by altering cell–cell and cell–matrix adhesion. J Cell Biol. 2018;217:4314‐4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Collin C, Moll R, Kubicka S, Ouhayoun JP, Franke WW. Characterization of human cytokeratin 2, an epidermal cytoskeletal protein synthesized late during differentiation. Exp Cell Res. 1992;202:132‐141. [DOI] [PubMed] [Google Scholar]
- 79. Bloor BK, Tidman N, Leigh IM, et al. Expression of keratin K2e in cutaneous and oral lesions: association with keratinocyte activation, proliferation, and keratinization. Am J Pathol. 2003;162:963‐975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Nuutila K, Siltanen A, Peura M, et al. Human skin transcriptome during superficial cutaneous wound healing. Wound Repair Regen. 2012;20:830‐839. [DOI] [PubMed] [Google Scholar]
- 81. Stojadinovic O, Pastar I, Nusbaum AG, Vukelic S, Krzyzanowska A, Tomic‐Canic M. Deregulation of epidermal stem cell niche contributes to pathogenesis of nonhealing venous ulcers. Wound Repair Regen. 2014;22:220‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Schutzius G, Kolter C, Bergling S, et al. BET bromodomain inhibitors regulate keratinocyte plasticity. Nat Chem Biol. 2021;17:280‐290. doi: 10.1038/s41589-020-00716-z [DOI] [PubMed] [Google Scholar]
- 83. Mercado N, Schutzius G, Kolter C, et al. IRF2 is a master regulator of human keratinocyte stem cell fate. Nat Commun. 2019;10:4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Miki M, Yasuoka S, Tsutsumi R, et al. Human airway trypsin‐like protease enhances interleukin‐8 synthesis in bronchial epithelial cells by activating protease‐activated receptor 2. Arch Biochem Biophys. 2019;664:167‐173. [DOI] [PubMed] [Google Scholar]
- 85. Zhao N, Huang GE, Guo L, Lu SH. ECRG1, a novel candidate of tumor suppressor gene in the esophageal carcinoma, triggers a senescent program in NIH3T3 cells. Exp Biol Med. 2006;231:84‐90. [DOI] [PubMed] [Google Scholar]
- 86. Fernandez C, Burgos A, Morales D, et al. TMPRSS11a is a novel age‐altered, tissue specific regulator of migration and wound healing. FASEB J. 2021;35:e21597. [DOI] [PubMed] [Google Scholar]
- 87. Chen WYJJ, Rogers AA. Recent insights into the causes of chronic leg ulceration in venous diseases and implications on other types of chronic wounds. Wound Repair Regen. 2007;15:434‐449. doi: 10.1111/j.1524-475X.2007.00250.x [DOI] [PubMed] [Google Scholar]
- 88. Grey JE, Enoch S, Harding KG. Venous and arterial leg ulcers. Br Med J. 2006;332:347‐350. doi: 10.1136/bmj.332.7537.347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ligi D, Mosti G, Croce L, Raffetto JD, Mannello F. Chronic venous disease—Part I: inflammatory biomarkers in wound healing. Biochim Biophys Acta. 2016;1862:1964‐1974. [DOI] [PubMed] [Google Scholar]
- 90. Grey JE, Harding KG, Enoch S, Harding KG. Pressure ulcers. BMJ. 2006;332:472‐475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Bhattacharya S, Mishra R, Bhattacharya S. Pressure ulcers: current understanding and newer modalities of treatment. Indian J Plast Surg. 2015;48:4‐16 (Medknow Publications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Petit F, Sears KE, Ahituv N. Limb development: a paradigm of gene regulation. Nat Rev Genet. 2017;18:245‐258. doi: 10.1038/nrg.2016.167 [DOI] [PubMed] [Google Scholar]
- 93. Brem H, Stojadinovic O, Diegelmann RF, et al. Molecular markers in patients with chronic wounds to guide surgical debridement. Mol Med. 2007;13:30‐39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Suply T, Hannedouche S, Carte N, et al. A natural ligand for the orphan receptor GPR15 modulates lymphocyte recruitment to epithelia. Sci Signal. 2017;10. [DOI] [PubMed] [Google Scholar]
