

Abstract
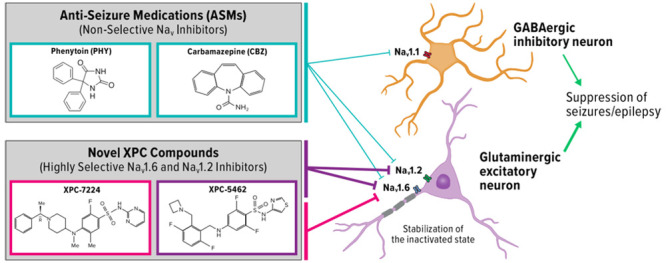
Voltage-gated sodium channel (NaV) inhibitors are used to treat neurological disorders of hyperexcitability such as epilepsy. These drugs act by attenuating neuronal action potential firing to reduce excitability in the brain. However, all currently available NaV-targeting antiseizure medications nonselectively inhibit the brain channels NaV1.1, NaV1.2, and NaV1.6, which potentially limits the efficacy and therapeutic safety margins of these drugs. Here, we report on XPC-7724 and XPC-5462, which represent a new class of small molecule NaV-targeting compounds. These compounds specifically target inhibition of the NaV1.6 and NaV1.2 channels, which are abundantly expressed in excitatory pyramidal neurons. They have a > 100-fold molecular selectivity against NaV1.1 channels, which are predominantly expressed in inhibitory neurons. Sparing NaV1.1 preserves the inhibitory activity in the brain. These compounds bind to and stabilize the inactivated state of the channels thereby reducing the activity of excitatory neurons. They have higher potency, with longer residency times and slower off-rates, than the clinically used antiseizure medications carbamazepine and phenytoin. The neuronal selectivity of these compounds is demonstrated in brain slices by inhibition of firing in cortical excitatory pyramidal neurons, without impacting fast spiking inhibitory interneurons. XPC-5462 also suppresses epileptiform activity in an ex vivo brain slice seizure model, whereas XPC-7224 does not, suggesting a possible requirement of Nav1.2 inhibition in 0-Mg2+- or 4-AP-induced brain slice seizure models. The profiles of these compounds will facilitate pharmacological dissection of the physiological roles of NaV1.2 and NaV1.6 in neurons and help define the role of specific channels in disease states. This unique selectivity profile provides a new approach to potentially treat disorders of neuronal hyperexcitability by selectively downregulating excitatory circuits.
Keywords: epilepsy, sodium channel, pharmacology, biophysics, precision medicine, antiseizure medication
Introduction
In the central nervous system (CNS), voltage-gated sodium channels (NaV) control initiation and propagation of action potentials (APs) in neurons.1−3 These properties establish NaV channels as attractive pharmacological targets for disorders of hyperexcitability, such as arrhythmias, epilepsy, and pain. Here, we introduce new pharmacological tools that enable a dissection of the function of NaV1.6 vs NaV1.2 and will help to define the role of each channel type in a variety of disease states. We have previously reported the antiseizure activity of a selective inhibitor of NaV1.6.4 Like NaV1.6, NaV1.2 is also preferentially expressed in excitatory circuits in the CNS. In this report, we introduce two pharmacological agents that are very similar in inhibition of NaV1.6, but dramatically different as inhibitors of NaV1.2. This will enable in vivo studies to evaluate the impact of NaV1.2 inhibition.
In the adult CNS, four NaV subtypes are highly expressed: NaV1.1, NaV1.2, NaV1.3, and NaV1.6.5 NaV1.2 and NaV1.6 are the major subtypes found in excitatory neurons, whereas NaV1.1 is predominantly expressed in inhibitory interneurons.6 Several lines of evidence from mouse and human studies now support the parsing of NaV subtypes into excitatory and inhibitory networks, including many studies indicating that gain-of-function (GOF) in NaV1.2 and NaV1.6 and loss-of-function (LOF) in NaV1.1 are linked to epilepsy, a disorder of hyperexcitability.6−15 The selective expression of NaV subtypes in excitatory and inhibitory circuits rationalizes a selective pharmacological approach in disorders of excitability that targets the excitatory networks while sparing the inhibitory networks.
NaV channels are heteromultimeric proteins composed of large ion conducting α-subunits and smaller auxiliary β-subunits.16 The α-subunit is made up of a single transcript that encodes four 6-transmembrane segment domains. Each one of these four structural domains can be divided into two functional subdomains known as the voltage-sensing domain (VSD) and the pore domain (PD). These two functional domains are connected through the intracellular S4–S5 linker to control the gating. NaV channels cycle through three basic gating states: rest, open, and inactivated. During depolarizations of sufficient magnitude, outward movement of VSDI-III translate changes in transmembrane potential into channel activation through an electromechanical coupling process to the activation gate at the inner pore.17−19 The channels then rapidly enter a fast inactivated state, mediated by the outward movement of VSD-IV. VSD-IV is specialized in its function in controlling inactivation with a hyperpolarized voltage dependence and a slightly slower transition rate to the UP state than VSDI-III and has been shown to be necessary and sufficient for inactivation.17,20 The hyperpolarized voltage dependence of VSD-IV compared to VSDI-III also enables the channels to enter inactivated states without channels opening, known as closed or steady-state inactivation.21 Inactivation is one of the mechanisms by which channel availability varies across different resting membrane potentials between neuronal types and subcompartments of the cell and during neuronal activity.22 S4 movement during inactivation triggers an interaction of the hydrophobic motif (IFM) in the DIII-IV linker with another region of the channel.23 Structural studies have established the basis of inactivation as an allosteric mechanism where the IFM motif binds into the space between S6 and S4–S5 and translates into a constriction that closes the permeation pathway.24,25 Importantly, at typical CNS neuronal resting membrane potentials (RMP) of around −70 to −80 mV the channels will be distributed between resting and inactivated states as RMP is often close to the voltage at which channels are equally distributed (inactivation V0.5).26
Most small molecules that are known to inhibit NaV channels have been nonselective, a function of the pore-binding site where key residues are highly conserved across the paralogues.27−31 These molecules have found wide-ranging clinical efficacy in disorders of excitability, including pain, arrhythmia, and epilepsy. Compounds such as carbamazepine (CBZ) and phenytoin (PHY) however, have been associated with narrow safety margins. Side-effects can appear at doses that overlap with therapeutic doses and may result from overinhibition of NaV currents and/or off-target interactions.32−34 The mechanism by which NaV channels are inhibited by these molecules is multifactorial and includes physical occlusion of the ion conductance,29 stabilization of inactivated states,30 and immobilization of voltage-sensors.35
More recently, a new class of selective NaV-inhibiting small molecules were identified that interact with the extracellular part of VSD-IV and exploit the sequence diversity among the paralogues at this site.36 These compounds bind to the activated state of VSD-IV and make an important electrostatic interaction with the fourth arginine residue on VSD-IV-S4 that traps the VSD-IV in the UP state and thus maintains the channel in a nonconducting, inactivated state.37,38 Initial compounds of this type potently inhibited NaV1.2, NaV1.6, and NaV1.7 and were highly selective against NaV1.1 and NaV1.5. Moreover, these compounds had very low CNS penetration. We have now discovered a new CNS penetrant class of VSD-IV-targeting compounds with an unprecedented selectivity profile, targeting NaV1.6 channels alone,4 or NaV1.6 and NaV1.2 simultaneously for use in epilepsy.39 Rodent seizure efficacy data indicate a greater therapeutic index results from NaV1.6 selective inhibition compared with the nonselective antiseizure medications (ASMs), CBZ and PHY, supporting the potential of increased safety margins for selective compounds.4
Here, we describe the comparative molecular pharmacology of the NaV1.6 selective XPC-7224 and the dual inhibitor of NaV1.6 and NaV1.2, XPC-5462, to the clinically used nonselective NaV-blocking ASMs, PHY and CBZ. We characterize several important biophysical and pharmacological differences between these compounds. The XPC compounds bind distinct sites of the channel, have exquisite selectivity for excitatory NaV subtypes, and display radically different kinetic profiles and state dependence of potency when compared with CBZ and PHY. The proposed mechanism for seizure reduction of this pharmacological profile is then demonstrated in patch-clamp recordings from excitatory and inhibitory neurons in adult mice and tested in multielectrode array (MEA) seizure model in acute brain slices. By exploring the differences in selectivity, kinetics, and state dependence, we propose that steady-state inhibition from resting membrane potentials is mechanistically sufficient for reducing excitability in CNS neurons and suppressing seizure-like activity in brain slices.
Results
XPC-7224 and XPC-5462 Are Molecularly Selective Inhibitors of NaV1.6 and NaV1.6/1.2, Respectively
State dependence is a common feature of small molecule NaV channel inhibitors, where compounds interact more strongly with the inactivated state of the channel.30,40 For instance, traditional local anesthetics target the inner pore cavity to inhibit inactivated or open states of the channel with greater potency than closed/rested states. This is also true for ASMs, such as PHY and CBZ (Figure 1A) that target the same binding site.27,41 The increased potency is proposed to result from a favorable structural configuration in the pore cavity in the inactivated state that increases binding affinity.42
Figure 1.
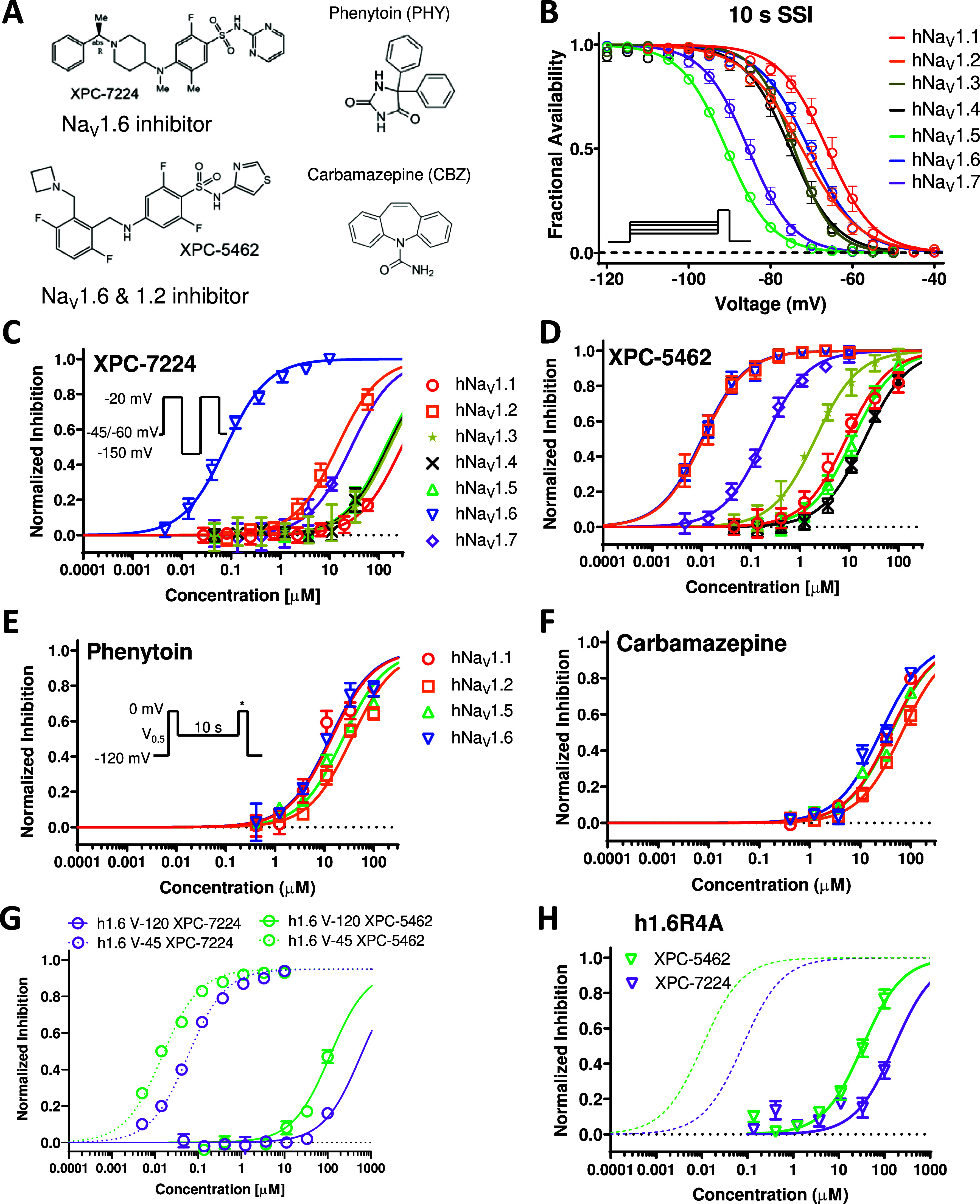
Comparative pharmacology of molecularly selective state dependent inhibitors compared with nonselective ASMs. (A) Structures of XPC compounds and classic pore-blocking ASMs, PHY and CBZ. (B) Normalized inactivation curves showing different voltage dependence of inactivation across human NaV subtypes hNaV1.1 (n = 11 cells), hNaV1.2 (n = 12 cells), hNaV1.5 (n = 63 cells), hNaV1.6 (n = 13 cells), and hNaV1.7 (n = 8 cells) (C) Potency of XPC-7224 plotted as fractional inhibition of different channel subtypes and fitted with a Hill equation, n = 3–11 cells per concentration. (D) Potency of XPC-5462, n = 3–11 cells per concentration. (E, F) Potency of the pore blocking ASMs measured using a protocol to capture compounds with fast off rates, n = 3–9 cells per concentration. (G) Membrane holding voltage dependence of potency for XPC-7224 (n = 3–5 cells per concentration) and XPC-5462 (n = 3–7 cells per data point). (H) Potencies for h1.6R4A channels for XPC-7224 (n = 3–6 cells per concentration) and XPC-5462 (n = 3–7 cells per concentration).
