

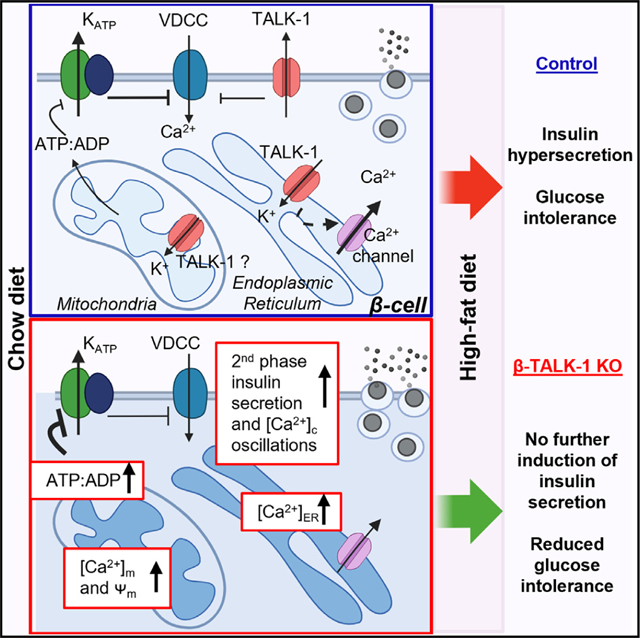
SUMMARY
Mitochondrial Ca2+ ([Ca2+]m) homeostasis is critical for β-cell function and becomes disrupted during the pathogenesis of diabetes. [Ca2+]m uptake is dependent on elevations in cytoplasmic Ca2+ ([Ca2+]c) and endoplasmic reticulum Ca2+ ([Ca2+]ER) release, both of which are regulated by the two-pore domain K+ channel TALK-1. Here, utilizing a novel β-cell TALK-1-knockout (β-TALK-1-KO) mouse model, we found that TALK-1 limited β-cell [Ca2+]m accumulation and ATP production. However, following exposure to a high-fat diet (HFD), ATP-linked respiration, glucose-stimulated oxygen consumption rate, and glucose-stimulated insulin secretion (GSIS) were increased in control but not TALK1-KO mice. Although β-TALK-1-KO animals showed similar GSIS before and after HFD treatment, these mice were protected from HFD-induced glucose intolerance. Collectively, these data identify that TALK-1 channel control of β-cell function reduces [Ca2+]m and suggest that metabolic remodeling in diabetes drives dysglycemia.
In brief
Graff et al. show that β-cells without TALK-1 channels have increased mitochondrial calcium and ATP production but reduced diabetogenic stress-induced changes in metabolism. Mice with β-cell TALK-1 ablation show protection from diet-induced glucose intolerance without elevated insulin secretion. This suggests that alterations in β-cell metabolism and/or insulin secretion drive dysglycemia.
Graphical Abstract
INTRODUCTION
Glucose-stimulated insulin secretion (GSIS) is coupled to β-cell cytoplasmic Ca2+ ([Ca2+]c) influx through voltage-dependent Ca2+ channels (VDCCs) and Ca2+ release from the endoplasmic reticulum (ER).1–8 β-cell insulin secretion occurs in a pulsatile manner from the pancreas due to synchronous oscillations in electrical excitability and [Ca2+]c influx.9 The kinetics and amplitude of ER Ca2+ ([Ca2+]ER) release contribute to islet β-cell Ca2+ oscillations and are controlled by the ER-localized two-pore domain (K2P) channel TALK-1, which provides a K+ countercurrent that maintains the electrical driving force for [Ca2+]ER efflux.10 TALK-1 augmentation of [Ca2+]ER release during glucose stimulation also activates the Ca2+-sensitive plasma membrane K+ current Kslow, which increases the inter-burst interval between [Ca2+]c oscillations and reduces [Ca2+]c oscillation frequency.10,11
As pancreatic insulin pulsatility is disrupted in type 2 diabetes mellitus (T2DM) and contributes to insulin resistance,12,13 this may involve changes in TALK-1 activity. Importantly, a non-synonymous gain-of-function (GOF) polymorphism in the KCNK16 (rs1535500) gene encoding TALK-1 has been associated with an increased risk for T2DM,14,15 and this may increase T2DM susceptibility by reducing β-cell Ca2+ influx and insulin secretion.16 Furthermore, another GOF mutation in TALK-1 is associated with maturity-onset diabetes of the young (MODY), which reduces β-cell glucose-stimulated Ca2+ influx, decreases [Ca2+]ER storage, and blunts GSIS.17 Although this suggests important roles for TALK-1 in the control of islet function, the β-cell-specific role(s) of TALK-1 have not been elucidated.
TALK-1 control of [Ca2+]ER and [Ca2+]c would be predicted to modulate mitochondrial Ca2+ ([Ca2+]m) handling and metabolism. While [Ca2+]c is a major source of [Ca2+]m uptake, [Ca2+]ER also contributes to [Ca2+]m handling through Ca2+ microdomains located at junctions between the ER and mitochondria, termed the mitochondrial-associated membrane (MAM).18 The high [Ca2+]c at the MAM is critical for [Ca2+]m uptake for two reasons; the mitochondrial Ca2+ uniporter (MCU) has a low affinity to Ca2+, and glucose-stimulated [Ca2+]m levels reach higher concentrations (≥12 μm)19 than those of [Ca2+]c (~1 μm). Therefore, TALK-1 control of the driving force for [Ca2+]ER release and thus [Ca2+]ER storage may regulate [Ca2+]m. TALK-1 may also regulate [Ca2+]m through its influence on [Ca2+]c. Interestingly, elevations in [Ca2+]c cause a reduction in cytosolic ATP through increased cellular workload as well as changes in [Ca2+]m control of Ca2+-dependent metabolic enzymes (e.g., dehydrogenases and the aspartate-glutamate carrier [AGC1]).20–23 Additionally, [Ca2+]m handling is dependent on K+ channel control of the inner mitochondrial membrane potential (ψm).24 While it is not known if TALK-1 directly impacts ψm, another K2P channel, TASK-3, localizes to the inner mitochondrial membrane, where it hyperpolarizes ψm.25 This suggests that TALK-1 may play important role(s) in regulating β-cell mitochondrial function and metabolism.
Here, we examined how TALK-1 controls β-cell mitochondrial function and glucose homeostasis under physiological and diabetic conditions. Our data determined that TALK-1 activity limited β-cell [Ca2+]m levels and ATP production. Under diabetogenic stress induced by high-fat diet (HFD), TALK-1 activity enhanced β-cell mitochondrial respiration and GSIS but, surprisingly, promoted glucose intolerance. Interestingly, β-cell-specific TALK-1-knockout (β-TALK-1-KO) animals showed similar GSIS before and after HFD treatment; however, these mice were protected from HFD-induced glucose intolerance. These data suggest that TALK-1-mediated alterations in β-cell function under diabetic conditions negatively impact glucose homeostasis.
RESULTS
TALK-1 channels regulate β-cell Vm, [Ca2+]c oscillations, and [Ca2+]ER
To determine how β-cell TALK-1 controls Ca2+ handling and glucose homeostasis, we generated a β-TALK-1-KO mouse model (Figures S1A–S1C). β-cell TALK-1 ablation was confirmed via specific loss of TALK-1 immunostaining in insulin-positive β-cells (Figure 1A). β-cell loss of TALK-1 resulted in reduction of whole-cell K2P current density (Figures 1B and 1C). Deletion of TALK-1 increased β-cell plasma membrane potential (Vm) plateau fraction under high glucose (11 mM glucose [G], control: 0.285 ± 0.047 vs. β-TALK-1-KO: 0.471 ± 0.057; p = 0.02583; Figures 1D and 1E); these β-TALK-1-KO Vm changes were equivalent to those observed in global TALK-1-KO β-cells.16 β-TALK-1-KO islets also showed an increase in islet [Ca2+]c oscillation frequency (Ca2+ peaks/min, control: 0.331 ± 0.026 vs. β-TALK-1-KO: 0.508 ± 0.048; p = 0.002; Figures 1F, 1G, and S2A–S2D). ER-localized TALK-1 channels provide a K+ countercurrent that enhances [Ca2+]ER release and slows [Ca2+]c oscillation frequency10; thus, we next examined if β-TALK-1-KO islets have perturbed [Ca2+]ER handling. Similar to TALK-1-KO islets,16 ablation of β-cell TALK-1 resulted in increased [Ca2+]ER storage, likely due to reduced K+ flux into the ER and blunted electrical driving force for [Ca2+]ER release10 (normalized [Ca2+]c, control: 96.635 ± 2.928 vs. β-TALK-1-KO: 116.835 ± 4.650; p = 0.00017; Figures 1H and 1I). These data show that TALK-1 controls β-cell Ca2+ handling through enhancement of Vm plateau fraction and [Ca2+]ER release.
Figure 1. TALK-1 channels regulate β-cell Vm, [Ca2+]c oscillations, and [Ca2+]ER.

(A) Representative immunofluorescence images of control (top) and β-TALK-1-KO (bottom) mouse pancreas sections stained for insulin (red), TALK-1 (green), somatostatin (magenta), and nuclei (blue). Scale bar: 25 μM.
(B) Representative whole-cell current recordings in β-cells from control mice (blue) or β-TALK-1-KO mice (red) utilizing a voltage clamp, in response to a ramp from −40 to +60 mV
(C) Average current density (pA/pF) at the specified membrane voltage in β-cells from control mice (blue; n = 4 mice) or β-TALK-1-KO mice (red; n = 5 mice).
