

Abstract
Despite recognizing the devastating consequences of metastasis, we are not yet able to effectively treat cancer that has spread to vital organs. The inherent complexity of genomic alterations in late-stage cancers, coupled with numerous heterotypic interactions that occur between tumour and stromal cells, represent fundamental challenges in our quest to understand and control metastatic disease. The incorporation of genomic and other systems level approaches, as well as technological breakthroughs in imaging and animal modelling, have galvanized the effort to overcome gaps in our understanding of metastasis. Future research carries with it the potential to translate the wealth of new knowledge and conceptual advances into effective targeted therapies.
Metastasis is the most deadly feature of cancer, accounting for greater than 90% of cancer-related mortality1–3. The clinical manifestation of metastatic lesions is the end result of a treacherous journey that few tumour cells are capable of completing, including local invasion and intravasation, survival in the circulation, homing and extravasation into the parenchyma of distant organs, and adaptation to the new environment and outgrowth of secondary lesions1,3–5. Although tumour cells that are shed from the primary tumour disseminate throughout the body, they tend to colonize select organs, with characteristically different periods of latency and efficiency depending on tumour type or subtype1,5. Steven Paget’s century-old ‘seed and soil’ hypothesis likened tumour cells to ‘seeds’ that are systemically distributed, but that only inhabit particular environments, or ‘soils’, which are supportive to their sustained growth6. Despite the intellectual clarity of this hypothesis, understanding the exact molecular and cellular basis of the complex events that facilitate cancer metastasis has been difficult.
We are now in an era in which systems level data are being generated at unprecedented rates, due, in large part, to a revolution in technology. Moreover, superior imaging modalities and experimental models afford the ability to explore the dynamic intricacies of cancer metastasis at a resolution that has not been achieved before (TIMELINE). We have begun to reveal fundamental concepts that underlie the development of metastasis: the lineage relationship between primary tumours and metastatic colonies7–10; the genetic properties of metastatic tumour cells that facilitate their interaction with the host stroma11,12; the unique functional contributions of different stromal components11,13–16; the organ-tropism of different cancer types and subtypes17–20; and the in vivo cellular and signalling dynamics during the different steps of metastasis21–24. With these newly acquired insights into the ‘black box’ of metastasis, the discovery of effective metastasis-targeting agents that specifically attack cancer cells and interrupt their communication with the microenvironment may soon be on the horizon. In this prospective Timeline article, we discuss possible new avenues for investigation that could lead to the development of novel approaches to prevent and to treat metastatic disease.
Timeline |. The major technological breakthroughs and conceptual advances in cancer metastasis research.
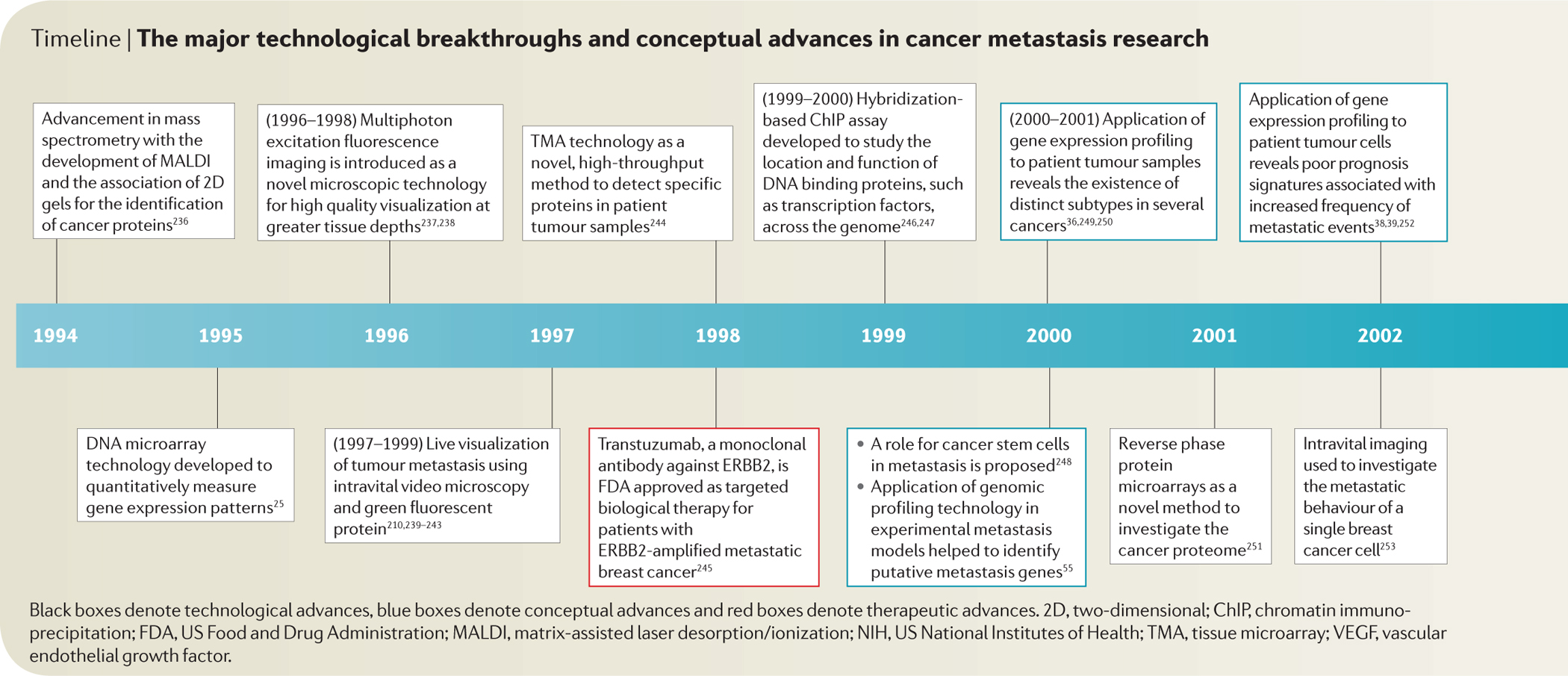
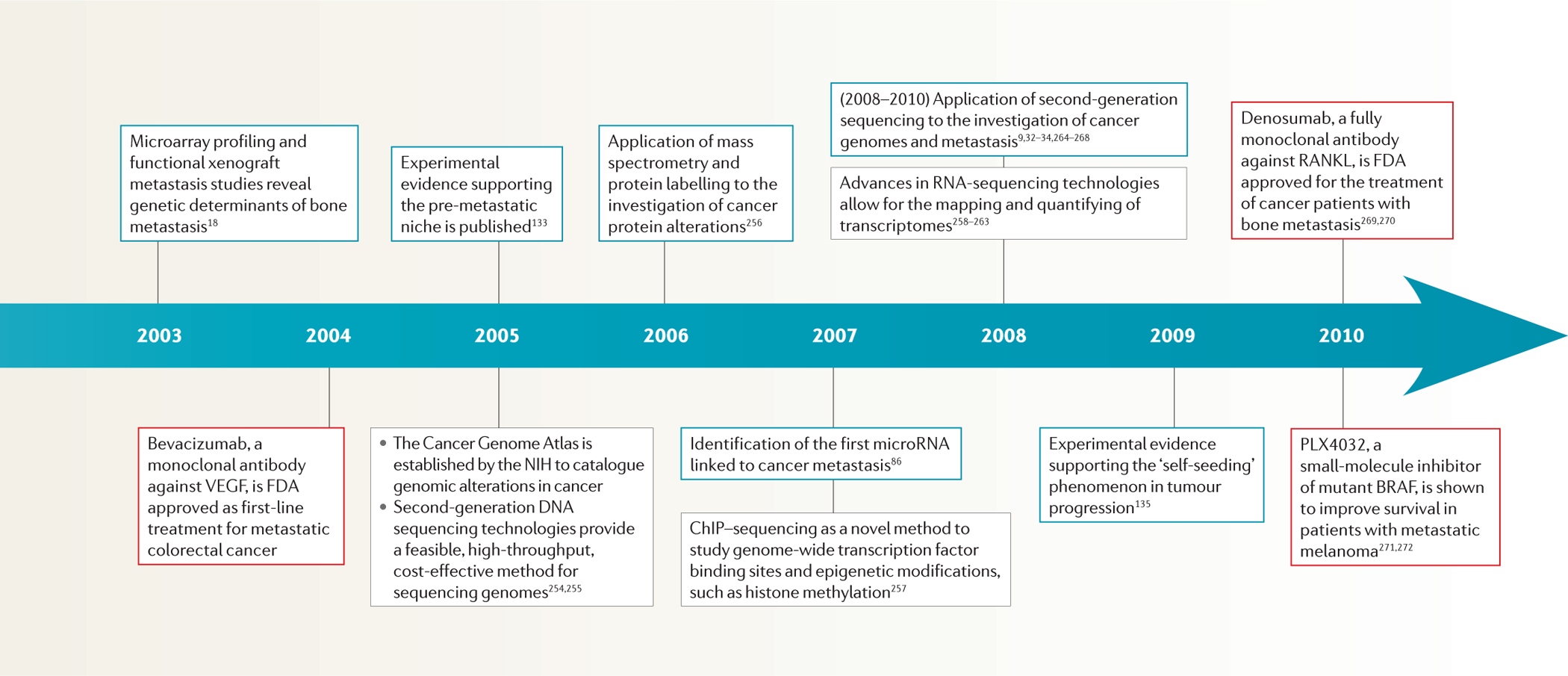
Black boxes denote technological advances, blue boxes denote conceptual advances and red boxes denote therapeutic advances. 2D, two-dimensional; ChIP, chromatin immunoprecipitation; FDA, US Food and Drug Administration; MALDI, matrix-assisted laser desorption/ionization; NIH, US National Institutes of Health; TMA, tissue microarray; VEGF, vascular endothelial growth factor.
Characterizing metastatic seeds
Fundamental to understanding cancer progression is the identification of the distinguishing features that endow certain tumour cells with metastatic capabilities. Characterizing such features enables the discovery of potential diagnostic and prognostic biomarkers, as well as targeted therapeutic agents. Genomic profiling25, second-generation sequencing26, proteomics27,28 and other systems level analytical techniques have dramatically accelerated the effort to comprehensively characterize metastatic tumour cells and to understand their natural history of evolution from primary tumours.
Molecular genealogy of metastasis.
It is generally agreed that cells acquire metastatic properties in the primary tumour, although there is an ongoing debate surrounding both the extent to which metastatic potential can be determined at this early stage in cancer progression and the lineage relationship between the primary tumour and metastatic lesions29. The linear and parallel models of metastatic progression are at the centre of this discussion (BOX 1). High-quality genomic studies have begun to explore the genetic and temporal relationship between primary lesions and metastatic lesions, the results of which will undoubtedly help us to resolve many important questions surrounding metastatic evolution8,30.
Box 1 |. Linear and parallel progression models.
According to the linear progression model, primary tumour cells undergo successive rounds of mutation and selection35, giving rise to a biologically heterogeneous cellular population in which a subset of malignant clones have accumulated genetic alterations, necessary for metastasis197,198. The metastatic capabilities may be developed at the primary site as a by-product of the selective pressures, or may further evolve after the tumour cells reach the secondary organs. Pioneering work by Isaiah Fidler in the 1970s demonstrated that only a subset of pre-existing cells in a heterogeneous primary tumour can successfully metastasize199. From a clinical perspective, a direct correlation between tumour size and frequency of metastatic events200,201, in addition to the reduction of metastatic risk by the surgical resection of tumours that are <2cm in size, also support the linear progression model.
By contrast, the parallel progression model argues that tumour cells may disseminate very early in malignant progression, colonize multiple secondary sites at different times and ultimately accumulate genetic changes independently from those incurred by the primary tumour8. Select studies comparing the growth rates of primary tumours and their secondary lesions concluded that metastases were too large to be initiated during advanced stages of cancer progression202,203. A more recent report provides evidence supporting the early dissemination of metastatic tumour cells in transgenic mouse models of breast cancer204; however, the competence of these disseminated tumour cells in forming secondary lesions is currently under investigation30. As such, these two competing, but not mutually exclusive, paradigms provide a conceptual basis for the investigation of metastatic evolution, and have important implications in the prevention and treatment of metastatic disease8.
DNA copy number analysis of metastatic prostate cancers has indicated a common clonal origin in most cases, although additional subclonal alterations were also observed in metastases31. Consistently, a comparison between whole-genome sequences of a basal-like primary breast tumour and its corresponding brain metastasis has shown that, although copy number alterations and overall mutational spectra within the genome were not significantly different, examining the prevalence of specific mutations revealed that a subset of cancer cells from the primary tumour were preferentially enriched in the metastatic lesion9. Genomic sequencing analyses of pancreatic cancer lesions also suggested that metastatic lesions are clonal in nature, and probably require additional driver mutations that are not found in the primary tumour, and also that certain alterations are linked to organ-specific metastasis32,33. A recent study extended the metastasis genealogy investigation to the single-cell level, revealing that a single clonal expansion formed the bulk of the primary tumour and seeded the metastases10. Collectively, these studies suggest that metastases are spawned from a late clonal expansion of the primary tumour, and that such clonal cells may be additionally endowed with essential metastatic capabilities. However, we still have little grasp on exactly how long it takes for these metastatic clonal populations to emerge. Quantitative analysis of the mutation range in different stages of colorectal cancer suggested that the time required for an invasive tumour to develop metastasis (<2 years) is much shorter than the time needed for a benign tumour to evolve into advanced cancer34. In pancreatic cancer, the timing between these stages of cancer progression followed a comparable pattern, although it was shown that 5 years lapsed before a non-metastatic founder cell acquired metastatic abilities33. These findings imply that a fairly small number of rate-liming mutations are required for advanced tumours to gain metastatic competence.
