

Abstract
Interleukin-12 (IL-12) and interleukin-23 (IL-23), which belong to the IL-12 family of cytokines, have a key role in intestinal homeostasis and inflammation and are implicated in the pathogenesis of inflammatory bowel disease. Upon their secretion by antigen-presenting cells, they exert both pro-inflammatory and anti-inflammatory receptor-mediated effects. An increased understanding of these biological effects, particularly the pro-inflammatory effects mediated by IL-12 and IL-23, has led to the development of monoclonal antibodies that target a subunit common to IL-12 and IL-23 (p40; targeted by ustekinumab and briakinumab), or the IL-23-specific subunit (p19; targeted by risankizumab, guselkumab, brazikumab and mirikizumab). This Review provides a summary of the biology of the IL-12 family cytokines IL-12 and IL-23, discusses the role of these cytokines in intestinal homeostasis and inflammation, and highlights IL-12- and IL-23-directed drug development for the treatment of Crohn’s disease and ulcerative colitis.
Introduction
Inflammatory bowel disease (IBD) is a complex gastrointestinal disorder arising from an inappropriate immune reaction against environmental factors, including gut microbiota, in genetically susceptible individuals1. A growing understanding of the underlying disease pathogenesis has resulted in the development of agents that target inflammatory components of the disease process, which has revolutionized IBD care2. For over a decade, the principal target of monoclonal antibodies in IBD was tumour necrosis factor (TNF). Despite the revolutionary nature of TNF inhibitors, 10–30% of patients in clinical trials and practice do not respond, and 23–46% require dose intensification beyond 12 weeks of therapy, a surrogate for loss-of-response to these agents3. Agents with novel modes of action were therefore required for treatment of IBD. The discovery of the interleukin-23 (IL-23) receptor (encoded by IL23R) as an IBD susceptibility locus4 and of the importance of the IL-12 family of cytokines in intestinal inflammation led to the development of biological agents that target IL-12 and/or IL-23 (ref. 5). This Review focuses on the IL-12 family cytokines IL-12 and IL-23, discussing their role in intestinal homeostasis and inflammation — including in IBD — and highlighting drug development related to the IL-12 and IL-23 axes.
IL-12 and IL-23 cytokine biology
IL-12 is comprised of two protein subunits, p40 and p35, linked by a disulfide bond. Initially discovered over 30 years ago as a protein released by a human lymphoblastoid cell line and capable of activating interferon-γ (IFNγ) production by natural killer (NK) cells and T cells, a clear role for IL-12 in driving T helper 1 (TH1) cell responses was subsequently established6–9.
IL-23 was identified a decade after the discovery of IL-12 (ref. 10). Phenotypic differences between mice deficient for either the IL-12 p40 or p35 subunit, specifically in their ability to clear bacterial infections10,11, led to the hypothesis that p40 might pair with subunits other than p35 to exert its antimicrobial effects. A sequence database search for homologues to p35 identified p19 as a protein that associated with p40 and formed the unique cytokine IL-23 (ref. 10). In contrast to IL-12, IL-23 was first described as having an effect on memory T cells but not on naive T cells10. Subsequently, IL-23 was shown to promote expansion and maintenance, but not differentiation, of T helper 17 (TH17) cells12. These effector T cells, which are characterized by production of IL-17A, IL-17F, IL-22, IL-26, TNF and in some cases IFNγ, are involved in immune responses to bacteria and fungi and have been recognized as key mediators in autoimmunity13.
Both IL-12 (p35 and p40) and IL-23 (p19 and p40) are produced by antigen-presenting cells (APCs), including dendritic cells and macro phages, in response to early innate signals14,15 (Figs. 1 and 2). IL-12 production by dendritic cells is also driven by the CD40 ligand on T cells16 and is modulated by cytokines such as IFNγ, IL-18, granulocyte–macrophage colony-stimulating factor (GM-CSF), IL-4 and by IL-12 itself15,17,18. Signals that also promote IL-12 production (including CD40 stimulation), certain cytokines, and bacteria or viruses can lead to production of IL-23 by APCs19–21. Studies support the ability of intestinal epithelial cells (IECs) to produce monomeric IL-23p19 under inflammatory and mitogenic signals22. Secretion of IL-23 by IECs has also been reported in a murine model of colitis23.
Fig. 1 |. Cellular sources, target cells, signalling and downstream effects of IL-12.
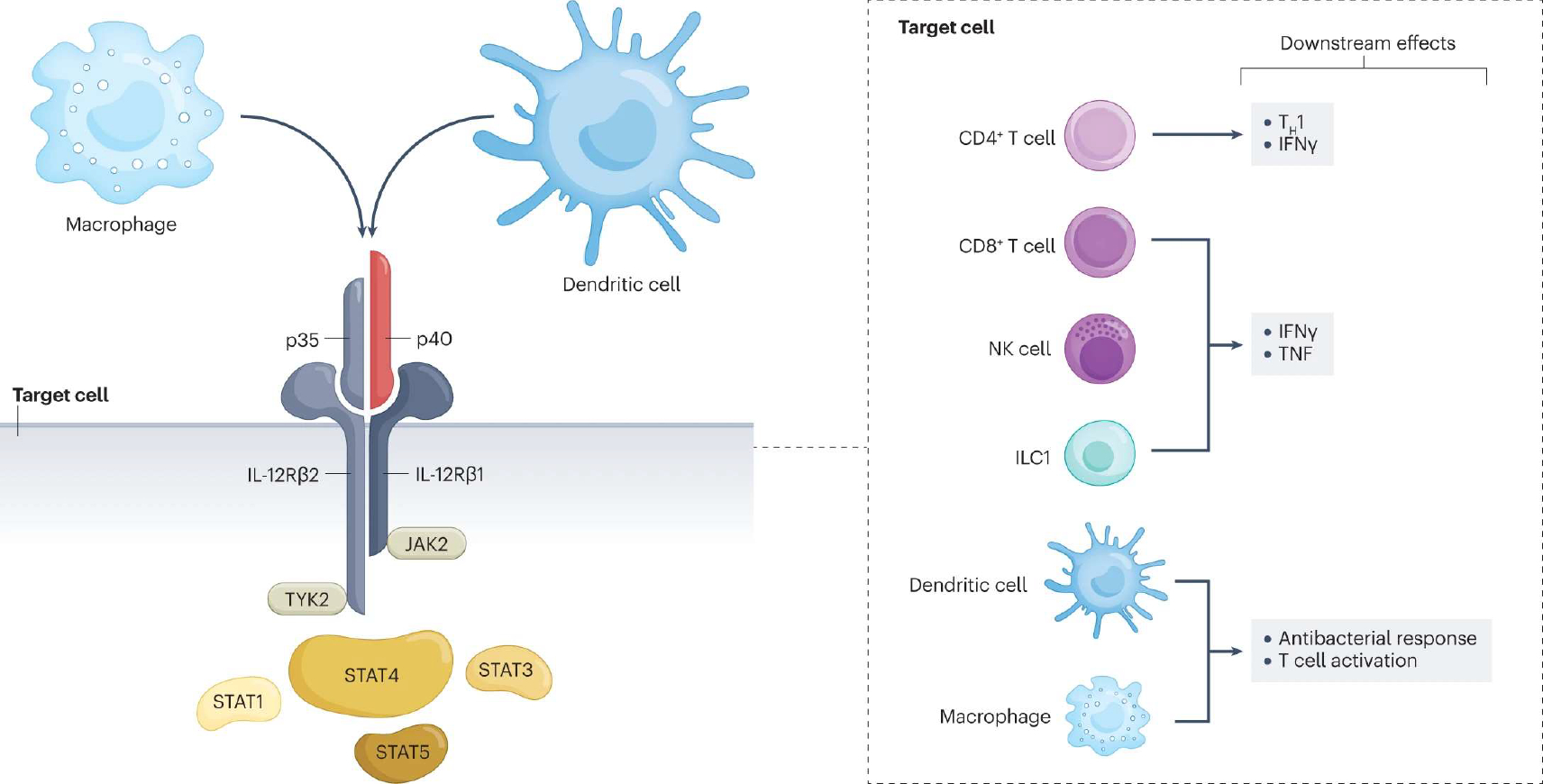
Interleukin-12 (IL-12) is a heterodimeric cytokine comprised of p40 (also part of the IL-23 dimer) and p35 subunits. It is produced by macrophages and dendritic cells. The receptor for IL-12 is composed of two different subunits, IL-12Rβ1 and IL-12Rβ2, which undergo conformational changes upon binding to IL-12 and bring into proximity two cytoplasmic tyrosine kinases, the Janus kinase 2 (JAK2) and tyrosine kinase 2 (TYK2), which are essential for downstream signalling of IL-12. JAKs trans/autophosphorylate each other and the receptor. Receptor phosphorylation enables binding and phosphorylation of signal transducers and activators of transcription (STATs), mainly STAT4. Phosphorylated STATs dimerize and translocate to the nucleus, where they regulate gene transcription. Different cell types express the IL-12 receptor on their membrane and are therefore targets for IL-12. Depending on the cell target, IL-12 exerts a variety of downstream effects. In naive CD4+ T cells, STAT4 signalling together with T-bet induce differentiation towards the T helper 1 (TH1) cell phenotype and production of interferon-γ (IFNγ). In CD8+ T cells, natural killer (NK) cells and group 1 innate lymphoid cells (ILC1s), IL-12 induces IFNγ and tumour necrosis factor (TNF) release. Finally, IL-12 signalling on dendritic cells and macrophages amplifies the antibacterial response and T cell activation.
Fig. 2 |. Cellular sources, target cells, signalling and downstream effects of IL-23.
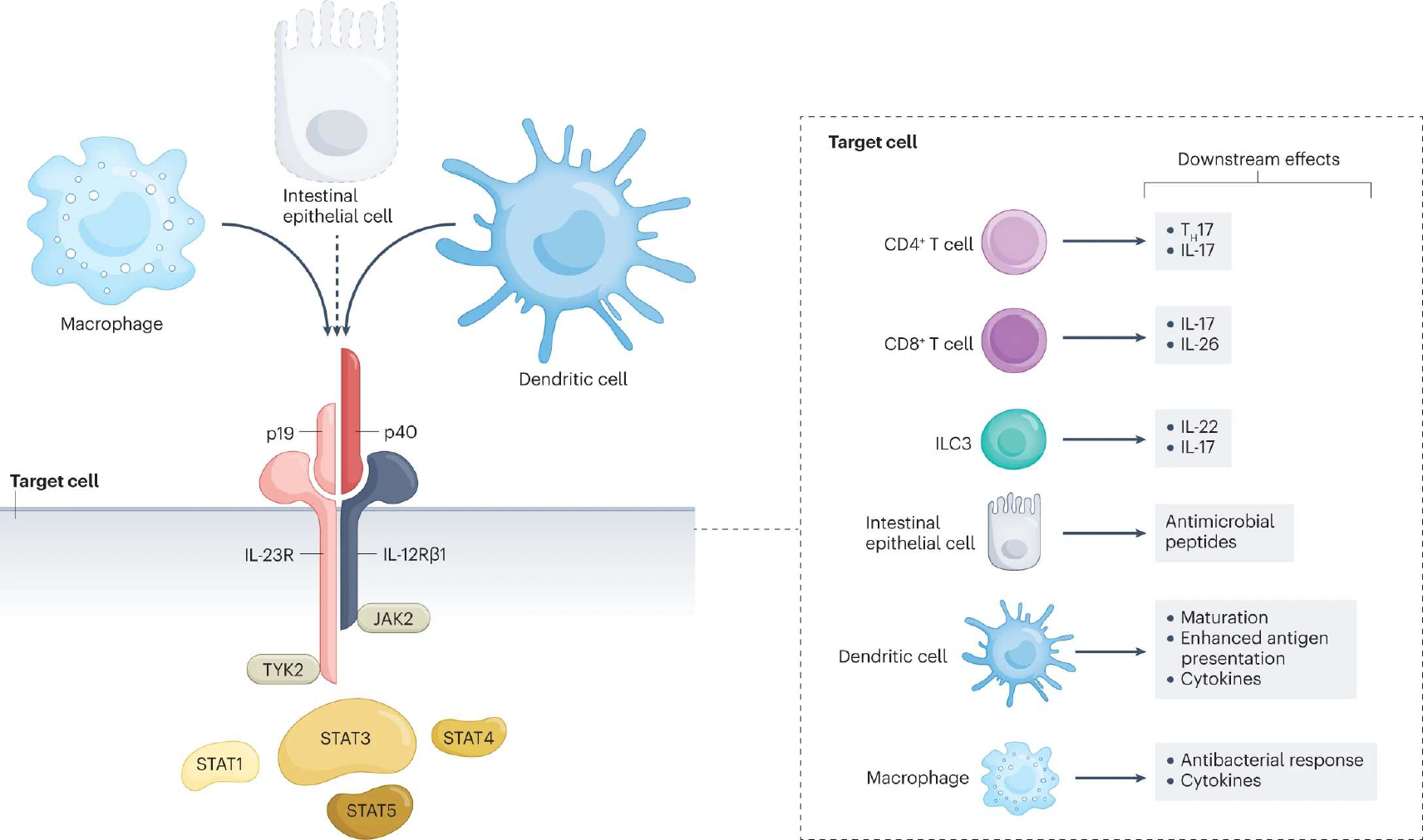
Interleukin-23 (IL-23) is a heterodimeric cytokine composed of p40 (also part of the IL-12 dimer) and p19 subunits. Like IL-12, and in response to similar stimuli (microbial signals, cytokines and co-stimulatory T cell ligands), IL-23 is produced by macrophages and dendritic cells. Some publications5–7 also show production of IL-23 by intestinal epithelial cells (dashed line, will need further confirmatory evidence). The IL-23 receptor is comprised of the IL-12Rβ1 and the IL-23R chains. Binding of IL-23 induces a conformational change that brings two cytoplasmic tyrosine kinases, Janus kinase 2 (JAK2) and tyrosine kinase 2 (TYK2), into proximity. JAKs trans/autophosphorylate each other and the receptor. Receptor phosphorylation enables binding and phosphorylation of signal transducers and activators of transcription (STATs), predominantly the STAT3 transcription factor. Phosphorylated STATs dimerize and translocate to the nucleus, where they regulate gene transcription. The IL-23 receptor is expressed on numerous cell types, including lymphoid cells, specifically CD4+ T helper 17 (TH17) and CD8+ T cells8. Innate lymphoid cells (ILCs), such as ILC3, also express the receptor and readily produce IL-22 and IL-17 cytokines in response to IL-23 stimulation. In IECs, IL-23 induces the expression of antimicrobial peptides. Furthermore, dendritic cells and macrophages respond to IL-23 stimulation by secreting a variety of cytokines. In addition, when the IL-23 receptor is engaged, dendritic cells demonstrate enhanced maturation and antigen presentation, and macrophages show an increased antibacterial response.