- 95. Chen C, Wu N, Duan Q, et al. C10orf99 contributes to the development of psoriasis by promoting the proliferation of keratinocytes. Sci Rep. 2018;8:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sezin T, Kempen L, Meyne LM, Mousavi S, Zillikens D, Sadik CD. GPR15 is not critically involved in the regulation of murine psoriasiform dermatitis. J Dermatol Sci. 2019;94:196‐204. [DOI] [PubMed] [Google Scholar]
- 97. Li Q, Guo Y, Chen F, Liu J, Jin P. Stromal cell‐derived factor‐1 promotes human adipose tissue‐derived stem cell survival and chronic wound healing. Exp Ther Med. 2016;12:45‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Abnormal connective tissue and angiogenesis in chronic wounds and WHCs. Histopathological evaluation (HE) of a representative chronic wound resection (PU, patient 50 025) is illustrated. (a) HE staining of a transversal section through a PU. Indicated by the long arrow is separation between wound border (right) and wound bed (left). A necrotic region is marked by arrow 1, a region with acanthosis by arrow 2. Granulation tissue is present in the dermal compartment. Insets B and C correspond to magnifications shown in B and C, respectively. (b) Magnified inset B from wound bed region. Evident are severe necrosis (ulceration), marked accumulation of inflammatory cells (neutrophils [arrow 3], macrophages, lymphocytes), granulation tissue formation, haemorrhages (arrow 4), blood vessels (angiogenesis) with activated endothelial cells (arrows 5), activated fibroblasts (arrows 6), and vascular fibrinoid necrosis and vasculitis (arrow 7). (c) Magnified inset C from wound border region. Manifestations of acanthosis (arrow 8) marked mixed inflammatory neutrophil, macrophage and lymphocyte infiltrates (arrow 9), haemorrhages (arrows 10), and granulation tissue formation (arrow 11) are visible.
Supplementary Figure 2. Quantification of protein markers in epidermal and dermal layers of peri‐wound regions or intact skin by qIHC analysis. An example is shown for Ki67 (intact skin), CD31 (WHC), KRT5 (WHC) and MPO (PU), each. CD31 and KRT5 expression was normalized to the regions of interest (ROIs) and represented as staining per area. MPO expression was normalized to total cell numbers. Ki67 expression was normalized to dermal ROIs or epidermal epithelial length. Left column, respective marker staining; right column, overlay image of marker‐positive nuclei or area. Bar lengths correspond to 50 μm for Ki67, 200 μm for CD31, 500 μm for KRT5, and 50 μm for MPO.
Supplementary Figure 3. Heatmap visualizing the most enriched pathways observed from downregulated genes attained through wound subregion versus intact skin comparisons.
Supplementary Figure 4. Comparison of chronic wounds versus intact skin gene expression data from our study to non‐healing versus healing and of hard‐to‐heal chronic VLUs gene signatures. (A) All top 15 upregulated and downregulated genes in the edge and bed of non‐healing VLUs listed in a published non‐healing versus healing signature dataset 60 (Tables 1–4) that were detected in our chronic wounds versus intact skin dataset are shown. (B) All 15 genes from the 25 gene signature 61 that were identified in our chronic wounds versus intact skin dataset are shown. Not identified: ARP2, BMP15, CAR1, CREB1l, IL17C, PEDF, RON, TEM4, TEM7R, WAVE2. *logFC ≤±2, **logFC ≤±3, ***logFC <±10 and adj p value <0.05; ns, non‐significant. The fold changes and significance values presented were derived from comparisons between chronic wounds (wound bed and border) and intact skin.
