

Abstract
Immune cell signaling is largely regulated by protein phosphorylation. Stimulation of toll-like receptors (TLRs) by pathogen-associated ligands drives the cascade of immune response, which can be influenced by differences in phosphoprotein abundance. Therefore, the analysis of phosphorylation signatures at a global level is central to understanding the complex and integrated signaling in macrophages upon pathogen attack. Here, we describe a mass spectrometry-based approach to identify and quantify phosphoproteome changes in response to the stimulation of TLR2, TLR4, and TLR7 with immune-response inducing ligands in cultured immune cells. This review will focus on the TLR stimulation of mouse macrophages as an example; however, the technique is applicable to any immortalized immune cell and any soluble stimuli. The methodology includes protocols for metabolic labeling of immune cells (stable isotope labeling of amino acids in cell culture, i.e., SILAC); ligand-initiated stimulation of immune receptors followed by cell lysis; in-solution trypsin digestion of proteins and enrichment of the resulting peptide mix for collecting phosphopeptides, which are then analyzed by high-resolution LC-MS/MS (liquid-chromatography tandem mass spectrometry).
Basic Protocol 1: SILAC labeling of mouse macrophages
Basic Protocol 2: Stimulation, cell lysis and Western Blotting
Basic Protocol 3: Trypsin digestion, fractionation and phosphopeptide enrichment
Basic Protocol 4: Quantitative mass spectrometry
Alternate Protocol: Culturing SILAC-labeled cells from frozen mouse macrophages cells
Keywords: cell signaling, innate immunity, mass spectrometry, phosphoproteomics
INTRODUCTION
Innate immune system comprises of Toll-like receptors (TLRs), which are pattern recognition receptors and act as the first line of defense from invading pathogens such as bacteria, fungus, and virus. About a dozen different types of TLRs are found in mammals and each recognizes a distinct set of molecular patterns (PAMPs) (Kang & Lee, 2011). Molecularly speaking, TLRs are transmembrane proteins found on the cell surface and have an extracellular region (Leucine-rich repeat) that detects pathogen molecules. Intracellular TLRs are found in the membrane of endosomes and recognize microbial components, which have been broken down by phagocytosis (Kagan, 2012). Upon binding to pathogen-derived ligands (e.g., lipopolysaccharide of Gram-negative bacteria and viral nucleic acids), TLRs get activated and rapidly trigger a cascade of intracellular signals generating inflammatory mediators that cause an immune response (O’Neill, Golenbock, & Bowie, 2013; Takeda & Akira, 2004). The signaling pathway is tightly controlled through stepwise recruitment of protein complexes and changes in their post-translational modifications (PTMs). Phosphorylation is one such regulatory PTM involved in signal transduction mechanism. This highly dynamic modification occurs on serine, threonine and tyrosine amino acids and enables cells to regulate many physiological processes including innate and adaptive immune system activation. Phosphorylation events are not restricted to only canonical TLR pathways but are involved in transcriptional regulation during early host response indicating multiple pathway crosstalk (Du et al., 2010). Thus, it is important to study phosphoprotein signatures in immune signaling at a global level as a step towards characterizing macrophage response to complex stimuli from pathogens.
This article discusses in detail the protocols for mass spectrometry-based phosphoproteome analysis in cultured mouse macrophages that are stimulated with TLR ligands. These protocols may be adapted for other immortalized immune cells and stimuli. The advancement in mass spectrometry instrumentation and data processing software has enabled accurate measurement of proteins and their PTMs, both qualitatively and quantitatively. Stable isotope labeling with amino acids in cell culture (SILAC) allows samples to be labeled and mixed prior to any processing for unbiased quantitative comparison of phosphopeptide abundance across a range of experimental treatments (Weintz et al., 2010). The first two protocols (Basic Protocols 1 and 2) describe SILAC of immortalized mouse macrophages and toll-like receptor stimulation using three different TLR ligands, i.e., lipopolysaccharide (LPS), for TLR4; Pam3CSK4, an analog of the immunologically active N-terminal portion of bacterial lipoprotein, for TLR2/TLR1; and an imidazoquinolinamine drug resiquimod, or R848, for TLR7). The cells are then lysed to examine the TLR activation using SDS-PAGE and Western Blotting. Basic Protocol 3 describes in-solution trypsin digestion of the cell lysates and followed by strong cation exchange (SCX) chromatography. The SCX fractions are subjected to phosphopeptide enrichment based on SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) technique prior to the identification of phosphopeptides using liquid chromatography coupled to mass spectrometry (LC-MS/MS), as described in Basic Protocol 4. Overall, this approach enables global analysis of the phosphoproteome changes during innate immunity activation by TLR signaling.
An overview of the pipeline described herein is presented in Figure 1.
Figure 1.
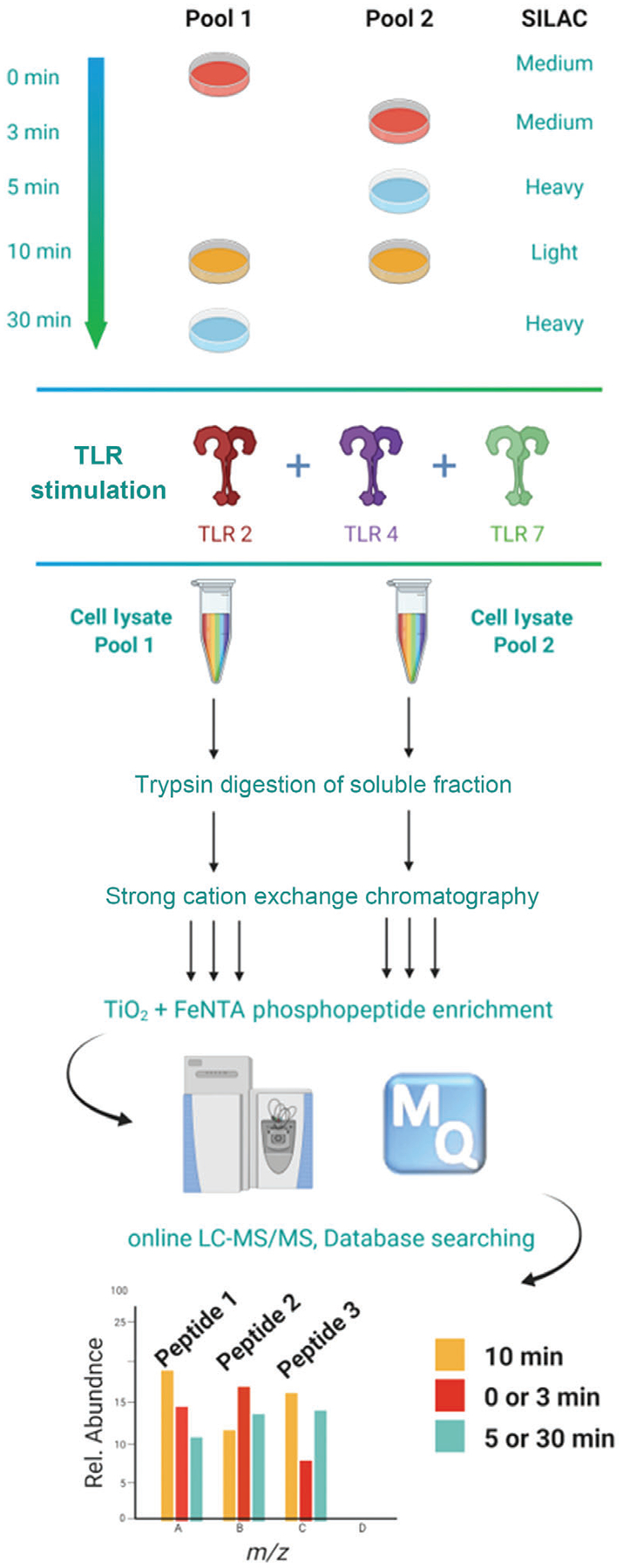
Experimental design for global phosphoproteome analysis of TLR-activated macrophages. Mouse macrophages are SILAC labeled with normal or heavy/medium arginine and lysine amino acids resulting in three states distinguishable by their mass. Each cell population is either left untreated or stimulated for 3, 5, 10, or 30 min. The 10 min stimulation time point is included in both pools to serve as a common reference point. Cell lysates are pooled (each containing all three isotopes at different time points), enzymatically digested, and fractionated by SCX. The phosphopeptides are then enriched by SMOAC methodology followed by LC−MS/MS analysis. The mass shift introduced by the SILAC amino acids results in triplet peaks (i.e., the same peptide from three different time points) with the relative intensities equal to the relative abundance of the peptide.
BASIC PROTOCOL 1
SILAC LABELING OF MOUSE MACROPHAGES
This protocol involves metabolic labeling of cells by culturing them in three different SILAC-labeled growth media (heavy, medium, light). The triple-labeling allows for the direct comparison of three different experimental conditions in one MS run. In the time-course study described here (see Fig. 1), the results from two triple-labeled sample pools with a common reference condition are combined to obtain a five timepoint result. A procedure to grow immortalized mouse macrophages from frozen stock is also discussed in the Alternate Protocol. The number of cells to start with depends on the availability, this protocol is optimized for cell numbers ranging from 10 to 30 million.