Selective NaV channel VSD-IV-targeting compounds are highly state-dependent, where high affinity binding is dependent upon an interaction with the UP state of VSD-IV that controls the inactivated state.37 For accurate measurement of subtype molecular selectivity in voltage-clamp experiments, it is vital to control the channel’s state and apply a transmembrane voltage that ensures equivalent state occupancy for compound binding. To establish this, we assessed the steady-state availability of the subtypes and found V0.5 and slope parameters (Figure 1B and Table S1). Next, concentration responses were measured for the compounds XPC-7224, XPC-5462, PHY, and CBZ at holding potentials where channels were maintained at fully inactivated potentials based on the fractional availability of the NaV channels (Figure 1B). The selectivity profile of the NaV1.6 compound XPC-7224 indicates that the IC50 for NaV1.6 was 0.078 μM (95% CI 0.072–0.085 μM), which was >100-fold more potent than all other NaV subtypes tested (Figure 1C). The inhibitory selectivity profile for a distinct chemical series, exemplified by XPC-5462, demonstrates equipotent inhibition of NaV1.2 and NaV1.6 with IC50 values in the low nanomolar range (NaV1.2 IC50 = 0.0109 μM [95% CI 0.00968–0.0122 μM] and NaV1.6 IC50 = 0.0103 μM [95% CI 0.00921–0.0115 μM]) (Figure 1D and Table 1).
Table 1. Potency of NaV Channel Subtype Inhibition of Compounds.
| XPC-7724 |
XPC-5462 |
phenytoin |
carbamazepine |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 | 95% CI | N (cells) | IC50 | 95% CI | N (cells) | IC50 | 95% CI | N (cells) | IC50 | 95% CI | N (cells) | |
| hNaV1.1 | 294 | 255–354 | 71 | 8.75 | 7.53–10.2 | 62 | 13.8 | 9.59–20.1 | 26 | 39.3 | 33.3–46.7 | 41 |
| hNaV1.2 | 14.3 | 12.2–16.9 | 32 | 0.0109 | 0.00968–0.0122 | 49 | 35.2 | 2.1–44.3 | 26 | 67.4 | 57.3–79.6 | 35 |
| hNaV1.3 | 167 | 117–268 | 45 | 206 | 1.85–2.30 | 63 | ||||||
| hNaV1.4 | 153 | 123–197 | 41 | 20.8 | 19.1–22.7 | 42 | ||||||
| hNaV1.5 | 138 | 103–197 | 15 | 11.9 | 10.4–13.5 | 27 | 23.8 | 19.2–29.6 | 23 | 41.1 | 35.3–47.8 | 39 |
| hNaV1.6 | 0.078 | 0.0718–0.0848 | 70 | 0.0103 | 0.000921–0.0115 | 50 | 13 | 10.1–16.9 | 29 | 25.6 | 21.1–30.8 | 45 |
| hNaV1.7 | 27.7 | 20.4–40.1 | 45 | 0.192 | 0.175–0.211 | 54 | ||||||
| mNaV1.6 | 0.13 | 0.103–0.164 | 30 | 0.0137 | 0.0112–0.0169 | 30 | ||||||
The potency of these compounds on mouse NaV1.6 channels was also found to be very similar to human channels suggesting no orthologue difference (Figure S1 and Table 1). The fully inactivated state protocol was not used with PHY and CBZ, because with these compounds’ inhibition is removed during the 20 or 60 ms hyperpolarization to −150 mV that is used to recover channel availability in the fully inactivated state protocol. Therefore, for PHY and CBZ, potency was tested by holding cells at the empirically determined inactivation V0.5 potential for each cell for 10 s before a test pulse, thereby allowing measurement of inhibition promoted by the high-affinity binding to the inactivated state. PHY and CBZ were much lower in potency and effectively nonselective with IC50 values within 3-fold for CNS channels and NaV1.5 (Figure 1E,F, Table 1). These results establish the molecular selectivity and increased potency of this new class of XPC compounds relative to those of traditional NaV targeting ASMs.
XPC-7224 and XPC-5462 Potency Are Highly Inactivated State-Dependent
We next tested the XPC compounds’ potency at a membrane potential of −120 mV, where the channels were fully available at rest. The potency for NaV1.6 was reduced by >1000-fold from 0.078 μM to >100 μM for XPC-7224 (n = 31 cells) and from 0.010 μM to >100 μM (n = 32 cells) for XPC-5462, indicating very strong preference for interacting with the inactivated state of the channel (Figure 1G).
High-Potency Inhibition by XPC-7224 and XPC-5462 Depends on the Fourth Positively Charged Residue on the VSD-IV-S4 (R1626) in hNaV1.6
A previous study using X-ray crystallography in NaV1.7 determined that an aryl sulfonamide (GX-674) compound binds the VSD-IV segment.37 This interaction between VSD-IV and the compound is mediated via the negatively charged sulfonamide group of GX-674 and the fourth positively charged arginine side chain (R4) on VSD-IV-S4 (R1608).37 Functional studies also demonstrated that neutralizing the R4 charge with alanine mutagenesis (h1.7-R1608A) led to loss of high-affinity binding.36,37 These XPC selective inhibitors are also aryl sulfonamides (Figure 1A) but differ substantially in chemical scaffold from previously developed NaV1.7 targeting compounds. To test if the corresponding NaV1.6 VSD-IV-S4 arginine side chain (R1626) comprises part of the high-affinity binding site for these compounds, we tested NaV1.6 (R1626A). The inactivation gating of the mutant was found to be more stable with an ∼20 mV left-shifted V0.5 compared with WT (Figure S2). The R1626A point mutation resulted in a > 1000-fold decrease in potency with an IC50 of 166 μM (95% CI 120–239 μM, n = 29 cells) and 33.6 μM (95% CI 27.1–41.8 μM, n = 34 cells) for XPC-7224 and XPC-5462, respectively (Figure 1H). This positively charged residue on hNaV1.6 VSD-IV-S4, therefore, comprises a critical part of the high-affinity binding interaction for these compounds. These findings highlight the similarities between XPC compounds and the GX-674 interaction with NaV1.7, establishing the molecular basis for the strong state-dependent binding of XPC-7224 and XPC-5462 to hNaV1.6.
XPC-7224 and XPC-5462 Delay Recovery from Inactivation
Previous studies have established that recovery from inactivation must occur before channels can reopen and the time dependence of this process reflects the return of VSD-IV to the down state.43 We measured the concentration dependence of the recovery from inactivation of NaV1.6 in the presence of the compounds and found that the compounds introduced a slow component for channel recovery (Tslow) (Figure 2A–D). XPC-7224 and XPC-5462 had a Tslow of ∼20 s, PHY had a Tslow of ∼3 s, while CBZ did not exhibit a slower component in the recovery rate. The Tslow fraction of recovery increased as the compound concentration increased. Figure 2E shows a plot of the concentration dependence of Tslow and indicates that it did not vary significantly with concentration. As the ligand unbinding rate (koff) is independent of concentration, these data support that Tslow reflects the unbinding rate of the compounds at −120 mV, which is rate limiting the return of VSD-IV back to the resting state. As no change was detected for CBZ, this suggests that the unbinding rate for this compound was much faster than the rate of recovery of the channel from inactivation.
Figure 2.
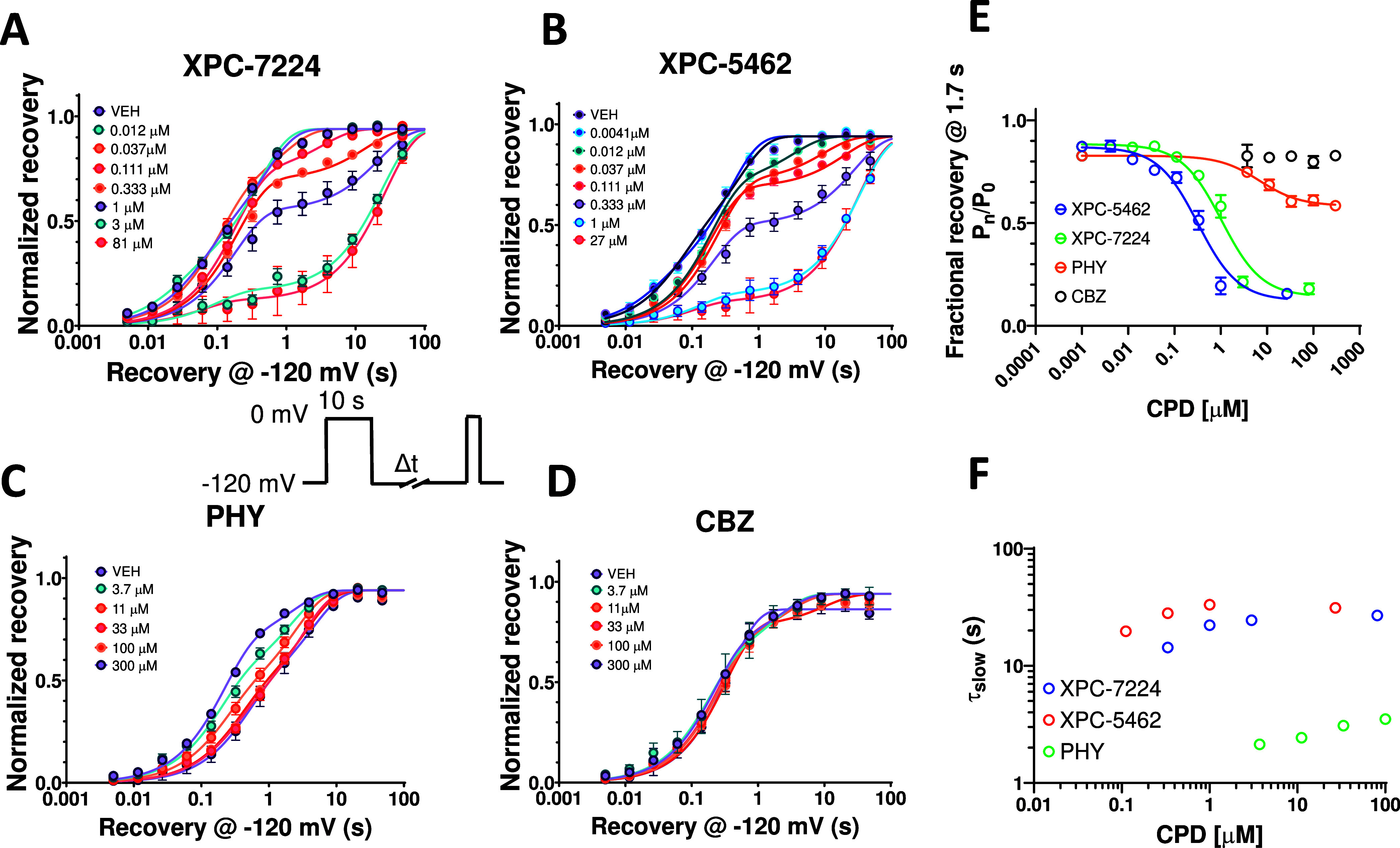
Recovery from inactivation is limited by compound unbinding. Plots of normalized recovery from inactivation in NaV1.6 channels induced by a 10s prepulse to 0 mV in the presence of different concentrations of compounds at −120 mV measured using the protocol shown in inset. (A) XPC-7224, n = 4–12 cells per concentration (B) XPC-5462, n = 3–8 cells per concentration (C) PHY, n = 6–17 cells per concentration (D) CBZ, n = 3–13 cells per concentration. (E) Time constants of the slow component of recovery induced by compound binding against concentration of compound measured at 1.7 s. (F) Plot of the fractional recovery after 1.7 s against compound concentration for XPC-7224 (n = 78 cells), XPC-5462 (n = 63 cells), and PHY (n = 109 cells). Mean data were fit with a double exponential function.