Overall, these genomic studies seem to disagree with the parallel progression hypothesis that metastatic cells arise from early intermediate cells with genetic and phenotypic characteristics that differ from their primary tumour; they instead favour the linear progression model of metastasis35. A limitation of most of these studies is the use of cancer specimens from late-stage patients whose aggressive primary tumours and metastases were obtained within a relatively short time frame. Further investigations using retrospective or prospective samples that are collected in more diverse clinical scenarios, such as metastases that develop following prolonged latency, from large cohorts of patients are needed to provide a comprehensive view of the evolutionary dynamics of metastasis for different cancers. The insight we gain will guide the selection of treatment and will assist the design of preventive and therapeutic strategies.
Classification and prognosis.
An important task in cancer management is to identify early stage tumours that are at a high risk of metastatic spread. Gene expression profiling of breast cancer samples revealed the existence of distinct subtypes, which not only define the biological diversity of breast cancer36, but which also engender meaningful prognostic and predictive value for patients37–39. The molecular underpinning for the hetero geneity in clinical prognosis of different tumour subtypes remains largely unclear and will be a major focus of future research. Distinct metastatic potentials observed in these cancer subtypes may simply reflect the cumulative effect of a diverse range of initiating and contributing oncogenic mutations. In a different, but not mutually exclusive, scenario, the same oncogenic insults may take place in different cell lineages, such as in basal versus luminal epithelial cells40–42, or during different stages of differentiation, such as adult tissue stem cells versus differentiated progenitor cells, and this may ultimately influence the malignant and metastatic potential of the resulting tumours. Indeed, the introduction of identical genetic elements into two independent normal human mammary epithelial cells led to tumour xenografts with distinct lung metastatic properties43. Such findings strongly suggest that the pre-existing cellular programmes among distinct epithelial cell types can influence the ultimate cancer phenotype irrespectively of subsequent genetic changes. Accordingly, exploring the role of adult tissue stem cells and stem cell-like tumour-initiating cells in the development and maintenance of metastatic capabilities is becoming an active area of investigation7,44. To date, transgenic model systems that introduce specific genetic elements into individual cell types of a particular tissue have been used to identify the cell of origin for several cancers45–47. For example, the inactivation of Rb1 and Trp53 in distinct pulmonary cell types recently showed that neuroendocrine and, to a lesser degree, alveolar type 2 cells are responsible for the development of small-cell lung cancer48. The picture is less clear in basal cell carcinoma, as two separate transgenic models revealed different cells of origin in the skin epidermis49,50. These seemingly incongruent results may be reconciled if each model were shown to represent a distinct clinical subtype of basal cell carcinoma. Future research harnessing the power of sophisticated animal models of tumour initiation and progression will be integral to studying the relationship between the cell of origin and the metastatic abilities of different cancer subtypes. As we explore the diversity of cell lineages and cellular hierarchy during organ development, tissue homeo stasis and oncogenesis, we will better understand the cellular contexts that allow the acquisition of metastatic functions.
Beyond the genetic and cellular heterogeneity of tumour cells, the inherent diversity in the host genetic background has also been recognized to influence metastatic risk. A pioneering study showed that an identical oncogenic event in the mammary tissue of mice with different genetic backgrounds led to the development of mammary tumours with similar primary tumour properties but with distinct lung metastasis proclivities51. Linkage analysis further mapped several genomic loci that modify metastatic potential in mice52–54. Future studies may extend this area of research to human patients with the application of genome-wide association study (GWAS) methods. By understanding the relative influence of a patient’s genetic background on their risk of developing metastatic disease, we will be better prepared to implement appropriate therapeutic options early on in treatment.
Identifying drivers of metastasis.
As prognostic gene signatures were being discovered, gene expression profiling was concomitantly being applied to identify functional drivers of metastasis (TABLE 1). This area of research was built on decades of groundbreaking work by Isaiah Filder, Fred Miller and many others who focused on deriving organ-specific metastatic sublines of tumour cells through in vivo selection or clonal expansion. Comparing the expression profiles of highly metastatic cells with their weakly metastatic counterparts from an isogenic background allowed for the efficient and unbiased identification of candidate regulators of metastasis, including metastasis-promoting18–20,55–57 and metastasis-suppressing genes58–60. Furthermore, gain-of-function or loss-of-function genomic screens, cross-species integrated genomic analyses and computational reanalysis of genomic profiling data have also led to the identification of functional mediators of metastasis with direct clinical relevance61–64 (TABLE 1).
Table 1 |.
Select general and organ-specific metastasis genes uncovered by novel approaches
| Genes | Function | Approach | Refs |
|---|---|---|---|
| Bone metastasis | |||
| IL11 and CTGF | TGFβ target genes that functionally promote metastasis tothe bone | Genomic profiling of bone-metastatic variants after in vivo selection in experimental mouse model | 18 |
| ADAMTS1 and MMP1 | Metalloproteinases that proteolytically release EGF-like ligands from tumour cells that downregulate OPG levels in osteoblasts through EGFR signalling | Genomic profiling of bone-metastatic variants after in vivo selection in experimental mouse model | 220 |
| JAGGED1 | Increases IL-6 production from osteoblasts to promote tumour growth, and stimulates osteoclast differentiation and activity | Expression profiling of metastatic variant cell lines and prognostic analysis of human clinical data | 142 |
| SRC | Promotes survival of DTCs in the bone microenvironment by inducing AKT-dependent survival cues to the inflammatory cytokines CXCL12 and TRAIL | Signalling pathway signature analysis of clinical profiling data | 221 |
| NF-κB* | Transcription factor that facilitates breast cancer bone metastasis through the osteoclast-promoting activity of GM-CSF | Surveillance of signalling pathway activity in bone metastatic cells | 222 |
| Lung metastasis | |||
| EREG, COX2, MMP1 and MMP2 | Act collectively to promote angiogenesis, intravasation and extravasation | Genomic profiling analysis of lung-tropic variants | 223 |
| ANGPTL4 | Promotes extravasation by disrupting endothelial integrity | Genomic profiling analysis of lung-tropic variants and analysis of signalling pathway activity in clinical lung metastasis | 224 |
| ERP5 | Promotes migration and metastasis that is dependent on ERBB2 and PI3K signalling | Forward genetic screen using an orthotopic mouse model of breast cancer | 58 |
| MTDH | Promotes chemoresistance and adhesion to the lung endothelium to facilitate metastasis | Phage display screening of lung-homing peptides and mathematical analysis of clinical genomic data set | 62,225 |
| ID1 | Promotes tumour-initiating function in lung metastasis | Genomic profiling of lung-tropic variants from in vivo selection | 20,226 |
| TNC | Promotes lung colonization by engaging the Notch and WNT developmental signalling pathways in the formation of a metastasis niche | Gene expression profiling of lung-tropic variants and immunostaining of lung metastases from patients with breast cancer | 164 |
| KLF17 | Inhibits lung metastasis by suppressing EMT | RNAi screen in a mouse model of lung metastasis | 227 |
| VEGFR1 ‡ | BMD haematopoietic progenitor cells expressing VEGFR1 establish the pre-metastatic niche in lung metastasis | Molecular profiling of stromal cells in pre-metastatic niche | 134 |
| S100A8*‡ and S100A9‡ | Inflammatory proteins expressed by the lung which recruit myeloid cells to establish the premetastatic niche | Gene expression profiling of lungs primed by primary tumours | 138,139 |
| CCL5 ‡ | BMD mesenchymal progenitor cells secrete CCL5, stimulating primary breast cancer cells to metastasize to the lung | Cytokine profiling of the co-culture of BMD mesenchymal progenitors and breast cancer cells | 13 |
| MMP9 ‡ | Expressed by lung endothelial cells and macrophages on induction by primary tumours expressing VEGFR1 tyrosine kinase | Genetic knockout and transgenic mouse modelling combined with in vitro tissue analysis | 228 |
| CXCL12‡ and CCL21‡ | Ligands found in the lung that bind to their respective chemokine receptors expressed by breast cancer cells | Gene expression surveillance of breast cancer cells combined with functional in vivo studies | 229 |
| Brain metastasis | |||
| ST6GALNAC5, COX2 and HB-EGF | Mediators of breast cancer metastasis that promote extravasation across the BBB and colonization of the brain | Genomic profiling of brain-tropic sublines derived from in vivo selection | 19 |
| MIF, IL8 and PAI1 | Tumour-derived factors that activate astrocytes in the brain and induce expression of inflammatory mediators | Cytokine profiling of tumour and astrocyte cultures | 141 |
| STAT3 | Promotes melanoma invasion, angiogenesis and metastasis to the brain | Signalling pathway analysis of in vivo selected bone metastatic variants | 230 |
| IL6, IL1B and TNF | Astrocyte-derived cytokines associated with the development of brain metastasis | Cytokine profiling of tumour and astrocyte cultures | 141 |
| General metastasis | |||
| LEF1 and HOXB9 | WNT and TCF target genes that promote metastasis to bone and brain through chemotactic invasion | Clinical data set analysis of signalling pathway activities | 163 |
| CXCR4 and CCR7 | Chemokine receptors expressed on breast cancer cells that promote chemotaxis and metastasis | Gene expression surveillance of breast cancer cells with different metastatic abilities | 229 |
| RHOC | Promotes tumour invasion and metastasis | Expression profiling of isogenic melanoma cell lines with different metastatic abilities | 56 |
| TWIST | EMT-promoting transcription factor | Expression profiling of mammary tumour cell lines with different metastatic abilities | 57 |
| MACC1 | Promotes proliferation, invasion and metastasis of colon cancer through HGF | Genome-wide expression analysis of primary and metastatic colon cancer | 231 |
| NEDD9 | Promotes melanoma migration, invasion and metastasis by interacting with focal adhesion kinase | Genomic copynumber analysis in inducible transgenic mice | 63 |
| LKB1 | Inactivation promotes lung cancer metastasis when combined with oncogenic KRAS, possibly by activation of SRC, P13K, and MEK1 and MEK2 | Transgenic mouse models and clinical analysis of lung cancer; integrative genomic and proteomic studies using in vivo lung cancer models | 64,65 |
| LOXL1 | Cell-autonomous role in promoting breast cancer invasion; systemic expression primes pre-metastatic niche for lung, liver and brain metastasis by recruiting CD11B+ myeloid cells | Gene expression profiling of hypoxia-induced genes and functional in vivo studies | 232,233 |
BBB, blood-brain barrier; BMD, bone marrow-derived; COX2, cyclooxygenase 2; DTC, disseminated tumour cell; EGF, epidermal growth factor; EGFR, EGF receptor; EMT, epithelial to mesenchymal transition; GM-CSF, granulocyte-macrophage colony-stimulating factor; IL, interleukin; KLF17, Kruppel-like factor 17; MACC1, metastasis associated in colon cancer 1; MMP1, matrix metalloproteinase 1; MTGH, metadherin; RNAi, RNA interference; STAT3, signal transducer and activator of transcription 3; TGFβ, transforming growth factor-β; TNF, tumour necrosis factor; TNC, tenascin C; VEGFR, vascular endothelial growth factor receptor.
Nuclear factor-κB (NF-κB) is a transcription factor that can be formed by a number of different proteins.
Indicates stromal genes involved in metastasis.
As highlighted above, advances in massively parallel sequencing technology have vastly improved our capacity to more comprehensively uncover genomic changes that underlie cancer development and progression65,66. Next-generation sequencing of matched primary tumours and metastasis or isogenic cell lines with different metastatic abilities, together with the characterization of transcriptomic and epigenomic alterations in cancer through RNA sequencing and chromatin immunoprecipitation (ChIP)–sequencing, respectively, will help to reveal previously unidentified genomic disturbances that potentially contribute to malignant progression. The considerable challenge that remains is the functional interpretation of these genetic and epigenetic changes. Considering the magnitude of alterations found in cancer genomes, this task seems daunting; however, sophisticated computational algorithms that are currently being explored, such as those that use sequence-based and structure-based predictive features (such as PolyPhen2), should aid in distinguishing the driver mutations from the passenger mutations and will probably reduce candidate gene lists by appreciable amounts67–69. Relevant genomic alterations should be translated into gene expression and protein level phenotypes, which can be predicted using computational models, but which will ultimately rely on fundamental molecular and biochemical approaches. Following characterization, these molecular phenotypes can be placed into the context of subcellular networks and pathways with an emphasis on functional consequences (discussed below). Finally, the altered cellular phenotypes should be connected to metastasis-promoting features and validated using clinically relevant experimental model systems. Ultimately, harnessing the power of next-generation sequencing will certainly facilitate our understanding of metastatic evolution.
Proteomics.
Direct exploration of protein-level variations using modern proteomic techniques27,28 has emerged as another powerful tool in the investigation of cancer metastasis. A key advantage of the proteomic approach is the ability to examine the biochemical, cellular and sometimes even organismal phenotypic states rather than genotypic expression patterns. Mass spectrometry can systematically analyse several thousands of proteins with quantitative precision through the combination of stable isotope labelling by amino acids in cell culture (SILAC)-based proteomic techniques and advanced bioinformatics. In recent years, quantitative proteomics has been applied to identify proteins that are differentially expressed in separate cellular compartments, such as those found in the membrane and/orsecreted by the cell (the subset of the proteome known as the secretome) of highly metastatic versus weakly metastatic cells70–74, although the functional importance of such proteins has not been thoroughly investigated. Furthermore, comparing the proteome of plasma, serum and/or urine from patients with metastatic cancer with those from patients with localized disease can aid in the recognition of prognostic biomarkers, a subset of which may be drivers of cancer progression and thus strong candidates for therapeutic intervention75. These approaches have not entered mainstream medicine owing to current shortcomings and inherent challenges such as the low sensitivity and specificity of existing cancer biomarkers76; a poor understanding of the biological and pathological importance of protein dynamics (changes in protein quantity and post-transcriptional modifications, for example); limited access to high-quality biospecimens; a lack of standardized methodology in discovery and validation studies; the inadequate incorporation of biomarker assessment in current clinical trial designs; and insufficient collaboration among proteomic, biological and clinical teams, as well as institutions.