Materials
Cell culture
Immortalized macrophages [immortalized in the lab from the primary macrophages according to the protocol from Eicke Latz lab (De Nardo, Kalvakolanu, & Latz, 2018)] or other immune cells (e.g., RAW 264.7 monocyte/macrophage from BALB/c strain, cat. no. ATCC TIB-71)
500 ml Dulbecco’s modified Eagle’s medium with stable glutamine deficient in l-arginine and l-lysine (DMEM)
50 ml fetal bovine serum per SILAC group, i.e., for each of the light, medium and heavy media solutions
5 ml of glutamate (from 200 mM stock solution) (per SILAC group) 10 ml HEPES buffer (from 1 M stock solution)
[4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid] (per SILAC group)
1 ml 500 × l-Proline (per SILAC group)
l-arginine.HCl labeled with 13C6 (Arg6) and 13C6 15N4 (Arg10) (Cambridge isotope laboratories), 1000 × stock solution (see recipe)
l-lysine.2HCl labeled with 2D4 (Lys4) and 13C6 15N2 (Lys8) (Cambridge isotope laboratories), 1000 × stock solution (see recipe)
Arg0 and Lys0 (Sigma Aldrich), 1000 × stock solution (see recipe)
Phosphate-buffered saline (PBS) buffer (1 ×) (MilliporeSigma, cat. no. P5493)
2 mM EDTA (MilliporeSigma, cat. no. E6758)
Trypan blue dye solution (MilliporeSigma, cat. no. 93595)
Equipment
10-cm tissue culture grade plate (FischerScientific, cat. no. 08-772E)
CO2 incubator (Thermo Scientific)
Bottle-top vacuum filter system (pore size 0.22 μm, Corning)
15-ml polypropylene tubes
Pipettes (1 ml, 2 ml, 20 μl)
Centrifuge (Eppendorf)
Cell slide
96-well TC-treated plate (Corning, cat. no. CLS3595)
Automatic cell counting instrument
NOTE: All solutions and equipment coming into contact with cells must be sterile, and proper sterile technique should be used accordingly.
Preparation of the SILAC media
-
1
Thaw glutamate, L-proline, and amino acid aliquots at room temperature.
-
2
To the 500 ml DMEM bottle, add prescribed amounts of glutamate, FBS, HEPES, l-proline, 0.5 ml of 1000× Arg0 stock, and 0.5 ml of 1000× Lys0 stock
The DMEM medium now contains 10% FBS, 20 mM HEPES, 2 mM glutamate, 0.398 mM l-Arg.HCl, and 0.798 mM l-Lys.2HCL. The molarities of labeled and unlabeled amino acids stay same for all SILAC versions (heavy, medium, light), i.e., 0.398 mM for l-Arg and 0.798 for l-Lys.
-
3
Repeat the same for preparing medium medium (glutamate + FBS + HEPES + l-proline + 0.5 ml each of Arg4 and Lys6) and heavy medium (glutamate + HEPES + FBS + l-proline 0.5 ml each of Arg10 and Lys8).
-
4
Filter the solution into a 500-ml medium bottle through 0.22-μm filter with the help of vacuum. Swirl briefly to mix.
-
5
Label the three media containers as light, medium, and heavy and store for up to a month at 4°C.
Incorporation of labeled amino acids in macrophages
-
6
Grow immortalized macrophages in DMEM on sterile tissue-culture plates in a humidified CO2 incubator at 37°C and passage until cells reach 80%−90% confluency. Steps on how to start a culture from frozen aliquots of immortalized macrophages are summarized in the Alternate Protocol.
The cell density should be approximately 10 × 106 viable cells/ml. Cell viability should be >95%.
Expand the cells in heavy, medium, and light SILAC media for at least five passages as follows
-
7
Aspirate the old medium above the cell layer using vacuum and release cellular matrix from the plastic with 2 ml of PBS buffer (1×) mixed with 2 mM EDTA.
-
8
Spread the buffer in the plate and let it sit for a few minutes. Meanwhile prepare a new tissue culture (TC) grade plate by adding 10 ml fresh media (light, medium or heavy) to it and put it aside until needed later.
-
9
Pipette the PBS buffer up-down in the plate using a 1-ml pipette to detach the cells. Turn the plate 90° and repeat the process.
The goal is to remove the hazy layer from the plastic surface and get it into the solution while being gentle to the cells. The macrophages might not survive if scraped instead of pipetted.
-
10
Transfer the cloudy solution into a 15-ml tube and centrifuge for 5 min at 400 × g, 20°C.
-
11
Remove the tube from the centrifuge and aspirate the PBS buffer. Resuspend the cell pellet in 1 ml of fresh DMEM medium.
2 ml medium should be used for a 15-cm plate.
-
12
Pipette up-down to break the pellet and homogenize the solution.
-
13
Prepare the cell slide by taking the thin plastic cover off and exposing the sample hole.
-
14
Transfer 20 μl of the Trypan blue dye into one of the wells in a 96-well plate. Add 20 μl of the homogenized cell matrix to the 20 μl dye in the 96-well plate. Pipette up-down to mix.
-
15
Transfer 20 μl of the above mixture on the cell slide and count the live cells using the automatic cell counter.
-
16
Calculate the volume required for 3 × 106 cells for the next passage. Transfer that volume into the plate prepared in step 8 for each of the light, medium, and heavy media. After 48 hr, check the plate for the condition of cells.
-
17
Passage at least five times for full incorporation of the isotopes into the cells.
The doubling time of cells in SILAC media is longer than in rich medium; thus, passage every 3–4 days.
-
18
Confirm label incorporation by analyzing an aliquot of each condition using mass spectrometry. Greater than 95% labeling is acceptable for further experimentation. See Figure 2.
-
19
One day before TLR activation, discard the nonadherent cells and plate 25 million cells on a 10-cm tissue culture grade plate for each treatment and time point.
-
20
Ninety minutes prior to TLR activation, remove the nonadherent cells and add freshly equilibrated SILAC media to the cells.
Figure 2.
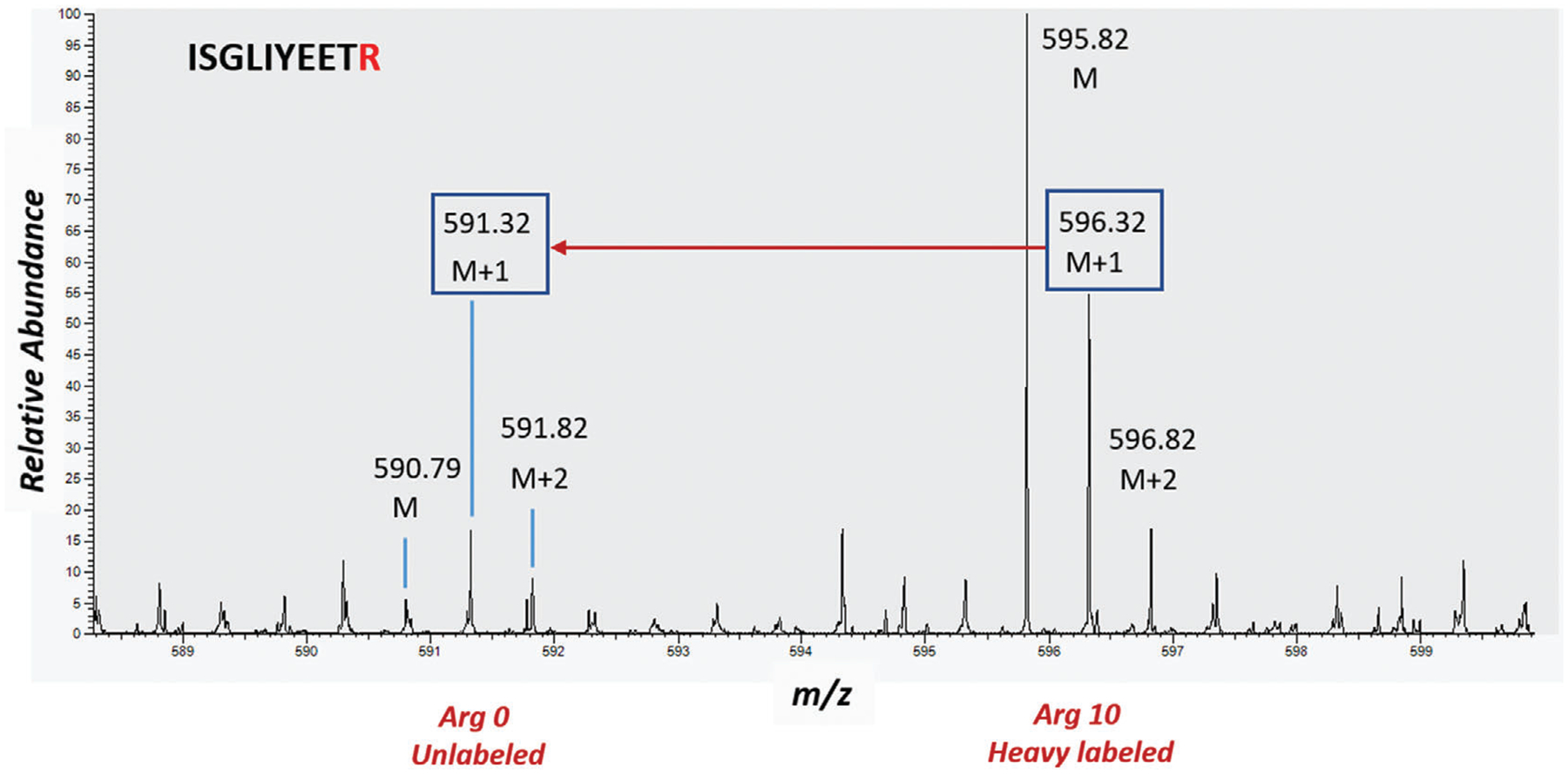
The spectrum shows SILAC labeling efficiency for an arginine containing peptide ISGLIYEETR (theoretical mass = 1190.62910 Da). The isotope cluster for this doubly charged peptide is annotated as M, M+1, M+2. The difference between the labeled (Arg 10) and unlabeled (Arg 0), i.e., 5 m/z units is shown by the red arrow. A comparison of relative abundances of the two clusters confirms the labeling efficiency of 90%−95%.