(D) Representative Vm recordings from β-cells recorded in intact mouse islets stimulated with 11 mM glucose from control (top trace) and β-TALK-1-KO (bottom trace) mice.
(E) Plateau fraction of β-cell electrical excitability in control islets (blue; n = 8 cells) and β-TALK-1-KO islets (red; n = 7 cells). Plateau fraction was calculated as the ratio of time spent in active phase to the period of bursting electrical activity in 11 mM glucose.
(F) Representative 2nd-phase [Ca2+]c oscillations in control and β-TALK-1-KO islets in 11 mM glucose (G) measured using Fura-2, AM.
(G) Average [Ca2+]c oscillation frequency (peaks/min) of control islets (blue; n = 44 islets from 3 mice) and β-TALK-1-KO islets (red; n = 44 islets from 3 mice).
(H) Representative [Ca2+]c recordings in control islet cells and β-TALK-1-KO islet cells in 2 mM G, 0 mM Ca2+, and 125 μM diazoxide stimulated with 50 μM CPA.
(I) Area under the curve (normalized [Ca2+]c) of [Ca2+]ER release response upon 50 μM CPA stimulation in control (blue; n = 242 cells from 3 mice) and β-TALK-1-KO (red; n = 115 cells from 3 mice).
Data are represented as mean ± SEM. Statistical significance was determined using unpaired two-tailed t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
β-TALK-1-KO islets exhibit altered mitochondrial Ca2+ and glucose-dependent changes in Ψm
It is well established that islet ATP oscillates and is out of phase with glucose-stimulated Ca2+ oscillations.26–28 Therefore, we next sought to determine if TALK-1 regulation of [Ca2+]c oscillation frequency alters ATP kinetics. For this, the ATP/ADP ratio was measured using the ratio-metric sensor PercevalHR, and [Ca2+]c was simultaneously monitored using Fura Red AM.29 Indeed, ATP oscillated out of phase with [Ca2+]c in both control and TALK-1-deficient islets (Figures 2A–2C). This suggests that TALK-1 control of β-cell [Ca2+]c oscillation frequency could impact islet metabolic oscillations.
Figure 2. β-TALK-1-KO islets exhibit altered mitochondrial Ca2+ and glucose-dependent changes in ψm.

(A and B) Representative ATP/ADP (gray) and [Ca2+]c (black) oscillations measured using PercevalHR and Fura Red AM (top), respectively, in control islets (left) and β-TALK-1-KO islets (right) in 9 mM G. Representative heatmaps (bottom) showing out-of-phase ATP/ADP and Ca2+ oscillations in control islets (left) and β-TALK-1-KO islets (right).
(C) Average correlation between ATP/ADP and Ca2+ oscillations in control islets (blue; n = 31 islets from 3 mice) and β-TALK-1-KO islets (red; 26 islets from 3 mice) in 9 mM G.
(D) Representative [Ca2+]m responses in β-cells expressing the single-wavelength [Ca2+]m sensor Cepia2mt in control islets (blue) and β-TALK-1-KO islets (red).
(E–G) Normalized [Ca2+]m in 1 mM G (E), normalized [Ca2+]m in 11 mM G (F), and the transient percentage of increase in [Ca2+]m in response to 11 mM G (G) in control islets (blue; n = 4 mice) and β-TALK-1-KO islets (red; n = 4 mice). Normalization was performed on islets isolated and imaged on identical days to prevent differences in expression of Cepia2mt impacting the mitochondrial Ca2+ signal.
(H) Representative [Ca2+]m recordings in β-cells expressing FRET-based [Ca2+]m indicator 4mtD3mCerulean3–16 in control islets (blue) and β-TALK-1-KO islets (red).
(I) Average basal [Ca2+]m in control islets (blue; n = 38 islets) and β-TALK-1-KO islets (red; n = 44 islets) in 1 mM G.
(J) Representative islet rhodamine 123 (Rh 123) fluorescence as a measure of ψm in control islets (blue) and β-TALK-1-KO islets (red) recorded in 3 and 17 mM G.
(K) Glucose-induced drop in Rh 123 fluorescence (minimum Rh 123 intensity) in 17 mM G in control and β-TALK-1-KO islets (n = 4 mice/genotype).
(L) Fold change in Rh 123 fluorescence in response to 2 μM FCCP stimulation compared to 17 mM G.
(M) Representative single-cell PercevalHR fluorescence in islets from control (blue) and β-TALK-1-KO mice (red).
(N) Area under the curve of glucose-induced ATP/ADP of data shown in (M) (n = 76–101 islets from 3 mice/genotype).
Data are represented as mean ± SEM. Statistical significance was determined using unpaired two-tailed t test; *p < 0.05, **p < 0.01, and ***p < 0.001.
Importantly, both β-cell [Ca2+]c and [Ca2+]ER control [Ca2+]m levels, which regulate metabolism and downstream signaling required for physiological GSIS. Thus, we next examined if TALK-1 modulation of β-cell Ca2+ handling impacts [Ca2+]m. This was accomplished utilizing a genetically encoded [Ca2+]m indicator, Cepia2mt,30 expressed specifically in β-cells from a rat-insulin promoter.31 Interestingly, β-TALK-1-KO islets had increased [Ca2+]m compared to controls in both low (1 mM) and high (11 mM) glucose (1 mM G: 1.205 ± 0.083 fold change; p = 0.049; 11 mM G: 1.172 ± 0.0650 fold change; p = 0.038; Figures 2D–2F, S2E, and S2F). Initially, glucose stimulation caused a transient increase in β-cell [Ca2+]m, which is likely due to [Ca2+]m influx through the MCU following depolarization-induced [Ca2+]c entry.32 Interestingly, although β-TALK-1-KO islets have increased basal [Ca2+]m that would be predicted to reduce the driving force of Ca2+ into mitochondria, the transient glucose-stimulated increase in β-cell [Ca2+]m was equivalent between β-TALK-1-KO and control islets (percentage of change, control: 22.048% ± 5.036% vs. β-TALK-1-KO: 15.526% ± 2.280%; p = 0.28; Figure 2G). Following an initial glucose-induced [Ca2+]m spike, β-TALK-1-KO and control islets showed prolonged reduction of β-cell [Ca2+]m below baseline; this is presumably due to the activation of the mitochondrial Na+/Ca2+ exchanger (NCLX) that is responsible for Na+ uptake and [Ca2+]m release, which occurs following Vm depolarization-induced cytoplasmic Na+ entry.33 These observations were further confirmed using a β-cell-specific [Ca2+]m fluorescence resonance energy transfer (FRET)-based indicator, 4mtD3mCerulaean3–16,34 that showed an elevation in β-TALK-1-KO [Ca2+]m similar to that observed with Cepia2mt (normalized [Ca2+]m, control: 1.267 ± 0.009 vs. β-TALK-1-KO: 1.328 ± 0.0126; p = 0.0001; Figures 2H and 2I). Furthermore, β-TALK-1-KO islets exhibited increased basal and glucose-stimulated [Ca2+]m without changes in [Ca2+]m uptake and release. Thus, TALK-1 limits the β-cell [Ca2+]m levels under stimulatory as well as non-stimulatory glucose conditions. This suggests that either TALK-1-mediated reduction of [Ca2+]ER sets [Ca2+]m or that TALK-1 directly controls [Ca2+]m.
If TALK-1 were to directly impact [Ca2+]m, it would require mitochondrial localization. Interestingly, TALK-1 protein was detected in isolated mitochondria from HEK293T cells overexpressing TALK-1 channels (Figure S3A). Furthermore, in β-cells, TALK-1 partially colocalizes with the mitochondrial-localized OXPHOS protein complexes (Figures S3B–S3D, Video S1). This suggests that TALK-1 might not only control [Ca2+]m through regulation of β-cell Ca2+ handling but also potentially through modulation of Ψm. Therefore, we next assessed if TALK-1 impacts β-cell ψm utilizing the potentiometric dye rhodamine 123 (Rh 123). TALK-1 deletion resulted in increased β-cell ψm hyperpolarization following glucose stimulation compared to controls (Figures 2J and 2K). Moreover, addition of the mitochondrial uncoupler carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP; 2 μM) led to greater ψm depolarization in β-TALK-1-KO islets, indicating increased respiratory capacity following β-cell TALK-1 deletion (Figure 2L). This is further supported by enhanced glucose-stimulated ATP/ADP levels in β-TALK-1-KO islets as measured by PercevalHR (Figures 2M and 2N). Taken together, these data indicate that TALK-1 serves as a novel regulator of β-cell mitochondrial function. Future studies are required to determine the exact mechanism of TALK-1 modulation of β-cell ψm and [Ca2+]m.
β-cell TALK-1 deficiency increases mitochondrial ATP production
Glucose-stimulated [Ca2+]m influx activates mitochondrial dehydrogenases and aspartate/glutamate shuttle (AGC1), which stimulate mitochondrial respiration and ATP synthesis.20–22,32,35–37 Therefore, we investigated how β-cell TALK-1 controls mitochondrial respiration by monitoring the oxygen consumption rate (OCR; Figures 3A and 3B). On a control chow diet, β-TALK-1-KO islets showed increased ATP production compared to control islets (pMol/min, control: 23.658 ± 1.498 vs. β-TALK-1-KO: 29.374 ± 1.431; p = 0.013; Figure 3C). However, there was no difference in basal respiration, spare respiratory capacity, proton leak, or coupling efficiency between β-TALK-1-KO and control islets (Figures 3D–3H). Exposure to an HFD led to equivalent increases in spare respiratory capacity and to proton leak in islets from both control and β-TALK-1-KO mice. However, only islets from HFD-fed control mice showed elevated levels of ATP production and glucose-stimulated OCR; these levels are comparable to ATP and glucose-stimulated OCR levels in islets from chow-fed β-TALK-1-KO mice. This suggests that TALK-1 activity may impart an adaptive mechanism to increase specific components of β-cell mitochondrial function in response to metabolic stress (e.g., ATP production and glucose-stimulated OCR).