Despite the multifaceted hurdles that riddle the path of proteomics en route to routine clinical use, the potential of proteomics to revolutionize metastasis research and patient care cannot be underestimated. At present, proteomic studies have yet to fulfil their potential of providing the functional insight that has been achieved by classical genomic studies, and therefore proteomic studies represent an area of investigation that warrants greater effort and resources in the coming years. Future studies will need to evaluate biomarkers for pre-cancerous lesions, localized disease and/or micrometastases in an effort to support early diagnosis and prognosis: these molecules may be very different from those that are found in advanced disease. Proteomic studies of microdissected clinical specimens should reveal intricate signalling networks that exist between the tumour and stromal compartment at a level that is unattainable by genomic profiling and sequencing studies alone. Ultimately, a multidisciplinary and systems approach will provide the most insightful and comprehensive understanding of cancer metastasis.
Towards systems biology of metastasis.
The application of genetic and molecular biology techniques to the investigation of cancer metastasis has yielded the vast majority of discoveries over the past few decades. As such, cancer metastasis has largely been explained through reductionism; that is, it has been defined by individual genetic disturbances and their resultant cellular phenotypes. Considering the complex nature of metastasis, a more holistic approach to its investigation, perhaps through systems biology, seems to be essential (FIG. 1).
Figure 1 |. Research strategies for understanding the molecular basis of cancer metastasis: from reductionism to systems biology.
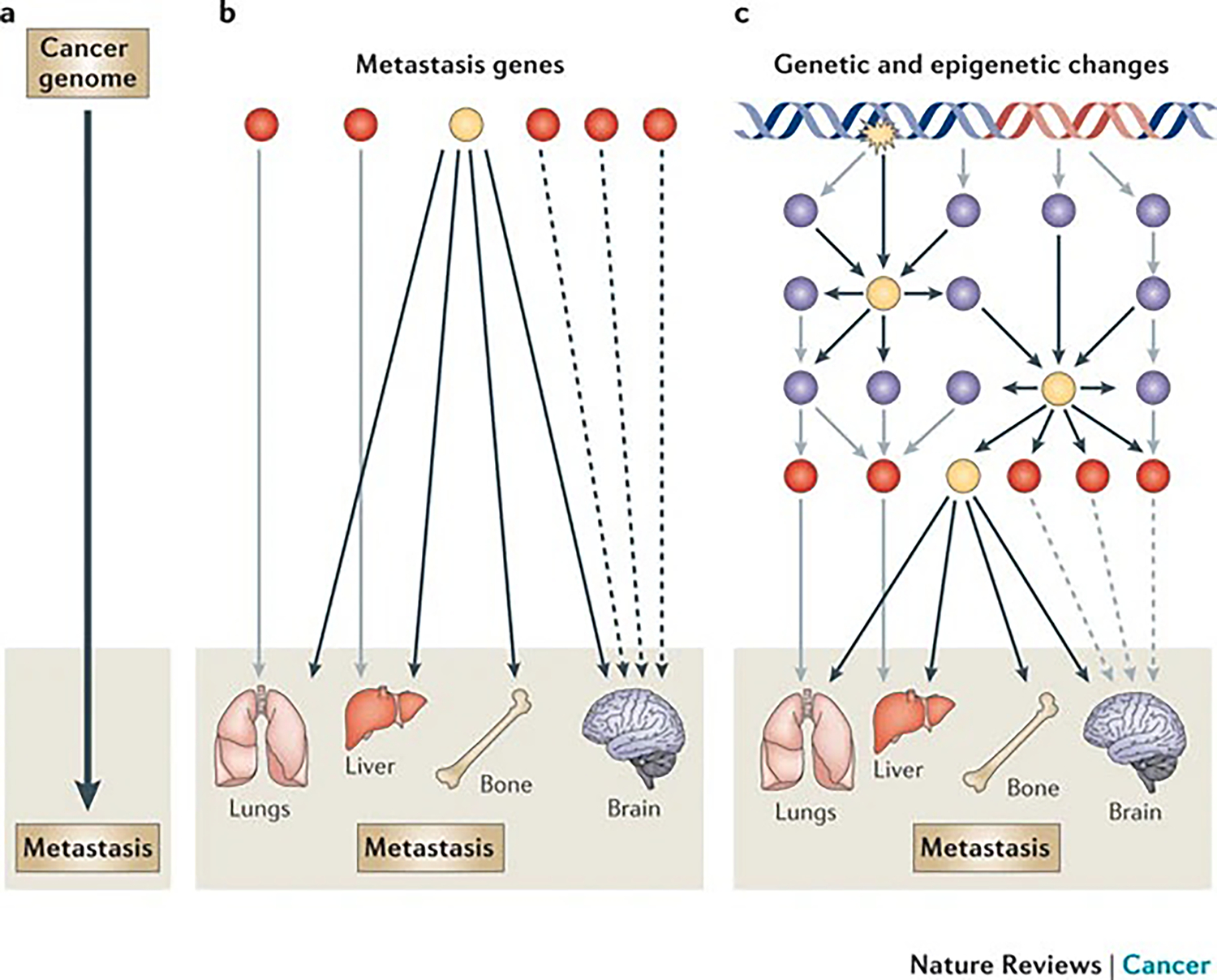
The evolving states of cancer, including metastasis, are reflected in dynamically changing expression patterns of the genes and proteins within the cancer cells. Metastasis research has generally relied on the linear approach of gene-to-trait mapping, which links the metastasis genotype with its corresponding metastatic phenotype (part a). More complex linear linkages may include multivalent relationships, in which one gene (or group of genes) can have functions in multiple metastasis phenotypes (pleiotropy), and a metastasis tissue tropism can be exerted by many independent genes or gene groups (redundancy) (part b). However, gene interactions are influenced by their context, which often cannot be captured by traditional one-gene-one-trait approaches. Therefore, metastatic behaviour should be considered as the consequence of the collective action of individual metastasis genes through nonlinear interactions (part c). Understanding the nature of these network level interactions and identifying crucial nodes of functional control will pave the way towards rational therapeutic design for metastatic breast cancer. Red circles represent genes or groups of genes that mediate tissue-specific metastasis of breast cancer. Blue circles represent regulators of metastasis genes. Yellow circles represent key functional nodes of metastasis regulation networks and are prime targets for therapeutic development. The black, grey and dashed arrows indicate different pathways.
Signalling pathways in cancer metastasis have been extensively studied at the level of individual proteins or as a linear cascade of proteins but they have been less frequently evaluated through a network approach. High-throughput data can be extracted from and annotated on the basis of comprehensive genomic, transcriptomic and proteomic interrogation of experimental or clinical samples65,77. The information we gather from these large-scale techniques can be used to develop network maps, interactomes, ensemble descriptions and gene expression modules with the assistance of advanced computational algorithms78. These network models can be validated, using a myriad of experimental methods, ranging from basic biochemistry looking at protein–protein interactions to sophisticated molecular real-time imaging examining signalling pathway activity in live tissue79. Importantly, the response of these networks to stimuli, which can now be measured through multiplex technologies such as multiparameter flow cytometry, and selective perturbations of individual components using small interfering RNA (siRNA) or drug compounds, is integral to establishing functional network models28. Ultimately, these multifunctional models can be used to discover novel signalling proteins, predict therapeutic response to selective inhibitors and uncover resistance mechanisms. For example, the drug–gene–phenotype Connectivity Map approach80 was successfully used to identify the mTOR inhibitor rapamycin as an effective agent for overcoming dexamethasone resistance in acute lymphoblastic leukaemia.
Two active areas of investigation in cancer metastasis that could benefit greatly from a systems biology approach are micro-RNA (miRNA) and epigenetic regulation. Of note, these cellular programmes are not independent of each other, as DNA methylation has been shown to silence miRNA activity81–83 and miRNAs have been shown to affect epigenetic changes84,85. miRNAs have predominately been implicated in cancer metastasis through their regulation of the early steps in tumour progression, such as migration and invasion86–88, but more recently through their effect on later stages, such as colonization89. miRNAs are commonly linked to their metastatic functions by regulating individual genes that are involved in metastasis. However, miRNAs, like transcription factors, function as master regulators that can simultaneously control the expression of several hundred genes and also affect dramatic shifts in cellular phenotype90,91. As such, mapping out individual genes that are targeted by miRNAs and other non-coding RNAs92 may not faithfully represent the extent of their influence. Instead, the breadth of genes regulated by an miRNA can be collected using genomic and proteomic approaches and subsequently translated into ensemble data sets with the help of computational algorithms that effectively group specific genes into functional modules. These modules can further be used to generate network models that will optimally reflect a more comprehensive picture of the cellular and behavioural functions that are affected by an miRNA and can ultimately provide a better insight into how miRNAs may affect cancer metastasis. Once these networks are established, the discovery of future cancer-promoting genes and their regulation by miRNAs can be placed in context.
Epigenetics has been implicated in cancer progression through its regulation of tumour initiation, stem-cell properties and, rather intriguingly, chronic inflammation85,93–96. Similar to miRNAs, epigenetic regulation modulates a broad range of coordinated genome-wide expression changes, but does so at a different level from conventional genome aberrations that are associated with cancer progression. The direct contribution of epigenetics to cancer metastasis is fairly unexplored, but we are now starting to make headway with the support of high-throughput genomic studies. For example, genome-wide methylation analysis of paired colorectal cancer primary tumour and liver metastasis specimens demonstrated differences in DNA methylation status in advanced cancer at a global and individual gene level when compared with localized disease97. However, the relationship between metastasis-related epigenetic differences and corresponding changes in gene expression or cellular function has not been defined. Using computational algorithms98, a systems level approach can help to incorporate the unique epigenetic-mediated gene expression and cellular changes in the context of previously defined genomic and proteomic alterations that are associated with metastasis. A systems level understanding of regulatory programmes that govern metastatic behaviour will ultimately need to be integrated within the context of tumour–stromal interactions that occur at different stages of tumour progression.
Cellular heterogeneity of metastasis
Understanding the contribution of stromal cells to cancer metastasis is essential to fulfil the promise of improved therapy, as foreseen by the seed and soil hypothesis. Here, we base our discussion on some of the key conceptual advances that have been made in the past decade regarding the role of stromal cells and cellular dynamics during different phases of metastasis. For more in-depth coverage of these topics see REFS 5,11,99.
Effect of the primary tumour microenvironment on metastasis.
Although the ability of non-neoplastic stromal cells to promote tumour proliferation, invasion, survival and chemoresistance is well known14,15,100–106, we are only now beginning to recognize their function in the development of cancer metastasis. It should not come as a surprise that tumour angiogenesis was among the initial findings that supported a role for stromal cells in cancer metastasis; the poor vascular integrity of newly synthesized blood vessels within the tumour allows for the escape of malignant cells with the potential of distant spread. As mediators of tumour angiogenesis were uncovered, targeted therapies against these molecules were designed and among the first to achieve clinical application for the control of late-stage metastatic disease107–109. Although anti-vascular endothelial growth factor (VEGF) therapy was approved to combat metastatic disease in patients110, recent studies in mice paradoxically revealed an increased risk of metastasis associated with this therapy111,112, which underscores the peril of oversimplifying the potential effect of targeting the tumour stroma. These preclinical findings corroborate results from recent clinical trials showing no overall survival benefit for the VEGF inhibitor bevacuzimab in various cancers113–115, bringing its therapeutic value into question116 and serving as a sobering reminder to consider the unexpected consequence of anticancer therapy, particularly in regards to metastasis.
In more recent years, we have witnessed important roles for additional primary tumour stromal cell types in cancer metastasis. Elegant studies combining intravital imaging and mouse modelling have convincingly demonstrated a pro-metastatic role of tumour-associated macrophages through colony stimulating factors (CSFs) and epidermal growth factor (EGF) signalling117,118. More recent reports have implicated additional pathological and molecular mechanisms that mediate crosstalk between tumour cells and macro phages, ultimately influencing cancer metastasis119,120. Leukocytes and other immune cells have been recognized as crucial regulators of primary tumour growth and metastasis121–123. Mesenchymal cells that reside in breast tissue have also been shown to affect the metastatic behaviour of breast cancer cells through CCL5 signalling13. Moreover, a recent report showed that mesenchymal cells can influence the metastatic behaviour of neuroendocrine small-cell lung cancer. Most intriguingly, the mesenchymal cells and neuroendocrine cells were descendants of the same progenitor cell, which exploited RAS signalling to generate functional intra-tumoural heterogeneity124. These studies have established stromal cells as important regulators of metastasis through their ability to influence cancer cell functions such as chemotaxis and invasion, as well as microenvironment properties, such as vessel integrity and the presence of immunological cells. Despite these advances, we have yet to discover many of the molecular components that facilitate communication between tumour cells and individual stromal cells of the primary tumour. We need a better working knowledge of the paracrine signalling network that mediates these molecular interactions — an area of research that will be facilitated by advanced proteomics — as these interactions are key components in our systems level understanding of metastasis. There is also limited insight into how multiple stromal components concomitantly associate with tumour cells. For example, what is the significance of inter-stromal crosstalk between different lineages of stromal cells in malignant progression? How does stromal heterogeneity synergize with tumour heterogeneity to encourage metastatic spread? Importantly, advances in molecular imaging and microscopy have opened new avenues of investigation for better examining the contributions of cellular heterogeneity in cancer progression (BOX 2).