ALTERNATE PROTOCOL
CULTURING SILAC LABELED CELLS FROM FROZEN MOUSE MAROPHAGES CELLS
In case of starting a culture from frozen aliquots of immortalized mouse macrophages, this protocol can be used.
Materials
Cell culture
Frozen immortalized macrophages or other immune cells (e.g., RAW 264.7 monocyte/macrophage from BALB/c strain, cat. no. ATCC TIB-71)
500 ml Dulbecco’s modified Eagle’s medium with stable glutamine deficient in l-arginine and l-lysine (DMEM)
50 ml fetal bovine serum (per SILAC group)
5 ml 100 × glutamate (per SILAC group)
10 ml HEPES (20 mM) (per SILAC group)
1 ml 500 × l-Proline (per SILAC group)
l-arginine HCl labeled with 13C6 (Arg6) and 13C6 15N4 (Arg10) (Cambridge isotope laboratories), 1000 × solution (see recipe)
l-lysine 2HCl labeled with 2D4(Lys4) and 13C6 15N2 (Lys8) (Cambridge isotope laboratories), 1000 × solution (see recipe)
Arg0 and Lys0 (Sigma Aldrich), 1000 × solution (see recipe)
PBS buffer (1 ×) (MilliporeSigma, cat. no. P5493)
2 mM EDTA (MilliporeSigma, cat. no. E6758)
Trypan blue dye solution (MilliporeSigma, cat. no. 93595)
Equipment
10-cm tissue culture grade plate (FischerScientific, cat. no. 08-772E)
CO2 incubator (Thermo Scientific)
Bottle-top vacuum filter system (pore size 0.22 μm, Corning)
15-ml conical polypropylene tubes
Centrifuge (Eppendorf)
Incorporation of labeled amino acids in macrophages
Prepare the SILAC media as described in Basic Protocol 1 (steps 1–5).
Thaw an aliquot of frozen Immortalized macrophages in a water bath at 37°C.
Warm an appropriate volume of the media (DMEM) (9 ml per frozen aliquot) as well in a water bath at 37°C for half an hour.
Pipette 9 ml of the appropriate (heavy, medium, or light SILAC media) into a 15-ml conical tube and add the thawed cell solution to it.
Mix the contents of the 15-ml tube and centrifuge for 5 min at 400 × g, 20°C.
Meanwhile pipette 10 ml fresh medium into a new 10-cm cell-culture plate (TC grade) and let it sit until needed later. Label the plate lid with date and SILAC label type.
Remove the tube from the centrifuge and aspirate the medium above the cell pellet. Be careful not to disturb the pellet.
Transfer 1 ml of the fresh medium from the cell-culture plate into the 15-ml tube, pipette up and down to break the pellet, and transfer everything back to the plate.
Put the plate in CO2 incubator for 24 hr. It may take 2–3 days for the cells to attach and the plate to be fully confluent.
Once most of the cells are attached, aspirate off the dead floating cells and proceed starting from step 2 in Basic Protocol 1.
BASIC PROTOCOL 2
STIMULATION, CELL LYSIS, AND WESTERN BLOTTING
Three different TLR ligands, namely, LPS, for TLR4; Pam3CSK4, an analog of the immunologically active N-terminal portion of bacterial lipoprotein, for TLR2/TLR1; and an imidazoquinolinamine drug resiquimod, or R848, for TLR7 can be used for macrophage stimulation to study phosphoprotein signatures in different TLRs. A time course-based study allows identification of the mechanisms of TLR action as per their exposure duration. The completion of stimulation can be confirmed by the presence of certain phosphorylated proteins (p-P105, p-P65, and p-JNK) in the Western Blot.
Materials
Stimulation ligands
100 ng/ml LPS solution in DMEM media (lipopolysaccharide from Salmonella Minnesota/R595, Enzo Life Sciences, cat. no. ALX-581-008-L001)
1 μM solution of Resiquimod /R848 (Enzo Life Sciences, cat. no. ALX-420-038-M005)
1 μM solution of Pam3CSK4 (InvivoGen, cat. code tlrl-pms)
Lysis buffer
PBS (1×) ice cold
80 μl Protease inhibitor cocktail (Roche) (see recipe)
80 μl Phosphatase inhibitor cocktail (MilliporeSigma) (see recipe)
16 μl 1 M dithiothreitol (DTT) in water (see recipe)
8 ml Pierce™ IP lysis buffer (Thermo Scientific, cat. no. 87787)
Pierce™ BCA Protein Assay Kit (Thermo Scientific, cat. no. 23225)
Polyacrylamide gel electrophoresis (PAGE)
10% Mini-PROTEAN® TGX™ Precast Protein Gels (BioRad, cat. no. 4561035)
Loading buffer containing NuPAGE™ LDS sample buffer (2×) and β-mercaptoethanol (see recipe)
MOPS buffer 1× (see recipe)
Protein ladder (BioRad) (see recipe)
Blank solution (1× loading buffer)
Western Blot
iBlot 2 Transfer Stacks, nitrocellulose, mini (Thermo Scientific, cat. no. IB23002) TBS/T, i.e., TBS with 0.1% TWEEN 20 (see recipe)
Blocking solution made from Omni milk powder in TBS/T (see recipe) Ponceau S staining solution (Cell Signaling Technology, cat. no. 59803) Primary antibody cocktail (αP-P105, αP-JNK, αP-P65, αP-P38) (see recipe) Secondary antibody (anti-rabbit horseradish peroxidase) (see recipe) Substrate solution (peroxide with luminol) (see recipe)
Equipment
Sample incubator at 4°C
CO2 incubator (Thermo Scientific)
Ice
Pipettes
Nanodrop instrument e.g., NanoDrop™ Lite Spectrophotometer (Thermo Scientific, cat. no. ND-LITE)
iBlot (membrane electro-transfer by Invitrogen)
Plate rocker
Scissors
Plastic tray
ChemiDoc MP imaging system (BioRad)
15-ml polypropylene tubes
Microcentrifuge tubes, 1.5 ml
95°C sample incubator
Centrifuge (Eppendorf)
LPS stimulation and lysis
-
1
Prior to stimulation, prepare the three TLR ligand solutions in SILAC media (separately for each SILAC condition) as per the following concentrations: 100 ng/ml LPS in, 1 μM Resiquimod or 1 μM Pam3Cys, each in heavy, medium, and light media (nine solutions in total).
-
2
Remove the media from the SILAC-labeled cells. Add 10 ml of the ligand solution per 10-cm dish of cells at different time points–0, 3, 5, 10, and 30 min as per the scheme shown in Figure 1, each time keeping the plate in incubator (37°C) after stimulation.
The suggested time point range is only an example for this kind of study. Depending on the purpose of the study in question, appropriate time points should be chosen.
-
3
For each ligand, stimulate cells in two 10-cm plates for 10 min in light medium (unlabeled) to serve as a reference point. Add the ligand to two of the medium-labeled plates at 0 and 3 min and two of the heavy-labeled plates at 5 and 30 min.
-
4
After time point zero, put the plate on ice and aspirate the medium.
-
5
Add ice-cold 1× PBS (2 ml per plate), swirl and aspirate the PBS.
-
6
Add 1 ml lysis buffer containing protease and phosphatase inhibitor cocktails to the plate and incubate for 15 min at 4°C while rocking the plate.
-
7
Put the plate back on ice and lift the cells off the plate by pouring the lysis buffer repeatedly. Minimize the froth by pouring slowly. Transfer the cell matrix into a 15-ml tube.
-
8
Centrifuge for 10 min at 1000 × g, 4°C. Transfer the supernatant into fresh 15-ml tubes leaving the debris behind. Centrifuge again at max speed for 10 min, 4°C, to fully precipitate the insoluble proteins.
-
9
Transfer the clear supernatants into fresh 15-ml tubes. The lysate can be stored at −20°C until further use.
The frozen lysate can be used for phosphoproteome analysis as an alternative to starting from the cell culture.
-
10
From the supernatant, transfer 50 μl into a 1.5-ml microcentrifuge tube and add 50 μl of 2× NuPAGE loading buffer. Repeat for rest of the samples. Heat the samples at 95°C for 10 min and spin quickly to bring the condensed liquid into the solution.
PAGE
-
11
Remove the comb from the cassette and put the cassette with gradient gel (4%−12%) in the gel box (smaller side facing inside).
Handle the polyacrylamide gel according to the manufacturer’s instructions.
-
12
First fill the inner chamber with MOPS buffer, make sure nothing is leaking to the outside chamber. Then, fill the outer chamber as well up to the half level mark.
-
13
Add blank, ladder, samples (10 μl each) in that order. For protein samples, do not load more than 10 μg per well (measure protein concentration using a nanodrop or BCA assay). Fill any unused wells with the blank to avoid the lanes running into each other. Run the gel for 45 min at 200 V. During that time prepare the blocking solution for Western Blot.