The biological effects of IL-12 and IL-23 are receptor-mediated. The IL-12 receptor (IL-12R) is comprised of two chains, IL-12Rβ1 and IL-12Rβ2, that bind to p40 and p35, respectively24 (Fig. 1). In addition to its well known expression on lymphoid cells, IL-12R is also expressed by macrophages and dendritic cells25,26. Whereas the IL-23 p40 subunit also binds to IL-12Rβ1, the p19 subunit associates with the IL-23R chain to drive intracellular signalling mediated by IL-23 (ref. 27) (Fig. 2). In addition to lymphoid cells (specifically, T cells and type 3 innate lymphoid cells (ILC3s)), IL-23R is also found on monocytes, macrophages and dendritic cells27,28. The IL-23R chain is not expressed by naive T cells, and IL-23, in contrast to IL-12, preferentially activates memory T cells10,29. In T cells, IL-23R is expressed in response to induction of the retinoid-related orphan receptor-γt (RORγt)-dependent transcriptional activity30.
Both IL-12 and IL-23 receptors are associated with and activate tyrosine kinase 2 (TYK2) and Janus kinase 2 (JAK2); however, receptor-mediated signalling occurs via distinct pathways that lead to independent immunological responses24,31 (Figs. 1 and 2). In vitro studies using cells derived from humans and mice have demonstrated that the signal transducer and activator of transcription (STAT) family members STAT1, STAT3, STAT4 and STAT5 can all be phosphorylated in response to binding of IL-12 and IL-23 to their receptors, although STAT4 is predominantly phosphorylated in response to IL-12 receptor binding32, and IL-23 responses are mediated predominantly by STAT3-dependent signalling27,33. However, the relative contribution of the different STAT family members in IL-12- and IL-23-mediated signalling can vary in different cell types34.
IL-12, IL-23 and gut homeostasis
Constitutive IL-12/IL-23 p40 promoter activity and IL-23p19 protein expression has been detected in the terminal ileum of unchallenged mice, suggesting that the small intestine is a critical, functionally relevant location for these cytokines under homeostatic conditions35. Production of IL-12 and IL-23 by APCs seems to be mainly triggered by Toll-like receptor signalling in the antigen-rich reservoir of the gut34,36,37. Stimulation of IL-12R on APCs promotes T cell activation26,38 and antibacterial responses39,40, and IL-23 has been shown to promote cytokine secretion and antimicrobial responses in human macrophages34,41 (Table 1). Hence, both IL-12 and IL-23 can shape immune responses independent of their activation of IFNγ and IL-17 responses42.
Table 1 |.
Role of IL-23 in mucosal homeostasis and disease
| Cell type | Pathway and/ or signalling molecules | Effect |
|---|---|---|
| Innate immune system | ||
| Type 3 innate lymphoid cell | IL-23R/STAT5 | Cell activation Cytokine production (IL-17A, IL-22) Antimicrobial effects |
| Granulocyte | IL-23R | Cell activation Pro-inflammatory cytokine production |
| Natural killer/intraepithelial lymphocytes | IL-23R | Cytotoxicity Cell activation Pro-inflammatory cytokine production |
| Adaptive immune system | ||
| Treg Cells, Teff cells, TH17 cells |
IL-23R RORγt (Teff cells) |
Suppression of Treg cells Proliferation and expansion of TH17 cells Production of IL-6 and TH17 cytokines |
| Intestinal epithelial cells | IL-22 Regenerating gene family proteins | Increased barrier function Antimicrobial effects |
Interleukin-23 (IL-23) exerts effects on both innate and adaptive immune cells. In type 3 innate lymphoid cells, granulocytes, intraepithelial lymphocytes and natural killer cells, IL-23 induces cell activation and cytokine production. Additional effects of IL-23 are related to adaptive immune cells, such as regulatory T (Treg) cells, and effector T (Teff) cells, such as T helper 17 (TH17) cells. In this context, IL-23 suppresses Treg cells, whereas it activates TH17 cells. Furthermore, this cytokine regulates barrier function and production of antimicrobial peptides in intestinal epithelial cells. IL-23R, IL-23 receptor; RORγt, retinoid-related orphan receptor-γt; STAT, signal transducer and activator of transcription.
TH17 cells, a subset of T cells, are also present in the small intestine under homeostatic conditions43, where they have been shown in mouse and human studies to undergo IL-23-mediated activation44–46, produce IL-17A and IL-17F, and exert inflammatory effects upon antigen challenge. This cell subset can also have an immunosuppressive function under homeostatic conditions, and in this case are referred to as regulatory TH17 cells43. IL-17A exerts direct effects on gut IECs by controlling the production of tight junction proteins and molecules that limit permeability of the epithelial barrier and preserve gut barrier function in vivo47. In mice, loss of the IL-17R adaptor protein, ACT1, suppressed these IL-17A protective effects, and γδ T cells were identified as a source of IL-23-independent IL-17A production in this setting47. In addition, mice deficient for IL-17 (Il17−/−) and those with a conditional deletion of the IL-17R adaptor protein ACT1 in epithelial cells had diminished colonic barrier function48. These findings might at least partially explain the lack of efficacy of the anti-IL-17 antibody secukinumab in patients with Crohn’s disease49.
IL-23 stimulation of colonic ILC3 cells activates STAT5 and the production of IL-22, a cytokine that is crucial for STAT3 activation in IECs and that has been associated with mucosal healing in mouse models of colitis50–52. Although ILC3 cells can produce TH17 cytokines, such as IL-17A and IL-22 (refs. 53,54), and belong to the lymphoid lineage, they lack CD3+ T cell receptors on their surface and are therefore activated via T cell-receptor-independent mechanisms. Gut-resident CX3CR1+ macrophages have been implicated as important sources of IL-23-dependent ILC3 activation, and IL-23 can cooperate with other cytokines to activate ILC3s55–57. Conditional knockout mice carrying a deletion in IL23R specifically in IECs have attenuated mucosal IL-22 mRNA levels and exhibit dysbiosis52.
Although a role for IL-23 in mucosal homeostasis is evident, a functional role for IL-12, which is a well established inducer of mucosal TH1 cell responses and IFNγ production, in the healthy intestine is less well understood. Interestingly, mice deficient in IL-12 p35 or p40 do not display spontaneous gut pathology, which potentially supports the concept that IL-12 is not essential to maintain homeostatic conditions in the intestine58,59.
IL-12, IL-23 and IBD
Multiple different genes within the IL-23–TH17 pathway have been associated with altered risk for both Crohn’s disease and ulcerative colitis. The most notable of these associations is a variant of IL23R that reduces risk of development of IBD by approximately two-fold in individuals of European ancestry4. The IL23RR381Q protective variant results in a loss-of-function of IL-23R, with reduced STAT3 signalling and TH17-mediated cell responses upon exposure to IL-23 (refs. 60–62). Studies have demonstrated that in human macrophages, autocrine and/or paracrine IL-23 promotes secretion of multiple other inflammatory cytokines (TNF, IL-1β, IL-6, IL-8 and IL-10)34, thereby providing another mechanism through which IL-23 might be contributing to intestinal inflammation. The IBD protective IL23RR381Q variant leads to a reduction of these cytokines in macrophages34. Interestingly, the capacity of IL-23 to induce responses in human macrophages requires dynamic recycling of IL-23R34, which might have implications when designing certain therapeutic approaches. There are multiple splice forms of IL-23R, and some63 but not all studies34 have shown that the IL23RR381Q variant results in increased expression of soluble IL-23R, which could then function as a decoy to reduce IL-23 responses. In addition, some in vitro studies have shown reduced protein stability of the IL23RR381Q variant64, whereas another has not34. Multiple other common and rare IBD genetic associations in the IL23R gene region have been identified4,65,66. In some cases, studies have gone on to examine mechanisms by which specific non-coding variants modulate IL-23R expression67. Notably, there is substantial heterogeneity in the IL23R gene across ancestries (for example, East Asian compared with European)68–70. Variants in regions containing genes that are associated with the IL-23 and TH17 cell pathways and that confer altered risk for IBD have also been identified, including variants in the cytokine subunits (IL12B (the p40 subunit)), signalling pathways (JAK2, TYK2, STAT3/STAT5, STAT4), transcription factors (RORC) and cell surface molecules (CCR6)66,71. Taken together, these genetic associations further highlight the importance of the IL-23–TH17 pathway in IBD pathogenesis.
As previously mentioned, the IL-12 and IL-23 pathways are important in host defence. Consistent with the loss-of-function phenotype, macrophages from IL23RR381Q IBD-protected carriers demonstrate less effective microbial clearance relative to macrophages from wild-type homozygous (IL23RR381/R381) carriers41. Although a higher overall risk of infection in IL23RR381Q carriers has not yet been demonstrated, one study reported an increased frequency of active pulmonary tuberculosis72, suggesting that IL23RR381Q carriers might be at greater risk of infection in regions with endemic tuberculosis. Another study found increased intestinal microbial diversity and richness, and increased frequency of select phylotypes, in IL23RR381Q carriers73, which might also theoretically contribute mechanistically to protection from development of Crohn’s disease.
The benefits of either blockade or deletion of the shared IL-12 and IL-23 p40 subunit in reducing intestinal inflammation have been demonstrated in multiple models of experimental colitis (for example, IL10−/−, adoptive T cell transfer, and 2,4,6-trinitrobenzenesulfonic acid)37,74–77. IL-12 expression is elevated in lamina propria mononuclear cells from patients with Crohn’s disease, and IL-12 can then promote lamina propria T cell inflammatory responses78,79. The results of p40 blockade in experimental animal studies have been corroborated in human studies. Reduced intestinal lamina propria inflammatory cytokines (IL-12p70, IL-23, IFNγ and TNF), including those expressed by T cells (IL-17 and IL-6) were found in patients with Crohn’s disease treated with antibodies targeting the shared p40 subunit compared with patients treated with placebo80, and a concentration-dependent reduction in the frequency of circulating T follicular helper cells has also been observed in patients with Crohn’s disease treated with antibodies to the shared p40 subunit81.