Box 2 |. Imaging cancer dynamics.
Currently, the mainstay objective of imaging modalities in the clinical setting is to characterize the extent of metastatic disease in cancer patients, which has been greatly refined by the integration of diverse macroscopic imaging modalities. Positron emission tomography (PET) has been combined with computed tomography (CT) imaging to improve our ability to stage metastatic disease and to monitor response to treatment205,206. The simultaneous use of multiple isotope-labelled probes, such as the combination of 18fluoride and 18fluorodeoxy glucose (18FDG), in conjunction with hybrid imaging modalities, has further enhanced our ability to detect metastatic disease207. Moreover, cancer-specific and organ-specific molecular probes, such as those used in the detection of melanoma using PET and liver metastasis using magnetic resonance imaging (MRI), respectively, hold great promise in augmenting the resolution with which we can detect systemic cancer spread208,209.
At the bench, we have come a long way from direct visualization of tumour cell invasion using green fluorescent protein210. For example, direct visual evidence of specific cell lineages, such as cancer stem cells, in metastasis can be obtained using high-resolution, non-invasive imaging211. The combination of a mammary imaging window and a photo-switchable fluorescent protein has been used to investigate the influence of spatially and functionally distinct tumour microenvironments on cancer cell invasion and intravasation212,213. The simultaneous observation of distinct stromal cells as they interact in the primary tumour can be achieved through multiphoton microscopy, which has the benefits of enhanced tissue depth and multiple colour channels21,23,214.
Probing beyond the cellular level, there is now evidence that molecular imaging will provide functional readouts of subcellular biological processes, such as protein–protein interactions215. These modalities can be used to visualize real-time in vivo subcellular events that regulate tumour metastasis. For example, non-invasive imaging techniques together with tumour cells genetically engineered to provide functional readouts of a signalling pathway can be used to test targeted anti-metastatic agents in preclinical metastasis models216. Importantly, these powerful tools have been integrated with computational programs to provide quantitative readouts in addition to high-resolution, real-time qualitative impressions. In the future, the sophistication of molecular imaging may also translate into the clinical setting to provide instant readout and direct visualization of biological processes that promote tumour metastasis217.
Tumour cells in transit.
Essential to cancer metastasis is the ability of primary tumour cells to enter the vasculature and to use these fluid ‘highways’ as a means to reach distant organs. Seminal studies by James Ewing highlighted the influence of the vascular anatomy on the ultimate destination for metastatic tumour cells3. Although we now understand that the vasculature alone does not explain the pattern and distribution of metastasis, the ability of tumour cells to endure substantial stress in transit remains a poorly understood aspect of metastasis. Even more, we have yet to characterize how this selection pressure affects the subsequent metastatic behaviour of surviving cells. Platelets have been recognized as an important blood component that protects circulating tumour cells (CTCs) and promotes their metastatic colonization125. Nevertheless, knowledge regarding the cellular and molecular mechanisms that allow tumour cells to survive and adapt within circulation remains scarce, owing in part to the inherent difficulty in isolating and analysing CTCs. Recent technical and conceptual advances have helped overcome these limitations.
The most effective strategies in isolating tumour cells from the circulation have relied on antibody-based epitope capture methods126. The application of microfluidic rare cell detection approaches has improved CTC purity and yield in recent years127. Although the technical challenges of detecting CTCs remain, rapid advances in bioengineering are rendering these hurdles surmountable. We already appreciate the clinical value of CTCs, as recent studies have associated their presence in the bone marrow with poor prognosis in patients with breast cancer128,129. On the basis of these findings, the detection of CTCs has been incorporated into the international tumour staging systems130 and endorsed through recent recommendations on tumour markers made by the American Society on Clinical Oncology (ASCO)131.
Despite their clear prognostic importance, the diagnostic value of CTCs is largely unknown and fairly unexplored. We have not defined whether CTCs can be reliably detected before the development of metastatic disease. Our ability to diagnose and to manage prostate cancer would clearly benefit from this understanding: the surveillance and molecular analysis of CTCs at regular intervals in patients with localized prostate cancer could help to distinguish indolent disease from aggressive disease and could ideally inform subsequent treatment decisions. Moreover, the ability of CTCs to predict clinical response to therapy would also help to guide disease management, as shown by examples in breast cancer and non-small-cell lung cancer127,132. As such, many of the advantages and strategies of clinical proteomic and next-generation sequencing discussed above can be adapted to the evaluation of CTCs.
There are also many unresolved questions concerning the biology of cancer metastasis that may reveal their answers in the exploration of CTCs. For example, how does the genomic and proteomic landscape of CTCs compare with their corresponding primary tumour or metastasis? Are there unique gene expression changes in CTCs that are not found in the primary tumour or metastasis? Do these expression patterns implicate independent signalling pathways or cellular phenotypes? Are these expression changes associated with stem-cell properties, epithelial to mesenchymal transition (EMT) and mesenchymal to epithelial transition (MET), or other metastasis-promoting functions? Can these genomic or expression patterns be exploited for drug development? The answers to these questions will improve our understanding of metastatic evolution and will resolve different schools of thought with respect to cancer progression, such as the linear versus parallel progression models (BOX 1).
Tumour–stromal interactions in distant organs.
The colonization and outgrowth of tumour cells in a secondary organ is often considered the rate-limiting, as well as the most poorly delineated, step in the metastatic cascade. We have begun to elucidate the basis of metastatic colonization by characterizing the functional involvement of the tumour stromal cells of the secondary site5. Considering the vast area of research that is encompassed by this subtopic, we focus on the salient advances that may help to direct future research.
The emerging concepts of the premetastatic niche99,133 and metastatic selfseeding134,135 have challenged our traditional view of metastasis and have stimulated new avenues of research (FIG. 2). Metastasis is no longer considered a unidirectional flow between primary tumours and distant organs. The pre-metastatic niche model shows that, preceding the arrival of disseminated tumour cells (DTCs), bone marrow-derived haematopoietic stem cells are mobilized by tumour-derived factors and are recruited to the secondary site where they negotiate a more hospitable microenvironment to foster the survival and expansion of metastatic lesions133. According to the self-seeding hypothesis, metastatic tumour cells can also return to the primary site, accelerating the growth and malignant evolution of primary tumours (FIG. 2). Further investigation using experimental and clinical models will help to define the precise role and mechanism of these events in metastasis.
Figure 2 |. Evolving view of the dynamic relationship between the primary tumour and metastasis.
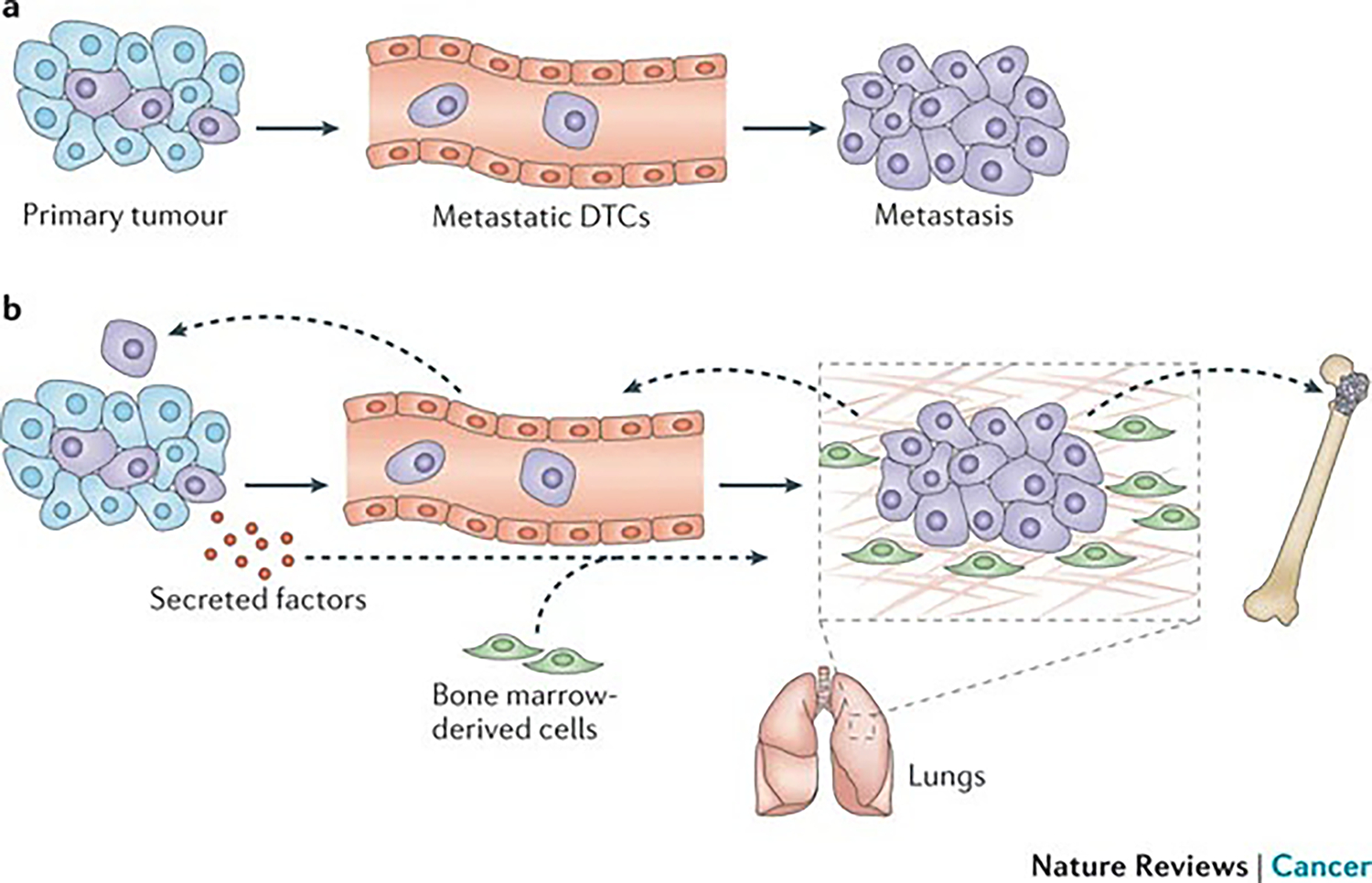
a | The traditional view of cancer metastasis in which primary tumour cells escape their site of origin, travel in a unidirectional path away from the primary site and ultimately colonize distant organs to give rise to systemic disease is shown. b | A dynamic view of cancer metastasis in which bone marrow-derived cells are mobilized by tumour-derived inflammatory factors and prime distant sites of metastasis to form the pre-metastatic niche is shown. Disseminated tumour cells (DTCs) (either from the primary tumour or from metastases) that have been selected with enhanced malignancy can colonize distant organs, as well as repopulate the primary site through the phenomenon of self-seeding.
Inflammatory cytokines have emerged as crucial mediators of the pre-metastatic niche and self-seeding133,135–137. The interplay between colonizing tumour cells and the microenvironment of the secondary organ also seems to involve inflammatory cytokines, exposing these molecules as prime targets for therapeutic intervention. Recent studies have shown that the tumour-induced secretion of interleukin-6 (IL-6) by stromal cells that reside in the bone and brain facilitates metastatic colonization138–141. Interestingly, inflammatory signals frequently affect tumour cells by altering their epigenetic regulatory programme and by conferring cancer-promoting properties142, which highlights a previously identified area of research that could benefit from a systems biology approach. For example, an epigenetic switch that is initiated by the oncoprotein SRC was sustained by an inflammatory regulatory network involving IL-6 and nuclear factor-κB (NF-κB) signalling in tumour progression85. Intriguingly, an independent report showed that SRC signalling was preferentially activated in latent breast cancer cells and mediated pro-survival responses to inflammatory cytokines that supported bone metastasis. Associating these findings within the context of many others, we recognize that inflammation-induced genetic and epigenetic changes may contribute to the survival of disseminated cancer cells and may represent a principal topic of future metastasis research and therapeutic development.
After surviving the adjustment to the secondary site, tumour cells must sustain their growth to develop overt metastases. Developmental pathways have emerged as important players in tumour progression and metastasis143. Although transforming growth factor-β (TGFβ)18,144,145, bone morphogenetic protein (BMP)146–151, WNT152–157 and Hedgehog158–160 signalling have all been shown to influence bone metastasis, the Notch pathway has recently joined the ranks through a stroma-dependent mechanism that was susceptible to pharmacological inhibition140. In lung cancer, cell-autonomous activation of the WNT–TCF transcriptional programme was shown to promote brain and bone metastasis through the actions of LEF1 and HOXB9 (REF. 161). Furthermore, both the Notch and WNT pathways were recently shown to participate in generating a viable metastatic niche for lung-colonizing breast cancer cells through the actions of the extracellular matrix protein tenascin C162. It is important to note that, similar to their pleiotropic function during cell fate decisions in metazoan organisms, developmental signalling pathways often regulate multiple metastasis genes with diverse functions that facilitate organ-specific metastasis, and therefore developmental signalling pathways represent key regulatory nodes in the metastasis network. A case in point is the TGFβ pathway, which activates the expression of prometastasis genes VEGF, angiopoietin-like 4 (ANGPTL4), JAGGED1, matrix metalloproteinase 1 (MMP1), CXCR4, parathyroid hormone-like hormone (PTHRP), IL6 and connective tissue growth factor (CTGF). The perspective of metastasis as a developmental programme that has gone awry will continue to be a major topic of research with important therapeutic implications.