Western blot
-
14
After the gel finishes running, pour out the buffer and take the cassette out. Crack open the cassette and cut out the top and bottom portions. Transfer the gel to the plastic tray and rinse with nanopure water 2–3 times.
-
15
Transfer The proteins onto a nitrocellulose membrane with semi-dry gel transfer instrument (iBlot 2 by Invitrogen). The arrangement is as follows: bottom set -> gel -> filter paper -> top set -> felt with metal strip.
Make sure to use the roller to remove bubbles that might inhibit the transfer.
-
16
Take the membrane out, cut the corners that are not needed with scissors. Put the membrane in a clean tray and add Ponceau stain solution. Rock on the shaker for a minute.
-
17
Pour the solution back into the bottle to be reused later. Ponceau stain can be used for years without getting spoiled.
-
18
Rinse the membrane with water until the red color goes away. Image the membrane using ChemiDoc MP imaging system by BioRad.
-
19
Put the membrane back into the tray and add enough blocking solution (10 ml) to cover the membrane. Rock the tray for 1 hr.
The blocking agent blocks the non-specific antibody binding and reduces the background signal.
-
20
Discard the blocking solution and rinse twice, each time with TBS/T to get rid of it.
-
21
Add primary antibody cocktail (stored on ice) over the membrane and rock overnight at 4°C.
-
22
The next day, pour back the primary antibody solution into the original tube and store at 4°C for future use. The same solution can be reused for years.
-
23
Wash the membrane with TBS/T buffer by rocking at room temperature for 5 min–repeat four times, discard the buffer. During these washes, also rock the secondary antibody and blocking solutions at room temperature (to be used in the next steps).
-
24
Block again with omni milk (10 ml) and rock for 10 min at room temperature.
-
25
Discard the milk and add secondary antibody (5 μl in 5 ml blocking solution), rock at room temperature for 1 hr.
-
26
Discard the antibody solution and wash the membrane with TBS/T buffer by rocking at room temperature for 5 min–repeat 4 times. Discard the buffer.
-
27
While the fourth wash is going on, prepare the substrate solution, vortex briefly and let sit for 5 min.
-
28
After the fourth wash, discard TBS/T and add the substrate solution dropwise over the membrane covering the full surface area. Cover the tray with foil and let the reaction proceed for 5 min.
-
29
Image the membrane using ChemiDoc MP imaging system by BioRad. (Pixel size 3 × 3; exposure time–auto optimal).
The membrane should be imaged within 30 min from the start of the substrate reaction.
-
30
Visualize the ladder with colorimetric image and the samples using chemiluminescence. A sample western blot confirming TLR activation is shown in Figure 3. The presence of bands corresponding to p-P105, p-P65, and p-JNK proteins at 120, 65, 54, and 46 kDa, respectively demonstrate the activation of TLR pathway by the TLR ligand (LPS) used in the experiment.
-
31
For phosphoproteomic analysis, combine in a microcentrifuge tube the light, medium, and heavy-labeled aliquots at a 1:1:1 ratio by weight using 1.3 mg total protein from each of the samples as follows: 0, 10 and 30 min samples together and 3, 5, and 10 min samples together, creating two separate pools (Fig. 1). The 10 min overlapping time point can be used as a reference for comparison across all time points between the two sample pools.
Figure 3.
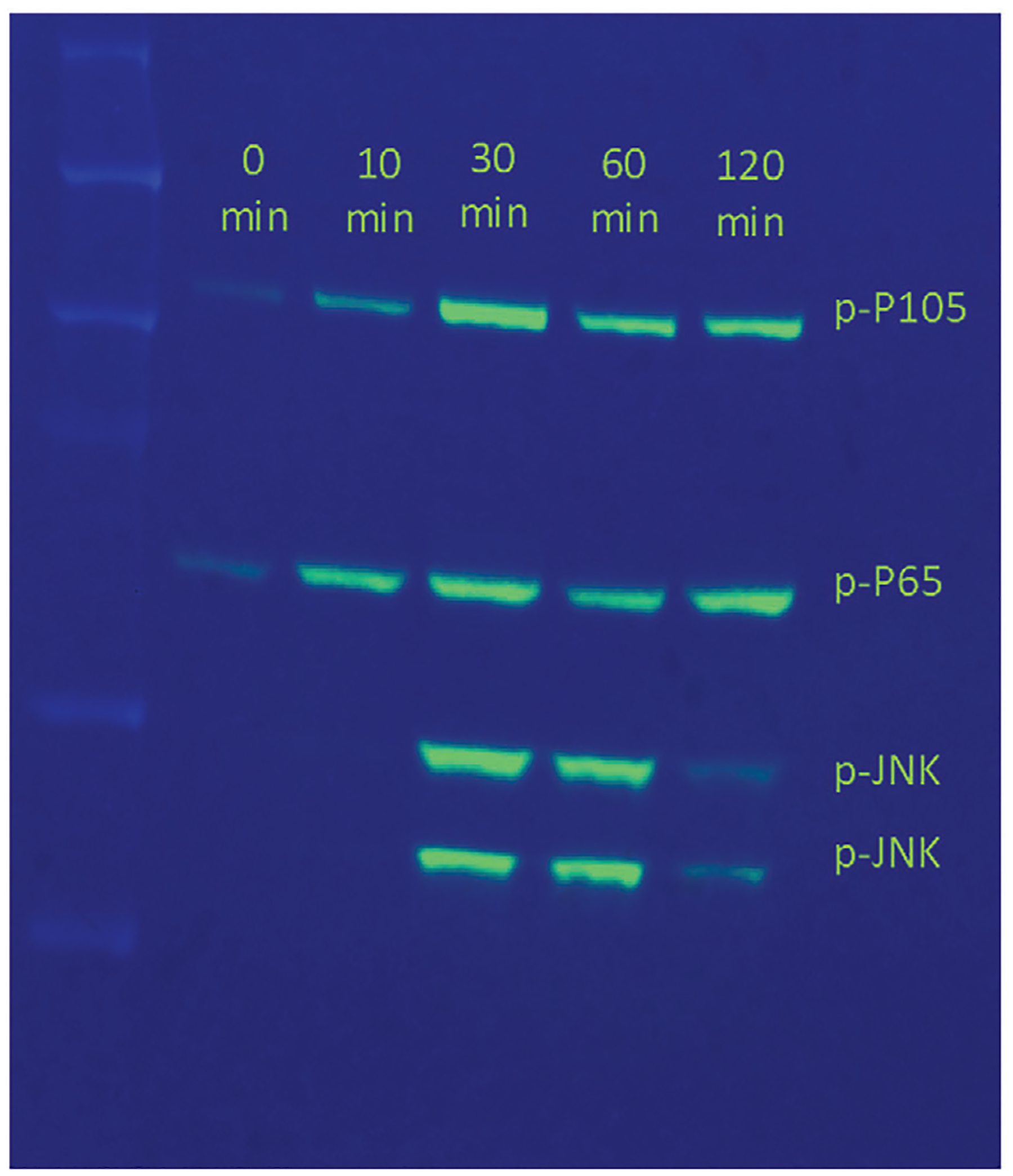
Stimulation of immortalized macrophages in the SILAC labeled media. Western Blot using the antibodies against phospho-p105, phospho-p65, and phospho-JNK. Equal amounts of total protein from cell lysates at times 0, 10, 30, 60, and 120 min after stimulation with LPS is loaded on the SDS-PAGE.
BASIC PROTOCOL 3
TRYPSIN DIGESTION, FRACTIONATION, AND PHOSPHOPEPTIDE ENRICHMENT
At this step, cells lysate pools containing heavy, medium, and light cultures as described in the previous protocol are subjected to protein reduction, alkylation, and digestion. The resulting peptide mix is then desalted and fractionated using strong cation exchange (SCX) chromatography to spread sample complexity over several fractions. SCX separates the peptides according to their charge state and allows the fractions to be subsequently enriched for phosphopeptides separately, thereby gaining a better phosphoproteome coverage. SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) is an effective strategy for phosphopeptide enrichment that involves TiO2-based enrichment immediately followed by Fe-NTA-based purification and affords higher amount of phosphopeptides compared to a single enrichment in isolation.
Materials
Digestion and desalting reagents
1 M stock of Dithiothreitol (DTT) or Tris(2-carboxyethyl) phosphine hydrochloride (TCEP. HCl) in 100 mM ammonium acetate or ammonium bicarbonate, pH 8–9 (see recipe)
0.8 M stock of Iodoacetamide (IAA) or Chloroacetamide (CAA) in 100 mM ammonium acetate or ammonium bicarbonate, pH 8–9 (see recipe)
Sequencing grade modified trypsin (Promega, cat. no. V5111)
100 mM ammonium acetate or ammonium bicarbonate, pH 8–9
Glacial Acetic acid (MilliporeSigma, cat. no. AX0074-6)
100% Acetonitrile (ACN)
0.1% Acetic acid
75% (v/v) acetonitrile (ACN) in 0.1% (v/v) acetic acid
Fractionation (strong cation exchange)
HPLC fractionation solvent A: 5 mM KH2PO4 (pH 2.7), 25% ACN (see recipe) HPLC fractionation solvent B: 5 mM KH2PO4 (pH 2.7), 500 mM KCl, 25% ACN (see recipe)
Phosphopeptide enrichment
High-Select™ TiO2 Phosphopeptide Enrichment Kit (Thermo Scientific, cat. no. A32993)
High-Select™ Fe-NTA Phosphopeptide Enrichment Kit (Thermo Scientific, cat. no. A32992)
0.1% (v/v) Formic acid, LC-MS grade
LC-MS grade water
Equipment
Microcentrifuge tubes, 1.5 ml
Low protein binding microcentrifuge tubes, 2 ml
pH strips
Vortex mixer
Sample Incubator at 37°C
Oasis HLB 1 cc Vac Cartridge (Waters, cat. no. WAT094225)
Vacuum manifold
Vacuum evaporator centrifuge, e.g., Savant SpeedVac Concentrator SPD 111V
PolySULPHOETHYL A SCX column (4.6 mm i.d. × 20 cm length, 5 μm particle size, 200 Å pore size) (PolyLC INC., cat. no. 204SE05)
Liquid chromatography system e.g., ÄKTA protein purification system (Cytiva).