We quantified the concentration dependence of the fraction Tslow by using the recovery at 1.7 s, a time-point that best separates Tfast and Tslow where unbound channels have recovered and bound ones have not (Figure 2F). This representation of the fraction that Tslow varied with concentration and was analyzed with the Hill equation to give IC50 values of 1.04 μM (95% CI 0.836–1.29 μM, n = 78 cells) and 0.322 μM (95% CI 0.252–0.413 μM, n = 63 cells) for XPC-7224 and XPC-5462, respectively, and 6.94 μM (95% CI 3.10–14.2 μM, n = 109 cells) for PHY. Interestingly, these IC50 values are lower than the potency for the fully inactivated-state shown in Table 1 and Figure 1 for XPC-7224 and XPC-5462 but they are similar to the PHY IC50. These data indicate that XPC compounds require more than 10 s to fully equilibrate with the inactivated states of the channel whereas PHY equilibrated within 10 s. This suggests that XPC compounds have slower binding kinetics than PHY.
XPC-7224 and XPC-5462 Have Slower Binding Kinetics Compared with PHY and CBZ
Equilibrium IC50 measurements are commonly used as a proxy for the dissociation constant (Kd), assuming that inhibition is tightly coupled to binding. Equilibrium measurements, however, yield no information about the kinetics of binding, which may play an important role in the physiological activity of a drug and are determined by intrinsic chemical rates of association and dissociation (kon and koff). kon and koff are proportional to the transitional state energy barriers encountered by a ligand when engaging or dissociating from the binding site and, therefore, give information about what is driving potency; the rate of binding or unbinding or a combination of both.3 We measured the kinetics of binding to NaV1.6 channels using a protocol that tracks the inhibition over 100 s of holding at the V0.5 (Figure 3A). The potencies for the compounds are measured at the end of 100 s at the V0.5 (Figure 3B). The mean normalized rate of inhibition was observed at approximately 3 times the IC50 of each compound to enable comparison of the kinetics at a concentration that is proportional to the equilibrium IC50 (Figure 3C). The mean data were fit with a single exponential function to give the following Tobs values; XPC-5462:36.8 s (95% CI 34.9–38.8 s, n = 69 cells), XPC-7224:84 s (95% CI 62.1–125 s, n = 65 cells), CBZ: 0.29 s (95% CI 0.182–0.419 s, n = 52 cells), and PHY: 1.86 s (95% CI 1.67–2.06 s, n = 56 cells). CBZ and PHY are >100× and > ∼20× faster, respectively, to equilibrate than XPC compounds, highlighting a profound kinetic distinction between the mechanisms of action of traditional pore-blockers and our XPC compounds.
Figure 3.
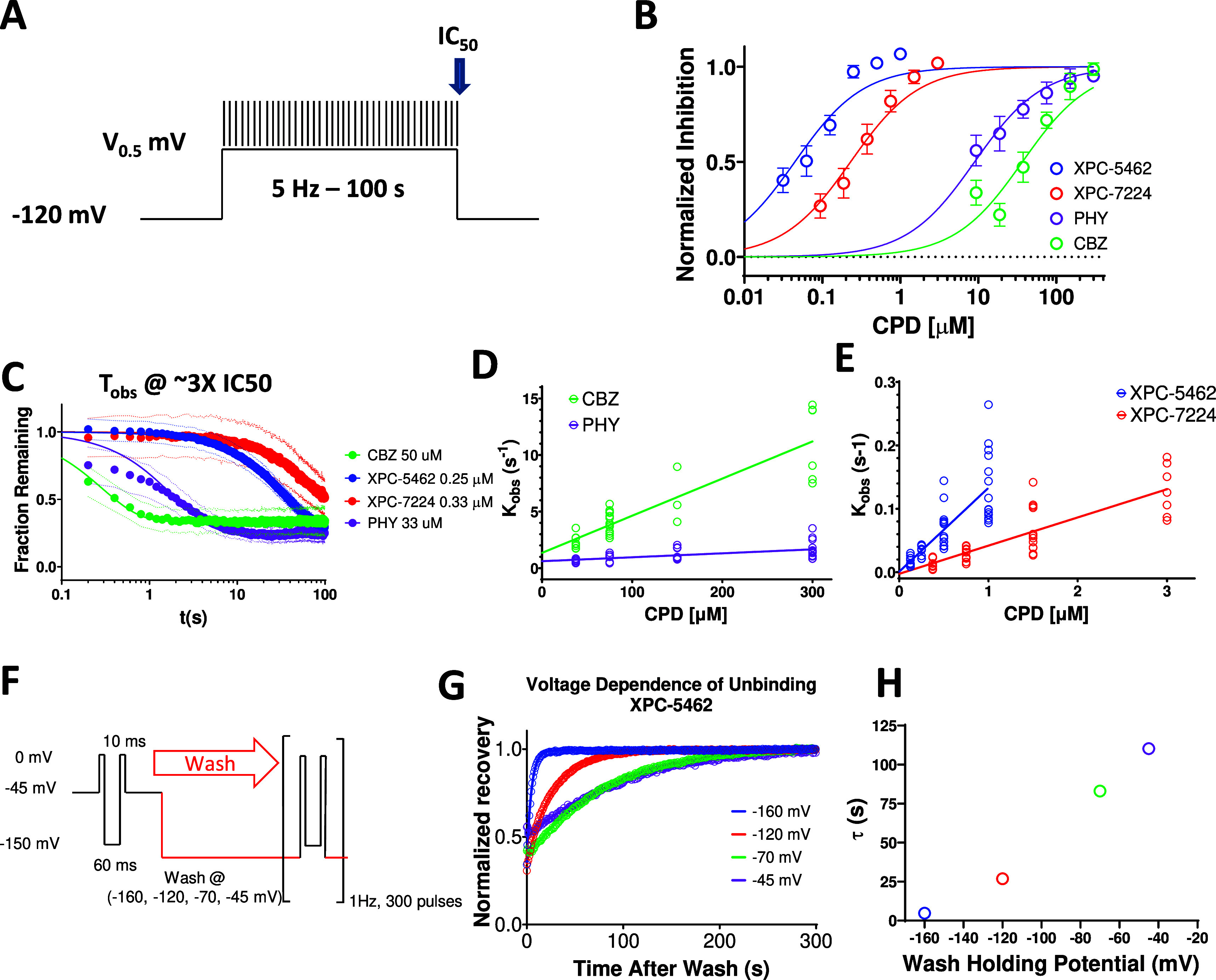
VSD-IV-binding XPC compounds display slower kinetics than typical pore binding ASMs. (A) Protocol used to establish kinetics of compound equilibration at a V0.5 holding potential where voltage is stepped to the V0.5 for 100 s while applying a test pulse at 5 Hz to assess the rate of inhibition. (B) Plot of normalized concentration response fit with the Hill equation to calculate IC50 values for NaV1.6 inhibition for XPC-5462 (n = 7 cells), XPC-7224 (n = 3 cells), PHY (n = 4 cells), and CBZ (n = 3 cells). (C) Normalized equilibration rates of compounds at approximately 3× the IC50 concentrations, dots represent 95% CI (n = 3–7 cells per compound). (D, E) Plot of Kobs against concentration fit with an equation for a straight line to give kon for CBZ (n = 3–13 cells per concentration), PHY (n = 7–14 cells per concentration), XPC-7724 (n = 7–12 cells per concentration), and XPC-5462 (n = 8–15 cells per concentration). (F) Protocol used to assess the voltage dependence of the unbinding rate in which the compound is equilibrated with the CPD in the inactivated state at −45 mV before simultaneously washing the CPD while stepping the voltage. (G) Plot of normalized fractional recovery against time for different holding voltages fit with single exponential functions. (H) Time constants of the recovery rate plotted against membrane voltage during the wash.
By assessing the Tobs over a wider range of increasing concentrations starting at approximately the IC50 (Figure S3), the kon for each compound can be estimated.44 We found that Tobs was concentration-dependent (Figure 3D,E), as expected from a pseudo-first-order kinetic reaction scheme in which the rate of relaxation following a concentration change is described by Kobs = kon[CPD] + koff where [CPD] is the concentration of XPC and Kobs = 1/Tobs. By plotting the Kobs against concentration, we obtained fitted parameters for kon, which for XPC-5462 was 1.32 × 105 M–1 s–1 (95% CI 1.02–1.62 × 105 M–1 s–1), XPC-7224 0.445 × 105 M–1 s–1 (95% CI 0.344–0.546 × 105 M–1 s–1), CBZ 0.328 × 105 M–1 s–1 (95% CI 0.263–0.393 × 105 M–1 s–1) and PHY 0.0349 × 105 M–1 s–1 (95% CI 0.0183–0.0515 × 105 M–1 s–1). Interestingly, the kon rates for CBZ and PHY were similar to or slower than the kon for XPC-7224 and XPC-5462 suggesting that the greater potency of XPC compounds is driven by more stable binding and longer residence times (Figure 3C). Therefore, the kinetic differences at potency matched concentrations are driven by a combination of slower koff and largely the lower concentrations of XPC compounds which reduces the rate of equilibration by mass action (kon*[CPD]).
Off Rates Are Voltage-Dependent and Reflect Compound Unbinding
As the membrane voltage is not constant in the neuronal physiological environment, we sought to understand the effect of membrane voltage on koff. To assess koff directly, we equilibrated the compounds with the channels at a holding-potential of −45 mV with test-pulses every 10 s for 5 min (Figure 3F). Next, the membrane voltage was simultaneously clamped to a negative holding potential, and the compound solution was exchanged for the control solution. The experiment was executed with XPC-5462 at a concentration close to the IC50 value (20 nM), to ensure that the wash step fully cleared all residual compound from the microfluidic chamber housing each cell. The recovery of current after washing with XPC-5462 was normalized to the recovery of control cell currents that were on the same plate but only exposed to vehicle and had accumulated some inactivation over the −45 mV holding period (Figure 3G). The normalized recovery traces were fit with single exponential functions to extract Toff at different membrane holding-potentials (Figure 3H). The more negative membrane potentials had the effect of accelerating XPC-5462 unbinding (Figure 3H). As the downward force on VSD-IV-S4 is increased with hyperpolarization, the high-affinity binding site becomes deformed, causing the compound to dissociate.