Figure 3. β-cell TALK-1 deficiency increases mitochondrial ATP production.

(A and B) Average OCR profiles measured in control islets (A) and β-TALK-1-KO islets (B) from mice fed either a chow diet or an HFD using the Agilent Seahorse XF Cell Mito Stress Test Kit. Islets were consecutively treated with 20 mM G, 4.5 μM oligomycin, 1 μM FCCP, and 2.5 μM rotenone/antimycin A (Rot/AA).
(C) ATP production rate was calculated by subtracting the minimal rate after oligomycin injection from the last rate measurement before oligomycin injection.
(D) Glucose-stimulated OCR response was calculated by subtracting basal respiration rate from maximal OCR in response to 20 mM G.
(E) Basal respiration rate was calculated by subtracting non-mitochondrial oxygen consumption from last rate measurement before first injection.
(F) Spare respiratory capacity is measured as the ratio of basal respiration to maximal respiration after FCCP injection (*100).
(G) Proton leak was calculated by subtracting non-mitochondrial oxygen consumption from minimum OCR after oligomycin addition.
(H) Coupling efficiency is the ratio of ATP-production coupled respiration to basal respiration (*100).
Data are represented as mean ± SEM. n = 11 wells from 3 control mice (chow diet), n = 16 wells from 3 β-TALK1-KO mice (chow diet), n = 35 wells from 4 control mice (HFD), and n = 43 wells from 7 β-TALK1-KO mice (HFD). Statistical significance was determined by one-way ANOVA, *p < 0.05 and **p < 0.01.
TALK-1-mediated alterations of β-cell function in response to HFD promote glucose intolerance
Because TALK-1 alters β-cell Ca2+ handling and mitochondrial function, we next examined glucose tolerance in β-TALK-1-KO animals. On a chow diet, female β-TALK-1-KO mice showed a modest improvement in glucose tolerance compared to controls (Figures 4A and 4B). Following exposure to HFD, β-TALK-1-KO male and female mice had significantly enhanced glucose tolerance compared to controls (males intraperitoneal glucose tolerance test [i.p. GTT] area under the curve [AUC], control: 19,722.5 ± 784.285 vs. β-TALK-1-KO: 12,635 ± 2,116.806; p = 0.011; females i.p. GTT AUC, control: 9,071.25 ± 486.511 vs. β-TALK-1-KO: 6,837 ± 663.226; p = 0.036; Figures 4C and 4D). However, HFD-fed β-TALK-1-KO mice did not have improved fasting glycemia as observed previously in HFD-fed global TALK-1-KO mice,16 which may indicate that the previously identified role for TALK-1 in glucagon secretion may set fasting glucose levels. This indicates that TALK-1 in β-cells controls glucose homeostasis under conditions of elevated blood glucose. Furthermore, the data suggest that TALK-1 regulation of β-cell Ca2+ handling and mitochondrial function reduces whole-body glucose clearance, especially under conditions of increased insulin demand.
Figure 4. Loss of β-cell TALK-1 improves glucose tolerance in response to HFD despite reduced insulin secretion.

(A–D) Glucose excursion profiles of control mice (blue) and β-TALK-1-KO mice (red) during intraperitoneal glucose tolerance test (i.p. GTT) (2 mg dextrose/g body weight) performed pre- and post-HFD feeding. Both male and female β-TALK-1-KO mice showed a trend toward an increase in glucose tolerance on a chow diet (A, males; B, females) and a significantly improved glucose tolerance post-HFD feeding (C, males: 2 week HFD; D, females: 5 week HFD).
(E) Static glucose-stimulated insulin secretion from control islets (blue) and β-TALK-1-KO islets (red) from mice fed a chow diet at the specified glucose concentrations.
(F) Dynamic insulin secretion (glucose stimulated over basal) measured at the indicated time points and glucose concentrations from control islets (blue) and β-TALK-1-KO islets (red) from mice fed a chow diet.
(G) Area under the curve of 2nd-phase glucose-stimulated insulin secretion (30–75 min).
(H) Static glucose-stimulated insulin secretion from control islets (blue) and β-TALK-1-KO islets (red) from mice fed an HFD at the specified glucose concentrations.
(I) Plasma insulin levels at 0, 10, and 20 min in HFD-fed control mice (blue) and β-TALK-1-KO mice (red) in response to an i.p. glucose injection (2 mg dextrose/g body weight).
(J) Representative 2nd-phase [Ca2+]c oscillations in control and β-TALK-1-KO islets in 11 mM G measured using Fura-2, AM in mice fed a 10 week HFD.
(K) Average [Ca2+]c oscillation frequency (peaks/min); n = islets from 3 mice/genotype.
Data are represented as mean ± SEM. (A and C) Males: n = 5 β-TALK-1-KO mice; n = 4 control mice. (B and D) Females: n = 4–5 β-TALK-1-KO mice; n = 4–5 control mice. (E) n = 4 β-TALK-1-KO mice; n = 4 control mice. (F) n = 3 β-TALK-1-KO mice; n = 3 control mice. (H and I) n = 5 β-TALK-1-KO mice; n = 5 control mice.
*p < 0.05, **p < 0.01, and ***p < 0.001.
We next assessed if improved glucose tolerance in β-TALK-1-KO mice was due to enhanced GSIS. β-TALK-1-KO islets exhibit enhanced GSIS on chow diet (pMol/L/islet: 9 mM G, control: 38.119 ± 10.316 vs. β-TALK-1-KO: 74.535 ± 4.773; p = 0.033; 11 mM G, control: 88.742 ± 10.201 vs. β-TALK-1-KO: 161.709 ± 15.673; p = 0.018; Figures 4E and S6A–S6D) compared to controls, which likely improves glucose tolerance. Furthermore, dynamic analyses of GSIS indicated a higher 2nd phase of insulin secretion in β-TALK-1-KO islets compared to controls (Figures 4F and 4G), which is consistent with the observed increase in Ca2+ oscillation frequency upon TALK-1 deletion. Interestingly, following exposure to a 10 week HFD, β-TALK-1-KO islets showed reduced insulin secretion compared to controls (pMol/L/islet: 11 mM G, control: 402.375 ± 4.889 vs. β-TALK-1-KO: 184.585 ± 4.936; p ≤ 0.0001; Figure 4H). Consistent with a lack of enhanced β-TALK-1-KO islet GSIS following HFD feeding, glucose-stimulated ψm hyperpolarization and ATP/ADP levels as well as insulin secretion when Ca2+ was clamped were similar to islets from chow fed β-TALK-1-KO mice (Figure S4). Moreover, HFD-fed β-TALK-1-KO mice and global TALK-1-KO mice showed reduced fasting as well as glucose-stimulated serum insulin compared to controls (Figures 4I and S5A). However, β-cell ablation of TALK-1 did not impact islet glucagon secretion in these HFD-fed mice (Figure S5B). Surprisingly, HFD-fed β-TALK-1-KO and control mice had equivalent insulin tolerance (Figure S6E). However, loss of β-cell TALK-1 protected mice from increased fat mass and decreased lean mass following exposure to HFD feeding without impacting weight gain (Figures S6F and S6G). Finally, TALK-1-mediated alterations in insulin secretion and glucose homeostasis did not alter β-cell proliferation and mass or expression of markers of oxidative stress (8-OhdG) and dedifferentiation (ALDH1A3) following HFD exposure (Figures S7 and S8).38,39 Taken together, this suggests that TALK-1-mediated amplification of basal and GSIS under metabolic stress may negatively impact glucose tolerance but independently of an overt reduction in insulin sensitivity.
DISCUSSION
In the present study, we identified that β-cell ablation of TALK-1 increased [Ca2+]m and ATP-coupled respiration. [Ca2+]m is set by [Ca2+]c and [Ca2+]ER; thus, TALK-1-mediated reductions in [Ca2+]c and [Ca2+]ER storage likely limit [Ca2+]m levels. As [Ca2+]m results in ψm depolarization, TALK-1-mediated constraint of β-cell [Ca2+]m levels may initially enhance the rate of ψm depolarization during [Ca2+]m entry. Furthermore, although the exact role of mitochondrial-localized TALK-1 channels remains to be determined, these channels may decrease [Ca2+]m by ψm hyperpolarization similar to TASK-3 K2P channels.25 Because [Ca2+]m serves critical roles in mitochondrial respiration such as activating Ca2+-dependent dehydrogenases and AGC1,20 changes in [Ca2+]m impact energy production. For example, β-cells with reduced [Ca2+]m show decreased ATP production such as β-cells without MCU-mediated [Ca2+]m uptake or β-cells with greater NCLX-mediated [Ca2+]m release.32,33 Additionally, β-cells with elevated [Ca2+]m show higher ATP production such as with NCLX knockdown.33 Similarly, we found that β-cells with TALK-1 had both lower [Ca2+]m and ATP production compared to β-cells without TALK-1. Interestingly, [Ca2+]m was diminished in TALK-1-expressing β-cells under non-stimulatory glucose conditions where [Ca2+]c is low, suggesting that TALK-1 control of [Ca2+]ER or its direct modulation of cm limits [Ca2+]m. However, we observe similar glucose-mediated changes in [Ca2+]m in the presence and absence of TALK-1, potentially due to an equivalent 1st-phase glucose-stimulated [Ca2+]c influx. Indeed, β-cell [Ca2+]m increases in response to elevated [Ca2+]c and exhibits glucose-stimulated oscillations that follow [Ca2+]c.40 Thus, [Ca2+]m is likely not only impacted by TALK-1 control of β-cell [Ca2+]ER but also may respond to the TALK-1-mediated reduction of [Ca2+]c oscillation frequency. This is supported by our data showing β-cell ATP/ADP oscillations were out of phase with [Ca2+]c. Together, these data established that islet-restricted TALK-1 channels impact β-cell [Ca2+]m levels and thus contribute to metabolism. Future studies are required to understand the exact mechanism of how TALK-1 activity impacts [Ca2+]m handling and respiration.