Nanodrop instrument e.g., NanoDrop™ Lite Spectrophotometer (Thermo Scientific, cat. no. ND-LITE)
Ultrasonic bath
NOTE: There are other choices in addition to Oasis HLB for C-18 based peptide desalting columns e.g., SepPak C18 1 cc Vac Cartridge (Waters, cat. no. WAT023590) and Millipore ZipTip Pipette Tips (Millipore Sigma, cat. no. Z720070). Appropriate selection should be made based on the sample amount.
Trypsin digestion
-
1
Reduce cysteines in the combined lysate samples (pool 1 and pool 2 from step 31, Basic Protocol 2) using DTT or TCEP.HCl buffer at a final concentration of 2 mM. Vortex briefly to homogenize the mixture. Incubate the samples for 1 hr at 56°C.
TCEP.HCl is slightly preferable over DTT as it is odorless, reactive at a broad pH range and more resistant to oxidation from air. Reducing agent can be used at concentrations ranging from 2–10 mM. Avoid temperatures lower than 37°C and higher than 60°C where urea-based carbamylation of lysines and protein N-termini can occur.
-
2
Cool the samples at room temperature prior to adding IAA or CAA buffer to a final concentration of 4 mM. Incubate the samples for 1 hr in dark (cover the samples with foil or store in a laboratory drawer) at room temperature to alkylate the reduced cysteines (S-carbamidomethylation).
IAA/CAA alkylation prevents the formation of disulfide bonds. This reagent is unstable and light sensitive, hence should be prepared fresh right before its use. The reagent can be used at concentrations ranging from 4–20 mM. Normally, its concentration in the solution is double that of the reducing agent.
-
3
Dilute the alkylated samples to 2 M urea using 100 mM ammonium acetate or ammonium bicarbonate, pH 8–9.
-
4
Add trypsin at an enzyme-to-substrate ratio of 1:100 (w/w) and digest the samples by incubating overnight at 37°C.
-
5
Quench the digestion reaction by adding enough (to reduce pH to 2) glacial acetic acid to reduce the pH below 2.
-
6
Clear the peptide solution by centrifugation for 10 min at 14,000 × g, 4°C. Recover the clear supernatant in a fresh 1.5-ml tube leaving any residue behind.
-
7
Next, condition the Oasis HLB column for desalting the peptide mix by washing with 1 ml of ACN, followed by 0.1% acetic acid.
Peptides must be desalted before LC/MS analysis to remove salts and urea from the digestion buffer. Manufacturer’s protocol for desalting is available on the product page for Oasis HLB column and can be followed as such. A vacuum manifold is needed for offline desalting in this manner.
-
8
Load the tryptic peptides on the column followed by washing with 3 ml of 0.1% acetic acid.
-
9
Elute the peptides from the column with 1 ml of 75% (v/v) ACN/ 0.1% (v/v) acetic acid and collect in a fresh 1.5-ml tube.
-
10
Dry the eluted peptides by vacuum centrifugation.
Avoid evaporating the solvent to complete dryness as this might prevent the reconstitution of peptides into the solution later.
Fractionation and phosphopeptide enrichment
-
11
Resuspend the dried peptides in 500 μl of HPLC fractionation solvent A.
-
12
Inject the peptide mix onto the PolySULPHOETHYL A SCX column. SCX chromatography can be carried out on a benchtop chromatography system such as ÄKTA protein purification system (Cytiva).
-
13
Elute the peptides at a flow rate of 0.3 ml/min using the following gradient: 0% HPLC Fractionation solvent B for 2 ml, 0%−14% B for 33 ml, 14%−100% B for 1 ml, and 100% B held for 4 ml.
-
14
Collect 2-ml fractions in low protein binding microcentrifuge tubes and desalt the peptides as described above in step 7 of the previous section.
-
15
Dry the desalted peptides by vacuum centrifugation.
-
16
Measure the peptide amounts at 205-nm using a nanodrop instrument. Gather at least 500 μg (minimum amount for loading) per sample for phosphopeptide enrichment as per the SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) protocol (see the application note in “Internet Resources”). The overview of the phosphopeptide enrichment workflow is shown in Figure 4.
-
17
Resuspend the dried SCX fractions in 100 μl of the binding buffer provided with the High-Select TiO2 Phosphopeptide enrichment kit (Thermo Fisher Scientific) and proceed according to manufacturer’s instructions. Save all the flow-through and wash fractions along the process for the second enrichment. Combine them all and dry under vacuum.
-
18
Elute the phosphopeptides in 100 μl of the supplied elution buffer. Dry the TiO2 eluent under vacuum.
-
19
Enrich the TiO2 wash/flow-through fractions again using the High-Select Fe-NTA Phosphopeptide enrichment kit (Thermo Fisher Scientific) according to manufacturer’s instructions and dry the eluent under vacuum.
-
20
Reconstitute the dried phosphopeptides in 10–15 μl of 0.1% formic acid and incubate in an ultrasonic water bath for 10 min at room temperature.
-
21
Centrifuge the samples for 10 min at 14,000 × g, 20°C, to pellet any insoluble particles. Transfer the clear supernatant containing phosphopeptides to a fresh tube without disturbing the pellet. Peptides are now ready for LC-M/MS analysis.
Figure 4.
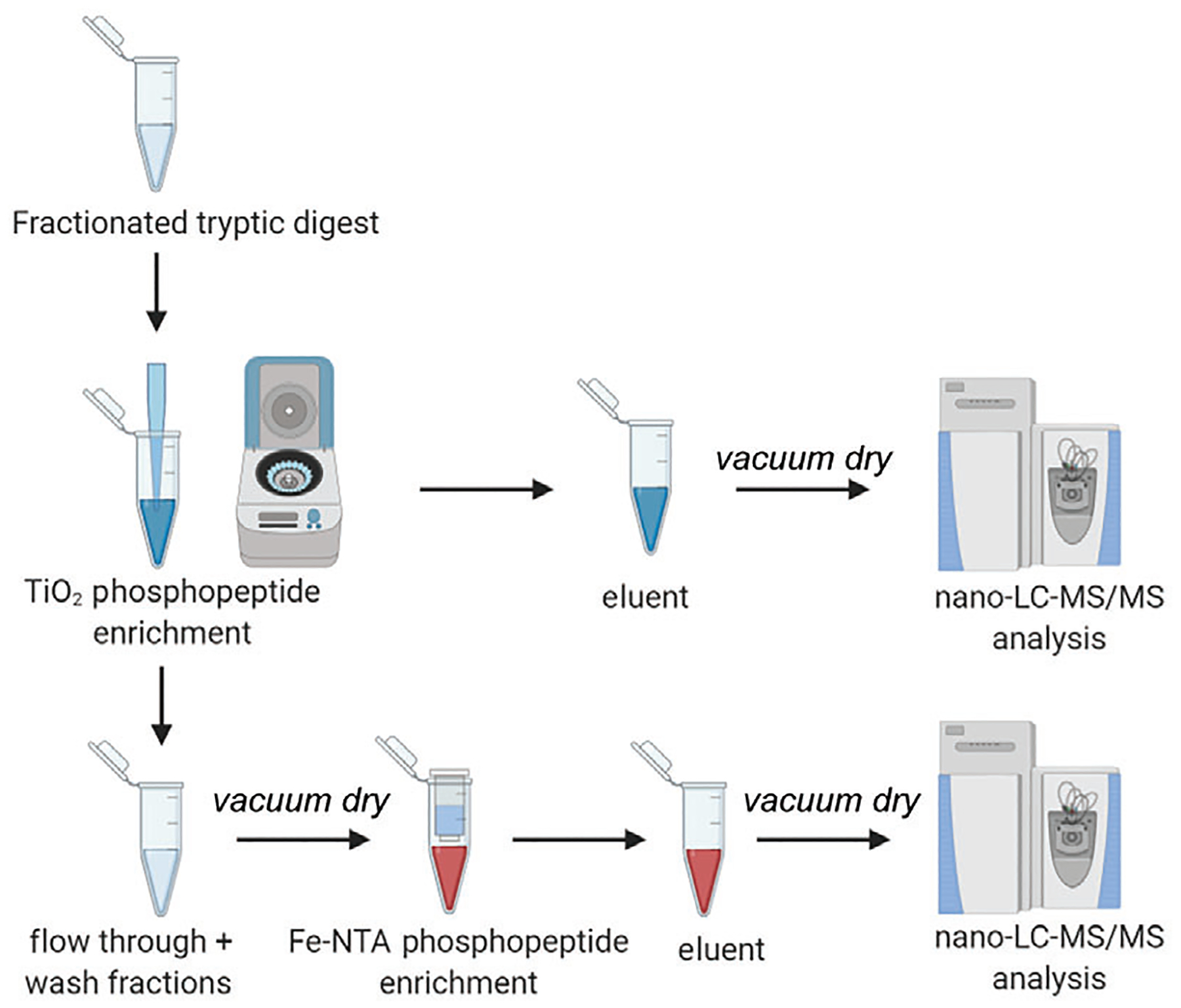
Experimental workflow of SMOAC strategy for Phosphopeptide enrichment of fractionated tryptic peptides. Phosphopeptides are first enriched using Titanium dioxide (TiO2) chromatography. The eluent is collected and vacuum dried for MS/MS analysis. The wash fractions and flow through from TiO2 are pooled together and subjected to a second enrichment with Fe-NTA (Ferric nitrilo triacetate), the eluent from which is collected separately for MS/MS analysis.