Membrane Potential Modulates the Apparent Potency of XPC-7224 and XPC-5462 Through Controlling the State of the VSD-IV
To examine the membrane voltage dependence of inhibition of NaV1.6 over a physiologically accessible voltage range, the compound’s apparent potency (IC50, app) was measured at voltages where channel inactivation rapidly changes.41 Potency was assessed by increasing the voltage in a stepwise manner from −90 to −70 with 180 s intervals at each voltage (Figure 4A). The voltage dependence of inactivation before compound application had a V0.5 of −76.5 mV (95% CI −77.0 to −75.9 mV) and slope of 5.76 (95% CI 5.06–6.54, n = 46 cells) (Figure 4B). To find the membrane potential dependence of IC50, app the normalized mean fractional inhibition from the last (180th) pulse as a function of compound concentration was fit with a Hill equation. The strong membrane voltage dependence of XPC-7224 and XPC-5462 potency varied over 20 mV from −90 to −70 mV by 100-fold, from a low μM IC50, app to the nM range (Figure 4C,D, Table S2). For comparison, the curves for the fully inactivated state potency held at −45 mV is shown in dashed lines for XPC-7224 and XPC-5462. The membrane voltage dependence of IC50, app for PHY and CBZ is lower with only a 10-fold difference between −90 mV and −70 mV (Figure 4E,F).
Figure 4.
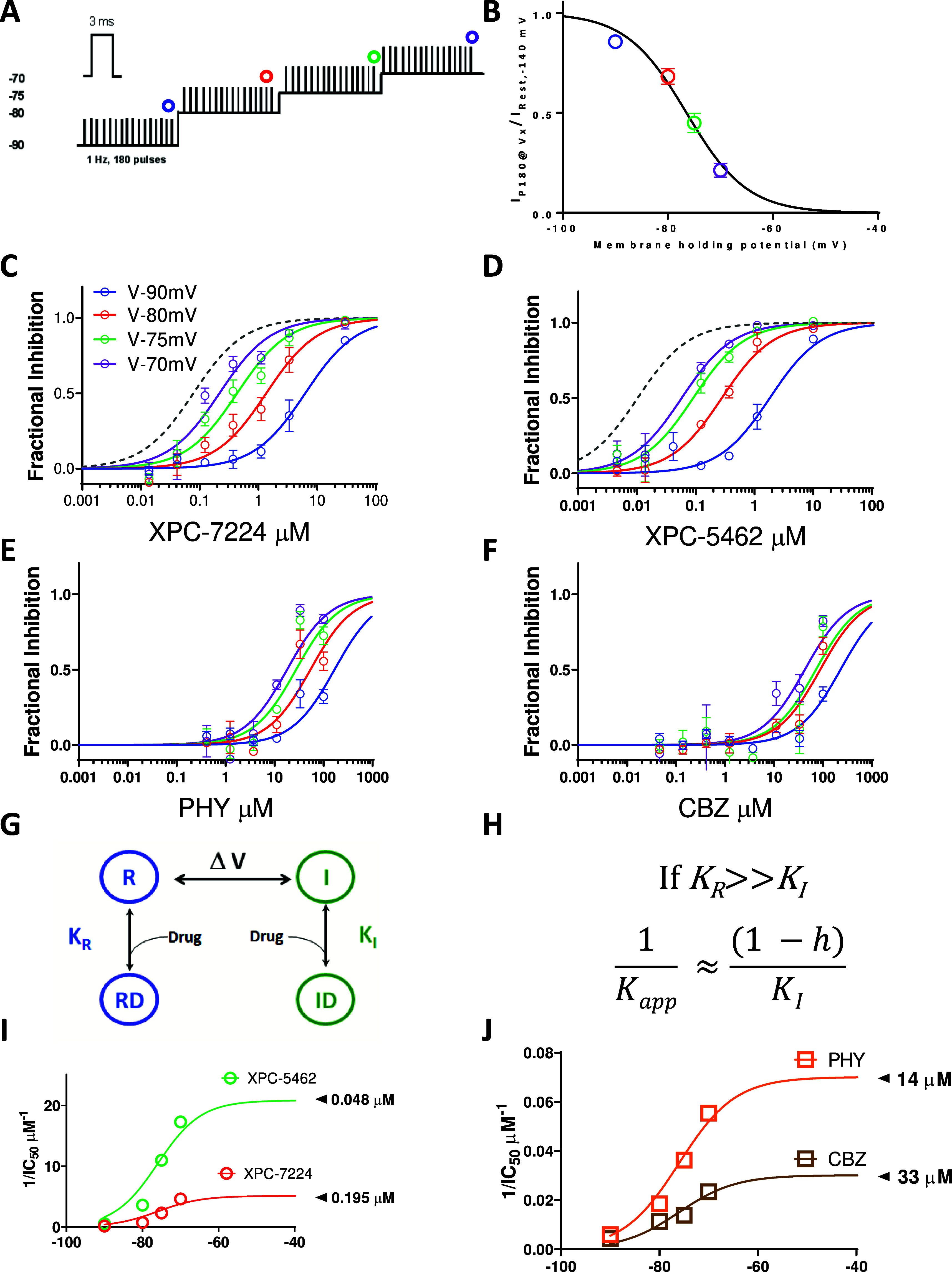
State dependence of hNaV1.6 inhibition. (A) Protocol used to assess state dependence of potencies. 180, 3 ms pulses to 0 mV were applied at 1 Hz at each holding potential. (B) Voltage dependence of inactivation in the protocol shown previously. After 3 min at each holding potential the availability of channels was measured and normalized to resting state availability and plotted against voltage (n = 46 cells) (C–F) Normalized fractional inhibition of channels measured at the 180th pulse at different voltages as indicated for XPC-7224 (n = 4–7 cells per concentration), XPC-5462 (n = 3–8 cells per concentration), PHY (n = 3–10 cells per concentration), and CBZ (n = 3–9 cells per concentration). In gray dashed line is the curve fit from fully inactivated NaV1.6 channels held at −45 mV (Figure 1). (G) Four-state scheme used to model interaction of compounds with inactivated or resting state channels. (H) Model prediction for variation of potency with state occupancy. (I, J) Plots of 1/IC50 fitted with the model.
To test if the shift in potency correlated with the fractional availability of the inactivated state, we tested a four-state binding model that assumed binding affinity to the inactivated state was much higher than the resting state (Figure 4G,H).41 The model predicts that the steady-state affinity (Kapp) will vary proportionally to the fraction of channels in the inactivated state according to the equation in Figure 4H where h represents the probability of inactivation curve from Figure 4B. The data are well fit by this equation and the projected maximal potencies (Figure 4I,J) are close to the inactivated state IC50’s previously described (Table 1). These data suggest that the potency of the compounds is directly determined by the proportion of channels in the inactivated state and is indirectly controlled by the membrane voltage.
XPC-7224 and XPC-5462 Inhibit Sodium Currents Independently of High Frequency Firing
To assess the use dependence of our VSD-binders, we used a protocol that measured use dependence from the resting states and from a partially inactivated state (empirically derived V0.5), which better represents neuronal RMP (Figure 5A). The mean V0.5 for all cells was −62.6 ± 0.269 mV, n = 177. The normalized current inhibition at first (steady resting-state inhibition) and 500th pulse (use-dependent inhibition) in each of the four compounds from the −120 mV holding potential show that there is minimal inhibition at any concentration due to the previously described state dependence (Figure 5B). Figure 5C shows the partially inactivated steady-state and use-dependent potency from a V0.5 holding-potential. The IC50 for steady-state inhibition from a V0.5 holding potential (P1) for XPC-7224 was 0.196 μM (95% CI 0.154–0.250 μM, n = 49 cells); XPC-5462 0.0376 μM (95% CI 0.0305–0.0462 μM, n = 41 cells), PHY 12.1 μM (95% CI 10.3–14.2 μM, n = 32 cells), and CBZ 17.0 μM (95% CI 13.4–21.7 μM, n = 25 cells). For UDB (P500) from V0.5 the IC50 for XPC-7224 was 0.197 μM (95% CI 0.152–0.258 μM); XPC-5462 0.0334 μM (95% CI 0.0261–0.0426 μM), PHY 6.80 μM (95% CI 5.67–8.14 μM) and CBZ 16.2 μM (95% CI 11.9–21.9 μM). The results indicate that neither the pore-blocking PHY nor our VSD-blockers have a large difference in the steady-state vs use-dependent potency from either −120 mV or the V0.5. This suggests that the VSD-IV-targeting XPC compounds might act primarily through interacting with and inhibiting the steady state rather than increasing inhibition over short high-frequency bursts. Given that selective VSD-binders show efficacy in animal models4 these data suggest that use-dependent block is not required for inhibition of hyperexcitability from moderate membrane voltages.
Figure 5.
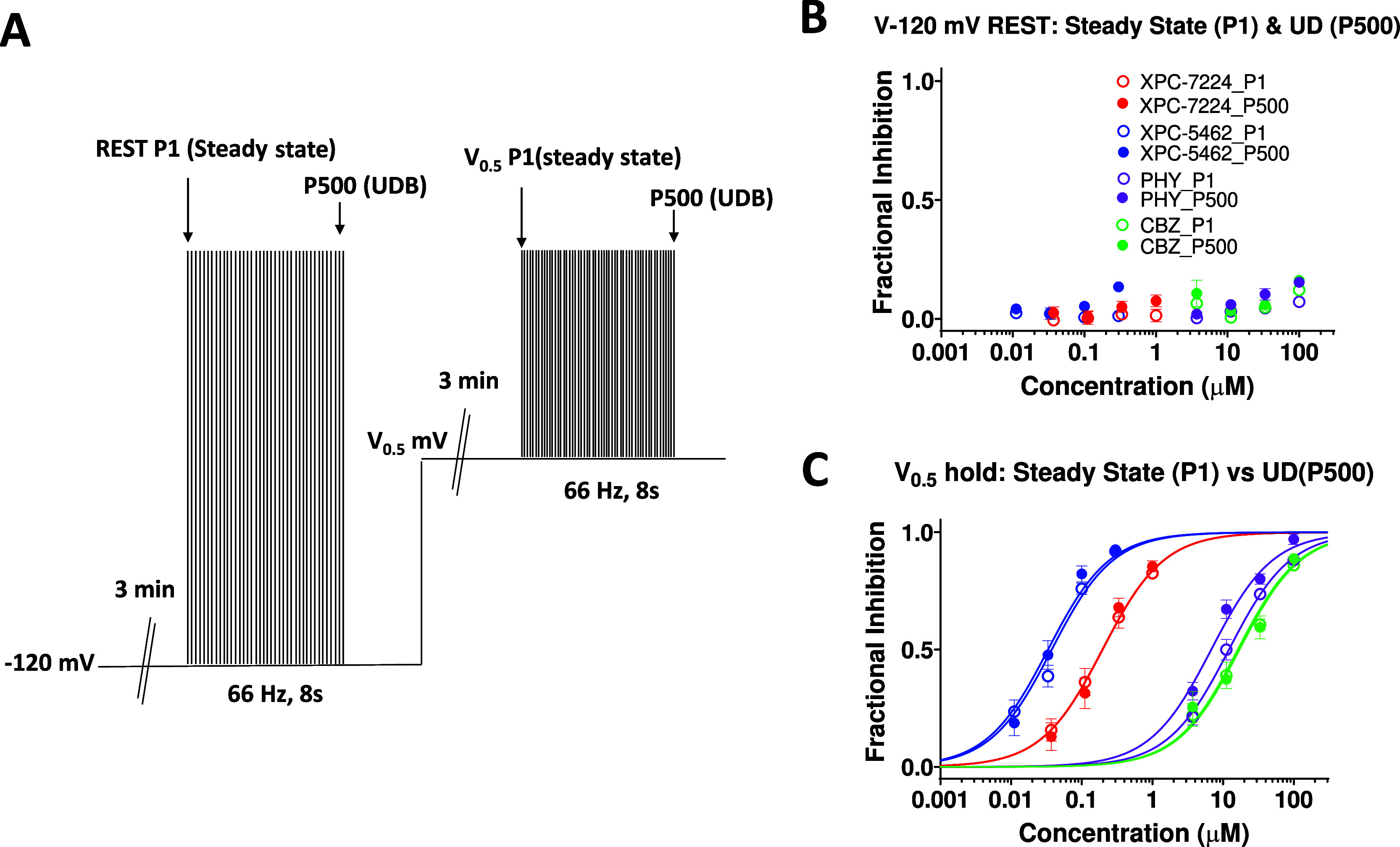
Steady-state inhibition from resting membrane potentials is sufficient for inhibition. (A) Protocol used to assess steady-state and use-dependent inhibition from −120 mV or the empirically derived V0.5. (B, C) Plot of concentration response of normalized inhibition at either the first pulse (steady state) or the 500th pulse (use dependent) from a holding potential of −120 mV (B) or V0.5 (C). n = 4–15 cells per concentration.