Alterations in TALK-1 activity would also be predicted to change the setpoint of β-cell [Ca2+]m. For example, GOF TALK-1 channels have been shown to reduce [Ca2+]ER and [Ca2+]c levels, and this would be predicted to diminish β-cell [Ca2+]m levels. Moreover, if TALK-1 GOF channels localize to mitochondria, they would be predicted to significantly hyperpolarize ψm. Thus, our data suggest that GOF polymorphisms and mutations in TALK-1 associated with T2DM and MODY, respectively, may cause β-cell dysfunction in part through reduced [Ca2+]m and respiration. Reactive oxygen species (e.g., singlet oxygen) also increase TALK-1 channel activity,41 and thus prolonged β-cell reactive oxygen species (ROS) levels observed during the pathogenesis of diabetes42 could limit [Ca2+]m and respiration through TALK-1 activation.20,43,44 As metabolic stress initially increases ATP production, it would be predicted to elevate β-cell [Ca2+]m. This may be due to alterations in β-cell [Ca2+]c entry and [Ca2+]ER release that occur in response to diabetic conditions to meet the increasing insulin demands,9,45–48 which presumably increases the chemical driving force for β-cell [Ca2+]m uptake. As β-cells without TALK-1 channels have high [Ca2+]m concentrations, this may limit further [Ca2+]m uptake under diabetogenic conditions. Furthermore, mitophagy is increased by elevated [Ca2+]m and also protects β-cells from inflammatory stress49–51; this suggests that TALK-1 may limit mitophagy by reducing [Ca2+]m. Future studies are required to characterize the precise role of TALK-1 in altering [Ca2+]m during diabetes pathogenesis and evaluate whether selective TALK-1 inhibitors could be used to mitigate mitochondrial dysfunction in diabetes.
Our results reveal that TALK-1 is required for metabolic-stress-induced increases in β-cell mitochondrial ATP production and insulin secretion. Many studies have suggested that HFD-induced increases in ATP synthesis and GSIS are required to maintain euglycemia in the presence of increasing insulin resistance.44,52,53 Moreover, perturbations in mitochondrial function can lead to overt diabetes due to impaired oxidative metabolism, ATP production, Ca2+ handling, and GSIS.54 However, our studies in β-TALK-1-KO mice show that loss of TALK-1-mediated enhancement in ATP production and GSIS under metabolic stress protects against glucose intolerance. This suggests that the changes in β-cell mitochondrial function and resulting elevation in insulin secretion in response to diabetic conditions negatively impact peripheral insulin demand. The results also suggest that therapeutically inhibiting islet TALK-1 channels could improve β-cell mitochondrial function, reduce hyperglycemia, and decrease insulin demand in patients with diabetes.
Limitations of the study
(1) This study does not establish how TALK-1 influences [Ca2+]m. Future investigations are required to determine if TALK-1 modulation of [Ca2+]c, [Ca2+]ER, and/or mitochondrial-localized TALK-1 impacts β-cell [Ca2+]m, ψm, and ATP production. (2) This study does not determine how β-cell TALK-1 decreased glucose tolerance on an HFD. Future glucose clamp studies are needed to elucidate how TALK-1 control of diet-induced alterations in GSIS and pulsatile insulin secretion kinetics impact peripheral insulin sensitivity, insulin demand, and glucose uptake. (3) This study does not establish if or how the TALK-1-mediated changes in metabolism contribute to β-cell insulin secretion.
STAR+METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, David Jacobson (david.a.jacobson@vanderbilt.edu).
Materials availability
Plasmids and viruses generated in this study will be provided upon request.
Data and code availability
All data required to evaluate the conclusions in the manuscript are present in the paper and/or the supplemental information.
This paper does not report original code.
All additional information needed to reanalyze the data detailed in this manuscript is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
All mice used for the studies were 12- to 18-week-old, age- and gender-matched, bred in-house on a C57BL/6J background. Animals were handled in compliance with guidelines approved by the Vanderbilt University Animal Care and Use Committee protocols (#M1600063–01). β-cell specific deletion of TALK-1 was achieved by crossing mice harboring a floxed Kcnk16 gene (loxP-flanked exons 2 and 3; targeted ES cells-Kcnk16tm1a(EUCOMM)Hmgu were used to generate the floxed Kcnk16 mouse model at the Vanderbilt University Genome Editing Resource) with mice expressing Cre recombinase from the start codon of Insulin1 (Ins1) gene (Ins1-Cre mice; Stock No: 026801; The Jackson Laboratory).55 Floxed Kcnk16 mice were used as controls for all studies.
METHOD DETAILS
Immunofluorescence
Mouse pancreata were fixed in 4% paraformaldehyde and embedded with paraffin. Rehydrated 5-μm sections were stained with primary antibodies against insulin (dilution 1:500; Dako, Santa Clara, CA, USA), somatostatin (dilution 1:250; sc-55565, Santa Cruz Biotechnology Inc., Dallas, TX, USA), and TALK-1 (dilution 1:175; APC-170, Alomone Labs, Jerusalem, Israel; or dilution 1:175; Prestige Antibodies Novus Biologicals #NBP1–83071) followed by secondary antibodies (dilution 1:500; anti-guinea pig Alexa Fluor 647, dilution 1:500; anti-mouse Alexa Fluor 488, and dilution 1:500; anti-rabbit Alexa Fluor 546). β-cell proliferation in response to HFD feeding was quantified by immunostaining for Ki-67 (dilution 1:300; ab15580, Abcam) and utilizing CytoNuclear FL v1.2 algorithm macros generated in HALO software (Indica Labs) to count insulin and Ki-67 double-positive cells. Dedifferentiation and oxidative stress in response to HFD exposure was monitored by immunostaining for the corresponding markers ALDH1A3 and 8OHdG utilizing rabbit ALDH1A3 (Novus Biologicals, NBP2–15339 at 1:100 dilution) and mouse 8OHdG (Abcam, ab62623 at 1:100 dilution) antibodies. For mitochondrial colocalization study, rehydrated 5-μm sections underwent antigen retrieval in a Citrate-based antigen unmasking buffer (H-3300, Vector Laboratories, Newark, CA, USA) and were stained with primary antibodies against TALK-1 (dilution 1:175; Prestige Antibodies Powered by Atlas Antibodies, Stockholm, Sweden) and total OXPHOS protein complexes (Abcam, Cambridge, United Kingdom). Nuclei were stained using ProLong Gold Antifade Mountant (Catalog no. P10144, Invitrogen, Waltham, MA, USA) supplemented with Hoechst 33342. Sections were imaged either with a Nikon Eclipse TE2000-U microscope, a Zeiss LSM 780 confocal microscope, Zeiss LSM 880 confocal microscope with Airyscan platform, or fluorescent ScanScope (Aperio).
Islet and β-cell isolation
Islets were isolated from mouse pancreata by collagenase P (Roche, Basel, Switzerland) digestion and density gradient centrifugation.56 The common bile duct was clamped at the junction with intestine, the duct was cannulated with a 30 G x 1/2 needle (BD PrecisionGlide, Needle, 305106) and the pancreas was distended with 2 mL of islet buffer (1× HBSS (from ThermoFisher 14185), 10 mM HEPES, 1mM MgCl2, 1.25 mM CaCl2, 5 mM glucose, pH 7.4) containing 0.4 mg/mL collagenase P (Roche 11249002001) and 0.5 mg BSA (Sigma-A8806). After excision, inflated pancreas was placed into a 15 mL conical vial of HBSS/BSA/collagenase solution, shaken using a wrist action shaker in a water bath (Burrel, Model 75) at 37°C for 10 min, followed by 5–10× shakes by hand, transfer to 50 mL conical, halt collagenase P reaction with 25 mL cold islet buffer, centrifuge for 1 min at 1800 RPM, resuspended the pellet in 40 mL fresh islet buffer, and wash two more times with cold islet buffer. The islets were either hand-picked or further separated with a Histopaque (Sigma 10771) gradient. Following isolation, islets were either dispersed into cell clusters or single cells with trituration in 0.05% trypsin (Invitrogen 25300) or maintained as whole islets. Cells were cultured in RPMI1640(Invitrogen 11875) with 15% FBS(Invitrogen 26140), 100IU/mL penicillin, and 100 mg/mL streptomycin (Invitrogen 15140) in a humidified incubator at 37°C with an atmosphere of 95% air and 5% CO2.