A clear role for IL-23 in promoting intestinal inflammation has also been demonstrated. Transgenic expression of IL-23p19 in mice results in severe intestinal inflammation82. On the other hand, blockade or deletion of either IL-23p19 (and not IL-12p35) or IL-23R in experimental models of colitis reduces inflammation37,76,83,84. It is presumed that this occurs because of a reduction in pathogenic TH17 cells. However, IL-23 can also promote inflammation through TH17-cell-independent mechanisms, including reducing regulatory T cells and increasing ILC3 responses85,86. Environmental factors (for example, food dyes) can also contribute to IL-23-driven intestinal inflammation87,88. In vivo studies have shown that while IL-12/IL-23p40 and IL-23p19 can lead to similar degrees of local intestinal inflammation in experimental models of colitis, IL-12/IL-23p40 may preferentially contribute to systemic immune activation48,76,77. Another study has shown that these two cytokines can act in a temporally sequential biphasic manner. Eftychi et al. demonstrated in mice that develop spontaneous colitis triggered by death of IECs that IL-12 promoted inflammation in response to intestinal epithelial barrier damage and exposure to bacteria in the early stages of disease, and that IL-23-dependent responses drove more chronic pathology as the mice aged89.
As previously described, IL-23 can mediate different roles in distinct cell subsets in the intestine. As such, IL-23 can mediate both inflammatory (via pathogenic TH17 cells, innate lymphoid cells and macrophages)85 and protective (epithelial cells, antimicrobial pathways and downregulation of TH1 cells)52,90–92 effects, which might have implications for different responses among patients to therapeutic blockade of this pathway. An increased understanding of the various effects of IL-23 and IL-12 (for example, cell-specific regulation, kinetics and immunological context) might ultimately enable improved design of therapeutic targeting.
IL-12- and IL-23-directed therapy for IBD
Drugs targeting IL-12/IL-23 currently approved or in clinical development for the treatment of IBD are fully human monoclonal antibodies directed against either the p40 subunit (ustekinumab and briakinumab) common to IL-12 and IL-23, or the IL-23-specific p19 subunit (risankizumab, guselkumab, brazikumab and mirikizumab). Designs and outcomes for trials representing the latest phase of clinical development for these agents are detailed in Supplementary Tables 1 and 2 and briefly summarized below. Recently published subgroup and/or post hoc analyses are also described below.
Moderate-to-severe Crohn’s disease
Ustekinumab.
Ustekinumab was approved for the treatment of moderate-to-severe Crohn’s disease on the basis of the UNITI induction (UNITI-1 and UNITI-2) and maintenance (IM-UNITI) trials93 (Supplementary Table 1). A substudy of the UNITI trials evaluated the efficacy of ustekinumab in inducing and maintaining endoscopic healing94. The primary outcome, mean change from baseline in the Simple Endoscopic Score for Crohn’s Disease (SES-CD), was significantly greater at week 8 in patients treated with ustekinumab (n = 155) compared with patients treated with placebo (n = 97) (−2.8 versus −0.7, P = 0.012). Although the mean change in SES-CD was numerically greater at week 44 with ustekinumab, it was not significantly greater than placebo (−2.5 versus −1.9, P = 0.176)94. Histological outcomes were also assessed using data from the UNITI programme. A significant reduction in the mean Global Histologic Disease Activity Score (GHAS) from baseline to week 8 was observed with ustekinumab (10.4 ± 7.0 to 7.1 ± 5.9, P < 0.001), but not with placebo (from 9.2 ± 6.4 to 7.8 ± 6.2, P = 0.193); the reduction in overall GHAS among those receiving ustekinumab and placebo was similar at week 44 (ref. 95).
The phase III SEAVUE trial96 was a randomized head-to-head trial of ustekinumab (~6 mg kg−1 intravenously at baseline followed by 90 mg subcutaneously every 8 weeks; n = 191) or adalimumab (160 mg and 80 mg subcutaneously at baseline and week 2, respectively, followed by 40 mg subcutaneously every 2 weeks; n = 195) for patients naive to treatment with biologics. The primary endpoint, clinical remission (Crohn’s Disease Activity Index (CDAI) score <150) at week 52, was achieved by 65% of patients treated with ustekinumab and 61% of patients treated with adalimumab (difference = 4.0%; 95% CI −5.5% to 13.5%; P = 0.417)96. Endoscopic response and remission rates were not statistically different at week 52 (Supplementary Table 1).
The efficacy of ustekinumab using either a treat-to-target or standard-of-care (dose frequency based on European Union summary of product characteristics (every 8 or 12 weeks)) strategy was explored in the phase IIIb STARDUST study. Patients who achieved a 70-point reduction in baseline CDAI score at week 16 with ustekinumab induction therapy (single 6 mg kg−1 intravenous dose at week 0 followed by 90 mg subcutaneously at week 8) were randomized into the treat-to-target (n = 220) or standard-of-care (n = 221) groups. Similar proportions of patients achieved the primary outcome of endoscopic response (≥50% reduction in SES-CD score from baseline) at week 48 using a treat-to-target or standard-of-care strategy (37.7% versus 29.9%, P = 0.0933; non-responder imputation)97.
Risankizumab.
Risankizumab was approved for the treatment of moderate-to-severe Crohn’s disease by the US Food and Drug Administration (FDA) on the basis of the results of three pivotal phase III trials (ADVANCE98 and MOTIVATE98 (induction) and FORTIFY99 (maintenance)). Treatment with risankizumab (600 mg or 1,200 mg intravenously) at weeks 0, 4 and 8 was significantly superior to placebo for the co-primary outcome of clinical remission (CDAI <150 for sites in the USA; average daily stool frequency ≤2.8 and abdominal pain ≤1 and not worse than baseline for sites in locations other than the USA) and endoscopic response (decrease in SES-CD >50% from baseline or ≥2-point reduction from baseline in SES-CD score for patients with isolated ileal disease and baseline SES-CD ≥4) at week 12 in both the ADVANCE (CDAI remission: 600 mg, 45% (152/336); 1,200 mg, 42% (141/339); placebo, 25% (43/175); stool frequency and abdominal pain score clinical remission: 600 mg, 43% (146/336); 1,200 mg, 41% (139/339); placebo, 22% (38/175); and endoscopic response: 600 mg, 40% (135/336); 1,200 mg, 32% (109/339); placebo, 12% (21/175)) and MOTIVATE induction studies (CDAI remission: 600 mg, 42% (80/191), 1,200 mg, 40% (77/191); placebo, 20% (37/187); stool frequency and abdominal pain score clinical remission: 600 mg, 35% (66/191); 1,200 mg, 40% (76/191); placebo, 19% (36/187); and endoscopic response: 600 mg, 29% (55/191); 1,200 mg, 34% (65/191); placebo, 11% (21/187))98, with no apparent benefit to higher doses observed with short-term treatment (Supplementary Table 1). Patients who responded to 12 weeks of risankizumab induction therapy in ADVANCE and MOTIVATE were re-randomized to subcutaneous treatment with 180 mg or 360 mg risankizumab, or placebo, every 8 weeks99. The proportion of patients achieving the co-primary endpoint of clinical remission and endoscopic response (both outcomes as defined previously) at 52 weeks was significantly higher compared with placebo in the risankizumab 360-mg group (CDAI remission: 52% (74/141) versus 41% (67/164) for placebo; stool frequency and abdominal pain score clinical remission: 52% (73/141) versus 40% (65/164) for placebo; and endoscopic response: 47% (66/141) versus 22% (36/164) for placebo), whereas risankizumab 180 mg was significantly superior to placebo for the outcomes of CDAI < 150 remission (55% (87/157) versus 41% (67/164)) and endoscopic response (47% (74/157) versus 22% (36/164))99 (Supplementary Table 1). Importantly, both risankizumab doses were significantly superior to placebo for the outcomes of endoscopic remission (SES-CD ≤4 and ≥2 point reduction versus baseline with no individual subscore greater than 1) and deep remission (CDAI < 150 and endoscopic remission) at week 52 (ref. 99) (Supplementary Table 1).
Endoscopic outcomes in response to risankizumab induction and maintenance treatment were numerically higher in patients who were intolerant to or had an inadequate response to conventional therapies (aminosalicylates, oral locally acting steroids, systemic steroids (prednisone or equivalent), and immunomodulators) compared with those who were intolerant to or had an inadequate response to approved biologic therapies (infliximab, adalimumab, certolizumab pegol, natalizumab, vedolizumab and/or ustekinumab)100. Risankizumab was also significantly more effective than placebo in inducing and maintaining endoscopic outcomes in the overall patient population. Specifically, patients receiving 600 mg intravenous risankizumab every 4 weeks in the ADVANCE and MOTIVATE trials (n = 527) had higher rates of endoscopic response (36.1% versus 11.6%), endoscopic remission (22.4% versus 6.6%), and ulcer-free endoscopy (SES-CD ulcerated surface subscore of 0 in patients with SES-CD ulcerated surface subscore ≥1 at baseline; 18.5% versus 5.8%) compared with placebo (n = 362) at week 12 (P < 0.001 for all comparisons)100. Similar trends were observed in a subanalysis of the FORTIFY maintenance trial: patients receiving 360 mg subcutaneous risankizumab (n = 141) every 8 weeks had higher rates of endoscopic response (46.5% versus 22.0%), endoscopic remission (39.1% versus 12.8%), ulcer-free endoscopy (30.5% versus 10.5%), and deep remission (29.1% versus 10.4%) compared with placebo (n = 164) at week 52 (P < 0.001 for all comparisons)100.
In subgroup analyses of the phase III trials examining the relationship between disease location and response to risankizumab treatment, patients with colonic and ileal colonic Crohn’s disease treated with risankizumab achieved significantly higher rates of the co-primary and composite endpoints of clinical remission and endoscopic response, and endoscopic remission at weeks 12 and 52, as well as sustained (week 12 and week 52) endoscopic remission at week 52 compared with patients treated with placebo disease101. Patients with ileal disease had lower rates relative to patients with colonic and ileal colonic disease for nearly all outcomes assessed at both weeks 12 and 52, although analyses at the latter timepoint were limited by a small number of patients with ileal disease (n = 15).
Guselkumab.
In the phase II GALAXI 1 trial, patients were randomly assigned to either intravenous treatment with 200 mg (n = 61), 600 mg (n = 63) or 1,200 mg (n = 61) guselkumab at weeks 0, 4 and 8; 6 mg kg−1 intravenous ustekinumab at week 0 and 90 mg subcutaneous ustekinumab at week 8 (n = 63); or intravenous placebo (n = 61). At week 12, significantly greater reductions from baseline CDAI (the primary endpoint) were reported in all guselkumab dose groups compared with the placebo group (Supplementary Table 1). Significant differences at week 12 were also observed for all guselkumab dose groups for the outcomes of clinical response, clinical remission, Patient-Reported Outcome-2 (PRO-2) remission, clinical biomarker response and endoscopic response102,103. In a treat-straight-through maintenance study design, patients randomized during induction to guselkumab 200 mg intravenously received 100 mg subcutaneously every 8 weeks, while those who received induction with either 600 mg or 1,200 mg intravenously received 200 mg subcutaneously every 4 weeks. Rates of CDAI clinical remission at week 48 ranged from 57.4% to 73.0%104.
Brazikumab.
Patients were randomized to treatment with 700 mg intravenous brazikumab (n = 59) or placebo (n = 60) at weeks 0 and 4, followed by open-label 210-mg subcutaneous brazikumab (n = 52) every 4 weeks from week 12 onwards in a phase IIa study. The primary end point, which was clinical response at week 8, was achieved in 49.2% of patients treated with brazikumab compared with 26.7% of patients treated with placebo (absolute difference 22.5%; 95% CI 5.6–39.5%, P = 0.010)105.
Mirikizumab.
Patients in the phase II SERENITY trial were randomized to treatment with 200 mg (n = 31), 600 mg (n = 32) or 1,000 mg (n = 64) intravenous mirikizumab, or placebo (n = 64), at weeks 0, 4 and 8. At week 12, rates of endoscopic response were significantly greater in the 600-mg and 1,000-mg mirikizumab groups compared with placebo106 (Supplementary Table 1). Patients who received mirikizumab and achieved ≥1 point improvement in SES-CD score at week 12 were re-randomized to either continue their intravenous treatment assignment (IV-C) or to 300 mg mirikizumab subcutaneously every 4 weeks. Endoscopic response rates at week 52 were 58.5% and 58.7% in the IV-C and subcutaneous groups, respectively. Furthermore, of those with endoscopic response at week 12, 69.6% and 66.7% in the IV-C and subcutaneous groups, respectively, achieved endoscopic response at week 52 (ref. 107).