Inhibiting NaV1.6 or NaV1.6 and 1.2 Selectively Suppresses Excitatory Cortical Neurons Ex Vivo
To test the cellular selectivity of these compounds, we recorded the intrinsic excitability of both excitatory and inhibitory neurons in layer 5 of the somatosensory cortex in brain slices. The selective inhibitor of NaV1.6, XPC-7224 and the dual inhibitor of NaV1.6 and NaV1.2, XPC-5462, at 500 and 150 nM, respectively, suppressed action potential firing in excitatory neurons, but spared inhibitory interneurons (Figure 6A,B,D,E). In contrast, 100 μM nonselective pore-targeting inhibitor CBZ inhibited both excitatory and inhibitory cells (Figure 6C,F). We chose the concentrations of compounds to be approximately 3-fold higher than the potency determined at −70 mV (Table S2), which is −77.6 mV with liquid junction potential (LJP) corrected, to approximately match LJP corrected RMP in these neurons (pyramidal cell RMP was −78.4 ± 2.00 mV, n = 13; fast spiking interneuron RMP was −77.7 ± 1.00 mV, n = 10). These data confirm that the NaV1.6 and 1.6/1.2 dual-targeting molecularly selective inhibitors act as specific excitatory neuron inhibitors.
Figure 6.

NaV1.6- and NaV1.6 and 1.2-targeting compounds selectively suppress action potentials in cortical excitatory pyramidal cells. Representative voltage traces from current clamp recordings of cortical neurons (A–C) and fast spiking interneurons (E, F) from selected depolarizing current injections before and after 10 min of incubation with the stated concentration of compounds. Middle panels show input–output plots of the number of APs fired normalized to the maximum number of APs of the vehicle control in the same cell against depolarizing current injection magnitude. Right panels show plots of the absolute maximum number of APs from the range of current injections in vehicle or compound. (A) XPC-7224 at 500 nM significantly inhibited AP firing from pyramidal cells (n = 3) but not fast spiking interneurons (n = 3) (D). (B) XPC-5462 at 150 nM significantly inhibited AP firing from pyramidal cells (n = 4) but not fast spiking interneurons (n = 3–4) (E). (C) CBZ at 100 μM significantly inhibited AP firing from pyramidal cells (n = 4–6) and fast spiking interneurons (n = 3) (B). Statistical significance between vehicle and compound number of APs at each current injection was tested using a two-way ANOVA followed by Bonferroni multiple comparisons test (*P <0.05). Student’s two-way paired t tests were used to test the significance of the maximal number of APs fired by each cell in vehicle or compound (*P <0.05, **P <0.01).
Suppression of AP Firing from CA1 Pyramidal Cells Is Modulated by Membrane Voltage
As XPC-7224 and XPC-5462 are state-dependent inhibitors (Figure 4) we hypothesized that they should suppress neuronal excitation more at depolarized membrane potentials. To test this hypothesis ex vivo, we assessed the suppression of AP firing in pyramidal neurons while modulating the membrane potential. Both XPC-7224 (500 nM) and XPC-5462 (150 nM) inhibited AP firing elicited by current injection in CA1 pyramidal neurons, as shown in Figure S4A(i,ii), B and E(i,ii), F, although interestingly with a smaller effect than in neocortical pyramidal neurons (Figure 6). The depolarization of membrane voltage decreased AP number, as shown in Figure S4A(iii), D, E(iii), H consistent with our hypothesis that the two compounds stabilized larger fractions of NaV channels in inactivated states upon depolarization.
NaV1.6 and NaV1.2 Suppression Suppressed Ex Vivo Seizure-Like Activity
Finally, we tested the efficacy of these compounds in suppressing neocortical ex vivo seizure-like events using an MEA to assess the effects of selective inhibition of excitatory neurons across an intact network (Figure S5). We found that both XPC-5462 and CBZ significantly reduced all epileptiform discharges (Figure S5E,G). XPC-7224 had a trending but nonsignificant effect (P = 0.09) on total epileptiform discharges in the 0 Mg2+ model (Figure S5E) but no clear effect in the 4-AP model. These data demonstrate that the isoform specific block of NaV1.2 and NaV1.6 channels together can significantly limit network seizure-like activity induced by 0 Mg2+ or 4-AP.
Discussion
In this study, we provide detailed comparative pharmacological and mechanistic characterizations of XPC-7224 and XPC-5462, which represent a new class of selective NaV inhibitors. The motivation for developing such a class of compounds was to improve the efficacy and reduce the common adverse event profile of common nonselective NaV inhibiting ASM drugs.32 Recently, NBI-921352, a molecule with a similar selectivity profile to XPC-7224 that was created at Xenon, has been shown to be efficacious and have improved tolerability in rodent seizure models when compared with nonselective NaV inhibitors.4 Neurocrine Biosciences are currently developing selective NaV inhibitors as potential treatments for seizure disorders.45,46
Our previous work with NBI-921352 highlighted the selectivity and in vivo pharmacological profile of this class of compounds but did not provide a detailed characterization of the biophysics and pharmacology. As XPC-7224 shares the same chemical scaffold as NBI-921352 it is likely that the mechanisms expounded here will be broadly applicable across that chemical scaffold. XPC-5462 has a scaffold sufficiently different from XPC-7224 to impart a different selectivity profile to include NaV1.2 activity. As we have shown here, however, the two compounds are almost identical in mechanistic profile aside from their selectivity; they interact with the same binding site (Figure 1H) and are similar in potency (Figure 1C,D, Table 1), kinetics (Figure 3C), and membrane voltage dependence (Figure 4). These pharmacological similarities suggest that XPC-7224 and XPC-5462 can be considered to have the same molecular mechanism of action.
There are several critical features that differentiate these VSD-IV-targeting compounds from the NaV-targeting ASMs currently in use. Most strikingly, the molecular selectivity for NaV1.6 or dual selectivity for NaV1.2 and 1.6 of these compounds position them as a unique class of NaV channel inhibitors that could translate into a differentiated therapy for epilepsy. The >1000-fold selectivity for the cardiac isoform NaV1.5 reduces the cardiac risk profile significantly. The sparing of NaV1.1 by both compounds will alleviate the possible liabilities of suppressing inhibitory interneuron activity. This is highly desirable as loss of NaV1.1 conductance is the pathophysiological mechanism driving hyperexcitability in early infantile onset epileptic encephalopathy 6 or Dravet syndrome.6,47 In addition to selectivity, the potency range (<200 nM) of these compounds is at least one order of magnitude higher than CBZ, PHY and other putative NaV-targeting ASMs such as lamotrigine and lacosamide that are in the 10–100 μM range.48,49
The discovery of NaV1.6- and NaV1.2-targeting compounds was achieved via a progressive medicinal chemistry driven evolution of aryl sulfonamide scaffolds that originally targeted NaV1.7 and NaV1.3.39 The binding mode of early aryl sulfonamides has been structurally elucidated via a cocrystallization of NaV channels with the ligand and found to be a pocket in the extracellular aqueous cleft at the top of the activated state of VSD-IV, which controls NaV channel inactivation.37,38 To assess whether XPC-7224 and XPC-5462 engage the same site as the NaV1.7-targeting compounds, we neutralized R1626 in NaV1.6, the critical VSD-IV-S4 arginine residue for aryl sulfonamide binding identified from the Nav1.7 crystal structure, to alanine. NaV1.6 R1626A displayed a > 1000-fold drop in potency for both XPC-7224 and XPC-5462, confirming this key binding interaction was conserved by the new scaffold.
We examined how tightly the XPC compounds were bound to the VSD-IV to stabilize the up state by measuring the rate of recovery from inactivation at −120 mV in the presence of compound. Recovery from inactivation requires the transitioning of the channel back to the resting state in which all voltage-sensors are in the down position.43 We found that XPC-7224 and XPC-5462 introduced a prominent slower second component of recovery that was not strongly concentration dependent but the fraction of slowly recovering current was concentration dependent (Figure 2). The simplest explanation is that the fraction of channels bound at VSD-IV increased with concentration and that the recovery rate of bound channels reflected the dissociation rate of the compound at −120 mV. Pore-targeting NaV inhibitors also stabilize inactivated states and slow recovery from inactivation.35,41 We saw a clearly resolvable slow component of recovery introduced by PHY, but not CBZ, indicating the CBZ dissociation rate was faster than recovery from inactivation. The time constants of the Tslow and the potency of the fraction of Tslow contribution tracked with the potency of the compounds in the order XPC-5462 > XPC-7224 > PHY. These data demonstrate a striking slowing of dissociation rates and increase in the residency time for the XPC compounds on channels compared with PHY and CBZ.
Equilibration rates are a function of on-rate and concentration as well as off-rates according to Kobs = kon*[CPD] + koff.3 A faster kon and/or koff, therefore, increases equilibration rates. By comparing the equilibration rates at concentrations 3-fold higher relative to the IC50, which are similar to the EC50 brain concentrations found to suppress seizures in MES rodent models,4,50 we found that these XPC compounds had a much slower equilibration rate than CBZ and PHY (Figure 3C). Surprisingly, the XPC compounds had faster kon rates than the pore-binding compounds, despite the slower equilibration rate at 3-fold the IC50 (Figure 3F). This suggests that a faster koff and mass action are mainly responsible for faster equilibration rates of CBZ and PHY than those of XPC compounds. We also found that the koff was accelerated by hyperpolarization, consistent with the destabilization of compound binding to the VSD-IV UP state (Figure 3H–J).
Prior experiments have demonstrated that these compounds have a strong preference for binding the inactivated state of VSD-IV and that the dissociation rate is dependent on the membrane voltage (Figure 3G). Consequently, there is a strong dependence of potency on the membrane voltage (Figure 4A–F). By holding the membrane voltage at increasingly depolarized potentials, the potency increased dramatically for the XPC compounds compared with PHY and CBZ. We found that the variation in potency was well fit with a four-state model where the binding affinity to the inactivated state was considered much greater than to the resting state (Figure 4G–J), like previous classic studies on NaV inhibitors.41 This demonstrates that the relationship between potency and membrane voltage is dependent upon the proportion of channels inactivated rather than a direct voltage dependence (Figure 4D). An important consequence of this dependency is that compound apparent potency will vary dependent upon the distribution of membrane potential in a neuron, channel subtype, and subcellular localization of channels.26,51
It has been hypothesized that use-dependent block may be a desirable feature of an inhibitor molecule for disorders of hyperexcitability including epilepsy. The rapid spiking and repeated depolarizations of the membrane that occur during hyperexcitability favor the inactivated state of the channel. Interestingly, the XPC compounds did not display a significant increase in potency on NaV1.6 after high-frequency stimulation when the membrane voltage was held at the V0.5 (−70.2 mV with LJP correction) (Figure 5). This suggests that with the XPC compounds steady-state inhibition was sufficient to allow the suppression of hyperexcitability before seizure-dependent spiking and depolarization occurs. Indeed, even with the more rapidly equilibrating PHY, inhibition only increases ∼17% after high-frequency firing. This is consistent with the prediction from the four-state model in Figure 4 that the steady-state apparent potency can only increase 2-fold when moving from a V0.5 potential to a fully inactivated potential, which would give an approximate ∼20% increase in inhibition at nonsaturating concentrations (Figure 5). These data suggest that use dependence may not be necessary for efficacy of these NaV-targeting ASMs.