Supplementary Table 1. Patient source and characteristics of wound and intact skin samples analysed in this study. Columns, from left to right. Wound type: LU (leg ulcer), FU (foot ulcer), PU (pressure ulcer) and WHC (wound healing complications) resections, Intact (intact skin) samples served as controls. Patient: Patient identifier. Gender, F (female) and M (male). Race: Cauc. (Caucasian), Asian, NS (Not specified). Age: Age (years) of patient/donor at the time of resection. Disease: The following diseases were reported as ongoing at the time of resection; CV (cardiovascular disease), HT (hypertension), PD (pulmonary disease), RD (renal disease), SA (seasonal allergy), ND (neurological disease), SD (skin disease, other), DB (diabetes), GI (gastrointestinal disease), CA (cancer), OD (other disease), DA (drug allergy). Anatomical location: Body location from which wound and intact skin resections were performed. Vis: Visits V1 or V2, first or second patient visit during which resections were performed. Ulcer size: Dimension of ulcers, in cm; NA, not indicated. Ulcer stage: Stage 2, 3 or 4 according to scale; NA, not indicated. Wound age: Defined as the duration between original wound diagnosis (determined by patient history at the time of first presentation at the department of plastic surgery) and wound resection. Wound subregion: Numbers indicate quantities of sub‐sections derived from each Bed (wound bed), Border (wound border) and Peri (peri‐wound) section, respectively, that were used for RNA‐Seq. Intact: Numbers indicate quantities of intact skin specimen per donor resection used for RNA‐Seq. Stippled boxes denote not applicable.
Supplementary Table 2. Histopathological assessment of chronic wound, WHC and intact skin specimen. HE stained sections of human LU, FU, PU, WHC and intact skin specimen were examined by a pathology expert. Left column, skin specimen examined; middle column, wound subregions analysed; right column, histopathological findings. NA, not applicable.
Supplementary Table 3. Comparative gene expression analysis results of aggregated wound bed, border and peri‐wound samples compared to intact skin. Table of genes, fold changes and Benjamini‐Hochberg adjusted p‐values.
Supplementary Table 4. GSEA (pathway enrichment) results generated from the gene expression results from pairwise comparisons between wound subregions and intact skin. The full list of pairwise, wound subregion comparisons; differentially expressed genes with relevant statistics are reported.
Supplementary Table 5. Intra‐patient CV values: Generated to determine wound subregion and intact skin variability, to determine the reproducibility of samples derived from each patient. The table includes the patient identifier, wound subregion/intact skin, wound subregion mean RPKM, SD, variance and covariance values.
Supplementary Table 6. Inter‐patient CV values: Generated to determine wound subregion and intact skin agnostic of patient. The table includes mean RPKM, SD, variance, and covariance values for each wound subregion.
Supplementary Table 7. Scaled median values of gene‐signatures of cell types representing different areas of wound healing biology, including inflammation, epithelial or endothelial biology across subregions, generated using xCell.
Supplementary Table 8. Caret and randomForest R packages feature selection, intersection results. Gene lists shown were attained by comparing each subregion or intact skin against the others. Genes highlighted in blue are shown in Figure 7b as an exploration of the discriminatory power of the selected gene/transcript as examples of individual effect.
Supplementary Table 9. Hypergeometric testing for pathway enrichment results generated from comparing PU vs. LU gene expression upregulated values. The table includes gene set/pathway identifier, enrichment score, geneset/pathway name and the list of PU vs. LU DE genes associated with the named pathway/geneset.
Supplementary Table 10. Hypergeometric testing for pathway enrichment results generated from comparing PU vs. LU gene expression down‐regulated values. The table includes gene set/pathway identifier, enrichment score, gene set/pathway name and the list of PU vs. LU DE genes associated with the named pathway/gene set.
Supplementary Table 11. GSEA (pathway enrichment) Upregulated results generated from the gene expression results from pairwise comparisons between ulcer type and intact skin. The full list of pairwise, ulcer type comparisons‐differentially expressed genes with relevant statistics are reported.
Supplementary Table 12. GSEA Down‐regulated results generated from the gene expression results from pairwise comparisons between ulcer type and intact skin. The full list of pairwise, wound ulcer type comparisons‐differentially expressed genes with relevant statistics are reported.
Supplementary Table 13. Comparative gene expression analysis results generated from pairwise comparisons of the different ulcer types. The ulcer types comprise of aggregated wound bed, border and peri‐wound samples compared with intact skin. The results include the contrast, gene name, fold changes and adjusted p‐values.
Data Availability Statement
All processed or analysed data are available as supplementary files. Raw, anonymised data may be made available upon reasonable request due to ethical reasons.