Briakinumab.
In a phase IIb study, patients were randomized to treatment with 400 mg (n = 45) or 700 mg (n = 139) intravenous briakinumab or placebo (n = 46) at 0, 4 and 8 weeks (Supplementary Table 1). At week 6, there was no significant difference in the proportion of patients achieving clinical remission in either the 400-mg or 700-mg briakinumab treatment groups compared with the placebo group. This trial was terminated by the sponsor owing to lack of efficacy108.
A discussion on potential future positioning and use of these agents in particular patient populations based on current available data appears later.
Moderate-to-severe ulcerative colitis
Ustekinumab.
In the phase III UNIFI trial, patients were randomized to receive induction treatment with 130 mg (n = 320) or ~6 mg kg−1 (n = 322) intravenous ustekinumab (n = 322) or placebo109. At week 8, the proportion of patients who achieved clinical remission (total score of ≤2 on the Mayo Score and no subscore >1 on any of the four Mayo Score components) was significantly greater in the 130 mg and ~6 mg kg−1 ustekinumab groups compared with placebo (Supplementary Table 2). At week 8, clinical responders to ustekinumab were re-randomized to receive 90 mg subcutaneous ustekinumab every 8 or 12 weeks or placebo. The proportion of patients receiving ustekinumab every 8 or 12 weeks in clinical remission at week 44 was significantly greater compared with placebo109. Endoscopic and histological outcomes analysed in the UNIFI trial also significantly favoured ustekinumab treatment over placebo during both the induction and maintenance phases of the trial and are described in detail in Supplementary Table 2. In long-term follow-up of patients who were week 8 or 16 responders (n = 428) and received ustekinumab maintenance therapy, those who achieved histo-endoscopic mucosal healing (Mayo Endoscopic Subscore ≤1 and neutrophil infiltration in <5% of crypts, no crypt destruction, and no erosions, ulcerations or granulation tissue based on the Geboes Score; n = 116 (26.5%)) after induction had higher rates of long-term (weeks 92 and 152) symptomatic and corticosteroid-free symptomatic remission than those who had endoscopic (n = 30 (6.8%)) or histological (n = 106 (24.2%)) improvement alone after induction. Although rates of both remission outcomes decreased between weeks 92 and 152 in patients who achieved either endoscopic or histological improvement alone, patients with histo-endoscopic mucosal healing maintained symptomatic remission over the same time period110.
Mirikizumab.
The phase III LUCENT-1 trial evaluated the efficacy of mirikizumab induction therapy (300 mg intravenously every 4 weeks, n = 868) compared with placebo (n = 294). At week 12, the mirikizumab group had significantly higher clinical remission rates compared with the placebo group (24.4% versus 13.3%, P = 0.00006). Endoscopic remission and histologic-endoscopic mucosal improvement rates were also significantly higher at week 12 in the mirikizumab group than in the placebo group111 (Supplementary Table 2).
Guselkumab.
In the phase IIb QUASAR study, patients were randomized to treatment with 200 mg (n = 101) or 400 mg (n = 107) intravenous guselkumab or placebo (n = 105) at weeks 0, 4 and 8. The primary outcome, which was clinical response at week 12, was achieved by 61.4% and 60.7% of patients in the guselkumab 200 mg and 400 mg groups, respectively, compared with 27.6% of those in the placebo group (P < 0.001 for both comparisons to placebo). Significantly higher rates of endoscopic improvement, histo-endoscopic mucosal improvement, and endoscopic normalization were also achieved in both guselkumab dose groups compared with placebo at week 12 (ref. 112) (Supplementary Table 2).
Given the favourable safety profile of the IL-12 and IL-23 agents (see next section), combining anti-IL-12 or anti-IL-23 agents with one or more biological agents and/or small molecules might be possible. The phase II VEGA study was the first trial to combine two biologics for the treatment of IBD, comparing the efficacy of guselkumab and golimumab combination therapy with monotherapy with either agent113 (Supplementary Table 2). Patients were randomized to golimumab 200 mg subcutaneously at week 0, 100 mg subcutaneously at week 2 and then every 4 weeks (n = 72); guselkumab 200 mg intravenously at weeks 0, 4 and 8 (n = 71); or the combination of golimumab and guselkumab at the same doses as in the monotherapy arms (n = 71), and followed to week 12. All patients were naive to TNF inhibitors as well as to ustekinumab and anti-IL-23 antibodies. The primary endpoint, which was clinical response at week 12, was achieved by 61.1% of patients in the golimumab monotherapy group and 74.6% of patients in the guselkumab monotherapy group compared with 83.1% in the combination group (P = 0.003 compared with golimumab alone and P = 0.215 compared with guselkumab alone). The proportions of patients with endoscopic improvement, endoscopic normalization, histological remission, both histological remission and endoscopic improvement, and both histological remission and endoscopic normalization were higher in the combination group compared with either the guselkumab or golimumab monotherapy groups (Supplementary Table 2).
A discussion on potential future positioning and use of these agents in particular patient populations based on currently available data appears later.
Safety of targeting IL-12 and IL-23
Most adverse effects reported in randomized controlled trials (RCTs) of agents targeting IL-12/IL-23 were mild, non-serious and did not require treatment discontinuation. Serious adverse effects, infections (including serious infections) and malignancies reported in phase II and III RCTs and open-label extensions of these trials in Crohn’s disease and ulcerative colitis (ustekinumab only) are shown in Table 2 and do not appear to differ compared with placebo treatment.
Table 2 |.
Serious adverse events, infections and malignancies reported in maintenance and/or open-label extension trials
| Agent | Serious adverse events | Infections | Malignancies |
|---|---|---|---|
| Ustekinumab | |||
| UNITI-IM93 (phase III CD) | Placebo: 20/133 (15%) 90mg every 12weeks: 16/106 (12.1%) 90mg every 8weeks: 13/131 (9.9%) |
Any Placebo: 66/133 (49.6%) 90mg every 12weeks: 61/106 (46.2%) 90mg every 8weeks: 63/131 (48.1%) Serious Placebo: 3/111 (2.3%) 90mg every 12weeks: 7/106 (5.3%) 90mg every 8weeks: 3/131 (2.3%) |
Basal cell carcinoma Placebo: 1 90mg every 8weeks: 1 |
| UNIFI109 (phase III UC) | Placebo: 17/175 (9.7%) 90mg every 12weeks: 13/172 (7.6%) 90mg every 8weeks: 15/176 (8.5%) |
Any Placebo: 81/175 (46.3%) 90mg every 12weeks: 58/172 (33.7%) 90mg every 8weeks: 86/176 (48.9%) Serious Placebo: 4/175 (2.3%) 90mg every 12weeks: 6/172 (3.5%) 90mg every 8weeks: 3/176 (1.7%) |
Excluding non-melanoma skin cancer Placebo: 0 90mg every 12 weeks: 1/172 (0.6%) 90mg every 8 weeks: 1/176 (0.6%) |
| SEAVUE96 (phase III CD)a | Adallmumab: 32/195 (16.4%) Ustekinumab: 25/191 (13.1%) |
Any Adallmumab: 79/195 (40.5%) Ustekinumab: 65/191 (34.0%) Serious Adalimumab: 5/195 (2.6%) Ustekinumab: 4/191 (2.1%) |
Adalimumab: 1/195 (0.5%, basal cell carcinoma) Ustekinumab: 0 |
| Risankizumab | |||
| M15–993 (ref. 129) (phase II CD)b | Period 2: 18 (62.5) Period 3: 9 (20.8) Periods 1–3: 46 (42.2) |
Any Period 2: 37 (128.5) Period 3: 32 (73.9) Periods 1–3: 107 (98.1) Serious Period 2: 1 (3.5) Period 3: 1 (2.3) Periods 1–3: 5 (4.6) |
None |
| M15–989 (ref. 156) and M16–000 (ref. 156) (phase II and III open-label extension CD)c | 23 (35.4%); 24.6 | Any 48 (73.8%); 112 Serious 6 (9.2%); 4.2 |
None |
| FORTIFY99 (phase III CD)d | 360mg: 21.0 180mg: 19.5 Placebo: 19.3 |
Serious 360mg: 6.0 180mg: 3.0 Placebo: 5.0 |
Not reported |
| Guselkumab | |||
| GALAXI104 (phase II CD)e | ≥1 serious adverse events 200mg IV followed by 100mg SC: 6 (8.2%) 600 mg IV followed by 200 mg SC: 5 (6.8%) 1,200mg IV followed by 200mg SC: 5 (6.8%) |
≥1 infection 200 mg IV followed by 100 mg SC: 25 (34.2%) 600mg IV followed by 200mg SC: 30 (41.1%) 1,200mg IV followed by 200 mg SC: 25 (34.2%) ≥1serious infection 200mg IV followed by 100mg SC: 2 (2.7%) 600mg IV followed by 200mg SC: 2 (2.7%) 1,200mg IV followed by 200mg SC: 1 (1.4%) |
Not reported |
CD, Crohn’s disease; IV, intravenous; SC, subcutaneous; UC, ulcerative colitis.
Adalimumab and ustekinumab administered at approved doses.
Period 2 (week 26, n=101); period 3 (week 52, n = 62); periods 1–3 (weeks 12–52, n=115); data expressed as number of events per 100 patient-years.
184weeks (n = 65, 167 patient-years); data expressed as number of patients (%); events per 100 patient-years.
52 weeks; data expressed as exposure adjusted event rates per 100 patient-years.
48 weeks.
As the first agent in this class approved for the treatment of various autoimmune disorders, including IBD, the most safety data available are for ustekinumab. In a pooled safety analysis of results from phase II and phase III studies (1,733 patient-years of follow-up), the number of patients with serious adverse events (27.50 (95% CI 23.45–32.04) versus 21.23 (95% CI 19.12–23.51)), infections (80.31 (95% CI 73.28–87.84) versus 64.32 (95% CI 60.60–68.21)), serious infections (5.53 (95% CI 3.81–7.77) versus 5.02 (95% CI 4.02–6.19)), and malignancies excluding nonmelanoma skin cancer (0.17 (95% CI 0.00–0.93) versus 0.40 (95% CI 0.16–0.83)) were similar between placebo and ustekinumab114. These results are further supported by data from the Psoriasis Longitudinal Assessment and Registry (PSOLAR) showing no increased risk for serious infections or malignancy with 12,472 patient-years of follow-up for ustekinumab115, as well as by data from observational ‘real world’ ustekinumab studies116. In their systematic review and meta-analysis, Honap and colleagues116 found a total of 498 adverse events reported in 2,977 patients (16.7%) for a pooled estimate of incidence rate of 13.5 (95% CI 9.6–18.6). Rates of serious infection (69 out of 1,749; 3.9%) and serious adverse events (86 out of 1,534; 5.6%) were low and comparable to those reported in the IM-UNITI RCT (5.6% versus 9.9–12.1% and 3.9% versus 2.3–5.3% real-world versus trials, respectively).
The phenotypes of patients with various genetic mutations or defects in the IL-12 and IL-23 pathway might also provide insights into hypothetical safety consequences associated with targeting this pathway. As previously discussed, IL23RR381Q carriers do not seem to be more vulnerable to infection generally; however, individuals with rare, significant loss-of-function mutations in other genetic components of the IL-23–TH17 pathway, including those common to IL-12 and IL-23 such as p40 and IL12βR1, demonstrate increased susceptibility to some infections (such as Salmonella, Candida and tuberculosis)117,118. As also previously noted, experimental animal models have demonstrated that IL-23 is required for regulation of responses to resident (for example, segmented filamentous bacteria) and pathogenic (for example, Listeria monocytogenes) bacteria119–123. Importantly, IL-23 compensates for IL-12 deficiency in both mice and humans during infectious challenge (for example, Mycobacterium and Salmonella Enteritidis)117,124. Despite these findings, as supported by the evidence described previously, increased susceptibility to infection has not yet been observed in patients with IBD treated with IL-12 and IL-23 neutralizing antibodies.
Although long-term safety data for many compounds are still accumulating, the available evidence from RCTs and real-world data suggests that neutralizing IL-12 and/or IL-23 is a safe strategy for the treatment of IBD.