We next tested the ability of the compounds to impair intrinsic excitability in the more physiological environment of somatosensory cortex neurons within a brain slice. As previously noted, NaV1.6 and NaV1.2 are dominantly expressed in excitatory neurons, whereas NaV1.1 is mainly expressed in inhibitory GABAergic neurons.6 Inhibition of either NaV1.6 alone with XPC-7224 or NaV1.6 and NaV1.2 together with XPC-5462 led to reduction of intrinsic excitability of cortical pyramidal cells while sparing fast spiking inhibitory neurons (Figure 6A,B). This suggests the molecular selectivity of the XPC compounds does confer cellular selectivity. We propose that this property is highly desirable because it leaves the native inhibitory neuronal system intact to assist in maintaining the excitation-inhibition balance. In contrast, the nonselective NaV ASM CBZ suppressed excitability in both excitatory and inhibitory neurons (Figure 6C), which we believe may lead to reduced safety margins.4 In addition, we demonstrated an increased level of AP suppression in CA1 pyramidal neurons with depolarization (Figure S4) supporting the translatability of our in vitro state-dependence experiments (Figure 4). This property would be considered desirable in suppressing hyperexcitability of excitatory neurons receiving excessive depolarizing excitatory input.
To further profile the effects of the compounds, we evaluated efficacy in two ex vivo seizure models, where spontaneous seizure-like activity is evoked by either removing extracellular Mg2+ from the slice or adding 4-AP. In both models, comparable effects were found with XPC-5462 and CBZ providing robust suppression of frequency of seizure-like activity and epileptiform discharges (Figure S5D–S5G). Surprisingly, NaV1.6 inhibition alone with XPC-7224 did not significantly reduce frequency or discharges, although there was a trend toward reduction of discharges in 4-AP. The differential profile of XPC-7224 and XPC-5462 suggests that in these ex vivo models, there may be a requirement for inhibition of NaV1.2 channels to suppress seizure-like activity. We have previously shown selective NaV1.6 inhibition is efficacious at preventing seizures in vivo with other compounds, including XPC-7224.4,50 The reason for the lack of an effect of XPC-7224 in ex vivo seizure models is not clear to us and requires further investigation. We speculate that to suppress ex vivo seizure-like activity may require a greater fractional inhibition of the total excitatory cell NaV current, which is made up of both NaV1.2 and NaV1.6, and NaV1.2 has a higher relative abundance.52
In conclusion, this study provides a detailed comparative pharmacological characterization of a novel class of selective NaV channel inhibitors. We show that XPC-7224 and XPC-5462 have distinct molecular selectivity profiles targeting either NaV1.6 or both NaV1.6 and NaV1.2. The compounds cause inhibition through the same mechanism of binding to VSD-IV and stabilization of the inactivated state of the channel, which cannot reset until the compound dissociates. This leads to a strong state and, therefore, membrane potential dependence of potency. The kinetics of these compounds are profoundly different from that of the existing NaV-targeting ASMs, CBZ and PHY, with slower equilibration rates driven by slow dissociation and longer residence times on the channels. We found the fractional inhibition of the NaV current to be similar at V0.5 potentials compared with fully inactivated channels, suggesting that these compounds might exert their activity from steady-state inhibition rather than a use-dependent block. This was confirmed through the inhibition of intrinsic neuronal excitability in brain slices from resting membrane potentials, where only excitatory neurons were inhibited by the XPC compounds. In ex vivo seizure models, only XPC-5462 and CBZ demonstrated a strong seizure suppression, indicating the importance of NaV1.2 inhibition in those models. It is unclear at this point the translational impact to the clinic of the ex vivo seizure models but it is important to highlight that selective NaV1.6 inhibition has already been demonstrated to be effective in rodent seizure models4 and even that NaV1.6 inhibition may be the primary driver of in vivo efficacy.50 Finally, we propose that this new class of selective NaV-targeting compounds will lead to increased clinical efficacy with a higher threshold to adverse side-effects because NaV1.1-mediated inhibitory neuronal excitability is unperturbed.
Materials and Methods
Cell Lines
Stably transfected FreeStyle 293 F cells (Thermo Fisher), used for electrophysiology experiments up to passage 30, were maintained in DMEM (Gibco Invitrogen) containing 10% fetal bovine serum and 0.5 mg/mL Geneticin (G418) at 37 °C with 5% CO2.
Animals
Male and female CF-1 (Charles River) were used in this study (age 3–5 weeks). Mice are housed in individually ventilated cages in a 12 h light, 12 h dark lighting regime. Animals received food and water ad libitum. All animal handling and experimentation involving animals were conducted by following approved protocols according to the guidelines of the Canadian Council on Animal Care.
Electrophysiology
Transfecting Cell Lines
Stable cell lines were transfected with an expression vector containing the full-length cDNA coding for a specific human sodium channel α-subunit using Lipofectamine (Thermo Fisher). The NaV1.x stable cell lines and accessory constructs used correspond to the GenBank accession numbers: Human NaV1.1 (NM_006920); human NaV1.2 (NM_021007); human NaV1.5 (NM_198056); human NaV1.6 (NM_014191); mouse NaV1.6 (NM_001077499); human NaV1.7 (NM_002977); human NaV1.3 (NM_0069220). The human NaV β1 subunit (NM_199037) was coexpressed in all cell lines. Human NaV1.6 channels were also coexpressed with human FHF2B (NM_033642) to increase the functional expression. Human NaV1.2 channels were also coexpressed with Contactin 1 (NM_001843) to increase functional expression. Cells were used in automated patch-clamp experiments 24 h postinduction using Doxycycline (Sigma-Aldrich).
NaV Channel Automated Qube Planar Patch-Clamp Assays
Patch-clamp assays were conducted using the Qube 384 (Sophion) automated voltage-clamp platform, using single hole plates. Data were collected using the Qube 384 (Sophion) automated voltage-clamp platform using single hole plates. To measure inactivated state inhibition, the membrane potential was maintained at a voltage where the inactivation is complete. For each NaV channel subtype, the Vh used to quantify compound inhibition were as follows: NaV1.6 (−45 mV), NaV1.1 (45 mV), NaV1.2 (45 mV), NaV1.3 (45 mV), NaV1.5 (−60 mV), and NaV1.7 (−60 mV). The voltage was briefly repolarized to a negative voltage (150 mV) for 20 ms for NaV1.5, NaV1.7, NaV1.3 or for 60 ms for NaV1.1, NaV1.2, and NaV1.6 to allow recovery from fast inactivation, followed by a test pulse to −20 or 0 mV for 10 ms to quantify the compound inhibition. The repolarization step allows compound-free channels to recover from fast inactivation, but compound-bound channels remain inhibited during the subsequent test step. The Qube is an automated electrophysiology instrument that is blinded to cell selections and experimentation, and selection is performed in a randomized manner. All subsequent data filtering and analysis is performed with automated filters that are applied to the entire data set from a given Qube run. Appropriate filters for minimum seal resistance were applied (typically >500 MΩ membrane resistance and >3 pF for capacitance), and series resistance was compensated at 100%. The pulse protocols were run at 1 Hz for hNaV1.7, hNaV1.5, hNaV1.3, and hNaV1.4 or at 0.04 Hz for NaV1.6, NaV1.1, and NaV1.2.
To construct concentration response curves, baseline currents were established after 20 min in vehicle (0.5% DMSO). Full inhibition response amplitudes were determined by adding tetrodotoxin (TTX, 300 nM) or tetracaine for NaV1.5 (10 μM) to each well at the end of the experiment. Compounds were then exposed at a single concentration for 20 min. One-sixth of every experimental plate was dedicated to vehicle-only wells that enabled correction for nonspecific drift (i.e., rundown) of the signal in each experiment. For all channel subtypes, inhibition by the compound reached the steady state within 20 min of incubation.
The current inhibition values (I(CPD)) were normalized to both the vehicle (Icontrol) and the full response defined by supramaximal TTX (ITTX) or tetracaine (for NaV1.5) addition responses according to eq 1:
| 1 |
This normalized inhibition was then further normalized to the span of the assay to account for the run-down seen in cells exposed to vehicle alone for 20 min as follows:
| 2 |
where:
Inorm, span = the current response normalized to within the span of the assay.
Inorm(CPD) = the normalized response in the presence of compound.
Inorm(VEH) = the normalized response in the absence of compound.
This normalization ensures that the data ranges were between 0 and 1, and there is no rundown in the plots. The normalized data from all cell recordings at a concentration were grouped together and plotted with GraphPad Prism 8, and IC50 values were calculated for the grouped data using the following version of the Hill equation:
| 3 |
,where:
Y is the fraction of sodium current blocked in the presence of the compound.
[CPD] = the concentration of compound.
IC50 = the IC50 concentration.
RD = the “rundown” of sodium current in vehicle alone, which is equal to 0 in this case, as the inhibition has already been normalized to the span.
The Hill slope was fixed to 1. The normalization of other Qube protocols was performed in the same way. The 95% CI for the IC50 from the fitted curve to the mean data were reported and N = the total number of cells that were used in the fit unless otherwise noted.
Recovery from inactivation was measured with a test-pulse to 0 mV following a depolarizing prepulse to 0 mV for 10 s from a holding potential of −120 mV. The mean normalized recovery of currents from a holding-potential of −120 mV in the compound was plotted and fit with a biexponential function.
Qube experiments were all performed at 27 °C ± 2 °C.
The recording solutions for NaV1.1, NaV1.2, NaV1.3, and NaV1.6 cell line studies contained: intracellular solution (ICS): 5 mM NaCl, 10 mM CsCl, 120 mM CsF, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES (4 (2 hydroxyethyl)-1-piperazineethanesulfonic acid buffer), 10 mM EGTA (ethylene glycol tetraacetic acid); adjusted to pH 7.2 with CsOH. Extracellular solution (ECS): 140 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES; adjusted to pH 7.4 with NaOH. Liquid junction potential (LJP) calculated for these solutions was 7.6 mV and voltages are not corrected unless stated in text. Solutions with a reversed Na+ gradient were used for NaV1.5 and NaV1.7 studies since they improved the technical success and the number of cells passing the quality control filters and inspection. The improvement resulted from the combination of an increased seal resistance and better voltage clamp of the large currents in our NaV 1.5 and NaV1.7 cell lines, which reduced any incidence of ringing due to series resistance compensation. ICS: 120 mM NaF, 10 mM CsCl, 0.1 mM CaCl2, 2 mM MgCl2, 10 mM HEPES, 10 mM EGTA; adjusted to pH 7.2 with CsOH. ECS: 1 mM NaCl, 139 mM CholineCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM HEPES; adjusted to pH 7.4 with NaOH. Osmolarity in all ICS and ECS solutions was adjusted with glucose to 300 and 310 mOsm/kg, respectively.
Brain Slice Patch Clamp Electrophysiology–Cortical Recordings
Parasagittal cortical brain slices were prepared from 3- to 5-week-old CF-1 mice from standard procedures adapted from previously published methods.53 Briefly, the mouse was deeply anesthetized with isoflurane and decapitated. The brain was removed and placed into chilled artificial cerebrospinal fluid (aCSF) solution containing (in mM): 125 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 2 MgCl2, 10 d-glucose, pH 7.3, osmolarity adjusted to ∼306 mOsm using sucrose. All solutions were saturated with 95% w/w O2 and 5% w/w CO2 constantly perfused with 95% w/w O2/5% CO2. Parasagittal slices with a thickness of 400 μm were prepared using a vibratome (Ted Pella, Inc.). Following sectioning, the slices were placed in a holding chamber and incubated in a water bath at 34 °C for 15 min.