BASIC PROTOCOL 4
QUANTITATIVE MASS SPECTROMETRY
Following completion of Basic Protocols 1 through 3, the samples are ready to be analyzed by bottom-up tandem mass spectrometry. Peptides are first injected on an online nanoflow chromatography system for separation using a reverse phase column with an appropriate solvent gradient (see Table 1). Separated peptides then enter the MS instrument to be fragmented into smaller peptides and MS/MS spectra are acquired in a data-dependent manner. The RAW output files are processed on a data processing platform compatible with the output format. A variety of MS data analysis software and search engines are commercially available. The platform should be chosen based on the MS experiment design and complexity e.g., MaxQuant is more suitable for SILAC experiments while Proteome Discoverer or Byonic are good choices for a relatively simple qualitative proteomics experiment. The output from MaxQuant can be visualized with Perseus application. Statistical analysis is often required post processing, if not already offered by the data processing software.
Table 1.
Chromatography and MS Instrument Acquisition Settings
| Sample volume | 10 μl |
| Stationary phase | Acclaim PepMap C-18 100 Å, 75 μm, 25 cm, 2 μm |
| LC solvent A | 100% H2O, 0.1% formic acid |
| LC solvent B | 80% ACN, 0.1% formic acid |
| LC gradient | 2.5%−8% B in 3 min |
| 8%−28% B in 52 min | |
| 28%−50% B in 10 min | |
| 50%−99% B in 4 min | |
| LC flow rate | 300 nL/min |
| Mass spectrometer | Orbitrap Fusion™ Lumos™ Tribrid™ |
| Method type | Data dependent MS2 |
| Spray voltage | 2.1 kV |
| MS1 detector | Orbitrap |
| MS1 scan range | 400–1500 m/z |
| MS1 resolution | 120,000 @ 200 m/z |
| MS1 AGC target | 4e5 |
| MS1 maximum IT | 50 ms |
| MS2 detector | Orbitrap |
| MS2 resolution | 30,000 @ 200 m/z |
| MS2 fixed first mass | 100 m/z |
| Isolation window | 0.8 m/z |
| MS2 AGC target | 5e4 |
| MS2 maximum IT | 54 ms |
| Activation type/collision energy | HCD 30%−35% |
| Minimum signal req. | 2e4 |
| Dynamic exclusion | 60 s |
Materials
Mass Spectrometry
Solvent A: 0.1% (v/v) formic acid
Solvent B: 80% Acetonitrile (ACN) in 0.1% formic acid (v/v)
Phosphopeptide samples from step 21, Basic Protocol 3
Equipment
Nanoflow HPLC system e.g., UltiMate 3000 RSLCnano (Thermo Scientific)
Reversed phase chromatography column e.g., Acclaim PepMap 100 C-18 (Thermo Scientific)
Electrospray ionization (ESI) capable ion trap tandem mass spectrometer e.g., LTQ-Orbitrap Velos™ or Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer (Thermo Scientific)
Quantitative mass spectrometry
-
1
Inject the phosphopeptides (1 μg/10 μl injection) onto the nano-LC system coupled to the mass spectrometer. Run the samples in technical duplicates of three. A topten instrument method can be used in data-dependent manner to automatically cycle between Orbitrap full scan and ion trap MS/MS.
All technical details using Thermo Orbitrap Fusion™ Lumos™ Tribrid™ mass spectrometer as an example are described in the Table 1.
Data processing
-
2
Analyze the RAW files using instrument-output compatible software such as Proteome Discoverer (Thermo Scientific) or vendor-neutral platforms such as MaxQuant (Tyanova, Temu, & Cox, 2016) or Byonic™ (Protein Metrics). Include the MaxQuant contaminant.fasta database when using this platform. The data processing parameters are described in Table 2. The spectrum of a phosphopeptide precursor ion from mouse RNA-binding protein Raly is shown as an example in Figure 5. The loss of a phosphate group (94.9714 Da) from serine residue is reflected by the prominent peak of doubly charged (y10–P) ion at 527.7754 m/z.
PTM score algorithm in MaxQuant allows phosphorylation site identification in a peptide sequence. Phosphopeptide ratios of stimulated versus unstimulated cells can be calculated using normalized signals for each time point.
Table 2.
Data Processing Parameters
| Platform | MaxQuant | Proteome Discoverer 2.4 |
| Search algorithm | Andromeda | SequestHT/Mascot |
| Database | Fasta File of full proteome of Arabidopsis thaliana (Mouse-ear cress) by UniProtKB; Taxon identifier 3702 | Fasta File of full proteome of Arabidopsis thaliana (Mouse-ear cress) by UniProtKB; Taxon identifier 3702 |
| Enzyme | Trypsin (full) | Trypsin (full) |
| Multiplicity/max. labeled | 3 | 3 |
| Missed cleavages | 2 | 2 |
| Min. peptide length | 3 | 3 |
| Peptides for quantification | Unique + razor | Unique + razor |
| Fragment mass tolerance | 20 ppm (First search) 4.5 ppm (Main search) | 0.02 Da |
| Neutral losses | Water, ammonia | Water, ammonia |
| Static modifications | Carbamidomethyl (C) | Carbamidomethyl (C) |
| Dynamic modifications | Oxidation (M) Acetylation (N-term) Phosphorylation (S,T,Y) | Oxidation (M) Acetylation (N-term) Phosphorylation (S,T,Y) |
| Target FDR | <1% | <1% |
Figure 5.
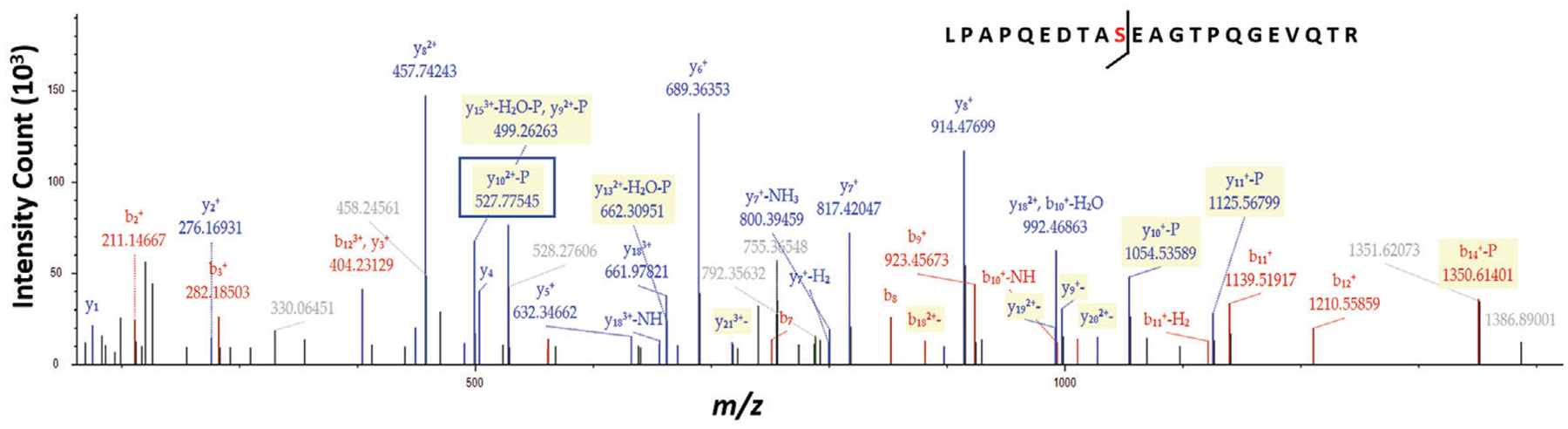
MS/MS fragment spectrum of the phosphopeptide LPAPQEDTASEAGTPQGEVQTR from RNA-binding protein Raly (Q64012) in mouse. The serine residue at position 10 is phosphorylated, which is confirmed by the presence of doubly charged y ion that lost a phosphate group (boxed and highlighted in yellow).
REAGENTS AND SOLUTIONS
Blocking solution
Prepare blocking agent for Western Blotting by dissolving 2.5 g milk, non-fat, dry, Omniblok™ (americanBio, cat. no. AB10109-00100) in 50 ml of 1× TBS/T (TBS with 0.1% TWEEN 20). Prepare fresh for each use, do not store.
Dithiothreitol (DTT) solution
Prepare a fresh stock of 1 M DL-Dithiothreitol (DTT; MilliporeSigma, cat. no. D9779) by dissolving 154.25 mg of the product in 1 ml of nanopure water or appropriate buffer. Dilute as needed. Store the 1M aliquots for up to a year at −20°C.