Clinical translation
As most metastatic cancers are inoperable, systemic treatments using chemotherapeutic or targeted therapy is often the only option to slow tumour growth or to relieve metastasis-associated morbidity. Although agents that target tumour-specific pathways have been, and will continue to be, major components of anti-metastasis therapy, treatment strategies targeting the tumour microenvironment of the secondary organs will greatly augment our ability to treat late-stage cancer.
Tumour cell-targeted therapy.
The ultimate goal in the treatment of metastatic cancer is to achieve sustained disease remission, which in theory would require the eradication of all cancer cells, regardless of their systemic distribution. With this in mind, genes and pathways that have crucial roles in primary tumour growth and metastasis are ideal targets for therapeutic inventions, as they are likely to show efficacy in reducing both tumour burden and metastasis risk. For example, rigorous research has shown that oncogenic BRAF signalling has an important role in the pathogenesis of malignant melanoma163–167, and this prompted clinical trials for specific and potent inhibitors of mutant BRAF, the initial results of which suggest dramatic efficacy in the treatment of metastatic malignant melanoma168,169. Despite substantial successes in targeted cancer therapy, the chronic problem that will continue to plague the future of targeted therapeutics is drug resistance. The mechanisms underlying resistance to targeted therapy are under active investigation170–178. Overcoming drug resistance will depend on these characterizations, as well as on using the knowledge that we have gained from previously defined effective strategies, such as second-generation ABL inhibitors179–181. Similar to the story of first-generation ABL kinase inhibitors in chronic myeloid leukaemia, the effects of BRAF inhibitors, although profound, are only temporary owing to resistance. Two recent studies182,183 elucidate the potential mechanisms that underlie resistance to BRAF inhibitors and thus provide evidence encouraging novel avenues for rational drug design of second-generation BRAF inhibitors. As new therapies for cancer metastasis are discovered, the field investigating drug resistance will grow dramatically.
Targeting the tumour microenvironment.
As discussed above, the cellular microenvironment of the secondary site has a crucial role in facilitating cancer metastasis. Often, the influence of the microenvironment, and the cellular and molecular adaptations undertaken by tumour cells to successfully colonize a secondary organ, alter metastatic cells in ways that render them resistant to cell-autonomous therapies that effectively treat their corresponding primary tumour184,185. Moreover, even when well-accepted cell-autonomous mechanisms of drug resistance are defined, as in the case of BCR-ABL, alternative methods of drug resistance that are driven by the microenvironment are still largely at work186. Experimental mouse models have shown the therapeutic inadequacies of targeted agents in treating metastatic lesions187,188. These observations and findings collectively support the rationale for targeting the microenvironment of the metastatic lesion in conjunction with targeting the tumour cells directly to better treat metastatic disease.
Our progress in treating bone metastasis by targeting molecules that are found in the tumour microenvironment, such as RANKL189,190, is a direct consequence of the insight gained from systematically dissecting the intricate molecular and cellular crosstalk between tumour cells and bone stromal cells. Elucidating the homeostatic balance between osteoclasts and osteoblasts in the bone microenvironment has been paramount in establishing a contextual basis for understanding bone metastasis and developing targeted therapies17. In addition, the characterization of the haematopoietic niche191 and its interactions with bone cells is likely to help us to understand the potential existence of metastasis niches that have been speculated to facilitate the survival and expansion of DTCs in bone. Thus, a sound framework of normal homeostatic mechanisms can improve our ability to understand and target tumour–stromal interactions in metastasis.
Future directions
We have made significant progress over the past decade in harnessing new technology and research tools to piece together an intellectual framework for understanding cancer metastasis. Moving forwards, efforts should be focused on closing some of the major gaps in areas of metastasis research that have important implications for therapeutic development. For example, the best window of opportunity to control metastatic disease may be the time period between metastatic seeding and the clinical detection of overt metastasis, as this time period represents an occasion when tumour cells are likely to be vulnerable to therapeutic agents and patients are expected to be in an optimal physical condition to endure treatment. However, despite our increasing knowledge about metastatic colonization, we still hold little understanding of how metastatic tumour cells behave as solitary disseminated entities, particularly at crucial junctures in their dynamic existence, such as during the establishment of micrometastases, activation from latency and response to therapeutic regimens. We will need to resolve tumour–stromal dynamics at the single cell level using advanced imaging technology, so that the fate of individual tumour cells and the different lineages of stromal cells, as well as their molecular interactions, can be traced during disease progression and drug treatment. The understanding that we gain from these studies could facilitate the generation of clinically relevant cancer models with the potential to guide the direction of drug development.
A systems biology approach for identifying linker components or key regulatory nodes that connect different functional networks and processes may help us to integrate the components of tumour progression with the individual steps of the metastatic cascade, leading to a more fluid, interdependent depiction of metastasis (FIG. 1). Network modelling can be correlative and therefore considered a descriptive science that often requires further investigation to establish causal relationships. For systems biology to thrive, there will need to be a shift in culture, especially from the funding and peer-reviewed perspective, to encourage a more global understanding of disease.
Systems level investigations will undoubtedly reveal many possible directions for therapeutic exploration. A potential bottleneck is the requirement to functionally validate individual candidate genes cost-effectively and efficaciously in clinically relevant animal models192–196 (TABLE 2). Xenograft models, which have played a predominant part in metastasis research, need to be supplemented with robust transgenic and knockout mouse models. Furthermore, humanized mouse models in which components of mouse stroma are replaced with human counterparts are proving to be valuable tools. By integrating the insight gained from these distinct animal models, we will improve our ability to characterize candidate metastasis genes. In particular, we will be able to understand their normal physiological roles, define their precise functions during different stages of tumour progression and evaluate their effect on therapeutic targeting. Extensive preclinical validation of candidate metastasis genes using robust animal models will increase the success rate of developing effective anti-metastasis agents while minimizing the risk of unexpected adverse side effects.
Table 2 |.
Mouse model systems used to investigate metastasis
| Mouse model | Advantages | Disadvantages |
|---|---|---|
| Xenograft models | • Can accurately model tumour cell dissemination and metastatic colonization using experimental metastasis or spontaneous metastasis models • Human tumour cells derived from patients can be used • Multiple genetic manipulations can be used • They are amenable to in vivo real-time imaging with reporter genes |
• Mice lack an intact immune system • Late-stage cancer cells are often required • There is some species-specific incompatibility • Established cancer cell lines that may evolve with passage in vitro are often used |
| Isograft or allograft models | • Can accurately model tumour dissemination and metastatic colonization • Mice have an intact immune system • Multiple genetic manipulations can be investigated • The models are amenable to in vivo real-time imaging with reporter genes |
• Only mouse-derived cancer cells can be used • The resection of the primary tumour is often required before the development of metastases • It is difficult to randomize groups owing to variable primary tumour size and presence of local invasion • There is limited organ tropism |
| Germline traditional transgenic models | • Provide an accurate model of initiation, local invasion and the early steps of metastasis • Can be used for drug testing and development • Mice have an intact immune system • Can be used to analyse the potential effect of drug targeting on normal physiology |
• Studies are often limited to lymph node and lung metastasis; these models do not develop bone metastases • There is a long lead-time in primary tumour and metastasis development • The development of metastatic lesions is asynchronous, making them difficult to track • There is incomplete penetrance of genetic modification • A tremendous amount of time, labour and resources are needed to generate mice with multiple genetic manipulations • A genetic mutation is usually present in all tissues, making it difficult to tease out cell-autonomous from non-cell-autonomous effects and initiation from progression and metastasis |
| Germline conditional transgenic models | • Conditional expression (tissue-specific and temporally restricted expression) of gene of interest (GOI) • The use of certain conditional promoters enables sporadic expression of GOI rather than activity in all tissue cells |
• Similar disadvantages as a traditional transgenic germline model |
| Humanized metastasis models234–237 | • The orthotopic injection of human tumour cells allows the evaluation of every step in metastasis • Engrafting a human tissue at the metastasis site allows the investigation of human-specific tumour–stroma interactions • Overcome limitations in transgenic mouse models that do not metastasize to bone |
• Gene expression analysis of tumour versus stroma is more difficult based on limitations discussed above • Models lack an intact immune system |
We also need to renovate the design of clinical trials to expedite the development and approval of anti-metastasis therapies. Owing to the considerable financial burden of testing therapeutic agents, clinical trials are seldom designed to evaluate anti-metastasis therapy in the setting of early stage cancer, which may represent a point in disease progression when metastasis might be preventable. As such, many effective anti-metastasis agents that are already US Food and Drug Administration (FDA)-approved for other indications in the United States have not benefited cancer patients who are at risk for metastasis. Reliable and specific biomarkers that reflect an accurate readout of disease progression and efficacy of therapeutic targeting should be implemented in clinical trial design. This will improve our ability to measure the effectiveness of drug targeting, select the optimal patient population for therapeutic intervention and gain accelerated regulatory approval. Finally, clinical trial design should incorporate standardized procedures for the collection of patient tumour samples from localized and metastatic disease at distinct clinical stages, as well as during different points in disease management (such as pre-therapy and post-therapy). These invaluable specimens, including their associated molecular profiles, should be deposited in publically available tumour and data banks to facilitate multidisciplinary research and collaborations. With concerted effort from basic researchers, clinical investigators, drug developers and regulatory agencies, considerable improvements in the management of metastatic cancer may be within reach. We foresee a future in which patients at a high risk of metastasis will be reliably identified using molecular profiles of their primary tumour and CTCs. Effective cocktails of drugs with minimal adverse side effects will be tailored to prevent metastatic recurrence in individual patients. Even for patients with advanced cancer, the diagnosis of metastasis will no longer carry the label of a terminal illness, but will rather be acknowledged as another complex chronic condition that can be effectively controlled with a large arsenal of effective therapeutic agents.
Acknowledgements
The authors would like to thank members of their laboratory, particularly M. A. Blanco, for helpful discussions and critical comments on this manuscript. They apologize to those colleagues whose work is not cited owing to space limitations. The authors’ research is funded by the Brewster Foundation, Champalimaud Foundation, American Cancer Society, Komen for the Cure, New Jersey Commission on Cancer Research, the US Department of Defense and the US National Institutes of Health (R01CA134519 and R01CA141062).
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Contributor Information
Nilay Sethi, Department of Molecular Biology, Washington Road, LTL 255, Princeton University, Princeton, New Jersey 08544, USA.; Robert Wood Johnson Medical School, Piscataway, New Jersey 08544, USA.
Yibin Kang, Department of Molecular Biology, Washington Road, LTL 255, Princeton University, Princeton, New Jersey 08544, USA.; Genomic Instability and Tumour Progression Program, Cancer Institute of New Jersey, New Brunswick, New Jersey 08903, USA.