IL-12 and IL-23 therapies: precision medicine
Molecular predictors
Biomarkers that predict response before treatment might identify patients who are more likely to benefit and reduce time associated with cycling through ineffective therapeutic interventions. Patients with Crohn’s disease who are unresponsive to TNF inhibitors had a significant upregulation of genes associated with IL-23R-dependent pathways compared with responders125. Furthermore, upregulation of IL-23p19, IL-23R, IL-17A and associated downstream phosphorylated STAT3 was observed in patients with non-response compared with responders. These results suggest that patients with non-response to TNF inhibitors might be good candidates for IL-23-targeted therapy125, although this is not always supported by clinical evidence98,107,126.
Few studies have investigated biomarkers predictive of response to IL-12 and/or IL-23 inhibition. A higher pre-treatment serum concentration of IL-22 was associated with a higher likelihood of response to brazikumab treatment in patients with Crohn’s disease105. However, pre-treatment expression levels of IL-22-responsive gene transcripts in colonic biopsy samples from patients with Crohn’s disease was not predictive of response to ustekinumab57. The association between baseline faecal microbiota composition and diversity and therapeutic response to ustekinumab in the CERTIFI study has also been analysed127. Patients with Crohn’s disease in remission were distinguishable from those with active disease 6 weeks after treatment according to baseline microbiota composition and clinical data (area under the curve 0.844; specificity 0.831, sensitivity 0.774). The median baseline community diversity in patients in remission was 1.7 times higher than in patients with active disease after treatment. Microbiota diversity increased over the 22 weeks of the study, in parallel with disease improvement, in patients with a response to ustekinumab, but not in those who were unresponsive to treatment. These baseline differences and changes in faecal microbiota in response to therapy suggest the potential for a noninvasive biomarker to initiate or monitor ustekinumab treatment. Validation in an external cohort and demonstration of an ustekinumab-specific signature is necessary given that the observations might simply reflect a milder disease phenotype and a higher probability of response.
Despite the lack of strong evidence for predictive biomarkers, IL-17 and IL-22 (cytokines downstream of IL-23) have been identified as pharmacodynamic biomarkers of IL-23 inhibition in RCTs. Patients with ulcerative colitis treated with mirikizumab had a reduction in IL-22 and IL-17 plasma concentrations from baseline to week 12 in a phase II RCT128. Similarly, IL22 gene expression was significantly reduced from baseline to week 12 in ileal biopsy samples from patients with Crohn’s disease treated with risankizumab compared with placebo129.
Clinical predictors
Few studies have identified clinical or demographic parameters associated with response to ustekinumab (reviewed elsewhere130). Post-hoc analyses for baseline clinical predictors of response to IL-12 and/or IL-23 inhibition have been conducted for several RCTs targeting IL-23 (discussed previously), but no significant findings have been reported. Indeed, inconsistent patterns of response to mirikizumab or risankizumab based on previous biologic exposure have been reported98,107,126. As it relates to predictors of treatment failure, bowel frequency and >2 previous biologic exposures were positively associated with time to ustekinumab dose intensification from every 8 weeks to every 4 or 6 weeks in a retrospective cohort study of 108 patients with ulcerative colitis131. Perianal disease, higher Harvey–Bradshaw Index scores, and opioid use were identified as predictors of failure to achieve remission in response to ustekinumab dose intensification in a retrospective cohort study of 123 patients with Crohn’s disease132.
Pharmacological predictors.
Differences in sex, body weight, serum albumin concentration or inflammatory burden can partially explain the inter-individual and intra-individual variability in drug clearance that is observed with intravenously or subcutaneously administered biologics targeting IL-12 and/or IL-23 (refs. 133,134). Immunogenicity has not been identified as a pharmacologically or clinically relevant factor given the low rates observed to date135, and therefore the use of concomitant immunomodulators to influence drug exposure or treatment outcomes might not be an important consideration for biologics that target IL12 and/or IL-23, unlike for those that target TNF136. Evaluation of the relationship between drug concentrations and outcomes has therefore emerged as a primary focus. An exposure–response relationship has been observed for ustekinumab137–139 but has not yet been demonstrated in a prospective interventional study. Previous studies in patients with Crohn’s disease treated with ustekinumab137,138 showed that higher serum ustekinumab concentrations during induction treatment were associated with clinical, endoscopic and biomarker (CRP and faecal calprotectin)-based outcomes at the end of induction and during maintenance therapy. Similar results were observed for patients with ulcerative colitis, including an association between ustekinumab139 serum concentrations and histological improvement. An association between serum risankizumab concentrations and CDAI response and remission, as well as endoscopic response, was observed in patients with Crohn’s disease129. However, no difference in serum mirikizumab concentrations was observed between responders and non-responders during induction and maintenance therapy107. To mitigate the effect of patient demographics and disease characteristics on mirikizumab drug clearance and exposure, Sandborn and colleagues128 employed a unique approach that adjusted induction dosing based on actual serum concentrations. Serum mirikizumab concentrations were therefore similar in patients who required dose adjustments to those who did not in both the 50-mg and the 200-mg groups, and the clinical response and remission rates were similar128. Future interventional studies are required to confirm drug exposure levels to optimize efficacy across the patient population for all biologic therapies140.
Positioning anti-IL-12/IL-23 agents in IBD
The anti-IL-12/IL-23p40 agent ustekinumab is the only drug in this class currently approved for the treatment of both Crohn’s disease and ulcerative colitis, and risankizumab, an anti-IL-23p19 agent, has also been approved for the treatment of Crohn’s disease. Other anti-IL-23p19 agents (brazikumab, mirikizumab and guselkumab) are in late-stage development and are expected to be commercially available in the foreseeable future141–143. Positioning these agents in treatment algorithms and/or appropriate patient selection will be critical. Risankizumab demonstrated superior efficacy to ustekinumab for the treatment of psoriasis144, and a similar phase III trial comparing both drugs in patients with Crohn’s disease with previous TNF inhibitor treatment is currently ongoing (NCT04524611).
Data from head-to-head trials might provide information not only on appropriate patients for specific compounds on a population level, but also shed light on positioning among the various classes. However, similar clinical and endoscopic remission rates were observed in the SEAVUE trial, which compared ustekinumab with adalimumab monotherapy in biologic-naive patients with Crohn’s disease, with higher infection rates observed in the adalimumab group145.
The current paucity of head-to-head trials and lack of robust molecular markers requires reliance on indirect comparisons. Network meta-analyses have investigated the efficacy and safety of biologics and small molecules in moderate-to-severe Crohn’s disease and ulcerative colitis. For patients with Crohn’s disease who failed treatment with TNF inhibitors, Singh and colleagues proposed IL-23-targeted therapy (ustekinumab and risankizumab) as the proposed mechanism of action, with risankizumab demonstrating superiority over the anti-α4β7 integrin antibody vedolizumab for induction of clinical remission146. In patients refractory to TNF inhibitors, IL-23 has a marked effect on shaping the immune landscape of the inflamed intestine. Specifically, IL-23 controls expansion of apoptosis-resistant intestinal TNFR2+IL-23R+ T cells, leading to molecular resistance to TNF inhibitor therapy in Crohn’s disease. These findings identify IL-23 as a suitable molecular target in patients with IBD refractory to TNF inhibitor therapy125.
The highest rates of clinical remission and endoscopic improvement in patients with ulcerative colitis previously treated with TNF inhibitors were observed with ustekinumab and tofacitinib, as reported by Singh and colleagues147. A separate network meta-analysis ranked ustekinumab highest among biologics for achieving the outcomes of clinical response and remission and endoscopic response in the same patient population148. The lowest total number of adverse events was observed for ustekinumab. In another study, ustekinumab was ranked highest for the outcome of endoscopic improvement in a network meta-analysis of outcomes in patients with ulcerative colitis naive to biologic therapy, was significantly superior to adalimumab and vedolizumab, and ranked second (after tofacitinib) for induction of endoscopic improvement in patients with previous exposure to biologic therapy149. Although translation of these data into clinical practice would ideally be further supported by validation in head-to-head clinical trials, these data provide relevant interim information to help guide clinical decision-making.
Ustekinumab was effective for treatment of extraintestinal manifestations, particularly arthralgia, psoriatic arthritis, psoriasis, pyoderma gangrenosum, and erythema nodosum in a systematic review and meta-analysis encompassing 254 patients with IBD and extraintestinal manifestations. No efficacy was observed in axial spondyloarthritis150. These agents might thus represent an important and safe alternative to TNF inhibitors, which are the primary therapeutic option for this indication. Indeed, TNF inhibitors can paradoxically induce or worsen psoriatic skin lesions in 1.6–2.7% of patients with IBD, with infliximab most frequently associated with these reactions (52.6–62.5% of reported cases)151. Switching to anti-IL-12/IL-23 agents has been reported as effective (and should be considered) when withdrawal of TNF inhibitors is warranted due to lesion severity or insufficient response to topical treatment151,152.
Other patients who might benefit from anti-IL-12/IL-23 treatments are those for whom safety is a primary consideration, including older people and those with malignancy or infection. Although favourable safety outcomes were observed in the ustekinumab UNITI and IM-UNITI trials (including up to 5 years of follow-up data in the long-term extension of IM-UNITI)153, the average age of participants was 38 and age stratification was not performed93. However, no significant differences in infection rates (5.2% versus 7.7%, P = 0.7), infusion reactions (2.6% versus 6.4%, P = 0.77) or postsurgical complications (P = 0.99) by age category were observed in a retrospective study comparing ustekinumab in patients with Crohn’s disease aged ≥65 years (n = 39) to those <65 years (n = 78)154. Similarly, safety data from patients with psoriasis ≥65 years of age reported no concerning safety signals, further supporting the potential for ustekinumab in this population155.
Future evidence-based recommendations for positioning of anti-IL-12/IL-23 therapies for the treatment of patients with IBD should be based on additional RCT data, including data from trials that directly compare the efficacy and safety of these agents with other classes of therapies.
Conclusions
A wealth of experimental data from in vitro, animal model and human genetic association studies support a pivotal role for IL-23 in the pathogenesis of IBD and other immune-mediated diseases. These data are supported clinically by the efficacy observed in pivotal controlled trials of agents targeting IL-23 (and in some cases IL-12) for the treatment of Crohn’s disease and ulcerative colitis, and which has led to the approval of ustekinumab for the treatment of both forms of IBD. There is evidence (discussed previously) suggesting that targeting IL-23 clinically might have deleterious effects on the maintenance of intestinal epithelial barrier integrity and/or microbial clearance, but infection rates, including rates of serious infections, do not seem to be increased with treatments targeting IL-23, although most long-term and real-world data supporting this observation have been accumulated for ustekinumab. To conclude that these observations are generalizable to the therapeutic class, longer-term data are needed for other agents under investigation. Nevertheless, the safety profile of these agents is encouraging: ustekinumab is approved as the first-line treatment for moderately-to-severely active Crohn’s disease and ulcerative colitis, and risankizumab was most recently approved by the FDA in June 2022 for the treatment of moderately-to-severely active Crohn’s disease. Again, whether this positioning will be consistent across all investigational agents in this class depends on the results of ongoing controlled trials. Precision medicine research should aim to provide a deeper understanding of the mechanism of action of these agents and to better define potential complementary and/or synergistic effects with other biologic or small-molecule therapies. This knowledge will further facilitate the appropriate positioning of these therapies and guide therapeutic decision-making based on the molecular backgrounds of individual patients. Late-phase clinical trial data also support the potential use of these therapies in specific patient populations, including older people, and those with previous malignancy, higher infection risk or psoriasis (including those with TNF inhibitor-mediated onset). Head-to-head studies and clinical tools that integrate potential clinical and biological predictors of response to various therapeutic agents will further enable personalized medicine-based treatment decisions.
Supplementary Material
Key points.
IL-12 and IL-23, which are members of the IL-12 family of cytokines, have a key role in intestinal homeostasis and inflammation, including in inflammatory bowel disease.
Multiple IL-12- and/or IL-23-neutralizing antibodies have been tested in immune-mediated diseases, including Crohn’s disease and ulcerative colitis.
In addition to demonstrated efficacy for clinical, endoscopic and histological outcomes, targeting IL-12 and/or IL-23 is a safe treatment strategy.
The exact positioning of such antibodies in current treatment algorithms will be influenced by ongoing head-to-head trials and evaluation of predictive molecular markers.
Acknowledgements
The authors would like to thank L. M. Shackelton and S. Donegan for critical technical review and editing. B.V. is supported by the Clinical Research Fund (KOOR) at the University Hospitals Leuven and the Research Council at KU Leuven. N.V.C. is supported in part by the NIDDK-funded San Diego Digestive Diseases Research Center (P30 DK120515).