Gel loading buffer
Prepare loading buffer for PAGE by first diluting 2-mercaptoethanol (Thermo Scientific, cat. no. 35602BID) 1:4 (v/v) with water and then preparing a 1:1 mix (v/v) of the diluted 2-mercaptoethanol and 4× NuPAGE™ LDS sample buffer (Thermo Scientific, cat. no. NP0007). Make fresh each time. Store the stock solutions indefinitely at room temperature.
Iodoacetamide (IAA) solution
Prepare a fresh stock of 0.8 M iodoacetamide (IAA: MilliporeSigma, cat. no. I1149) by dissolving 147.96 mg of the product in 1 ml of appropriate buffer. Do not store.
KH2PO4 (5 mM; pH 2.7), 25% ACN
Weigh appropriate amount of potassium phosphate monobasic (KH2PO4; MilliporeSigma, cat. no. P0662) to prepare 5 mM solution of required volume and dissolve in HPLC grade water containing 25% (v/v) acetonitrile (ACN), adjust pH to 2.7 using phosphoric acid. Store up to 1 week at room temperature.
KH2PO4 (5 mM; pH 2.7), 500 mM KCl, 25% ACN
Weigh appropriate amounts of potassium phosphate monobasic (KH2PO4; MilliporeSigma, cat. no. P0662) and potassium chloride, molecular biology grade (KCl; MilliporeSigma, cat. no. 529552) to prepare a solution that is 5 mM in KH2PO4 and 500 mM in KCl for the required volume and dissolve in HPLC grade water containing 25% (v/v) ACN, adjust pH to 2.7 using phosphoric acid. Store up to 1 week at room temperature.
MOPS buffer, 1×
-
Prepare 1× solution from the 20× stock NuPAGE™ MOPS SDS Running Buffer (Thermo Scientific, cat. no. NP0001) by diluting with nanopure water. Prepare fresh each time.
The 20× stock can be stored indefinitely at room temperature.
Phosphatase inhibitor cocktail
Dissolve one tablet PhosSTOP (MilliporeSigma, cat. no. 4906845001) in 1 ml of nanopure water or 1 ml of 100 mM phosphate buffer, pH 7.0, for making a 10× stock solution. Stock solution is stable for 1 month at 2°C–8°C, or at least 6 months at −30°C.
Primary antibody cocktail
Primary antibodies:
Phospho-p38 MAPK (Thr180/Tyr182) Antibody (Cell Signaling Technology, cat. no. 9211)
Phospho-SAPK/JNK (Thr183/Tyr185) Antibody (Cell Signaling Technology, cat. no. 9251)
Phospho-NF-κB p65 (Ser536) Antibody (Cell Signaling Technology, cat. no. 3031)
-
Phospho-NF-κB p105 (Ser933) (18E6) Rabbit mAb (Cell Signaling Technology, cat. no. 4806)
To prepare primary antibody mix, first prepare 1× TBS, 0.1% TWEEN-20 with 5% (w/v) bovine serum albumin (BSA; Cell Signaling Technology, cat. no. 9998) and 0.02% sodium azide (NaN3). Add each of the four primary antibodies (see above) at 1:1000 dilution (v/v) (refer to the product sheet of the antibody used to figure out the appropriate dilution ratio). Store this cocktail mix up to 1 year at 4°C. Store pure antibody stock solutions at −20°C.
Protease inhibitor cocktail
Dissolve one cOmplete™ Mini EDTA-free Protease Inhibitor Cocktail (Roche, cat. no. 04693159001) tablet in 1 ml of nanopure water (18.2 megohm ionic purity) or 1 ml of 100 mM phosphate buffer, pH 7.0, for making a 10× stock solution. Stock solution is stable for 1–2 weeks at 2°C-8°C, or for at least 12 weeks at −30°C.
Protein ladder
-
Prepare protein ladder by mixing 2 μl of the Precision Plus Protein™ Unstained Protein Standards, Strep-tagged recombinant, 1 ml (BioRad, cat. no. 1610363)with 8 μl of gel loading buffer. Prepare fresh each time.
Store the stock at −20°C until expiration date.
Secondary antibody
-
Anti-rabbit IgG, horseradish peroxidase (HRP)-linked Antibody (Cell Signaling Technology, cat. no. 7074)
Secondary antibody stock is supplied at 20,000×, dilute this to 1000× with 1× TBS (no TWEEN). Combine 5 μl of 1000× solution with 4995 μl of the blocking solution to reach 1× concentration for the secondary antibody. Rock for 1 hr at room temperature before using. Prepare the 1× solution fresh each time, 1000× stock solution of the secondary antibody can be stored at 4°C.
SILAC isotope stock solutions
l-arginine.HCl labeled with 13C6 (Arg6) (Cambridge Isotope Laboratories, cat. no. CLM-2265-H-PK)
l-arginine.HCl labeled with 13C6 15N4 (Arg10) (Cambridge Isotope Laboratories, cat. no. CNLM-539-H-PK)
l-lysine.2HCl labeled with 2D4 (Lys4) (Cambridge Isotope Laboratories, cat. no. DLM-2640-PK)
l-lysine.2HCl labeled with 13C6 15N2 (Lys8) (Cambridge Isotope Laboratories, cat. no. CNLM-291-H-PK), 1000× solution
Unlabeled L-arginine.HCl (Arg0) (Cambridge Isotope Laboratories, cat. no. ULM-8347-PK) Also available at MilliporeSigma
-
Unlabeled L-lysine.2HCl (Lys0) (Cambridge Isotope Laboratories, cat. no. ULM-8766-PK). Also available at MilliporeSigma
Preparation starting from 1 g vial of each of the isotopes: Add the required amount of water in the product vial itself to prepare 0.398 M solution of Arg isotopes and 0.798 M solution of Lys isotopes. Alternatively, a required amount of isotope can be weighed to prepare a different volume of solution with the stated molarities. At this point you will have six different isotope solutions. Heat the solutions briefly to make sure the amino acids are fully dissolved. Vortex and transfer 1 ml into 1.5-ml tubes. Label these as 1000× stock solutions. The final concentration of isotopes needed in the SILAC DMEM media is 0.398 mM for Arg isotopes and 0.798 mM for Lys isotopes. The 1000× stocks can be stored at −30°C for a few years.
Substrate solution
-
Clarity™ Western ECL Substrate (BioRad, cat. no. 1705060)
The kit contains 100 ml peroxide reagent and 100 ml luminol/enhancer reagent. Mix the two detection reagents in 1:1 ratio (v/v) to prepare the substrate solution for reporter enzyme (HRP) in the secondary antibody. Prepare the substrate solution fresh each time. Store the kit up to 1 year at room temperature.
TBS with 0.1% TWEEN 20
Prepare 1× TBS/0.1% TWEEN 20 by diluting 100 ml of Tris Buffered Saline with TWEEN 20 (20×) (BioRad, cat. no. BUF028)with 1.9 L of nanopure water. Store at room temperature until expiration date.
COMMENTARY
Background Information
The techniques discussed in this article present design and execution of mass spectrometry-based assays to understand and measure phosphorylation changes during immune cell signaling. The discovery-based approach allows the observation of temporal phosphoproteomic responses to stimulation of distinct TLRs. These studies can help model a biological system from a phosphorylation based signaling perspective (Manes et al., 2015), as well as can easily be modified for any complex signaling network. Phosphoproteomic profiling of stimulated macrophages has revealed that TLR responses vary both in terms of major modification targets and phosphoproteome dynamics (Sjoelund, Smelkinson, & Nita-Lazar, 2014). Therefore, a global, quantitative analysis of phosphorylation as a molecular signature is a critical step towards our understanding of innate immune activation caused by different pathogens.
There are other chemical and enzymatic labeling techniques available for relative quantitation with mass spectrometry, e.g., Isotope-Coded Affinity Tagging (ICAT), Dimethyl labeling, 18O labeling, labeling with isobaric tags (tandem mass tags) (Anand, Samuel, Ang, Keerthikumar, & Mathivanan, 2017). While these allow for higher order multiplexing and comparison of multiple experimental conditions, certain practical difficulties, e.g., incomplete incorporation into proteolytic peptides, inability to use primary amine containing buffers, and retention time shifts of deuterated peptides in reversed-phase chromatography, limit their application. SILAC, on the other hand is a metabolic labeling method where label incorporation occurs at the earliest possible moment in the sample preparation process (Hoedt, Zhang, & Neubert, 2019; Ong & Mann, 2007). This makes SILAC less prone to errors, unintended biases in measurement and hence is the preferred method for quantitative comparison of phosphopeptides by mass spectrometry. For MS data processing, numerous software solutions are available that are specific to the instrument compatibility and processing functionality (Chen, Hou, Tanner, & Cheng, 2020). It is important to choose an appropriate bioinformatic platform as per the quantitation method in question and the analytical requirements. The data volume and complexity also require the use of statistical approaches for meaningful interpretations of the results. Some of the analyses pertinent to phosphoproteomics include time series clustering of phosphorylation sites, gene ontology and pathway enrichment analyses, and signaling pathway analysis. In Basic Protocol 3, tryptic digestion of proteins is described for in-solution conditions. However, the overall protocol allows for in-gel digestion (Shevchenko, Tomas, Havlis, Olsen, & Mann, 2006) as well (a simple replacement keeping rest of the protocols unchanged) for cases where in-solution digestion might cause a loss of low-abundance proteins. Since the percentage of phosphorylated peptides is much less in a pool of unmodified peptides, the enrichment is imperative for their detection by mass spectrometry. Most commonly employed techniques for phosphopeptide enrichment are immobilized metal affinity chromatography (IMAC) (Thingholm & Larsen, 2016) and metal oxide affinity chromatography (MOAC) (Leitner, 2010). These techniques are complementary in terms of enrichment specificity, retaining mono and multi-phosphopeptides and capturing serine versus threonine versus tyrosine phosphorylation. Coupling the two enrichments in tandem, i.e., TiO2 followed by Fe-NTA chromatography can deepen the phosphoproteome coverage. Hence, we suggest SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) (Thermo Scientific), where peptides not bound to TiO2 are enriched further with Fe-NTA instead of being discarded as the flow-through. In addition, strong cation exchange chromatography can simplify the proteome and partially enrich acidic phosphopeptides as well. For a comprehensive review on phosphopeptide enrichment strategies, see (Low et al., 2020).