References
- 1.Fidler IJ The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature Rev. Cancer 3, 453–458 (2003). [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Chambers AF, Groom AC & MacDonald IC Dissemination and growth of cancer cells in metastatic sites. Nature Rev. Cancer 2, 563–572 (2002). [DOI] [PubMed] [Google Scholar]
- 4.Gupta GP & Massague J Cancer metastasis: building a framework. Cell 127, 679–695 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Nguyen DX, Bos PD & Massague J Metastasis: from dissemination to organ-specific colonization. Nature Rev. Cancer 9, 274–284 (2009). [DOI] [PubMed] [Google Scholar]
- 6.Paget S The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 8, 98–101 (1989). [PubMed] [Google Scholar]
- 7.Chaffer CL & Weinberg RA A perspective on cancer cell metastasis. Science 331, 1559–1564 (2011). [DOI] [PubMed] [Google Scholar]
- 8.Klein CA Parallel progression of primary tumours and metastases. Nature Rev. Cancer 9, 302–312 (2009). [DOI] [PubMed] [Google Scholar]
- 9.Ding L et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature 464, 999–1005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Navin N et al. Tumour evolution inferred by single-cell sequencing. Nature 472, 90–94 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Joyce JA & Pollard JW Microenvironmental regulation of metastasis. Nature Rev. Cancer 9, 239–252 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nguyen DX & Massague J Genetic determinants of cancer metastasis. Nature Rev. Genet. 8, 341–352 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Karnoub AE et al. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 449, 557–563 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Bhowmick NA, Neilson EG & Moses HL Stromal fibroblasts in cancer initiation and progression. Nature 432, 332–337 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orimo A et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 121, 335–348 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Scheel C et al. Paracrine and autocrine signals induce and maintain mesenchymal and stem cell States in the breast. Cell 145, 926–940 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weilbaecher KN, Guise TA & McCauley LK Cancer to bone: a fatal attraction. Nature Rev. Cancer 11, 411–425 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kang Y et al. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 3, 537–549 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Bos PD et al. Genes that mediate breast cancer metastasis to the brain. Nature 459, 1005–1009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Minn AJ et al. Genes that mediate breast cancer metastasis to lung. Nature 436, 518–524 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahai E Illuminating the metastatic process. Nature Rev. Cancer 7, 737–749 (2007). [DOI] [PubMed] [Google Scholar]
- 22.Kienast Y et al. Real-time imaging reveals the single steps of brain metastasis formation. Nature Med. 16, 116–122 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Kedrin D et al. Intravital imaging of metastatic behavior through a mammary imaging window. Nature Methods 5, 1019–1021 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Condeelis J & Segall JE Intravital imaging of cell movement in tumours. Nature Rev. Cancer 3, 921–930 (2003). [DOI] [PubMed] [Google Scholar]
- 25.Schena M, Shalon D, Davis RW & Brown PO Quantitative monitoring of gene expression patterns with a complementary DNA microarray. Science 270, 467–470 (1995). [DOI] [PubMed] [Google Scholar]
- 26.Bentley DR et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 456, 53–59 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olsen JV et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 127, 635–648 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Irish JM et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell 118, 217–228 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Hynes RO Metastatic potential: generic predisposition of the primary tumor or rare, metastatic variants-or both? Cell 113, 821–823 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Kouros-Mehr H et al. GATA-3 links tumor differentiation and dissemination in a luminal breast cancer model. Cancer Cell 13, 141–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu W et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nature Med. 15, 559–565 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Campbell PJ et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature 467, 1109–1113 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yachida S et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature 467, 1114–1117 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jones S et al. Comparative lesion sequencing provides insights into tumor evolution. Proc. Natl Acad. Sci. USA 105, 4283–4288 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cairns J Mutation selection and the natural history of cancer. Nature 255, 197–200 (1975). [DOI] [PubMed] [Google Scholar]
- 36.Perou CM et al. Molecular portraits of human breast tumours. Nature 406, 747–752 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Sorlie T et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl Acad. Sci. USA 98, 10869–10874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van ‘t Veer LJ et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature 415, 530–536 (2002). [DOI] [PubMed] [Google Scholar]
- 39.van de Vijver MJ et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 347, 1999–2009 (2002). [DOI] [PubMed] [Google Scholar]
- 40.Wang X et al. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature 461, 495–500 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lim E et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nature Med. 15, 907–913 (2009). [DOI] [PubMed] [Google Scholar]
- 42.Molyneux G et al. BRCA1 basal-like breast cancers originate from luminal epithelial progenitors and not from basal stem cells. Cell Stem Cell 7, 403–417 (2010). [DOI] [PubMed] [Google Scholar]
- 43.Ince TA et al. Transformation of different human breast epithelial cell types leads to distinct tumor phenotypes. Cancer Cell 12, 160–170 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Li F, Tiede B, Massague J & Kang Y Beyond tumorigenesis: cancer stem cells in metastasis. Cell Res. 17, 3–14 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Johnson RA et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466, 632–636 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barker N et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457, 608–611 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Goldstein AS, Huang J, Guo C, Garraway IP & Witte ON Identification of a cell of origin for human prostate cancer. Science 329, 568–571 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutherland KD et al. Cell of origin of small cell lung cancer: inactivation of Trp53 and rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Youssef KK et al. Identification of the cell lineage at the origin of basal cell carcinoma. Nature Cell Biol. 12, 299–305 (2010). [DOI] [PubMed] [Google Scholar]
- 50.Wang GY, Wang J, Mancianti ML & Epstein EH Jr. Basal cell carcinomas arise from hair follicle stem cells in Ptch1+/− mice. Cancer Cell 19, 114–124 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lifsted T et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int. J. Cancer 77, 640–644 (1998). [DOI] [PubMed] [Google Scholar]
- 52.Hunter K, Welch DR & Liu ET Genetic background is an important determinant of metastatic potential. Nature Genet. 34, 23–24(2003). [DOI] [PubMed] [Google Scholar]
- 53.Hunter KW et al. Predisposition to efficient mammary tumor metastatic progression is linked to the breast cancer metastasis suppressor gene Brms1. Cancer Res. 61, 8866–8872 (2001). [PubMed] [Google Scholar]
- 54.Park YG et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nature Genet. 37, 1055–1062 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clark EA, Golub TR, Lander ES & Hynes RO Genomic analysis of metastasis reveals an essential role for RhoC. Nature 406, 532–535 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Yang J et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 (2004). [DOI] [PubMed] [Google Scholar]
- 57.Gumireddy K et al. In vivo selection for metastasis promoting genes in the mouse. Proc. Natl Acad. Sci. USA 104, 6696–6701 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cook LM, Hurst DR & Welch DR Metastasis suppressors and the tumor microenvironment. Semin. Cancer Biol. 21, 113–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith SC & Theodorescu D Learning therapeutic lessons from metastasis suppressor proteins. Nature Rev. Cancer 9, 253–264 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Steeg PS et al. Evidence for a novel gene associated with low tumor metastatic potential. J. Natl Cancer Inst. 80, 200–204 (1988). [DOI] [PubMed] [Google Scholar]
- 61.Hu G et al. MTDH activation by 8q22 genomic gain promotes chemoresistance and metastasis of poor-prognosis breast cancer. Cancer Cell 15, 9–20 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kim M et al. Comparative oncogenomics identifies NEDD9 as a melanoma metastasis gene. Cell 125, 1269–1281 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Ji H et al. LKB1 modulates lung cancer differentiation and metastasis. Nature 448, 807–810 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Carretero J et al. Integrative genomic and proteomic analyses identify targets for Lkb1-deficient metastatic lung tumors. Cancer Cell 17, 547–559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Meyerson M, Gabriel S & Getz G Advances in understanding cancer genomes through second-generation sequencing. Nature Rev. Genet. 11, 685–696 (2010). [DOI] [PubMed] [Google Scholar]
- 66.Alkan C, Coe BP & Eichler EE Genome structural variation discovery and genotyping. Nature Rev. Genet. 12, 363–376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kaminker JS, Zhang Y, Watanabe C & Zhang Z CanPredict: a computational tool for predicting cancer-associated missense mutations. Nucleic Acids Res. 35, W595–W598 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ng PC & Henikoff S Predicting deleterious amino acid substitutions. Genome Res. 11, 863–874 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Adzhubei IA et al. A method and server for predicting damaging missense mutations. Nature Methods 7, 248–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leth-Larsen R et al. Metastasis-related plasma membrane proteins of human breast cancer cells identified by comparative quantitative mass spectrometry. Mol. Cell. Proteomics 8, 1436–1449 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yao H et al. Identification of metastasis associated proteins in human lung squamous carcinoma using two-dimensional difference gel electrophoresis and laser capture microdissection. Lung Cancer 65, 41–48 (2009). [DOI] [PubMed] [Google Scholar]
- 72.Li DJ et al. Identification of 14–3-3 sigma as a lymph node metastasis-related protein in human lung squamous carcinoma. Cancer Lett. 279, 65–73 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Xue H et al. Identification of serum biomarkers for colorectal cancer metastasis using a differential secretome approach. J. Proteome Res. 9, 545–555 (2010). [DOI] [PubMed] [Google Scholar]
- 74.Luque-Garcia JL et al. Differential protein expression on the cell surface of colorectal cancer cells associated to tumor metastasis. Proteomics 10, 940–952 (2010). [DOI] [PubMed] [Google Scholar]
- 75.Sreekumar A et al. Metabolomic profiles delineate potential role for sarcosine in prostate cancer progression. Nature 457, 910–914 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 76.Kulasingam V & Diamandis EP Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nature Clin. Pract. Oncol. 5, 588–599 (2008). [DOI] [PubMed] [Google Scholar]
- 77.Petricoin EF, Zoon KC, Kohn EC, Barrett JC & Liotta LA Clinical proteomics: translating benchside promise into bedside reality. Nature Rev. Drug Discovery 1, 683–695 (2002). [DOI] [PubMed] [Google Scholar]
- 78.Bandyopadhyay S et al. A human MAP kinase interactome. Nature Methods 7, 801–805 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chan CT, Paulmurugan R, Reeves RE, Solow-Cordero D & Gambhir SS Molecular imaging of phosphorylation events for drug development. Mol. Imaging Biol. 11, 144–158 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lamb J et al. The Connectivity Map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 81.Pichiorri F et al. Downregulation of p53-inducible microRNAs 192,194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 18, 367–381 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 82.Bueno MJ et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell 13, 496–506 (2008). [DOI] [PubMed] [Google Scholar]
- 83.Fazi F et al. Epigenetic silencing of the myelopoiesis regulator microRNA-223 by the AML1/ETO oncoprotein. Cancer Cell 12, 457–466 (2007). [DOI] [PubMed] [Google Scholar]
- 84.Varambally S et al. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 322, 1695–1699 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iliopoulos D, Hirsch HA & Struhl K An epigenetic switch involving NF-κB, Lin28, Let-7 MicroRNA, and IL6 links inflammation to cell transformation. Cell 139, 693–706 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ma L, Teruya-Feldstein J & Weinberg RA Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature 449, 682–688 (2007). [DOI] [PubMed] [Google Scholar]
- 87.Tavazoie SF et al. Endogenous human microRNAs that suppress breast cancer metastasis. Nature 451, 147–152 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nicoloso MS, Spizzo R, Shimizu M, Rossi S & Calin GA MicroRNAs-the micro steering wheel of tumour metastases. Nature Rev. Cancer 9, 293–302 (2009). [DOI] [PubMed] [Google Scholar]
- 89.Korpal M et al. Direct targeting of Sec23a by miR-200s influences cancer cell secretome and promotes metastatic colonization. Nature Med. 17, 1101–1108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baek D et al. The impact of microRNAs on protein output. Nature 455, 64–71 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lim LP et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433, 769–773 (2005). [DOI] [PubMed] [Google Scholar]
- 92.Gupta RA et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 464, 1071–1076 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Brower V Epigenetics: unravelling the cancer code. Nature 471, S12–S13 (2011). [DOI] [PubMed] [Google Scholar]
- 94.Esteller M Epigenetics in cancer. N. Engl. J. Med. 358, 1148–1159 (2008). [DOI] [PubMed] [Google Scholar]
- 95.Jones PA & Baylin SB The epigenomics of cancer. Cell 128, 683–692 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Niwa T & Ushijima T Induction of epigenetic alterations by chronic inflammation and its significance on carcinogenesis. Adv. Genet. 71, 41–56 (2010). [DOI] [PubMed] [Google Scholar]