TALK-1 whole-cell currents
A whole-cell patch-clamp technique was utilized to record TALK-1 currents in single β-cells using an Axopatch 200B amplifier with pCLAMP10 software. A Digidata 1440 was used to digitize currents that were low-pass-filtered at 1 kHz and sampled at 10 kHz. Cells were washed with the extracellular buffer [a modified Krebs-Ringer-HEPES buffer (KRHB)] containing (mM) 119.0 NaCl, 2.0 CaCl2, 4.7 KCl, 25.0 HEPES, 1.2 MgSO4, 1.2 KH2PO4, and 11 mM glucose (pH 7.4 with NaOH). For specific examination of K2P channels, KATP channels were blocked with tolbutamide (100 μM), voltage-gated K+ channels were blocked with tetraethylammonium (TEA; 10 mM), and Ca2+ was removed from the extracellular buffer to prevent activation of Ca2+-activated K+ channels.10,57 Patch electrodes (3–5 MU) were backfilled with intracellular solution (IC) containing (mM) 140.0 KCl, 1.0 MgCl2, 10.0 EGTA, 10.0 HEPES, and 4.0 Mg-ATP (pH 7.25 with KOH). β-cell Vm was ramped from −120 mV to +60 mV from a holding potential of −80 mV to generate K2P currents. The whole-cell currents were analyzed using ClampFit (Molecular Devices) and Excel (Microsoft Corp., Redmond, WA, USA).
β-cell Vm recordings
A perforated patch-clamp technique was employed to record β-cell Vm using an Axopatch 200B amplifier with pCLAMP10 software. Whole islets were washed with KRHB with (mM) 119.0 NaCl, 2.0 CaCl2, 4.7 KCl, 25.0 HEPES, 1.2 MgSO4, 1.2 KH2PO4 (adjusted to pH 7.4 with NaOH) supplemented with 11 mM glucose and incubated in KRHB for 20 min at 37°C, 5% CO2. Patch electrodes (3–5 MΩ) were back-filled with IC containing (mM) 140.0 KCl, 1.0 MgCl2, and 5.0 HEPES (adjusted to pH 7.2 with KOH) supplemented with 20 μg/mL amphotericin B. β-cell Vm recordings were analyzed using ClampFit (Molecular Devices) and Excel (Microsoft Corp., Redmond, WA, USA).
Intracellular Ca2+ imaging
Islets were incubated for 25 min in RPMI supplemented with 2 μM Fura-2, AM (Molecular Probes), followed by incubation in KRHB with 2 mM glucose for 20 min. Fura-2, AM fluorescence (Ratio 340Ex/380Ex-535Em; F340/F380) was measured every 5 s as an indicator of intracellular Ca2+ using a Nikon Eclipse Ti2 microscope equipped with a Photometrics Prime 95B 25mm sCMOS Camera.58 For [Ca2+]c measurements, β-cell glucose-stimulated Ca2+ influx was triggered with KRHB supplemented with 11 mM glucose. For [Ca2+]ER measurements, islets were perfused in KRHB buffer containing 2 mM glucose, 100 μM diazoxide, without extracellular Ca2+. Fura-2, AM fluorescence was monitored as an indicator of [Ca2+]ER release mediated through blockade of the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) with 50 μM CPA (Alomone Labs, Jerusalem, Israel).6 For all measurements, the cells were perifused at a flow rate of 2 mL/min at 37°C. Ex; Excitation wavelength (nm), Em; Emission wavelength (nm).
ATP/ADP measurements
Mouse islets were transduced with the genetically encoded ratio-metric ATP/ADP sensor Perceval-HR (Addgene plasmid #49082; http://n2t.net/addgene:49082; RRID:Addgene_49082) expressed under the control of CMV promoter in an adenoviral format (~3*105 PFU) in RPMI for 4 h at 37°C, 5% CO2. For experiments for simultaneous measurement of [Ca2+]c and ATP/ADP, 2 days post transfection, transduced islets were loaded and incubated with 2.5 μM Fura-Red AM (Catalog no. F3021, Invitrogen, Waltham, MA, USA) in RPMI supplemented with 2 mM glucose for 30 min. Islets were then washed and incubated in KRHB containing 9 mM glucose for 20 min at 37°C, 5% CO2. ATP/ADP and [Ca2+]c oscillations were simultaneously monitored using a Nikon Eclipse Ti2 microscope equipped with a Photometrics Prime 95B 25mm sCMOS Camera. Additionally, ATP/ADP alone was monitored using a similar experimental setup at glucose concentrations indicated in the figure legends. The following excitation (Ex) and emission (Em) filters and ratios were used for the experiments: Fura-Red, 430/24Ex and 500/20Ex, 630/60Em (Ratio F430/F500) and Perceval-HR, 430/24Ex and 500/20Ex, 535/30Em (Ratio F500/F430).
Lentivirus production
To measure [Ca2+]m specifically in islet β-cells, single-wavelength [Ca2+]m sensor Cepia2mt30 and FRET-based [Ca2+]m sensor 4mtD3mCerulean3–1634 were cloned downstream of a rat insulin promoter (RIP) in a lentiviral plasmid.17 These plasmids were then packaged into lentiviruses in HEK293FT cells. Briefly, HEK293FT cells were cultured in DMEM GlutaMax-I with 10% FBS, 100IU/mL penicillin, and 100 mg/mL streptomycin to 50–70% confluency in 100 mm dishes 24 h before transfection. On the day of transfection, HEK293FT cells were incubated in DMEM GlutaMax-I with 10% Heat-Inactivated FBS, with 70 μM chloroquine for 10 min, following which DNA mixtures (diluted in 470 μl of 2X HBS) were added dropwise onto the cells. The DNA mixtures consisted of: 46.7 μl of 2.5M CaCl2, 18.65 μg lentiviral plasmid, 13.9 μg packaging plasmid (pCMV-dR7.74psPAX2; psPAX2 was a gift from Didier Trono (Addgene plasmid #12260; http://n2t.net/addgene:12260; RRID:Addgene_12260), and 5.56 μg envelope plasmid (pMD2.G was a gift from Didier Trono (Addgene plasmid #12259; http://n2t.net/addgene:12259; RRID:Addgene_12259).17 After ~7 h, transfection media was replaced with fresh DMEM GlutaMax-I culture media. Lentivirus-containing media was harvested 72 h later, centrifuged at 300 × g for 5 min, and filtered through 0.45 μm filters to remove cell debris. Filtered lentivirus-containing media was aliquoted and frozen at −80°C before use to transduce mouse islet cells.17
Mitochondrial Ca2+ imaging
To measure β-cell [Ca2+]m, whole islets or partially dispersed islets were transduced with 3rd-generation lentiviruses (~10 multiplicity of infection) expressing Cepia2mt or 4mtD3mCerulean3–16 plasmids for ~6 h and cultured in RPMI with 11 mM glucose for 48 h–72 h. For [Ca2+]m imaging, cells were incubated in 1 mM glucose KRHB for 20 min and then imaged under conditions detailed in the figures. Cepia2mt measurements were performed using a Zeiss LSM780 confocal microscope with a 20X objective every 5 s using 480 Ex and 535 Em filters. 4mtD3mCerulean3–16 measurements were performed using a Nikon Eclipse Ti2 microscope equipped with a Photometrics Prime 95B 25mm sCMOS Camera with a 20X objective; FRET was measured using 410Ex-480Em/535Em filters (Ratio F535/F480).34 For all measurements, the cells were perifused at a flow rate of 2 mL/min.
Mitochondrial membrane potential recordings
To quantify changes in ψm, whole islets were loaded with the potentiometric dye Rhodamine 123 (Rh 123; 10 μM) for 30 min in KRHB supplemented with 1 mM glucose. Following incubation, islets were washed with KRBH and Rh 123 fluorescence was imaged every 15 s under conditions detailed in the figures using a Nikon Eclipse Ti2 microscope equipped with a Photometrics Prime 95B 25mm sCMOS Camera (480Ex/535Em filters).
Seahorse assays
Seahorse experiments were performed using an Agilent Seahorse XFe96 with XFe96 Spheroid Microplates and XFe96 Spheroid Flux Pak (Catalog no. 102905–100).59 Whole islets were isolated from mice that had either been on a standard chow diet or a high-fat diet (HFD; D12492; 60% kcal% fat; Research Diets, Inc.) and incubated over-night in RPMI supplemented with 0.5% BSA. 15 mouse islets were carefully seeded into the bottom of the wells of a CellTak coated XF96 spheroid plate containing 170 μL/well of warm assay medium (Seahorse XF base medium minimal DMEM, supplemented with 2 mM glucose and 0.1% FBS). The plate was then allowed to incubate for 1 h in a 37°C incubator without CO2. The sensor cartridge was prepped according to the protocol (Agilent 102960–000) and the assay was run with the Agilent Seahorse XF Cell Mito Stress Test Kit (Catalog no. 103015–100). After measuring basal OCR, glucose (20 mM), oligomycin (4.5 mM), carbonyl cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP) (1 mM), and rotenone/antimycin A (Rot/AA; 2.5 mM) were sequentially added at indicated time points to modulate mitochondrial OCR.
Glucose homeostasis and body composition
Mice were placed on either a standard chow diet or a high-fat diet (HFD; D12492; 60% kcal% fat; Research Diets, Inc.). Glucose tolerance test (GTT) and insulin tolerance test (ITT) were performed as follows. Briefly, mice were fasted for 5–6 h and then intraperitoneally (i.p.) injected with either 2 mg dextrose/g body weight or 0.75 UI human recombinant insulin/kg body weight (catalog no. 12585014, Gibco). Blood obtained from the tail vein was monitored for glucose using a CONTOUR NEXT blood glucose meter (Bayer Healthcare) were then taken at the indicated time points.16 Body composition in HFD-fed mice was assessed utilizing the LF50 Body Composition Mice Analyzer (Bruker E1400005_01) following manufacturer’s instructions.