Alimentiv Translational Research Consortium (ATRC)
Silvio Danese10,11, Geert D’Haens12,13, Lars Eckmann14, William A. Faubion15, Brian G. Feagan16, Vipul Jairath16, Christopher Ma17, Saurabh Mehandru4,18, Julian Panes3, Florian Rieder19,20, William J. Sandborn14, Mark S. Silverberg21, Marisol Veny3, Severine Vermeire1,2 & Stefania Vetrano22,23
10Gastroenterology and Endoscopy, IRCCS Ospedale San Raffaele, Milan, Italy. 11University Vita-Salute San Raffaele, Milan, Italy. 12Amsterdam UMC, Amsterdam Medical Center, University of Amsterdam, Department of Gastroenterology and Hepatology, Amsterdam, Netherlands. 13Metabolism Research Institute, Amsterdam UMC, University of Amsterdam, Amsterdam, Netherlands. 14Division of Gastroenterology, University of California San Diego, La Jolla, CA, USA. 15Division of Gastroenterology and Hepatology, Mayo Clinic, Rochester, MN, USA. 16Departments of Medicine and Epidemiology and Biostatistics, Western University, London, Ontario, Canada. 17Division of Gastroenterology and Hepatology, Departments of Medicine and Community Health Sciences, University of Calgary, Calgary, Alberta, Canada. 18Precision Institute of Immunology, Icahn School of Medicine at Mount Sinai, New York, NY, USA. 19Department of Gastroenterology, Hepatology and Nutrition, Digestive Diseases and Surgery Institute, Cleveland Clinic Foundation, Cleveland, OH, USA. 20Department of Inflammation and Immunity, Lerner Research Institute, Cleveland Clinic Foundation, Cleveland, OH, USA. 21Mount Sinai Hospital IBD Centre, University of Toronto, Toronto, Ontario, Canada. 22Department of Biomedical Sciences, Humanitas University, Pieve Emanuele, Milan, Italy. 23Laboratory of Gastrointestinal Immunopathology, IRCCS Humanitas Research Hospital — IRCCS, Rozzano, Milan, Italy.
Footnotes
Competing interests
Alimentiv Inc. is an academic gastrointestinal contract research organization (CRO), operating under the Alimentiv Health Trust. Alimentiv Inc. provides comprehensive clinical trial services, precision medicine offerings, and centralized imaging solutions for endoscopy, histopathology, and other imaging modalities. The beneficiaries of the Alimentiv Health Trust are the employees of the enterprises it holds. None of the authors is a beneficiary of the Alimentiv Health Trust. B.V., N.V.C., G.D’H., B.G.F., V.J., C.M., W.J.S. and A.S. are consultants to Alimentiv Inc. and have a primary academic appointment; they do not hold equity positions or shares in Alimentiv Inc. B.V. reports research support from AbbVie, Biora Therapeutics, Pfizer, Sossei Heptares and Takeda; speaker’s fees from Abbvie, Biogen, Bristol Myers Squibb, Celltrion, Chiesi, Falk, Ferring, Galapagos, Janssen, MSD, Pfizer, R-Biopharm, Takeda, Truvion and Viatris; and consultancy fees from Abbvie, Alimentiv, Applied Strategic, Atheneum, Biora Therapeutics, Bristol Myers Squibb, Galapagos, Guidepont, Inotrem, Inotrem, Ipsos, Janssen, Mylan, Progenity, Sandoz, Sosei Heptares, Takeda Tillots Pharma and Viatris. A.S. reports research grants from Roche-Genentech, Abbvie, GSK, Scipher Medicine, Alimentiv Inc, Boehringer Ingelheim and Origo Biopharma; consulting fees from Genentech, GSK, Pfizer, HotSpot Therapeutics, Alimentiv, Origo Biopharma and Boxer Capital. B.E.S. reports research grants from Takeda, Pfizer, Theravance Biopharma R&D and Janssen; consulting fees from 4D Pharma, Abivax, Abbvie, Alimentiv, Allergan, Amgen, Arena Pharmaceuticals, AstraZeneca, Bacainn Therapeutics, Boehringer-Ingelheim, Boston Pharmaceuticals, Bristol-Myers Squibb, Calibr, Capella Bioscience, Celgene, Celltrion Healthcare, ClostraBio, Enthera, F.Hoffmann-La Roche, Ferring, Galapagos, Gilead, GlaxoSmithKline, GossamerBio, Immunic, Index Pharmaceuticals, Innovation Pharmaceuticals, Ironwood Pharmaceuticals, Janssen, Kaleido, Kallyope, Lilly, MiroBio, Morphic Therapeutic, Oppilan Pharma, OSE Immunotherapeutics, Otsuka, Palatin Technologies, Pfizer, Progenity, Prometheus Biosciences, Prometheus Laboratories, Protagonist Therapeutics, Q32 Bio, Redhill Biopharma, Rheos Medicines, Salix Pharmaceuticals, Seres Therapeutics, Shire, Sienna Biopharmaceuticals, Sun Pharma, Surrozen, Takeda, Target PharmaSolutions, Teva Branded Pharmaceutical Products R&D, Thelium, Theravance Biopharma R&D, TLL Pharma, USWM Enterprises, Ventyx Biosciences, Viela Bio, Vivante Health and Vivelix Pharmaceuticals; and holds stock in Vivante Health and Ventyx Biosciences. M.F.N. reports consulting fees from MSD Sharp & Dohme GmbH, PPM Services, IFM Therapeutics, Sterna Biologicals, Boehringer Ingleheim GmbH and Co. KG, Janssen Cilag GmbH, Pentax Europe GmbH, Takeda, Amgen GmbH, Pfizer Pharma, Falk Foundation e.v., Abbvie and Celgene. N.V.C. reports research grants and personal fees from R-Biopharm, Takeda and UCB; and personal fees from Alimentiv, Inc (formerly Robarts Clinical Trials, Inc), Celltrion and Prometheus. These activities were all outside the submitted work. C.A. and H.L. declare no competing interests. The competing interests of the Alimentiv Translational Research Consortium Member Authors are listed in Supplementary Box 1.
Additional information
Peer review information Nature Reviews Gastroenterology & Hepatology thanks Giovanni Monteleone and Herbert Tilg for their contribution to the peer review of this work.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41575-023-00768-1.
References
- 1.Chang JT Pathophysiology of inflammatory bowel diseases. N. Engl. J. Med. 383, 2652–2664 (2020). [DOI] [PubMed] [Google Scholar]
- 2.Baumgart DC & Le Berre C Newer biologic and small-molecule therapies for inflammatory bowel disease. N. Engl. J. Med. 385, 1302–1315 (2021). [DOI] [PubMed] [Google Scholar]
- 3.Roda G, Jharap B, Neeraj N & Colombel JF Loss of response to anti-TNFs: definition, epidemiology, and management. Clin. Transl. Gastroenterol. 7, e135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duerr RH et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314, 1461–1463 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Verstockt B, Van Assche G, Vermeire S & Ferrante M Biological therapy targeting the IL-23/IL-17 axis in inflammatory bowel disease. Expert. Opin. Biol. Ther. 17, 31–47 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Kobayashi M et al. Identification and purification of natural killer cell stimulatory factor (NKSF), a cytokine with multiple biologic effects on human lymphocytes. J. Exp. Med. 170, 827–845 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hsieh CS et al. Development of TH1 CD4+ T cells through IL-12 produced by Listeria-induced macrophages. Science 260, 547–549 (1993). [DOI] [PubMed] [Google Scholar]
- 8.Manetti R et al. Natural killer cell stimulatory factor (interleukin 12 [IL-12]) induces T helper type 1 (Th1)-specific immune responses and inhibits the development of IL-4-producing Th cells. J. Exp. Med. 177, 1199–1204 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Seder RA, Gazzinelli R, Sher A & Paul WE Interleukin 12 acts directly on CD4+ T cells to enhance priming for interferon gamma production and diminishes interleukin 4 inhibition of such priming. Proc. Natl Acad. Sci. USA 90, 10188–10192 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oppmann B et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13, 715–725 (2000). [DOI] [PubMed] [Google Scholar]
- 11.Brombacher F et al. IL-12 is dispensable for innate and adaptive immunity against low doses of Listeria monocytogenes. Int. Immunol. 11, 325–332 (1999). [DOI] [PubMed] [Google Scholar]
- 12.Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ & Gurney AL Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J. Biol. Chem. 278, 1910–1914 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Tesmer LA, Lundy SK, Sarkar S & Fox DA Th17 cells in human disease. Immunol. Rev. 223, 87–113 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pirhonen J, Matikainen S & Julkunen I Regulation of virus-induced IL-12 and IL-23 expression in human macrophages. J. Immunol. 169, 5673–5678 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Verreck FA et al. Human IL-23-producing type 1 macrophages promote but IL-10-producing type 2 macrophages subvert immunity to (myco)bacteria. Proc. Natl Acad. Sci. USA 101, 4560–4565 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cella M et al. Ligation of CD40 on dendritic cells triggers production of high levels of interleukin-12 and enhances T cell stimulatory capacity: T–T help via APC activation. J. Exp. Med. 184, 747–752 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wesa A & Galy A Increased production of pro-inflammatory cytokines and enhanced T cell responses after activation of human dendritic cells with IL-1 and CD40 ligand. BMC Immunol. 3, 14 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma X et al. The interleukin 12 p40 gene promoter is primed by interferon gamma in monocytic cells. J. Exp. Med. 183, 147–157 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luque-Martin R et al. IFN-gamma drives human monocyte differentiation into highly proinflammatory macrophages that resemble a phenotype relevant to psoriasis. J. Immunol. 207, 555–568 (2021). [DOI] [PubMed] [Google Scholar]
- 20.Shi Q et al. PGE2 elevates IL-23 production in human dendritic cells via a cAMP dependent pathway. Mediat. Inflamm. 2015, 984690 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geyer CE et al. C-reactive protein controls IL-23 production by human monocytes. Int. J. Mol. Sci. 22, 11638 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lim KS et al. Inflammatory and mitogenic signals drive interleukin 23 subunit alpha (IL23A) secretion independent of IL12B in intestinal epithelial cells. J. Biol. Chem. 295, 6387–6400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macho-Fernandez E et al. Lymphotoxin beta receptor signaling limits mucosal damage through driving IL-23 production by epithelial cells. Mucosal Immunol. 8, 403–413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moschen AR, Tilg H & Raine T IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 16, 185–196 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Schwarz E & Carson WE III Analysis of potential biomarkers of response to IL-12 therapy. J. Leukoc. Biol. 112, 557–567 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grohmann U et al. Positive regulatory role of IL-12 in macrophages and modulation by IFN-gamma. J. Immunol. 167, 221–227 (2001). [DOI] [PubMed] [Google Scholar]
- 27.Parham C et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rβ1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 168, 5699–5708 (2002). [DOI] [PubMed] [Google Scholar]
- 28.Awasthi A et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J. Immunol. 182, 5904–5908 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frucht DM IL-23: a cytokine that acts on memory T cells. Sci. STKE 2002, pe1 (2002). [DOI] [PubMed] [Google Scholar]
- 30.Ivanov II et al. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 126, 1121–1133 (2006). [DOI] [PubMed] [Google Scholar]
- 31.Glassman CR et al. Structural basis for IL-12 and IL-23 receptor sharing reveals a gateway for shaping actions on T versus NK cells. Cell 184, 983–999.e924 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thierfelder WE et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 382, 171–174 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Floss DM et al. Identification of canonical tyrosine-dependent and non-canonical tyrosine-independent STAT3 activation sites in the intracellular domain of the interleukin 23 receptor. J. Biol. Chem. 288, 19386–19400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun R, Hedl M & Abraham C IL23 induces IL23R recycling and amplifies innate receptor-induced signalling and cytokines in human macrophages, and the IBD-protective IL23R R381Q variant modulates these outcomes. Gut 69, 264–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Becker C et al. Constitutive p40 promoter activation and IL-23 production in the terminal ileum mediated by dendritic cells. J. Clin. Invest. 112, 693–706 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fuss IJ et al. Both IL-12p70 and IL-23 are synthesized during active Crohn’s disease and are down-regulated by treatment with anti-IL-12 p40 monoclonal antibody. Inflamm. Bowel Dis. 12, 9–15 (2006). [DOI] [PubMed] [Google Scholar]