Critical Parameters
It is critical that the experimental steps after stimulation (Basic Protocols 3 and 4) are carried out in a swift manner (it should take around 3 days to get the samples ready for MS runs) considering the labile nature of phosphorylation as a PTM; pH and temperature are also key factors in keeping this PTM intact. The solutions and equipment coming into contact with cells must be sterile; make sure there is no bacterial growth. The suggested time point range in Basic Protocol 2 is just an example and should be adjusted depending on the purpose of the study. Protease and phosphatase inhibitors must be added to the lysis buffer to preserve the phosphoproteome during and after cell lysis. Make lysis buffer fresh for each experiment. Additionally, due to volatile nature of certain buffers (containing ammonia) and solvents (ACN), preparing fresh buffers is essential. Compatibility of buffers, salts, detergent, protease, and phosphatase inhibitors with LC-MS/MS is also important. To reduce the contamination of the MS instrument, all reagents utilized in the sample preparation should be of HPLC grade. At the end of Basic Protocol 2, the nitrocellulose membrane should be imaged within 30 min from the start of the substrate reaction. During vacuum centrifugation, avoid evaporating the solvent to complete dryness as this might prevent the reconstitution of peptides back into the solution. Peptides must be desalted offline post digestion and prior to LC/MS analysis to remove salts and urea from the sample. When it comes to quantitative mass spectrometry, it must be ensured that the instrument is properly calibrated using a calibrant that molecularly resembles the analyte under investigation and that the threshold settings are optimized for detecting phosphopeptides.
Troubleshooting
See Table 3 for commonly encountered problems and their solutions.
Table 3.
Troubleshooting
| Problem | Possible cause | Solution |
|---|---|---|
| Inadequate MS data | Inefficient SILAC labeling | Check label incorporation by mass spectrometry on an aliquot of labeled sample before further experimentation, calculate % of unlabeled peptides (should be less than 5%) |
| Low phosphoproteome coverage | Cell lysis not performed properly |
|
| Low enrichment specificity | Retention of non-phosphorylated peptides during enrichment | Increase the number of washing steps during SMOAC enrichment |
| Unexpectedly low number of identifications post processing MS data | MS instrument not optimized, bioinformatic treatment missing crucial parameters | Carefully optimize data acquisition parameters for phosphopeptide detection on the mass spectrometer and double check all the data processing parameters for the software run |
Understanding Results
Labeling efficiency in cells when checked by mass spectrometry should be ≥95%. This can be visually confirmed by comparing the relative peak intensities of the same peptide in both the labeled and unlabeled forms; the labeled version should be almost 100% in intensity (see Fig. 2). TLR stimulation and the occurrence of immune signaling cascade is reflected in the Western Blot. The presence of bands corresponding to phosphoproteins p-P105, p-P65, and p-JNK proteins at 120, 65, 54, and 46 kDa, respectively in the Western Blot demonstrate the activation of TLR pathway by the TLR ligand. Depending on the amount of starting material (grams of protein), and considering optimum instrument performance, the number of unique phosphosites identified with this protocol should range between hundreds and single digit thousands.
Time Considerations
SILAC labeling in cell culture takes roughly 2 weeks to ensure >95% label incorporation. TLR stimulation time depends on the study design; lysing of cell PAGE require 1 hr each. Western Blotting needs overnight treatment with the primary antibody; therefore, it is advisable to perform this step towards the end of the day. Reduction and alkylation steps require 2 hr total; digestion happens overnight. In time limited situations, 4-hr digestion is recommended. SCX fractionation and phosphopeptide enrichment take 1 full day. As soon as the samples are ready, LC-MS/MS should be performed. It is not advisable to freeze the samples at this point. Mass spectrometric data acquisition depends on the number of samples, replicate runs and gradient times. Data analysis is dependent on the processing software chosen, computer performance and number of samples; it may take anywhere between days and weeks.
Acknowledgments
The authors would like to thank Dr. Nathan Manes for providing the data for the phosphopeptide spectrum. This research was supported by the Intramural Research Program of NIAID, NIH.
Footnotes
Application note for sequential enrichment of Metal Oxide Affinity Chromatography (SMOAC) phosphopeptide enrichment.
Literature Cited
- Anand S, Samuel M, Ang CS, Keerthikumar S, & Mathivanan S (2017). Label-based and label-free strategies for protein quantitation. Methods in Molecular Biology, 1549, 31–43. doi: 10.1007/978-1-4939-6740-7_4. [DOI] [PubMed] [Google Scholar]
- Chen C, Hou J, Tanner JJ, & Cheng J (2020). Bioinformatics methods for mass spectrometry-based proteomics data analysis. International Journal of Molecular Sciences, 21(8), 2873. doi: 10.3390/ijms21082873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Nardo D, Kalvakolanu DV, & Latz E (2018). Immortalization of murine bone marrow-derived macrophages. Methods in Molecular Biology, 1784, 35–49. doi: 10.1007/978-1-4939-7837-3_4. [DOI] [PubMed] [Google Scholar]
- Du R, Long J, Yao J, Dong Y, Yang X, Tang S, … Chen X (2010). Subcellular quantitative proteomics reveals multiple pathway crosstalk that coordinates specific signaling and transcriptional regulation for the early host response to LPS. Journal of Proteome Research, 9(4), 1805–1821. doi: 10.1021/pr900962c. [DOI] [PubMed] [Google Scholar]
- Hoedt E, Zhang G, & Neubert TA (2019). Stable isotope labeling by amino acids in cell culture (SILAC) for quantitative proteomics. In Woods AG & Darie CC (Eds.), Advancements of mass spectrometry in biomedical research (pp. 531–539). Cham: Springer International Publishing. [Google Scholar]
- Kagan JC (2012). Signaling organelles of the innate immune system. Cell, 151(6), 1168–1178. doi: 10.1016/j.cell.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JY, & Lee JO (2011). Structural biology of the Toll-like receptor family. Annual Review of Biochemistry, 80, 917–941. doi: 10.1146/annurev-biochem-052909-141507. [DOI] [PubMed] [Google Scholar]
- Leitner A (2010). Phosphopeptide enrichment using metal oxide affinity chromatography. TrAC Trends in Analytical Chemistry, 29(2), 177–185. doi: 10.1016/j.trac.2009.08.007. [DOI] [Google Scholar]
- Low TY, Mohtar MA, Lee PY, Omar N, Zhou H, & Ye M (2020). Widening The Bottleneck Of Phosphoproteomics: Evolving strategies for phosphopeptide enrichment. Mass Spectrometry Reviews, n/a(n/a). doi: 10.1002/mas.21636. [DOI] [PubMed] [Google Scholar]
- Manes NP, Angermann BR, Koppenol-Raab M, An E, Sjoelund VH, Sun J, … Nita-Lazar A (2015). Targeted Proteomics-driven computational modeling of macrophage S1P chemosensing. Molecular and Cellular Proteomics, 14(10), 2661–2681. doi: 10.1074/mcp.M115.048918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LAJ, Golenbock D, & Bowie AG (2013). The history of Toll-like receptors — redefining innate immunity. Nature Reviews Immunology, 13(6), 453–460. doi: 10.1038/nri3446. [DOI] [PubMed] [Google Scholar]
- Ong S-E, & Mann M (2007). Stable isotope labeling by amino acids in cell culture for quantitative proteomics. In Sechi S (Ed.), Quantitative proteomics by mass spectrometry (pp. 37–52). Totowa, NJ: Humana Press. [DOI] [PubMed] [Google Scholar]
- Shevchenko A, Tomas H, Havlis J, Olsen JV, & Mann M (2006). In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nature Protocols, 1(6), 2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- Sjoelund V, Smelkinson M, & Nita-Lazar A (2014). Phosphoproteome profiling of the macrophage response to different toll-like receptor ligands identifies differences in global phosphorylation dynamics. Journal of Proteome Research, 13(11), 5185–5197. doi: 10.1021/pr5002466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, & Akira S (2004). TLR signaling pathways. Seminars in Immunology, 16(1), 3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- Thingholm TE, & Larsen MR (2016). Phosphopeptide enrichment by immobilized metal affinity chromatography. In von Stechow L (Ed.), Phospho-proteomics: Methods and protocols (pp. 123–133). New York, NY: Springer New York. [DOI] [PubMed] [Google Scholar]
- Tyanova S, Temu T, & Cox J (2016). The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nature Protocols, 11(12), 2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- Weintz G, Olsen JV, Frühauf K, Niedzielska M, Amit I, Jantsch J, … Lang R (2010). The phosphoproteome of toll-like receptor-activated macrophages. Molecular Systems Biology, 6, 371. doi: 10.1038/msb.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]