Isolation of mitochondria and immunoblotting
293FT cells were grown to ~90% confluence in Dulbecco’s Modified Eagle Media (DMEM) GlutaMax-I (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, Gibco), 100 IU•ml—1 penicillin (Gibco), and 100 mg•ml—1 streptomycin (Gibco) (complete media (CM)) at 37°C, 5% CO2 in 100 mm tissue culture dishes. Cells were transfected for 36 h with 10 μg of pcDNA3.1 expressing TALK-1 T3-FLAG and 10 μg of pcDNA3.1 expressing 3XHA-EGFP-OMP25 using 32.5 μL Lipofectamine 3000 and 40 μL P3000 in a total volume of 500 μL of antibiotic-free Opti-MEM I Reduced Serum Medium (Thermo Fisher Scientific). Mitochondria isolatation60: cells were washed once with 10 mL of 1× phosphate buffered saline (PBS) and gently scrapped into 1 mL of KPBS buffer (mM; 136 KCl and 10 KH2PO4, pH 7.25) on ice and centrifuged at 1,000g for 2 min at 4°C. The supernatant was removed, cells were fully resuspended in 1mL KPBS and transferred to a 2 mL tissue grinder (Catalog no. 357422, WHEATON). Cells were homogenized with 25 strokes of the plunger and centrifuged at 1,000g for 2 min at 4°C. The supernatant was applied to pre-washed anti-HA magnetic beads (Thermo Fisher Scientific) which was followed by a 3.5 min incubation on an end-over-end rotator at 4°C. Beads were then collected on the magnet and the overlying solution was thoroughly aspirated. The beads were gently washed twice with KPBS, resuspended in 100 μL lysis buffer (mM; 50 Tris-HCl, pH 7.4, 150 NaCl, 1 EDTA, 1% (v/v) Triton X-100, and protease inhibitor) and incubated on ice for 10 min after pipetting to homogeneity. Beads were collected on a magnet and the overlying solution was transferred to a new 1.5-mL tube using a gel-loading tip. The solution was centrifuged at 17,000g for 10 min at 4°C and the supernatant (mitochondrial fraction) was transferred to a new 1.5-mL tube and stored at −20°C for immunoblotting applications. For western blotting, the samples were thawed on ice, combined 3:1 with 4× Laemmli buffer (Bio-Rad), and incubated 10 min at 95°C. The protein samples were cooled to RT and 2 μg of each were resolved on a 4–20% Mini-PROTEAN TGX Precast Protein Gel (Bio-Rad) at 100 mV for 2 h. The proteins were transferred to a nitrocellulose membrane for 90 min at 100 mV at 4°C. The membrane was blocked in 3% dry milk powder (Catalog no. M17200, RPI) in 1× TBS with 0.1% Tween 20 (blocking solution) for 1 h at room temperature. The membrane was then incubated overnight with 8 μg/mL mouse anti-FLAG M2 antibody (catalog no. F3165, Sigma Aldrich) in blocking solution. The membrane was washed four times (10 min/wash) in 1× TBS with 0.1% Tween 20 and incubated 45 min with 1:2500 goat anti-mouse IgG (H + L), HRP conjugate secondary (Catalog no. W4021, Promega) in blocking solution with 0.1% Tween 20 at RT. The membrane was washed four times (10 min/wash) in 1× TBS with 0.1% Tween 20 and once (5 min) in 1× PBS. FLAG tagged TALK-1 protein bands were visualized using SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific). After imaging the membrane was washed in PBS for 10 min, incubated in Restore Western blot stripping buffer (Catalog no. 21059, Thermo Fisher Scientific) for 30 min, and washed for 10 min in PBS. It was then re-blocked and re-probed and imaged as explained above, first with 1:500 rabbit anti-VDAC (Catalog no. 4661, Cell Signaling Technology) and 1:2500 goat anti-rabbit IgG (H + L), HRP conjugate secondary (Catalog no. W4011, Promega) then with 1:1000 rabbit anti-calreticulin (Catalog no. 12238, Cell Signaling Technology) and 1:2500 goat anti-rabbit IgG (H + L), HRP conjugate secondary, and lastly with 1:2000 mouse anti-GAPDH (Catalog no. MA5–15738, Thermo Fisher Scientific) and 1:2500 goat anti-mouse IgG (H + L), HRP conjugate secondary. VDAC, calreticulin, and GAPDH protein bands were visualized as explained above.
Insulin and glucagon secretion assays
For serum insulin measurements, mice were fasted for ~6 h and then intraperitoneally injected with 2mg dextrose/g body weight. Tail blood samples were collected in Microvette CB 300 K2 EDTA tubes (Catalog no. 16.444.100, Sarstedt) at the indicated time points. Serum insulin was measured via radioimmunoassay by the Vanderbilt University Hormone Assay and Analytical Services Core (Nashville, TN, USA).
For in vitro static insulin and glucagon secretion assays, islets were isolated from mice fed a standard chow diet or an HFD and were incubated over-night in RPMI supplemented with 0.5 mg/mL BSA. On the day of experiment, islets were equilibrated in DMEM containing 0.5 mg/mL BSA, 0.5 mM CaCl2 and 10.0 mM HEPES (DMEM*) supplemented with 10% FBS and 5.5 mM glucose for 1 h at 37°C, 5% CO2. ~20 islets/well were picked into 400 μL DMEM* without FBS at glucose concentrations specified in the figures in 24-well plate(s) and insulin or glucagon secretion was measured over 1 h at 37°C, 5% CO2. Supernatants were then collected, supplemented with protease inhibitor (Catalog no. 4693132001; Roche, Basel, Switzerland), and stored at −20°C until analysis. Insulin secretion was measured by the Vanderbilt University Hormone Assay and Analytical Services Core (Nashville, TN, USA) via radioimmunoassay. Glucagon secretion was measured using Mouse Glucagon ELISA kit (Catalog no. 10–1281-01, Mercodia Inc).
Dynamic insulin secretion was measured from islets chambers loaded with 75 islets/chamber (BioRep Technologies) and perifused at a flow rate of 1 mL/min in response to glucose stimulations indicated in the figure legends. The effluent was collected at 3-min intervals and insulin concentration in each perifusion fraction was measured by an insulin ELISA assay (Catalog no. 10–1249-01, Mercodia Inc).
QUANTIFICATION AND STATISTICAL ANALYSIS
Functional data were analyzed using Axon Clampfit (Molecular Devices), GraphPad Prism 8 (GraphPad Software Inc.), or Excel (Microsoft Corp., Redmond, WA, USA) and presented as mean ± standard error (SE) for the specified number of samples (N). Statistical significance was determined using two-tailed t-tests or one-way ANOVA as appropriate. p-value %0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Guinea-pig anti-insulin | Dako | Cat#A0564; RRID:AB_10013624 |
| Mouse anti-somatostatin | Santa Cruz Biotechnology | Cat#sc-55565; RRID:AB_831726 |
| Rabbit anti-TALK-1 | Novus Biologicals | Cat#NBP1-83071; RRID:AB_11021713 |
| Rabbit anti-TALK-1 | Alomone Labs | Cat#APC-170; RRID:AB_2756754 |
| Rabbit anti-Ki67 | Abcam | Cat#ab15580; RRID:AB_443209 |
| Rabbit anti-ALDH1A3 | Novus Biologicals | Cat#NBP2-15339; RRID:AB_2665496 |
| Mouse anti-8OHdG | Abcam | Cat#ab62623; RRID:AB_940049 |
| Mouse anti-FLAG M2 | Sigma Aldrich | Cat#F3165; RRID:AB_259529 |
| Rabbit anti-VDAC | Cell Signaling Technology | Cat#4661; RRID:AB_10557420 |
| Rabbit anti-calreticulin | Cell Signaling Technology | Cat#12238; RRID:AB_2688013 |
| Mouse anti-GAPDH | Thermo Fisher Scientific | Cat# MA5-15738; RRID:AB_10977387 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Collagenase P | Roche | Cat#11249002001 |
| Histopaque | Sigma | Cat#10771 |
| HBSS | Invitrogen | Cat#14185 |
| RPMI | Invitrogen | Cat#11875 |
| DMEM | Invitrogen | Cat# 10569044 |
| Bovine Serum Albumin | Sigma | Cat#A8806 |
| Fetal Bovine Serum | Invitrogen | Cat#26140 |
| HEPES | Sigma | Cat#h3375 |
| Magnesium chloride hexahydrate | Fisher | Cat#AC41341-5000 |
| Magnesium sulfate heptahydrate | Sigma | Cat#63140 |
| Potassium chloride | Sigma | Cat#p4504 |
| Potassium phosphate monobasic | Sigma | Cat#60218 |
| Sodium chloride | Sigma | Cat#s7653 |
| Sodium phosphate dibasic dihydrate | Sigma | Cat#7907 |
| Tolbutamide | MP biomedicals | Cat#190274 |
| Triton X-100 non-ionic | Roache | Cat#10789704001 |
| Trypsin, 0.05% (1X) with EDTA 4Na | Invitrogen | Cat#25300 |
| Versene | Invitrogen | Cat#15040 |
| Adenosine 5’-triphosphate magnesium salt | Sigma | Cat#A9187 |
| Amphotericin B solubilized. | Sigma | Cat#a9528 |
| LB Broth | Sigma | Cat#l3022 |
| Fura-2,AM | Molecular Probes | Cat#F1221 |
| Rhodamine 123 | Thermo Fisher Scientific | Cat#R302 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Agilent Seahorse XF Cell Mito Stress Test Kit | Agilent Technologies | Cat#103015-100 |
| Ultrasensitive Mouse Insulin ELISA | Mercodia Inc | Cat#10-1249-01 |
| Mouse Insulin ELISA | Alpco Ltd | 80-INSMS-E01 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| 293FT Cell Line | Thermo Fisher Scientific | Cat#R70007 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Targeted ES cells-Kcnk16tm1a(EUCOMM)Hmgu were used to generate the Kcnk16 fl/fl mouse model at Vanderbilt University | EUCOMM IMPC | https://www.mousephenotype.org/data/alleles/MGI:1921821/tm1a(EUCOMM)Hmgu/ |
| Ins1 Cre mice; B6(Cg)-Ins1tm1.1(cre)Thor/J | (Thorens et al.)55; Obtained from the Jackson Laboratory | Strain #026801 |
| C57Bl/6J mice | The Jackson Laboratory | Strain #:000664 |
|
| ||
| Recombinant DNA | ||
|
| ||
| pMD2.G | a gift from Didier Trono, obtained from Addgene |
Cat #12259 |
| psPAX2 | a gift from Didier Trono, obtained from Addgene | Cat #12260 |
| RIP-Cepia2mt | (Suzuki et al.)30; Obtained from Addgene | Cat #58218 |
| RIP-4mtD3mCerulean3-16 | (Greotti et al.)34 | |
| CMV-PercevalHR | This paper and Addgene | Cat #49082 |
| pcDNA3.1-TALK-1 T3-FLAG | This paper | |
| pcDNA3.1-3XHA-EGFP-OMP25 | This paper and Addgene | Cat#83356 |
|
| ||
| Software and algorithms | ||
|
| ||
| GraphPad Prism v9.0.0 | Graphpad Software | https://www.graphpad.com/ |
| Clampfit 10.7 Software | Molecular Devices | https://www.moleculardevices.com/products/axon-patch-clamp-system |
| Biorender | Biorender | https://www.biorender.com/ |
| Nikon NIS-Elements | Nikon Healthcare | https://www.microscope.healthcare.nikon.com/ |
Highlights.