- 37.Kullberg MC et al. IL-23 plays a key role in Helicobacter hepaticus-induced T cell-dependent colitis. J. Exp. Med. 203, 2485–2494 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grohmann U et al. IL-12 acts directly on DC to promote nuclear localization of NF-κB and primes DC for IL-12 production. Immunity 9, 315–323 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Yang R et al. IL-12 + IL-18 cosignaling in human macrophages and lung epithelial cells activates cathelicidin and autophagy, inhibiting intracellular mycobacterial growth. J. Immunol. 200, 2405–2417 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xing Z, Zganiacz A & Santosuosso M Role of IL-12 in macrophage activation during intracellular infection: IL-12 and mycobacteria synergistically release TNF-α and nitric oxide from macrophages via IFN-γ induction. J. Leukoc. Biol. 68, 897–902 (2000). [PubMed] [Google Scholar]
- 41.Sun R & Abraham C IL23 promotes antimicrobial pathways in human macrophages, which are reduced with the IBD-protective IL23R R381Q variant. Cell Mol. Gastroenterol. Hepatol. 10, 673–697 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bastos KR et al. What kind of message does IL-12/IL-23 bring to macrophages and dendritic cells? Microbes Infect. 6, 630–636 (2004). [DOI] [PubMed] [Google Scholar]
- 43.Esplugues E et al. Control of TH17 cells occurs in the small intestine. Nature 475, 514–518 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Monteleone I, Sarra M, Pallone F & Monteleone G Th17-related cytokines in inflammatory bowel diseases: friends or foes? Curr. Mol. Med. 12, 592–597 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Punkenburg E et al. Batf-dependent Th17 cells critically regulate IL-23 driven colitis-associated colon cancer. Gut 65, 1139–1150 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Huber S et al. Th17 cells express interleukin-10 receptor and are controlled by Foxp3− and Foxp3+ regulatory CD4+ T cells in an interleukin-10-dependent manner. Immunity 34, 554–565 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee JS et al. Interleukin-23-independent IL-17 production regulates intestinal epithelial permeability. Immunity 43, 727–738 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maxwell JR et al. Differential roles for interleukin-23 and interleukin-17 in intestinal immunoregulation. Immunity 43, 739–750 (2015). [DOI] [PubMed] [Google Scholar]
- 49.Hueber W et al. Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: unexpected results of a randomised, double-blind placebo-controlled trial. Gut 61, 1693–1700 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pickert G et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 206, 1465–1472 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bauche D et al. IL-23 and IL-2 activation of STAT5 is required for optimal IL-22 production in ILC3s during colitis. Sci. Immunol. 5, eaav1080 (2020). [DOI] [PubMed] [Google Scholar]
- 52.Aden K et al. Epithelial IL-23R signaling licenses protective IL-22 responses in intestinal inflammation. Cell Rep. 16, 2208–2218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Powell N et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology 149, 456–467.e15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peng V, Jaeger N & Colonna M Innate lymphoid cells and inflammatory bowel disease. Adv. Exp. Med. Biol. 1365, 97–112 (2022). [DOI] [PubMed] [Google Scholar]
- 55.Bauche D et al. LAG3+ regulatory T cells restrain interleukin-23-producing CX3CR1+ gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity 49, 342–352 (2018). [DOI] [PubMed] [Google Scholar]
- 56.Rankin LC & Arpaia N Treg cells: a LAGging hand holds the double-edged sword of the IL-23 axis. Immunity 49, 201–203 (2018). [DOI] [PubMed] [Google Scholar]
- 57.Powell N et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut 69, 578–590 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simmons CP et al. Impaired resistance and enhanced pathology during infection with a noninvasive, attaching–effacing enteric bacterial pathogen, Citrobacter rodentium, in mice lacking IL-12 or IFN-gamma. J. Immunol. 168, 1804–1812 (2002). [DOI] [PubMed] [Google Scholar]
- 59.Zundler S & Neurath MF Interleukin-12: functional activities and implications for disease. Cytokine Growth Factor Rev. 26, 559–568 (2015). [DOI] [PubMed] [Google Scholar]
- 60.Sarin R, Wu X & Abraham C Inflammatory disease protective R381Q IL23 receptor polymorphism results in decreased primary CD4+ and CD8+ human T-cell functional responses. Proc. Natl Acad. Sci. USA 108, 9560–9565 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pidasheva S et al. Functional studies on the IBD susceptibility gene IL23R implicate reduced receptor function in the protective genetic variant R381Q. PLoS ONE 6, e25038 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Di Meglio P et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 6, e17160 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yu RY, Brazaitis J & Gallagher G The human IL-23 receptor rs11209026 A allele promotes the expression of a soluble IL-23R-encoding mRNA species. J. Immunol. 194, 1062–1068 (2015). [DOI] [PubMed] [Google Scholar]
- 64.Sivanesan D et al. IL23R (interleukin 23 receptor) variants protective against inflammatory bowel diseases (IBD) display loss of function due to impaired protein stability and intracellular trafficking. J. Biol. Chem. 291, 8673–8685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beaudoin M et al. Deep resequencing of GWAS loci identifies rare variants in CARD9, IL23R and RNF186 that are associated with ulcerative colitis. PLoS Genet. 9, e1003723 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jostins L et al. Host–microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zwiers A et al. Cutting edge: a variant of the IL-23R gene associated with inflammatory bowel disease induces loss of microRNA regulation and enhanced protein production. J. Immunol. 188, 1573–1577 (2012). [DOI] [PubMed] [Google Scholar]
- 68.Liu JZ et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brant SR et al. Genome-wide association study identifies African-specific susceptibility loci in African Americans with inflammatory bowel disease. Gastroenterology 152, 206–217 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Huang C et al. Characterization of genetic loci that affect susceptibility to inflammatory bowel diseases in African Americans. Gastroenterology 149, 1575–1586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barrett JC et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 40, 955–962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ben-Selma W & Boukadida J IL23R(Arg381Gln) functional polymorphism is associated with active pulmonary tuberculosis severity. Clin. Vaccin. Immunol. 19, 1188–1192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zakrzewski M et al. IL23R-protective coding variant promotes beneficial bacteria and diversity in the ileal microbiome in healthy individuals without inflammatory bowel disease. J. Crohns Colitis 13, 451–461 (2019). [DOI] [PubMed] [Google Scholar]
- 74.Neurath MF, Fuss I, Kelsall BL, Stuber E & Strober W Antibodies to interleukin 12 abrogate established experimental colitis in mice. J. Exp. Med. 182, 1281–1290 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Davidson NJ et al. IL-12, but not IFN-γ, plays a major role in sustaining the chronic phase of colitis in IL-10-deficient mice. J. Immunol. 161, 3143–3149 (1998). [PubMed] [Google Scholar]
- 76.Hue S et al. Interleukin-23 drives innate and T cell-mediated intestinal inflammation. J. Exp. Med. 203, 2473–2483 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Uhlig HH et al. Differential activity of IL-12 and IL-23 in mucosal and systemic innate immune pathology. Immunity 25, 309–318 (2006). [DOI] [PubMed] [Google Scholar]
- 78.Monteleone G et al. Interleukin 12 is expressed and actively released by Crohn’s disease intestinal lamina propria mononuclear cells. Gastroenterology 112, 1169–1178 (1997). [DOI] [PubMed] [Google Scholar]
- 79.Monteleone G, Parrello T, Luzza F & Pallone F Response of human intestinal lamina propria T lymphocytes to interleukin 12: additive effects of interleukin 15 and 7. Gut 43, 620–628 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mannon PJ et al. Anti-interleukin-12 antibody for active Crohn’s disease. N. Engl. J. Med. 351, 2069–2079 (2004). [DOI] [PubMed] [Google Scholar]
- 81.Globig AM et al. Ustekinumab inhibits T follicular helper cell differentiation in patients with Crohn’s disease. Cell Mol. Gastroenterol. Hepatol. 11, 1–12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wiekowski MT et al. Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J. Immunol. 166, 7563–7570 (2001). [DOI] [PubMed] [Google Scholar]
- 83.Yen D et al. IL-23 is essential for T cell-mediated colitis and promotes inflammation via IL-17 and IL-6. J. Clin. Invest. 116, 1310–1316 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Elson CO et al. Monoclonal anti-interleukin 23 reverses active colitis in a T cell-mediated model in mice. Gastroenterology 132, 2359–2370 (2007). [DOI] [PubMed] [Google Scholar]
- 85.Buonocore S et al. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 464, 1371–1375 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Izcue A et al. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28, 559–570 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Chen L et al. Diet modifies colonic microbiota and CD4+ T-cell repertoire to induce flares of colitis in mice with myeloid-cell expression of interleukin 23. Gastroenterology 155, 1177–1191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.He Z et al. Food colorants metabolized by commensal bacteria promote colitis in mice with dysregulated expression of interleukin-23. Cell Metab. 33, 1358–1371 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Eftychi C et al. Temporally distinct functions of the cytokines IL-12 and IL-23 drive chronic colon inflammation in response to intestinal barrier impairment. Immunity 51, 367–380 (2019). [DOI] [PubMed] [Google Scholar]
- 90.Cox JH et al. Opposing consequences of IL-23 signaling mediated by innate and adaptive cells in chemically induced colitis in mice. Mucosal Immunol. 5, 99–109 (2012). [DOI] [PubMed] [Google Scholar]
- 91.Becker C et al. Cutting edge: IL-23 cross-regulates IL-12 production in T cell-dependent experimental colitis. J. Immunol. 177, 2760–2764 (2006). [DOI] [PubMed] [Google Scholar]
- 92.Aychek T et al. IL-23-mediated mononuclear phagocyte crosstalk protects mice from Citrobacter rodentium-induced colon immunopathology. Nat. Commun. 6, 6525 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Feagan BG et al. Ustekinumab as induction and maintenance therapy for Crohn’s disease. N. Engl. J. Med. 375, 1946–1960 (2016). [DOI] [PubMed] [Google Scholar]
- 94.Rutgeerts P et al. Efficacy of ustekinumab for inducing endoscopic healing in patients with Crohn’s disease. Gastroenterology 155, 1045–1058 (2018). [DOI] [PubMed] [Google Scholar]
- 95.Li K et al. Effects of ustekinumab on histologic disease activity in patients with Crohn’s disease. Gastroenterology 157, 1019–1031 (2019). [DOI] [PubMed] [Google Scholar]
- 96.Sands BE et al. Ustekinumab versus adalimumab for induction and maintenance therapy in biologic-naive patients with moderately to severely active Crohn’s disease: a multicentre, randomised, double-blind, parallel-group, phase 3b trial. Lancet 399, 2200–2211 (2022). [DOI] [PubMed] [Google Scholar]
- 97.Danese S et al. Treat to target versus standard of care for patients with Crohn’s disease treated with ustekinumab (STARDUST): an open-label, multicentre, randomised phase 3b trial. Lancet Gastroenterol. Hepatol. 7, 294–306 (2022). [DOI] [PubMed] [Google Scholar]
- 98.D’Haens G et al. Risankizumab as induction therapy for Crohn’s disease: results from the phase 3 ADVANCE and MOTIVATE induction trials. Lancet 399, 2015–2030 (2022). [DOI] [PubMed] [Google Scholar]
- 99.Ferrante M et al. Risankizumab as maintenance therapy for moderately to severely active Crohn’s disease: results from the multicentre, randomised, double-blind, placebo-controlled, withdrawal phase 3 FORTIFY maintenance trial. Lancet 399, 2031–2046 (2022). [DOI] [PubMed] [Google Scholar]
- 100.Ferrante M et al. OP25 Patients with moderate to severe Crohn’s disease with and without prior biologic failure demonstrate improved endoscopic outcomes with risankizumab: results from phase 3 induction and maintenance trials. J. Crohns Colitis 16, i027–i028 (2022). [Google Scholar]