Following a 60 min incubation at room temperature, a brain slice was selected and placed on the stage of an upright microscope (SliceScope Pro 2000, Scientifica). The slice was constantly perfused with room temperature aCSF, containing 0.1% DMSO as a vehicle control, and oxygenated with 95% O2/5% CO2. The slice was visualized using bright-field microscopy, and a healthy neuron was selected from neocortical layer 5. Whole-cell configuration was achieved with a pipet (bath resistance 4–6 MΩ) containing internal solution. Stimulation was applied in current-clamp mode and consisted of a series of 1000 ms square pulses, beginning at −20 pA and increasing by +20 pA increments (3000 ms between pulses). The internal solution contained (in mM): 120 KMeSO4, 20 KCl, 10 HEPES, 4 Na2ATP, 2 MgCl2, 0.3 Tris-GTP, 0.2 EGTA, pH 7.2–7.3 using KOH or HCl and osmolarity of 285–295 mOsm. LJP calculated for these solutions was +12.7 mV. Experiments were performed at room temperature 20–22 °C.
Brain Sice Patch Clamp Electrophysiology – CA1 Recordings
Brain slices of hippocampal regions were obtained from 3- to 5-week-old C57BL/6 male mice and were prepared with a Vibratome (Leica, VT 1200) at 0 °C. Slicing solution contained (mM): 216 sucrose, 2.5 KCl, 1.25 NaH2PO4, 10 MgSO4, 0.5 CaCl2, 11 glucose, 26 NaHCO3 (pH 7.2–7.4; 305–315 mOsm), constantly bubbled with carbogen. Coronal slices including hippocampal regions were 300 μm thick, prepared with a advancing speed of 0.14 mm/s and a vibration range of 1.2 mm. Slices were then transferred to bath solution (artificial cerebral spinal fluid, aCSF) containing (in mM): 129 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 10 glucose, 26 NaHCO3; 300–310 mOsm, pH: 7.2–7.4, constantly bubbled with carbogen, and recovered for 30 min at 32–34 °C.
For electrophysiology, glass pipettes were pulled with a P-2000 puller (Sutter Instrument) to generate tips with a resistance of 4–6 MΩ. Pipette solution contained (in mM): 128 K-Gluconate, 9 HEPES, 0.5 MgCl2, 8 NaCl, 0.1 EGTA, 14 Tris2-phosphocreatine, 4 Na2-ATP, 0.3 tris-GTP; ∼ 290 mOsm, pH: 7.2–7.25. Bath solution (aCSF) contained (in mM): 129 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1 MgSO4, 2 CaCl2, 10 glucose, 26 NaHCO3; 300–310 mOsm, pH: 7.2–7.4, constantly bubbled with carbogen. Electrophysiological data were acquired using Multiclamp 700B amplifiers (Molecular Devices) at 32–34 °C. Data were acquired at 100 kHz and filtered at 50 kHz. Pipette capacitance was compensated by 90% of the fast capacitance measured under Gigahm seal conditions in voltage-clamp prior to establishing a whole-cell configuration. During current clamp, series resistance was <25 MΩ in all recordings and the bridge was balanced by >70%. Liquid junction potential was corrected after data were obtained. Action potential was counted with a threshold of −20 mV.
Once the recordings in vehicle were completed, and while still holding the patch on the same neuron, the bath solution was changed from 0.1% DMSO in aCSF to compound in aCSF. The slice was incubated in circulating compound for 10 min before repeating the series of square pulse stimulations. Working stock solutions were prepared in DMSO at a concentration of 20 mM.
All data analysis was done offline using ClampFit 10.7 (Molecular Devices). Data are presented as a mean ± SEM. For each sweep, the number of evoked APs was counted and plotted as a function of current injection (beginning with −20 pA) and then normalized to the maximum vehicle response. These generated “input/output” (or “I–O”) curves demonstrating the relationship between the stimulus and AP frequency.
Multielectrode Array (MEA) Recordings
Male and female CF-1 mice between 4 and 8 weeks were used. Mice were anesthetized with isoflurane before being euthanized by cervical dislocation. Brains were then removed and stored in cold cutting solution (in mM): 3 MgCl2; 126 NaCl; 26 NaHCO3; 3.5 KCl; 1.26 NaH2PO4; 10 glucose. 350 μm horizontal sections containing somatosensory cortex were made, using a Leica VT1200 vibratome (Nussloch, Germany). Slices were then transferred to a holding chamber and incubated for 1–2 h at room temperature in artificial CSF (ACSF) containing (in mM): 2 CaCl2; 1 MgCl2; 126 NaCl; 26 NaHCO3; 3.5 KCl; 1.26 NaH2PO4; 10 glucose. All of the solutions were bubbled continuously to saturate with carboxygen (95% O2 and 5% CO2).
MEA recordings were performed on the 3Brain BioCAM DupleX system (Switzerland), using the 3Brain Accura HD-MEA chips with 4096 electrodes at a pitch of 60 μm. Slices were placed onto the electrodes with a harp placed on top to keep the slice gently pressed down to the recording electrodes. Slices were perfused continuously with artificial cerebrospinal fluid (aCSF) and a zero Mg2+ aCSF or 100 μM 4-aminopyridine was used to induce epileptiform activity over a 50 min period. Before removal of Mg2+ or addition of 4-aminopyridine, slices were perfused for 10 min in either aCSF with 0.1% DMSO or compound, respectively. Recordings were obtained from the entire slice. Experiments were performed at 33–36 °C. The solutions were perfused at the rate of 5.0 mL/min. Signals were sampled at 10 kHz with a high-pass filter at 2 Hz. Analysis of seizure-like events and epileptiform activity was done using the Xenon LFP Analysis Platform.54 Electrophysiological recordings were analyzed from the entire neocortical region of the brain slice, sampling every 120 μm. Epileptiform activity was considered seizure-like events when it had high-frequency rhythmic bursts associated with high-frequency signals lasting at least 10 s. All pathological discharges observed from the recordings were considered epileptiform discharges for analysis purposes.
Quantification and Statistical Analysis
Quantification of data was done on GraphPad Prism 8 or ClampFit 10.7 (Molecular Devices) as indicated. Quantitative data are presented as mean ± SEM. The number of cells or brain slices tested is reported in the figure legends or tables where appropriate. Significance was tested using two-way ANOVA followed by Bonferroni multiple comparisons tests or using Student’s two-way paired t tests as indicated in the figure legends.
Safety
No new or significant hazards or risks were associated with the reported work.
Acknowledgments
Editorial services were provided by Prescott Medical Communications Group (Chicago, IL), with support from Neurocrine Biosciences, Inc.
Glossary
Abbreviations
- AP
action potential
- ASM
antiseizure medication
- CBZ
carbamazepine
- CNS
central nervous system
- GOF
gain-of-function
- LOF
loss-of-function
- MEA
multielectrode array
- Nav
voltage-gated sodium channel
- PHY
phenytoin
- RMP
resting membrane potential
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acschemneuro.3c00757.
Table of steady-state inactivation properties of NaV subtypes, table of membrane voltage dependence of compound potency, concentration response curves displaying potency of XPC-5462 and XPC6624 on mouse NaV1.6, steady-state inactivation curves for NaV1.6 WT vs NaV1.6 (R1626A) mutant, concentration-dependence graphs for kinetic evaluation of compounds, additional results on suppression of AP firing from hippocampal CA1 pyramidal cells, multielectrode array results of compound effects in an ex vivo model of ictogenesis (PDF)
Author Contributions
The experiments in this report were conceptualized by S.G., J.J., R.P., W.Y., C.C., and J.E. Experiments were conducted and analyzed by S.G., A.W., N.S., M.S., R.P., S.T., M.W., W.Y., and M.G. Cell lines and mutants were created by J.M. and R.D. Medicinal Chemistry expertise was provided by T.F. P.R. provided supervisory input for M.G. F.S. provided supervisory input for W.Y. All authors had access to data. S.G. wrote the manuscript; W.Y., M.G., F.S., and J.J. contributed to the writing and editing of the manuscript. All work conducted for this manuscript was under the supervision of S.G. and J.J. All authors approved the final manuscript draft for submission.
Xenon Pharmaceuticals, Inc. funded this study and participated in the study design, research, analysis, data collection, interpretation of data, reviewing, and approval of the publication. MG was supported by a Mitacs Accelerate fellowship (IT10714) and a Natural Sciences and Engineering Research Council of Canada (NSERC) Canada Graduate scholarship (535333–2019). PR received funding from NSERC (RGPIN03920).
The authors declare the following competing financial interest(s): S.G., N.S., A.W., W.Y., R.P., M.S., S.T., J.M., M.W., R.D., T.F., C.C., J.E., and J.J. are current or former full-time employees of Xenon Pharmaceuticals, Inc. and have equity in the company. F.S. and W.Y. are full-time employees of Neurocrine Biosciences, Inc. and have equity in the company. M.G. was supported by a Mitacs Accelerate fellowship in partnership with Xenon Pharmaceuticals (IT10714).
Originally published ASAP February 15, 2024; Abstract graphic revised March 5, 2024.