β-cell TALK-1 channels limit mitochondrial Ca2+accumulation
Ablation of β-cell TALK-1 increased ATP production and insulin secretion
TALK-1 promotes HFD-induced changes in β-cell oxidative phosphorylation and GSIS
β-TALK-1-KO animals are protected from glucose intolerance on a high-fat diet
ACKNOWLEDGMENTS
Processing of paraffin-embedded pancreata was performed by The Vanderbilt Translational Pathology Shared Resource (2P30CA068485-14). Slide scanning was performed using the Islet and Pancreas Analysis Core (DK20593). The FRET-based sensor 4mtD3mCerulean3-16 was generously donated by Dr. Tullio Pozzan, University of Padua, Italy. Total-OXPHOS antibody cocktail was kindly donated by Dr. Maureen Gannon, Vanderbilt University. Confocal microscopy was performed using the Vanderbilt Cell Imaging Shared Resource (DK020593). D.A.J. was supported by NIH grants R01DK097392, R01DK129340, and R01DK115620. S.M.G. was supported by F31 DK118855 and the Vanderbilt Molecular Endocrinology Training Program (METP; 5T32DK07563). N.C.V. was supported by 1F31 DK109625 and the Vanderbilt METP (5T32DK07563). J.R.D. was supported by the Initiative for Maximizing Student Development at Vanderbilt (T32GM139800) and the Vanderbilt METP (5T32DK07563).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2024.113673.
REFERENCES
- 1.Grodsky GM, Batts AA, Bennett LL, Vcella C, McWilliams NB, and Smith DF (1963). Effects of Carbohydrates on Secretion of Insulin from Isolated Rat Pancreas. Am. J. Physiol 205, 638–644. [DOI] [PubMed] [Google Scholar]
- 2.Coore HG, and Randle PJ (1964). Regulation of insulin secretion studied with pieces of rabbit pancreas incubated in vitro. Biochem. J 93, 66–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grodsky GM, and Bennett LL (1966). Cation requirements for insulin secretion in the isolated perfused pancreas. Diabetes 15, 910–913. [DOI] [PubMed] [Google Scholar]
- 4.Milner RD, and Hales CN (1967). The role of calcium and magnesium in insulin secretion from rabbit pancreas studied in vitro. Diabetologia 3, 47–49. [DOI] [PubMed] [Google Scholar]
- 5.Henquin JC (2000). Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49, 1751–1760. [DOI] [PubMed] [Google Scholar]
- 6.Fridlyand LE, Tamarina N, and Philipson LH (2010). Bursting and calcium oscillations in pancreatic beta-cells: specific pacemakers for specific mechanisms. Am. J. Physiol. Endocrinol. Metab 299, E517–E532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beauvois MC, Merezak C, Jonas JC, Ravier MA, Henquin JC, and Gilon P. (2006). Glucose-induced mixed [Ca2+]c oscillations in mouse beta-cells are controlled by the membrane potential and the SERCA3 Ca2+-ATPase of the endoplasmic reticulum. Am. J. Physiol. Cell Physiol 290, C1503–C1511. [DOI] [PubMed] [Google Scholar]
- 8.Bertram R, Sherman A, and Satin LS (2007). Metabolic and electrical oscillations: partners in controlling pulsatile insulin secretion. Am. J. Physiol. Endocrinol. Metab 293, E890–E900. [DOI] [PubMed] [Google Scholar]
- 9.Gilon P, Chae HY, Rutter GA, and Ravier MA (2014). Calcium signaling in pancreatic beta-cells in health and in Type 2 diabetes. Cell Calcium 56, 340–361. [DOI] [PubMed] [Google Scholar]
- 10.Vierra NC, Dadi PK, Milian SC, Dickerson MT, Jordan KL, Gilon P, and Jacobson DA (2017). TALK-1 channels control beta cell endoplasmic reticulum Ca(2+) homeostasis. Sci. Signal 10, eaan2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, Houamed K, Kupershmidt S, Roden D, and Satin LS (2005). Pharmacological properties and functional role of Kslow current in mouse pancreatic beta-cells: SK channels contribute to Kslow tail current and modulate insulin secretion. J. Gen. Physiol 126, 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lang DA, Matthews DR, Burnett M, and Turner RC (1981). Brief, irregular oscillations of basal plasma insulin and glucose concentrations in diabetic man. Diabetes 30, 435–439. [DOI] [PubMed] [Google Scholar]
- 13.Satin LS, Butler PC, Ha J, and Sherman AS (2015). Pulsatile insulin secretion, impaired glucose tolerance and type 2 diabetes. Mol. Aspect. Med 42, 61–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cho YS, Chen CH, Hu C, Long J, Ong RTH, Sim X, Takeuchi F, Wu Y, Go MJ, Yamauchi T, et al. (2011). Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat. Genet 44, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DIAbetes Genetics Replication And Meta-analysis DIAGRAM Consortium; Go MJ, Zhang W, Below JE, Gaulton KJ, et al. ; Asian Genetic Epidemiology Network Type 2 Diabetes AGEN-T2D Consortium; South Asian Type 2 Diabetes SAT2D Consortium; Mexican American Type 2 Diabetes MAT2D Consortium; Type 2 Diabetes Genetic Exploration by Nex-generation sequencing in muylti-Ethnic Samples T2D-GENES Consortium; and Mahajan A. (2014). Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat. Genet 46, 234–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vierra NC, Dadi PK, Jeong I, Dickerson M, Powell DR, and Jacobson DA (2015). Type 2 Diabetes-Associated K+ Channel TALK-1 Modulates beta-Cell Electrical Excitability, Second-Phase Insulin Secretion, and Glucose Homeostasis. Diabetes 64, 3818–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Graff SM, Johnson SR, Leo PJ, Dadi PK, Dickerson MT, Nakhe AY, McInerney-Leo AM, Marshall M, Zaborska KE, Schaub CM, et al. (2021). A KCNK16 mutation causing TALK-1 gain of function is associated with maturity-onset diabetes of the young. JCI Insight 6, e138057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giacomello M, Drago I, Bortolozzi M, Scorzeto M, Gianelle A, Pizzo P, and Pozzan T. (2010). Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of storeoperated Ca2+ channels. Mol. Cell 38, 280–290. [DOI] [PubMed] [Google Scholar]
- 19.Bermont F, Hermant A, Benninga R, Chabert C, Jacot G, Santo-Domingo J, Kraus MRC, Feige JN, and De Marchi U. (2020). Targeting Mitochondrial Calcium Uptake with the Natural Flavonol Kaempferol, to Promote Metabolism/Secretion Coupling in Pancreatic beta-cells. Nutrients 12, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merrins MJ, Corkey BE, Kibbey RG, and Prentki M. (2022). Metabolic cycles and signals for insulin secretion. Cell Metabol. 34, 947–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Denton RM, and McCormack JG (1980). On the role of the calcium transport cycle in heart and other mammalian mitochondria. FEBS Lett. 119, 1–8. [DOI] [PubMed] [Google Scholar]
- 22.Rutter GA (1990). Ca2(+)-binding to citrate cycle dehydrogenases. Int. J. Biochem 22, 1081–1088. [DOI] [PubMed] [Google Scholar]
- 23.McCormack JG, Longo EA, and Corkey BE (1990). Glucose-induced activation of pyruvate dehydrogenase in isolated rat pancreatic islets. Biochem. J 267, 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Szewczyk A, Jarmuszkiewicz W, and Kunz WS (2009). Mitochondrial potassium channels. IUBMB Life 61, 134–143. [DOI] [PubMed] [Google Scholar]