- 97.Ju HX et al. Distinct profiles of epigenetic evolution between colorectal cancers with and without metastasis. Am. J. Pathol. 178, 1835–1846 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raghavan K, Ruskin HJ, Perrin D, Goasmat F & Burns J Computational micromodel for epigenetic mechanisms. PLoS ONE 5, e14031 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Psaila B & Lyden D The metastatic niche: adapting the foreign soil. Nature Rev. Cancer 9, 285–293 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bissell MJ & Radisky D Putting tumours in context. Nature Rev. Cancer 1, 46–54 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bierie B & Moses HL Tumour microenvironment: TGFβ: the molecular Jekyll and Hyde of cancer. Nature Rev. Cancer 6, 506–520 (2006). [DOI] [PubMed] [Google Scholar]
- 102.Wiseman BS & Werb Z Stromal effects on mammary gland development and breast cancer. Science 296, 1046–1049 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Shekhar MP, Pauley R & Heppner G Host microenvironment in breast cancer development: extracellular matrix-stromal cell contribution to neoplastic phenotype of epithelial cells in the breast. Breast Cancer Res. 5, 130–135 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tlsty TD & Hein PW Know thy neighbor: stromal cells can contribute oncogenic signals. Curr. Opin. Genet. Dev. 11, 54–59 (2001). [DOI] [PubMed] [Google Scholar]
- 105.Quante M et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell 19, 257–272 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Tlsty TD & Coussens LM Tumor stroma and regulation of cancer development. Annu. Rev. Pathol. 1, 119–150 (2006). [DOI] [PubMed] [Google Scholar]
- 107.Sessa C, Guibal A, Del Conte G & Ruegg C Biomarkers of angiogenesis for the development of antiangiogenic therapies in oncology: tools or decorations? Nature Clin. Pract. Oncol. 5, 378–391 (2008). [DOI] [PubMed] [Google Scholar]
- 108.Reinacher-Schick A, Pohl M & Schmiegel W Drug insight: antiangiogenic therapies for gastrointestinal cancers-focus on monoclonal antibodies. Nature Clin. Pract. Gastroenterol. Hepatol. 5, 250–267 (2008). [DOI] [PubMed] [Google Scholar]
- 109.Banerjee S, Dowsett M, Ashworth A & Martin LA Mechanisms of disease: angiogenesis and the management of breast cancer. Nature Clin. Pract. Oncol. 4, 536–550 (2007). [DOI] [PubMed] [Google Scholar]
- 110.Miller K et al. Paclitaxel plus bevacizumab versus paclitaxel alone for metastatic breast cancer. N. Engl. J. Med. 357, 2666–2676 (2007). [DOI] [PubMed] [Google Scholar]
- 111.Ebos JM et al. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell 15, 232–239 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Paez-Ribes M et al. Antiangiogenic therapy elicits malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell 15, 220–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Robert NJ et al. RIBBON-1: randomized, double-blind, placebo-controlled, phase III trial of chemotherapy with or without bevacizumab for first-line treatment of human epidermal growth factor receptor 2-negative, locally recurrent or metastatic breast cancer. J. Clin. Oncol. 29, 1252–1260 (2011). [DOI] [PubMed] [Google Scholar]
- 114.Miles DW et al. Phase III study of bevacizumab plus docetaxel compared with placebo plus docetaxel for the first-line treatment of human epidermal growth factor receptor 2-negative metastatic breast cancer. J. Clin. Oncol. 28, 3239–3247 (2010). [DOI] [PubMed] [Google Scholar]
- 115.Miles D et al. Disease course patterns after discontinuation of bevacizumab: pooled analysis of randomized phase III trials. J. Clin. Oncol. 29, 83–88 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Gonzalez-Angulo AM, Hortobagyi GN & Ellis LM Targeted therapies: peaking beneath the surface of recent bevacizumab trials. Nature Rev. Clin. Oncol. 8, 319–320 (2011). [DOI] [PubMed] [Google Scholar]
- 117.Wyckoff J et al. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 64, 7022–7029 (2004). [DOI] [PubMed] [Google Scholar]
- 118.Condeelis J & Pollard JW Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124, 263–266 (2006). [DOI] [PubMed] [Google Scholar]
- 119.Rolny C et al. HRG inhibits tumor growth and metastasis by inducing macrophage polarization and vessel normalization through downregulation of PlGF. Cancer Cell 19, 31–44 (2011). [DOI] [PubMed] [Google Scholar]
- 120.Chen J et al. CCL18 from tumor-associated macrophages promotes breast cancer metastasis via PITPNM3. Cancer Cell 19, 541–555 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Erez N & Coussens LM Leukocytes as paracrine regulators of metastasis and determinants of organ-specific colonization. Int. J. Cancer 128, 2536–2544 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.DeNardo DG et al. CD4+ T cells regulate pulmonary metastasis of mammary carcinomas by enhancing protumor properties of macrophages. Cancer Cell 16, 91–102 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Schmid MC et al. Receptor tyrosine kinases and TLR/IL1Rs unexpectedly activate myeloid cell PI3Kγ, a single convergent point promoting tumor inflammation and progression. Cancer Cell 19, 715–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Calbo J et al. A functional role for tumor cell heterogeneity in a mouse model of small cell lung cancer. Cancer Cell 19, 244–256 (2011). [DOI] [PubMed] [Google Scholar]
- 125.Gay LJ & Felding-Habermann B Contribution of platelets to tumour metastasis. Nature Rev. Cancer 11, 123–134 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Allard WJ et al. Tumor cells circulate in the peripheral blood of all major carcinomas but not in healthy subjects or patients with nonmalignant diseases. Clin. Cancer Res. 10, 6897–6904 (2004). [DOI] [PubMed] [Google Scholar]
- 127.Nagrath S et al. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature 450, 1235–1239 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Slade MJ & Coombes RC The clinical significance of disseminated tumor cells in breast cancer. Nature Clin. Pract. Oncol. 4, 30–41 (2007). [DOI] [PubMed] [Google Scholar]
- 129.Braun S et al. A pooled analysis of bone marrow micrometastasis in breast cancer. N. Engl. J. Med. 353, 793–802 (2005). [DOI] [PubMed] [Google Scholar]
- 130.Singletary SE, Greene FL & Sobin LH Classification of isolated tumor cells: clarification of the 6th edition of the American Joint Committee on Cancer Staging Manual. Cancer 98, 2740–2741 (2003). [DOI] [PubMed] [Google Scholar]
- 131.Harris L et al. American Society of Clinical Oncology 2007 update of recommendations for the use of tumor markers in breast cancer. J. Clin. Oncol. 25, 5287–5312 (2007). [DOI] [PubMed] [Google Scholar]
- 132.Maheswaran S et al. Detection of mutations in EGFR in circulating lung-cancer cells. N. Engl. J. Med. 359, 366–377 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kaplan RN et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the pre-metastatic niche. Nature 438, 820–827 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Comen E, Norton L & Massague J Clinical implications of cancer self-seeding. Nature Rev. Clin. Oncol. 8, 369–377 (2011). [DOI] [PubMed] [Google Scholar]
- 135.Kim MY et al. Tumor self-seeding by circulating cancer cells. Cell 139, 1315–1326 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hiratsuka S, Watanabe A, Aburatani H & Maru Y Tumour-mediated upregulation of chemoattractants and recruitment of myeloid cells predetermines lung metastasis. Nature Cell Biol. 8, 1369–1375 (2006). [DOI] [PubMed] [Google Scholar]
- 137.Hiratsuka S et al. The S100A8-serum amyloid A3-TLR4 paracrine cascade establishes a pre-metastatic phase. Nature Cell Biol. 10, 1349–1355 (2008). [DOI] [PubMed] [Google Scholar]
- 138.Ara T & Declerck YA Interleukin-6 in bone metastasis and cancer progression. Eur. J. Cancer 46, 1223–1231 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Seike T et al. Interaction between lung cancer cells and astrocytes via specific inflammatory cytokines in the microenvironment of brain metastasis. Clin. Exp. Metastasis 28, 13–25 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sethi N, Dai X, Winter CG & Kang Y Tumor-derived JAGGED1 promotes osteolytic bone metastasis of breast cancer by engaging notch signaling in bone cells. Cancer Cell 19, 192–205 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Ara T et al. Interleukin-6 in the bone marrow microenvironment promotes the growth and survival of neuroblastoma cells. Cancer Res. 69, 329–337 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Cabodi S & Taverna D Interfering with inflammation: a new strategy to block breast cancer self-renewal and progression? Breast Cancer Res. 12, 305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sethi N & Kang Y Dysregulation of developmental pathways in bone metastasis. Bone 48, 16–22 (2011). [DOI] [PubMed] [Google Scholar]
- 144.Guise TA Molecular mechanisms of osteolytic bone metastases. Cancer 88, 2892–2898 (2000). [DOI] [PubMed] [Google Scholar]
- 145.Yin JJ et al. TGF-β signaling blockade inhibits PTHrP secretion by breast cancer cells and bone metastases development. J. Clin. Invest. 103, 197–206 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Buijs JT et al. Bone morphogenetic protein 7 in the development and treatment of bone metastases from breast cancer. Cancer Res. 67, 8742–8751 (2007). [DOI] [PubMed] [Google Scholar]
- 147.Buijs JT et al. TGF-β and BMP7 interactions in tumour progression and bone metastasis. Clin. Exp. Metastasis 24, 609–617 (2007). [DOI] [PubMed] [Google Scholar]
- 148.Buijs JT et al. BMP7, a putative regulator of epithelial homeostasis in the human prostate, is a potent inhibitor of prostate cancer bone metastasis in vivo. Am. J. Pathol. 171, 1047–1057 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Dai J et al. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 65, 8274–8285 (2005). [DOI] [PubMed] [Google Scholar]
- 150.Feeley BT et al. Overexpression of noggin inhibits BMP-mediated growth of osteolytic prostate cancer lesions. Bone 38, 154–166 (2006). [DOI] [PubMed] [Google Scholar]
- 151.Katsuno Y et al. Bone morphogenetic protein signaling enhances invasion and bone metastasis of breast cancer cells through Smad pathway. Oncogene 27, 6322–6333 (2008). [DOI] [PubMed] [Google Scholar]
- 152.Bu G et al. Breast cancer-derived Dickkopf1 inhibits osteoblast differentiation and osteoprotegerin expression: implication for breast cancer osteolytic bone metastases. Int. J. Cancer 123, 1034–1042 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Chen G et al. Up-regulation of Wnt-1 and β-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer 101, 1345–1356 (2004). [DOI] [PubMed] [Google Scholar]
- 154.Dai J et al. Prostate cancer induces bone metastasis through Wnt-induced bone morphogenetic protein-dependent and independent mechanisms. Cancer Res. 68, 5785–5794 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Hall CL, Bafico A, Dai J, Aaronson SA & Keller ET Prostate cancer cells promote osteoblastic bone metastases through Wnts. Cancer Res. 65, 7554–7560 (2005). [DOI] [PubMed] [Google Scholar]
- 156.Oshima T et al. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood 106, 3160–3165 (2005). [DOI] [PubMed] [Google Scholar]
- 157.Tian E et al. The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N. Engl. J. Med. 349, 2483–2494 (2003). [DOI] [PubMed] [Google Scholar]
- 158.Pratap J et al. Runx2 transcriptional activation of Indian Hedgehog and a downstream bone metastatic pathway in breast cancer cells. Cancer Res. 68, 7795–7802 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Sterling JA et al. The hedgehog signaling molecule Gli2 induces parathyroid hormone-related peptide expression and osteolysis in metastatic human breast cancer cells. Cancer Res. 66, 7548–7553 (2006). [DOI] [PubMed] [Google Scholar]
- 160.Zunich SM et al. Paracrine sonic hedgehog signalling by prostate cancer cells induces osteoblast differentiation. Mol. Cancer 8, 12 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Nguyen DX et al. WNT/TCF signaling through LEF1 and HOXB9 mediates lung adenocarcinoma metastasis. Cell 138, 51–62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Oskarsson T et al. Breast cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Med. 17, 867–874 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Davies H et al. Mutations of the BRAF gene in human cancer. Nature 417, 949–954 (2002). [DOI] [PubMed] [Google Scholar]
- 164.Huntington JT et al. Overexpression of collagenase 1 (MMP-1) is mediated by the ERK pathway in invasive melanoma cells: role of BRAF mutation and fibroblast growth factor signaling. J. Biol. Chem. 279, 33168–33176 (2004). [DOI] [PubMed] [Google Scholar]
- 165.Klein RM & Aplin AE Rnd3 regulation of the actin cytoskeleton promotes melanoma migration and invasive outgrowth in three dimensions. Cancer Res. 69, 2224–2233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Old WM et al. Functional proteomics identifies targets of phosphorylation by B-Raf signaling in melanoma. Mol. Cell 34, 115–131 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Arozarena I et al. Oncogenic BRAF induces melanoma cell invasion by downregulating the cGMP-specific phosphodiesterase PDE5A. Cancer Cell 19, 45–57 (2011). [DOI] [PubMed] [Google Scholar]
- 168.Flaherty KT et al. Inhibition of mutated, activated BRAF in metastatic melanoma. N. Engl. J. Med. 363, 809–819 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Bollag G et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature 467, 596–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Turke AB et al. Preexistence and clonal selection of MET amplification in EGFR mutant NSCLC. Cancer Cell 17, 77–88 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Guix M et al. Acquired resistance to EGFR tyrosine kinase inhibitors in cancer cells is mediated by loss of IGF-binding proteins. J. Clin. Invest. 118, 2609–2619 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Engelman JA et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 316, 1039–1043 (2007). [DOI] [PubMed] [Google Scholar]
- 173.Poulikakos PI & Rosen N Mutant BRAF melanomas-dependence and resistance. Cancer Cell 19, 11–15 (2011). [DOI] [PubMed] [Google Scholar]
- 174.Poulikakos PI, Zhang C, Bollag G, Shokat KM & Rosen N RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464, 427–430 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Hatzivassiliou G et al. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464, 431–435 (2010). [DOI] [PubMed] [Google Scholar]
- 176.Heidorn SJ et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140, 209–221 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Martin RW, Connell PP & Bishop DK The Yin and Yang of treating BRCA-deficient tumors. Cell 132, 919–920 (2008). [DOI] [PubMed] [Google Scholar]
- 178.Edwards SL et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 451, 1111–1115 (2008). [DOI] [PubMed] [Google Scholar]
- 179.Shah NP et al. Multiple BCR-ABL kinase domain mutations confer polyclonal resistance to the tyrosine kinase inhibitor imatinib (STI571) in chronic phase and blast crisis chronic myeloid leukemia. Cancer Cell 2, 117–125 (2002). [DOI] [PubMed] [Google Scholar]
- 180.Shah NP et al. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science 305, 399–401 (2004). [DOI] [PubMed] [Google Scholar]
- 181.Burgess MR, Skaggs BJ, Shah NP, Lee FY & Sawyers CL Comparative analysis of two clinically active BCR-ABL kinase inhibitors reveals the role of conformation-specific binding in resistance. Proc. Natl Acad. Sci. USA 102, 3395–3400 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Nazarian R et al. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature 468, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Johannessen CM et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968–972 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Karlou M, Tzelepi V & Efstathiou E Therapeutic targeting of the prostate cancer microenvironment. Nature Rev. Urol. 7, 494–509 (2010). [DOI] [PubMed] [Google Scholar]