Supplementary Material
References
- Bean B. P. The action potential in mammalian central neurons. Nat. Rev. Neurosci. 2007, 8 (6), 451–465. 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Hodgkin A. L.; Huxley A. F. A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 1952, 117 (4), 500–544. 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B.. Ion Channels of Excitable Membranes; Oxford University Press, Incorporated, 1992. [Google Scholar]
- Johnson J. P.; Focken T.; Khakh K.; Tari P. K.; Dube C.; Goodchild S. J.; Andrez J. C.; Bankar G.; Bogucki D.; Burford K.; et al. NBI-921352, a first-in-class, NaV1.6 selective, sodium channel inhibitor that prevents seizures in Scn8a gain-of-function mice, and wild-type mice and rats. Elife 2022, 11, e72468 10.7554/eLife.72468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trimmer J. S.; Rhodes K. J. Localization of voltage-gated ion channels in mammalian brain. Annu. Rev. Physiol. 2004, 66, 477–519. 10.1146/annurev.physiol.66.032102.113328. [DOI] [PubMed] [Google Scholar]
- Yu F. H.; Mantegazza M.; Westenbroek R. E.; Robbins C. A.; Kalume F.; Burton K. A.; Spain W. J.; McKnight G. S.; Scheuer T.; Catterall W. A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9 (9), 1142–1149. 10.1038/nn1754. [DOI] [PubMed] [Google Scholar]
- Sanders S. J.; Campbell A. J.; Cottrell J. R.; M?ller R. S.; Wagner F. F.; Auldridge A. L.; Bernier R. A.; Catterall W. A.; Chung W. K.; Empfield J. R.; et al. Progress in Understanding and Treating SCN2A-Mediated Disorders. Trends Neurosci. 2018, 41 (7), 442–456. 10.1016/j.tins.2018.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 317–338. 10.1146/annurev-pharmtox-011112-140232. [DOI] [PubMed] [Google Scholar]
- Gardella E.; Mo̷ller R. S. Phenotypic and genetic spectrum of SCN8A-related disorders, treatment options, and outcomes. Epilepsia 2019, 60 (3), S77–S85. 10.1111/epi.16319. [DOI] [PubMed] [Google Scholar]
- Hammer M. F.; Wagnon J. L.; Mefford H. C.; Meisler M. H.. SCN8A-Related Epilepsy with Encephalopathy. In GeneReviews((R)), Adam; M., P; Ardinger H. H.; Pagon R. A.; Wallace S. E.; Bean L. J. H.; Gripp K. W.; Mirzaa G. M.; Amemiya A., Eds., 1993. [Google Scholar]
- Johannesen K. M.; Gardella E.; Encinas A. C.; Lehesjoki A. E.; Linnankivi T.; Petersen M. B.; Lund I. C. B.; Blichfeldt S.; Miranda M. J.; Pal D. K.; et al. The spectrum of intermediate SCN8A-related epilepsy. Epilepsia 2019, 60 (5), 830–844. 10.1111/epi.14705. [DOI] [PubMed] [Google Scholar]
- Kearney J. A.; Plummer N. W.; Smith M. R.; Kapur J.; Cummins T. R.; Waxman S. G.; Goldin A. L.; Meisler M. H. A gain-of-function mutation in the sodium channel gene Scn2a results in seizures and behavioral abnormalities. Neuroscience 2001, 102 (2), 307–317. 10.1016/S0306-4522(00)00479-6. [DOI] [PubMed] [Google Scholar]
- Begemann A.; Acuna M. A.; Zweier M.; Vincent M.; Steindl K.; Bachmann-Gagescu R.; Hackenberg A.; Abela L.; Plecko B.; Kroell-Seger J.; et al. Further corroboration of distinct functional features in SCN2A variants causing intellectual disability or epileptic phenotypes. Mol. Med. 2019, 25 (1), 6. 10.1186/s10020-019-0073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara T.; Tsurubuchi Y.; Agarwala K. L.; Ito M.; Fukuma G.; Mazaki-Miyazaki E.; Nagafuji H.; Noda M.; Imoto K.; Wada K.; et al. A missense mutation of the Na+ channel alpha II subunit gene Na(v)1.2 in a patient with febrile and afebrile seizures causes channel dysfunction. Proc. Natl. Acad. Sci. U. S. A. 2001, 98 (11), 6384–6389. 10.1073/pnas.111065098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Shalom R.; Keeshen C. M.; Berrios K. N.; An J. Y.; Sanders S. J.; Bender K. J. Opposing Effects on NaV1.2 Function Underlie Differences Between SCN2A Variants Observed in Individuals With Autism Spectrum Disorder or Infantile Seizures. Biol. Psychiatry 2017, 82 (3), 224–232. 10.1016/j.biopsych.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W. A. Voltage-gated sodium channels at 60: Structure, function and pathophysiology. J. Physiol. 2012, 590 (11), 2577–2589. 10.1113/jphysiol.2011.224204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda B.; Bezanilla F. Tracking voltage-dependent conformational changes in skeletal muscle sodium channel during activation. J. Gen. Physiol. 2002, 120 (5), 629–645. 10.1085/jgp.20028679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong C. M.; Bezanilla F. Inactivation of the sodium channel. II. Gating current experiments. J. Gen. Physiol. 1977, 70 (5), 567–590. 10.1085/jgp.70.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F.; Armstrong C. M. Inactivation of the sodium channel. I. Sodium current experiments. J. Gen. Physiol. 1977, 70 (5), 549–566. 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capes D. L.; Goldschen-Ohm M. P.; Arcisio-Miranda M.; Bezanilla F.; Chanda B. Domain IV voltage-sensor movement is both sufficient and rate limiting for fast inactivation in sodium channels. J. Gen. Physiol. 2013, 142 (2), 101–112. 10.1085/jgp.201310998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean B. P. Sodium channel inactivation in the crayfish giant axon. Must channels open before inactivating?. Biophys. J. 1981, 35 (3), 595–614. 10.1016/S0006-3495(81)84815-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mickus T.; Jung H.; Spruston N. Properties of slow, cumulative sodium channel inactivation in rat hippocampal CA1 pyramidal neurons. Biophys. J. 1999, 76 (2), 846–860. 10.1016/S0006-3495(99)77248-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West J. W.; Patton D. E.; Scheuer T.; Wang Y.; Goldin A. L.; Catterall W. A. A cluster of hydrophobic amino acid residues required for fast Na(+)-channel inactivation. Proc. Natl. Acad. Sci. U. S. A. 1992, 89 (22), 10910–10914. 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X.; Li Z.; Zhou Q.; Shen H.; Wu K.; Huang X.; Chen J.; Zhang J.; Zhu X.; Lei J.; et al. Structure of the human voltage-gated sodium channel Nav1.4 in complex with beta1. Science 2018, 362, 6412. 10.1126/science.aau2486. [DOI] [PubMed] [Google Scholar]
- Shen H.; Zhou Q.; Pan X.; Li Z.; Wu J.; Yan N. Structure of a eukaryotic voltage-gated sodium channel at near-atomic resolution. Science 2017, 355, 6328. 10.1126/science.aal4326. [DOI] [PubMed] [Google Scholar]
- Hu W.; Tian C.; Li T.; Yang M.; Hou H.; Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat. Neurosci. 2009, 12 (8), 996–1002. 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]
- Ragsdale D. S.; McPhee J. C.; Scheuer T.; Catterall W. A. Common molecular determinants of local anesthetic, antiarrhythmic, and anticonvulsant block of voltage-gated Na+ channels. Proc. Natl. Acad. Sci. U. S. A. 1996, 93 (17), 9270–9275. 10.1073/pnas.93.17.9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale D. S.; Avoli M. Sodium channels as molecular targets for antiepileptic drugs. Brain Res. Brain Res. Rev. 1998, 26 (1), 16–28. 10.1016/S0165-0173(97)00054-4. [DOI] [PubMed] [Google Scholar]
- Li Z.; Jin X.; Wu T.; Huang G.; Wu K.; Lei J.; Pan X.; Yan N. Structural Basis for Pore Blockade of the Human Cardiac Sodium Channel Nav 1.5 by the Antiarrhythmic Drug Quinidine*. Angew. Chem., Int. Ed. Engl. 2021, 60 (20), 11474–11480. 10.1002/anie.202102196. [DOI] [PubMed] [Google Scholar]
- Ragsdale D. S.; McPhee J. C.; Scheuer T.; Catterall W. A. Molecular determinants of state-dependent block of Na+ channels by local anesthetics. Science 1994, 265 (5179), 1724–1728. 10.1126/science.8085162. [DOI] [PubMed] [Google Scholar]
- Ghovanloo M. R.; Choudhury K.; Bandaru T. S.; Fouda M. A.; Rayani K.; Rusinova R.; Phaterpekar T.; Nelkenbrecher K.; Watkins A. R.; Poburko D.; et al. Cannabidiol inhibits the skeletal muscle Nav1.4 by blocking its pore and by altering membrane elasticity. J. Gen. Physiol. 2021, 153, 5. 10.1085/jgp.202012701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokken K.; Fairley P.. Sodium Channel Blocker Toxicity. In StatPearls, 2022. [PubMed] [Google Scholar]
- Iorga A.; Horowitz B. Z.. Phenytoin Toxicity. In StatPearls, 2022. [PubMed] [Google Scholar]
- Al Khalili Y.; Sekhon S.; Jain S.. Carbamazepine Toxicity. In StatPearls, 2022. [PubMed] [Google Scholar]
- Hanck D. A.; Nikitina E.; McNulty M. M.; Fozzard H. A.; Lipkind G. M.; Sheets M. F. Using lidocaine and benzocaine to link sodium channel molecular conformations to state-dependent antiarrhythmic drug affinity. Circ. Res. 2009, 105 (5), 492–499. 10.1161/CIRCRESAHA.109.198572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormack K.; Santos S.; Chapman M. L.; Krafte D. S.; Marron B. E.; West C. W.; Krambis M. J.; Antonio B. M.; Zellmer S. G.; Printzenhoff D.; et al. Voltage sensor interaction site for selective small molecule inhibitors of voltage-gated sodium channels. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (29), E2724–2732 10.1073/pnas.1220844110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahuja S.; Mukund S.; Deng L.; Khakh K.; Chang E.; Ho H.; Shriver S.; Young C.; Lin S.; Johnson J. P. Jr; et al. Structural basis of Nav1.7 inhibition by an isoform-selective small-molecule antagonist. Science 2015, 350 (6267), aac5464. 10.1126/science.aac5464. [DOI] [PubMed] [Google Scholar]
- Li X.; Xu F.; Xu H.; Zhang S.; Gao Y.; Zhang H.; Dong Y.; Zheng Y.; Yang B.; Sun J.; et al. Structural basis for modulation of human NaV1.3 by clinical drug and selective antagonist. Nat. Commun. 2022, 13 (1), 1286. 10.1038/s41467-022-28808-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Focken T.; Burford K.; Grimwood M. E.; Zenova A.; Andrez J. C.; Gong W.; Wilson M.; Taron M.; Decker S.; Lofstrand V.; et al. Identification of CNS-Penetrant Aryl Sulfonamides as Isoform-Selective NaV1.6 Inhibitors with Efficacy in Mouse Models of Epilepsy. J. Med. Chem. 2019, 62 (21), 9618–9641. 10.1021/acs.jmedchem.9b01032. [DOI] [PubMed] [Google Scholar]
- Bean B. P.; Cohen C. J.; Tsien R. W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983, 81 (5), 613–642. 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. C.; Bean B. P. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 1994, 46 (4), 716–725. [PubMed] [Google Scholar]
- Nau C.; Wang G. K. Interactions of local anesthetics with voltage-gated Na+ channels. J. Membr. Biol. 2004, 201 (1), 1–8. 10.1007/s00232-004-0702-y. [DOI] [PubMed] [Google Scholar]
- Kuo C. C.; Bean B. P. Na+ channels must deactivate to recover from inactivation. Neuron 1994, 12 (4), 819–829. 10.1016/0896-6273(94)90335-2. [DOI] [PubMed] [Google Scholar]
- Bankar G.; Goodchild S. J.; Howard S.; Nelkenbrecher K.; Waldbrook M.; Dourado M.; Shuart N. G.; Lin S.; Young C.; Xie Z.; et al. Selective NaV1.7 Antagonists with Long Residence Time Show Improved Efficacy against Inflammatory and Neuropathic Pain. Cell Rep. 2018, 24 (12), 3133–3145. 10.1016/j.celrep.2018.08.063. [DOI] [PubMed] [Google Scholar]
- Neurocrine Biosciences . Study to Evaluate NBI-921352 as Adjunctive Therapy in Subjects With SCN8A Developmental and Epileptic Encephalopathy Syndrome (SCN8A-DEE); ClinicalTrials.gov, 2021.
- Neurocrine Biosciences . A Study to Investigate How Effective, Safe and Tolerable the Drug NBI-921352 is When Used With Anti-seizure Medications in Adults With Focal Onset Seizures; ClinicalTrials.gov, 2021.
- Catterall W. A.; Kalume F.; Oakley J. C. NaV1.1 channels and epilepsy. J. Physiol. 2010, 588 (11), 1849–1859. 10.1113/jphysiol.2010.187484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo C. C.; Lu L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurones. Br. J. Pharmacol. 1997, 121 (6), 1231–1238. 10.1038/sj.bjp.0701221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo S.; Bean B. P. Lacosamide Inhibition of Nav1.7 Voltage-Gated Sodium Channels: Slow Binding to Fast-Inactivated States. Mol. Pharmacol. 2017, 91 (4), 277–286. 10.1124/mol.116.106401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J.; Focken T.; Tari P. K.; Dube C.; Goodchild S. J.; Andrez J. C.; Bankar G.; Burford K.; Chang E.; Chowdhury S.; et al. NaV1.6 inhibition drives the efficacy of voltage-gated sodium channel inhibitors to prevent electrically induces seizures in both wild type and Scn8aN1768D/+ gain of function mice. BioRxIV 2023, 10.1101/2023.08.03.551823. [DOI] [Google Scholar]
- Hu W.; Bean B. P. Differential Control of Axonal and Somatic Resting Potential by Voltage-Dependent Conductances in Cortical Layer 5 Pyramidal Neurons. Neuron 2018, 99 (6), 1355. 10.1016/j.neuron.2018.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wang H. G.; Pitt G. S.; Liu Z. J. Direct Observation of Compartment-Specific Localization and Dynamics of Voltage-Gated Sodium Channels. J. Neurosci. 2022, 42, 5482–5498. 10.1523/JNEUROSCI.0086-22.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai C.; Abe Y.; Westenbroek R. E.; Scheuer T.; Catterall W. A. Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (30), E3139–3148 10.1073/pnas.1411131111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevan A.; Codadu N. K.; Parrish R. R. Xenon LFP Analysis Platform Is a Novel Graphical User Interface for Analysis of Local Field Potential From Large-Scale MEA Recordings. Front. Neurosci. 2022, 16, 904931. 10.3389/fnins.2022.904931. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.