- 101.Bossuyt P et al. OP40 Efficacy of risankizumab induction and maintenance therapy by baseline Crohn’s disease location: post hoc analysis of the phase 3 ADVANCE, MOTIVATE, and FORTIFY studies. J. Crohns Colitis 16, i048 (2022). [Google Scholar]
- 102.Sandborn WJ et al. The efficacy and safety of guselkumab induction therapy in patients with moderately to severely active Crohn’s disease: week 12 interim analyses from the phase 2 GALAXI 1 study. U. Eur. Gastroenterol. 8, 64 (2020). [Google Scholar]
- 103.Sandborn WJ et al. Guselkumab for the treatment of Crohn’s disease: Induction results from the Phase 2 GALAXI-1 study. Gastroenterology 10.1053/j.gastro.2022.01.047 (2022). [DOI] [PubMed] [Google Scholar]
- 104.Danese S et al. OP24 Clinical efficacy and safety of guselkumab maintenance therapy in patients with moderately to severely active Crohn’s disease: week 48 analyses from the phase 2 GALAXI 1 study. J. Crohns Colitis 16, i026–i027 (2022). [Google Scholar]
- 105.Sands BE et al. Efficacy and safety of MEDI2070, an antibody against interleukin 23, patients with moderate to severe Crohn’s disease: a phase 2a study. Gastroenterology 153, 77–86 (2017). [DOI] [PubMed] [Google Scholar]
- 106.Sands BE et al. 1003 — Efficacy and safety of mirikizumab (LY3074828) in a phase 2 study of patients with Crohn’s disease. Gastroenterology 156, S216 (2019). [DOI] [PubMed] [Google Scholar]
- 107.Sands BE et al. Efficacy and safety of mirikizumab after 52-weeks maintenance treatment in patients with moderate-to-severe Crohn’s disease. Gastroenterology 160, S37 (2021). [Google Scholar]
- 108.Panaccione R et al. Briakinumab for treatment of Crohn’s disease: results of a randomized trial. Inflamm. Bowel Dis. 21, 1329–1340 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Sands BE et al. Ustekinumab as induction and maintenance therapy for ulcerative colitis. N. Engl. J. Med. 381, 1201–1214 (2019). [DOI] [PubMed] [Google Scholar]
- 110.Leong R et al. DOP55 Long-term outcomes after histologic-endoscopic mucosal healing: results from the UNIFI study in ulcerative colitis. J. Crohns Colitis 16, i102–i103 (2022). [Google Scholar]
- 111.D’Haens G et al. OP26 Efficacy and safety of mirikizumab as induction therapy in patients with moderately to severely active ulcerative colitis: results from the phase 3 LUCENT-1 study. J. Crohns Colitis 16, i028–i029 (2022). [PMC free article] [PubMed] [Google Scholar]
- 112.Dignass A et al. OP23 The efficacy and safety of guselkumab induction therapy in patients with moderately to severely active ulcerative colitis: phase 2b QUASAR study results through week 12. J. Crohns Colitis 16, i025–i026 (2022). [PMC free article] [PubMed] [Google Scholar]
- 113.Sands BE et al. OP36 Efficacy and safety of combination induction therapy with guselkumab and golimumab in participants with moderately-to-severely active ulcerative colitis: results through week 12 of a phase 2a randomized, double-blind, active-controlled, parallel-group, multicenter, proof-of-concept study. J. Crohns Colitis 16, i042–i043 (2022). [PMC free article] [PubMed] [Google Scholar]
- 114.Sandborn WJ et al. Safety of ustekinumab in inflammatory bowel disease: pooled safety analysis of results from phase 2/3 studies. Inflamm. Bowel Dis. 27, 994–1007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Fiorentino D et al. Risk of malignancy with systemic psoriasis treatment in the Psoriasis Longitudinal Assessment Registry. J. Am. Acad. Dermatol. 77, 845–854 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Honap S et al. Effectiveness and safety of ustekinumab in inflammatory bowel disease: a systematic review and meta-analysis. Dig. Dis. Sci. 67, 1018–1035 (2022). [DOI] [PubMed] [Google Scholar]
- 117.Martinez-Barricarte R et al. Human IFN-γ immunity to mycobacteria is governed by both IL-12 and IL-23. Sci. Immunol. 3, eaau6759 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Teng MW et al. IL-12 and IL-23 cytokines: from discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 21, 719–729 (2015). [DOI] [PubMed] [Google Scholar]
- 119.Meeks KD, Sieve AN, Kolls JK, Ghilardi N & Berg RE IL-23 is required for protection against systemic infection with Listeria monocytogenes. J. Immunol. 183, 8026–8034 (2009). [DOI] [PubMed] [Google Scholar]
- 120.Abraham C & Cho J Interleukin-23/Th17 pathways and inflammatory bowel disease. Inflamm. Bowel Dis. 15, 1090–1100 (2009). [DOI] [PubMed] [Google Scholar]
- 121.Shih VF et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc. Natl Acad. Sci. USA 111, 13942–13947 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gaffen SL, Jain R, Garg AV & Cua DJ The IL-23–IL-17 immune axis: from mechanisms to therapeutic testing. Nat. Rev. Immunol. 14, 585–600 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Patel DD & Kuchroo VK Th17 cell pathway in human immunity: lessons from genetics and therapeutic interventions. Immunity 43, 1040–1051 (2015). [DOI] [PubMed] [Google Scholar]
- 124.Schulz SM et al. Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. J. Immunol. 181, 7891–7901 (2008). [DOI] [PubMed] [Google Scholar]
- 125.Schmitt H et al. Expansion of IL-23 receptor bearing TNFR2+ T cells is associated with molecular resistance to anti-TNF therapy in Crohn’s disease. Gut 68, 814–828 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Sandborn W et al. OP303 efficacy and safety of mirikizumab (LY3074828) in patients with moderate-to-severe ulcerative colitis in a phase 2 study. U. Eur. Gastroenterol. J. 6, A119–A120 (2018). [Google Scholar]
- 127.Doherty MK et al. Fecal microbiota signatures are associated with response to ustekinumab therapy among Crohn’s disease patients. mBio 9, e02120–02117 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sandborn WJ et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology 158, 537–549 (2020). [DOI] [PubMed] [Google Scholar]
- 129.Feagan BG et al. Induction therapy with the selective interleukin-23 inhibitor risankizumab in patients with moderate-to-severe Crohn’s disease: a randomised, double-blind, placebo-controlled phase 2 study. Lancet 389, 1699–1709 (2017). [DOI] [PubMed] [Google Scholar]
- 130.Gisbert JP & Chaparro M Predictors of primary response to biologic treatment [anti-TNF, vedolizumab, and ustekinumab] in patients with inflammatory bowel disease: from basic science to clinical practice. J. Crohns Colitis 14, 694–709 (2020). [DOI] [PubMed] [Google Scholar]
- 131.Dalal RS et al. Predictors and outcomes of ustekinumab dose intensification in ulcerative colitis: a multicenter cohort study. Clin. Gastroenterol. Hepatol. 10.1016/j.cgh.2021.03.028 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Dalal RS, Njie C, Marcus J, Gupta S & Allegretti JR Predictors of ustekinumab failure in Crohn’s disease after dose intensification. Inflamm. Bowel Dis. 27, 1294–1301 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lefevre PLC, Shackelton LM & Vande Casteele N Factors influencing drug disposition of monoclonal antibodies in inflammatory bowel disease: implications for personalized medicine. BioDrugs 33, 453–468 (2019). [DOI] [PubMed] [Google Scholar]
- 134.Wang Z et al. Population pharmacokinetic-pharmacodynamic model-based exploration of alternative ustekinumab dosage regimens for patients with Crohn’s disease. Br. J. Clin. Pharmacol. 88, 323–335 (2022). [DOI] [PubMed] [Google Scholar]
- 135.Bots SJ et al. Anti-drug antibody formation against biologic agents in inflammatory bowel disease: a systematic review and meta-analysis. BioDrugs 35, 715–733 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Adedokun OJ et al. Pharmacokinetics and exposure response relationships of ustekinumab in patients with Crohn’s disease. Gastroenterology 154, 1660–1671 (2018). [DOI] [PubMed] [Google Scholar]
- 137.Verstockt B et al. Ustekinumab exposure-outcome analysis in Crohn’s disease only in part explains limited endoscopic remission rates. J. Crohns Colitis 13, 864–872 (2019). [DOI] [PubMed] [Google Scholar]
- 138.Hanzel J et al. Peak concentrations of ustekinumab after intravenous induction therapy identify patients with Crohn’s disease likely to achieve endoscopic and biochemical remission. Clin. Gastroenterol. Hepatol. 19, 111–118 (2021). [DOI] [PubMed] [Google Scholar]
- 139.Adedokun OJ et al. Ustekinumab pharmacokinetics and exposure response in a phase 3 randomized trial of patients with ulcerative colitis. Clin. Gastroenterol. Hepatol. 18, 2244–2255 (2020). [DOI] [PubMed] [Google Scholar]
- 140.Alsoud D, Vermeire S & Verstockt B Monitoring vedolizumab and ustekinumab drug levels in patients with inflammatory bowel disease: hype or hope. Curr. Opin. Pharmacol. 55, 17–30 (2020). [DOI] [PubMed] [Google Scholar]
- 141.D’Haens GR et al. 775a risankizumab induction therapy in patients with moderate-to-severe Crohn’s disease with intolerance or inadequate response to conventional and/or biologic therapy: results from the phase 3 ADVANCE study. Gastroenterology 161, e28 (2021). [Google Scholar]
- 142.Sands BE et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with Crohn’s disease. Gastroenterology 158, 537–549 (2021). [DOI] [PubMed] [Google Scholar]
- 143.Danese S et al. OP28 The effect of guselkumab induction therapy on early clinical outcome measures in patients with Moderately to Severely Active Crohn’s Disease: Results from the phase 2 GALAXI 1 study. J. Crohns Colitis 15, S027–S028 (2021). [Google Scholar]
- 144.Strober B et al. Efficacy of risankizumab in patients with moderate-to-severe plaque psoriasis by baseline demographics, disease characteristics and prior biologic therapy: an integrated analysis of the phase III UltIMMa-1 and UltIMMa-2 studies. J. Eur. Acad. Dermatol. Venereol. 34, 2830–2838 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Irving PM et al. OP02 Ustekinumab versus adalimumab for induction and maintenance therapy in moderate-to-severe Crohn’s disease: the SEAVUE study. J. Crohns Colitis 15, S001–S002 (2021). [Google Scholar]
- 146.Singh S et al. Comparative efficacy and safety of biologic therapies for moderate-to-severe Crohn’s disease: a systematic review and network meta-analysis. Lancet Gastroenterol. Hepatol. 6, 1002–1014 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Singh S, Murad MH, Fumery M, Dulai PS & Sandborn WJ First- and second-line pharmacotherapies for patients with moderate to severely active ulcerative colitis: an updated network meta-analysis. Clin. Gastroenterol. Hepatol. 18, 2179–2191 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Burr NE, Gracie DJ, Black CJ & Ford AC Efficacy of biological therapies and small molecules in moderate to severe ulcerative colitis: systematic review and network meta-analysis. Gut 10.1136/gutjnl-2021-326390 (2021). [DOI] [PubMed] [Google Scholar]
- 149.Lasa JS, Olivera PA, Danese S & Peyrin-Biroulet L Efficacy and safety of biologics and small molecule drugs for patients with moderate-to-severe ulcerative colitis: a systematic review and network meta-analysis. Lancet Gastroenterol. Hepatol. 7, 161–170 (2022). [DOI] [PubMed] [Google Scholar]
- 150.Guillo L, D’Amico F, Danese S & Peyrin-Biroulet L Ustekinumab for extra-intestinal manifestations of inflammatory bowel disease: a systematic literature review. J. Crohns Colitis 15, 1236–1243 (2021). [DOI] [PubMed] [Google Scholar]
- 151.Li SJ, Perez-Chada LM & Merola JF TNF inhibitor-induced psoriasis: proposed algorithm for treatment and management. J. Psoriasis Psoriat. Arthritis 4, 70–80 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Puig L, Morales-Munera CE, Lopez-Ferrer A & Geli C Ustekinumab treatment of TNF antagonist-induced paradoxical psoriasis flare in a patient with psoriatic arthritis: case report and review. Dermatology 225, 14–17 (2012). [DOI] [PubMed] [Google Scholar]
- 153.Sandborn WJ et al. Five-year efficacy and safety of ustekinumab treatment in Crohn’s disease: the IM-UNITI trial. Clin. Gastroenterol. Hepatol. 20, 578–590 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Garg R et al. Real-world effectiveness and safety of ustekinumab in elderly Crohn’s disease patients. Dig. Dis. Sci. 10.1007/s10620-021-07117-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hayashi M et al. Efficacy and safety of ustekinumab treatment in elderly patients with psoriasis. J. Dermatol. 41, 974–980 (2014). [DOI] [PubMed] [Google Scholar]
- 156.Ferrante M et al. Long-term safety and efficacy of risankizumab treatment in patients with Crohn’s disease: results from the phase 2 open-label extension study. J. Crohns Colitis 15, 2001–2010 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.