- 185.Emmenegger U & Kerbel RS Cancer: chemotherapy counteracted. Nature 468, 637–638 (2010). [DOI] [PubMed] [Google Scholar]
- 186.Williams RT, den Besten W & Sherr CJ Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. 21, 2283–2287 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Francia G et al. Comparative impact of trastuzumab and cyclophosphamide on HER-2-positive human breast cancer xenografts. Clin. Cancer Res. 15, 6358–6366 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Man S et al. Antitumor effects in mice of low-dose (metronomic) cyclophosphamide administered continuously through the drinking water. Cancer Res. 62, 2731–2735 (2002). [PubMed] [Google Scholar]
- 189.Fizazi K et al. Denosumab versus zoledronic acid for treatment of bone metastases in men with castration-resistant prostate cancer: a randomised, double-blind study. Lancet 377, 813–822 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Stopeck AT et al. Denosumab compared with zoledronic acid for the treatment of bone metastases in patients with advanced breast cancer: a randomized, double-blind study. J. Clin. Oncol. 28, 5132–5139 (2010). [DOI] [PubMed] [Google Scholar]
- 191.Lymperi S, Ferraro F & Scadden DT The HSC niche concept has turned 31. Has our knowledge matured? Ann. N. Y Acad. Sci. 1192, 12–18 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Premsrirut PK et al. A rapid and scalable system for studying gene function in mice using conditional RNA interference. Cell 145, 145–158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ellwood-Yen K et al. Myc-driven murine prostate cancer shares molecular features with human prostate tumors. Cancer Cell 4, 223–238 (2003). [DOI] [PubMed] [Google Scholar]
- 194.Sweet-Cordero A et al. An oncogenic KRAS2 expression signature identified by cross-species gene-expression analysis. Nature Genet. 37, 48–55 (2005). [DOI] [PubMed] [Google Scholar]
- 195.Graeber TG & Sawyers CL Cross-species comparisons of cancer signaling. Nature Genet. 37, 7–8 (2005). [DOI] [PubMed] [Google Scholar]
- 196.Francia G, Cruz-Munoz W, Man S, Xu P & Kerbel RS Mouse models of advanced spontaneous metastasis for experimental therapeutics. Nature Rev. Cancer 11, 135–141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Vogelstein B et al. Genetic alterations during colorectal-tumor development. N. Engl. J. Med. 319, 525–532 (1988). [DOI] [PubMed] [Google Scholar]
- 198.Foulds L The experimental study of tumor progression: a review. Cancer Res. 14, 327–339 (1954). [PubMed] [Google Scholar]
- 199.Fidler IJ & Lieber S Quantitative analysis of the mechanism of glucocorticoid enhancement of experimental metastasis. Res. Commun. Chem. Pathol. Pharmacol. 4, 607–613 (1972). [PubMed] [Google Scholar]
- 200.Koscielny S et al. Breast cancer: relationship between the size of the primary tumour and the probability of metastatic dissemination. Br. J. Cancer 49, 709–715 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Kinouchi T et al. Impact of tumor size on the clinical outcomes of patients with Robson State I renal cell carcinoma. Cancer 85, 689–695 (1999). [DOI] [PubMed] [Google Scholar]
- 202.Collins VP, Loeffler RK & Tivey H Observations on growth rates of human tumors. Am. J. Roentgenol. Radium Ther. Nucl. Med. 76, 988–1000 (1956). [PubMed] [Google Scholar]
- 203.Friberg S & Mattson S On the growth rates of human malignant tumors: implications for medical decision making. J. Surg. Oncol. 65, 284–297 (1997). [DOI] [PubMed] [Google Scholar]
- 204.Husemann Y et al. Systemic spread is an early step in breast cancer. Cancer Cell 13, 58–68 (2008). [DOI] [PubMed] [Google Scholar]
- 205.Ellis MJ et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA 302, 774–780 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ben-Haim S & Ell P 18F-FDG PET and PET/CT in the evaluation of cancer treatment response. J. Nucl. Med. 50, 88–99 (2009). [DOI] [PubMed] [Google Scholar]
- 207.Iagaru A et al. Novel strategy for a cocktail 18F-fluoride and 18F-FDG PET/CT scan for evaluation of malignancy: results of the pilot-phase study. J. Nucl. Med. 50, 501–505 (2009). [DOI] [PubMed] [Google Scholar]
- 208.McCann TE et al. Molecular imaging of tumor invasion and metastases: the role of MRI. NMR Biomed. 12 Dec 2010. (doi: 10.1002/nbm.1590). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Ren G et al. Melanin-targeted preclinical PET imaging of melanoma metastasis. J. Nucl. Med. 50, 1692–1699 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Chishima T et al. Cancer invasion and micrometastasis visualized in live tissue by green fluorescent protein expression. Cancer Res. 57, 2042–2047 (1997). [PubMed] [Google Scholar]
- 211.Liu H et al. Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl Acad. Sci. USA 107, 18115–18120 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hatta K, Tsujii H & Omura T Cell tracking using a photoconvertible fluorescent protein. Nature Protoc. 1, 960–967 (2006). [DOI] [PubMed] [Google Scholar]
- 213.Gligorijevic B, Kedrin D, Segall JE, Condeelis J & van Rheenen J Dendra2 photoswitching through the Mammary Imaging Window. J. Vis. Exp. 5 Jun 2009. (doi: 10.3791/1278). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Ewald AJ, Werb Z & Egeblad M Dynamic, long-term in vivo imaging of tumor-stroma interactions in mouse models of breast cancer using spinning-disk confocal microscopy. Cold Spring Harb. Protoc. 2011, pdb.top97 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Massoud TF, Paulmurugan R & Gambhir SS A molecularly engineered split reporter for imaging protein-protein interactions with positron emission tomography. Nature Med. 16, 921–926 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Korpal M et al. Imaging transforming growth factor-β signaling dynamics and therapeutic response in breast cancer bone metastasis. Nature Med. 15, 960–966 (2009). [DOI] [PubMed] [Google Scholar]
- 217.Wistuba II, Gelovani JG, Jacoby JJ, Davis SE & Herbst RS Methodological and practical challenges for personalized cancer therapies. Nature Rev. Clin. Oncol. 8, 135–141 (2011). [DOI] [PubMed] [Google Scholar]
- 218.Lu X et al. ADAMTS1 and MMP1 proteolytically engage EGF-like ligands in an osteolytic signaling cascade for bone metastasis. Genes Dev. 23, 1882–1894 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Zhang XH et al. Latent bone metastasis in breast cancer tied to Src-dependent survival signals. Cancer Cell 16, 67–78 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Park BK et al. NF-κB in breast cancer cells promotes osteolytic bone metastasis by inducing osteoclastogenesis via GM-CSF. Nature Med. 13, 62–69 (2007). [DOI] [PubMed] [Google Scholar]
- 221.Gupta GP et al. Mediators of vascular remodelling co-opted for sequential steps in lung metastasis. Nature 446, 765–770 (2007). [DOI] [PubMed] [Google Scholar]
- 222.Padua D et al. TGFβ primes breast tumors for lung metastasis seeding through angiopoietin-like 4. Cell 133, 66–77 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Brown DM & Ruoslahti E Metadherin, a cell surface protein in breast tumors that mediates lung metastasis. Cancer Cell 5, 365–374 (2004). [DOI] [PubMed] [Google Scholar]
- 224.Gupta GP et al. ID genes mediate tumor reinitiation during breast cancer lung metastasis. Proc. Natl Acad. Sci. USA 104, 19506–19511 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Gumireddy K et al. KLF17 is a negative regulator of epithelial-mesenchymal transition and metastasis in breast cancer. Nature Cell Biol. 11, 1297–1304 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Hiratsuka S et al. MMP9 induction by vascular endothelial growth factor receptor-1 is involved in lung-specific metastasis. Cancer Cell 2, 289–300 (2002). [DOI] [PubMed] [Google Scholar]
- 227.Muller A et al. Involvement of chemokine receptors in breast cancer metastasis. Nature 410, 50–56 (2001). [DOI] [PubMed] [Google Scholar]
- 228.Xie TX et al. Activation of stat3 in human melanoma promotes brain metastasis. Cancer Res. 66, 3188–3196 (2006). [DOI] [PubMed] [Google Scholar]
- 229.Stein U et al. MACC1, a newly identified key regulator of HGF-MET signaling, predicts colon cancer metastasis. Nature Med. 15, 59–67 (2009). [DOI] [PubMed] [Google Scholar]
- 230.Erler JT et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Erler JT et al. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440, 1222–1226 (2006). [DOI] [PubMed] [Google Scholar]
- 232.Kuperwasser C et al. A mouse model of human breast cancer metastasis to human bone. Cancer Res. 65, 6130–6138 (2005). [DOI] [PubMed] [Google Scholar]
- 233.Yonou H et al. Establishment of a novel species- and tissue-specific metastasis model of human prostate cancer in humanized non-obese diabetic/severe combined immunodeficient mice engrafted with human adult lung and bone. Cancer Res. 61, 2177–2182 (2001). [PubMed] [Google Scholar]
- 234.Nemeth JA et al. Severe combined immunodeficient-hu model of human prostate cancer metastasis to human bone. Cancer Res. 59, 1987–1993 (1999). [PubMed] [Google Scholar]
- 235.Shtivelman E & Namikawa R Species-specific metastasis of human tumor cells in the severe combined immunodeficiency mouse engrafted with human tissue. Proc. Natl Acad. Sci. USA 92, 4661–4665 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Rasmussen HH, Mortz E, Mann M, Roepstorff P & Celis JE Identification of transformation sensitive proteins recorded in human two-dimensional gel protein databases by mass spectrometric peptide mapping alone and in combination with microsequencing. Electrophoresis 15, 406–416 (1994). [DOI] [PubMed] [Google Scholar]
- 237.Centonze VE & White JG Multiphoton excitation provides optical sections from deeper within scattering specimens than confocal imaging. Biophys. J. 75, 2015–2024 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Xu C, Zipfel W, Shear JB, Williams RM & Webb WW Multiphoton fluorescence excitation: new spectral windows for biological nonlinear microscopy. Proc. Natl Acad. Sci. USA 93, 10763–10768 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kan Z & Liu TJ Video microscopy of tumor metastasis: using the green fluorescent protein (GFP) gene as a cancer-cell-labeling system. Clin. Exp. Metastasis 17, 49–55 (1999). [DOI] [PubMed] [Google Scholar]
- 240.Scherbarth S & Orr FW Intravital videomicroscopic evidence for regulation of metastasis by the hepatic microvasculature: effects of interleukin-1α on metastasis and the location of B16F1 melanoma cell arrest. Cancer Res. 57, 4105–4110 (1997). [PubMed] [Google Scholar]
- 241.Chambers AF et al. Steps in tumor metastasis: new concepts from intravital videomicroscopy. Cancer Metastasis Rev. 14, 279–301 (1995). [DOI] [PubMed] [Google Scholar]
- 242.MacDonald TJ, Tabrizi P, Shimada H, Zlokovic BV & Laug WE Detection of brain tumor invasion and micrometastasis in vivo by expression of enhanced green fluorescent protein. Neurosurgery 43, 1437–1443 (1998). [DOI] [PubMed] [Google Scholar]
- 243.Farina KL et al. Cell motility of tumor cells visualized in living intact primary tumors using green fluorescent protein. Cancer Res. 58, 2528–2532 (1998). [PubMed] [Google Scholar]
- 244.Kononen J et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nature Med. 4, 844–847 (1998). [DOI] [PubMed] [Google Scholar]
- 245.Monoclonal antibody approved for metastatic breast cancer. Oncology 12, 1727 (1998). [PubMed] [Google Scholar]
- 246.Ren B et al. Genome-wide location and function of DNA binding proteins. Science 290, 2306–2309 (2000). [DOI] [PubMed] [Google Scholar]
- 247.Blat Y & Kleckner N Cohesins bind to preferential sites along yeast chromosome III, with differential regulation along arms versus the centric region. Cell 98, 249–259 (1999). [DOI] [PubMed] [Google Scholar]
- 248.Reya T, Morrison SJ, Clarke MF & Weissman IL Stem cells, cancer, and cancer stem cells. Nature 414, 105–111 (2001). [DOI] [PubMed] [Google Scholar]
- 249.Alizadeh AA et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511 (2000). [DOI] [PubMed] [Google Scholar]
- 250.Virtaneva K et al. Expression profiling reveals fundamental biological differences in acute myeloid leukemia with isolated trisomy 8 and normal cytogenetics. Proc. Natl Acad. Sci. USA 98, 1124–1129 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Paweletz CP et al. Reverse phase protein microarrays which capture disease progression show activation of pro-survival pathways at the cancer invasion front. Oncogene 20, 1981–1989 (2001). [DOI] [PubMed] [Google Scholar]
- 252.Ramaswamy S, Ross KN, Lander ES & Golub TR A molecular signature of metastasis in primary solid tumors. Nature Genet. 33, 49–54 (2003). [DOI] [PubMed] [Google Scholar]
- 253.Wang W et al. Single cell behavior in metastatic primary mammary tumors correlated with gene expression patterns revealed by molecular profiling. Cancer Res. 62, 6278–6288 (2002). [PubMed] [Google Scholar]
- 254.Margulies M et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Shendure J et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005). [DOI] [PubMed] [Google Scholar]
- 256.Domon B & Aebersold R Mass spectrometry and protein analysis. Science 312, 212–217 (2006). [DOI] [PubMed] [Google Scholar]
- 257.Barski A et al. High-resolution profiling of histone methylations in the human genome. Cell 129, 823–837 (2007). [DOI] [PubMed] [Google Scholar]
- 258.Nagalakshmi U et al. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Wilhelm BT et al. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature 453, 1239–1243 (2008). [DOI] [PubMed] [Google Scholar]
- 260.Mortazavi A, Williams BA, McCue K, Schaeffer L & Wold B Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nature Methods 5, 621–628 (2008). [DOI] [PubMed] [Google Scholar]
- 261.Lister R et al. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cloonan N et al. Stem cell transcriptome profiling via massive-scale mRNA sequencing. Nature Methods 5, 613–619 (2008). [DOI] [PubMed] [Google Scholar]
- 263.Marioni JC, Mason CE, Mane SM, Stephens M & Gilad Y RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 18, 1509–1517 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lee W et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature 465, 473–477 (2010). [DOI] [PubMed] [Google Scholar]
- 265.Ley TJ et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 456, 66–72 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Mardis ER et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Shah SP et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 461, 809–813 (2009). [DOI] [PubMed] [Google Scholar]
- 268.Pleasance ED et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463, 191–196 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Barton MK Denosumab an option for patients with bone metastasis from breast cancer. CA Cancer J. Clin. 61, 135–136 (2011). [DOI] [PubMed] [Google Scholar]
- 270.Lipton A & Goessl C Clinical development of antiRANKL therapies for treatment and prevention of bone metastasis. Bone 48, 96–99 (2011). [DOI] [PubMed] [Google Scholar]
- 271.Vultur A, Villanueva J & Herlyn M BRAF inhibitor unveils its potential against advanced melanoma. Cancer Cell 18, 301–302 (2010). [DOI] [PubMed] [Google Scholar]
- 272.Kim T, Kim J & Lee MG Inhibition of mutated BRAF in melanoma. N. Engl. J. Med. 363, 2261 (2010). [DOI] [PubMed] [Google Scholar]