- 25.Yao J, McHedlishvili D, McIntire WE, Guagliardo NA, Erisir A, Coburn CA, Santarelli VP, Bayliss DA, and Barrett PQ (2017). Functional TASK-3-Like Channels in Mitochondria of Aldosterone-Producing Zona Glomerulosa Cells. Hypertension 70, 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li J, Shuai HY, Gylfe E, and Tengholm A. (2013). Oscillations of submembrane ATP in glucose-stimulated beta cells depend on negative feedback from Ca(2+). Diabetologia 56, 1577–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Merrins MJ, Poudel C, McKenna JP, Ha J, Sherman A, Bertram R, and Satin LS (2016). Phase Analysis of Metabolic Oscillations and Membrane Potential in Pancreatic Islet beta-Cells. Biophys. J 110, 691–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McKenna JP, Ha J, Merrins MJ, Satin LS, Sherman A, and Bertram R. (2016). Ca2+ Effects on ATP Production and Consumption Have Regulatory Roles on Oscillatory Islet Activity. Biophys. J 110, 733–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tantama M, Martıńez-Franç ois JR, Mongeon R, and Yellen G. (2013). Imaging energy status in live cells with a fluorescent biosensor of the intracellular ATP-to-ADP ratio. Nat. Commun 4, 2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, and Iino M. (2014). Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun 5, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Bender A, Wang P, Karakose E, Inabnet WB, Libutti SK, Arnold A, Lambertini L, Stang M, Chen H, et al. (2017). Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat. Commun 8, 767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Georgiadou E, Haythorne E, Dickerson MT, Lopez-Noriega L, Pullen TJ, da Silva Xavier G, Davis SPX, Martinez-Sanchez A, Semplici F, Rizzuto R, et al. (2020). The pore-forming subunit MCU of the mitochondrial Ca(2+) uniporter is required for normal glucose-stimulated insulin secretion in vitro and in vivo in mice. Diabetologia 63, 1368–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nita II, Hershfinkel M, Fishman D, Ozeri E, Rutter GA, Sensi SL, Khananshvili D, Lewis EC, and Sekler I. (2012). The mitochondrial Na+/Ca2+ exchanger upregulates glucose dependent Ca2+ signalling linked to insulin secretion. PLoS One 7, e46649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Greotti E, Fortunati I, Pendin D, Ferrante C, Galla L, Zentilin L, Giacca M, Kaludercic N, Di Sante M, Mariotti L, et al. (2019). mCerulean3-Based Cameleon Sensor to Explore Mitochondrial Ca(2+) Dynamics In Vivo. iScience 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutter GA (2001). Nutrient-secretion coupling in the pancreatic islet beta-cell: recent advances. Mol. Aspect. Med 22, 247–284. [DOI] [PubMed] [Google Scholar]
- 36.De Marchi U, Thevenet J, Hermant A, Dioum E, and Wiederkehr A. (2014). Calcium co-regulates oxidative metabolism and ATP synthase-dependent respiration in pancreatic beta cells. J. Biol. Chem 289, 9182–9194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rutter GA, Burnett P, Rizzuto R, Brini M, Murgia M, Pozzan T, Tavaré JM, and Denton RM (1996). Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proc. Natl. Acad. Sci. USAUSA 93, 5489–5494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim-Muller JY, Fan J, Kim YJR, Lee SA, Ishida E, Blaner WS, and Accili D. (2016). Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic beta cells in diabetic mice. Nat. Commun 7, 12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Richter C, Park JW, and Ames BN (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USAUSA 85, 6465–6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tarasov AI, Semplici F, Li D, Rizzuto R, Ravier MA, Gilon P, and Rutter GA (2013). Frequency-dependent mitochondrial Ca(2+) accumulation regulates ATP synthesis in pancreatic beta cells. Pflugers Arch. 465, 543–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Duprat F, Girard C, Jarretou G, and Lazdunski M. (2005). Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J. Physiol 562, 235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robertson RP (2004). Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J. Biol. Chem 279, 42351–42354. [DOI] [PubMed] [Google Scholar]
- 43.Chareyron I, Christen S, Moco S, Valsesia A, Lassueur S, Dayon L, Wollheim CB, Santo Domingo J, and Wiederkehr A. (2020). Augmented mitochondrial energy metabolism is an early response to chronic glucose stress in human pancreatic beta cells. Diabetologia 63, 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roat R, Rao V, Doliba NM, Matschinsky FM, Tobias JW, Garcia E, Ahima RS, and Imai Y. (2014). Alterations of pancreatic islet structure, metabolism and gene expression in diet-induced obese C57BL/6J mice. PLoS One 9, e86815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gonzalez A, Merino B, Marroquı L, Ñ eco P, Alonso-Magdalena P, Caballero-Garrido E, Vieira E, Soriano S, Gomis R, Nadal A, and Quesada I. (2013). Insulin hypersecretion in islets from diet-induced hyperinsulinemic obese female mice is associated with several functional adaptations in individual beta-cells. Endocrinology 154, 3515–3524. [DOI] [PubMed] [Google Scholar]
- 46.Rose T, Efendic S, and Rupnik M. (2007). Ca2+-secretion coupling is impaired in diabetic Goto Kakizaki rats. J. Gen. Physiol 129, 493–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ravier MA, Sehlin J, and Henquin JC (2002). Disorganization of cytoplasmic Ca(2+) oscillations and pulsatile insulin secretion in islets from ob/obmice. Diabetologia 45, 1154–1163. [DOI] [PubMed] [Google Scholar]
- 48.Roe MW, Philipson LH, Frangakis CJ, Kuznetsov A, Mertz RJ, Lancaster ME, Spencer B, Worley JF 3rd, and Dukes ID (1994). Defective glucose-dependent endoplasmic reticulum Ca2+ sequestration in diabetic mouse islets of Langerhans. J. Biol. Chem 269, 18279–18282. [PubMed] [Google Scholar]
- 49.Fex M, Nicholas LM, Vishnu N, Medina A, Sharoyko VV, Nicholls DG, Spégel P, and Mulder H. (2018). The pathogenetic role of beta-cell mitochondria in type 2 diabetes. J. Endocrinol 236, R145–R159. [DOI] [PubMed] [Google Scholar]
- 50.Twig G, Elorza A, Molina AJA, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, et al. (2008). Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 27, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidarala V, Pearson GL, Parekh VS, Thompson B, Christen L, Gingerich MA, Zhu J, Stromer T, Ren J, Reck EC, et al. (2020). Mitophagy protects beta cells from inflammatory damage in diabetes. JCI Insight 5, e141138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kanno A, Asahara SI, Masuda K, Matsuda T, Kimura-Koyanagi M, Seino S, Ogawa W, and Kido Y. (2015). Compensatory hyperinsulinemia in high-fat diet-induced obese mice is associated with enhanced insulin translation in islets. Biochem. Biophys. Res. Commun 458, 681–686. [DOI] [PubMed] [Google Scholar]
- 53.Ye J. (2021). Mechanism of insulin resistance in obesity: a role of ATP. Front. Med 15, 372–382. [DOI] [PubMed] [Google Scholar]
- 54.Adam J, Ramracheya R, Chibalina MV, Ternette N, Hamilton A, Tarasov AI, Zhang Q, Rebelato E, Rorsman NJG, Martín-Del-Río R, et al. (2017). Fumarate Hydratase Deletion in Pancreatic beta Cells Leads to Progressive Diabetes. Cell Rep. 20, 3135–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Thorens B, Tarussio D, Maestro MA, Rovira M, Heikkilä E, and Ferrer J. (2015). Ins1(Cre) knock-in mice for beta cell-specific gene recombination. Diabetologia 58, 558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dickerson MT, Dadi PK, Butterworth RB, Nakhe AY, Graff SM, Zaborska KE, Schaub CM, and Jacobson DA (2020). Tetraspanin-7 regulation of L-type voltage-dependent calcium channels controls pancreatic beta-cell insulin secretion. J. Physiol 598, 4887–4905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vierra NC, Dickerson MT, Jordan KL, Dadi PK, Katdare KA, Altman MK, Milian SC, and Jacobson DA (2018). TALK-1 reduces delta-cell endoplasmic reticulum and cytoplasmic calcium levels limiting somatostatin secretion. Mol. Metabol 9, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zaborska KE, Dadi PK, Dickerson MT, Nakhe AY, Thorson AS, Schaub CM, Graff SM, Stanley JE, Kondapavuluru RS, Denton JS, and Jacobson DA (2020). Lactate activation of alpha-cell KATP channels inhibits glucagon secretion by hyperpolarizing the membrane potential and reducing Ca(2+) entry. Mol. Metabol 42, 101056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Taddeo EP, Stiles L, Sereda S, Ritou E, Wolf DM, Abdullah M, Swanson Z, Wilhelm J, Bellin M, McDonald P, et al. (2018). Individual islet respirometry reveals functional diversity within the islet population of mice and human donors. Mol. Metabol 16, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen WW, Freinkman E, and Sabatini DM (2017). Rapid immunopurification of mitochondria for metabolite profiling and absolute quantification of matrix metabolites. Nat. Protoc 12, 2215–2231. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data required to evaluate the conclusions in the manuscript are present in the paper and/or the supplemental information.
This paper does not report original code.
All additional information needed to reanalyze the data detailed in this manuscript is available from the lead contact upon request.